

Abstract
The family GH77 contains 4-α-glucanotransferase acting on α-1,4-glucans, known as amylomaltase in prokaryotes and disproportionating enzyme in plants. A group of bacterial GH77 members, represented by amylomaltases from Escherichia coli and Corynebacterium glutamicum, possesses an N-terminal extension that forms a distinct immunoglobulin-like fold domain, of which no function has been identified. Here, in silico analysis of 100 selected sequences of N-terminal domain homologues disclosed several well-conserved residues, among which Tyr108 (E. coli amylomaltase numbering) may be involved in α-glucan binding. These N-terminal domains, therefore, may represent a new type of starch-binding domain and define a new CBM family. This hypothesis is supported by docking of maltooligosaccharides to the N-terminal domain in amylomaltases, representing the four clusters of the phylogenetic tree.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13205-021-02787-8.
Keywords: Amylomaltase, Family GH77, N-terminal domain, Starch-binding domain, CBM families, Conserved sequence regions, Evolutionary relatedness
Introduction
In the sequence-based classification system of carbohydrate-active enzymes, the CAZy database (https://www.cazy.org/; Lombard et al. 2014), glycoside hydrolase family 77 (GH77) is a monospecific family of 4-α-glucanotransferases (EC 2.4.1.25) (Janecek and Gabrisko 2016). In Archaea and Bacteria, these enzymes are also known as amylomaltases (Terada et al. 1999; Kaper et al. 2005; Godany et al. 2008; Mehboob et al. 2016, 2020), while they are named disproportionating enzymes (DPEs; or just D-enzymes) in plants (Takaha et al. 1993; Wattebled et al. 2003). Currently, family GH77 contains more than 12,000 sequences (Lombard et al. 2014), the vast majority (> 11,800) are from bacteria; while the remaining members are distributed between Archaea (~ 70) and Eucarya, i.e. plants and green algae (~ 90). In total, so far 20 enzymes of GH77 are characterised experimentally and crystal structures are solved of 6 bacterial amylomaltases and 2 plant DPEs (Lombard et al. 2014).
At the higher hierarchical level, GH77 together with the main α-amylase family GH13 and family GH70 of circularly permuted glucansucrases constitutes the so-called α-amylase clan GH-H (Kuriki and Imanaka 1999; MacGregor et al. 2001; van der Maarel et al. 2002; Janecek et al. 2014; Lombard et al. 2014; Janecek and Gabrisko 2016; Gangoiti et al. 2018). The GH77 members, such as the GH13 and GH70 enzymes, employ a retaining reaction mechanism and adopt the α-amylase-type (β/α)8-barrel (TIM-barrel) catalytic domain (Kuriki and Imanaka 1999; Janecek et al. 2014). Similar to TIM-barrels from the α-amylase family GH13, family GH77 has a protruding domain B inserted between strand β3 and helix α3 (Matsuura et al. 1984; Vujicic-Zagar et al. 2010). However, the GH77 TIM-barrel contains additional insertions following other β-strands (MacGregor et al. 2001; Janecek and Gabrisko 2016). Overall, the tertiary structure of GH77 is composed of two main domains, the catalytic TIM-barrel and domain B, the latter comprising three auxiliary subdomains known as B1, B2 and B3 (Przylas et al. 2000b). While subdomain B1 corresponds to domain B in GH13 and B3 may play the role of domain C in GH13, the subdomain B2 is unique to GH77 and has no counterpart identified in GH13 and GH70 (Matsuura et al. 1984; MacGregor et al. 2001; Vujicic-Zagar et al. 2010; Janecek and Gabrisko 2016). Notably, the lack of an antiparallel β-sandwich domain C succeeding the catalytic TIM-barrel is the main feature distinguishing GH77 4-α-glucanotransferases from the other clan GH-H members in GH13 and GH70 (Przylas et al. 2000b). The primary structures of 4-α-glucanotransferases of GH77 have 4–7 conserved sequence regions (CSRs), CSR-I–CSR-VII, in common with GH13 and GH70 (Janecek 2002; Janecek and Gabrisko 2016). The catalytic machinery of GH77 enzymes consists of a triad of aspartic acid, glutamic acid and aspartic acid localised at the C-terminal ends of strands β4 (Asp; catalytic nucleophile), β5 (Glu; proton donor) and β7 (Asp; transition-state stabiliser), respectively. These were first observed for GH77 as Asp293, Glu340 and Asp395 in the crystal structure of amylomaltase from Thermus aquaticus (Przylas et al. 2000a, b). Currently, seven additional structures are available in GH77, namely of amylomaltases from Aquifex aeolicus [Protein Data Bank (PDB): 1TZ7], Thermus thermophilus (Barends et al. 2007), Thermus brockianus (Jung et al. 2011), Escherichia coli (Weiss et al. 2015) and Corynebacterium glutamicum (Joo et al. 2016) and two DPEs from Arabidopsis thaliana (O’Neill et al. 2015) and potato (Imamura et al. 2020).
In general, enzymes of GH77 catalyse transfer of a glucan chain from one α-1,4-glucan to extend another α-1,4-glucan or produce a cyclic α-1,4-glucan from a single linear α-1,4-glucan chain (Takaha et al. 1993; Terada et al. 1999; Wattebled et al. 2003; Kaper et al. 2005; Godany et al. 2008; Mehboob et al. 2016). The degree of polymerization (DP) of the latter products, often referred to as cycloamylose, is 17 or higher; thus these cyclic α-1,4-glucans are much larger than the α-, β- and γ-cyclodextrins of DP 6, 7 and 8, respectively, produced by cyclodextrin glucanotransferases of GH13 (Fujii et al. 2005a, b, 2007; Srisimarat et al. 2012; van der Maarel and Leemhuis 2013; Roth et al. 2017; Tumhom et al. 2017).
In addition to catalytic domains of carbohydrate-active enzymes, CAZy classifies carbohydrate-binding modules (CBMs) (Boraston et al. 2004). CBMs are functionally and structurally independent domains without enzymatic activity, which by binding carbohydrates can support the function of catalytic domains. In amylolytic enzymes, these modules are known as starch-binding domains (SBDs) or less frequently as glycogen-binding domains, and in principle, they contribute by binding α-glucans, for example on starch granules (Janecek et al. 2011). Nowadays, CAZy categorises 88 CBM families (Lombard et al. 2014), among which 15 are considered as SBDs; CBM20, 21, 25, 26, 34, 41, 45, 48, 53, 58, 68, 69, 74, 82, and 83 (Janecek et al. 2019). Members belonging to these individual SBD CBM families, with the exception of family CBM74 (Valk et al. 2016), are approximately 100 amino acid residues long (Janecek et al. 2011, 2019; Carvalho et al. 2015; Armenta et al. 2017).
As it was demonstrated previously, amylomaltases from borreliae may be a rather unique group within family GH77 exhibiting unusual amino acid residues at functionally important positions in individual CSRs (Godany et al. 2008; Kuchtova and Janecek 2015). Otherwise, most of the typical GH77 4-α-glucanotransferases are covered by the prokaryotic Thermus-like amylomaltases (Przylas et al. 2000a, b; Barends et al. 2007; Jung et al. 2011) and the eukaryotic DPE1s (O’Neill et al. 2015; Imamura et al. 2020). These enzymes are usually ~ 500 residues long, however, part of the GH77 family is formed by longer prokaryotic and eukaryotic sequences (Kuchtova and Janecek 2015). Thus, in Eucarya, the DPE2s possess a ~ 140 residues long insertion between the catalytic nucleophile and proton donor and have two SBDs in tandem of family CBM20 preceding the TIM-barrel domain (Lloyd et al. 2004; Steichen et al. 2008; Kuchtova and Janecek 2015; Janecek et al. 2019). In addition, in Bacteria, a group of amylomaltases differs from the Thermus-like GH77 (Przylas et al. 2000a, b; Barends et al. 2007; Jung et al. 2011) by a ~ 190 residues long N-terminal extension (Kuchtova and Janecek 2015; Janecek et al. 2019). Currently, this group is best represented by amylomaltases from Escherichia coli (Pugsley and Dubreuil 1988; Weiss et al. 2015) and Corynebacterium glutamicum (Srisimarat et al. 2011; Joo et al. 2016) both with available crystal structures where part of the N-terminal extension adopts an immunoglobulin-like fold similar to those seen in structure-determined SBD families (Janecek et al. 2019). Although especially the C. glutamicum amylomaltase has been extensively studied from the structure/function point of view (Srisimarat et al. 2012; Rachadech et al. 2015; Nimpiboon et al. 2016a, b; Tumhom et al. 2017, 2018), no function has been assigned to the N-terminal domain from both E. coli and C. glutamicum GH77 enzymes (Weiss et al. 2015; Joo et al. 2016; Janecek et al. 2019). Recently, the 4-α-glucanotransferase from Bifidobacterium longum was characterised (Jeong et al. 2020), which according to its sequence also belongs to the group having the N-terminal extension.
The need to expand fundamental knowledge on ancillary domains in family GH77 and clan GH-H motivates the present bioinformatics investigation of the unusual N-terminal domains, that are candidates of a novel SBD CBM family and seen in crystal structures of GH77 amylomaltases from E. coli and C. glutamicum. The approach involves search for and retrieval of a relevant group of bacterial amylomaltases containing N-terminal extensions homologous to those found in the E. coli and C. glutamicum GH77 enzymes. The comparison includes docking trials with a series of α-1,4-oligoglucosides, to the two determined and the two modelled three-dimensional structures of the four phylogenetically distinguished new putative N-terminal SBDs in GH77.
Materials and methods
Sequence collection
There is a huge number of GH77 members in the CAZy database (currently > 12,000 sequences; Lombard et al. 2014; https://www.cazy.org/), the two GH77 amylomaltases with solved crystal structures from Escherichia coli (Pugsley and Dubreuil 1988; Weiss et al. 2015) and Corynebacterium glutamicum (Srisimarat et al. 2011; Joo et al. 2016), possessing the mutually homologous N-terminal extension, were chosen as the main representatives in the present study. All GH77 sequences exhibiting resemblance with the N-terminal module of these two enzymes were collected from CAZy, because protein BLAST searches (Altschul et al. 1990; https://blast.ncbi.nlm.nih.gov/) using these E. coli and C. glutamicum N-terminal domains as queries failed to provide meaningful results. A preliminary set of 682 sequences was obtained by browsing family GH77 data from CAZy, the main criterion being a sequence length of 650–750 residues. The sequences were aligned by the programme Clustal-Omega available at the European Bioinformatics Institute’s server (Sievers et al. 2011; https://www.ebi.ac.uk/Tools/msa/clustalo/) and following an initial alignment, 194 sequences were eliminated for one or more of three reasons: (i) they did not contain a domain homologous to the N-terminal domain of amylomaltases from E. coli and C. glutamicum; (ii) they did not contain the complete catalytic machinery; and (iii) they significantly disrupted the multiple alignment. This resulted in 488 sequences of bacterial GH77 amylomaltases with a convincing N-terminal domain homologous to those found in E. coli and C. glutamicum amylomaltases. One hundred sequences (including E. coli and C. glutamicum amylomaltases as well as the third experimentally characterised amylomaltase from B. longum having the N-terminal extension homologous to that present in the two former enzymes) were finally selected for in-depth analysis (Table S1) ensuring the widest possible taxonomical diversity and a minimum length of ~ 70 residues of the predicted N-terminal domain.
All amino acid sequences were retrieved from UniProt (UniProt Consortium 2017; https://www.uniprot.org/) and/or GenBank (Benson et al. 2018; https://www.ncbi.nlm.nih.gov/genbank/) databases. The sequence boundaries for the N-terminal domains in amylomaltases from E. coli and C. glutamicum were retrieved from their tertiary structures (Weiss et al. 2015; Joo et al. 2016), and those of other full-length GH77 amylomaltase sequences were defined based on sequence alignment with the E. coli and C. glutamicum enzymes.
Sequence comparison and evolutionary analysis
The alignment of the selected 100 sequences of the N-terminal module (Table S1) was performed using the programme Clustal-Omega (Sievers et al. 2011; https://www.ebi.ac.uk/Tools/msa/clustalo/). Only a subtle manual tuning was necessary to maximise similarities considering the best-conserved residues and those potentially involved in carbohydrate binding based on inspection of the two three-dimensional structures (Weiss et al. 2015; Joo et al. 2016).
The evolutionary tree was calculated from the final sequence alignment of all 100 N-terminal domains including all gaps as a maximum-likelihood tree using the WAG substitution model (Whelan and Goldman 2001) and the bootstrapping procedure with 500 bootstrap trials (Felsenstein 1985) implemented in the MEGA-X package (Kumar et al. 2018). The tree was displayed with the programme iTOL (Letunic and Bork 2007; https://itol.embl.de/).
Sequence logos of five CSRs defined within the alignment were created using the WebLogo3 online server (Crooks et al. 2004; http://weblogo.threeplusone.com/). Four sequence logos were calculated—one for each of the four clusters identified in the evolutionary tree.
Comparison of tertiary structures and molecular docking
The coordinates of the GH77 template amylomaltases from E. coli (Weiss et al. 2015) and C. glutamicum (Joo et al. 2016) were retrieved from the Protein Data Bank (PDB; Berman et al. 2000; https://www.rcsb.org/) under the PDB codes 4S3Q (4S3R) and 5B68, respectively. The structural data were modified to contain only the N-terminal domain of the two enzymes by cutting out the remaining parts of their structures from the original PDB files based on the literature information (Weiss et al. 2015; Joo et al. 2016).
Reflecting the preliminary evolutionary distribution of all 100 GH77 amylomaltases into four groups (Table S1), structures of the N-terminal domain of two other amylomaltases from Kushneria marisflavi (Yun and Bae 2018; UniProt: A0A240US28) and Pelotomaculum thermopropionicum (Kosaka et al. 2008; UniProt: A5D1W1) were modelled using the fold recognition Phyre2 server (Kelley and Sternberg 2009; http://www.sbg.bio.ic.ac.uk/~phyre2/). The K. marisflavi and P. thermopropionicum amylomaltases represent two clusters additional to the two ones containing the experimentally characterised amylomaltases from E. coli and C. glutamicum.
All molecular docking trials were performed by the program CB-Dock (Liu et al. 2020; http://clab.labshare.cn/cb-dock/) that utilises the AutoDock Vina (Trott and Olson 2010; http://vina.scripps.edu/) with all parameters used as default. The structures of the N-terminal domains from E. coli (Weiss et al. 2015) and C. glutamicum (Joo et al. 2016) amylomaltases as well as the structural models from homologous domains in K. marisflavi (Yun and Bae 2018) and P. thermopropionicum (Kosaka et al. 2008) amylomaltases were docked with maltose (G2), maltotriose (G3), maltotetraose (G4) and β-cyclodextrin (β-CD). Three-dimensional structures of the ligands were retrieved from the PubChem database (Kim et al. 2019; https://pubchem.ncbi.nlm.nih.gov/) and converted into PDB coordinates by the SMILES programme (Weininger et al. 1988; https://cactus.nci.nih.gov/translate/). The resulting complexes of individual structures with bound maltooligosaccharides were displayed using the UCSF Chimera programme (Pettersen et al. 2004).
Results and discussion
Family GH77 has attracted a special interest not only as a member of the α-amylase clan GH-H (MacGregor et al. 2001; Lombard et al. 2014; Janecek and Gabrisko 2016), but also due to the fact that GH77 amylomaltases from borreliae contain unique substitutions in their amino acid sequence, especially the presence of a lysine two residues before the catalytic nucleophile instead of an arginine otherwise invariant throughout clan GH-H (Godany et al. 2008; Kuchtova and Janecek 2015). This difference was first observed in the amylomaltase from Borrelia burgdorferi (Machovic and Janecek 2003). The present study, however, focuses on a different feature of quite a large group of bacterial GH77 amylomaltases, namely a unique N-terminal extension comprising a separate domain (Fig. 1) that adopts an immunoglobulin-like fold, which is also characteristic for SBD CBM families (Janecek et al. 2019). The first in silico analysis predicting this domain in family GH77 (Kuchtova and Janecek 2015) was later confirmed by the crystal structures of amylomaltases from E. coli (Weiss et al. 2015) and C. glutamicum (Joo et al. 2016), having this N-terminal extension as opposed to the typical Thermus-like amylomaltases (Fig. 1). Notably, the N-terminal extension has two separated parts, the so-called N-terminal domain adopting an immunoglobulin-like fold and considered a potential novel SBD, which immediately precedes the catalytic TIM-barrel, and also a smaller domain N1 situated at the very N-terminus of the protein (Fig. 1).
Fig. 1.

Domain arrangement of selected amylomaltases of family GH77. The enzymes from E. coli (a) and C. glutamicum (b) represent the group of bacterial amylomaltases with N-terminal extensions, a part of which, indicated as the “N-terminal domain”, constitutes a presumed CBM, i.e. a novel SBD. Individual domains or subdomains are coloured as follows: domain N1—grey; N-terminal domain (the potential novel SBD CBM family)—red; catalytic TIM-barrel domain A—blue; subdomains B1, B2 and B3—green, cyan and yellow, respectively. The residues of the catalytic triad of family GH77 (and hence of the entire clan GH-H)—aspartic acid, glutamic acid and aspartic acid, positioned on strands β4, β5 and β7, respectively, of the TIM-barrel, are shown as red diamonds. For comparison, the domain arrangement of a typical bacterial Thermus-like GH77 amylomaltase from Thermus aquaticus (c) is also shown
Sequence analysis and evolutionary relationships
The amino acid sequence alignment of the N-terminal domains of the 100 collected GH77 amylomaltases (Fig. 2), despite the obvious overall homology, also shows two larger distinguishable groups (Table S1): (i) sequences No. 1–49 represented by the amylomaltase from E. coli (Weiss et al. 2015); and (ii) sequences No. 50–100 represented by the C. glutamicum enzyme (Joo et al. 2016). These groups clearly differ by the length of their N-terminal domain, being 70–80 residues for the E. coli-like and ~ 100 residues for the C. glutamicum-like group (Fig. 2).
Fig. 2.

Amino acid sequence alignment of the N-terminal domains of GH77 amylomaltases. The sequence order from the top corresponds to their appearance in the evolutionary tree (Fig. 3). Four proteins, representing four potential clusters in the tree (distinguished by different colours), are marked by an asterisk, i.e. amylomaltases from Kushneria marisflavi (blue), Escherichia coli (red), Pelotomaculum thermopropionicum (magenta) and Corynebacterium glutamicum (green). Each protein is labelled by the UniProt accession number and the name of the organism. The five suggested conserved sequence regions (CSR-1–CSR-5) are indicated above the alignment. The best-conserved aromatic position corresponding to Tyr108 in amylomaltase from E. coli and the two invariant residues (the glycine succeeding the Tyr108 and the proline at the end of the domain) are signified by dollar and hash symbols, respectively, below the alignment. The colour code for the selected residues: W, yellow; F, Y—blue; V, L, I—green; D, E—red; R, K—cyan; H—brown; C—magenta; G, P—black
The evolutionary tree (Fig. 3) shows that the selected 100 sequences (Fig. 2), in fact, segregate into four clusters: (i) sequences No. 1–17 (blue group in the tree); (ii) 18–49 (red); (iii) 50–64 (magenta); and (iv) 65–100 (green). In addition to the two best characterised amylomaltases, i.e. from E. coli (No. 49; Weiss et al. 2015) and C. glutamicum (No. 80; Joo et al. 2016)—representing sequences No. 18–49 (red group; covering mostly Gammaproteobacteria) and No. 65–100 (green group; mostly Actinobacteria), respectively, the hypothetical amylomaltases from K. marisflavi (No. 8; Jun and Bae 2018; UniProt: A0A240US28) and P. thermopropionicum (No. 64; Kosaka et al. 2008; UniProt: A5D1W1) were chosen from the two remaining groups of sequences No. 1–17 (blue; covering mostly Proteobacteria) and No. 50–64 (magenta; mostly Firmicutes and Alphaproteobacteria). It is of note, that the third experimentally characterised amylomaltase from B. longum having the N-terminal domain (No. 69; Jeong et al. 2020) is positioned in the cluster represented by C. glutamicum amylomaltase, thus reflecting its actinobacterial origin (Fig. 3). In conclusion, each of the two large groups recognised in the multiple alignment (Fig. 2) is formed by two evolutionarily independent clusters. This division unambiguously observed in the evolutionary tree (Fig. 3) is kept throughout the present study (Table S1).
Fig. 3.

Evolutionary tree of the N-terminal domains of GH77 amylomaltases. The tree displays the relatedness of 100 unique non-redundant GH77 sequences (Table S1) containing the N-terminal domain corresponding to an immunoglobulin-like fold observed in crystal structures of E. coli (Pugsley and Dubreuil 1988; Weiss et al. 2015) and C. glutamicum amylomaltases (Srisimarat et al. 2011; Joo et al. 2016). Each protein is labelled by the UniProt accession number and the name of the organism. Four proteins, representing four clusters illustrated on the tree and each distinguished by different colours, are marked by an asterisk, i.e. amylomaltases from Kushneria marisflavi (blue), Escherichia coli (red), Pelotomaculum thermopropionicum (magenta) and Corynebacterium glutamicum (green). The tree is based on the alignment of the N-terminal domains (Fig. 2)
Aromatic residues, phenylalanine, tyrosine and/or tryptophan are usually responsible for binding α-glucans in SBDs classified in various CBM families (Janecek et al. 2011, 2019). Therefore, a thorough inspection of the 100 aligned N-terminal domains was performed to identify the best-conserved residues with special attention to conserved aromatic residues. Indeed, aromatic residues are conserved at several positions in the four identified clusters, e.g. Tyr76 and Trp78 in the group represented by the amylomaltase from E. coli (Fig. 2 shows these as Tyr25 and Trp27 in the E. coli sequence; No. 49) and Phe133 in the group represented by the C. glutamicum amylomaltase (Fig. 2 shows this as Phe63 in the C. glutamicum sequence; No. 80). These three positions, however, are not conserved in all four groups. Overall, the only widely, albeit not invariantly conserved aromatic position corresponds to Tyr108 of the E. coli amylomaltase (Fig. 2, position Tyr57 in the E. coli sequence; No. 49). Remarkably, this tyrosine is invariant in three of the four groups, i.e. those containing E. coli, K. marisflavi and P. thermopropionicum amylomaltases. In the fourth group containing the C. glutamicum amylomaltase, it is substituted in 18 of 36 cases by tryptophan (Trp143) (Fig. 2, position Trp73 in C. glutamicum; No. 80), although, e.g. the third experimentally characterised of this set B. longum amylomaltase contains a tyrosine in that position (Fig. 2, position Tyr65 in B. longum; No. 69). The Tyr108 in E. coli amylomaltase and corresponding aromatic residues in all the sequences support the hypothesis that the N-terminal domain is involved in α-glucan binding and defines a new SBD.
It is worth mentioning, however, that this Tyr108 can hardly be related to two functional tyrosines (Tyr54 and Tyr101) from the catalytic domain identified in the Thermus aquaticus amylomaltase contributing to the second α-glucan binding site (Fujii et al. 2007). Moreover, both Tyr54 and Tyr101 do not represent invariantly conserved residues in the family GH77, as demonstrated by a previous in silico study comparing more than 400 sequences of GH77 amylomaltases (Kuchtova and Janecek 2015).
Regarding fully invariant residues, N-terminal domains of bacterial GH77 amylomaltases only have two, Gly107 and Pro128 (E. coli amylomaltase numbering) of which the former precedes the potential α-glucan binding Tyr108, while the latter is at the C-terminal residue of the domain (Fig. 2; positions Gly56 and Pro77, respectively, in E. coli; No. 49). Glycine and proline residues are both significant on the so-called consensus sequence of the first known SBD (Svensson et al. 1989; Janecek and Sevcik 1999), belonging to the current family CBM20 (Janecek et al. 2011, 2019; Lombard et al. 2014).
In an effort to focus attention on segments potentially bearing residues involved in α-glucan binding, the five best-conserved short stretches were proposed to constitute conserved sequence regions (CSRs) of the N-terminal domains in bacterial GH77 amylomaltases as follows (Fig. 2; E. coli amylomaltase sequence and numbering): (i) CSR-1—55_PNVMVYTSG; (ii) CSR-2—66_MPMVVE; (iii) CSR-3—80_LTTE; (iv) CSR-4—105_PEGYHTLT; and (v) CSR-5—121_HCRVIVAP. Identification of CSRs has become typical for catalytic domains to emphasise key residues involved in activity and/or substrate specificity. This is particularly important for large and polyspecific GH families, such as in the individual α-amylase families GH13, GH57, GH119 and GH126 (Janecek 2002; Blesak and Janecek 2012, 2013; Janecek and Kuchtova 2012; Janecek et al. 2014; Janecek and Gabrisko 2016; Kerenyiova and Janecek 2020a, b). It also makes sense, however, to establish CSRs for putative SBDs. Among all known SBDs, currently classified in 15 CBM families in CAZy (Lombard et al. 2014), residues from the best-conserved regions usually belong to one or in some cases to two binding sites (Janecek et al. 2011, 2019).
In agreement with four clusters being found in the evolutionary tree of N-terminal domains from bacterial GH77 enzymes (Figs. 2 and 3), sequence logos covering 35 positions for the five proposed CSRs were created for each cluster (Fig. 4). All logos contain the invariant Gly107 and Pro128, at CSR-4 position 22 and CSR-5 position 35, respectively (E. coli amylomaltase numbering), plus the potentially α-glucan binding Tyr108 at CSR-4 position 23. The tripeptide GYH in CSR-4 that is invariant in two of the four evolutionary clusters, containing E. coli and P. thermopropionicum amylomaltases, respectively (Fig. 4b, c), is one of the best-conserved stretches in the N-terminal domain. The other highly conserved region is found at the last five residues, L/I/V-A/I-V/I-A/T-P, at CSR-5 positions 31–35. At the start of the logo, conserved proline residues (CSR-1; positions 1–2) are seen except in the first position in the logo of the group of P. thermopropionicum amylomaltase (Fig. 4c). Concerning additional positions of interest, Glu83 (E. coli amylomaltase numbering; CSR-3 position 19) is almost invariantly conserved and deserves attention.
Fig. 4.

Sequence logos of N-terminal domains from individual clusters of family GH77 bacterial amylomaltases. The four groups (for details, see Table S1) are represented by a Kushneria marisflavi (Proteobacteria; ~ 70 residues long; 17 sequences); b Escherichia coli (Gammaproteobacteria; ~ 80 residues long; 32 sequences); c Pelotomaculum thermopropionicum (Firmicutes and Alphaproteobacteria ~ 100 residues long; 15 sequences); and d Corynebacterium glutamicum (Actinobacteria; ~ 90 residues long; 36 sequences). CSR-1, residues 1–9; CSR-2, residues 10–15; CSR-3, residues 16–19; CSR-4, residues 20–27; CSR-5, residues 28–35. The aromatic residue, marked by the red asterisk, corresponding to Tyr108 and Trp143 in E. coli and C. glutamicum amylomaltases, respectively, is potentially involved in stacking interactions with α-glucans. All other residues involved in hydrogen bonding contacts with docked α-glucans (G2, G3, G4 and β-CD; for details, see Table 1) are indicated by black asterisks
Importantly, the sequence logos identify positions suitable to discriminate the four clusters by showing positions illustrating their individual uniqueness. This is a key attribute of CSRs, hence of sequence logos, which are well known and widely utilised in the α-amylase families mentioned above to distinguish subfamilies and/or enzyme specificities (Janecek 2002; Blesak and Janecek 2012, 2013; Janecek and Kuchtova 2012; Janecek et al. 2014; Janecek and Gabrisko 2016; Kerenyiova and Janecek 2020a, b). Notably, it was described for a single SBD CBM family (Janecek et al. 2019), CBM41 composed of two genuine groups mutually distinguished by a characteristic sequence pattern of three essential aromatic residues, “W‐W‐∼10aa‐W” and “W‐W‐∼30aa‐W”, implying that the position of the third tryptophan in the pattern is not shared by the two groups (Janecek et al. 2017). Similarly, only the C. glutamicum amylomaltase group has an invariantly conserved histidine, His90 in CSR-2 position 15 (Fig. 4d). Another example of interest is the well-conserved cysteine in the E. coli amylomaltase group (Cys122; Fig. 4b; CSR-5 position 29).
Tertiary structure comparison and molecular docking
The amylomaltases having the N-terminal domain are not the only family GH77 members that possess extra sequence compared to the canonical Thermus-like amylomaltases of just the catalytic TIM-barrel domain and a few inserted subdomains (Fig. 1). In addition to amylomaltases from borreliae that may have single unique substitutions even in functionally important positions, but still within the otherwise conserved basic domain arrangement (Machovic and Janecek 2003; Godany et al. 2008; Kuchtova and Janecek 2015; Janecek and Gabrisko 2016), in the Eucarya, the DPE2 version exists as a typical GH77 amylomaltase with a ~ 140 residues insertion between the catalytic nucleophile and proton donor in the TIM-barrel which, moreover, is preceded by two SBDs of CBM20 (Lloyd et al. 2004; Steichen et al. 2008; Kuchtova and Janecek 2015; Janecek et al. 2019). Unfortunately, no three-dimensional structure is available for DPE2 and the structural fold of the ~ 140-residue insertion is not known, neither is its potential function (Steichen et al. 2008), nor if it is essential for activity (Ruzanski et al. 2013). Remarkably, however, the function of DPE2 with the two CBM20s in Arabidopsis thaliana was effectively retained by replacement with amylomaltase from E. coli (Ruzanski et al. 2013). In that light, it may be relevant that the N-terminal domain in the E. coli enzyme (Fig. 1) has an immunoglobulin-like fold typical of SBDs (Janecek et al. 2019), and can be speculated to act as an SBD.
The two crystal structures from E. coli (Weiss et al. 2015) and C. glutamicum (Joo et al. 2016) describe amylomaltases with the investigated N-terminal domain (Fig. 1). In neither case was the structure of the N-terminal domain obtained in complex with an α-glucan. Since the thorough phylogenetic analysis revealed four clusters (Fig. 3), models for structural comparison were made of N-terminal domains of two hypothetical amylomaltases of K. marisflavi and P. thermopropionicum from the other two clusters. In agreement with the closer evolutionary relatedness observed on the one hand for the E. coli and K. marisflavi groups and on the other for the C. glutamicum and P. thermopropionicum groups (Fig. 3), N-terminal domains of K. marisflavi and P. thermopropionicum were modelled using those of amylomaltases from E. coli (PDB code: 4S3R; Weiss et al. 2015) and C. glutamicum (PDB: 5B68; Joo et al. 2016), respectively, as templates. Notably, none of the established SBD CBM families (Janecek et al. 2019) with available three-dimensional structures were identified as a suitable template.
To get an idea of how an α-glucan binds if the N-terminal domain acted as an SBD, maltose (G2), maltotriose (G3), maltotetraose (G4) and β-cyclodextrin (β-CD) were docked. Since the SBDs from various CBM families have already been demonstrated to retain their binding abilities even if being separated from their catalytic domains, i.e. they can preserve the binding also in an isolated form (for a review, see Janecek et al. 2019), the docking was in each case performed with extracted N-terminal domains. Although it may not reflect the situation in real amylomaltases completely, for the purpose of the present study it has been considered sufficient.
Not to bias the results, docking by CB-Dock does not require to focus the ligand on a target place in the protein (Liu et al. 2020), i.e. on the presumed aromatic binding residue Tyr108 in amylomaltase from E. coli and its counterparts, Trp143, Tyr121 and Tyr146 from C. glutamicum, K. marisflavi and P. thermopropionicum, respectively. Importantly, using all four maltooligosaccharides, there should be a single α-glucan-binding site in every studied N-terminal domain corresponding to each other (cf. Figures 5and 6). Nevertheless, it seems that at least Tyr121 and Tyr146 from K. marisflavi and P. thermopropionicum amylomaltases have not been recognised as making direct hydrogen bond contacts with all four ligands—Tyr121 may be involved with G2, G4 and β-CD, whereas Tyr 146 appears to be involved just with G2 (Table 1). Note, Table 1 summarises only the residues involved in hydrogen bond contacts. Importantly, the aromatic residues—Tyr108, Trp143, Tyr121 and Tyr146—may interact with respective ligands also via stacking interactions (cf. Figs. 5 and 6). Results of all individual docking trials are summarised in Table 1. Notably, these complexes indicated an appropriate binding energy ranging from the best of − 5.9 kJ/mol for maltotriose bound to the N-terminal domain from K. marisflavi amylomaltase to − 3.7 kJ/mol for the maltose complex of the P. thermopropionicum N-terminal domain (Table 1). Interestingly, in addition to the aromatic position represented by Tyr108 (E. coli amylomaltase numbering) that might interact by stacking onto glucose moieties, a few other residues were involved in hydrogen bond formation with docked α-glucans (Table 1), corresponding, e.g. to the positions of Glu83 (CSR-3), Thr110 (CSR-4), Thr112 (CSR-4) and Arg123 (CSR-5) in E. coli amylomaltase (Fig. 4).
Fig. 5.
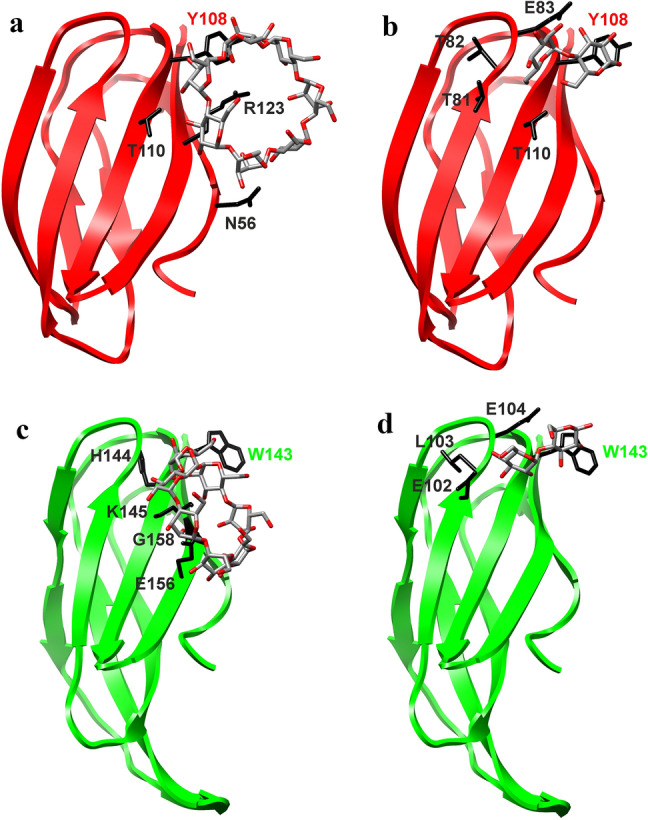
Visualisation of molecular docking experiments with real structures. The N-terminal domain of family GH77 amylomaltases from a, b Escherichia coli (PDB: 4S3Q; Weiss et al. 2015) and c, d Corynebacterium glutamicum (PDB: 5B68; Joo et al. 2016) with docked β-cyclodextrin (a, c) and maltose (b, d). Side-chains of residues potentially involved in carbohydrate binding are displayed (in black) and labelled accordingly (for details, see Table 1). The best-conserved aromatic residue, i.e. Tyr108 of the E. coli amylomaltase and Trp143 of the C. glutamicum counterpart (cf. Figure 2) and possibly involved in stacking interactions with glucose moieties, is specifically colour-highlighted
Fig. 6.
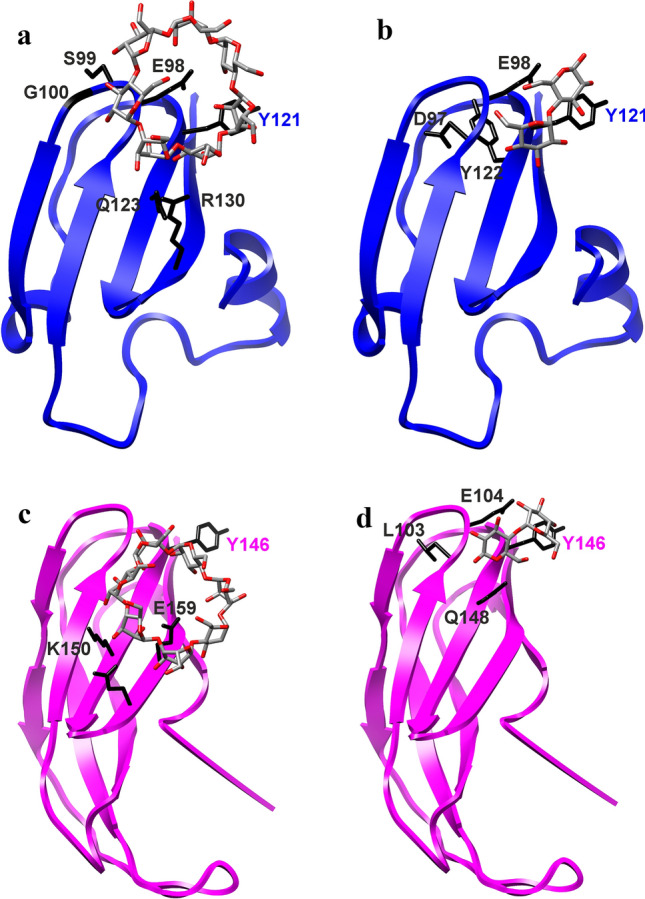
Visualisation of molecular docking experiments with modelled structures. The models of the N-terminal domain of family GH77 amylomaltases from: a, b Kushneria marisflavi (UniProt: A0A24OU528; template: 4S3R; Weiss et al. 2015) and c, d Pelotomaculum thermopropionicum (UniProt: A5D1W1; template: 5B68; Joo et al. 2016) with docked β-cyclodextrin (a, c) and maltose (b, d). Side-chains of residues potentially involved in carbohydrate binding are displayed (in black) and labelled accordingly (for details, see Table 1). The best-conserved aromatic residue, i.e. Tyr121 of the K. marisflavi amylomaltase and Tyr146 of the P. thermopropionicum counterpart (cf. Figure 2) and possibly involved in stacking interactions with glucose moieties, is also highlighted, regardless of whether it is involved or not in hydrogen bond contacts during the docking trials (Table 1)
Table 1.
Characteristics of docking trials of the N-terminal domain of four selected amylomaltasesa
| N-terminal domain | G2 | G3 | G4 | β-CD |
|---|---|---|---|---|
| E. coli | Vina Score: − 4.6 | Vina Score: − 5.1 | Vina Score: − 5.8 | Vina Score: − 5.0 |
| T81, T82, E83, Y108, T110 × 2 | T82, E83, E84, Y108, T110 × 2, R123 × 2 | T81 × 2, E83, Q87, Y108 × 2, H109, T110, T112 | N56 × 2, Y108, T110 × 2, R123 × 3 | |
| C. glutamicum | Vina Score: − 4.3 | Vina Score: − 4.6 | Vina Score: − 5.1 | Vina Score: − 4.7 |
| E102 × 2, L103 × 2, E104, W143 | E102 × 2, L103, E104 × 2, W143, H144 × 2, K145 × 3, G158 | E102, L103 × 2, E104, W143, H144, E156, G158 | W143, H144 × 2, K145 × 2, E156 × 3, G158 | |
| K. marisflavi | Vina Score: − 4.7 | Vina Score: − 5.9 | Vina Score: − 4.8 | Vina Score: − 4.3 |
| D97, E98, Y121, Y122 × 2 | P71, I96, D97, Y122 × 2, R130 × 3 | I96, D97, E98 × 3, Y121, R130 × 2, | E98 × 2, S99, G100 × 2, Y121, Q123 × 3, R130 | |
| P. thermopropionicum | Vina Score: − 3.7 | Vina Score: − 3.8 | Vina Score: − 4.6 | Vina Score: − 4.2 |
| L103, E104 × 3, Y146, Q148 | S78 × 2, K107 × 2, G141, L142 | S78, E104, N105 × 2, K107, L142, P143, W144 × 2 | K150 × 3, Q157, E159 |
aThe results of docking maltose (G2), maltotriose (G3), maltotetraose (G4) and β-cyclodextrin (β-CD) on the structure of the hypothesised CBM and putative novel SBD of four representative family GH77 amylomaltases. The structures of potential CBMs in amylomaltases from Escherichia coli (Weiss et al. 2015; PDB: 4S3Q) and Corynebacterium glutamicum (Joo et al. 2016; PDB: 5B68) were extracted from their structural coordinates, whereas the counterparts from Kushneria marisflavi (UniProt: A0A240US28) and Pelotomaculum thermopropionicum (UniProt: A5D1W1) were obtained by homology modelling using the fold recognition approach performed on the Phyre2 server (Kelley and Sternberg 2009). Vina Score means the binding energy (kJ/mol) for the individual complexes. The residues represent the residues with numbering from the N-terminus of a given amylomaltase involved in the hydrogen bond contact with the respective oligosaccharide; for some of the residues, there are more bond contacts (indicated by “ × 2” or “ × 3”). Note, not all of them belong to the five CSRs (Figs. 2 and 4). The complexes with maltose and β-CD are displayed in Fig. 5 for the two experimentally determined structures and in Fig. 6 for two modelled structures
There are two positions of interest—Gly107 and Pro128 (E. coli amylomaltase numbering), which are conserved invariantly (Fig. 2). However, with regard to their eventual involvement in the α-glucan binding, neither the glycine, nor the proline here corresponding to Gly107 and Pro128, respectively, has been found as involved in binding α-glucans in the docking trials (Fig. 4 and Table 1). As far as other glycine and proline residues are concerned, there are a few of them that might be interacting with the tested α-glucans (Table 1). Although some of these residues are positioned within the identified CSRs (cf. Figs. 2 and 4) and some of them are located outside these regions, obviously, there seems to be no tendency in preserving their potential binding function.
Each N-terminal domain thus exhibits potential to act as an SBD with at least one starch-binding site as seen to be the case in the currently established SBD CBM families (Janecek et al. 2019). Admittedly, docking of a given α-glucan resulted in slightly different arrangements of binding residues within the same indicated single binding site (Table 1). It should be noted that to see different residues in a binding site of a potential SBD (a CBM in general) may not be too surprising since in an SBD (CBM) there typically should be one or two main binding residues; the overall binding being then helped by different surrounding residues depending on a given particular case (Penninga et al. 1996; Sorimachi et al. 1997; Janecek et al. 2017, 2019). In the present case of the N-terminal domain of GH77 amylomaltases, this residue is suggested to be the Tyr108 of E. coli amylomaltase (and its counterparts in homologous amylomaltases). What is, however, most important is the fact that each of the four different maltooligosaccharides (G2, G3, G4 and β-CD) was docked in each N-terminal domain (4 different bacterial origins) within the mutually corresponding single potential binding site (cf. Figs. 5 and 6; Table 1).
Conclusions
The present study provides an in silico analysis of the N-terminal domain from 100 selected bacterial amylomaltases classified in family GH77. This domain is predicted to function as a type of SBD that would define a novel CBM family. From the evolutionary point of view, these GH77 amylomaltases are divided into four clusters, roughly reflecting bacterial phyla and classes as follows: (i) Gammaproteobacteria; (ii) Proteobacteria; (iii) Firmicutes and Alphaproteobacteria; and (iv) Actinobacteria, illustrated by amylomaltases from E. coli, K. marisflavi, P. thermopropionicum and C. glutamicum, respectively. The conserved Tyr108 of E. coli amylomaltase and its counterparts throughout the four phylogenetic clusters are proposed as the key residue responsible for α-glucan binding. Based on a careful sequence comparison including definition of CSRs coupled with docking of linear maltooligosaccharides and β-CD, a few additional residues are predicted to belong to the starch-binding site. All candidate residues identified in the present study should be among the first targets for future mutational analysis. The experimental work to confirm the starch-binding role of this N-terminal domain has been initiated.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work was financially supported by the Grant No. 2/0146/21 from the Slovak Grant Agency VEGA and by Grant No. 6108-00476B from Independent Research Fund Denmark ∣ Natural Sciences (FNU).
Abbreviations
- CBM
Carbohydrate-binding module
- β-CD
β-Cyclodextrin
- CSR
Conserved sequence region
- DPE
Disproportionating enzyme
- G2
Maltose
- G3
Maltotriose
- G4
Maltotetraose
- GH
Glycoside hydrolase
- PDB
Protein Data Bank
- SBD
Starch-binding domain
Author contributions
FM collected data, analysed results, prepared figures and contributed to writing the manuscript; MSM and BS contributed to interpreting results and writing the manuscript; SJ designed the study, contributed to collecting data, analysed and interpreted results, prepared figures and wrote the manuscript. All the authors contributed to discussion of the research and approved the final version of the manuscript.
Declarations
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Armenta S, Moreno-Mendieta S, Sanchez-Cuapio Z, Sanchez S, Rodriguez-Sanoja R. Advances in molecular engineering of carbohydrate-binding modules. Proteins. 2017;85:1602–1617. doi: 10.1002/prot.25327. [DOI] [PubMed] [Google Scholar]
- Barends TR, Bultema JB, Kaper T, van der Maarel MJEC, Dijkhuizen L, Dijkstra BW. Three-way stabilization of the covalent intermediate in amylomaltase, an α-amylase-like transglycosylase. J Biol Chem. 2007;282:17242–17249. doi: 10.1074/jbc.M701444200. [DOI] [PubMed] [Google Scholar]
- Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Ostell J, Pruitt KD, Sayers EW. GenBank. Nucleic Acids Res. 2018;46:D41–D47. doi: 10.1093/nar/gkx1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesak K, Janecek S. Sequence fingerprints of enzyme specificities from the glycoside hydrolase family GH57. Extremophiles. 2012;16:497–506. doi: 10.1007/s00792-012-0449-9. [DOI] [PubMed] [Google Scholar]
- Blesak K, Janecek S. Two potentially novel amylolytic enzyme specificities in the prokaryotic glycoside hydrolase α-amylase family GH57. Microbiology. 2013;159:2584–2593. doi: 10.1099/mic.0.071084-0. [DOI] [PubMed] [Google Scholar]
- Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J. 2004;382:769–781. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CC, Phan NN, Chen Y, Reilly PJ. Carbohydrate-binding module tribes. Biopolymers. 2015;103:203–214. doi: 10.1002/bip.22584. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Fujii K, Minagawa H, Terada Y, Takaha T, Kuriki T, Shimada J, Kaneko H. Protein engineering of amylomaltase from Thermus aquaticus with random and saturation mutageneses. Biologia. 2005;60(Suppl. 16):97–102. doi: 10.1128/AEM.71.10.5823-5827.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Minagawa H, Terada Y, Takaha T, Kuriki T, Shimada J, Kaneko H. Use of random and saturation mutageneses to improve the properties of Thermus aquaticus amylomaltase for efficient production of cycloamyloses. Appl Environ Microbiol. 2005;71:5823–5827. doi: 10.1128/AEM.71.10.5823-5827.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Minagawa H, Terada Y, Takaha T, Kuriki T, Shimada J, Kaneko H. Function of second glucan binding site including tyrosines 54 and 101 in Thermus aquaticus amylomaltase. J Biosci Bioeng. 2007;103:167–173. doi: 10.1263/jbb.103.167. [DOI] [PubMed] [Google Scholar]
- Gangoiti J, Pijning T, Dijkhuizen L. Biotechnological potential of novel glycoside hydrolase family 70 enzymes synthesizing α-glucans from starch and sucrose. Biotechnol Adv. 2018;36:196–207. doi: 10.1016/j.biotechadv.2017.11.001. [DOI] [PubMed] [Google Scholar]
- Godany A, Vidova B, Janecek S. The unique glycoside hydrolase family 77 amylomaltase from Borrelia burgdorferi with only catalytic triad conserved. FEMS Microbiol Lett. 2008;284:84–91. doi: 10.1111/j.1574-6968.2008.01191.x. [DOI] [PubMed] [Google Scholar]
- Imamura K, Matsuura T, Nakagawa A, Kitamura S, Kusunoki M, Takaha T, Unno H. Structural analysis and reaction mechanism of the disproportionating enzyme (D-enzyme) from potato. Protein Sci. 2020;29:2085–2100. doi: 10.1002/pro.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janecek S. How many conserved sequence regions are there in the α-amylase family? Biologia. 2002;57(Suppl. 11):29–41. [Google Scholar]
- Janecek S, Gabrisko M. Remarkable evolutionary relatedness among the enzymes and proteins from the α-amylase family. Cell Mol Life Sci. 2016;73:2707–2725. doi: 10.1007/s00018-016-2246-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janecek S, Kuchtova A. In silico identification of catalytic residues and domain fold of the family GH119 sharing the catalytic machinery with the α-amylase family GH57. FEBS Lett. 2012;586:3360–3366. doi: 10.1016/j.febslet.2012.07.020. [DOI] [PubMed] [Google Scholar]
- Janecek S, Sevcik J. The evolution of starch-binding domain. FEBS Lett. 1999;456:119–125. doi: 10.1016/s0014-5793(99)00919-9. [DOI] [PubMed] [Google Scholar]
- Janecek S, Svensson B, MacGregor EA. Structural and evolutionary aspects of two families of non-catalytic domains present in starch and glycogen binding proteins from microbes, plants and animals. Enzyme Microb Technol. 2011;49:429–440. doi: 10.1016/j.enzmictec.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Janecek S, Svensson B, MacGregor EA. α-Amylase: an enzyme specificity found in various families of glycoside hydrolases. Cell Mol Life Sci. 2014;71:1149–1170. doi: 10.1007/s00018-013-1388-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janecek S, Majzlova K, Svensson B, MacGregor EA. The starch-binding domain family CBM41—an in silico analysis of evolutionary relationships. Proteins. 2017;85:1480–1492. doi: 10.1002/prot.25309. [DOI] [PubMed] [Google Scholar]
- Janecek S, Marecek F, MacGregor EA, Svensson B. Starch-binding domains as CBM families—history, occurrence, structure, function and evolution. Biotechnol Adv. 2019;37:107451. doi: 10.1016/j.biotechadv.2019.107451. [DOI] [PubMed] [Google Scholar]
- Jeong DW, Jeong HM, Shin YJ, Woo SH, Shim JH. Properties of recombinant 4-α-glucanotransferase from Bifidobacterium longum subsp. longum JCM 1217 and its application. Food Sci Biotechnol. 2020;29:667–674. doi: 10.1007/s10068-019-00707-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo S, Kim S, Seo H, Kim KJ. Crystal structure of amylomaltase from Corynebacterium glutamicum. J Agric Food Chem. 2016;64:5662–5670. doi: 10.1021/acs.jafc.6b02296. [DOI] [PubMed] [Google Scholar]
- Jung JH, Jung TY, Seo DH, Yoon SM, Choi HC, Park BC, Park CS, Woo EJ. Structural and functional analysis of substrate recognition by the 250s loop in amylomaltase from Thermus brockianus. Proteins. 2011;79:633–644. doi: 10.1002/prot.22911. [DOI] [PubMed] [Google Scholar]
- Kaper T, Talik B, Ettema TJ, Bos H, van der Maarel MJEC, Dijkhuizen L. Amylomaltase of Pyrobaculum aerophilum IM2 produces thermoreversible starch gels. Appl Environ Microbiol. 2005;71:5098–5106. doi: 10.1128/AEM.71.9.5098-5106.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kerenyiova L, Janecek S. A detailed in silico analysis of the amylolytic family GH126 and its possible relatedness to family GH76. Carbohydr Res. 2020;495:108082. doi: 10.1016/j.carres.2020.108082. [DOI] [PubMed] [Google Scholar]
- Kerenyiova L, Janecek S. Extension of the taxonomic coverage of the family GH126 outside Firmicutes and in silico characterization of its non-catalytic terminal domains. 3 Biotech. 2020;10:420. doi: 10.1007/s13205-020-02415-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, Zaslavsky L, Zhang J, Bolton EE. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47:D1102–D1109. doi: 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka T, Kato S, Shimoyama T, Ishii S, Abe T, Watanabe K. The genome of Pelotomaculum thermopropionicum reveals niche-associated evolution in anaerobic microbiota. Genome Res. 2008;18:442–448. doi: 10.1101/gr.7136508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchtova A, Janecek S. In silico analysis of family GH77 with focus on amylomaltases from borreliae and disproportionating enzymes DPE2 from plants and bacteria. Biochim Biophys Acta. 2015;1854:1260–1268. doi: 10.1016/j.bbapap.2015.05.009. [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriki T, Imanaka T. The concept of the α-amylase family: structural similarity and common catalytic mechanism. J Biosci Bioeng. 1999;87:557–565. doi: 10.1016/s1389-1723(99)80114-5. [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- Liu Y, Grimm M, Dai WT, Hou MC, Xiao ZX, Cao Y. CB-Dock: a web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol Sin. 2020;41:138–144. doi: 10.1038/s41401-019-0228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd JR, Blennow A, Burhenne K, Kossmann J. Repression of a novel isoform of disproportionating enzyme (stDPE2) in potato leads to inhibition of starch degradation in leaves but not tubers stored at low temperature. Plant Physiol. 2004;134:1347–1354. doi: 10.1104/pp.103.038026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGregor EA, Janecek S, Svensson B. Relationship of sequence and structure to specificity in the α-amylase family of enzymes. Biochim Biophys Acta. 2001;1546:1–20. doi: 10.1016/s0167-4838(00)00302-2. [DOI] [PubMed] [Google Scholar]
- Machovic M, Janecek S. The invariant residues in the α-amylase family: just the catalytic triad. Biologia. 2003;58:1127–1132. [Google Scholar]
- Matsuura Y, Kusunoki M, Harada W, Kakudo M. Structure and possible catalytic residues of Taka-amylase A. J Biochem. 1984;95:697–702. doi: 10.1093/oxfordjournals.jbchem.a134659. [DOI] [PubMed] [Google Scholar]
- Mehboob S, Ahmad N, Rashid N, Imanaka T, Akhtar M. Pcal_0768, a hyperactive 4-α-glucanotransferase from Pyrobacculum calidifontis. Extremophiles. 2016;20:559–566. doi: 10.1007/s00792-016-0850-x. [DOI] [PubMed] [Google Scholar]
- Mehboob S, Ahmad N, Munir S, Ali R, Younas H, Rashid N. Gene cloning, expression enhancement in Escherichia coli and biochemical characterization of a highly thermostable amylomaltase from Pyrobaculum calidifontis. Int J Biol Macromol. 2020;165:645–653. doi: 10.1016/j.ijbiomac.2020.09.071. [DOI] [PubMed] [Google Scholar]
- Nimpiboon P, Kaulpiboon J, Krusong K, Nakamura S, Kidokoro S, Pongsawasdi P. Mutagenesis for improvement of activity and thermostability of amylomaltase from Corynebacterium glutamicum. Int J Biol Macromol. 2016;86:820–828. doi: 10.1016/j.ijbiomac.2016.02.022. [DOI] [PubMed] [Google Scholar]
- Nimpiboon P, Krusong K, Kaulpiboon J, Kidokoro S, Pongsawasdi P. Roles of N287 in catalysis and product formation of amylomaltase from Corynebacterium glutamicum. Biochem Biophys Res Commun. 2016;478:759–764. doi: 10.1016/j.bbrc.2016.08.021. [DOI] [PubMed] [Google Scholar]
- O’Neill EC, Stevenson CE, Tantanarat K, Latousakis D, Donaldson MI, Rejzek M, Nepogodiev SA, Limpaseni T, Field RA, Lawson DM. Structural dissection of the maltodextrin disproportionation cycle of the Arabidopsis plastidial disproportionating enzyme 1 (DPE1) J Biol Chem. 2015;290:29834–29853. doi: 10.1074/jbc.M115.682245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penninga D, van der Veen BA, Knegtel RM, van Hijum SA, Rozeboom HJ, Kalk KH, Dijkstra BW, Dijkhuizen L. The raw starch binding domain of cyclodextrin glycosyltransferase from Bacillus circulans strain 251. J Biol Chem. 1996;271:32777–32784. doi: 10.1074/jbc.271.51.32777. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;13:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Przylas I, Terada Y, Fujii K, Takaha T, Saenger W, Sträter N. X-ray structure of acarbose bound to amylomaltase from Thermus aquaticus. Implications for the synthesis of large cyclic glucans. Eur J Biochem. 2000;267:6903–6913. doi: 10.1046/j.1432-1033.2000.01790.x. [DOI] [PubMed] [Google Scholar]
- Przylas I, Tomoo K, Terada Y, Takaha T, Fujii K, Saenger W, Sträter N. Crystal structure of amylomaltase from Thermus aquaticus, a glycosyltransferase catalysing the production of large cyclic glucans. J Mol Biol. 2000;296:873–886. doi: 10.1006/jmbi.1999.3503. [DOI] [PubMed] [Google Scholar]
- Pugsley AP, Dubreuil C. Molecular characterization of malQ, the structural gene for the Escherichia coli enzyme amylomaltase. Mol Microbiol. 1988;2:473–479. doi: 10.1111/j.1365-2958.1988.tb00053.x. [DOI] [PubMed] [Google Scholar]
- Rachadech W, Nimpiboon P, Naumthong W, Nakapong S, Krusong K, Pongsawasdi P. Identification of essential tryptophan in amylomaltase from Corynebacterium glutamicum. Int J Biol Macromol. 2015;76:230–235. doi: 10.1016/j.ijbiomac.2015.02.035. [DOI] [PubMed] [Google Scholar]
- Roth C, Weizenmann N, Bexten N, Saenger W, Zimmermann W, Maier T, Sträter N. Amylose recognition and ring-size determination of amylomaltase. Sci Adv. 2017;3:e1601386. doi: 10.1126/sciadv.1601386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzanski C, Smirnova J, Rejzek M, Cockburn D, Pedersen HL, Pike M, Willats WG, Svensson B, Steup M, Ebenhöh O, Smith AM, Field RA. A bacterial glucanotransferase can replace the complex maltose metabolism required for starch to sucrose conversion in leaves at night. J Biol Chem. 2013;288:28581–28598. doi: 10.1074/jbc.M113.497867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorimachi K, Le Gal-Coëffet MF, Williamson G, Archer DB, Williamson MP. Solution structure of the granular starch binding domain of Aspergillus niger glucoamylase bound to β-cyclodextrin. Structure. 1997;5:647–661. doi: 10.1016/s0969-2126(97)00220-7. [DOI] [PubMed] [Google Scholar]
- Srisimarat W, Powviriyakul A, Kaulpiboon J, Krusong K, Zimmermann W, Pongsawasdi P. A novel amylomaltase from Corynebacterium glutamicum and analysis of the large-ring cyclodextrin products. J Incl Phenom Macrocycl Chem. 2011;70:369–375. doi: 10.1007/s10847-010-9890-5. [DOI] [Google Scholar]
- Srisimarat W, Kaulpiboon J, Krusong K, Zimmermann W, Pongsawasdi P. Altered large-ring cyclodextrin product profile due to a mutation at Tyr-172 in the amylomaltase of Corynebacterium glutamicum. Appl Environ Microbiol. 2012;78:7223–7228. doi: 10.1128/AEM.01366-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steichen JM, Petty RV, Sharkey TD. Domain characterization of a 4-α-glucanotransferase essential for maltose metabolism in photosynthetic leaves. J Biol Chem. 2008;283:20797–20804. doi: 10.1074/jbc.M803051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson B, Jespersen H, Sierks MR, MacGregor EA. Sequence homology between putative raw-starch binding domains from different starch-degrading enzymes. Biochem J. 1989;264:309–311. doi: 10.1042/bj2640309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaha T, Yanase M, Okada S, Smith SM. Disproportionating enzyme (4-α-glucanotransferase; EC 2.4.1.25) of potato. Purification, molecular cloning, and potential role in starch metabolism. J Biol Chem. 1993;268:1391–1396. doi: 10.1016/S0021-9258(18)54088-6. [DOI] [PubMed] [Google Scholar]
- Terada T, Fujii K, Takaha T, Okada S. Thermus aquaticus ATCC 33923 amylomaltase gene cloning and expression and enzyme characterization: production of cycloamylose. Appl Environ Microbiol. 1999;65:910–915. doi: 10.1128/AEM.65.3.910-915.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumhom S, Krusong K, Pongsawasdi P. Y418 in 410s loop is required for high transglucosylation activity and large-ring cyclodextrin production of amylomaltase from Corynebacterium glutamicum. Biochem Biophys Res Commun. 2017;488:516–521. doi: 10.1016/j.bbrc.2017.05.078. [DOI] [PubMed] [Google Scholar]
- Tumhom S, Krusong K, Kidokoro SI, Katoh E, Pongsawasdi P. Significance of H461 at subsite +1 in substrate binding and transglucosylation activity of amylomaltase from Corynebacterium glutamicum. Arch Biochem Biophys. 2018;652:3–8. doi: 10.1016/j.abb.2018.06.002. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk V, Lammerts van Bueren A, van der Kaaij RM, Dijkhuizen L. Carbohydrate-binding module 74 is a novel starch-binding domain associated with large and multidomain α-amylase enzymes. FEBS J. 2016;283:2354–2368. doi: 10.1111/febs.13745. [DOI] [PubMed] [Google Scholar]
- van der Maarel MJEC, Leemhuis H. Starch modification with microbial α-glucanotransferase enzymes. Carbohydr Polym. 2013;93:116–121. doi: 10.1016/j.carbpol.2012.01.065. [DOI] [PubMed] [Google Scholar]
- van der Maarel MJEC, van der Veen B, Uitdehaag JC, Leemhuis H, Dijkhuizen L. Properties and applications of starch-converting enzymes of the α-amylase family. J Biotechnol. 2002;94:137–155. doi: 10.1016/s0168-1656(01)00407-2. [DOI] [PubMed] [Google Scholar]
- Vujicic-Zagar A, Pijning T, Kralj S, Lopez CA, Eeuwema W, Dijkhuizen L, Dijkstra BW. Crystal structure of a 117 kDa glucansucrase fragment provides insight into evolution and product specificity of GH70 enzymes. Proc Natl Acad Sci USA. 2010;107:21406–21411. doi: 10.1073/pnas.1007531107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattebled F, Ral JP, Dauvillee D, Myers AM, James MG, Schlichting R, Giersch C, Ball SG, D’Hulst C. STA11, a Chlamydomonas reinhardtii locus required for normal starch granule biogenesis, encodes disproportionating enzyme. Further evidence for a function of α-1,4 glucanotransferases during starch granule biosynthesis in green algae. Plant Physiol. 2003;132:137–145. doi: 10.1104/pp.102.016527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci. 1988;28:31–36. doi: 10.1021/ci00057a005. [DOI] [Google Scholar]
- Weiss SC, Skerra A, Schiefner A. Structural basis for the interconversion of maltodextrins by MalQ, the amylomaltase of Escherichia coli. J Biol Chem. 2015;290:21352–21364. doi: 10.1074/jbc.M115.667337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum likelihood approach. Mol Biol Evol. 2001;18:691–699. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
- Yun JH, Bae JW. Complete genome sequence of the halophile bacterium Kushneria marisflavi KCCM 80003T, isolated from seawater in Korea. Mar Genomics. 2018;37:35–38. doi: 10.1016/j.margen.2017.11.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.