

Abstract
Background
Diabetic myopathy involves hyperglycaemia and inflammation that causes skeletal muscle dysfunction; however, the potential cellular mechanisms that occur between hyperglycaemia and inflammation, which induces sarcopenia, and muscle dysfunction remain unknown. In this study, we investigated hyperglycaemia‐induced inflammation mediating high‐mobility group box 1 activation, which is involved in a novel form of cell death, pyroptosis, diabetic sarcopenia, atrophy, and adverse muscle remodelling. Furthermore, we investigated the therapeutic potential of bone morphogenetic protein‐7 (BMP‐7), an osteoporosis drug, to treat pyroptosis, and diabetic muscle myopathy.
Methods
C57BL6 mice were treated with saline (control), streptozotocin (STZ), or STZ + BMP‐7 to generate diabetic muscle myopathy. Diabetes was established by determining the increased levels of glucose. Then, muscle function was examined, and animals were sacrificed. Gastrocnemius muscle or blood samples were analysed for inflammation, pyroptosis, weight loss, muscle atrophy, and adverse structural remodelling of gastrocnemius muscle using histology, enzyme‐linked immunosorbent assay, immunohistochemistry, western blotting, and reverse transcription polymerase chain reaction.
Results
A significant (P < 0.05) increase in hyperglycaemia leads to an increase in inflammasome (high‐mobility group box 1, toll‐like receptor‐4, and nucleotide‐binding oligomerization domain, leucine‐rich repeat and pyrin domain containing protein 3) formation in diabetic muscle cells. Further analysis showed an up‐regulation of the downstream pyroptotic pathway with significant (P < 0.05) number of positive muscle cells expressing pyroptosis‐specific markers [caspase‐1, interleukin (IL)‐1β, IL‐18, and gasdermin‐D]. Pyroptotic cell death is involved in further increasing inflammation by releasing pro‐inflammatory cytokine IL‐6. Structural analysis showed the loss of muscle weight, decreased myofibrillar area, and increased fibrosis leading to muscle dysfunction. Consistent with this finding, BMP‐7 attenuated hyperglycaemia (~50%), pyroptosis, inflammation, and diabetic adverse structural modifications as well as improved muscle function.
Conclusions
In conclusion, we report for the first time that increased hyperglycaemia and inflammation involve cellular pyroptosis that induces significant muscle cell loss and adverse remodelling in diabetic myopathy. We also report that targeting pyroptosis with BMP‐7 improves diabetic muscle pathophysiology and muscle function. These findings suggest that BMP‐7 could be a potential therapeutic option to treat diabetic myopathy.
Keywords: Atrophy, Inflammation, Fibrosis, Muscle dysfunction
Introduction
Diabetic patients are more susceptible than healthy people in the development and progression of sarcopenia muscle atrophy. 1 Diabetes is a major metabolic disorder usually accompanied with hyperglycaemia, oxidative stress, and inflammation, leading to multiorgan diseases such as cardiomyopathy, nephropathy, neuropathy, periodontal disease, retinopathy, impaired wound healing, and skeletal muscle dysfunction. 1 , 2 , 3 , 4 , 5 Skeletal muscle myopathy, especially when developed due to sarcopenia and muscle atrophy, has been considered as a major pathophysiological feature of diabetes. 1 , 2 , 6 Despite this, little attention has been paid to diabetes‐induced muscle sarcopenia, atrophy, and progressed muscle dysfunction, although these factors significantly contribute to impaired quality of life, as well as increased morbidity and mortality in patients. The exact pathophysiological mechanisms involved in diabetic muscle dysfunction are complex and need further investigation.
Apoptosis, necrosis, and autophagy have been reported in skeletal muscle dysfunction in aged humans. 7 , 8 , 9 Various studies reported these types of cell deaths in diabetic muscle dysfunction and suggest that they are generally mediated through oxidative stress. 7 , 8 , 9 , 10 Recent studies indicate that diabetes involves peripheral inflammation with increased macrophages in the blood of diabetic patients 11 ; however, it remains unknown whether muscle tissue has increased inflammation that might trigger cellular mechanisms in the induction of diabetic sarcopenia, atrophy, and muscle dysfunction. An inflammation‐induced cell death, pyroptosis, has been reported in the gut and other organs involving infection. 12 Infection‐initiated inflammation that causes pyroptosis has been considered to be a major mechanism that leads to organ dysfunction. Recent evidence suggests that non‐cell dividing organs such as heart involve pyroptosis, which is caused by sterile inflammation. 13 Pyroptosis, distinct from apoptosis and necrosis, is initiated by inflammation mediated through microbial infection or damage‐associated molecular patterns (DAMPs). This leads to the formation of the inflammasome and a series of downstream activated pyroptosis markers such as caspase‐1, interleukin (IL)‐1β, and IL‐18. 14 , 15 It remains unknown whether diabetes‐induced muscle dysfunction involves inflammation‐induced pyroptosis in gastrocnemius muscle (GM) tissue.
Moreover, antioxidant and pharmacological agents such as myostatin and activin have been examined to treat skeletal muscle atrophy and dysfunction in disease conditions; however, these drugs have been reported to have off‐target effects, which raise concerns regarding their efficacy in patients. 16 , 17 Therefore, alternative strategies are needed to attenuate diabetes‐induced sarcopenia and atrophy. We recently reported that bone morphogenetic protein‐7 (BMP‐7), an osteogenic protein‐1, which belongs to the transforming growth factor β superfamily, inhibits apoptosis and inflammation in the diabetic heart. 18 Furthermore, we also demonstrated that BMP‐7 inhibits plaque formation, monocyte infiltration, and pro‐inflammatory cytokine secretion in atherosclerosis. 19 , 20 However, it remains unknown whether BMP‐7 attenuates inflammation‐induced pyroptosis, sarcopenia, skeletal muscle atrophy, and adverse remodelling.
This study will examine whether the presence of inflammation‐induced cell death, pyroptosis, leads to sarcopenia, skeletal muscle atrophy, and adverse muscle remodelling that causes skeletal muscle dysfunction in diabetic male and female mice. Furthermore, we investigated whether BMP‐7 would have a therapeutic effect on diabetes‐induced pyroptosis, sarcopenia, muscle atrophy, and muscle dysfunction in diabetic animals.
Materials and methods
Animal model and experimental design
All animal procedures in the current study were approved by the Institutional Animal Care and Use Committee of the University of Central Florida. We used a total of 48 C57BL/6J mice age 10 ± 2 weeks old, both male and female sexes, as depicted in Figure 1A. Mice were divided into three groups: control, streptozotocin (STZ), and STZ + BMP‐7 (n = 16; 8M + 8F/group). Animals were administered 200 mg/kg STZ via intraperitoneal injection, whereas recombinant mouse BMP‐7 (Bioclone, cat#PA‐0401) of 200 μg/kg body weight was administered via intravenous injection immediately after STZ injection. Additional injections of BMP‐7 were given on two successive days. Total cumulative dose of BMP‐7 given was 600 μg/kg body weight. Control animals were administered with saline injections. Body weight was measured prior to STZ injection and at the time of sacrifice. On Day 42 (D‐42), muscle function was tested, and animals were euthanized under 4% isoflurane followed by cervical dislocation. Blood samples were collected by heart puncture via exsanguination for enzyme‐linked immunosorbent assay (ELISA). GMs were collected, weighed, and stored at −80°C in RNA later for western blotting and gene expression studies, or 4% paraformaldehyde for histological staining.
Figure 1.
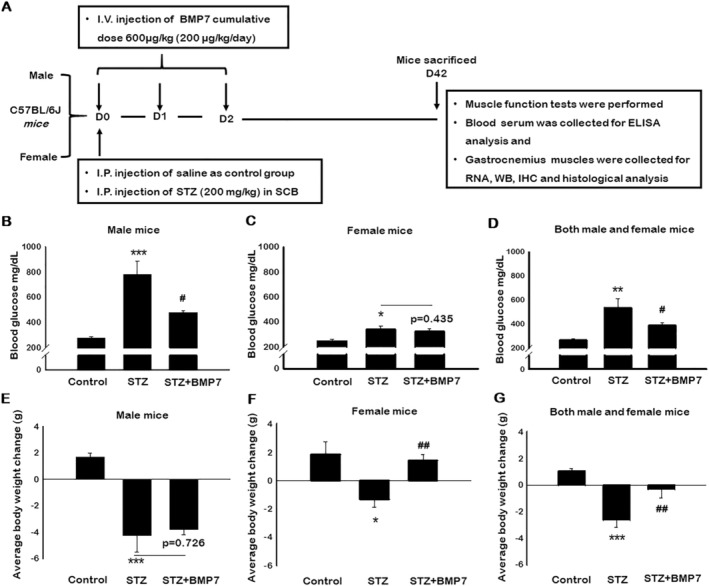
Bone morphogenetic protein‐7 (BMP‐7) treatment attenuates diabetes‐induced hyperglycaemia and improves diabetes induced weight loss. Schematic representation of injection schedule and study design (A). Streptozotocin (STZ)‐induced diabetes was confirmed with elevated glucose levels. Bar graphs represent elevated glucose levels in both male and female mice of STZ‐administered mice, whereas BMP‐7 treatment potentially reduced the glucose levels on D42 [B: Male mice (n = 5–6); C: Female mice (n = 6); D: Both male and female mice (n = 12–13)]. Similarly, histogram represents the gain in body weight (BW) upon BMP‐7 treatment after 42 days of STZ administration [E: Male mice (n = 5–8); F: Female mice (n = 5–8); G: Both male and female mice (n = 11–13)]. Error bars = mean ± standard error of the mean. One‐way ANOVA and Tukey tests were performed to assess statistical significance. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. control, #P < 0.05 and ##P < 0.01, vs. STZ.
Determination of blood glucose levels
Blood prick method was used to measure the random blood glucose levels at D‐42 using OneTouch Ultra Mini glucose metre. 18 Briefly, all mice were subjected for glucose test using 3–6 μL of blood collected via tail vein puncture.
Immunohistochemistry staining
Double immunohistochemistry (IHC) staining was performed as published by us. 18 , 21 GM tissue sections were deparaffinized followed by rehydration. Sections were blocked with 10% normal goat serum (Vector Labs). Following blocking, sections were stained for skeletal muscle myosin using anti‐myosin primary antibody and Alexa Fluor® 488 goat anti‐rabbit (Invitrogen, Carlsbad, CA) secondary antibody. After myosin staining, a second blocking step was performed prior to co‐staining with inflammasome primary antibodies (prepared in 10% GS) of high‐mobility group box 1 (HMGB1), toll‐like receptor‐4 (TLR4), and nucleotide‐binding oligomerization domain, leucine‐rich repeat and pyrin domain containing protein 3 (NLRP3). Sections were also stained for pyroptosis‐specific markers using caspase‐1, IL‐1β, IL‐18, and gasdermin‐D (GSDMD) as primary antibodies and Alexa flour® 568‐goat anti‐rabbit antibody as secondary antibody. Finally, the sections were washed, and the nuclei were stained with mounting medium containing 4′,6‐diamidino‐2‐phenylindole (DAPI; Vector Labs cat#H‐1200). Images (four to five fields/section) were taken at ×20 magnification using Keyence fluorescence microscope (Keyence, Itasca, IL) for quantification, and representative images were recorded at ×40 magnification. Quantitative data for pyroptotic cell death were calculated by dividing positive cells over total DAPI times 100 [(total cells+ve/total DAPI)*100], and SigmaPlot software was used for graphical representation. Details of antibodies were provided in Supporting Information, Table S1A.
cDNA synthesis and reverse transcription polymerase chain reaction
Total RNA was isolated from the GM tissue homogenate using Trizol™ (Invitrogen, Carlsbad, CA) and reverse transcribed into cDNA using the Superscript™ III First Strand Synthesis system (Invitrogen, Carlsbad, CA). Following cDNA synthesis, quantitative real‐time polymerase chain reaction (PCR) was performed using CFX96 C1000 Touch™ Thermal Cycler Multicolor Real‐Time PCR Detection System (Bio‐Rad, Hercules, CA) with SYBR Green (Invitrogen, Carlsbad, CA). PCR was carried out for pyroptotic initiator HMGB1, inflammasome marker NLRP3, pyroptotic markers (caspase‐1, IL‐1β, IL‐18, and GSDMD), and for muscle atrophy marker muscle RING‐finger protein‐1 (MuRF1). The used list of mouse‐specific primers is presented in Table S1B. Glyceraldehyde 3‐phosphate dehydrogenase was used as loading control. Quantitative PCR was performed with an initial step of denaturation at 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 20 s and 60°C for 20 s. Melt curves were established and normalized fold expression was calculated by using 2−∆∆Ct method.
Western blot analysis
Western blot was performed as reported previously. 18 , 21 GM tissue (15–20 mg) was homogenized using radioimmunoprecipitation lysis buffer, supernatant was collected after centrifugation, and protein concentration was estimated using Bio‐Rad protein assay. Protein samples (50–80 μg) were loaded and run on sodium dodecyl sulfate polyacrylamide gel electrophoresis (10% or 15%) for 90–120 min at 150 V. The gels were transblotted onto a polyvinylidene difluoride membrane using Mini Trans‐Blot Electrophoretic Transfer Cell (Bio‐Rad, Hercules, CA) for 60 min. Membranes were blocked with 5% non‐fat milk in 1× tris‐buffered saline with Tween at room temperature for 1 h and then incubated with primary antibodies (1:1000 v/v dilution) for HMGB1, NLRP3, GSDMD, and β‐actin (as a loading control) overnight at 4°C. Following primary antibody incubation, membranes were washed with 1× tris‐buffered saline with Tween and incubated with horseradish peroxidase‐conjugated goat anti‐rabbit IgG secondary antibody (1:1000 v/v dilution), for 1 h at room temperature. Finally, membranes were exposed to enhanced chemiluminescence reagent (Thermo Technologies, Rockford, IL) and the signal was detected using X‐ray films. Densitometric analysis was performed using ImageJ software on scanned X‐ray films. All protein band intensities were normalized to beta (β)‐actin and expressed as arbitrary units. Antibody details were given in Table S1A.
Enzyme‐linked immunosorbent assay
Blood samples were centrifuged at 775 g for 20 min. Serum was separated and stored at −80°C. Pro‐inflammatory cytokine levels of IL‐6 were analysed in mice serum samples using a sandwich ELISA kit (Ray Biotech, Inc., Norcross, GA, USA, cat#ELM‐IL6) following the supplier's protocol. Data were plotted in a bar graph using SigmaPlot software.
Histological analysis for atrophy and fibrosis
Haematoxylin and eosin staining
To evaluate myofibrillar loss, GM sections were stained with haematoxylin and eosin as we described previously. 21 , 22 Sections were deparaffinized, rehydrated, and subsequently stained with haematoxylin (Thermo Fisher Scientific; cat#7211), 1% acid alcohol (Poly Scientific R&D Corp; cat#S104), bluing reagent (Thermo Fisher Scientific; cat#73011), and eosin (Thermo Fisher Scientific; cat#7111) solutions. Nuclei were stained in blue/purple, and muscle cells were in pink. Images were recorded using Keyence microscope (Itasca, IL, USA). ImageJ software was used to quantify myofibrillar size (mm2) at ×20 magnification captured pictures, and data were represented in a bar graph using SigmaPlot software. Representative images were recorded at ×40 magnification.
Masson's trichrome staining
To determine the interstitial (IF) and vascular (VF) fibrosis, trichrome staining was performed as we described previously. 18 , 21 , 23 , 24 Using ImageJ, IF was determined by measuring the collagen deposition (blue) in six to eight areas/section to quantify the fibrotic area in mm2, while VF was determined by measuring total vessel area and fibrotic area by imageJ. Percent VF was calculated using (VF/total vessel area) × 100, and graphs were plotted using SigmaPlot software. Representative images were recorded at ×40 magnification.
Muscle function tests
To evaluate the effects of STZ‐induced diabetes with and without BMP‐7, we determined the muscle function using grip strength metre, 25 , 26 rotarod, 27 , 28 and weights test 29 on D‐42.
Grip strength test
The grip strength of the four limbs of the mouse (combined) was assessed using grip strength metre (Columbus Instruments, Columbus, OH) as described by Hakim et al. and Pasteuning‐Vuhman et al. 25 , 26 Mice were placed on the mesh bar, allowed to hold with four limbs, and gently pulled away from the mesh bar in a horizontal fashion. The force applied by the mouse to hold the mesh was recorded as peak force in grammes. The average grip force of six to nine grip strength measurements was calculated and normalized with body weight (g). Data were represented in a bar graph using SigmaPlot software.
Rotarod test
To test the mouse endurance, mice were placed on the rotarod apparatus (Columbus Instruments, Columbus, OH) following the protocol. 27 , 28 Briefly, mice were placed on the rotating rod with an initial speed of 4 rpm and gradually accelerated to 40 rpm over 5 min. Three trials were performed, and each trial lasted approximately 5 to 8 min, with a resting period of 20 min between each trial. Once the mouse had fallen, time and speed were recorded. The results of three trials were averaged and calculated using latency to fall/duration in seconds. Results were plotted as a bar graph using SigmaPlot software.
Weights test
Mice muscle strength was assessed by performing the weights test procedure as reported. 29 Mice were allowed to freely grasp the weights ranging from 15 to 65 g (15, 25, 35, 45, 50, 55, 60, and 65 g) for 3 s by holding the tail. Score [trial × time (TT)] was calculated by taking the number of links in the heaviest weight held for the full 3 s, multiplied by the time (s) it is held. Three independent trials were performed with 5 min rest, followed by average time (weight hold) in seconds calculated, and graph was plotted using SigmaPlot software.
Statistical analysis
Values are presented as mean ± standard error of the mean. To test the significance between the groups, statistical analyses were performed using Student's t‐test and one‐way analysis of variance followed by Tukey test using SigmaPlot software, with P < 0.05 considered as statistically significant.
Results
Bone morphogenetic protein‐7 treatment attenuates diabetes‐induced hyperglycaemia
Figure 1B shows a significantly (P < 0.05) elevated blood glucose levels in diabetic male mice, as compared with control, whereas BMP‐7 treatment significantly (P < 0.05; ~34%) attenuated STZ‐induced hyperglycaemia. However, female mice showed significantly (P < 0.05; Figure 1C) elevated glucose levels in diabetic mice compared with controls, but less than diabetic males (~2.2 times), whereas BMP‐7 treatment showed a non‐significant (~8%) reduction in glucose levels. The insignificant reduction in female mice with BMP‐7 treatment might be due to an insufficient elevation of glucose levels in females. In addition, we also noticed an increase in glucose levels of control male mice compared with females, but the data were statistically insignificant. Further, both male and female mice data showed significantly increased (P < 0.05; Figure 1D) hyperglycaemia in diabetic animals, whereas BMP‐7 treatment significantly (P < 0.05; ~27%) attenuated diabetes‐induced hyperglycaemia. Consistent with our previous studies, 18 BMP‐7 is able to attenuate diabetic hyperglycaemia.
Bone morphogenetic protein‐7 treatment improves diabetes‐induced weight loss
To evaluate the effect of BMP‐7 on weight loss in STZ‐induced diabetes, mice were weighed prior to the treatment as well as at the time of sacrifice (D‐42). A significant (P < 0.05) decrease in body weight was observed in STZ‐administered male mice as compared with the control (Figure 1E), while a non‐significant increase in body weight was observed upon BMP‐7 treatment in STZ‐administered male mice. Similarly, a significant reduction in body weight was observed in STZ‐administered female mice, whereas BMP‐7‐treated mice showed a significant gain in body weight (P < 0.05; Figure 1F). Moreover, STZ‐administered female mice demonstrated an interesting phenomenon of weight loss, which was approximately 50% less than male mice in STZ group, suggesting that the development and progression of diabetes is different in males and females. However, our male and female mice combined data showed a significant (P < 0.05) reduction in body weight in STZ‐administered mice, whereas a significant (P < 0.05) improvement in body weight was observed with BMP‐7 treatment (Figure 1G), suggesting that BMP‐7 treatment attenuated weight loss observed following diabetes development.
Bone morphogenetic protein‐7 treatment inhibits pyroptosis initiator high‐mobility group box 1
Immunohistochemistry staining was performed to evaluate the effects of BMP‐7 treatment on total number of positive cells for pyroptotic initiator HMGB1. In Figure 2i‐A, images demonstrated the presence of a higher number of HMGB1 positive cells in the STZ‐administered group (f–j) compared with control (a–e). Moreover, BMP‐7 treatment significantly (P < 0.05) reduced the HMGB1+ve cells (k–o). Our HMGB1 data in male and female mice showed a significant (P < 0.05; Figure 2i‐B and 2i‐C) increase in number of positive cells in diabetic group, and this increase was significantly (P < 0.05) attenuated with BMP‐7 treatment. Male animal diabetic changes on HMGB1 were similar in pattern as of female animals. Combined data of male and female mice showed a significant (P < 0.05; Figure 2i‐D) increase in the number of positive cells in diabetic animals, whereas a significant (P < 0.05) reduction was observed with BMP‐7 treatment.
Figure 2.
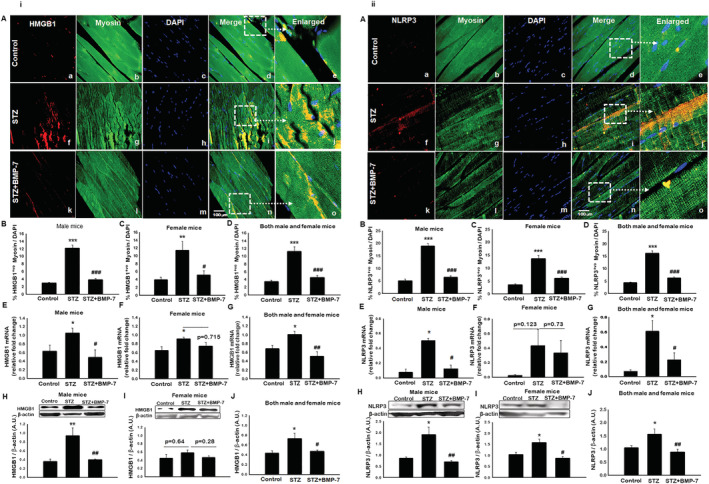
(i) Bone morphogenetic protein‐7 (BMP‐7) treatment inhibits pyroptosis initiator high‐mobility group box 1 (HMGB1). Hyperglycaemia‐induced HMGB1 can initiate the inflammasome activation and subsequent pyroptotic cascade results in pyroptotic cell death. Representative photomicrographs of muscle sections stained with myosin and inflammasome markers. Individual boxes in panel (A) show HMGB1+ve cells shown in red (a, f, k), muscle cells in green (b, g, l), DAPI in blue (c, h, m), and merged images (d, i, n). Scale bar = 100 μm. White dotted boxes and arrows indicate enlarged section of merged images (e, j, o). Quantitative analysis‐derived in bar graphs of HMGB1+ve muscle cells quantified over total DAPI [B: Male mice (n = 8); C: Female mice (n = 7–8); D: Both male and female mice (n = 16)]. Relative fold change in gene expression of HMGB1 [E: Male mice (n = 5–7); F: Female mice (n = 5); G: Both male and female mice (n = 11–12)]. Representative blot and densitometric analysis of HMGB1 [H: Male mice (n = 7); I: Female mice (n = 4–5); J: Both male and female mice (n = 13–14)]. One‐way ANOVA and Tukey tests were performed to assess statistical significance. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. control, #P < 0.05, ##P < 0.01, and ###P < 0.01 vs. STZ. (ii) BMP‐7 treatment decreases nucleotide‐binding oligomerization domain, leucine‐rich repeat and pyrin domain containing protein 3 (NLRP3) inflammasome formation in diabetes. HMGB1‐toll‐like receptor‐4 (TLR4) signalling initiates the NLRP3 inflammasome activation. Representative photomicrographs of muscle sections stained with myosin and inflammasome markers NLRP3. Individual boxes in panel (A) shows NLRP3+ve cells in red (a, f, k), muscle cells in green (b, g, l), DAPI in blue (c, h, m), and merged images (d, i, n). Scale bar = 100 μm. White dotted boxes and arrows indicate enlarged section of merged images (e, j, o). Quantitative analysis‐derived in bar graphs of NLRP3+ve muscle cells quantified over total DAPI [B: Male mice (n = 7–8); C: Female mice (n = 8); D: Both male and female mice (n = 15–16)]. Relative fold change in gene expression of [E: Male mice (n = 5–7); F: Female mice (n = 4–6); G: Both male and female mice (n = 9–11)]. Representative blot and densitometric analysis of NLRP3 [H: Male mice (n = 6–7); I: Female mice (n = 5–6); J: Both male and female mice (n = 14–15)]. One‐way ANOVA and Tukey tests were performed to assess statistical significance. *P < 0.05 and ***P < 0.001 vs. control, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. streptozotocin (STZ).
To strengthen our findings, we performed reverse transcription PCR (RT‐PCR) (gene) and western blot (protein) analysis. A significant (P < 0.05) increase in both gene (Figure 2i‐E) and protein expression (Figure 2i‐H) was observed in diabetic male mice as compared with controls, whereas a significant (P < 0.05) reduction was observed with BMP‐7 treatment. Female mice showed a significant (P < 0.05; Figure 2i ‐F) increase in HMGB1 gene expression as compared with control, whereas BMP‐7 treatment reduced the HMGB1 gene expression, but the data were statistically non‐significant. Western blot analysis data revealed an increase in HMGB1 protein expression in diabetic female mice compared with control (Figure 2i ‐I), but the results are not statistically significant. Additionally, a non‐significant reduction in HMGB1 protein expression was observed with BMP‐7 treatment. Noticeably, we observed that HMGB1 protein expression in diabetic female mice is less compared with male mice. Moreover, combined data of male and female mice showed a significant (P < 0.05; Figure 2i‐G and 2i‐J) increase in HMGB1 gene and protein expression in diabetic mice, which was significantly reduced upon BMP‐7 treatment, suggesting the efficacy of BMP‐7 in attenuating hyperglycaemia‐induced pyroptosis initiator HMGB1.
Bone morphogenetic protein‐7 treatment inhibits toll‐like receptor‐4 expression in diabetes
Published studies suggest the activation of intracellular signalling pathways via TLR4 in presence of DAMP protein HMGB1. 30 , 31 IHC staining was performed to evaluate whether TLR4 expression is up‐regulated in diabetes. We further evaluated effects of BMP‐7 on TLR4 expression. As shown in Figure S1A, representative photomicrographs specify the presence of higher number of +ve cells for TLR4 in diabetic animal group (f–j) as compared with controls (a–e). A significant reduction in the number of TLR4+ve cells (k–o) was observed upon BMP‐7 treatment, suggesting the potential role of BMP‐7 in the inhibition of TLR4 expression. Moreover, quantitative analysis for TLR4 (Figure S1B and S1C) in male and female mice showed a significant increase in number of positive cells in diabetes, whereas BMP‐7 treatment significantly diminished the TLR4 positive cells. Importantly, TLR4 positive cells in diabetic females were lesser (~22%) compared with males. Male and female mice combined data (Figure S1D) showed a significant increase in TLR4 in diabetes, whereas BMP‐7 treatment counteracted TLR4 expression, suggesting the inhibitory potency of BMP‐7.
Bone morphogenetic protein‐7 treatment decreases nucleotide‐binding oligomerization domain, leucine‐rich repeat and pyrin domain containing protein 3 inflammasome formation in diabetes
Nucleotide‐binding oligomerization domain, leucine‐rich repeat and pyrin domain containing protein 3 (NLRP3) is a downstream regulator as published by us and others. 21 , 32 , 33 , 34 Therefore, we evaluated whether HMGB1 initiation would lead to inflammasome formation with TLR4 and NLRP3. To identify the role of BMP‐7 in NLRP3 inflammasome formation, we performed IHC staining followed by gene and protein analysis. The fluorescent images (Figure 2ii‐A) showed significantly (P < 0.05) higher number of NLRP3 positive cells in STZ‐administered mice (f–j) as compared with control (a–e). However, a significant (P < 0.05) reduction in NLRP3+ve cells (k–o) was observed upon BMP‐7 treatment. Further quantitative analysis (Figure 2ii‐B and 2ii ‐C) for males and females showed a significant increase in number of NLRP3 positive cells in STZ‐administered mice, which was significantly reduced upon BMP‐7 treatment. IHC data for both male and female mice (P < 0.05; Figure 2ii‐D) showed a significant increase in NLRP3 positive cell in diabetes mice, and this increased number of positive cells was significantly (P < 0.05) decreased with BMP‐7 treatment.
Next, NLRP3 gene expression (RT‐PCR analysis) was significantly (P < 0.05; Figure 2ii‐E) increased in diabetic male mice as compared with control, whereas BMP‐7 treatment significantly (P < 0.05) reduced diabetes‐induced NLRP3 gene expression. However, female mice showed similar pattern (Figure 2ii‐F) as of males, but the data were not statistically significant.
Western blot analysis revealed a significant (P < 0.05) increase in NLRP3 protein expression in STZ‐administered male and female mice as shown in Figure 2ii‐H and 2ii‐I. BMP‐7 treatment significantly (P < 0.05; Figure 2ii‐H and 2ii‐I) decreased the NLRP3 protein expression. Further, we noticed that the increase in NLRP3 gene and protein expression in diabetes female mice is less compared with male mice, which is consistent with IHC data. Both male and female mice data showed a significant (P < 0.05; Figure 2ii‐G and 2ii‐J) increase in gene and protein expression in diabetes mice, whereas BMP‐7 treatment significantly decreased the inflammasome formation, suggesting its therapeutic efficacy in attenuation of NLRP3 inflammasome formation.
Bone morphogenetic protein‐7 treatment reduces pyroptosis cascade markers caspase‐1, interleukin‐β, and interleukin‐18
Toll‐like receptor‐4 expression and inflammasome formation leads to the activation of pyroptosis cascade such as caspase‐1 and downstream markers IL‐1β and IL‐18. 35 , 36 We first established presence of these markers in diabetic mice tissue and then determined the potential role of BMP‐7 on STZ‐induced pyroptotic cascade in GM tissue. Representative fluorescent images demonstrated significantly (P < 0.05) higher number of pyroptosis markers caspase‐1 (Figure 3i‐A), IL‐1β (Figure 3ii‐A), and IL‐18 (Figure 3iii‐A) +ve cells in STZ administered mice (f–j) as compared with control (a–e). Diabetic mice treated with BMP‐7 showed significantly (P < 0.05) reduced number of +ve cells (k–o) specific for pyroptosis markers caspase‐1, IL‐β, and IL‐18. Our pyroptotic cascade quantitative data for male and female mice showed a significant (P < 0.05; Figure 3i, 3ii, 3iii‐B and 3C) increase in number of positive cells in diabetic group and this increase was significantly (P < 0.05) reduced with BMP‐7 treatment. Combined data of male and female mice showed a significant (P < 0.05; Figure 3i‐D, 3ii‐D, and 3iii‐D) increase in pyroptosis markers in diabetic mice, whereas BMP‐7 treatment significantly (P < 0.05) reduced all pyroptosis‐specific markers.
Figure 3.
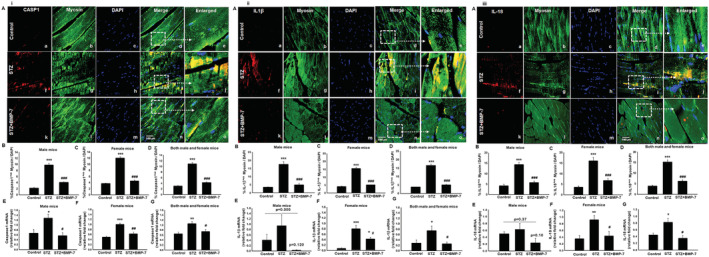
Bone morphogenetic protein‐7 (BMP‐7) treatment reduces pyroptosis cascade markers caspase1, interleukin (IL)‐β, and IL‐18. Activation of nucleotide‐binding oligomerization domain, leucine‐rich repeat and pyrin domain containing protein 3 (NLRP3) inflammasome furthers the pyroptosis cascade. Representative photomicrographs of muscle sections stained with myosin and pyroptosis markers (i: caspase‐1, ii: IL‐1β, and iii: IL‐18; Panel A). Individual boxes in panel (A) show caspase‐1+ve, IL‐1β+ve, and IL‐18+ve cells in red (a, f, k), muscle cells in green (b, g, l), DAPI in blue (c, h, m), and merged images (d, i, n). Scale bar = 100 μm. White dotted boxes and arrows indicate enlarged section of merged images (e, j, o). Quantitative analysis‐derived histogram of (i) caspase‐1+ve, (ii) IL‐1β+ve, and (iii) IL‐18+ve muscle cells quantified over total DAPI [B: Male mice (n = 7–8); C: Female mice (n = 8); D: Both male and female mice (n = 15–16)]. Relative fold change in gene expression of (i) caspase‐1 [E: Male mice (n = 5–6); F: Female mice (n = 4–5); G: Both male and female mice (n = 12)], (ii) IL‐1β [E: Male mice (n = 5); F: Female mice (n = 4–5); G: Both male and female mice (n = 10–11)], and (iii) IL‐18 [E: Male mice (n = 4–5); F: Female mice (n = 4–5); G: Both male and female mice (n = 8–10)]. One‐way ANOVA and Tukey tests were performed to assess statistical significance. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. control, #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. streptozotocin (STZ). Fold change in gene expression was represented as arbitrary units.
To strengthen our findings, we performed RT‐PCR (gene) analysis. A significant (P < 0.05) up‐regulation of caspase‐1 gene expression was observed in diabetic male (Figure 3i‐E) and female mice (Figure 3i‐F) as compared with control, whereas BMP‐7 treatment significantly down‐regulated caspase‐1 expression. However, an insignificant increase in downstream markers IL‐1β (Figure 3ii‐E) and IL‐18 (Figure 3iii‐E) was observed in diabetic male mice vs. control and a non‐significant decrease was observed upon BMP‐7 treatment. Whereas there was a significant (P < 0.05) increase in IL‐1β (Figure 3ii‐F) and IL‐18 (Figure 3iii‐F), gene expressions were observed in diabetic female mice while BMP‐7 treatment significantly (P < 0.05) reduced the gene up‐regulation.
Combined male and female mice data for gene analysis of caspase‐1 (Figure 3i‐G), IL‐1β (Figure 3ii‐G), and IL‐18 (Figure 3iii‐G) showed a significant (P < 0.05) increase in all the pyroptosis markers in diabetes mice, whereas BMP‐7 treatment significantly attenuated all pyroptosis markers. Additionally, we also noticed that except IL‐18, the increase in downstream marker gene expressions in diabetes females is less compared with males.
Bone morphogenetic protein‐7 treatment inhibits pyroptosis executioner gasdermin‐D
Recently, GSDMD has been considered as key pyroptosis executioner. 15 , 37 In Figure S2A, IHC images demonstrated the presence of a higher number of GSDMD positive cells in the diabetic group (f–j) as compared with control (a–e). Moreover, BMP‐7 treatment significantly (P < 0.05) reduced the GSDMD+ve cells (k–o). Our quantitative GSDMD data in male (Figure S2B) and female (Figure S2C) mice showed of significant (P < 0.05) increase in number of positive cells in diabetic group and this increase was significantly (P < 0.05) decreased with BMP‐7 treatment. Diabetic changes of female mice on GSDMD were similar in pattern as of males. Combined data of male and female mice showed a significant (P < 0.05; Figure S2D) increase in the number of positive cells in diabetic animals, whereas a significant (P < 0.05) reduction was observed with BMP‐7 treatment.
To strengthen our findings, we performed RT‐PCR (gene) and western blot (protein) analysis. A significant (P < 0.05) increase in GSDMD gene expression was observed in diabetic male (Figure S2E) and female (Figure S2F) mice as compared with controls, whereas BMP‐7 treatment significantly (P < 0.05) attenuated the GSDMD expression. Western blot analysis data revealed a significant increase in GSDMD protein expression in diabetic male (Figure S2H) and female (Figure S2I) mice compared with control, and this increase was significantly (P < 0.05) reduced upon BMP‐7 treatment. Remarkably, we observed that GSDMD gene and protein expressions in diabetic male mice are less compared with female mice. Both male and female mice data showed a significant (P < 0.05) increase in gene (Figure S2G) and protein (Figure S2J) expression in diabetes mice, whereas BMP‐7 treatment significantly inhibited the pyroptosis executioner GSDMD, suggesting its potency in pyroptosis attenuation.
Bone morphogenetic protein‐7 treatment inhibits diabetes‐induced pro‐inflammatory cytokine interleukin‐6
To understand pyroptosis‐associated pro‐inflammatory cytokine with or without BMP‐7 treatment, we performed ELISA in mice serum samples. As shown in Figure 4i, data analysis for male and female mice revealed a significant (P < 0.05; Figure 4i‐A and 4i‐B) increase in IL‐6 levels in diabetic mice, suggesting inflammatory response. However, a significant (P < 0.05; Figure 4i‐A and 4i‐B) decrease in IL‐6 levels was observed upon BMP‐7 treatment, suggesting the efficacy of BMP‐7 in immune regulation. Moreover, we observed an interesting phenomenon that increased levels of IL‐6 were lesser in diabetes female mice compared with male mice. Our results of male and female mice combined data showed a significant (P < 0.05; Figure 4i‐C) increase in IL‐6 levels in diabetic mice, whereas BMP‐7 treatment significantly (P < 0.05) attenuated the IL‐6 levels.
Figure 4.
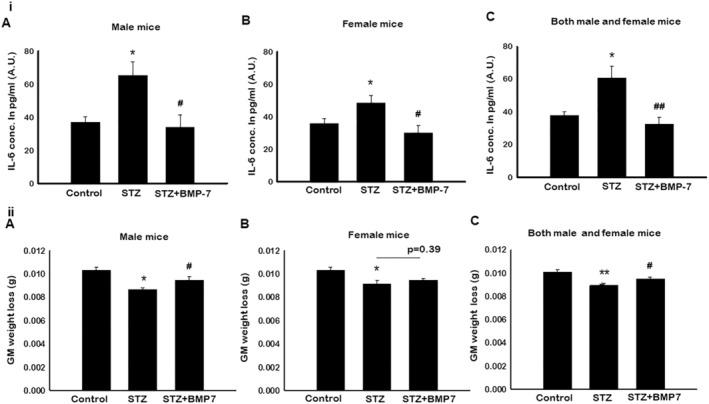
(i) Bone morphogenetic protein‐7 (BMP‐7) inhibits pro‐inflammatory cytokine interleukin (IL)‐6. Enzyme‐linked immunosorbent assay (ELISA) was performed to determine the serum levels of pro‐inflammatory cytokine IL‐6. Bar graphs represent elevated IL‐6 levels in (A) male mice, (B) female mice, and (C) both male and female mice of streptozotocin (STZ)‐administered mice, whereas BMP‐7 treatment potentially reduced the IL‐6 levels on D42. Error bars = mean ± standard error of the mean (SEM). One‐way ANOVA and Tukey tests were performed to assess statistical significance. *P < 0.05 vs. control, #P < 0.05 and ##P < 0.01 vs. STZ, n = 12–14 (both males and females), n = 5–7 (male mice), and n = 6–7 (female mice). (ii) BMP‐7 treatment improves diabetes‐induced sarcopenia. Bar graph demonstrating the ratio of muscle weight‐to‐body weight (in grammes) was significantly decreased in STZ administered on D42. (A: Male mice; B: Female mice; C: Both male and female mice), whereas BMP‐7 treatment significantly increased the muscle mass. Error bars = mean ± SEM. One‐way ANOVA and Tukey tests were performed to assess statistical significance. *P < 0.05 and ***P < 0.001 vs. control, #P < 0.05 vs. STZ, n = 11–16 (both males and females), n = 5–7 (male mice), and n = 5–7 (female mice).
Bone morphogenetic protein‐7 treatment improves diabetes‐induced sarcopenia
To determine the impact of BMP‐7 on GM mass after STZ administration, the ratio of muscle weight to body weight was calculated. Our data show a significantly (P < 0.05; Figure 4ii‐A) developed sarcopenia (decrease in the GM mass) in male mice following STZ treatment as compared with control. A significant improvement in sarcopenia was observed upon BMP‐7 treatment in male mice (P < 0.05; Figure 4ii‐A). Consistent with male mice, a significant (P < 0.05; Figure 4ii‐B) sarcopenia development was observed in STZ‐administered female mice, whereas treatment with BMP‐7 showed a trend of improved sarcopenia, but data were statistically non‐significant. Moreover, both male and female mice combined data (Figure 4ii‐C) showed a significant increase in developed sarcopenia in diabetic animals, which was attenuated with BMP‐7 treatment.
Bone morphogenetic protein‐7 treatment inhibits diabetes‐induced muscle atrophy
Histological staining (haematoxylin and eosin) was performed on GM tissue sections to determine the effect of BMP‐7 on muscle atrophy in diabetes. In Figure 5A, representative photomicrographs demonstrated a significant decrease in GM myocyte area, suggesting muscle atrophy (Figure 5A‐b) in STZ‐administered mice as compared with control (Figure 5A‐a). Following treatment with BMP‐7, a significant (P < 0.05; Figure 5A‐c) increase in GM myocyte area was observed, suggesting decrease in atrophy in diabetic mice. Further, quantitative analysis of male and female mice confirmed that the cell size was significantly (P < 0.05; Figure 5B and 5C) decreased in GM tissues of STZ received mice compared with control, whereas a significant (P < 0.05) increase in muscle cell size was observed with BMP‐7 treatment. Additionally, our male and female mice combined data showed significantly (P < 0.05; Figure 5D) decreased muscle myocyte area in diabetes mice, which was significantly increased upon BMP‐7 treatment (P < 0.05; Figure 5D). Furthermore, to confirm attenuated atrophy by BMP‐7 at the molecular level, we performed RT‐PCR analysis for MuRF1 gene expression, which is considered as a major atrophy gene in skeletal muscle. 38 In STZ‐administered male mice, MuRF1 gene expression was significantly (P < 0.05; Figure 5E) increased as compared with control, whereas BMP‐7 treatment significantly (P < 0.05) reduced STZ‐induced MuRF1 gene expression. Noticeably, the MuRF1 gene expression was not statistically significant in diabetes female mice as compared with control. The exact reason for this discrepancy compared with histological finding on atrophy is not known. However, BMP‐7 treatment significantly (P < 0.05; Figure 5F) diminished the MuRF1 expression in female diabetic mice. Both male and female mice data showed significantly (P < 0.05; Figure 5G) induced atrophy in diabetes animals, whereas BMP‐7 treatment attenuated the diabetes‐induced muscle atrophy gene MuRF1. This set of data suggests the presence of atrophy in diabetes, which is attenuated with BMP‐7.
Figure 5.
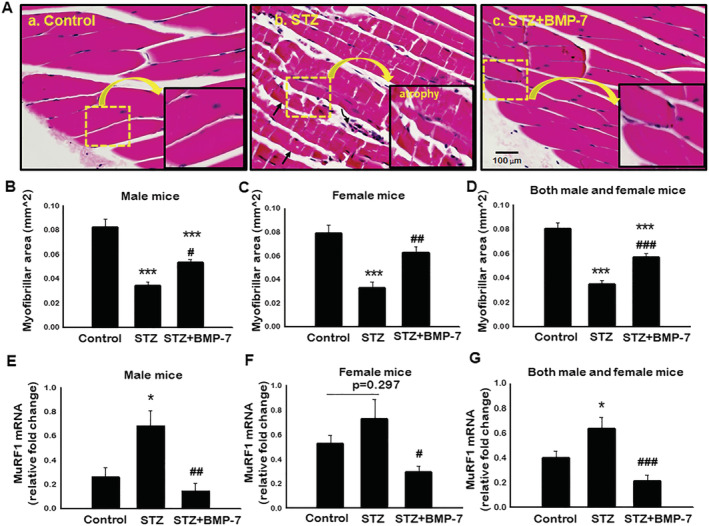
Bone morphogenetic protein‐7 (BMP‐7) treatment inhibits diabetes‐induced muscle atrophy. (A) Representative photomicrographs (×40) are sections stained with haematoxylin and eosin to detect atrophy in gastrocnemius muscle on Day 42 after streptozotocin (STZ) administration in control and experimental groups. Stained sections were quantified at ×20 magnification and magnified for visualization of atrophy. Bar graph represents quantitative analysis for muscle atrophy [B: Male mice (n = 7–8); C: Female mice (n = 8); D: Both male and female mice (n = 16)]. Scale bar = 100 μm. Relative fold change in MuRF1 gene expression in bar graph [E: Male mice (n = 6–7); F: Female mice (n = 4–6); G: Both male and female mice (n = 11–14)]. Error bars = mean ± standard error of the mean. One‐way ANOVA and Tukey tests were performed to assess statistical significance. *P < 0.05 and ***P < 0.001 vs. control, #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. STZ.
Bone morphogenetic protein‐7 treatment significantly reduces diabetes‐induced muscle fibrosis
Skeletal muscle fibrosis is an adverse remodelling mechanism that occurs in diabetes. 18 To determine whether BMP‐7 attenuates IF and VF in STZ‐induced GM, Masson's trichrome staining was performed to quantify the presence of collagen. Blue area in representative photomicrographs (Figure 6) demonstrated significantly (P < 0.05) increased IF (Figure 6A‐b) and VF (Figure 6B‐e) in STZ‐administered mice as compared with control (Figure 6A‐a and 6B‐d). Following BMP‐7 treatment, a significant (P < 0.05; Figure 6A‐c and 6B‐f) reduction in IF and VF was observed. Furthermore, our quantitative IF data were significantly increased (P < 0.05; Figure 6C–6E) in STZ‐induced diabetes mice compared with controls, whereas BMP‐7 treatment significantly (P < 0.05) reduced IF in both male and female mice. Our VF quantitative data were significantly (P < 0.05; Figure 6F–6H) increased in STZ‐administered male and female mice as compared with control, whereas BMP‐7 treatment statistically reduced VF in GM in diabetic males and females. These results suggest that reduction in collagen deposition and fibrosis further potentiate the therapeutic efficacy of BMP‐7 in diabetic animals.
Figure 6.
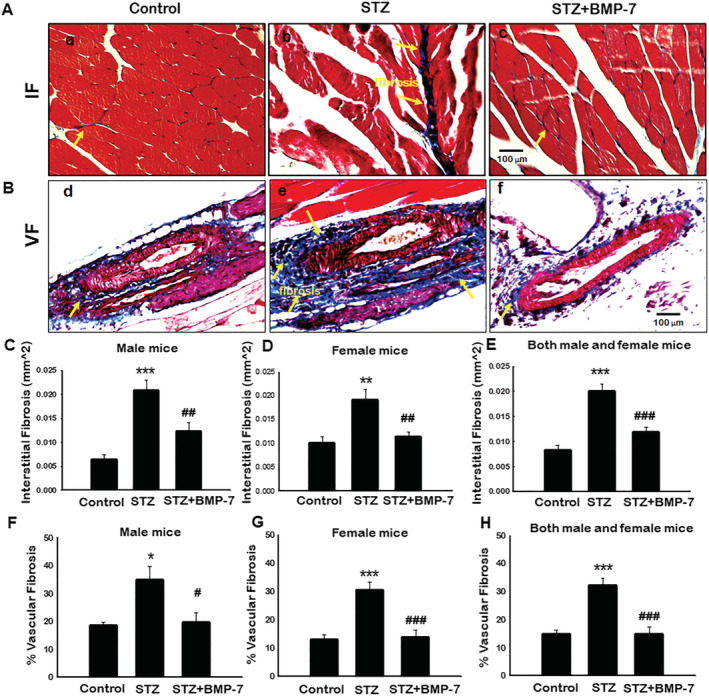
Bone morphogenetic protein‐7 (BMP‐7) treatment significantly reduces diabetes‐induced muscle fibrosis. Representative images of Masson's trichrome demonstrated interstitial (Panel A) and vascular fibrosis (Panel B) in gastrocnemius muscle on Day 42 after streptozotocin (STZ) administration in control and experimental groups. Stained sections were quantified at ×20 magnification and magnified for visualization of interstitial and vascular fibrosis. Quantitative analysis for intestinal fibrosis [C: Male mice (n = 7–8); D: Female mice (n = 7–8); E: Both male and female mice (n = 14–16)]. Percentage of vascular fibrosis quantified over the vessel area [F: Male mice (n = 5–6); G: Female mice (n = 6–8); H: Both male and female mice (n = 11–14)]. One‐way ANOVA and Tukey tests were performed to assess statistical significance. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. control, #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. STZ. Scale bar = 100 μm.
Bone morphogenetic protein‐7 treatment improves diabetes‐induced muscle dysfunction
To assess whether BMP‐7 treatment can improve the muscle function, animals were subjected to three different types of tests: (1) grip strength for four limbs, (2) rotarod test, and (3) weights test for forelimb muscle strength.
Grip strength analysis showed a significant (P < 0.05) increase in grip strength of control female mice (~12%) as compared with control males. Further, our data showed a significant (P < 0.05; Figure 7A and 7B) deficit in grip strength in diabetic male (~13%) and female (~11%) mice as compared with respective controls, whereas BMP‐7 treatment significantly (P < 0.05) improved the grip strength. Noticeably, we observed a significant (P < 0.05) improvement in grip strength in BMP‐7‐treated males (~29%) and females (~8%) as compared with diabetic mice. Combined data of male and female mice showed significantly (P < 0.05; Figure 7C) reduced grip strength in diabetic mice, which was significantly improved with BMP‐7 treatment.
Figure 7.
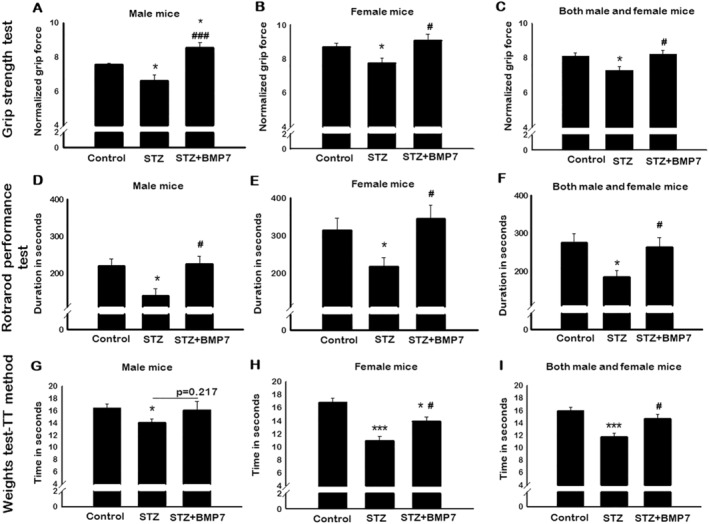
Bone morphogenetic protein‐7 (BMP‐7) treatment improves diabetes‐induced muscle dysfunction. BMP‐7 treatment improves muscle function under hyperglycaemic conditions. To evaluate this on Day 42 after streptozotocin (STZ) administration, animals were subjected to different muscle function tests. Bar graphs represent the quantification and analysis for four‐limb grip strength [A: Male mice (n = 6–7); B: Female mice (n = 6–7); C: Both male and female mice (n = 14–15)], rotarod performance test [D: Male mice (n = 5–6); E: Female mice (n = 5–7); F: Both male and female mice (n = 12–13)], and weights test‐TT method [G: Male mice (n = 5); H: Female mice (n = 6); I: Both male and female mice (n = 11–15)]. Error bars = mean ± standard error of the mean. One‐way ANOVA and Tukey tests were performed to assess statistical significance. *P < 0.05 and ***P < 0.001 vs. control, #P < 0.05 and ###P < 0.001 vs. STZ.
For the rotarod test, a significant (P < 0.05) increase in latency to fall was observed in female mice (~42%) as compared with control males. Then, a significant (P < 0.05; Figure 7D and 7E) decrease in muscle function performance, that is, the number of seconds stayed on a rotating rod (latency to fall) was observed in STZ‐administered male (~37%) and female (~30%) mice as compared with control. On the other hand, a significant (P < 0.05) improvement in diabetic mice muscle function was observed with BMP‐7 treatment, suggesting the potential impact of BMP‐7 treatment on muscle dysfunction. Our result of combined data for male and female mice showed a significant (P < 0.05, Figure 7F) decrease in muscle function in diabetic mice, whereas BMP‐7 treatment significantly (P < 0.05) improved the diabetes‐induced muscle dysfunction.
For the weights test, a significant (P < 0.05) decrease in forelimb muscle strength was observed in STZ‐administered male mice as compared with control (Figure 7G) using trial × time method. BMP‐7 treatment showed an increase in the gain of muscle strength, but data were statistically non‐significant. A significant (P < 0.05; Figure 7H) decrease in muscle strength was observed in STZ‐administered female mice as compared with control. Female mice lost 16% of muscle strength compared with male counterparts. Importantly, BMP‐7 treatment significantly restored the diabetes‐induced muscle dysfunction. Male and female mice combined data showed a significant (P < 0.05; Figure 7I) reduction in muscle strength in diabetic animals, whereas BMP‐7 treatment significantly (P<0.05; Figure 7I) improved the forelimb muscle strength. These results confirm that BMP‐7 significantly counteracts diabetes‐induced sarcopenia and atrophy further muscle dysfunction.
Discussion
Apoptosis and oxidative stress are key players in skeletal muscle sarcopenia, cachexia, and in adverse muscle remodelling (fibrosis and atrophy) in aging, cancer, and diabetes. 1 , 2 , 39 Recent data show that inflammation could be a triggering agent in the development and progression of sarcopenia and cachexia. 40 , 41 , 42 Moreover, in the diabetic condition, hyperglycaemia dominates in the blood stream as well as various diabetic organs such as muscle and causes diabetic muscle myopathy. 3 , 43 Therefore, hyperglycaemia and inflammation are detrimental in diabetic muscle myopathy. However, close association between hyperglycaemia and induced inflammation in diabetic muscle myopathy is unknown. Additional questions that further arise are as follows: (1) what the cellular mechanisms involved between the hyperglycaemia and inflammation are and (2) whether those cellular mechanisms are the key players that cause sarcopenia, fibrosis, and atrophy that lead to diabetic muscle dysfunction and chronic muscle myopathy. The current study is undertaken to establish the close link between hyperglycaemia and inflammation as well as the involved cellular level mechanisms that induce sarcopenia, fibrosis, and muscle dysfunction. Furthermore, we will provide strong evidence on the use of BMP‐7, an osteoporosis drug that attenuated induced hyperglycaemia, inhibited cellular mechanisms involved in the inflammatory process, inhibited sarcopenia, fibrosis, and improved muscle dysfunction.
In the present study, we developed an STZ‐induced diabetic mouse model that shows a significant increase in hyperglycaemia and weight loss. These data are consistent with previously published reports by us and others. 2 , 18 Recent data show that diabetic patients and animal models have increased hyperglycaemia associated with increased levels of inflammation. 44 , 45 Increase in hyperglycaemia has also been observed in lipopolysaccharide‐induced septic rat model. 46 This increase in hyperglycaemia and inflammation in the rat model is consistent with critically ill sepsis patients. 47 Increased sterile inflammation has been involved in the release of DAMPs such as HMGB1. In addition, HMGB1 has been observed in these patients and in the acute lung injury rat model, suggesting its association with inflammation. HMGB1 has been reported to be present in vascular smooth muscle cells, endothelial cells, and monocytes in normal homeostasis. 48 However, an increased level of HMGB1 has also been observed in various tissue trauma, ischaemia, and inflammatory diseases. 49 , 50 Consistent with these studies, we report in the present findings that HMGB1 was significantly increased in diabetic skeletal muscle cells. Moreover, we also report that increased HMGB1 in diabetic muscle myopathy induces sterile inflammation. To further understand the significance of increased HMGB1 in diabetic muscle cells, we investigated the presence of its receptor, TLR4. Interestingly, we observed significantly increased levels of TLR4 (Figure S1), confirming that the increase in HMGB1 is associated with increased levels of TLR4 that may initiate further cell signalling and sterile inflammation. Therefore, we investigated activation of inflammation‐induced cell death called pyroptosis in diabetic muscle myopathy. Pyroptosis is a newly investigated inflammation‐induced cell death that was reported to occur with bacterial infections. 51 , 52 , 53 Recently, we and others have reported that doxorubicin (anti‐cancer drug) induced cardiomyopathy, myocardial ischaemia, and cancer involve sterile inflammation, which induces pyroptosis. 12 , 21 , 32 However, it was never investigated whether increased levels of hyperglycaemia can induce the HMGB1 via TLR4 that triggers sterile inflammation and forms the NLRP3‐associated inflammasome and the downstream pathway of pyroptotic cell death in diabetic skeletal muscle.
We report that HMGB1–TLR4 signalling gives rise to NLRP3‐mediated inflammasome formation (Figure 2ii). This inflammasome formation has been reported in the in vitro doxorubicin‐treated H9C2 cells 32 and Sol8 cells, 54 Porphyromonas gingivalis‐stimulated macrophages, 55 as well as in vivo models of ischaemia reperfusion injury, 13 myocardial infarction, 56 dox‐induced cardiomyopathy, 21 and brain diseases. 57 Our data are in agreement with these findings on sterile inflammation and formed NLRP3 inflammasome as previously published in vitro and in vivo reports. 13 , 21 , 32 , 54 , 56 However, it would be interesting in the future studies to examine whether HMGB1–TLR4 complex formation contributes differently compared with the HMGB1–TLR4 signalling pathway in diabetic myopathy.
Moreover, these studies suggest that formed inflammasome triggers up‐regulation of pyroptosis markers caspase‐1, IL‐1β, and IL‐18 that cause pore formation in the cells and induce cell death pyroptosis. To confirm the role of NLRP3 inflammasome in muscle pyroptosis, we examined the cascade of pyroptotic cell death markers caspase‐1, IL‐1β, IL‐18, and GSDMD, using IHC, western blotting, and RT‐PCR techniques. We observed a significant increase in pyroptosis markers caspase‐1, IL‐1β, and IL‐18 (Figure 3), suggesting the presence of the new form of cell death pyroptosis in diabetic muscle cells. Our data on pyroptosis death markers are consistent with previously published reports on bacteria‐induced pyroptosis in macrophages and sterile inflammation‐induced pyroptosis in the heart, kidney, and brain. 21 , 58 , 59 Up‐regulation of caspase‐1 and release of IL‐1β mediated through caspase‐1 and a substrate of GSDMD, are commonly reported in bacterial‐induced pyroptosis in macrophages 60 ; however, it remains elusive whether GSDMD is up‐regulated to execute pyroptosis in sterile inflammation‐induced diabetic muscle cell pyroptosis. Our data shows an up‐regulation in diabetic muscle cells of the executor of pyroptosis, GSDMD (Figure S2), as reported by others in macrophages, which further confirms the presence of muscle cell pyroptosis. 61
Previous studies have shown that HMGB1 induces sterile inflammation in diseases such as septic shock, ischaemia, and various neurological disorders including Parkinson's, multiple sclerosis, and Alzheimer's. 48 , 62 , 63 , 64 , 65 , 66 , 67 However, to confirm whether HMGB1‐triggers inflammation in diabetic muscle myopathy, we studied the presence of inflammatory cytokine IL‐6. Data reported in this study confirm the significant increase in pro‐inflammatory cytokine IL‐6 (Figure 4i) and presence of HMGB1‐induced sterile inflammation. The increased level of IL‐6 is detrimental due to its association in the development and progression of diabetic cardiomyopathy, inflammatory disease, and lung dysfunction. 68 , 69 , 70 Therefore, our data on inflammatory IL‐6 are in agreement with these published reports on sterile inflammation.
Next, we determined the functional significance of pyroptosis and inflammation in the diabetic muscle. To establish that this form of cell death has significance in diabetic muscle myopathy, we determined muscle weight loss, sarcopenia, muscle cell atrophy, fibrosis, and muscle dysfunction. In the present study, we report that pyroptosis occurring in diabetic muscle cells shows a significant loss of muscle mass. Then, we examined whether loss of muscle mass leads to structural modification in the skeletal muscle such as atrophy and fibrosis and further contributes in sarcopenia development and progression. Noticeably, we observed a significant decrease in loss of myofibrillar area and increased levels of MuRF1 gene, suggesting the presence of muscle atrophy. Increased levels of HMGB1‐induced pyroptosis and inflammation‐mediated muscle loss give rise to myofibroblast cell proliferation that provides structural adverse remodelling and stiffness in the diseased organ; therefore, we found presence of IF and VF following pyroptosis in the diabetic skeletal muscle. These data strongly suggest that a series of pathological events such as pyroptosis, sarcopenia, and adverse muscle remodelling could lead to muscle dysfunction. Further, our muscle dysfunction data showed a significant decrease in muscle function and confirmed the functional significance of these series of pathological events in STZ‐induced diabetic muscle myopathy. Our muscle dysfunction data are consistent with earlier published studies, which implied that sarcopenia, muscle atrophy, and fibrosis are key contributors in skeletal muscle dysfunction. 39 , 71 , 72 , 73
Moreover, we mentioned above that HMBG1‐induced pyroptosis and sterile inflammation leading to sarcopenia, atrophy, and muscle dysfunction are the major contributors in the development and progression of diabetic muscle myopathy. Therefore, targeting HMGB1 directly or with a growth factor that can attenuate sterile inflammation and pyroptosis, which further attenuates a series of pathological events, could be a potential therapeutic agent to treat diabetic muscle myopathy, as there are limited options to treat this condition. We explored the potential of BMP‐7, a commonly used drug to treat osteoporosis in patients, and was examined in animal models of atherosclerosis, where it was found to polarize pro‐inflammatory M1 macrophages into anti‐inflammatory M2 macrophages and decrease atherosclerosis. 19 Moreover, BMP‐7 has also been used to treat inflammation and fibrosis in kidneys. 74 However, the role of BMP‐7 in the inhibition of HMGB1, pyroptosis, sarcopenia, fibrosis, atrophy, and muscle dysfunction in diabetic muscle myopathy has never been established. We treated STZ‐induced diabetic animals with BMP‐7 and examined the levels of hyperglycaemia, HMGB1, and sterile inflammation. Our data show a significant decrease in hyperglycaemia and sterile inflammation, which is consistent with our previously published report in diabetic cardiomyopathy. 18 Next, we examined levels of HMGB1 and its receptor, TLR4, which were significantly reduced along with the NLRP3 inflammasome and pyroptosis markers, suggesting that BMP‐7 targets HMGB1 and its downstream pathway of pyroptosis (Figures 2 and 3). These novel findings reported raise further questions on the involvement of cell signalling pathways. In this report, involvement of cell signalling pathways was not examined. However, we anticipate that this opens a new avenue for investigation in the future studies in this subject matter by our lab or others. Our fibrosis data show a significant reduction following BMP‐7 treatment. This reduced fibrosis following BMP‐7 treatment is in agreement with previously published report in kidney disease. 74 Interestingly, the present study shows that BMP‐7 treatment significantly reduces muscle atrophy and sarcopenia while improving diabetic muscle dysfunction (Figure 8), which could be a future therapeutic drug to treat diabetic patients suffering from muscle myopathy.
Figure 8.
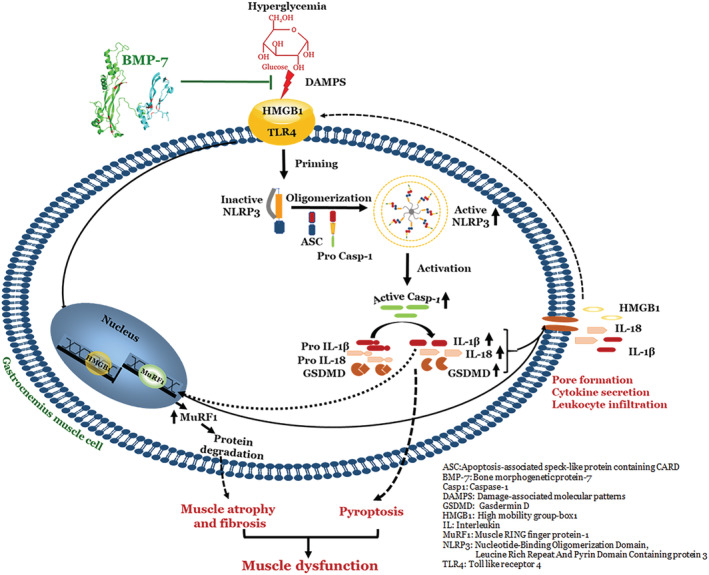
Schematic representation of overall study: Bone morphogenetic protein‐7 (BMP‐7) attenuates diabetes‐induced pyroptosis, muscle atrophy, and fibrosis.
Understanding the effects of sex differences on aging, disease development, and progression is a current area of investigation. Specifically, it has been identified that sex hormones cause the difference in anabolic and catabolic muscle rate in younger women who experience menopause. 75 Recent data suggest that in muscle regeneration, sex hormone testosterone acts via androgen receptor and enhances anabolic cell signalling in skeletal muscle cell. 76 Additionally, it has been observed that oestrogen, another active hormone that plays a role in skeletal muscle cell catabolic and anabolic processes, decreases muscle atrophy, oxidative stress, and inflammation in post‐menopausal women. 77 , 78 The current study does not investigate the role of testosterone or oestrogen in menopausal animals. This study suggests that female animals at their young age, between 10–12 weeks, show lower levels of hyperglycaemia, weight loss, inflammation, myofibrillar loss, and atrophy‐specific MuRF1 gene compared with male animals of the same age group. This set of data suggests that females have a better recovery rate to improve hyperglycaemia, atrophy, and weight loss in diabetes. However, the exact reason for this improved recovery in diabetic females is not well understood or investigated in the present study. Nevertheless, based on published reports, we state that oestrogen plays a pivotal role to decrease atrophy, oxidative stress, and inflammation, which is in agreement with our findings. Therefore, this report starts a new area of investigation to understand whether oestrogen directly initiates cell signalling and plays a role in the recovery of diabetic pathological alterations. Noticeably, we observed the same level of HMGB1–TLR4 signalling, inflammasome formation, and pyroptosis in male and female animals. Here, we could postulate that up‐regulated hyperglycaemia and inflammation triggered the process of pyroptosis, but oestrogen may not target the specific pathway of pyroptosis. This could be a potential reason that a difference in pyroptosis was not seen in these sex differences. Additionally, there could be more reasons that need further investigation to understand why the levels of pyroptosis are same in both sexes. Moreover, we observed that skeletal muscle function measurements with grip strength and rotarod tests were superior in diabetic female group compared with the male counterpart. As mentioned above, the decrease in hyperglycaemia, inflammation, and atrophy in female animals could be a potential reason to see improved muscle function. The effects of BMP‐7 on the improvement of hyperglycaemia and muscle function were the same in both sexes. Importantly, this study describes the association between hyperglycaemia‐induced pyroptosis through the initiation of HMGB1–TLR4 signalling, subsequent inflammasome activation, and downstream up‐regulation of pyroptosis in diabetic muscle cells. So, this study does not address the exact causative mechanistic role of BMP‐7 in the attenuation of hyperglycaemia and inflammatory cell death. Therefore, future studies are required to establish a causative mechanistic relationship on hyperglycaemia‐induced pyroptosis and its attenuation by BMP‐7 in diabetic muscle myopathy. Moreover, this study design determines the effect of BMP‐7 in STZ treatment and for 2 days after; therefore, future studies are warranted to examine whether BMP‐7 is effective in the developed acute and chronic stages of diabetic myopathy.
In conclusion, we report for the first time, to the best of our knowledge, that diabetes induces hyperglycaemia and inflammation, which triggers a cellular pathway mediated through up‐regulation of HMGB1 via TLR4 receptor. This in turn further initiates NLRP3 inflammasome and downstream pathway of pyroptosis, which is executed by GSDMD and pore formation. These pyroptotic cells release inflammatory cytokines IL‐18 and IL‐6 that further cause atrophy, adverse muscle remodelling, and muscle dysfunction. We also report that females recover much faster in hyperglycaemia, inflammation, atrophy, and muscle function. This study opens new areas of investigation: (1) what cell signalling mechanisms contribute to further pyroptosis, (2) how oestrogen plays a role in diabetes, and (3) whether oestrogen could be a future therapy for diabetes in females. Moreover, we provide the data and postulate that BMP‐7, which was never tested to treat HMGB1‐mediated pyroptosis, could be a future therapeutic agent to treat diabetes.
Author contributions
D.K.S. designed and supervised the study. A.N.C performed the experiments, analysed data, prepared figures, and drafted the manuscript. D.K.S revised the manuscript and approved the final version of the manuscript.
Conflict of interest
The authors declare no competing interests.
Funding
This study was supported in part by National Institutes of Health grant 1R01DK120866‐01 to D.K.S.
Supporting information
Figure S1. BMP‐7 Treatment Inhibits TLR4 Expression in Diabetes.
Figure S2: BMP‐7 Treatment Inhibits Pyroptosis Executioner GSDMD
Table S1: List of Antibodies and primers used for the study.
Acknowledgements
The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. Moreover, the authors would like to thank Zahra Tavakoli Dargani, Dr Ibrahim Elmadbouh, and Kaley Garner for technical assistance and Fatima Bianca Dessouki for proofreading of the manuscript.
Aluganti Narasimhulu C., and Singla D. K. (2021) Amelioration of diabetes‐induced inflammation mediated pyroptosis, sarcopenia, and adverse muscle remodelling by bone morphogenetic protein‐7, Journal of Cachexia, Sarcopenia and Muscle, 12, 403–420, 10.1002/jcsm.12662
References
- 1. Park SW, Goodpaster BH, Strotmeyer ES, Kuller LH, Broudeau R, Kammerer C, et al. Accelerated loss of skeletal muscle strength in older adults with type 2 diabetes: the health, aging, and body composition study. Diabetes Care 2007;30:1507–1512. [DOI] [PubMed] [Google Scholar]
- 2. Park SW, Goodpaster BH, Lee JS, Kuller LH, Boudreau R, de Rekeneire N, et al. Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care 2009;32:1993–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aragno M, Mastrocola R, Catalano MG, Brignardello E, Danni O, Boccuzzi G. Oxidative stress impairs skeletal muscle repair in diabetic rats. Diabetes 2004;53:1082–1088. [DOI] [PubMed] [Google Scholar]
- 4. Bloomgarden ZT. Diabetic nephropathy. Diabetes Care 2005;28:745–751. [DOI] [PubMed] [Google Scholar]
- 5. Bloomgarden ZT. Diabetic retinopathy and diabetic neuropathy. Diabetes Care 2007;30:760–765. [DOI] [PubMed] [Google Scholar]
- 6. Wang X, Hu Z, Hu J, Du J, Mitch WE. Insulin resistance accelerates muscle protein degradation: activation of the ubiquitin‐proteasome pathway by defects in muscle cell signaling. Endocrinology 2006;147:4160–4168. [DOI] [PubMed] [Google Scholar]
- 7. Frier BC, Noble EG, Locke M. Diabetes‐induced atrophy is associated with a muscle‐specific alteration in NF‐kappaB activation and expression. Cell Stress Chaperones 2008;13:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marzetti E, Leeuwenburgh C. Skeletal muscle apoptosis, sarcopenia and frailty at old age. Exp Gerontol 2006;41:1234–1238. [DOI] [PubMed] [Google Scholar]
- 9. Kruse R, Vind BF, Petersson SJ, Kristensen JM, Hojlund K. Markers of autophagy are adapted to hyperglycaemia in skeletal muscle in type 2 diabetes. Diabetologia 2015;58:2087–2095. [DOI] [PubMed] [Google Scholar]
- 10. Ábrigo J, Elorza AA, Riedel CA, Vilos C, Simon F, Cabrera D, et al. Role of oxidative stress as key regulator of muscle wasting during cachexia. Oxid Med Cell Longev 2018;2018:2063179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li YP, Chen Y, Li AS, Reid MB. Hydrogen peroxide stimulates ubiquitin‐conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am J Physiol Cell Physiol 2003;285:C806–C812. [DOI] [PubMed] [Google Scholar]
- 12. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 2009;7:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qiu Z, Lei S, Zhao B, Wu Y, Su W, Liu M, et al. NLRP3 inflammasome activation‐mediated pyroptosis aggravates myocardial ischemia/reperfusion injury in diabetic rats. Oxid Med Cell Longev 2017;2017:9743280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Man SM, Karki R, Kanneganti T‐D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev 2017;277:61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Man SM, Kanneganti T‐D. Gasdermin D: the long‐awaited executioner of pyroptosis. Cell Res 2015;25:1183–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, et al. Antioxidants prevent health‐promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A 2009;106:8665–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith RC, Lin BK. Myostatin inhibitors as therapies for muscle wasting associated with cancer and other disorders. Curr Opin Support Palliat Care 2013;7:352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Urbina P, Singla DK. BMP‐7 attenuates adverse cardiac remodeling mediated through M2 macrophages in prediabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 2014;307:H762–H772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Singla DK, Singla R, Wang J. BMP‐7 treatment increases M2 macrophage differentiation and reduces inflammation and plaque formation in Apo E‐/‐ mice. PloS one 2016;11:e0147897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rocher C, Singla DK. SMAD‐PI3K‐Akt‐mTOR pathway mediates BMP‐7 polarization of monocytes into M2 macrophages. PloS one 2013;8:e84009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Singla DK, Johnson TA, Tavakoli Dargani Z. Exosome treatment enhances anti‐inflammatory M2 macrophages and reduces inflammation‐induced pyroptosis in doxorubicin‐induced cardiomyopathy. Cell 2019;8:1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singla DK. Akt‐mTOR pathway inhibits apoptosis and fibrosis in doxorubicin‐induced cardiotoxicity following embryonic stem cell transplantation. Cell Transplant 2015;24:1031–1042. [DOI] [PubMed] [Google Scholar]
- 23. Singla DK, Lyons GE, Kamp TJ. Transplanted embryonic stem cells following mouse myocardial infarction inhibit apoptosis and cardiac remodeling. Am J Physiol Heart Circ Physiol 2007;293:H1308–H1314. [DOI] [PubMed] [Google Scholar]
- 24. Yan B, Singla RD, Abdelli LS, Singal PK, Singla DK. Regulation of PTEN/Akt pathway enhances cardiomyogenesis and attenuates adverse left ventricular remodeling following thymosin beta4 overexpressing embryonic stem cell transplantation in the infarcted heart. PloS one 2013;8:e75580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hakim CH, Li D, Duan D. Monitoring murine skeletal muscle function for muscle gene therapy. Methods Mol Biol 2011;709:75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pasteuning‐Vuhman S, Putker K, Tanganyika‐de Winter CL, Boertje‐Van Der Meulen JW, Van Vliet L, Overzier M, et al. Natural disease history of mouse models for limb girdle muscular dystrophy types 2D and 2F. PloS one 2017;12:e0182704‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beastrom N, Lu H, Macke A, Canan BD, Johnson EK, Penton CM, et al. mdx((5)cv) mice manifest more severe muscle dysfunction and diaphragm force deficits than do mdx Mice. Am J Pathol 2011;179:2464–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mandillo S, Heise I, Garbugino L, Tocchini‐Valentini GP, Giuliani A, Wells S, et al. Early motor deficits in mouse disease models are reliably uncovered using an automated home‐cage wheel‐running system: a cross‐laboratory validation. Dis Model Mech 2014;7:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deacon RM. Measuring the strength of mice. J Vis Exp 2013;2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Zoelen MA, Yang H, Florquin S, Meijers JC, Akira S, Arnold B, et al. Role of toll‐like receptors 2 and 4, and the receptor for advanced glycation end products in high‐mobility group box 1‐induced inflammation in vivo. Shock 2009;31:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Furlani D, Donndorf P, Westien I, Ugurlucan M, Pittermann E, Wang W, et al. HMGB‐1 induces c‐kit+ cell microvascular rolling and adhesion via both toll‐like receptor‐2 and toll‐like receptor‐4 of endothelial cells. J Cell Mol Med 2012;16:1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tavakoli Dargani Z, Singla DK. Embryonic stem cell‐derived exosomes inhibit doxorubicin‐induced TLR4‐NLRP3‐mediated cell death‐pyroptosis. Am J Physiol Heart Circ Physiol 2019;317:H460–H471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Luo B, Huang F, Liu Y, Liang Y, Wei Z, Ke H, et al. NLRP3 inflammasome as a molecular marker in diabetic cardiomyopathy. Front Physiol 2017;8:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Singh LP. The NLRP3 inflammasome and diabetic cardiomyopathy: editorial to: "Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model" by Beibei Luo et al. Cardiovasc Drugs Ther 2014;28:5–6. [DOI] [PubMed] [Google Scholar]
- 35. Miao EA, Rajan JV, Aderem A. Caspase‐1‐induced pyroptotic cell death. Immunol Rev 2011;243:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zeng C, Wang R, Tan H. Role of pyroptosis in cardiovascular diseases and its therapeutic implications. Int J Biol Sci 2019;15:1345–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shi J, Gao W, Shao F. Pyroptosis: gasdermin‐mediated programmed necrotic cell death. Trends Biochem Sci 2017;42:245–254. [DOI] [PubMed] [Google Scholar]
- 38. Bodine SC, Baehr LM. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin‐1. Am J Physiol Endocrinol Metab 2014;307:E469–E484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kalyani RR, Corriere M, Ferrucci L. Age‐related and disease‐related muscle loss: the effect of diabetes, obesity, and other diseases. Lancet Diabetes Endocrinol 2014;2:819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ebisui C, Tsujinaka T, Morimoto T, Kan K, Iijima S, Yano M, et al. Interleukin‐6 induces proteolysis by activating intracellular proteases (cathepsins B and L, proteasome) in C2C12 myotubes. Clin Sci (Lond) 1995;89:431–439. [DOI] [PubMed] [Google Scholar]
- 41. Tsujinaka T, Ebisui C, Fujita J, Kishibuchi M, Morimoto T, Ogawa A, et al. Muscle undergoes atrophy in association with increase of lysosomal cathepsin activity in interleukin‐6 transgenic mouse. Biochem Biophys Res Commun 1995;207:168–174. [DOI] [PubMed] [Google Scholar]
- 42. Dalle S, Rossmeislova L, Koppo K. The role of inflammation in age‐related sarcopenia. Front Physiol 2017;8:1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tsutsui H, Kinugawa S, Matsushima S, Yokota T. Oxidative stress in cardiac and skeletal muscle dysfunction associated with diabetes mellitus. J Clin Biochem Nutr 2011;48:68–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 2006;116:1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marques‐Vidal P, Schmid R, Bochud M, Bastardot F, Von Känel R, Paccaud F, et al. Adipocytokines, hepatic and inflammatory biomarkers and incidence of type 2 diabetes. the CoLaus study. PloS one 2012;7:e51768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hagiwara S, Iwasaka H, Hasegawa A, Koga H, Noguchi T. Effects of hyperglycemia and insulin therapy on high mobility group box 1 in endotoxin‐induced acute lung injury in a rat model. Crit Care Med 2008;36:2407–2413. [DOI] [PubMed] [Google Scholar]
- 47. Frankenfield DC, Oniert LA, Badellino MM, Wiles CE III, Bagley SM, Goodarzi S, et al. Correlation between measured energy expenditure and clinically obtained variables in trauma and sepsis patients. JPEN J Parenter Enteral Nutr 1994;18:398–403. [DOI] [PubMed] [Google Scholar]
- 48. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 2011;29:139–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Venereau E, Schiraldi M, Uguccioni M, Bianchi ME. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol Immunol 2013;55:76–82. [DOI] [PubMed] [Google Scholar]
- 50. Izuishi K, Tsung A, Jeyabalan G, Critchlow ND, Li J, Tracey KJ, et al. Cutting edge: high‐mobility group box 1 preconditioning protects against liver ischemia‐reperfusion injury. J Immunol 2006;176:7154–7158. [DOI] [PubMed] [Google Scholar]
- 51. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 2005;73:1907–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Obregon C, Dreher D, Kok M, Cochand L, Kiama GS, Nicod LP. Human alveolar macrophages infected by virulent bacteria expressing SipB are a major source of active interleukin‐18. Infect Immun 2003;71:4382–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sansonetti PJ, Phalipon A, Arondel J, Thirumalai K, Banerjee S, Akira S, et al. Caspase‐1 activation of IL‐1beta and IL‐18 are essential for Shigella flexneri‐induced inflammation. Immunity 2000;12:581–590. [DOI] [PubMed] [Google Scholar]
- 54. Tavakoli Dargani Z, Singla R, Johnson T, Kukreja R, Singla DK. Exosomes derived from embryonic stem cells inhibit doxorubicin and inflammation‐induced pyroptosis in muscle cells. Can J Physiol Pharmacol 2018;96:304–307. [DOI] [PubMed] [Google Scholar]
- 55. Fleetwood AJ, Lee MK, Singleton W, Achuthan A, Lee MC, O'Brien‐Simpson NM, et al. Metabolic remodeling, inflammasome activation, and pyroptosis in macrophages stimulated by porphyromonas gingivalis and its outer membrane vesicles. Front Cell Infect Microbiol 2017;7:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Takahashi M. NLRP3 inflammasome as a novel player in myocardial infarction. Int Heart J 2014;55:101–105. [DOI] [PubMed] [Google Scholar]
- 57. Voet S, Srinivasan S, Lamkanfi M, van Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med 2019;11:e10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Denes A, Coutts G, Lénárt N, Cruickshank SM, Pelegrin P, Skinner J, et al. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc Natl Acad Sci U S A 2015;112:4050–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lorenz G, Darisipudi MN, Anders H‐J. Canonical and non‐canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis. Nephrol Dial Transplant 2013;29:41–48. [DOI] [PubMed] [Google Scholar]
- 60. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin‐1β secretion. Cell Research 2015;25:1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang D, Qian J, Zhang P, Li H, Shen H, Li X, et al. Gasdermin D serves as a key executioner of pyroptosis in experimental cerebral ischemia and reperfusion model both in vivo and in vitro. J Neurosci Res 2019;97:645–660. [DOI] [PubMed] [Google Scholar]
- 62. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG‐1 as a late mediator of endotoxin lethality in mice. Science 1999;285:248–251. [DOI] [PubMed] [Google Scholar]
- 63. Kim JB, Choi JS, Yu YM, Nam K, Piao CS, Kim SW, et al. HMGB1, a novel cytokine‐like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci 2006;26:6413–6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Okuma Y, Liu K, Wake H, Zhang J, Maruo T, Date I, et al. Anti‐high mobility group box‐1 antibody therapy for traumatic brain injury. Ann Neurol 2012;72:373–384. [DOI] [PubMed] [Google Scholar]
- 65. Sasaki T, Liu K, Agari T, Yasuhara T, Morimoto J, Okazaki M, et al. Anti‐high mobility group box 1 antibody exerts neuroprotection in a rat model of Parkinson's disease. Exp Neurol 2016;275:220–231. [DOI] [PubMed] [Google Scholar]
- 66. Wang J, Hu X, Xie J, Xu W, Jiang H. Beta‐1‐adrenergic receptors mediate Nrf2‐HO‐1‐HMGB1 axis regulation to attenuate hypoxia/reoxygenation‐induced cardiomyocytes injury in vitro. Cell Physiol Biochem 2015;35:767–777. [DOI] [PubMed] [Google Scholar]
- 67. Fujita K, Motoki K, Tagawa K, Chen X, Hama H, Nakajima K, et al. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4‐MARCKS, is a potential therapeutic target for Alzheimer's disease. Sci Rep 2016;6:31895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Qu D, Liu J, Lau CW, Huang Y. IL‐6 in diabetes and cardiovascular complications. Br J Pharmacol 2014;171:3595–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tanaka T, Narazaki M, Kishimoto T. IL‐6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 2014;6:a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McGonagle D, Sharif K, O'Regan A, Bridgewood C. The role of cytokines including interleukin‐6 in COVID‐19 induced pneumonia and macrophage activation syndrome‐like disease. Autoimmun Rev 2020;19:102537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Eid AA, Ionescu AA, Nixon LS, Lewis‐Jenkins V, Matthews SB, Griffiths TL, et al. Inflammatory response and body composition in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001;164:1414–1418. [DOI] [PubMed] [Google Scholar]
- 72. Roubenoff R, Roubenoff RA, Cannon JG, Kehayias JJ, Zhuang H, Dawson‐Hughes B, et al. Rheumatoid cachexia: cytokine‐driven hypermetabolism accompanying reduced body cell mass in chronic inflammation. J Clin Invest 1994;93:2379–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Anker SD, Ponikowski PP, Clark AL, Leyva F, Rauchhaus M, Kemp M, et al. Cytokines and neurohormones relating to body composition alterations in the wasting syndrome of chronic heart failure. Eur Heart J 1999;20:683–693. [DOI] [PubMed] [Google Scholar]
- 74. Boon MR, van der Horst G, van der Pluijm G, Tamsma JT, Smit JW, Rensen PC. Bone morphogenetic protein 7: a broad‐spectrum growth factor with multiple target therapeutic potency. Cytokine Growth Factor Rev 2011;22:221–229. [DOI] [PubMed] [Google Scholar]
- 75. Hansen M. Female hormones: do they influence muscle and tendon protein metabolism? Proc Nutr Soc 2018;77:32–41. [DOI] [PubMed] [Google Scholar]
- 76. Vingren JL, Kraemer WJ, Ratamess NA, Anderson JM, Volek JS, Maresh CM. Testosterone physiology in resistance exercise and training: the up‐stream regulatory elements. Sports Med 2010;40:1037–1053. [DOI] [PubMed] [Google Scholar]
- 77. Hansen M, Skovgaard D, Reitelseder S, Holm L, Langbjerg H, Kjaer M. Effects of estrogen replacement and lower androgen status on skeletal muscle collagen and myofibrillar protein synthesis in postmenopausal women. J Gerontol A Biol Sci Med Sci 2012;67:1005–1013. [DOI] [PubMed] [Google Scholar]
- 78. Smith GI, Yoshino J, Reeds DN, Bradley D, Burrows RE, Heisey HD, et al. Testosterone and progesterone, but not estradiol, stimulate muscle protein synthesis in postmenopausal women. J Clin Endocrinol Metab 2014;99:256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. BMP‐7 Treatment Inhibits TLR4 Expression in Diabetes.
Figure S2: BMP‐7 Treatment Inhibits Pyroptosis Executioner GSDMD
Table S1: List of Antibodies and primers used for the study.