

Abstract
Atherosclerosis, a chronic comprehensive cardiovascular disease, is characterized by the lipid infiltration, formation of foam cells derived from macrophages and inflammation in the vessel wall. Substantial evidence confirms that the activity of autophagic bodies plays a pivot role in regulating cell deaths, but the mechanisms of autophagy to regulate the pyroptosis of macrophages in atherosclerosis remain unclear. In our study, we explored that ox-LDL decreased the cell viability and destroyed the integrity of cell membrane, resulting in the pyroptosis of THP-1 derived macrophages in a dose-dependent manner. Western blotting, qRT-PCR and ELISA also showed that chloroquine (CQ) could up-regulate the expression of p62 through impairing autophagy and induce the pyroptosis of macrophages treated by ox-LDL, as evidenced by the decrease of cell viability and membrane integrity, and the increase of pro-caspase-1, GSDMD, and proinflammatory factors IL-1β and IL-18. Further researches demonstrated that Nrf2, a nuclear factor activated by p62, was linked to macrophage pyroptosis. Overactivating or suppressing Nrf2/ARE signaling would correspondingly aggravate or alleviate pyroptosis, in which the level of p62 was regulated by Nrf2 feedback. Then, bioinformatic analysis verified that there was a close interaction between p62, Nrf2/ARE signaling proteins and pyroptosis-related proteins. Taken together, our results show that blocking autophagy promotes the pyroptosis of ox-LDL-treated macrophages via the p62/Nrf2/ARE axis, providing a novel therapeutic target for atherosclerosis.
Keywords: Atherosclerosis, Pyroptosis, Autophagy, Nrf2/ARE pathway
Introduction
Atherosclerosis (AS), one of the predominant causes of cardiovascular diseases, has high morbidity and mortality worldwide [27]. The main causes of atherosclerosis induced by smoking, hyperlipidemia and other factors are arterial vessels lesion, abnormal lipid metabolism and inflammation, which are manifested as the structural and functional abnormalities of vascular endothelial cells, smooth muscle cells and macrophages [3, 44]. Macrophages take in excess oxidized-low density lipoprotein (ox-LDL) to form foam cells, which play a key role in the early and advance stage of atherosclerosis. The death of foam cells can lead to the occurrence and expansion of extracellular lipid nuclei, which has an important effect on plaque rupture and thrombosis [45]. Therefore, macrophage death can predict the instability of plaques in atherosclerosis.
Inflammatory caspases are critical for inflammasome-mediated immunity/diseases. Caspase-1 is activated by the canonical inflammasomes, such as NLRP3, NAIP–NLRC4, AIM2 and Pyrin. Pro-caspase-1 is involved in the cleavage and maturation of IL-1β/18, and also triggers a form of programmed necrosis known as pyroptosis [38]. Activated caspase-1 cleaves the N-terminal domain and C-terminal domain of gasdermin D (GSDMD), executive protein of pyroptosis, releases the N-terminal domain with the activity of pore drilling on the phospholipid of the binding membrane, and then destroys the cell membrane to cause pyroptosis [5, 9]. Pyroptosis, a new caspase-1-dependent inflammatory modality of cell death, is characterized by cell swelling, pore formation and membrane rupture, resulting in massive leakage of cytoplasmic components [15]. Accumulating evidences have suggested that pyroptosis raise the risk of atherosclerotic plaque rupture, which undoubtedly implies a potential value of blocking pyroptosis in AS [33].
Autophagy is a dynamic regulatory system for maintaining the stability of intracellular environment, selectively degrading long-lived proteins and damaged organelles [7]. It affects cholesterol efflux and inflammation in atherosclerosis, and is also related to pyroptosis [31]. Ye et al. verified that autophagy blockage leads to inflammation and pyroptosis of endothelial cells (ECs) with ox-LDL treatment [51]. And another study demonstrated that autophagy activation can protect cells from pyroptosis, as confirmed by decreased expression of NLRP3, caspase-1, IL-1β, IL-18 and GSDMD, and that p62 is also involved [29]. What is more, autophagy deficiency results in increased susceptibility of vascular smooth muscle cells (SMCs) to cell death at an early stage of atherosclerosis [36], but how macrophage pyroptosis is regulated by autophagy in atherosclerosis remains unclear. p62/SQSTM1, a cargo receptor for autophagic degradation of ubiquitinated targets, is overstocked together with elevated level of autophagy barrier [19]. A generous amount of p62 blocks nuclear factor erytheroid-derived-2-like 2 (Nrf2) ubiquitination, transcriptionally activates Nrf2. And then the activated Nrf2 combines with the downstream antioxidant response element (ARE) such as heme oxygenase-1 (HO-1), initiating the Nrf2/ARE signaling pathway [26]. Serious studies suggest that Nrf2/ARE pathway has a protective effect on atherosclerosis and the resulting pyroptosis by inhibiting ROS formation [2, 17]. Conversely, recent reports have suggested that Nrf2/ARE pathway can aggravate atherosclerosis. Nrf2 continuously activated by p62, phosphorylates p62 and forms a positive circulation loop, which is related to tumor cell proliferation and stress injury [23, 28, 40].
In this study, we modeled the cells formed by macrophages that overloaded ox-LDL, combined autophagy with pyroptosis, and explored the effect of autophagy blockage on the pyroptosis of macrophages. According to the nonclassical pathway of p62 activating Nrf2, we further investigated whether Nrf2/ARE pathway influences this process, aiming at providing a new target for the prevention and treatment of atherosclerosis.
Materials and methods
Materials
THP-1 monocytes were purchased from the China Center for Type Culture Collection. Ox-LDL was purchased from Guangzhou Yiyuan (China), and CQ was from MedChemExpress (USA). Tert-butylhydroquinone (tBHQ) and ML385 are specific activator and inhibitor of Nrf2, respectively, which were obtained from TagerMor (China). Primary and secondary antibodies for pro-caspase-1, GSDMD, p62/SQSTM1, LC3II/I, Nrf2, HO-1, and β-actin were purchased from Abcam (UK).
Methods
Cell culture and treatment
THP-1 cells were cultured in RPMI1640 medium (Hyclone, USA) supplemented with 10% fetal bovine serum (FBS) (Hyclone) at 37°C in a humidified incubator with 5% CO2. Cells were transferred to 6-well or 96-well plates, and each well was primed with PMA (Sigma, USA) at a concentration of 100 ng/mL for 48 h to induce the differentiation of THP-1 cells into macrophages. After treatment with different concentrations of ox-LDL (0, 25, 50, 75, or 100 μg/mL) for 48 h, cells were collected for the subsequent experiment.
Cell viability
We used a cell counting kit 8 (CCK-8) assay obtained from (Dojindo, China) to detect the cell viability. Each group of cells were cultured in a 96 well-plate for 24 h, then 10 μL CCK-8 solution was added into each well and cultured for 3 h. Absorbance was measured at 450 nm wavelength.
Lactate dehydrogenase (LDH) release assay
CytoTox 96 non-Radioactive cytotoxicity assay kit was used to measure cytotoxicity, which reflected the integrity of cell membrane. After corresponding treatment of cells, the culture supernatant was collect and CytoTox 96 kit was used to determine the LDH level according to the manufacturer’s instructions (Abcam, UK). Cytotoxicity (%) = 100 × (Test Sample-Low Control)/(High Control-Low Control).
Enzyme-linked immunosorbent assay (ELISA)
The measurement of protein levels of IL-1β and IL-18 were conducted using the ELISA kit (Cloud-Clone Corp and Elabscience, China). After cells were exposed to the indicated treatments, the culture supernatants were harvested and the amount of mature IL-1β and IL-18 released was determined using the ELISA kit according to the manufacturer’s instructions.
Flow cytometry analysis (FCM)
After treatment, cells were washed with PBS contains 5 μg/mL Hoechst 33342 and 10 μg/mL propidium iodide (PI). Pyroptosis cells were detected in THP-1 macrophages by flow cytometry using FACSCanto IITM (BD, Biosciences, San Diego, CA, USA), and the number of pyroptosis cells was represented by the proportion of staining cells in the total cells.
Western blot analysis
After the indicated treatments, cells were lysed using RIPA lysis buffer (Beyotime) containing the protease inhibitor PMSF (Beyotime, China)and the protein concentration was determined using the BCA Protein Assay Kit (Solarbio, China). The extracted proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes for Western blot analysis. Membranes were incubated with primary antibodies overnight at 4°C and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h before being blocked with 5% nonfat milk for 1 h. Then, target proteins were visualized with an enhanced chemiluminescence agent. Finally, densitometric analysis was performed on the scanned images using Tanon Gis software. Protein strips with similar molecular weight were regenerated after treatment with membrane regeneration solution (Solarbio, China).
Quantitative real-time polymerase chain reaction (qRT-PCR)
The total RNA was extracted from the cells, by applying Transzol Up Plus RNA Kit (TransGen Biotech, China). Then, RNA from each group and 10 μL PCR reaction solution were mixed into 10 μL of the Quantscript RT Kit (TIANGEN Biotech, China) for reverse transcription. Then diluted cDNA was used for qRT-PCR using SuperReal PreMix Plus (SYBR Green) Kit (TIANGEN Biotech, China). And the primer was synthesized by Shanghai Yingjun Biotechnology Co., Ltd. The mRNA level of individual genes was normalized to β-actin, and relative expression was calculated using the ΔCT method.
All primers were obtained from Invitrogen: caspase-1 (Forward, 5’-GCACAAGACCTCTGACAGCA-3’; Reverse, 5’-TTGGGCAGTTCTTGGTATTC-3’), GSDMD (Forward, 5’-TGCTTGCCGTACTCCATTCCATC-3’; Reverse, 5’-AGTTCTGAAGAGCCTGCCTCCA-3’), p62 (Forward, 5’-CAGAGAAGCCCATGGACAG-3’; Reverse, 5’-AGCTGCCTTGTACCCACATC-3’), LC3 (Forward, 5’-GATGTCCGACTTATTCGAGAGC-3’; Reverse, 5’-TTGAGCTGTAAGCGCCTTCTA-3’), Nrf2 (Forward, 5’-TCAGCGACGGAAAGAGTATGA-3’; Reverse, 5’-CCACTGGTTTCTGACTGGATGT-3’), HO-1 (Forward, 5’-AAGACTGCGTTCCTGCTCAAC-3’, Reverse, 5’-AAAGCCCTACAGCAACTGTCG-3’), β-actin (Forward, 5’-AGCAGCATCGCCCCAAAGTT-3’; Reverse, 5’-GGGCACGA AGGCTCATCATT-3’).
Bioinformatic analyses of the protein-protein interaction network
The Search Tool for the Retrieval of Interacting Genes (STRING; http://string-db.org; version 10.5) was used to construct the protein-protein interaction (PPI) network. And Cytoscape software (version 3.6.1) was performed to plot the PPI network.
Statistical analysis
Data from triplicate individual experiments were processed using Prism software (GraphPad 8.0), which are showed as mean ± standard deviation (SD). Statistical analysis was performed by t-test or one-way analysis of variance. Differences were considered statistically significant when p < 0.05.
Results
ox-LDL induced THP-1 macrophages to pyroptosis in a concentration dependent manner
To explore the effect of ox-LDL on macrophage pyroptosis in vitro, we pretreated THP-1 macrophages with different concentrations of ox-LDL for 48 h. We found that ox-LDL (25_100 μg/mL) treatment induced the both mRNA and protein expressions of pro-caspase-1 and GSDMD, which can regulate pyroptosis, in a concentration-dependent manner (Fig. 1a, b, c). We also found that ox-LDL (25_100 μg/mL) treatment for 48 h resulted in a significant decrease in cell viability and increased LDH release in a concentration-dependent manner (Fig. 1d, e). In addition, the release of IL-1β and IL-18 in the supernatant increased following ox-LDL treatment (Fig. 1f, g). These results show that ox-LDL induces pyroptosis in a concentration-dependent manner in THP-1 macrophages. And the ox-LDL-induced pyroptosis was especially pronounced at the concentration of 75 μg/mL, so the exposure model of 75 μg/mL for 48 h was used in subsequent experiments.
Fig. 1.

ox-LDL induced THP-1 macrophages to pyroptosis in a concentration-dependent manner. Cells were pretreated with ox-LDL (0, 25, 50, 75, and 100 μg/mL) for 48 h. (a) Western blotting was performed to determine the level of pro-caspase-1 (p-casp1) and GSDMD. (b, c) Relative protein and mRNA expressions of two molecules were expressed as a percentage of β-actin. (d) Cell viability was measured using a CCK-8 detection kit. (e) Effects of different concentrations of ox-LDL induced LDH release were measured by a CytoTox 96 kit. (f, g) ELISA was used to determine the secretion of IL-1β and IL-18 in the supernatant. Values are expressed as the mean ± SEM (n = 3) of independent experiments at least 3 repeated measurements. *p < 0.05 and **p < 0.01 different from the control group
Autophagy blockage triggered pyroptosis and inflammation in macrophages exposed to ox-LDL
Although the effect of autophagy on pyroptosis has been elucidated, the role of this process in THP-1 macrophages is not yet clear. Based on the fact that CQ has been widely applied to inhibit autophagy and form autophagy blockage [13, 32, 35], we used CQ (50 μM) as an autophagy inhibitor to detect pyroptosis of THP-1 cells under the condition of autophagy blockage for 24 h. As described in Fig. 2a, b, overexpression of p62 and LC3II/I in ox-LDL-treated cells indicated that the degradation of autophagosome was slightly blocked. And after adding CQ, the levels of p62 and LC3II/I were further increased, which proved the formation of autophagy blockage (Fig. 2a, b). At the same time, the expression of pro-caspase-1 and GSDMD were all increased in the CQ-treated group as shown by western blotting and qRT-PCR (Fig. 2c). Compared with the ox-LDL-treated group, incubation of cells with CQ further reduced the cell viability (Fig. 2d). Moreover, LDH release also showed an upward trend (Fig. 2e). As previously described, pyroptosis is accompanied by secretion of pro-inflammatory factor IL-1β and IL-18 [18, 41]. By using ELISA, we found that the release of IL-1β and IL-18 was also further increased when autophagy blockage occurred (Fig. 2f, g). Next, the content of pyroptosis cells was measured by flow cytometry (Fig. 2h). From the ratio, we could see that on the basis of ox-LDL, the number of pyroptosis cells increased more obviously after adding CQ. These observations suggested that autophagy defect could promote macrophage pyroptosis and inflammation in the presence of ox-LDL.
Fig. 2.

Autophagy blockage triggered pyroptosis and inflammation in macrophages exposed to ox-LDL. Cells were divided into the following treatment groups: control, ox-LDL (75 μg/mL), and ox-LDL+CQ (50 μM). (a, b, c) p62, LC3II/I, p-casp1 and GSDMD levels were detected by Western blotting and qRT-PCR. (d) Cell viability and (e) LDH release were performed in each treatment group. (f, g) The levels of IL-1β and IL-18 were assessed by ELISA. (h) The percentage of pyroptosis by using FCM. Values are expressed as the mean ± SEM (n = 3) of independent experiments at least 3 repeated measurements. *p < 0.05 and **p < 0.01 different from the control group, #p < 0.05 and ##p < 0.01 different from the ox-LDL-treated group
Nrf2/ARE pathway was activated in ox-LDL-treated macrophages during autophagy blockage
When autophagy is blocked, p62 is accumulated in large quantities. After co-treatment with ox-LDL and CQ, western blotting showed that the expression level of LC3II/I and p62 increased, indicating that the autophagy blockage was formed (Fig. 3a, b, c). Recent studies have shown that the physical interaction between autophagic adaptor p62 and Nrf2 leads to increased transcriptional activity of Nrf2 [1, 4]. Similarly, our results showed the both protein and mRNA levels of HO-1 and Nrf2 were also significantly increased (Fig. 3d, e, f). Thus, our data indicated that autophagy blockage can active Nrf2, which is likely to be achieved through accumulation of p62.
Fig. 3.

Nrf2/ARE pathway was activated in ox-LDL-treated macrophages during autophagy blockage. (a) Western blotting in each of the following treatment groups: control, ox-LDL (75 μg/mL), and ox-LDL+CQ (50 μM). (b, c, d, e) Expression of p62, LC3II/I, HO-1 and Nrf2 under each groups. (f) mRNA levels of HO-1 and Nrf2 were showed by qRT-PCR. Values are expressed as the mean ± SEM (n = 3) of independent experiments at least 3 repeated measurements. *p < 0.05 and **p < 0.01 different from the control group, #p < 0.05 and ##p < 0.01 different from the ox-LDL-treated group
p62/Nrf2/ARE axis was involved in autophagy blockage-mediated aggregation of macrophage pyroptosis
To confirm that the presence of p62/Nrf2/ARE axis in macrophage pyroptosis mediated by autophagy blockage, the Nrf2 agonist tBHQ and inhibitor ML385 were applied. The expression of HO-1 and Nrf2 reflected the activation of Nrf2/ARE pathway. As shown in Fig. 4a, b, the levels of HO-1 and Nrf2 were promoted and inhibited in cells stimulated by tBHQ and ML385, respectively, compared with ox-LDL treated group. Meanwhile, we found that the expression of pro-caspase-1 and GSDMD was increased under the action of tBHQ, while the expression of these proteins showed opposite results in the ML385 treated group when CQ and ox-LDL exist together (Fig. 4c, d). Given that the expression of pro-caspase-1 and GSDMD were increased or decreased in the group treated with tBHQ or ML385, we hypothesized that p62 may be involved because p62 is a special activator of Nrf2 as well. As expected, Nrf2 overexpression upregulated the level of p62, thus inhibiting autophagy and further aggravating autophagy blockage (Fig. 4c, d). qRT-PCR also showed that the expression trend of mRNA was the same as that of protein (Fig. 4e). What is more, tBHQ treatment significantly depressed cell viability and exacerbated LDH release; however, ML385 treatment improved cell viability and LDH release induced by CQ following ox-LDL exposure (Fig. 4f, g). Furthermore, the secretion of IL-1β and IL-18 was also further increased or decreased affected by tBHQ or ML385 (Fig. 4h, i). The FCM data showed that the proportion of stained pyroptosis cells was also increased and decreased separately (Fig. 4j).
Fig. 4.
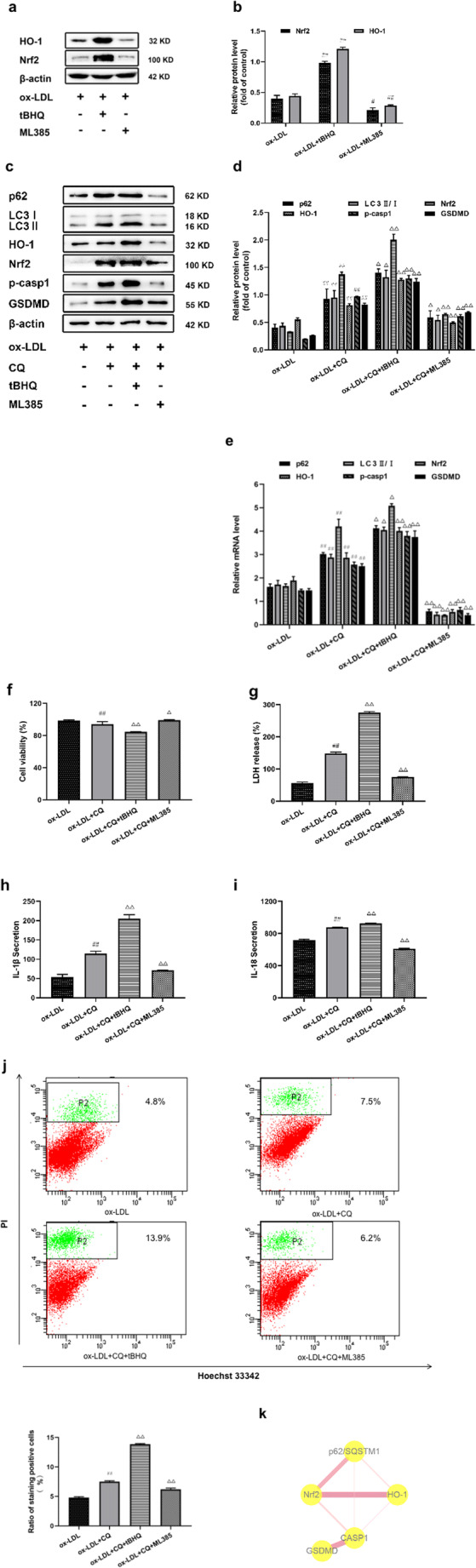
p62/Nrf2/ARE axis was involved in autophagy blockage-mediated aggregation of macrophage pyroptosis. In the presence of ox-LDL (75 μg/mL), cells were stimulated by a Nrf2 agonist (tBHQ) or inhibitor (ML385) respectively. Next, (a, b) HO-1 and Nrf2 protein level were measured by immunoblotting. Cells were pretreated with CQ for 24 h and then tBHQ (20 μM) or ML385 (5 μM) were added to the cells for an additional 24 h in the present of ox-LDL. (c, d, e) Relative protein and mRNA levels of p62, LC3II/I, p-casp1, GSDMD, HO-1 and Nrf2 determined by immunoblotting and PCR. (f) Cell viability and (g) LDH release were detected in the four groups. (h, i) IL-1β and IL-18 secretion were assessed by ELISA. (j) FCM was used to test the radio of stained cells. (k) A network of related proteins and their co-interaction was analyzed using STRING and cytoscape. The thickness of the line presents the degree of their relationships. Values are expressed as the mean ± SEM (n = 3) of independent experiments at least 3 repeated measurements. *p < 0.05 and **p < 0.01 different from the control group, #p < 0.05 and ##p < 0.01 different from the ox-LDL-treated group, and △p < 0.05 and △△p < 0.01 different from the ox-LDL+CQ-treated group
Construction of the PPI network and analysis of p62, Nrf2/ARE pathway proteins and pyroptosis related proteins were done using Cytoscape to show the relationship between these key proteins (Fig. 4k). The thickness and color depth of the edge were directly proportional to the combined degree. Table 1 showed that each of these proteins had different degree of interaction strength. Together, these findings portended that p62/Nrf2/ARE axis played a potent role in the deterioration of macrophage pyroptosis mediated by autophagy blockage.
Table 1.
Mutual combination score of p62, Nrf2/ARE pathway proteins and pyroptosis related proteins
| Node 1 | Node 2 | Node 1 accession | Node 2 accession | Combined score |
|---|---|---|---|---|
| Nrf2 | HO-1 | ENSP00000380252 | ENSP00000216117 | 0.996 |
| HO-1 | Nrf2 | ENSP00000216117 | ENSP00000380252 | 0.996 |
| GSDMD | CASP1 | ENSP00000433209 | ENSP00000433138 | 0.959 |
| CASP1 | GSDMD | ENSP00000433138 | ENSP00000433209 | 0.959 |
| SQSTM1 | Nrf2 | ENSP00000374455 | ENSP00000380252 | 0.825 |
| Nrf2 | SQSTM1 | ENSP00000380252 | ENSP00000374455 | 0.825 |
| Nrf2 | CASP1 | ENSP00000380252 | ENSP00000433138 | 0.524 |
| CASP1 | Nrf2 | ENSP00000433138 | ENSP00000380252 | 0.524 |
| SQSTM1 | HO-1 | ENSP00000374455 | ENSP00000216117 | 0.468 |
| HO-1 | SQSTM1 | ENSP00000216117 | ENSP00000374455 | 0.468 |
| HO-1 | CASP1 | ENSP00000216117 | ENSP00000433138 | 0.421 |
| CASP1 | HO-1 | ENSP00000433138 | ENSP00000216117 | 0.421 |
| SQSTM1 | CASP1 | ENSP00000374455 | ENSP00000433138 | 0.402 |
| CASP1 | SQSTM1 | ENSP00000433138 | ENSP00000374455 | 0.402 |
Discussion
Data gathered in this study indicate that autophagy blockage confirmed by the additional effect of CQ exacerbates ox-LDL induced THP-1 macrophage pyroptosis. Specifically, ox-LDL dose-dependently triggers the pyroptosis of macrophage, and inhibiting autophagy aggregate pyroptosis of macrophage treated by ox-LDL at 75 μg/mL, the optimal concentration for research. It is noteworthy that this study determined that this process has a bearing on accumulation of p62 and Nrf2/ARE pathway activation. Consequently, these results may support a novel effect for autophagy blockage in deteriorating pyroptosis resulting from ox-LDL induction in macrophage by activating the p62/Nrf2/ARE axis, which provides a potential therapeutic strategy for improving atherosclerosis.
Macrophage death in atherosclerosis is a marked feature of advanced plaques, and is the main contributor to necrotic core formation and plaque instability [46]. Forms of macrophage death include apoptosis, passive or accidental necrosis as well as other necrosis [34]. Among them, pyroptosis has been well-defined in recent years, an inflammatory form of regulatory cell death induced by caspase-1 activation [16]. Some reports showed that ox-LDL in atherosclerotic plaques activate inflammasomes, such as NLRP3, by breaking lysosomes, and then cathepsin is released, leading to the cleavage and activation of pro-caspase-1 [14, 22]. Later, activated caspase-1 participates in the shear of GSDMD and the rapid formation of plasma membrane pores, thus allowing cell swelling and membrane dissolution. Similarly, our studies showed that excessive ox-LDL with a concentration of more than 75 μg/mL can induce increased levels of pro-caspase-1, GSDMD, IL-1β and IL-18, decrease cell viability and impair cell membrane integrity, resulting in pyroptosis of THP-1 macrophages. Zheng et al. have found that both inflammasomes and caspase-1 deficiency decrease atherosclerosis in ApoE−/− mice [49]. Others have shown that the expression of caspase-1 in carotid plaques is higher than that in non-atherosclerotic arteries [39]. All those add credence to the view that pyroptosis that occurs in human macrophages may be implicated in necrotic core formation and lesion instability in advanced atherosclerotic plaque [48].
Although autophagy and pyroptosis are two independent patterns of cell death, Jiang et al. proved that there is an important feedback network between autophagy, NF-κB signaling pathway and LPS-induced pyroptosis [12]. Cardiovascular drugs that have been widely used in clinical practice, such as Carvedilol, an α-, β-blocker, are being tested to attenuate macrophage pyroptosis via autophagy activation [42]. Moreover, miR-103 encodes the gene related to the end of autophagy, which reduces the incidence of pyroptosis by regulating the autophagy [43]. Silencing NIX hastened ox-LDL-induced J774A.1 macrophage cells pyroptosis via mitochondrial autophagy [37] and some novel connections between autophagy and pyroptosis are new trends in recent research. In our previous study, we found that pyroptosis mediated by pro-caspase-1 and GSDMD is up-regulated by autophagy through CQ, an autophagy inhibitor. Beyond that, under the inhibitory effect of CQ on autophagy, the release of inflammatory cytokines IL-1β and IL-18 increased significantly, which further verified that activated caspase-1 promoted the transformation of pro-IL-1β and pro-IL-18 into their bioactive forms [47]. These data underscored that impaired autophagy intensified the pyroptosis of macrophages. Of note is that hydroxychloroquine has been widely used in the treatment of recent COVID-19 in recent years [11], so this study may bring a new inspiration for the clinical application of CQ in patients with atherosclerosis and COVID-19.
Multiple signal pathways related to pyroptosis regulated by autophagy have always been a hot spot for research, and in particular, the pathways involved in oxidative stress. Wang et al. reported that pretreatment with an autophagy inducer, Rapa, negatively mediated nuclear translocation of NF-κB p65, NLRP3 inflammasome activation and pyroptotic cell death through scavenging excess reactive oxygen species (ROS) [42]. Others have also shown that activation of NLRP3 inflammasome and pyroptosis can be suppressed by activated ROS-dependent autophagy in human umbilical vein endothelial cells (HUVECs) [20]. On the basis of them, we proposed whether Nrf2/ARE pathway, a well-known antioxidant pathway, is involved as well. In our follow-up experiments, we found that p62 expression increased, Nrf2 and its downstream active enzyme HO-1 increased, and the levels of pyroptosis-related proteins such as pro-caspase-1 and GSDMD also increased after macrophages were treated with an autophagy inhibitor based on the presence of ox-LDL. This is an in-depth test and verify our previous idea that as a result of autophagy defect, a large amount of autophagy substrate protein p62 is accumulated, which activates Nrf2, translocates it into the nucleus, and finally the phenomenon of pyroptosis is intensified. Nrf2/ARE pathway is abnormally activated by accumulated p62 in this study is similar to that found by Jiang et al. and V. Ashutosh Rao et al. [21].
While Nrf2/ARE signaling has been widely confirmed to play a key role in protecting the body from oxidative stress and inflammatory diseases [8], the latest reports indicated that overactivation of Nrf2 can promote liver cell inflammation, apoptosis and fibrosis, as well as tumor cell proliferation [30, 50]. And it is not uncommon to think that it causes atherosclerosis. Nrf2/ARE pathway has been shown to regulate the expression of scavenger receptor-B (CD36) in macrophages of ApoE−/− Nrf2+/+ mice, assist macrophages to absorb ox-LDL, and promote the formation of atherosclerotic plaques [6]. Kloska et al. transplanted the bone marrow of Nrf2−/− mice into ApoE−/− hypercholesterolemia mice, the degree of atherosclerotic lesions in the latter was significantly improved [24]. Barajas et al. also raised the high expression of Nrf2 promotes the formation of atherosclerotic lesions in a gender dependent manner [25]. Recent evidence suggested that KEAP1-Nrf2/ARE antioxidant pathway is closely related to autophagy, and they coordinate to affect the structure and function of effector cells in atherosclerosis. The characteristic mechanism is p62, an autophagy regulatory protein, can be phosphorylated in different manners. The binding affinity between phosphorylated p62 and KEAP1 increases, and subsequently induces the expression of NRF2 target genes [10]. Coincidentally, our findings on Nrf2/ARE pathway as an accelerator in autophagy blockage to promote ox-LDL-induced macrophage pyroptosis are in line with the foregoing. In order to prove this, we applied Nrf2/ARE pathway activator and inhibitor to carrying out the rescue test, and the observation that pro-caspase-1, GSDMD, IL-1β and IL-18 expression increased and decreased, respectively, further affirmed our results. The interaction network among p62, Nrf2, HO-1, GSDMD, caspase-1 proteins also strongly demonstrated the potential regulation of p62/Nrf2/ARE axis. It is worth noting that our data provide the first evidence that Nrf2 activated by p62 can feedback upregulates the expression of p62 and forms a vicious cycle of macrophage pyroptosis. This work greatly enriches the complex crosstalk between autophagy and Nrf2/ARE pathway, which may be a potential inducement of atherosclerotic plaque rupture.
In summary, as shown in Fig. 5, the blockage of autophagy makes the ox-LDL induced pyroptosis of THP-1 macrophages as well as IL-1β and IL-18 secretion more serious through the malignant activation between p62 and Nrf2/ARE pathway. However, if we can break the limitation of lack of verification at gene level and experiments conducted in vivo, our conclusion will be more complete and reliable, so this is what we will seek in the future research. Our findings raise the possibility that the p62/Nrf2/ARE axis is important for macrophage pyroptosis stimulated by ox-LDL during autophagy blockage, which may point out a new direction for the improvement of atherosclerosis and the clinical application of drugs that inhibit autophagy in atherosclerosis.
Fig. 5.
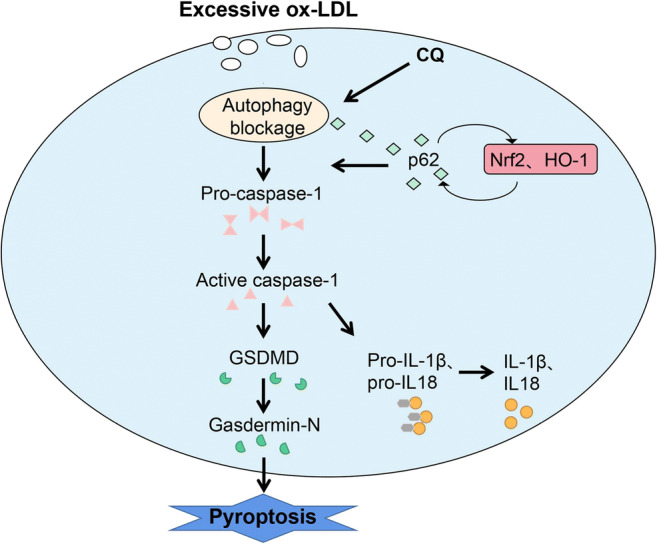
The proposed schematic of the p62/Nrf2/ARE axis regulated by autophagy blockage in pyroptosis of THP-1 macrophages. Excessive ox-LDL was ingested by macrophages and led to foam cell formation. Autophagy blockage activated the cleavage of pro-caspase-1 and GSDMD, promoted the release of IL-1β and IL-18, and eventually induced pyroptosis. Autophagy blockage induced by CQ triggered pyroptosis which was p62-dependent. The Nrf2/ARE pathway was activated by the accumulated p62, which increased the expression of Nrf2 and HO-1. Activated Nrf2 feedback promoted the accumulation of p62, leading to the aggravation of autophagy damage and pyroptosis
Abbreviations
- AS
Atherosclerosis
- ARE
Antioxidant response element
- CQ
Chloroquine
- caspase-1
Cysteinyl aspartate specific proteinase-1
- ox-LDL
Oxidized-low density lipoprotein
- ELISA
Enzyme-linked immunosorbent assay
- FCM
Flow cytometry analysis
- GSDMD
Gasdermin D
- HO-1
Heme oxygenase-1
- LDH
Lactate dehydrogenase
- Nrf2
Nuclear factor erytheroid-derived-2-like 2
- p62/SQSTM1
Sequestosome 1
- qRT-PCR
Quantitative real-time polymerase chain reaction
- tBHQ
Tert-butylhydroquinone
Author contribution
First author: do experiments, write articles, participate in part of the experimental design. Second, third, fourth and fifth Author: assist in completing the experiment. Corresponding author: participate in article revision, experimental design and supervision, financial support. The authors declare that all data were generated in-house and that no paper mill was used.
Funding
This study was supported by the grants from the National Natural Science Foundation of China (No. 81672084).
Declarations
Research involving Human Participants and/or Animals
This study does not involve human participants and/or animals.
Informed consent
All authors were informed and agreed to sign.
Conflict of interest
The authors declare no competing interests.
Footnotes
Key Points
Ox-LDL stimulated macrophage to pyroptosis in a concentration dependent manner.
Autophagy blockage aggravated pyroptosis of macrophage exposed by ox-LDL.
Autophagy blockage-mediated macrophage pyroptosis was deteriorated by p62/Nrf2/ARE axis.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Aito T, Ichimura Y, Taguchi K, Suzuki T, Mizushima T, Takagi K, Hirose Y, Nagahashi M, Iso T, Fukutomi T, Ohishi M, Endo K, Uemura T, Nishito Y, Okuda S, Obata M, Kouno T, Imamura R, Tada Y, Obata R, Yasuda D, Takahashi K, Fujimura T, Pi J, Lee MS, Ueno T, Ohe T, Mashino T, Wakai T, Kojima H, Okabe T, Nagano T, Motohashi H, Waguri S, Soga T, Yamamoto M, Tanaka K, Komatsu M. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun. 2016;7:12030. doi: 10.1038/ncomms12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.An YW, Jhang KA, Woo SY, Kang JL, Chong YH. Sulforaphane exerts its anti-inflammatory effect against amyloid-β peptide via STAT-1 dephosphorylation and activation of Nrf2/HO-1 cascade in human THP-1 macrophages. Neurobiol Aging. 2016;38:1–10. doi: 10.1016/j.neurobiolaging.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 3.Barajas B, Che N, Yin F, Rowshanrad A, Orozco LD, Gong KW, Wang X, Castellani LW, Reue K, Lusis AJ, Araujo JA. NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler Thromb Vasc Biol. 2011;31:58–66. doi: 10.1161/ATVBAHA.110.210906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartolini D, Dallaglio K, Torquato P, Piroddi M, Galli F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl Res. 2018;193:54–71. doi: 10.1016/j.trsl.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, Gomez GA, Holley CL, Bierschenk D, Stacey KJ, Yap AS, Bezbradica JS, Schroder K. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med. 2018;215:827–840. doi: 10.1084/jem.20172222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bozaykut P, Karademir B, Yazgan B, Sozen E, Siow RC, Mann GE, Ozer NK. Effects of vitamin E on peroxisome proliferator-activated receptor γ and nuclear factor-erythroid 2-related factor 2 in hypercholesterolemia-induced atherosclerosis. Free Radic Biol Med. 2014;70:174–181. doi: 10.1016/j.freeradbiomed.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 7.Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res. 2017;120:1812–1824. doi: 10.1161/CIRCRESAHA.117.311082. [DOI] [PubMed] [Google Scholar]
- 8.Chen QM, Maltagliati AJ. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol Genomics. 2018;50:77–97. doi: 10.1152/physiolgenomics.00041.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 10.Dodson M, Redmann M, Rajasekaran NS, Darley-Usmar V, Zhang J. KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem J. 2015;469:347–355. doi: 10.1042/BJ20150568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Funck-Brentano C, Salem JE (2020) Chloroquine or hydroxychloroquine for COVID-19: why might they be hazardous? Lancet 6736:1016–1017 [DOI] [PMC free article] [PubMed]
- 12.Gao Y, You X, Liu Y, Gao F, Zhang Y, Yang J, Yang C. Induction of autophagy protects human dental pulp cells from lipopolysaccharide-induced pyroptotic cell death. Exp Ther Med. 2020;19:2202–2210. doi: 10.3892/etm.2020.8475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonçalves RM, Agnes JP, Delgobo M, de Souza PO, Thomé MP, Heimfarth L, Lenz G, Moreira J, Zanotto-Filho A. Late autophagy inhibitor chloroquine improves efficacy of the histone deacetylase inhibitor SAHA and temozolomide in gliomas. Biochem Pharmacol. 2019;163:440–450. doi: 10.1016/j.bcp.2019.03.015. [DOI] [PubMed] [Google Scholar]
- 14.Grebe A, Hoss F, Latz E. NLRP3 Inflammasome and the IL-1 pathway in atherosclerosis. Circ Res. 2018;122:1722–1740. doi: 10.1161/CIRCRESAHA.118.311362. [DOI] [PubMed] [Google Scholar]
- 15.Guo M, Yan R, Ji Q, Yao H, Sun M, Duan L, Xue Z, Jia Y. IFN regulatory Factor-1 induced macrophage pyroptosis by modulating m6A modification of circ_0029589 in patients with acute coronary syndrome. Int Immunopharmacol. 2020;86:106800. doi: 10.1016/j.intimp.2020.106800. [DOI] [PubMed] [Google Scholar]
- 16.Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: its regulation and involvement in atherosclerosis. J Cell Physiol. 2018;233:2116–2132. doi: 10.1002/jcp.25930. [DOI] [PubMed] [Google Scholar]
- 17.Hu Q, Zhang T, Yi L, Zhou X, Mi M. Dihydromyricetin inhibits NLRP3 inflammasome-dependent pyroptosis by activating the Nrf2 signaling pathway in vascular endothelial cells. Biofactors. 2018;44:123–136. doi: 10.1002/biof.1395. [DOI] [PubMed] [Google Scholar]
- 18.Hughes MM, O'Neill L. Metabolic regulation of NLRP3. Immunol Rev. 2018;281:88–98. doi: 10.1111/imr.12608. [DOI] [PubMed] [Google Scholar]
- 19.Jeong SJ, Zhang X, Rodriguez-Velez A, Evans TD, Razani B. p62/SQSTM1 and selective autophagy in cardiometabolic diseases. Antioxid Redox Signal. 2019;31:458–471. doi: 10.1089/ars.2018.7649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang C, Jiang L, Li Q, Liu X, Zhang T, Dong L, Liu T, Liu L, Hu G, Sun X, Jiang L. Acrolein induces NLRP3 inflammasome-mediated pyroptosis and suppresses migration via ROS-dependent autophagy in vascular endothelial cells. Toxicology. 2018;410:26–40. doi: 10.1016/j.tox.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 21.Jiang T, Harder B, Rojo de la Vega M, Wong PK, Chapman E, Zhang DD. p62 links autophagy and Nrf2 signaling. Free Radic Biol Med. 2015;88:199–204. doi: 10.1016/j.freeradbiomed.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karasawa T, Takahashi M. Role of NLRP3 Inflammasomes in Atherosclerosis. J Atheroscler Thromb. 2017;24:443–451. doi: 10.5551/jat.RV17001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kavurma MM, Rayner KJ, Karunakaran D. The walking dead: macrophage inflammation and death in atherosclerosis. Curr Opin Lipidol. 2017;28:91–98. doi: 10.1097/MOL.0000000000000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kloska D, Kopacz A, Piechota-Polanczyk A, Nowak WN, Dulak J, Jozkowicz A, Grochot-Przeczek A. Nrf2 in aging - focus on the cardiovascular system. Vasc Pharmacol. 2019;112:42–53. doi: 10.1016/j.vph.2018.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Kobiyama K, Ley K. Atherosclerosis. Circ Res. 2018;123:1118–1120. doi: 10.1161/CIRCRESAHA.118.313816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 27.Ladapo JA, Goldfeld KS, Douglas PS. Projected morbidity and mortality from missed diagnoses of coronary artery disease in the United States. Int J Cardiol. 2015;195:250–252. doi: 10.1016/j.ijcard.2015.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee YA, Noon LA, Akat KM, Ybanez MD, Lee TF, Berres ML, Fujiwara N, Goossens N, Chou HI, Parvin-Nejad FP, Khambu B, Kramer E, Gordon R, Pfleger C, Germain D, John GR, Campbell KN, Yue Z, Yin XM, Cuervo AM, Czaja MJ, Fiel MI, Hoshida Y, Friedman SL. Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nat Commun. 2018;9:4962. doi: 10.1038/s41467-018-07338-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li MY, Zhu XL, Zhao BX, Shi L, Wang W, Hu W, Qin SL, Chen BH, Zhou PH, Qiu B, Gao Y, Liu BL. Adrenomedullin alleviates the pyroptosis of Leydig cells by promoting autophagy via the ROS-AMPK-mTOR axis. Cell Death Dis. 2019;10:489. doi: 10.1038/s41419-019-1728-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li T, Jiang D, Wu K. p62 promotes bladder cancer cell growth by activating KEAP1/NRF2-dependent antioxidative response. Cancer Sci. 2020;111:1156–1164. doi: 10.1111/cas.14321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang X, Wang C, Sun Y, Song W, Lin J, Li J, Guan X. p62/mTOR/LXRα pathway inhibits cholesterol efflux mediated by ABCA1 and ABCG1 during autophagy blockage. Biochem Biophys Res Commun. 2019;514:1093–1100. doi: 10.1016/j.bbrc.2019.04.134. [DOI] [PubMed] [Google Scholar]
- 32.Lin YC, Lin JF, Wen SI, Yang SC, Tsai TF, Chen HE, Chou KY, Hwang TI. Chloroquine and hydroxychloroquine inhibit bladder cancer cell growth by targeting basal autophagy and enhancing apoptosis. Kaohsiung J Med Sci. 2017;33:215–223. doi: 10.1016/j.kjms.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 33.Lu LQ, Tian J, Luo XJ, Peng J (2021) Targeting the pathways of regulated necrosis: a potential strategy for alleviation of cardio-cerebrovascular injury. Cell Mol Life Sci 78:63–67 [DOI] [PMC free article] [PubMed]
- 34.Martinet W, Coornaert I, Puylaert P, De Meyer G. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front Pharmacol. 2019;10:306. doi: 10.3389/fphar.2019.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, Coppes RP, Engedal N, Mari M, Reggiori F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–1455. doi: 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osonoi Y, Mita T, Azuma K, Nakajima K, Masuyama A, Goto H, Nishida Y, Miyatsuka T, Fujitani Y, Koike M, Mitsumata M, Watada H. Defective autophagy in vascular smooth muscle cells enhances cell death and atherosclerosis. Autophagy. 2018;14:1991–2006. doi: 10.1080/15548627.2018.1501132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng X, Chen H, Li Y, Huang D, Huang B, Sun D. Effects of NIX-mediated mitophagy on ox-LDL-induced macrophage pyroptosis in atherosclerosis. Cell Biol Int. 2020;44:1481–1490. doi: 10.1002/cbin.11343. [DOI] [PubMed] [Google Scholar]
- 38.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 39.Shi X, Xie WL, Kong WW, Chen D, Qu P. Expression of the NLRP3 inflammasome in carotid atherosclerosis. J Stroke Cerebrovasc Dis. 2015;24:2455–2466. doi: 10.1016/j.jstrokecerebrovasdis.2015.03.024. [DOI] [PubMed] [Google Scholar]
- 40.Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118:653–667. doi: 10.1161/CIRCRESAHA.115.306256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity. 2019;50:1352–1364. doi: 10.1016/j.immuni.2019.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Jiang L, Shi L, Yao K, Sun X, Yang G, Jiang L, Zhang C, Wang N, Zhang H, Wang Y, Liu X. Zearalenone induces NLRP3-dependent pyroptosis via activation of NF-κB modulated by autophagy in INS-1 cells. Toxicology. 2019;428:152304. doi: 10.1016/j.tox.2019.152304. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Song X, Li Z, Liu N, Yan Y, Li T, Sun W, Guan Y, Li M, Yang Y, Yang X, Liu B. MicroRNA-103 protects coronary artery endothelial cells against H2O2-induced oxidative stress via BNIP3-mediated end-stage autophagy and antipyroptosis pathways. Oxidative Med Cell Longev. 2020;2020:8351342. doi: 10.1155/2020/8351342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolf MP, Hunziker P (2020) Atherosclerosis: insights into vascular pathobiology and outlook to novel treatments. J Cardiovasc Transl Res 13:744–757 [DOI] [PubMed]
- 45.Xu H, Jiang J, Chen W, Li W, Chen Z. Vascular macrophages in atherosclerosis. J Immunol Res. 2019;2019:4354786. doi: 10.1155/2019/4354786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu YJ, Zheng L, Hu YW, Wang Q. Pyroptosis and its relationship to atherosclerosis. Clin Chim Acta. 2018;476:28–37. doi: 10.1016/j.cca.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 47.Yin Y, Li X, Sha X, Xi H, Li YF, Shao Y, Mai J, Virtue A, Lopez-Pastrana J, Meng S, Tilley DG, Monroy MA, Choi ET, Thomas CJ, Jiang X, Wang H, Yang XF. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler Thromb Vasc Biol. 2015;35:804–816. doi: 10.1161/ATVBAHA.115.305282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhaolin Z, Guohua L, Shiyuan W, Zuo W. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2019;52:e12563. doi: 10.1111/cpr.12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng F, Xing S, Gong Z, Mu W, Xing Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediat Inflamm. 2014;2014:507208. doi: 10.1155/2014/507208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Y, Wang K, Zhou Y, Li T, Yang M, Wang R, Chen Y, Cao M, Hu R. HEATR1 deficiency promotes pancreatic cancer proliferation and gemcitabine resistance by up-regulating Nrf2 signaling. Redox Biol. 2020;29:101390. doi: 10.1016/j.redox.2019.101390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zi Y, Yi-An Y, Bing J, Yan L, Jing T, Chun-Yu G, Fan P, Hao L, Jia-Ni T, Han-Jin H, Fei C, Xue-Bo L. Sirt6-induced autophagy restricted TREM-1-mediated pyroptosis in ox-LDL-treated endothelial cells: relevance to prognostication of patients with acute myocardial infarction. Cell Death Dis. 2019;5:88. doi: 10.1038/s41420-019-0168-4. [DOI] [PMC free article] [PubMed] [Google Scholar]