

Abstract
Three gene families in plants viz. Argonaute (AGOs), Dicer-like (DCLs) and RNA dependent RNA polymerase (RDRs) constitute the core components of small RNA mediated gene silencing machinery. The present study endeavours to identify members of these gene families in tea and to investigate their expression patterns in different tissues and various stress regimes. Using genome-wide analysis, we have identified 18 AGOs, 5 DCLs and 9 RDRs in tea, and analyzed their phylogenetic relationship with orthologs of Arabidopsis thaliana. Gene expression analysis revealed constitutive expression of CsAGO1 in all the studied tissues and stress conditions, whereas CsAGO10c showed most variable expression among all the genes. CsAGO10c gene was found to be upregulated in tissues undergoing high meristematic activity such as buds and roots, as well as in Exobasidium vexans infected samples. CsRDR2 and two paralogs of CsAGO4, which are known to participate in biogenesis of hc-siRNAs, showed similarities in their expression levels in most of the tea plant tissues. This report provides first ever insight into the important gene families involved in biogenesis of small RNAs in tea. The comprehensive knowledge of these small RNA biogenesis purveyors can be utilized for tea crop improvement aimed at stress tolerance and quality enhancement.
Subject terms: Abiotic, Biotic, Drought, Computational biology and bioinformatics
Introduction
Gene regulation in eukaryotes depends on post-transcriptional RNA interference mechanisms which is mediated by the action of the small RNAs (sRNAs). Gene silencing molecules like miRNAs and siRNAs are not only responsible for endogenous regulation of gene expression but are also involved in cross-kingdom mutualistic relations and interaction networks1. The use of RNAi technology by involving artificial miRNAs has also been an effective control measure against various biotic threats to plants1,2. Since RNA silencing mechanism is important for various regulatory aspects of plants, so a comprehensive understanding of the components of this machinery is needed. The RNA dependent RNA polymerases (RDRs) and Dicer-like proteins (DCLs) are directly involved in small RNA biogenesis, whereas Argonaute (AGO) constitutes a significant component of the RNA induced silencing complex (RISC)3. RDRs are responsible for the synthesis of dsRNAs using an RNA template, whereas DCLs are responsible for cleavage of the dsRNAs to form 21–24 nucleotide long functional small RNAs. These sRNAs, either miRNAs or any class of siRNAs, get incorporated into the RISC to drive the gene silencing machinery4. The sRNAs bind to specific AGO proteins and then guide the RISC to their corresponding target genes through complementary base pairing between target mRNA and the guide strand of the sRNA. This mode of gene regulation may be mediated by two approaches, viz. target mRNA cleavage or translational inhibition5.
The AGO proteins of plants and animals can be grouped into three types based on the nature of small RNAs with which they are associated. The first category of AGO proteins is known to interact predominantly with miRNAs and siRNAs, whereas the second category known as the PIWI proteins are exclusively found in animals which interact with PIWI-interacting RNAs (piRNAs). A third category of AGO proteins, which bind to secondary siRNAs, was reported in worms6. Several studies have suggested the presence of four typical domains in AGO proteins viz. N terminal domain (Argo-N), PAZ domain, MID domain and PIWI domain7. PAZ domain contains a nucleotide-binding pocket that anchors the two nucleotide 3′ overhangs of the small RNAs generated after RNase III-like activity of DCLs8. The PIWI domain exhibits extensive functional homology to RNase H and is known to impart ‘slicer’ activity of the AGO proteins9. The MID domain is known to bind the 5′ phosphates of small RNAs and anchors small RNAs onto the AGO proteins10. The Argo-N domain may facilitate the separation of the small RNA:target duplex after slicing by interrupting the duplex structure11. In addition to these domains, two linker domains viz. Argo-L1 and Argo-L2 may be present between the ArgoN-PAZ lobes and PAZ-Piwi lobes respectively. In plants, different species exhibit the presence of different numbers of AGOs in their genome. For instance, 10 AGOs have been reported in Arabidopsis, 13 in Citrus, maize and rice possess 17 and 19 AGOs respectively, whereas Saccharum has been reported to consist of 21 AGO genes in its genome12–14.
The DCLs are found to have six different conserved domains viz., DEAD-box helicase, Helicase C-terminal domain, a Dicer dimerization domain, PAZ, Ribonuclease-III and dsRNA binding motif. However, one or more domains mentioned above may be missing even in a functional DCL protein15. RDRs are represented by only one unique conserved domain in their sequence i.e., RNA-dependent RNA polymerase (RdRP)14.
Tea is popular as the most consumed non-alcoholic beverage all over the world as it provides numerous secondary metabolites that account for its rich taste and health benefits. Studies concerning miRNA-mediated regulation of gene expression in tea under various forms of biotic and abiotic stresses have been carried out16–21. The availability of annotated tea genomes has given a wider scope for understanding of genes associated with sRNA biogenesis and function. The tea genome size has been estimated to be about 2.94 Gb, assembled in 15 pseudo-chromosomes which anchor about 86.73% of the assembled sequences22. Such a considerable genome size corresponds to a large scale expansion of gene families. Identification of miRNAs and their putative target genes have well been facilitated by the availability of reference genome of tea for both the CSS (C. sinensis var. sinensis) and CSA (C. sinensis var. assamica) varieties. Differentially expressed miRNAs responsible for regulating the expression of genes related to biotic and abiotic stresses, accumulation of secondary metabolites and growth and development in tea, have also been reviewed recently23,24. Genome-wide analysis of the AGO, DCL and RDR gene families will decipher the diversity in these gene families and their function in this important commercial crop.
Results
Genome-wide identification and domain analysis of AGOs, DCLs and RDRs in C. sinensis
To perform genome-wide identification of the AGO, DCL and RDR gene families in C. sinensis, we obtained the Hidden Markov Model (HMM) profiles of the conserved domains and searched all the genes of C. sinensis present in Tea Plant Information Archive (TPIA) database for the presence of AGO, DCL and RDR specific conserved domains. Eighteen CsAGOs were identified after analysing against the pfam database for presence of the following AGO specific domains—Argo-N: N-terminal domain of AGO proteins; PAZ: a domain that anchors the 3′ end of the bound small RNA and Piwi_Ago-like: PIWI domain present in the C-terminal region. Similarly, five DCLs and twelve RDRs were identified in C. sinensis genome using HMM profiles of gene specific conserved domains viz., RNaseIII, PAZ and dsRNA binding motif (for DCLs) and RdRP (for RDRs) followed by analysis against the pfam database. Two genes (accession numbers TEA000774.1 and TEA010224.1) with RdRP domains were further discarded as their lengths were small i.e., 38 and 73 amino acids, respectively for considering them as functional and without any close phylogenetic relationship with other identified CsRDRs. Further, TEA007002.1 was also discarded due to anomalies in lengths of its genomic and coding sequences. Thus after assessing of structural integrity of the conserved domains, 18 CsAGOs, 5 CsDCLs and 9 CsRDRs have been identified in the tea genome. The identified genes were named according to the phylogenetic relationships exhibited by their corresponding protein sequences with AGOs, RDRs and DCLs of A. thaliana obtained from TAIR (Table 1 and Fig. 1). The multiple sequence alignment showed high sequence similarity between the protein sequences, particularly in conserved functional domain regions (Supplementary Figure S1a–c).
Table 1.
Properties of identified CsAGO, CsDCL and CsRDR genes.
| Sl. no. | Assigned ID | Accession | Location | Start (5′) | Stop (3′) | Strand | Transcript length | Protein length |
|---|---|---|---|---|---|---|---|---|
| 1 | CsAGO1 | TEA010617 | Scaffold2091 | 586,414 | 597,932 | + | 3603 | 1200 |
| 2 | CsAGO2/3a | TEA015241 | Scaffold409 | 653,585 | 647,587 | − | 3168 | 1055 |
| 3 | CsAGO2/3b | TEA015275 | Scaffold409 | 542,266 | 536,576 | − | 3219 | 1072 |
| 4 | CsAGO2/3c | TEA003283 | Scaffold622 | 1,919,612 | 1,908,711 | − | 2301 | 766 |
| 5 | CsAGO2/3d | TEA007610 | Scaffold1775 | 1,333,085 | 1,328,089 | − | 2751 | 916 |
| 6 | CsAGO2/3e | TEA014680 | Scaffold2339 | 505,271 | 495,573 | − | 3405 | 1134 |
| 7 | CsAGO2/3f | TEA023201 | Scaffold5319 | 119,575 | 104,830 | − | 3183 | 1060 |
| 8 | CsAGO4a | TEA006285 | Scaffold1609 | 1,048,204 | 1,065,155 | + | 2871 | 956 |
| 9 | CsAGO4b | TEA033162 | Scaffold371 | 591,571 | 577,022 | − | 2838 | 945 |
| 10 | CsAGO4c | TEA015455 | Scaffold17947 | 50,775 | 97,852 | + | 4386 | 1461 |
| 11 | CsAGO5a | TEA005678 | Scaffold3329 | 2,163,110 | 2,154,877 | − | 3372 | 1123 |
| 12 | CsAGO5b | TEA005529 | Scaffold2891 | 638,503 | 630,782 | − | 2919 | 972 |
| 13 | CsAGO5c | TEA021652 | Scaffold3619 | 1,616,391 | 1,626,257 | + | 3060 | 1019 |
| 14 | CsAGO6 | TEA023892 | Scaffold366 | 435,162 | 444,062 | + | 2706 | 901 |
| 15 | CsAGO7 | TEA019830 | Scaffold660 | 687,517 | 683,178 | − | 3084 | 1027 |
| 16 | CsAGO10a | TEA008616 | Scaffold347 | 412,492 | 400,307 | − | 2994 | 997 |
| 17 | CsAGO10b | TEA008114 | Scaffold736 | 2,967,158 | 2,956,963 | − | 2997 | 998 |
| 18 | CsAGO10c | TEA008720 | Scaffold2968 | 415,128 | 391,947 | − | 2802 | 933 |
| 19 | CsDCL1a | TEA021156 | Scaffold2220 | 297,568 | 282,536 | − | 4491 | 1496 |
| 20 | CsDCL1b | TEA023787 | Scaffold6409 | 125,218 | 129,705 | + | 2652 | 883 |
| 21 | CsDCL2 | TEA015697 | Scaffold3698 | 1,259,866 | 1,288,358 | + | 4104 | 1367 |
| 22 | CsDCL3 | TEA011352 | Scaffold4138 | 1,757,571 | 1,714,236 | − | 5202 | 1733 |
| 23 | CsDCL4 | TEA025380 | Scaffold423 | 1,226,694 | 1,153,631 | − | 4680 | 1559 |
| 24 | CsRDR1a | TEA021321 | Scaffold1968 | 1,006,049 | 991,064 | − | 3459 | 1152 |
| 25 | CsRDR1b | TEA029634 | Scaffold872 | 905,729 | 927,644 | + | 3609 | 1202 |
| 26 | CsRDR1c | TEA021085 | Scaffold2268 | 122,465 | 84,100 | − | 3492 | 1163 |
| 27 | CsRDR2 | TEA021724 | Scaffold1551 | 145,105 | 159,756 | + | 3450 | 1149 |
| 28 | CsRDR5a | TEA010218 | Scaffold4444 | 610,116 | 625,490 | + | 2481 | 826 |
| 29 | CsRDR5b | TEA031356 | Scaffold1203 | 903,839 | 921,291 | + | 1029 | 342 |
| 30 | CsRDR6a | TEA013845 | Scaffold2753 | 994,451 | 989,757 | − | 1977 | 658 |
| 31 | CsRDR6b | TEA018620 | Scaffold3982 | 713,824 | 732,349 | + | 3921 | 1306 |
| 32 | CsRDR6c | TEA018638 | Scaffold3982 | 773,264 | 779,257 | + | 3642 | 1213 |
Figure 1.

Phylogenetic trees showing relationships between (A) AGOs, (B) DCLs and (C) RDRs of C. sinensis and A. thaliana. The trees were constructed using the maximum likelihood method and a bootstrap replicate of 1000. The trees with the highest bootstrap support for each gene class have been shown here.
Phylogenetic classification of identified genes
To define the homology between the identified protein sequences, phylogenetic trees of the 18 CsAGOs, 5 CsDCLs and 9 CsRDRs were constructed along with designated AtAGOs, AtDCLs and AtRDRs found in TAIR using maximum likelihood (ML) approach. The resulting trees produced well-resolved phylogeny with high bootstrap support. It was evident that C. sinensis AGO family proteins can be classified into three major clusters, with Group-I and II comprising of seven CsAGOs each, whereas group- III comprised of four members (Fig. 1A). The best ML scoring rooted tree indicates that Group-III AGOs probably emerged earliest during evolution compared to Group-I and II. Maximum-likelihood based phylogenetic tree constructed for CsRDRs and the six designated AtRDRs showed the presence of two major clusters with Group-I represented by seven members of CsRDR family whereas only two CsRDR proteins present in Group-II (Fig. 1B). However, no such substantive groups or clusters were seen in the topological pattern of DCLs, which indicated a more or less parallel evolutionary trend for the DCL genes (Fig. 1C).
Evolutionary relationship between C. sinensis and other plant AGOs, DCLs and RDRs
To determine the evolutionary relationship between tea and other plants in terms of proteins involved in small RNA machinery, we comprehensively analysed the phylogeny between single orthologs of CsAGOs, CsDCLs and CsRDRs found in representative species of all plant lineages. For this objective, 61 orthologous protein sequences of CsAGO1 and CsDCL1a, and 58 orthologous sequences of CsRDR1a were identified from different species belonging to algae, bryophytes, lycophytes, monocots and dicots. These protein sequences harboured characteristic domains and motifs of AGO, RDR and DCL proteins (Supplementary Table S1a–c). All the listed plant species in Phytozome-12 were selected and NJ trees were constructed using the orthologous protein sequences with 5000 bootstrap replicates.
The resulting NJ tree obtained for AGO contained five major clusters with significant bootstrap values. The topology of the tree depicts lower plants as the ancestors of the AGO gene family as they have settled in the basal group-I. AGO protein of C. sinensis finds its place in group-V alongside majority of the eudicots including Arabidopsis (Supplementary Figure S2a). However, topology of the tree constructed for RDR proteins did not follow typical evolutionary pattern with the basal group comprising a mix of algae, bryophyte and eudicots (Supplementary Figure S2b). The presence of only one representative conserved domain across the complete peptide sequence of RDRs of all the plant lineages may be responsible for retrieving such a tree with low bootstrap support and undefined distribution of plant species across the clusters. Orthologs of DCL proteins in the considered plant species mostly exhibited parallel evolution and no well-defined clusters or groups can be identified based on chronology of plant kingdom evolution (Supplementary Figure S2c). This pattern is also similar to the evolutionary pattern of paralogous DCLs of tea, as described in the previous section.
The CsAGO, CsDCL and CsRDR gene families
In this study, eighteen AGO members with all the characteristic domains were found in the tea tree genome. The nucleotide length of these genes varied between 2301 bp (CsAGO2/3c) to 4386 bp (CsAGO4c), while their encoded protein lengths ranged between 766 (CsAGO2/3c) to 1461 (CsAGO4c) amino acid residues. On an average, CsDCL genes exhibited longer nucleotide lengths which ranged from 2652 bases in CsDCL1b to 5202 bases in CsDCL3. The nucleotide lengths of CsRDRs ranged from 1029 bp (CsRDR5b) to 3921 bp (CsRDR6b) with their corresponding peptide lengths ranging from 342 to1306 amino acid residues respectively. The AGO genes were mostly oriented on reverse strands with only 5 genes being positioned on the forward strand. Similarly two DCLs were located on the forward strand and 3 DCLs on the reverse strand. However, RDR genes were mostly were located on the forward strand with only three genes oriented on the reverse strand (Table 1).
The ProtParam tool analysis showed significant differences in molecular weights of AGO (ranging from 85.93 to 161.84 kDa), DCL (98.7 to 194.14 kDa) and RDR (38.8 to 148.03 kDa) proteins of C. sinensis. Most of the CsAGOs have a relatively high isoelectric point (pI) (theoretical pI > 9) except CsAGO4a, b and c. However pI values were comparatively lower in CsDCLs and CsRDRs with most of them exhibiting a theoretical pI value of less than 8. All the identified proteins have negative GRAVY (Grand Average of Hydropathicity) values which implies that genes of all the three families are non-polar or hydrophilic in nature. Comparatively, CsAGOs are typically more hydrophilic than the members of CsDCL and CsRDR families. Out of all the 32 enlisted proteins, only seven of them viz. CsAGO2/3d, CsDCL1a, CsDCL1b, CsRDR5a, CsRDR6a, CsRDR6b and CsRDR6c have an Instability index (II) value less than 40 and hence can be considered as stable proteins. All CsDCLs, four CsRDRs and only one CsAGO have more number of negatively charged residues (Asp + Glu) as compared to positively charged residues (Arg + Lys) (Table 2).
Table 2.
Physico-chemical properties of AGO, DCL and RDR proteins of C. sinensis.
| Proteins | Accession | Mol. wt. (kDa) | pI | (Asp + Glu) | (Arg + Lys) | Total atoms | II | Aliphatic index | GRAVY |
|---|---|---|---|---|---|---|---|---|---|
| CsAGO1 | TEA010617 | 132.077 | 9.39 | 110 | 144 | 18,503 | 51.63 | 77.02 | − 0.420 |
| CsAGO2/3a | TEA015241 | 116.347 | 9.28 | 106 | 136 | 16,285 | 42.51 | 74.53 | − 0.459 |
| CsAGO2/3b | TEA015275 | 117.989 | 9.31 | 108 | 139 | 16,498 | 40.27 | 73.90 | − 0.473 |
| CsAGO2/3c | TEA003283 | 85.930 | 9.62 | 78 | 112 | 12,090 | 45.08 | 77.58 | − 0.616 |
| CsAGO2/3d | TEA007610 | 102.060 | 9.25 | 96 | 122 | 14,350 | 39.24 | 82.30 | − 0.341 |
| CsAGO2/3e | TEA014680 | 128.096 | 9.08 | 123 | 149 | 18,033 | 44.60 | 83.82 | − 0.357 |
| CsAGO2/3f | TEA023201 | 119.975 | 9.20 | 112 | 140 | 16,868 | 43.81 | 79.84 | − 0.458 |
| CsAGO4a | TEA006285 | 106.596 | 8.86 | 101 | 116 | 14,976 | 45.69 | 81.43 | − 0.321 |
| CsAGO4b | TEA033162 | 105.464 | 8.72 | 103 | 113 | 14,860 | 41.39 | 84.63 | − 0.300 |
| CsAGO4c | TEA015455 | 161.840 | 6.17 | 179 | 164 | 22,703 | 46.11 | 87.45 | − 0.245 |
| CsAGO5a | TEA005678 | 126.351 | 9.44 | 116 | 156 | 17,797 | 43.40 | 81.32 | − 0.443 |
| CsAGO5b | TEA005529 | 108.701 | 9.45 | 95 | 130 | 15,311 | 45.44 | 80.90 | − 0.368 |
| CsAGO5c | TEA021652 | 114.239 | 9.58 | 106 | 145 | 16,048 | 53.75 | 76.04 | − 0.560 |
| CsAGO6 | TEA023892 | 100.718 | 9.44 | 86 | 117 | 14,236 | 46.52 | 85.13 | − 0.331 |
| CsAGO7 | TEA019830 | 116.723 | 9.24 | 100 | 128 | 16,424 | 53.88 | 82.76 | − 0.435 |
| CsAGO10a | TEA008616 | 112.132 | 9.27 | 101 | 134 | 15,758 | 44.30 | 79.69 | − 0.455 |
| CsAGO10b | TEA008114 | 112.097 | 9.31 | 101 | 135 | 15,766 | 43.33 | 79.90 | − 0.455 |
| CsAGO10c | TEA008720 | 105.275 | 9.07 | 94 | 116 | 14,816 | 43.98 | 84.95 | − 0.373 |
| CsDCL1a | TEA021156 | 166.298 | 5.83 | 199 | 172 | 23,364 | 39.34 | 88.95 | − 0.261 |
| CsDCL1b | TEA023787 | 98.697 | 5.65 | 117 | 99 | 13,868 | 38.41 | 88.78 | − 0.236 |
| CsDCL2 | TEA015697 | 154.501 | 6.30 | 157 | 143 | 21,736 | 44.52 | 94.44 | − 0.129 |
| CsDCL3 | TEA011352 | 194.137 | 7.28 | 205 | 204 | 27,337 | 44.59 | 91.96 | − 0.234 |
| CsDCL4 | TEA025380 | 176.031 | 6.15 | 195 | 173 | 24,662 | 42.60 | 86.45 | − 0.221 |
| CsRDR1a | TEA021321 | 131.820 | 5.56 | 158 | 133 | 18,458 | 45.01 | 84.66 | − 0.244 |
| CsRDR1b | TEA029634 | 137.362 | 7.71 | 149 | 151 | 19,243 | 41.42 | 82.79 | − 0.292 |
| CsRDR1c | TEA021085 | 113.126 | 8.32 | 148 | 155 | 18,696 | 41.96 | 84.49 | − 0.298 |
| CsRDR2 | TEA021724 | 129.666 | 6.98 | 136 | 134 | 18,198 | 41.78 | 86.27 | − 0.230 |
| CsRDR5a | TEA010218 | 94.037 | 6.17 | 108 | 100 | 13,173 | 36.80 | 84.13 | − 0.327 |
| CsRDR5b | TEA031356 | 38.794 | 7.61 | 41 | 42 | 5462 | 46.40 | 90.06 | − 0.143 |
| CsRDR6a | TEA013845 | 74.158 | 6.13 | 80 | 74 | 10,363 | 31.84 | 81.81 | − 0.309 |
| CsRDR6b | TEA018620 | 148.025 | 7.29 | 167 | 167 | 20,752 | 36.13 | 82.47 | − 0.337 |
| CsRDR6c | TEA018638 | 138.266 | 8.33 | 157 | 165 | 19,388 | 35.16 | 80.59 | − 0.377 |
Structure of genes and conserved motifs in their encoded proteins
The exon–intron organisation of the genes is portrayed to elucidate the structural diversity of the CsAGO, CsDCL and CsRDR family genes. The number of exons varied significantly among the CsAGO genes, with CsAGO4c comprising 37 exons whereas CsAGO7 and all paralogs of CsAGO2/3 comprising 3–5 exons only. The length of introns also varies among different CsAGOs. Most of the introns in CsAGO7 and paralogs of CsAGO2/3 are in intron phase-2 (i.e., disrupting a codon between its second and third bases), whereas in rest of the CsAGO genes most introns are in phase-0 (i.e., present between two separate triplet codons). Most of the genes comprised more than one type of introns, except for CsAGO2/3e which consisted of four phase-2 introns (Fig. 2A). Significant differences in terms of loss/gain of exons and their arrangements was observed among genes belonging to different phylogenetic sub-trees, which may further add an element of diversity in structure and functions of CsAGOs. Besides, CsAGOs comprise of more phase-2 introns than phase-0 introns, unlike their A. thaliana homologs, where phase-0 introns outnumbered phase-2 introns (Fig. 2B). Moreover, AtAGOs exhibited a similar pattern in distribution of exons in their structures according to the clusters formed by AtAGOs and CsAGOs in the phylogenetic tree. Distribution of exons among the members of CsDCL genes showed that exon numbers varied from 26 in CsDCL3 and CsDCL4 to 9 in CsDCL1b (Fig. 3A). The CsRDR genes consisted of very lesser number of exons ranging from 2 to 6, except for CsRDR5a and CsRDR5b, consisting of 17 and 10 exons respectively (Fig. 4A). The introns mainly belonged to phase-0 type among the CsDCL and CsRDR genes, similar to the distribution patterns of introns among their A. thaliana counterparts (Figs. 3B and 4B). Several differences in terms of loss or gain of introns, intron phases and their shuffling were observed among the genes thus adding structural and functional diversity to the members of the three gene families.
Figure 2.

Gene structures showing the organization of exons and introns, and associated intron phases [0, 1 and 2] of (A) CsAGO and (B) AtAGO genes. The NJ phylogenetic tree of CDS is shown on the left side of the figure.
Figure 3.

Gene structures showing the organization of exons and introns, and associated intron phases [0, 1 and 2] of (A) CsDCL and (B) AtDCL genes. The NJ phylogenetic tree of CDS is shown on the left side of the figure.
Figure 4.

Gene structures showing the organization of exons and introns, and associated intron phases [0, 1 and 2] of (A) CsRDR and (B) AtRDR genes. The NJ phylogenetic tree of CDS is shown on the left side of the figure.
The conserved motifs of the AGO, DCL and RDR proteins were detected using the online MEME server (Multiple Expectation Maximization for Motif Elicitation). For CsAGOs, eight motifs out of at least ten were part of known domains according to Pfam codes (Fig. 5A and Supplementary Table S2a). Motifs 1, 2, 3 and 9 are associated with Piwi domain, whereas motifs 4, 6, 7 and 10 represent Argo-L1, PAZ, Argo-N and Argo-L2 respectively. The functions or secondary associations of motifs 5 and 8 are still unknown. Conserved motif analysis of CsDCL proteins resulted in recognition of five motifs mapped to known domains. According to pfam annotation, motifs 2 and 7 represent parts of PAZ domain whereas motifs 1, 5 and 10 represent RNaseIII, Helicase C-terminal and DEAD box domains respectively (Fig. 5B and Supplementary Table S2b). Since RdRP is the only conserved domain present in the plant RDRs, most of the motifs identified in CsRDRs are parts of the RdRP domain, with motifs 4 and 10 not having any defined annotations in pfam (Fig. 5C and Supplementary Table S2c). The logos of the corresponding motifs have been presented in Supplementary Figure S3.
Figure 5.

Distribution of conserved motifs identified in proteins encoded by (A) CsAGOs, (B) CsDCLs and (C) CsRDRs. The motif index represents the corresponding motif number depicted in Supplementary Figure S3 and Supplementary Table S2 for motif annotation.
Potential miRNA target sites in the identified gene transcripts
miRNAs regulate key biological processes such as growth, signal transduction, response to stress etc. AGO, DCL and RDR proteins are themselves involved in miRNA biogenesis and thus identification of miRNA target sites in the transcripts of these gene families may help to elucidate any potential self-regulatory or feedback mechanisms in plant miRNA biogenesis. Target analysis using the set of all plant miRNAs deposited in miRBase was carried out with expect value (e-value) threshold of 2.0, which revealed three potential miRNA target sites in CsAGO2/3a, two such sites in CsAGO2/3c and one target site each in CsAGO4a, CsAGO5c, CsAGO10b, CsRDR1c, CsRDR6b and CsRDR6c. No putative target sites within the e-value cut-off of 2.0 could be detected in CsDCL genes. The identified miRNAs are located on the 3′ strand of the stem-loop hairpin precursors. The UPE (Unpaired energy) value varied from 8.597 (ath-miR5658) to 27.278 (bdi-miR169c-3p) (Supplementary Table S3). The UPE represents the relative energy required to open the miRNA secondary structure around its target mRNA and thus a lower value corresponds with a better chance of contact between miRNA and target mRNA.
Cis-acting regulatory elements
Various cis-acting regulatory elements were found in the promoter regions (2 kb upstream of translation start site) of the identified genes. Primarily TATA box, which is one of the major regulatory components and is present around 30 bases before the translation start site, has been detected in the upstream sequences of most of the genes. Common cis-acting enhancers and regulatory elements viz., CAAT box and A-box are also present in promoter regions of a number of genes. Other cis-acting elements detected in CsAGOs, CsDCLs and CsRDRs can be classified into four groups based on their functional properties viz. hormone responsive elements, stress and defence response, plant growth and development and light-responsive elements. The number of these elements detected in promoter regions of each gene has been shown in Fig. 6.
Figure 6.

Number of each cis-acting element in the promoter region (2 kb upstream of translation start site) of respective genes belonging to (A) CsAGO, (B) CsDCL and (C) CsRDR. The elements have been separated into four distinct groups (by using a blank column between two groups) based on their functional properties (categories from left to right—hormone responsive; stress and defence response; plant growth and development; light responsive).
dn/ds values of orthologs of AGOs, DCLs and RDRs in C. sinensis and A. thaliana
The dn/ds values were calculated for the orthologous genes pairs of CsAGO, CsDCL and CsRDR with those of A. thaliana (Table 3). The dn/ds values were found to be less than 1 for all the entries implying that purifying or stabililizing selection has been the major evolutionary mechanism in these genes25.
Table 3.
Synonymous and non-synonymous substitution rates of orthologous gene pairs.
| Gene name | A. thaliana gene-ID | S-sites | N-sites | ds | dn | dn/ds | Divergence time (MYA) |
|---|---|---|---|---|---|---|---|
| CsAGO1 | AT1G48410.1 | 730 | 2411 | 2.3054 | 0.1424 | 0.062 | 177.338 |
| CsAGO2/3a | AT1G31280.1 | 693.7 | 2270.3 | 3.016 | 0.4384 | 0.145 | 232.000 |
| CsAGO2/3b | AT1G31280.1 | 685 | 2288 | 2.9057 | 0.4463 | 0.154 | 223.515 |
| CsAGO2/3c | AT1G31280.1 | 500.5 | 1722.5 | 2.4394 | 0.5372 | 0.220 | 187.646 |
| CsAGO2/3d | AT1G31280.1 | 648.4 | 2054.6 | 2.3885 | 0.3841 | 0.161 | 183.731 |
| CsAGO2/3e | AT1G31280.1 | 687.3 | 2294.7 | 2.2713 | 0.4584 | 0.202 | 174.715 |
| CsAGO2/3f | AT1G31280.1 | 688 | 2249 | 2.0033 | 0.445 | 0.222 | 154.100 |
| CsAGO4a | AT2G27040.1 | 627.6 | 2036.4 | 3.2404 | 0.2366 | 0.073 | 249.262 |
| CsAGO4b | AT2G27040.1 | 595.4 | 2038.6 | 3.0558 | 0.1731 | 0.057 | 235.062 |
| CsAGO4c | AT2G27040.1 | 585.5 | 1937.5 | 2.9976 | 0.1655 | 0.055 | 230.585 |
| CsAGO5a | AT2G27880.1 | 620.4 | 2109.6 | 3.9804 | 0.2849 | 0.072 | 306.185 |
| CsAGO5b | AT2G27880.1 | 670.9 | 2188.1 | 3.8707 | 0.3082 | 0.080 | 297.746 |
| CsAGO5c | AT2G27880.1 | 634 | 2114 | 3.3537 | 0.3018 | 0.090 | 257.977 |
| CsAGO6 | AT2G32940.1 | 627.9 | 1982.1 | 1.9205 | 0.2331 | 0.121 | 147.731 |
| CsAGO7 | AT1G69440.1 | 689.4 | 2211.6 | 2.4501 | 0.2258 | 0.092 | 188.469 |
| CsAGO10a | AT5G43810.1 | 692.2 | 2229.8 | 3.1338 | 0.114 | 0.036 | 241.062 |
| CsAGO10b | AT5G43810.1 | 671.3 | 2250.7 | 3.4197 | 0.0963 | 0.028 | 263.054 |
| CsAGO10c | AT5G43810.1 | 654.4 | 2129.6 | 3.261 | 0.1854 | 0.057 | 250.846 |
| CsDCL1a | AT1G01040.1 | 1081 | 3326 | 2.356 | 0.141 | 0.060 | 181.231 |
| CsDCL1b | AT1G01040.1 | 656.7 | 1965.3 | 2.1638 | 0.1073 | 0.050 | 166.446 |
| CsDCL2 | AT3G03300.1 | 948.1 | 3008.9 | 2.1394 | 0.3262 | 0.152 | 164.569 |
| CsDCL3 | AT3G43920.1 | 1107.2 | 3542.8 | 2.4104 | 0.4073 | 0.169 | 185.415 |
| CsDCL4 | AT5G20320.1 | 1002 | 3279 | 2.1895 | 0.4017 | 0.183 | 168.423 |
| CsRDR1a | AT1G14790.1 | 736.8 | 2557.2 | 4.011 | 0.2683 | 0.067 | 308.538 |
| CsRDR1b | AT1G14790.1 | 737.1 | 2547.9 | 8.4387 | 0.2341 | 0.028 | 649.131 |
| CsRDR1c | AT1G14790.1 | 735.8 | 2558.2 | 3.4007 | 0.2312 | 0.068 | 261.592 |
| CsRDR2 | AT4G11130.1 | 819.4 | 2525.6 | 1.8328 | 0.2862 | 0.156 | 140.985 |
| CsRDR5a | AT2G19930.1 | 479.1 | 1623.9 | 3.5722 | 0.3541 | 0.099 | 274.785 |
| CsRDR5b | AT2G19930.1 | 224.9 | 786.1 | 14.5924 | 0.6591 | 0.045 | 1122.492 |
| CsRDR6a | AT3G49500.1 | 496.2 | 1477.8 | 2.7386 | 0.1925 | 0.070 | 210.662 |
| CsRDR6b | AT3G49500.1 | 842.3 | 2739.7 | 2.5387 | 0.2272 | 0.089 | 195.285 |
| CsRDR6c | AT3G49500.1 | 847.8 | 2734.2 | 2.5503 | 0.2152 | 0.084 | 196.177 |
Chromosomal location of CsAGO, CsDCL and CsRDR genes
The tea genome has been recently assembled into 15 pseudo-chromosomes. Information regarding physical location of each of the gene was obtained by a blast search using sequences of the genes and the pseudo-chromosomes. The 32 genes under consideration were found to be located in 12 pseudo-chromosomes (Fig. 7). It was observed that any of these genes were not present on chromosome numbers 1, 9 and 10. All the five CsDCLs were found to be present on separate chromosomes, whereas gene pairs like CsRDR5a/CsRDR5b, CsRDR1a/CsRDR1b and CsRDR6b/CsRDR6c exhibited the presence of these genes in close vicinity with each other. Similarly, presence of homologous genes on the same location was also observed in case CsAGO genes, such as homologous pair of CsAGO2/3e and CsAGO2/3f and close location of CsAGO2/3a, CsAGO2/3b and CsAGO2/3c. This suggests that these genes might have evolved as a result of tandem duplication, thus giving rise to homologous genes. Tandem duplication events are often considered as a major driving force for the evolution of novel biological functions.
Figure 7.

The distribution of AGO, DCL and RDR genes on pseudo-chromosomes of C. sinensis. Chromosome numbers have been indicated on the top of each chromosome. The position of each gene on the respective chromosome has been depicted in terms of kilobase-pairs by numbers beside each gene.
Expression analysis of AGO, DCL and RDR genes in different parts of tea plant
To get a perception of the steady-state expression of CsAGO, CsDCL and CsRDR genes, the transcriptomic RNA-seq data was utilized from a bioproject that had been deposited previously in NCBI Genbank with the accession number PRJNA230752. The generated RNA-seq data included transcriptome profiles of thirteen different tissue samples of tea plant26. The final expression data of the AGO, DCL and RDR genes obtained after analysis were log transformed and illustrated in a heatmap (Fig. 8). CsAGO10c, CsAGO5b, CsRDR1c and CsRDR5b showed relatively distinctive expression patterns as compared to all the other analysed genes. This is because of the significant differences in their level of accumulation in different tissues. CsAGO10c exhibits the most noticeable tissue specificity within the AGO gene family as it gets highly expressed in the buds such as apical bud and both early stage and later stage lateral buds. The expression level of this gene is also seen to be relatively high in agronomically important young tissues like one leaf and a bud and two leaves and a bud. In contrast, the expression level of CsAGO10c falls drastically in mature structures like old-leaf, mature leaf and stem. Such contrasting expression measures can also be seen to some extent in CsAGO5a which shows extremely low build-up in mature leaf and flower compared to other tissues. Transcript of CsAGO2/3f has not been detected in any of the tissues analysed in this project. Most of the CsRDR genes show varied expression levels in different tissues, with the most diverse array displayed by CsRDR5b. This gene is highly expressed in tissues like apical bud and lateral buds whereas on the other hand its expression falls drastically in flower and root tissues. Regarding the DCL gene family all the CsDCL genes show a relatively average expression level in all the analysed tissues with no clear distinguishable differences. Genes involved in sRNA biogenesis show maximum variability in their expression in the old-leaf as compared to other tissues. CsRDR1c is showed higher expression in the old-leaf tissue, while CsAGO10c, CsAGO6, CsRDR6a and CsRDR5a exhibit reduced expression.
Figure 8.
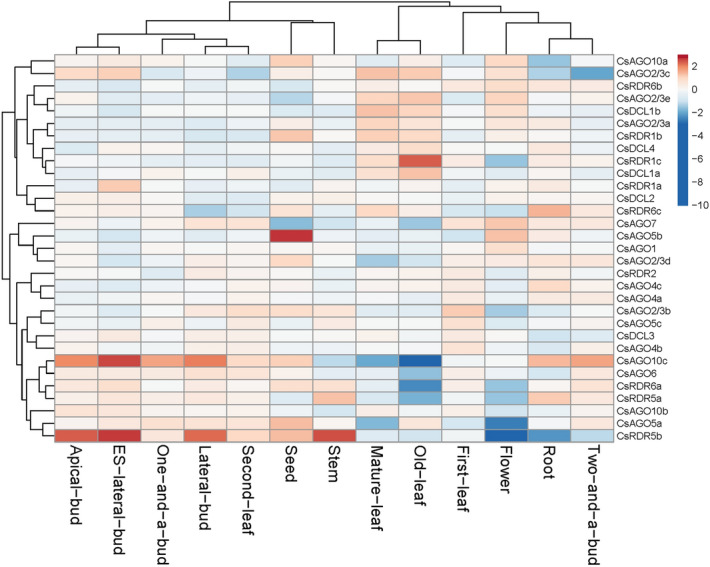
Normalized expression profiles of AGO, DCL and RDR genes of C. sinensis in different plant parts (left to right—apical bud, early-stage lateral bud, one leaf and a bud, lateral bud, second leaf, seed, stem, mature leaf, old leaf, first leaf, flower, root, two leaves and a bud).
Expression of CsAGO, CsDCL and CsRDR genes during biotic stress conditions
To envisage an overview of the differential gene expression pattern of AGO, DCL and RDR genes of C. sinensis in case of biotic stress, RNAseq data from two publicly available bioprojects were analysed. The generated transcriptomic data for the first bioproject (accession no. PRJNA439206) included the expression profiles of tea leaves and roots upon infection by Ectropis oblique27. In general, the expression contours of most of the genes were different in leaves and roots, for both infected and control tissues. For instance, CsAGO2/3a, CsAGO2/3d, CsAGO5b, CsAGO7, CsRDR6a, CsRDR6c and both paralogs of CsRDR5 were up-regulated in roots and down-regulated in leaves. However, when infected tissues were compared with the control samples, some changes were seen in expression levels of particular genes. CsAGO2/3a was highly expressed in the infected root sample as compared to the control. Correspondingly the expression level of CsAGO2/3a was also higher in E. oblique infected leaves than in the uninfected leaf sample. Another gene CsAGO5b shows greater expression levels in roots of uninfected plants than that of infected plant roots (Fig. 9A).
Figure 9.

(A) Normalized expression profiles of AGO, DCL and RDR genes of C. sinensis in roots and leaves upon E. oblique infection (ER and EL) and non-infected plant (CKR and CKL). (B) Normalized expression profiles of AGO, DCL and RDR genes of C. sinensis during different stages of blister blight disease in susceptible (SG) and resistant (RG) genotypes. Four distinct stages of infection as depicted are as follows—Spore inoculation (Inoculation); Spore germination (Germination); Haustorial development (Haustoria); Sporulation and secondary infection (Infection). (C) Normalized expression profiles of AGO, DCL and RDR genes of C. sinensis under different conditions of abiotic stress (CK- control; HT- high temperature; DT- drought; HD- high temperature and drought).
In a second bioproject (accession no. PRJNA306068), which has been used for analysing the expression of identified genes, RNAseq data was generated for tolerant and susceptible genotypes during blister blight disease development at four different stages of infection28. The most drastic differential expression was found in case of CsAGO10c and CsAGO2/3c. Out of all the analysed genes, CsAGO10c has the highest expression level in spore germination stage in the susceptible genotype and the lowest expression in the sporulation and secondary infection stage of the resistant genotype. CsAGO2/3c exhibits an expression pattern that is in contrast with that of CsAGO10c since expression of CsAGO2/3c is up-regulated in inoculation stage and down-regulated in the germination stage for both the genotypes (Fig. 9B). Most of the other genes however, exhibit average levels of expression changes in different infection stages.
Expression analysis of CsAGO, CsDCL and CsRDR genes in response to heat and drought stress
Differential expression trends of the members of aforesaid gene families were also analysed for a dataset associated with high temperature and drought treatments (accession no. PRJNA545401)29. Transcriptome data was used to generate the expression profiles of the CsAGOs, CsDCLs and CsRDRs in high temperature (HT), drought (DT), high temperature + drought (HD) and control (CK) conditions (Fig. 9C). CsAGO2/3f can be predicted to be an important drought responsive gene, since it was highly upregulated in the drought conditions as compared to control and HT treated samples. More specifically, expression level of CsAGO2/3f was even more in DT than in HD treated samples. Similarly, CsAGO5a shows increased expression in response to drought as its expression has been seen to be upregulated in DT and HD conditions. On the other hand, CsAGO2/3a exhibited a negative correlation with the onset of drought and showed downregulation in drought stress conditions. Furthermore, CsAGO10c has been found as one of the genes with significant level of differential expression and was considerably down-regulated in plants treated with simultaneous exposure of high temperature and drought.
Co-expression network analysis
To further understand the correlation among the AGO, RDR and DCL genes in terms of their expression, a positive correlation network analysis was carried out using the RNAseq data (Fig. 10). The co-expression network was constructed with Pearson’s correlation coefficient threshold of 0.5. Three gene pairs viz. CsAGO1/CsAGO7, CsRDR6b/CsAGO4a and CsRDR1b/CsRDR1c are found to be interacting and co-expressing independently. Exhibition of linear correlation was observed in expression patterns of CsDCL4, CsDCL1a, CsDCL1b and CsAGO2/3e. Two more conspicuous networks can be seen which show intensive cross-links among various genes. Besides, in some cases gene members belonging to the same phylogenetic clade have been found to be present in the same network with close relationship in their expression. These results suggest that various combinations of AGO, DCL and RDR may participate in different RNA interference pathways in C. sinensis.
Figure 10.

Co-expression networks of CsAGO, CsDCL and CsRDR genes showing positive correlation, based on combined expression data from various tissues and stress conditions.
qRT-PCR based expression analysis of CsAGO, CsDCL and CsRDR genes in various tissues
To validate the expression of AGO, DCL and RDR genes in various tissues in tea, we analysed the expression profiles of 7 CsAGOs, 5 CsDCLs and 7 CsRDRs by qRT-PCR, which were randomly selected from the total set of 32 genes. Since CsAGO2/3f exhibited interesting expression patterns in the high throughput analysis showing particularly drought-specific trends, primers were designed for this gene based on its CDS. However, even after repeated trials, CsAGO2/3f did not show any amplification in any of the analysed tissues. Five different tissues were considered for this analysis, viz. bunji bud, unopened bud, third leaf, flower bud and young stem of TV1 plants, which is a popular Indian tea cultivar. As shown in Fig. 11, the 19 analysed genes showed variable expression in five different tissues. CsAGO6, CsDCL2, CsDCL3, CsRDR5a and CsRDR6c did not show significant variation in their expression levels in different tissues, the pattern of which is mostly similar to the expression levels detected for these genes in the SRA data analysis (Fig. 8). Among rest of the genes, CsAGO1, CsAGO10a and CsDCL1b showed comparatively highest expression in unfolded apical buds relative to other analysed genes. The expression levels of CsDCL4 and CsRDR1a were remarkably higher in flower buds with respect to other tissues. CsDCL1a showed downregulation in all the other tissues with respect to bunji bud, whereas CsAGO1, CsAGO7 and CsRDR1b were expressed least in bunji bud. CsAGO7 was found to express more in third leaf as compared to other tissues, whereas CsAGO2/3a, CsAGO2/3d, and CsAGO10a exhibited minimal expression in third leaf tissue. Among all the analysed genes, a higher expression level in stem was displayed by CsAGO2/3a, CsRDR1b and CsRDR6a, whereas CsDCL1a had lowest expression in stem.
Figure 11.

qRT-PCR analysis showing the results of expression pattern of 19 considered genes in different tissues of TV1 cultivar of tea plant. The names of the genes are shown in the x-axis, and y-axis represents the fold changes of expression of the genes.
Discussion
RNA interference is an adaptable phenomenon that regulates the degree of accumulation of gene transcripts by sequence specific gene silencing machinery. Thus exploring the differential expression patterns of the core genes of this silencing machinery in different conditions becomes indispensable. Coordinated function of RDR-DCL-AGO genes is crucial for processing different classes of small RNAs, which indirectly makes them involved in regulation of diverse biological pathways30,31. Members of these three gene families are involved in biogenesis of sRNAs and effective silencing of their targets. For example, DCL1 is primarily involved in the biogenesis of microRNAs with no necessity of RDR proteins, whereas DCL2, 3 and 4 are mainly responsible for processing of siRNAs originating from long dsRNAs synthesized by the action of RDR proteins32,33. Moreover, DCL3 and DCL4 products are also known to have discrete functions, with the former known to be involved in RNA-directed-DNA-methylation (RdDM) pathway and latter being a component of the RNA interference apparatus34. In regard to the AGO gene family, AGO1 is generally the most prevalent member engaged in miRNA mediated gene silencing process. However in some cases other homologs of AGO genes also participate in completing the silencing machinery of various miRNAs. For example, miR390-AGO7 module is involved in the regulation of Auxin Response Factors (ARFs) and miR166-AGO10 module has been reported in development of shoot apical meristem35. Three gene families—AGOs, DCLs and RDRs—which function as the key components for biogenesis and action of the sRNAs in tea plants were identified in this study. Eighteen AGOs, five DCLs and nine RDRs were predicted in this study from tea plants with the number of genes being significantly higher than that in A. thaliana. However this increase in number of genes might be attributed to more number of chromosomes and bigger genome size of tea25. The expression pattern of the genes was also analysed in different tissues of the tea plant and also in response to biotic and abiotic stresses.
Structural organisation and gene expansion
The domain analysis in the CsAGO family revealed the presence of N-terminal domain, PAZ, Piwi, Mid domains and linker-1 and linker-2 domains. However, only the ArgoN, PAZ, Piwi and Argo-L2 domains were found to be present in all the identified AGOs whereas the Mid domain was absent in CsAGO7, CsAGO4c and five members of CsAGO2/3 clade except CsAGO2/3d. Linker-1 domain was absent only in CsAGO4a. A Glycine-rich-AGO1 specific domain was found in CsAGO1, which has already been reported in AGO1 proteins of many other plants such as Arabidopsis and Coffea36,37. In the DCL family CsDCL1a, CsDCL2, and CsDCL3 showed the presence of N-terminal and C-terminal domains of DEAD-box helicase, Dicer-dimerization domain, PAZ and RNase III domains. CsDCL1b however lacked the DEAD-box helicase and the Dicer-dimerization domains, whereas PAZ domain was absent in CsDCL4. A dsRNA binding motif was also detected in the protein sequences of CsDCL1a and CsDCL1b. The CsRDR proteins consisted of one conserved domain i.e. RdRP, which is truncated to some extent in CsRDR5a and CsRDR5b. Furthermore, a part of RNA recognition motif superfamily (RRM_SF) is present towards the upstream region of CsRDR2 protein. Presence of RRM_SF has also been reported in RDR2 protein of Salvia miltiorrhiza38. The gene copy numbers are greater than those of A. thaliana indicating that the genes might have undergone significant expansion through gene duplication events. Selection pressures leading to large-scale duplication events have also been observed for a number of stress responsive genes in tea22,39. Expansion of AGO and RDR gene families suggests a corresponding diversification of the gene function in tea40. This is also substantiated by tandem duplication events observed for homologous genes of CsRDRs and CsAGOs, which are present on the same chromosomal location.
AGOs, DCLs and RDRs as moderators of gene silencing
Different set of genes are regulated in distinct developmental stages of a plant in a tissue specific manner. CsAGO1 seems to be the most ubiquitously expressed gene in all the tissues and various stress conditions considered in this study. It is one of the main components of gene silencing machinery and is known to participate in the biogenesis of most of the conserved miRNAs and siRNAs in tea24. In our study CsAGO1 seems to be ubiquitously expressed in all the tissues and various stress conditions in tea. As stated earlier, CsAGO10c has been found to be the most dynamically regulated AGO gene according to the nature of the plant tissue in C. sinensis, and shows high accumulation in buds. Activity of miR166-AGO10 module is important for meristem formation in plants35. Expression of CsAGO10c is found to be higher in tissues undergoing active meristematic development such as buds, roots and seeds. Furthermore, CsAGO10c also exhibits significant potential in supplementing the establishment of Exobasidium vexans infection in tea to cause blister blight disease. The level of accumulation of CsAGO10c seems to be quite similar during inoculation, germination and haustorial development stages in both the resistant and susceptible genotypes. However at the final stage of sporulation and secondary infection, the expression of CsAGO10c is highly down-regulated in the resistant genotype, as compared to the susceptible one. During the E. oblique infestation in tea, expression levels of CsAGO2/3a, b and d were higher in effected roots and leaves as compared to control. On a similar trend, expression of AGO2 has been reported to be significantly induced by biotic stress in Capsicum annuum41. This indicates that AGO2 might be somehow involved in regulation of defense mechanisms in plants against pathogens. In case of drought stress in tea, expression level of CsAGO2/3a is reduced significantly compared to control. Interestingly, the 3′UTR of AGO2 has been validated as a target site for a drought responsive miRNA i.e. miR403 in Arabidopsis42. Downregulation of CsAGO2/3a in tea during drought and heat stress may result from any such interaction with a stress-induced miRNA, which warrants further investigation. In Arabidopsis, AGO2 and AGO3 have been reported to play substantial roles in antiviral defence and epigenetic pathway respectively, and both these genes show high amount of homology in their protein sequences43. CsAGO5a exhibits preferential accumulation in seed, while CsAGO5b mostly accumulates in both seed and flower which could be a probable result of active involvement of CsAGO5 in reproductive tissues. Higher expression of AGO5 has also been reported in Arabidopsis during all stages of flower and seed formation44.
DCL genes are important components for biogenesis of miRNAs and various classes of siRNAs. Even though plants have evolved four different groups of DCLs, these are said to have structurally diversified with overlapping functions45. The DCL genes in tea seemed to exhibit a more or less uniform expression levels in all the tissues, with the only notable observation being slightly higher expression of CsDCL1b in mature and old leaves and in flower. Potential role of DCL1 genes in inducing flowering has also been suggested earlier in Arabidopsis where dcl1/dcl3 mutants exhibited delay in flowering46.
RDRs participate in dsRNA synthesis for the biogenesis of siRNAs. We identified nine RDRs in our study representing four different homologous groups viz. RDR1, 2, 5 and 6. Expression of CsRDR genes showed significant degree of variability in different tissues and stress conditions. RDR2 has been reported to be actively involved in biogenesis of hc-siRNA, along with participation of AGO4 in the DNA methylation pathway in Arabidopsis47. In our study, expression levels of CsAGO4a and CsAGO4c are mostly similar to CsRDR2 in all the considered tissues of tea plant. CsRDR5a, CsRDR5b and CsRDR6c showed low expression levels in E. oblique infested samples, but were also found to be tissue specific showing significant differences in their expression. This suggests they might play critical roles under definite circumstances in plant growth and development or that their expression could be induced in response to specific environmental signals and during various stress conditions. The qRT-PCR results also showed variable expression patterns of the AGO, DCL and RDR genes in different tissues.
Functions of AGO, DCL and RDR proteins as components of silencing machinery in inducing resistance against abiotic and biotic stress has been studied extensively in various plants14,41,48. Moreover, miRNA mediated gene silencing is a crucial regulatory process of important agronomic traits of various crops. Hence, comprehensive knowledge about the regulatory potential of these three components of gene silencing machinery becomes important in the aspect of genetic improvement of an economically important crop such as tea.
Conclusion
Functional association between AGOs, DCLs and RDRs is responsible for supplementing gene regulatory functions like RNA interference and RdDM in eukaryotes. Evaluating the potential roles of these important gene families in a commercially important crop like tea certainly helps to engineer tea crop to enhance crop productivity and quality. In the present study, we have identified 18 AGOs, 5 DCLs and 9 RDRs in tea genome. Phylogenetic and structural analyses of these gene sequences show differences in arrangement of exons and introns, based on which they can be grouped into distinct clades. Even though the identified genes exhibit evolutionary expansion in tea, their expression patterns in various tissues and stress conditions indicate presence of overlapping functions among the paralogous members. Presence of stress hormone related promoter elements in their upstream region indicates the involvement of these genes in adaptation during stress condition in tea. The genes identified in this study can be used as potential targets for crop improvement for developing stress resistant tea cultivars.
Materials and methods
Identification of CsAGO, CsDCL and CsRDR gene family members
The reference genome, coding sequences (CDS) and peptide sequences of C. sinensis var. sinensis were downloaded for Tea Plant Information Archive (TPIA). In order to identify the AGO, DCL and RDR gene families, the alignment files of the PIWI, PAZ, RNaseIII and RdRP domains were downloaded from pfam database in Stockholm format from which the corresponding HMM profiles were built using the HMMER toolkit49,50. The tea peptide sequences were then searched for the presence of HMM-profiles of the conserved domains followed by subjecting the identified non-redundant proteins to domain analysis in batch CD search against the pfam and SMART databases with default cut-off parameters51. Peptide sequences of C. sinensis were also BLASTP searched against AGO, DCL and RDR protein sequences of A. thaliana to ensure that any putative genes of these three gene families are not left out from the analysis. Sequences containing N-terminal (pfam16486), PAZ (pfam02170) and PIWI (pfam02171) domains were recognized as AGO proteins. Linker and Mid domains however may or may not be present in all the identified genes. Similarly, tea peptides showing presence of RNase III domains were analysed in batch CD search against pfam and SMART database to detect the presence of all the conserved domains of DCL proteins viz., DEAD (pfam00270), Helicase-C (pfam00271), Dicer-dimer (pfam03368), PAZ (pfam02170), RNaseIII (pfam00636) and DSRM (pfam00035). For identification of RDRs, the peptides which exhibited the presence of RdRP domain were considered as putative RDRs of tea. The positions and structural integrity of the identified domains were also confirmed by biosequence analysis using Hidden Markov Models in HMMER database52. The identified genes were named according to their positions in phylogenetic trees which also included designated AGOs, DCLs and RDRs of A. thaliana53.
Characterization and physicochemical properties
Amino acid properties, physicochemical traits such as charge, molecular weight (g mol−1), aliphatic index, instability index (II), isoelectric point (pI), grand average of hydropathy (GRAVY) and other properties of the CsAGO, CsDCL and CsRDR proteins were calculated using the ProtParam tool in the ExPASy web server54.
Sequence alignments and phylogenetic analysis
Multiple sequence alignments for the predicted CsAGO, CsDCL and CsRDR proteins were performed using ClustalX 2.1 program with default settings, and viewed using GeneDoc software v1.0 (https://genedoc.software.informer.com)55,56. The identified conserved domain sites specific for AGO, DCL and RDR were manually checked and verified using the coordinates’ data of the conserved domains in each protein, obtained using the ‘hmmscan’ tool from the HMMER web server50. MEGA7 software (https://www.megasoftware.net) was used to carry out the evolutionary and phylogenetic analyses57. Preliminary trees for heuristic search were obtained by applying Neighbour Joining/BioNJ method to a matrix of pairwise distances estimated using Jones-Taylor-Tshorton (JTT) matrix-based model58. Final phylogenetic trees were constructed using Maximum Likelihood method based on the Jones-Taylor-Thorton (JTT) model using bootstrap of 1000 replicates. The trees were squared to scale, with number of substitutions per site represented by branch lengths. The phylogenetic analyses also included putative orthologous genes from other plant species, which were BLASTP searched and downloaded from Phytozome59 using CsAGO1, CsDCL1a and CsRDR1a encoded proteins as query. Neighbour Joining (NJ) trees using 5000 bootstrap replicates and JTT based model were constructed using the identified orthologs.
Prediction of gene structure, motifs and miRNA target sites
The structures of the AGO, DCL and RDR family genes showing exon–intron organization were determined based on alignments of their coding sequences with the corresponding genomic sequences, and an illustration was obtained using Gene Structure Display Server 2.060. The conserved motifs in the identified proteins were identified in MEME web server keeping the optimal motif width between 6 and 200, and the maximum number of different motifs as ten61. The discovered motifs were annotated with Pfam and hmmscan programs49,50. For miRNA target sites prediction within the CsAGO, CsDCL and CsRDR transcripts, sequences of identified transcripts were used as target gene input to the psRNATarget server62 and analysed against all the available plant miRNAs using an expect value threshold of 2.0 and maximum energy to un-pair the target site (UPE) up to 50 units.
Identification of cis-acting regulatory elements, chromosomal location and dn/ds calculation
The data about locations of the identified genes in different scaffolds of the genome were obtained from TPIA platform and 2000 bases upstream sequences were retrieved. The cis-acting elements present in these upstream promoter regions of AGO, DCL and RDR genes of C. sinensis were identified using PLANT CARE database63.To gather information about the chromosomal locations of each gene, the pseudo-chromosome sequences of tea genome available in TPIA, and the gene sequences were blasted, following which coordinates of each gene on the chromosomes were depicted on physical map of each chromosome using Mapchart v2.364.
Orthologous genes of the CsAGOs, CsDCLs and CsRDRs were identified in A. thaliana by using BLAST tool of Phytozome59. The best hit for each of the genes were designated as orthologous partners and rates of synonymous and non-synonymous substitutions were determined using the PAL2NAL utility65. The dn/ds ratio was calculated in order to assess the selection history and divergence time of the gene families. The divergence time (T) was calculated using the formula T = ds/(2λ) × 10−6 million years ago (MYA), where value of λ = 6.5 × 10–9 (Universal substitution rate)66,67. The pairwise alignment files for the protein sequences required as inputs in the PAL2NAL program were created using Clustal Omega68.
Analysis of AGO, DCL and RDR gene expression in tea
Transcriptome data from four different bioprojects submitted in NCBI were downloaded for in silico analysis of expression data of CsAGO, CsDCL and CsRDR genes. The details of the bioprojects are as follows: (i) To explore the expression patterns of these genes in different tissues of tea plant, the Illumina RNA-sequencing data of C. sinensis (L.) O. Kuntze cv. ‘Longjing 43’ was downloaded from GenBank archives (accession no. PRJNA230752)26. The SRA data of 13 different tissue samples viz. apical bud, early stage lateral bud, lateral bud, flower, seed, stem, root, mature leaf, old leaf, two and a bud, one and a bud, first leaf and second leaf were downloaded, from which the low quality reads and adapters were removed, and then mapped to the tea reference genome22. (ii) Data obtained from the bioproject with accession no. PRJNA439206 includes transcriptome profiles of leaves and roots of E. oblique infested plants along with the control samples27. (iii) The third dataset with accession no. PRJNA306068 represents blister blight infected leaf samples at four different stages of infection viz., spore inoculation, germination, haustoria development, and sporulation and secondary infection for both susceptible and resistant genotypes of tea28. (iv) Differential expression analysis of the said genes was also carried out for a particular study associated with abiotic stress, i.e. bioproject PRJNA545401. This dataset includes the RNA seq data for tea plants treated with high temperature and drought conditions29. An annotation file consisting of only the identified AGO, DCL and RDR genes with their respective gene-IDs was manually created to get the mapping and expression data of only these genes. The gene expression data was normalized by FPKM (fragments per kilobase per million) and the resulting FPKM values of genes were log2 transformed using edgeR and Trinity (R language-based) programs. The dispersion value threshold was set as 0.1, as the samples analysed were tissues belonging to same cultivar of tea and were highly similar in their genetic constitution. The heatmaps along with the expression clustering were generated and visualized using Clustvis—an R based online tool (https://biit.cs.ut.ee/clustvis/)69.
Gene co-expression network construction
To represent the co-expression profiles of the identified genes, we performed gene co-expression network analysis using the FPKM data generated for the gene expression evaluation using RNA-seq data. Cytoscape software version 3.7.2 (https://cytoscape.org) was used for this purpose where FPKM matrices of the gene expression were fed as inputs70.
qPCR validation of selected transcripts in tissues representing different developmental stages
To determine the expression of some of the members from the 3 different gene families, qRT-PCR analysis was carried out using the different tissue samples collected from young saplings of TV1 cultivar. Around 100 mg tissue was used to extract total RNA with Trizol reagent following manufacturer’s protocol (Invitrogen, USA). Quantity and quality of the purified DNA-free RNAs were determined using Nanodrop 1000 (Thermo scientific, USA) and agarose gel electrophoresis. The cDNA was prepared by using 2 μg of total extracted RNA using SuperScript III cDNA synthesis kit (Invitrogen, USA) following manufacturer’s protocol. The cDNA samples were diluted 40 times and then subjected to qRT-PCR. The diluted cDNAs were used for 25 μl PCR reactions using QuantiFast SYBR Green PCR Master Mix (Qiagen, India). The gene specific primers were designed manually for all the transcripts along with 18S rRNA (NCBI Genbank id: AF207029.1) as an internal control71. The primers used in this study are listed in Supplementary Table S4. Real-time PCR analysis was conducted as described previously72, using primer specific annealing temperatures. Two technical replicates from three individual biological replicates were considered for each experiment conducted. The relative expression analyses of the qRT-PCR results were expressed using the 2−ΔΔCT method73. Five different tissues viz. bunji bud, unfolded bud, young third leaf, unopened flower bud, and young stem of TV1 plants were considered to analyse the relative expression levels in various tissues.
Ethical approval
The authors have obtained permission to collect tea plant material for the experiment. The authors also declare that the experimental research work conducted in this study comply with relevant institutional, national, and international guidelines and legislation.
Supplementary Information
Acknowledgements
This work was supported by the funds from DST-SERB funded ECR Award Project (ECR 710/2017) granted by Govt. of India and facilities provided by the Gauhati University, Assam, India. The authors are thankful to DST, Govt. of India for providing DST-FIST Support to Department of Botany, Gauhati University, where this research work was carried out.
Author contributions
N.A. and D.B.K. conceptualized the study and designed experiments. D.B.K. wrote the manuscript, D.B.K., P.M.B. and B.D. carried out the bioinformatic analysis, analysed the data and prepared tables and figures, N.A. obtained funding and supervised this investigation. T.K.M. and S.C. carried out the qPCR analysis. N.A. and T.K.M. revised the manuscript. All authors read and approved the final paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-87991-5.
References
- 1.Gualtieri C, Leonetti P, Macovei A. Plant miRNA cross-kingdom transfer targeting parasitic and mutualistic organisms as a tool to advance modern agriculture. Front Plant Sci. 2020;11:930. doi: 10.3389/fpls.2020.00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bordoloi KS, Agarwala N. MicroRNAs in plant insect interaction and insect pest control. Plant Gene. 2021;25:100271. doi: 10.1016/j.plgene.2021.100271. [DOI] [Google Scholar]
- 3.Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pratt AJ, MacRae IJ. The RNA-induced silencing complex: A versatile gene-silencing machine. J. Biol. Chem. 2009;284(27):17897–17901. doi: 10.1074/jbc.R900012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu SR, Zhou JJ, Hu CG, Wei CL, Zhang JZ. MicroRNA-mediated gene silencing in plant defense and viral counter-defense. Front. Microbiol. 2017;8:1801. doi: 10.3389/fmicb.2017.01801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaucheret H. Plant ARGONAUTES. Trends Plant Sci. 2008;13(7):350–358. doi: 10.1016/j.tplants.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 7.Song JJ, Smith SK, Hannon GJ, Joshua-Tor L. Crystal structure of Argonaute and its implications for RISC slicer activity. Science. 2004;305:1434–1437. doi: 10.1126/science.1102514. [DOI] [PubMed] [Google Scholar]
- 8.Yan KS, Yan S, Farooq A, Han A, Zeng L, Zhou MM. Structure and conserved RNA binding of the PAZ domain. Nature. 2003;426:469–474. doi: 10.1038/nature02129. [DOI] [PubMed] [Google Scholar]
- 9.Parker JS, Roe SM, Barford D. Crystal structure of a PIWI protein suggests mechanisms for siRNA recognition and slicer activity. EMBO J. 2004;23(24):4727–4737. doi: 10.1038/sj.emboj.7600488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parker JS, Roe SM, Barford D. Structural insights into mRNA recognition from a PIWI domain-siRNA guide complex. Nature. 2005;434(7033):663–666. doi: 10.1038/nature03462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mallory A, Vaucheret H. Form, function, and regulation of ARGONAUTE proteins. Plant Cell. 2010;22(12):3879–3889. doi: 10.1105/tpc.110.080671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu D, Yang H, Zou C, Li WX, Xu Y, Xie C. Identification and functional characterization of the AGO1 ortholog in maize. J. Integr. Plant Biol. 2016;58(8):749–758. doi: 10.1111/jipb.12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabbione A, Daurelio L, Vegetti A, Talon M, Tadeo F, Dotto M. Genome-wide analysis of AGO, DCL and RDR gene families reveals RNA-directed DNA methylation is involved in fruit abscission in Citrus sinensis. BMC Plant Biol. 2019;19(1):401. doi: 10.1186/s12870-019-1998-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cui DL, Meng JY, Ren XY, Yue JJ, Fu HY, Huang MT, et al. Genome-wide identification and characterization of DCL, AGO and RDR gene families in Saccharum spontaneum. Sci. Rep. 2020;10:13202. doi: 10.1038/s41598-020-70061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Margis R, Fusaro AF, Smith NA, Curtin SJ, Watson JM, Finnegan EJ, Waterhouse PM. The evolution and diversification of Dicers in plants. FEBS Lett. 2006;580(10):2442–2450. doi: 10.1016/j.febslet.2006.03.072. [DOI] [PubMed] [Google Scholar]
- 16.Jeyaraj A, Liu S, Zhang X, Zhang R, Shangguan M, Wei C. Genome-wide identification of microRNAs responsive to Ectropis oblique feeding in tea plant (Camellia sinensis L.) Sci. Rep. 2017;7(1):13634. doi: 10.1038/s41598-017-13692-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeyaraj A, Wang X, Wang S, Liu S, Zhang R, Wu A, et al. Identification of regulatory networks of microRNAs and their targets in response to Colletotrichum gloeosporioides in tea plant (Camellia sinensis L.) Front. Plant Sci. 2019;10:1096. doi: 10.3389/fpls.2019.01096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu SC, Xu YX, Ma JQ, Wang WW, Chen W, Huang DJ, et al. Small RNA and degradome profiling reveals important roles for microRNAs and their targets in tea plant response to drought stress. Physiol. Plant. 2016;158:435–451. doi: 10.1111/ppl.12477. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Zhu X, Chen X, Song C, Zou Z, Wang Y, et al. Identification and characterization of cold-responsive microRNAs in tea plant (Camellia sinensis) and their targets using high-throughput sequencing and degradome analysis. BMC Plant Biol. 2014;14:271. doi: 10.1186/s12870-014-0271-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun P, Cheng C, Lin Y, Zhu Q, Lin J, Lai Z. Combined small RNA and degradome sequencing reveals complex microRNA regulation of catechin biosynthesis in tea (Camellia sinensis) PLoS ONE. 2017;12(2):e0171173. doi: 10.1371/journal.pone.0171173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo Y, Zhao S, Zhu C, Chang X, Yue C, Wang Z, et al. Identification of drought-responsive miRNAs and physiological characterization of tea plant (Camellia sinensis L.) under drought stress. BMC Plant Biol. 2017;17:211. doi: 10.1186/s12870-017-1172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia EH, Tong W, Hou Y, An Y, Chen L, Wu Q, et al. The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into its genome evolution and adaptation. Mol. Plant. 2020;13(7):1013–1026. doi: 10.1016/j.molp.2020.04.010. [DOI] [PubMed] [Google Scholar]
- 23.Krishnatreya DB, Agarwala N, Gill SS, Bandyopadhyay T. Understanding the role of miRNAs for improvement of tea quality and stress tolerance. J. Biotechnol. 2021;328:34–46. doi: 10.1016/j.jbiotec.2020.12.019. [DOI] [PubMed] [Google Scholar]
- 24.Jeyaraj A, Elango T, Li X, Guo G. Utilization of microRNAs and their regulatory functions for improving biotic stress tolerance in tea plant [Camellia sinensis (L.) O. Kuntze] RNA Biol. 2020;17(10):1365–1382. doi: 10.1080/15476286.2020.1774987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia EH, Tong W, Wu Q, Wei S, Zhao J, Zhang ZZ, et al. Tea plant genomics: Achievements, challenges and perspectives. Hortic. Res. 2020;7:7. doi: 10.1038/s41438-019-0225-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li CF, Zhu Y, Yu Y, Zhao QY, Wang SJ, Wang XC, et al. Global transcriptome and gene regulation network for secondary metabolite biosynthesis of tea plant (Camellia sinensis) BMC Genomics. 2015;16(1):560. doi: 10.1186/s12864-015-1773-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang H, Wang Y, Li L, Li F, He Y, Wu J, et al. Transcriptomic and phytochemical analyses reveal root-mediated resource-based defense response to leaf herbivory by Ectropis oblique in tea plant (Camellia sinensis) J. Agric. Food Chem. 2019;67:5465–5476. doi: 10.1021/acs.jafc.9b00195. [DOI] [PubMed] [Google Scholar]
- 28.Jayaswall K, Mahajan P, Singh G, Parmar R, Seth R, Raina A, et al. Transcriptome analysis reveals candidate genes involved in blister blight defense in tea (Camellia sinensis (L.) Kuntze) Sci. Rep. 2016;6:30412. doi: 10.1038/srep30412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding Y, Wang Y, Qiu C, Qian W, Xie H, Ding Z. Alternative splicing in tea plants was extensively triggered by drought, heat and their combined stresses. PeerJ. 2020;8:e8258. doi: 10.7717/peerj.8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borges F, Martienssen RA. The expanding world of small RNAs in plants. Nat. Rev. Mol. Cell Biol. 2015;16(12):727–741. doi: 10.1038/nrm4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bologna NG, Voinnet O. The diversity, biogenesis, and activities of endogenous silencing small RNAs in Arabidopsis. Annu. Rev. Plant Biol. 2015;65:473–503. doi: 10.1146/annurev-arplant-050213-035728. [DOI] [PubMed] [Google Scholar]
- 32.Nagano H, Fukudome A, Hiraguri A, Moriyama H, Fukuhara T. Distinct substrate specificities of Arabidopsis DCL3 and DCL4. Nucleic Acids Res. 2014;42(3):1845–1856. doi: 10.1093/nar/gkt1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haag JR, Ream TS, Marasco M, Nicora CD, Norbeck AD, Pasa-Tolic L, et al. In vitro transcription activities of Pol IV, Pol V, and RDR2 reveal coupling of Pol IV and RDR2 for dsRNA synthesis in plant RNA silencing. Mol. Cell. 2012;48(5):811–818. doi: 10.1016/j.molcel.2012.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie Z, Johansen LK, Gustafson AM, Kasschau KD, Lellis AD, Zilberman D, et al. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004;2(5):E104. doi: 10.1371/journal.pbio.0020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iki T. Messages on small RNA duplexes in plants. J. Plant Res. 2017;130:7–16. doi: 10.1007/s10265-016-0876-2. [DOI] [PubMed] [Google Scholar]
- 36.Bohmert K, Camus I, Bellini C, Bouchez D, Caboche M, Benning C. AGO1 defines a novel locus of Arabidopsis controlling leaf development. EMBO J. 1998;17(1):170–180. doi: 10.1093/emboj/17.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandes-Brum CN, Rezende PM, Ribeiro THC, Oleivera RR, Cardosco TCS, Amaral LR, et al. A genome-wide analysis of the RNA-guided silencing pathway in coffee reveals insights into its regulatory mechanisms. PLoS ONE. 2017;12(4):e0176333. doi: 10.1371/journal.pone.0176333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shao F, Lu S. Identification, molecular cloning and expression analysis of five RNA-dependent RNA polymerase genes in Salvia miltiorrhiza. PLoS ONE. 2014;9(4):e95117. doi: 10.1371/journal.pone.0095117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xia EH, Zhang HB, Sheng J, Li K, Zhang QJ, Kim C, et al. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol. Plant. 2017;10(6):866–877. doi: 10.1016/j.molp.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 40.Singh RK, Gase K, Baldwin IT, Pandey SP. Molecular evolution and diversification of the Argonaute family of proteins in plants. BMC Plant Biol. 2015;15:23. doi: 10.1186/s12870-014-0364-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qin L, Mo N, Muhammad T, Liang Y. Genome-wide analysis of DCL, AGO, and RDR gene families in pepper (Capsicum annuum L.) Int. J. Mol. Sci. 2018;19(4):1038. doi: 10.3390/ijms19041038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferdous J, Hussain S, Shi BJ. Role of microRNAs in plant drought tolerance. Plant Biotechnol. J. 2015;13(3):293–305. doi: 10.1111/pbi.12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Z, Liu X, Guo X, Wang XJ, Zhang X. Arabidopsis AGO3 predominantly recruits 24-nt small RNAs to regulate epigenetic silencing. Nat. Plants. 2016;2:16049. doi: 10.1038/nplants.2016.49. [DOI] [PubMed] [Google Scholar]
- 44.Kapoor M, Arora R, Lama T, Nijhawan A, Khurana JP, Tyagi AK, et al. Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA Polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genomics. 2008;9:451. doi: 10.1186/1471-2164-9-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henderson IR, Zhang X, Lu C, Johnson L, Meyers BC, et al. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat. Genet. 2006;38:721–725. doi: 10.1038/ng1804. [DOI] [PubMed] [Google Scholar]
- 46.Schmitz RJ, Hong L, Fitzpatrick KE, Amasino RM. DICER-LIKE 1 and DICER-LIKE 3 redundantly act to promote flowering via repression of FLOWERING LOCUS C in Arabidopsis thaliana. Genetics. 2007;176(2):1359–1362. doi: 10.1534/genetics.107.070649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kasschau KD, Fahlgren N, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, et al. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 2007;5(3):e57. doi: 10.1371/journal.pbio.0050057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Navarro L, Dunoyer P, Jay F, Arnold B, Dharmasiri N, Estelle M, et al. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science. 2006;312:436–439. doi: 10.1126/science.1126088. [DOI] [PubMed] [Google Scholar]
- 49.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:138–141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finn RD, Clements J, Eddy SR. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marchler-Bauer A, Bryant SH. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–W331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, Finn RD. HMMER web server: 2018 update. Nucleic Acids Res. 2018;46(W1):W200–W204. doi: 10.1093/nar/gky448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cao JY, Xu YP, Li W, Li SS, Rahman H, Cai XZ. Genome-wide identification of Dicer-Like, Argonaute, and RNA-Dependent RNA polymerase gene families in Brassica species and functional analyses of their Arabidopsis homologs in resistance to Sclerotinia sclerotiorum. Front. Plant Sci. 2016;7:1614. doi: 10.3389/fpls.2016.01614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 55.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettiganet PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 56.Nicholas, K.B. & Nicholas, H.B.J. Genedoc, a Tool for Editing and Annotating Multiple Sequence Alignments. Distributed by the author (1997).
- 57.Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kakar KU, Nawaz Z, Cui Z, Cao P, Jin J, Shu Q, et al. Evolutionary and expression analysis of CAMTA gene family in Nicotiana tabacum yielded insights into their origin, expansion and stress responses. Sci. Rep. 2018;8:10322. doi: 10.1038/s41598-018-28148-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo AY, Zhu QH, Chen X, Luo JC. GSDS: A gene structure display server. Hereditas. 2007;29:1023–1026. [PubMed] [Google Scholar]
- 61.Bailey TL, Johnson J, Grant CE, Noble WS. The MEME suite. Nucleic Acids Res. 2015;43(W1):W39–W49. doi: 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dai X, Zhao PX. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011;39:W155–W159. doi: 10.1093/nar/gkr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lescot M, Dehais P, Thijs G, Marchal K, Moreau Y, Peer YV, et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002;30(1):325–327. doi: 10.1093/nar/30.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Voorrips RE. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002;93(1):77–78. doi: 10.1093/jhered/93.1.77. [DOI] [PubMed] [Google Scholar]
- 65.Suyama M, Torrents D, Bork P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wei C, Yang H, Wang S, et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. 2018;115(18):E4151–E4158. doi: 10.1073/pnas.1719622115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bordoloi KS, Dihingia P, Krishnatreya DB, Agarwala N. Genome-wide identification, characterization and expression analysis of the expansin gene family under drought stress in tea (Camellia sinensis L.) Plant Sci. Today. 2021;8(1):32–44. doi: 10.14719/pst.2021.8.1.923. [DOI] [Google Scholar]
- 68.Sievers F, Higgins DG. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014;1079:105–116. doi: 10.1007/978-1-62703-646-7_6. [DOI] [PubMed] [Google Scholar]
- 69.Metsalu T, Vilo J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015;43(W1):W566–W570. doi: 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ma QP, Hao S, Chen X, Li HX. Validation of reliability for reference genes under various abiotic stresses in tea plant. Russ. J. Plant Physiol. 2016;63:423–432. doi: 10.1134/S1021443716030080. [DOI] [Google Scholar]
- 72.Chowrasia S, Panda AK, Rawal HC, Kaur H, Mondal TK. Identification of jumonjiC domain containing gene family among the Oryza species and their expression analysis in FL478, a salt tolerant rice genotype. Plant Physiol. Biochem. 2018;130:43–53. doi: 10.1016/j.plaphy.2018.06.031. [DOI] [PubMed] [Google Scholar]
- 73.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nature. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.