

Abstract
Genome-wide transcriptomic analyses in whole tissues reflect the aggregate gene expression in heterogeneous cell populations comprising resident and migratory cells, and they are unable to identify cell type–specific information. We used a computational method (population-specific expression analysis [PSEA]) to decompose gene expression in gingival tissues into cell type–specific signatures for 8 cell types (epithelial cells, fibroblasts, endothelial cells, neutrophils, monocytes/macrophages, plasma cells, T cells, and B cells). We used a gene expression data set generated using microarrays from 120 persons (310 tissue samples; 241 periodontitis affected and 69 healthy). Decomposition of the whole-tissue transcriptomes identified differentially expressed genes in each of the cell types, which mapped to biologically relevant pathways, including dysregulation of Th17 cell differentiation, AGE-RAGE signaling, and epithelial-mesenchymal transition in epithelial cells. We validated selected PSEA-predicted, differentially expressed genes in purified gingival epithelial cells and B cells from an unrelated cohort (n = 15 persons), each of whom contributed with 1 periodontitis-affected and 1 healthy gingival tissue sample. Differential expression of these genes by quantitative reverse transcription polymerase chain reaction corroborated the PSEA predictions and pointed to dysregulation of biologically important pathways in periodontitis. Collectively, our results demonstrate the robustness of the PSEA in the decomposition of gingival tissue transcriptomes and its ability to identify differentially regulated transcripts in particular cellular constituents. These genes may serve as candidates for further investigation with respect to their roles in the pathogenesis of periodontitis.
Keywords: gene expression, periodontitis, validation, reverse transcription polymerase chain reaction, epithelial cells, B cells
Introduction
Periodontitis is a chronic inflammatory disease that is associated with microbial dysbiosis and characterized by loss of connective tissue attachment and alveolar bone (Kinane et al. 2017). Although our understanding of the pathobiology of the disease has been significantly enhanced in the past 2 decades, the mapping of intra- and intercellular signaling pathways orchestrating the host response to bacterial dysbiosis is a work in progress (Ebersole et al. 2013; Cekici et al. 2014). Delineation of transcriptomic signatures in the gingival tissues at various disease stages has the potential to elucidate key molecular mechanisms underlying the initiation and progression of periodontitis (Demmer et al. 2008; Sawle et al. 2016). Genome-wide transcriptomic analyses in gingival tissues using microarray and RNA sequencing have been used to this end by our group and others (Demmer et al. 2008; Kebschull et al. 2013; Horie et al. 2016), reflecting the average level of gene expression in a mixed population of cells, including resident tissue components (e.g., epithelial cells, fibroblasts), as well as migratory cells responsible for immune surveillance and inflammatory responses (including polymorphonuclear neutrophils, monocytes/macrophages, and T and B cells, among others). However, the onset and progression of periodontitis are likely orchestrated by contributions of distinct cell populations whose interactions form complex molecular networks that underlie homeostatic or catabolic processes in the gingival tissues (Takayanagi 2005; Cekici et al. 2014). Analyses based on whole-tissue transcriptomes are not suited to identify cell type–specific information, because the expression of a particular gene can increase during the transition from health to disease in one cell type and decrease in another, but these opposing changes will remain largely undetected when only a net change is assessed (Heath et al. 2016). Another shortcoming of the assessment of aggregate fold changes of expression in mixed cell populations is that they cannot distinguish between true changes in expression and those resulting from dynamic fluctuations in the relative proportion of individual cell types that commonly occur during the transition to a pathological state (Newman et al. 2015).
To mitigate the shortcomings associated with whole-tissue transcriptomic signatures, a novel computational method was developed to decompose aggregate gene expression profiles in tissue samples that comprise a heterogeneous cellular composition (Kuhn et al. 2011). The method, termed population-specific expression analysis (PSEA), uses cell population–specific marker genes to generate individual population expression profiles in silico without a need for additional experimental steps such as fluorescence-activated cell sorting or laser-capture microdissection. The method can also account for expression changes that occur due to the differential abundance of particular cell types in each sample (Kuhn et al. 2011). In the present study, we first applied PSEA to transcriptomic data sets derived from gingival tissues harvested from states of gingival health or established periodontitis and identified genes that are differentially expressed specifically in each of 8 cell types that represent major constituents of the gingival tissues. Next, we used tissue dissociation and immune-magnetic bead purification methods to isolate epithelial cells and B cells from an independent set of gingival biopsies that were not involved in the above computational decomposition and performed quantitative reverse-transcription polymerase chain reaction (qRT-PCR) assays to validate the PSEA-predicted differential expression of selected genes in states of health and periodontitis. Our results demonstrate that PSEA can be successfully used in the decomposition of the human gingival transcriptome and can facilitate an understanding of the cell-specific molecular processes that occur in the gingival tissues during the course of periodontitis.
Methods
PSEA Analysis
Gene Expression Data Set
We used a gene expression data set in gingival tissues that we generated earlier using Affymetrix HG-U133Plus 2.0 microarrays (Papapanou et al. 2009), available through GSE16134. A detailed description of the data set is presented in the Appendix.
The PSEA Method
Decomposition of the gene expression signal into molecular subtypes was performed using the PSEA method (Kuhn et al. 2011). Briefly, probesets expressed in a given cell type were identified by assessing the linear dependence of their expression against the expression of marker probesets uniquely expressed in the particular cell type, among all cell types considered. Differential expression was identified by comparing the slope of healthy tissue samples with that of periodontitis-affected samples. For each given gene (probeset), a log2 fold change, a P value of expression in the cell type, and a P value for differential expression in the cell type were computed. Note that since the expression of each gene tested was plotted against that of the marker genes in the same sample, the number of cells of a given type was constant at each point, and differences in cell composition between health and disease did not confound the analyses.
Identification of Marker Genes
Marker genes were identified using Gene Expression Barcode 3.0 (McCall et al. 2014) that reports the probability that a given probeset is expressed in a particular cell type (see Appendix). To define markers, we identified probesets that had a probability of expression of 1 in a given cell type of interest and of 0 in each of the other cell types under consideration. These genes were derived by set-theoretic (Venn) operations on Barcode entries for each of the following 8 cell types: epithelial cells, endothelial cells (CD31+), fibroblasts, monocytes (CD14+), neutrophils, plasma cells, T cells (CD3+), and B cells (CD19+). Additional filtering steps of the marker probesets are described in the Appendix. The final list of the marker probesets used in the analyses is presented in Appendix Table 1.
Model Fitting
The expression of the 54,675 probesets in all samples was fitted to each of 1,208 linear models, each of which had 1 or more cell types expressed but only 1 cell type differentially expressed. The best model for each probeset was determined using the Akaike information criterion (AIC) units (Akaike 1973). Models within 2 AIC units of the best one were selected. Additional filtering of probesets resulted in their removal from differential expression analyses (Appendix).
Gene Set Overrepresentation Analysis
Differentially expressed genes for each cell type with P value <0.05 were included to identify potentially overrepresented processes according to KEGG and Wiki (WP) pathways, using overrepresentation analysis as implemented in gProfiler (Reimand et al. 2007). Pathways with a false discovery rate (FDR) ≤0.1 were reported.
Validation of PSEA-Predicted Genes
Validation of PSEA-predicted genes was performed in an independent set of gingival tissue samples (15 pairs of healthy and periodontitis-affected sites). After preparation of single cell suspensions and immunomagnetic separation of epithelial cells and B cells, selected PSEA-predicted genes were validated using qRT-PCR. Experimental details are presented in the Appendix.
Results
PSEA Analysis
Table 1 lists PSEA-decomposed, cell type–specific gene expression profiles that fulfilled all filtering steps mentioned above. These included 11 transcripts in epithelial cells, 4 in fibroblasts, 5 in endothelial cells, 13 in neutrophils, 6 in plasma cells, and 8 in B cells. No PSEA-decomposed transcripts fulfilled both the absolute log2 fold change differential expression of >0.4 or the CC = 1 filtering steps in monocytes/macrophages or in T cells. A more extensive list of PSEA-decomposed genes by cell type, irrespective of |log2 FC| or CC, is presented in Appendix Table 3. This list includes 29 transcripts in epithelial cells, 12 in fibroblasts, 13 in endothelial cells, 21 in neutrophils, 5 in monocytes/macrophages, 11 in plasma cells, 3 in T cells, and 13 in B cells.
Table 1.
PSEA-Decomposed, Cell Type–Specific Gene Expression Profiles That Fulfilled All Filtering Steps.
| Probe ID | Gene Symbol | Log2FC | Ref P Value | Diff P Value |
|---|---|---|---|---|
| Epithelial cells | ||||
| 210479_s_at | RORA | −0.67 | 1.1E-10 | 1.1E-07 |
| 205637_s_at | SH3GL3 | −0.43 | 2.1E-21 | 4.5E-07 |
| 227309_at | YOD1 | −0.45 | 3.5E-21 | 1.0E-06 |
| 211966_at | COL4A2 | −1.59 | 8.7E-04 | 3.2E-06 |
| 223895_s_at | EPN3 | −0.42 | 1.4E-13 | 3.3E-05 |
| 222190_s_at | C16orf58 | −1.46 | 3.4E-02 | 1.9E-03 |
| 225510_at | OAF | −0.57 | 2.2E-05 | 2.7E-03 |
| 226632_at | CYGB | −1.26 | 3.9E-02 | 5.7E-03 |
| 203085_s_at | TGFB1 | −0.69 | 2.1E-03 | 0.01 |
| 209216_at | WDR45 | −0.46 | 1.0E-04 | 0.01 |
| Fibroblasts | ||||
| 208872_s_at | REEP5 | 0.95 | 0.047 | 1.10E-03 |
| 219315_s_at | TMEM204 | −0.71 | 1.60E-04 | 6.80E-03 |
| 202828_s_at | MMP14 | 0.57 | 2.50E-03 | 0.012 |
| 208851_s_at | THY1 | −0.46 | 3.10E-09 | 3.50E-03 |
| Endothelial cells | ||||
| 225369_at | ESAM | −0.41 | 4.5E-26 | 5.7E-07 |
| 228339_at | ECSCR | −0.44 | 5.5E-24 | 3.2E-06 |
| 215535_s_at | AGPAT1 | −0.65 | 8.7E-08 | 1.0E-03 |
| 212494_at | TNS2 | −0.52 | 2.1E-07 | 6.0E-03 |
| 209166_s_at | MAN2B1 | −0.68 | 9.73E-05 | 0.01 |
| Neutrophils | ||||
| 226907_at | PPP1R14C | −1.14 | 5.7E-09 | 2.4E-10 |
| 223694_at | TRIM7 | −0.99 | 1.1E-07 | 1.9E-08 |
| 202428_x_at | DBI | −3.19 | 1.3E-02 | 1.3E-05 |
| 232116_at | GRHL3 | −0.74 | 2.7E-07 | 2.5E-05 |
| 227736_at | C10orf99 | −1.15 | 2.1E-04 | 4.5E-05 |
| 220013_at | EPHX3 | −0.95 | 3.2E-05 | 5.4E-05 |
| 204203_at | CEBPG | −0.39 | 5.2E-14 | 6.3E-05 |
| 230769_at | DENND2C | −0.97 | 7.9E-05 | 1.1E-04 |
| 214626_s_at | GANAB | 0.83 | 1.0E-02 | 1.1E-04 |
| 228587_at | FAM83G | −0.43 | 4.8E-12 | 2.2E-04 |
| 204616_at | UCHL3 | −0.61 | 3.1E-07 | 3.1E-04 |
| 209311_at | BCL2L2 | −1.2 | 3.1E-03 | 4.7E-04 |
| 201315_x_at | IFITM2 | 0.75 | 2.0E-02 | 1.2E-03 |
| 224615_x_at | HM13 | 0.55 | 1.0E-02 | 0.02 |
| Plasma cells | ||||
| 212890_at | SLC38A10 | −1.17 | 1.3E-11 | 1.4E-05 |
| 55093_at | CHPF2 | −1.3 | 1.4E-09 | 4.2E-05 |
| 206593_s_at | MED22 | −2.75 | 1.5E-03 | 2.6E-03 |
| 204158_s_at | TCIRG1 | −1.21 | 2.5E-04 | 0.02 |
| 200644_at | MARCKSL1 | −1.49 | 3.9E-03 | 0.04 |
| 202369_s_at | TRAM2 | −0.55 | 3.7E-08 | 0.05 |
| B cells | ||||
| 202539_s_at | HMGCR | −0.89 | 1.20E-09 | 4.00E-05 |
| 204552_at | INPP4A | 0.86 | 0.01 | 3.00E-03 |
| 210785_s_at | THEMIS2 | 0.85 | 0.02 | 6.00E-03 |
| 204912_at | IL10RA | 0.6 | 2.20E-04 | 6.00E-03 |
| 212712_at | CAMSAP1 | −0.89 | 9.30E-05 | 8.00E-03 |
| 220306_at | FAM46C | 0.83 | 0.02 | 0.01 |
| 1554252_a_at | CERS3 | −1.65 | 0.02 | 0.02 |
| 206896_s_at | GNG7 | 0.78 | 0.04 | 0.04 |
Filtering steps included: P value of presence of the gene in the cell type that is differentially expressed <0.05; P value of differential expression in periodontitis-affected versus healthy gingiva <0.05; |log2FC differential expression| >0.4, and confidence coefficient (CC) = 1.Log2FC: Log2-based fold change of expression in periodontitis-affected over healthy gingival tissues. Ref P value: P value for expression of the particular probe at the specific cell type. Diff P value: P value for the differential expression of the particular probe between periodontitis-affected and healthy gingival tissues at the specific cell type.
Gene Set Overrepresentation Analysis
Significantly (FDR ≤0.1) enriched KEGG and WP pathways by cell type, based on ≥2 PSEA-decomposed differentially expressed genes, are presented in Table 2. Observe that the input in these analyses included all probesets listed in Appendix Table 3. Five KEGG pathways (Th17 cell differentiation, AGE-RAGE signaling pathway in diabetes complications, relaxin signaling pathway, pathogenetic Escherichia coli infection, and inflammatory bowel disease) and 4 WP pathways (including epithelial to mesenchymal transition in colorectal cancer and cannabinoid receptor signaling) were identified as differentially enriched in epithelial cells in periodontitis-affected versus healthy gingival tissues (Fig. 1). VEGFA-VEGFR2 signaling and senescence and autophagy in cancer were among the significantly enriched pathways in fibroblasts. Fat digestion and absorption (KEGG) and sterol regulatory element-binding proteins (SREBP) signaling (WP) were significantly enriched in endothelial cells, and nuclear receptors meta-pathway (WP) and human complement system pathway (WP) were among those enriched in neutrophils. The protein processing in endoplasmic reticulum pathway (KEGG) was among those overrepresented in plasma cells, and the human cytomegalovirus infection pathway (KEGG) was among those enriched in B cells.
Table 2.
Overrepresented KEGG and WP Pathways Based on ≥2 PSEA-Decomposed Differentially Expressed Genes between Periodontitis-Affected and Healthy Gingival Tissues, by Cell Type.
| Pathway | Pathway ID | Adjusted P Value | Genes |
|---|---|---|---|
| Epithelial cells | |||
| Th17 cell differentiation | KEGG:04659 | 0.03031515 | RORA, MAPK13, TGFB1 |
| AGE-RAGE signaling pathway in diabetic complications | KEGG:04933 | 0.03031515 | COL4A2, MAPK13, TGFB1 |
| Relaxin signaling pathway | KEGG:04926 | 0.03701637 | COL4A2, MAPK13, TGFB1 |
| Pathogenic Escherichia coli infection | KEGG:05130 | 0.09286103 | OCLN, MAPK13, TUBB2A |
| Inflammatory bowel disease (IBD) | KEGG:05321 | 0.09286103 | RORA, TGFB1 |
| Epithelial to mesenchymal transition in colorectal cancer | WP:WP4239 | 0.00876677 | COL4A2, OCLN, MAPK13, TGFB1 |
| Cannabinoid receptor signaling | WP:WP3869 | 0.04737078 | MAPK13, CYP2C9 |
| Hepatitis C and hepatocellular carcinoma | WP:WP3646 | 0.06769704 | COL4A2, TGFB1 |
| Pathogenic Escherichia coli infection | WP:WP2272 | 0.06769704 | OCLN, TUBB2A |
| Fibroblasts | |||
| VEGFA-VEGFR2 signaling | WP:WP3888 | 0.04963475 | CALU, MMP14, PBXIP1 |
| Senescence and autophagy in cancer | WP:WP615 | 0.04963475 | IGFBP5, MMP14 |
| Endothelial cells | |||
| Fat digestion and absorption | KEGG:04975 | 0.05687131 | AGPAT1, SCARB1 |
| Sterol regulatory element-binding proteins (SREBP) signaling | WP:WP1982 | 0.060402348 | SEC24A, SCARB1 |
| Neutrophils | |||
| Human complement system | WP:WP2806 | 0.080752415 | SELPLG, LAMC1 |
| Nuclear receptors meta-pathway | WP:WP2882 | 0.080752415 | PPP1R14C, DBI, CYP2C9 |
| Plasma cells | |||
| Protein processing in endoplasmic reticulum | KEGG:04141 | 0.005626135 | WFS1, RRBP1, PREB |
| B cells | |||
| Human cytomegalovirus infection | KEGG:05163 | 0.071917892 | IL10RA, GNG7, TAPBP |
PSEA, population-specific expression analysis.
Figure 1.

KEGG and WP pathway analysis in epithelial cells. Significantly enriched pathways (false discovery rate of ≤0.1) based on ≥2 population-specific expression analysis (PSEA)–decomposed differentially expressed genes are presented, along with the adjusted P value, the −log10 of the adjusted P value, and the involved genes.
Experimental Validation of Top PSEA Assignments in Gingival Epithelial Cells and B Cells
We selected 2 predicted differentially expressed transcripts in epithelial cells (TGF-β and RORA) and B cells (CAMSAP1 and CERS3) for experimental validation based on high differential fold change, high level of confidence, and established association with a translated protein. The qRT-PCR analyses showed statistically significant lower expression levels of both TGF-β1 and RORA in epithelial cells isolated from periodontitis-affected versus healthy gingival tissues, consistent with the PSEA prediction (P < 0.05, for both matched and unmatched analysis; Fig. 2A–D). Validation in B cells involved only unmatched samples, as no pairs of healthy/periodontitis-affected samples from the same donor and of sufficient quality were available. We detected a significantly lower expression of CERS3 in B cells isolated from periodontitis-affected sites compared to healthy sites, as predicted (P < 0.05; Fig. 3A), but no statistically significant difference in the expression of CAMSAP1 could be detected (Fig. 3B).
Figure 2.
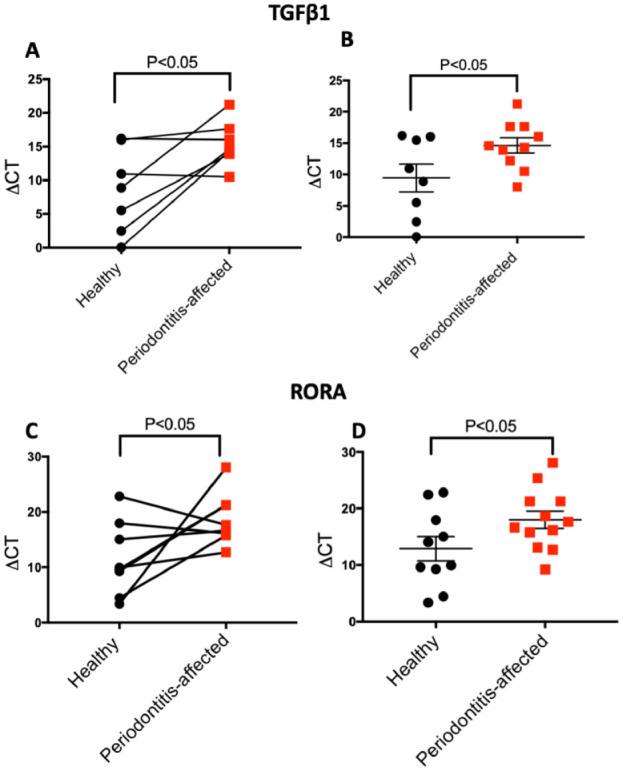
Validation of differential expression of population-specific expression analysis (PSEA)–predicted genes in isolated epithelial cells from gingival tissues. Relative expression levels (ΔCt values) of TGF-β1 assessed through quantitative reverse-transcription polymerase chain reaction (qRT-PCR) in matched (n = 7 pairs connected by horizontal lines; A) and nonmatched analyses (n = 18; B). Relative expression levels of RORA assessed through qRT-PCR in matched (n = 8 pairs by horizontal lines; C) and nonmatched analyses (n = 22; D). Data are presented as mean and standard error of mean; 18s was used as normalizer. P values are derived by 1-tailed t tests, for paired (A, C) and nonpaired observations (B, D). Note that higher ΔCt values indicate lower expression.
Figure 3.
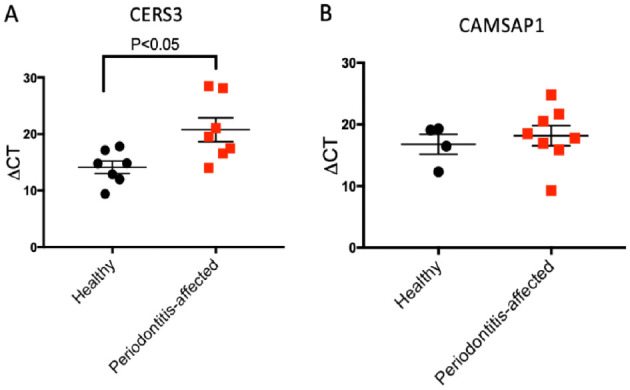
Validation of differential expression of population-specific expression analysis (PSEA)–predicted genes in isolated B cells from gingival tissues. (A) Relative expression levels of CERS3 (ΔCt values) assessed through quantitative reverse-transcription polymerase chain reaction (qRT-PCR) (n = 14). (B) Relative expression levels of CAMSAP1 (ΔCt values) assessed through qRT-PCR (n = 12). Data are presented as mean and standard error of mean. 18s was used as normalizer. P values are derived using 1-tailed t tests for nonpaired observations. Note that higher ΔCt values indicate lower expression.
Discussion
Deciphering molecular signatures that distinguish between healthy gingival tissues and those at different stages of periodontitis offers insights into the pathophysiology of the disease process and may, ultimately, identify potential therapeutic targets. However, detection of cellular-level perturbations based on whole-tissue transcriptomic analyses is challenging due to tissue heterogeneity and cellular population shifts during the transition from health to disease. To our knowledge, we applied for the first time a computational method, PSEA, to decompose whole gingival tissue transcriptomes into cell type–specific differential gene expression between periodontal health and periodontitis. Subsequently, we validated the PSEA computations by assessing the differential expression of specific genes in purified gingival epithelial cells and B cells derived from unrelated healthy or periodontitis-affected tissue samples using qRT-PCR. Our findings point to the utility of PSEA as an alternative to more labor-intensive and costly methodologies in transcriptomic studies of the pathobiology of periodontitis.
In recent years, several medium- or high-throughput technologies have been introduced to study specific cellular components in heterogeneous tissue samples, including single-cell and population-specific transcriptome analysis using qRT-PCR, RNA fluorescence in situ hybridization (RNA-FISH), RNA-seq, cDNA microarrays, and serial analysis of gene expression (SAGE) (Hunt-Newbury et al. 2007; Esumi et al. 2008; Deng et al. 2014). Alternatively, use of a computational method such as PSEA can help to decompose these aggregate signals into cell type–specific signatures while partly circumventing a number of technical difficulties associated with the above methodologies. However, a number of limitations associated with our study must be acknowledged. First, PSEA is inherently dependent on availability of cell markers previously identified; thus, potential inaccuracies in the specificity of the used markers may inevitably affect the metadata generated. It is conceivable that the relatively limited number of the available cell type–specific markers, in combination with the diversity of the composite cell populations in the gingival tissues, has limited our power to detect differentially expressed transcripts of low abundance or to accurately predict transcription in less populous cell types. Discovery of additional cell-specific markers and use of larger databases may address these shortcomings in future work. However, we emphasize that PSEA decomposition does not require consideration of every conceivable cell type in the tissues, provided that the models generated using the ultimately filtered probes have a good statistical fit (i.e., high R2 values), as was the case in our analyses (Appendix Table 3). Thus, the fact that we did not include less abundant cell types that occur in the gingiva in our models did not affect our inferences regarding genes differentially expressed in the 8 studied cell types.
In our validation experiments, we selected 2 PSEA-predicted differentially expressed genes, on the basis of maximum absolute fold change and high confidence coefficient, in 2 cell types that are highly prevalent in periodontal tissues (epithelial cells and B cells) and used pairs of healthy and periodontitis-affected gingival tissues from 15 de novo recruited individuals. As the cells of interest were dissociated and cryopreserved immediately, the distortion in the transcriptional profiles after tissue harvesting was kept to a minimum, as recently demonstrated in a comprehensive study (Guillaumet-Adkins et al. 2017). The amount of tissue harvested in each biopsy did not allow us to separate additional cell types, and the RNA obtained from each cell subset did not allow us to validate more than 2 genes in each. Thus, these experiments should be viewed as a “proof-of-principle” validation of the PSEA method in the context of gingival transcriptomes, rather than as specific verification of each predicted probe. Additional validation studies will be obviously necessary for other specific cell types and genes of interest.
The PSEA-predicted lower expression of TGF-β in epithelial cells in periodontitis-affected tissues was validated in purified epithelial cells from independent samples. Epithelial cells are the first line of defense against toxic stimuli and periodontal pathogens, orchestrate oral tissue homeostasis, and play crucial roles in the initiation of dysbiotic changes at the dento-gingival niche (Cekici et al. 2014). Epithelial cell–derived TGF-β plays a pivotal role in maintaining a balance between tolerance and immunity (Denney et al. 2015) and exerts its functions through activation of intracellular Smad2/3 proteins and suppression of inflammatory pathways. Concomitantly, TGF-β promotes expression of adhesion molecules and tight junction proteins such as claudin 1, which maintain epithelial barrier integrity (Howe et al. 2005). In intestinal epithelia, TGF-β is a potent inducer of epithelial cell margination, an essential process for tissue repair and wound healing (Troncone et al. 2018). Furthermore, TGF-β1 promotes differentiation of M2 macrophages, an anti-inflammatory subset that actively participates in tissue repair and homeostasis and attenuates the macrophage inflammatory response to bacterial products (Troncone et al. 2018). The disruption of the monocyte/macrophage phenotype and a significant shift toward proinflammatory polarization of macrophages have been recently reported to be associated with the pathogenesis of periodontal disease (Almubarak et al. 2020). The current data further point to the importance of epithelial TGF-β signaling in periodontitis.
RORA is another computationally predicted differentially expressed gene in gingival epithelium, the lower expression of which in states of health was also validated in epithelial cells isolated from unrelated gingival tissue samples. Earlier mechanistic studies in human monocytes showed that deletion of RORA leads to activation of the nuclear factor κB (NF-κB) signaling pathway and to downstream induction of proinflammatory cytokines such as TNF, IL-1β, and IL-6 at both the transcriptional and the protein levels (Nejati Moharrami et al. 2018). In the same study, RORA knockout cells were found to produce high levels of pro–IL-1β, even in the absence of lipopolysaccharide challenge. Corroborating these observations, studies using an intestinal epithelium-specific, RORA-deficient mouse model showed that RORA is crucial for maintaining intestinal homeostasis by attenuating NF-κB transcriptional activity and preventing inflammation (Oh et al. 2019). In an earlier study using reverse engineering approaches, we identified RORA as a master regulator of the transcriptional landscape in periodontitis (Sawle et al. 2016). Consistent with these observations, our finding of higher expression of RORA in epithelial cells from healthy gingiva highlights its importance as a potential molecular target for the restoration of epithelial homeostasis and attenuation of innate immunity in periodontitis.
Pathway enrichment analysis of genes predicted by PSEA to be differentially expressed in gingival epithelium showed enrichment of Th17 signaling, AGE-RAGE receptor signaling, and the epithelial-mesenchymal transition (EMT) signaling pathways. Earlier work has demonstrated the involvement of Th17 cells and their signature cytokine profiles in the pathogenesis of periodontitis (Gaffen and Hajishengallis 2008), whereas AGE-RAGE signaling plays a pivotal role in the pathogenesis of both periodontitis and diabetes mellitus (Lalla et al. 2000, 2001). Higher expression of the receptor for AGEs, RAGE, has been reported in periodontitis-affected gingival tissues and in the peripheral blood of patients with periodontitis when compared to periodontally healthy individuals; circulating soluble forms of RAGE were proposed as biomarkers for the presence and severity/extent of periodontitis (Detzen et al. 2019). Challenge of epithelial cells with AGEs was found to result in the phosphorylation of ERK, p38, and subsequent activation of NF-κB (Kido et al. 2020). The third significantly enriched pathway in epithelial cells (EMT) represents a cellular process characterized by changes in transcriptional and proteomic changes that result in the transdifferentiation of the epithelial phenotype to a mesenchymal phenotype. Indeed, PSEA-predicted differential expression of MAPK13, OCL, TGFB, and COL4A2 in epithelial cells, all of which are associated with EMT (Scanlon et al. 2013). These findings point to a possible mechanistic overlap between the transcriptional landscape of periodontitis and carcinogenesis, which is intriguing and warrants further investigation.
We also predicted and independently validated the downregulation of CERS3 in B cells isolated from periodontitis-affected tissue compared to healthy gingiva. CERS3 is a member of the ceramide synthetases protein family, and ceramide is an important signaling molecule in sphingolipid metabolism (Levy and Futerman 2010). Ceramides are present in the cytoplasm of host cells and play essential roles in orchestrating immune responses (Albeituni and Stiban 2019). Recently, a diminished expression of acid ceramidase in periodontal lesions as well as in Porphyromonas gingivalis–stimulated epithelial cells in vitro was reported (Azuma et al. 2018). Furthermore, overexpression of acid ceramidase in epithelial cells resulted in attenuation of the proinflammatory immune response and apoptosis in response to challenge by P. gingivalis, highlighting a possible anti-inflammatory role of ceramides in gingival tissue (Azuma et al. 2018).
Pathway analysis on predicted differentially expressed genes in fibroblasts showed enrichment of VEGFA-VEGFR2 and senescence and autophagy pathways. Activation of the VEGF/VEGFR2 axis has been reported in periodontal disease, and high angiogenesis activity in periodontal lesions was correlated with VEGF expression in the stroma (Vladau et al. 2016). Similar to other chronic inflammatory diseases, periodontitis has been associated with autophagic alterations (Zhuang et al. 2016). Increased levels of autophagy gene expression and high levels of mitochondrial reactive oxygen species production in peripheral blood mononuclear cells were observed in patients with periodontitis (Bullon et al. 2012). We also found that sterol regulatory element-binding protein signaling was among the dysregulated pathways in endothelial cells. SREBP1C is a key lipogenic transcription factor that regulates cholesterol and fatty acid metabolism and synthesis (Wang et al. 2015). Activation and higher levels of SREBP1C have been reported in periodontal disease–affected tissue in patients with diabetes (Kuo et al. 2016). Upregulation of SREBP1C was critical for induction of NLRP3, an inflammasome component, by high-glucose-treated P. gingivalis (Kuo et al. 2016). Convergence of these important pathways and their biological relevance to periodontal disease warrant further investigation.
Collectively, our results demonstrate the robustness of the PSEA in the decomposition of gingival tissue transcriptomes and its ability to identify differentially regulated transcripts in particular cellular constituents. These genes may serve as candidates for further investigation with respect to their roles in the pathogenesis of periodontitis.
Author Contributions
F. Momen-Heravi, contributed to design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; R.A. Friedman, contributed to design, data analysis, and interpretation, drafted and critically revised the manuscript; S. Albeshri, M. Kebschull, contributed to data acquisition, critically revised the manuscript; A. Sawle, contributed to data analysis, critically revised the manuscript; A. Kuhn, contributed to design, critically revised the manuscript; P.N. Papapanou, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, sj-pdf-1-jdr-10.1177_0022034520979614 for Cell Type–Specific Decomposition of Gingival Tissue Transcriptomes by F. Momen-Heravi, R.A. Friedman, S. Albeshri, A. Sawle, M. Kebschull, A. Kuhn and P.N. Papapanou in Journal of Dental Research
Acknowledgments
We thank Dr. Michael J. Zilliox for helpful correspondence regarding the Barcode.
Footnotes
A supplemental appendix to this article is available online.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from National Institutes of Health/National Institute of Dental and Craniofacial Research (DE015649, DE021820, and DE024735) and by an unrestricted gift from Colgate-Palmolive to P.N. Papapanou, as well as by the National Center for Advancing Translational Sciences (TR000040).
ORCID iDs: F. Momen-Heravi  https://orcid.org/0000-0003-4534-1450
https://orcid.org/0000-0003-4534-1450
References
- Akaike H. 1973. Information theory and the maximum likelihood principle. In: Petrov BN, Csäki F, editors. 2nd International Symposium on Information Theory. Budapest: Akademiai Kiàdo. [Google Scholar]
- Albeituni S, Stiban J. 2019. Roles of ceramides and other sphingolipids in immune cell function and inflammation. Adv Exp Med Biol. 1161:169–191. [DOI] [PubMed] [Google Scholar]
- Almubarak A, Tanagala KKK, Papapanou PN, Lalla E, Momen-Heravi F. 2020. Disruption of monocyte and macrophage homeostasis in periodontitis. Front Immunol. 11:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma MM, Balani P, Boisvert H, Gil M, Egashira K, Yamaguchi T, Hasturk H, Duncan M, Kawai T, Movila A. 2018. Endogenous acid ceramidase protects epithelial cells from Porphyromonas gingivalis–induced inflammation in vitro. Biochem Biophys Res Commun. 495(4):2383–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullon P, Cordero MD, Quiles JL, Ramirez-Tortosa Mdel C, Gonzalez-Alonso A, Alfonsi S, Garcia-Marin R, de Miguel M, Battino M. 2012. Autophagy in periodontitis patients and gingival fibroblasts: unraveling the link between chronic diseases and inflammation. BMC Med. 10:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cekici A, Kantarci A, Hasturk H, Van Dyke TE. 2014. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol 2000. 64(1):57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmer RT, Behle JH, Wolf DL, Handfield M, Kebschull M, Celenti R, Pavlidis P, Papapanou PN. 2008. Transcriptomes in healthy and diseased gingival tissues. J Periodontol. 79(11):2112–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Ramskold D, Reinius B, Sandberg R. 2014. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 343(6167):193–196. [DOI] [PubMed] [Google Scholar]
- Denney L, Byrne AJ, Shea TJ, Buckley JS, Pease JE, Herledan GM, Walker SA, Gregory LG, Lloyd CM. 2015. Pulmonary epithelial cell-derived cytokine TGF-β1 is a critical cofactor for enhanced innate lymphoid cell function. Immunity. 43(5):945–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detzen L, Cheng B, Chen CY, Papapanou PN, Lalla E. 2019. Soluble forms of the receptor for advanced glycation endproducts (rage) in periodontitis. Sci Rep. 9(1):8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole JL, Dawson DR, 3rd, Morford LA, Peyyala R, Miller CS, Gonzalez OA. 2013. Periodontal disease immunology: ‘double indemnity’ in protecting the host. Periodontol 2000. 62(1):163–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esumi S, Wu SX, Yanagawa Y, Obata K, Sugimoto Y, Tamamaki N. 2008. Method for single-cell microarray analysis and application to gene-expression profiling of gabaergic neuron progenitors. Neurosci Res. 60(4):439–451. [DOI] [PubMed] [Google Scholar]
- Gaffen SL, Hajishengallis G. 2008. A new inflammatory cytokine on the block: re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and iL-17. J Dent Res. 87(9):817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaumet-Adkins A, Rodriguez-Esteban G, Mereu E, Mendez-Lago M, Jaitin DA, Villanueva A, Vidal A, Martinez-Marti A, Felip E, Vivancos A, et al. 2017. Single-cell transcriptome conservation in cryopreserved cells and tissues. Genome Biol. 18(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath JR, Ribas A, Mischel PS. 2016. Single-cell analysis tools for drug discovery and development. Nat Rev Drug Discov. 15(3):204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie M, Yamaguchi Y, Saito A, Nagase T, Lizio M, Itoh M, Kawaji H, Lassmann T, Carninci P, Forrest AR, et al. 2016. Transcriptome analysis of periodontitis-associated fibroblasts by cage sequencing identified DLX5 and RUNX2 long variant as novel regulators involved in periodontitis. Sci Rep. 6:33666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe KL, Reardon C, Wang A, Nazli A, McKay DM. 2005. Transforming growth factor-beta regulation of epithelial tight junction proteins enhances barrier function and blocks enterohemorrhagic Escherichia coli O157:H7-induced increased permeability. Am J Pathol. 167(6):1587–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt-Newbury R, Viveiros R, Johnsen R, Mah A, Anastas D, Fang L, Halfnight E, Lee D, Lin J, Lorch A, et al. 2007. High-throughput in vivo analysis of gene expression in Caenorhabditis elegans. PLoS Biol. 5(9):e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebschull M, Guarnieri P, Demmer RT, Boulesteix AL, Pavlidis P, Papapanou PN. 2013. Molecular differences between chronic and aggressive periodontitis. J Dent Res. 92(12):1081–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kido R, Hiroshima Y, Kido JI, Ikuta T, Sakamoto E, Inagaki Y, Naruishi K, Yumoto H. 2020. Advanced glycation end-products increase lipocalin 2 expression in human oral epithelial cells. J Periodontal Res. 55(4):539–550. [DOI] [PubMed] [Google Scholar]
- Kinane DF, Stathopoulou PG, Papapanou PN. 2017. Periodontal diseases. Nat Rev Dis Primers. 3:17038. [DOI] [PubMed] [Google Scholar]
- Kuhn A, Thu D, Waldvogel HJ, Faull RL, Luthi-Carter R. 2011. Population-specific expression analysis (PSEA) reveals molecular changes in diseased brain. Nat Methods. 8(11):945–947. [DOI] [PubMed] [Google Scholar]
- Kuo HC, Chang LC, Chen TC, Lee KC, Lee KF, Chen CN, Yu HR. 2016. Sterol regulatory element-binding protein-1c regulates inflammasome activation in gingival fibroblasts infected with high-glucose-treated Porphyromonas gingivalis. Front Cell Infect Microbiol. 6:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalla E, Lamster IB, Feit M, Huang L, Spessot A, Qu W, Kislinger T, Lu Y, Stern DM, Schmidt AM. 2000. Blockade of rage suppresses periodontitis-associated bone loss in diabetic mice. J Clin Invest. 105(8):1117–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalla E, Lamster IB, Stern DM, Schmidt AM. 2001. Receptor for advanced glycation end products, inflammation, and accelerated periodontal disease in diabetes: mechanisms and insights into therapeutic modalities. Ann Periodontol. 6(1):113–118. [DOI] [PubMed] [Google Scholar]
- Levy M, Futerman AH. 2010. Mammalian ceramide synthases. IUBMB life. 62(5):347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MN, Jaffee HA, Zelisko SJ, Sinha N, Hooiveld G, Irizarry RA, Zilliox MJ. 2014. The gene expression barcode 3.0: improved data processing and mining tools. Nucleic Acids Res. 42(1):D938–D943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejati Moharrami N, Bjørkø y, Tande E, Ryan L, Espevik T, Boyartchuk V. 2018. RORα controls inflammatory state of human macrophages. PLoS One. 13(11):e0207374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. 2015. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 12(5):453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SK, Kim D, Kim K, Boo K, Yu YS, Kim IS, Jeon Y, Im SK, Lee SH, Lee JM, et al. 2019. RORα is crucial for attenuated inflammatory response to maintain intestinal homeostasis. Proc Natl Acad Sci U S A. 116(42):21140–21149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapanou PN, Behle JH, Kebschull M, Celenti R, Wolf DL, Handfield M, Pavlidis P, Demmer RT. 2009. Subgingival bacterial colonization profiles correlate with gingival tissue gene expression. BMC Microbiol. 9:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, Kull M, Peterson H, Hansen J, Vilo J. 2007. g: Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 35(Suppl 2):W193–W200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawle AD, Kebschull M, Demmer RT, Papapanou PN. 2016. Identification of master regulator genes in human periodontitis. J Dent Res. 95(9):1010–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon CS, Van Tubergen EA, Inglehart RC, D’Silva NJ. 2013. Biomarkers of epithelial-mesenchymal transition in squamous cell carcinoma. J Dent Res. 92(2):114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi H. 2005. Inflammatory bone destruction and osteoimmunology. J Periodontal Res. 40(4):287–293. [DOI] [PubMed] [Google Scholar]
- Troncone E, Marafini I, Stolfi C, Monteleone G. 2018. Transforming growth factor-β1/Smad7 in intestinal immunity, inflammation, and cancer. Front Immunol. 9:1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Viscarra J, Kim SJ, Sul HS. 2015. Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol. 16(11):678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladau M, Cimpean AM, Balica RA, Jitariu AA, Popovici RA, Raica M. 2016. VEGF/VEGFR2 axis in periodontal disease progression and angiogenesis: basic approach for a new therapeutic strategy. In Vivo. 30(1):53–60. [PubMed] [Google Scholar]
- Zhuang H, Ali K, Ardu S, Tredwin C, Hu B. 2016. Autophagy in dental tissues: a double-edged sword. Cell Death Dis. 7:e2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-jdr-10.1177_0022034520979614 for Cell Type–Specific Decomposition of Gingival Tissue Transcriptomes by F. Momen-Heravi, R.A. Friedman, S. Albeshri, A. Sawle, M. Kebschull, A. Kuhn and P.N. Papapanou in Journal of Dental Research