

Abstract
Background
Prolonged critically ill patients frequently develop debilitating muscle weakness that can affect both peripheral nerves and skeletal muscle. In‐depth knowledge on the temporal contribution of neural and muscular components to muscle weakness is currently incomplete.
Methods
We used a fluid‐resuscitated, antibiotic‐treated, parenterally fed murine model of prolonged (5 days) sepsis‐induced muscle weakness (caecal ligation and puncture; n = 148). Electromyography (EMG) measurements were performed in two nerve–muscle complexes, combined with histological analysis of neuromuscular junction denervation, axonal degeneration, and demyelination. In situ muscle force measurements distinguished neural from muscular contribution to reduced muscle force generation. In myofibres, imaging and biomechanics were combined to evaluate myofibrillar contractile calcium sensitivity, sarcomere organization, and fibre structural properties. Myosin and actin protein content and titin gene expression were measured on the whole muscle.
Results
Five days of sepsis resulted in increased EMG latency (P = 0.006) and decreased EMG amplitude (P < 0.0001) in the dorsal caudal tail nerve–tail complex, whereas only EMG amplitude was affected in the sciatic nerve–gastrocnemius muscle complex (P < 0.0001). Myelin sheath abnormalities (P = 0.2), axonal degeneration (number of axons; P = 0.4), and neuromuscular junction denervation (P = 0.09) were largely absent in response to sepsis, but signs of axonal swelling [higher axon area (P < 0.0001) and g‐ratio (P = 0.03)] were observed. A reduction in maximal muscle force was present after indirect nerve stimulation (P = 0.007) and after direct muscle stimulation (P = 0.03). The degree of force reduction was similar with both stimulations (P = 0.2), identifying skeletal muscle, but not peripheral nerves, as the main contributor to muscle weakness. Myofibrillar calcium sensitivity of the contractile apparatus was unaffected by sepsis (P ≥ 0.6), whereas septic myofibres displayed disorganized sarcomeres (P < 0.0001) and altered myofibre axial elasticity (P < 0.0001). Septic myofibres suffered from increased rupturing in a passive stretching protocol (25% more than control myofibres; P = 0.04), which was associated with impaired myofibre active force generation (P = 0.04), linking altered myofibre integrity to function. Sepsis also caused a reduction in muscle titin gene expression (P = 0.04) and myosin and actin protein content (P = 0.05), but not the myosin‐to‐actin ratio (P = 0.7).
Conclusions
Prolonged sepsis‐induced muscle weakness may predominantly be related to a disruption in myofibrillar cytoarchitectural structure, rather than to neural abnormalities.
Keywords: Sepsis, Muscle weakness, Muscle contraction, Neuropathy, Biomechanics
Introduction
Severe muscle wasting and weakness frequently develop in critically ill patients who stay in the intensive care unit (ICU) for an extended period. This ICU‐acquired weakness is associated with greater mortality and morbidity, both in‐hospital and after discharge. 1 , 2 Despite progress in intensive care medicine, efficient therapeutic interventions are still lacking.
Two main entities describe neuromuscular abnormalities in weak critically ill patients: critical illness myopathy (CIM) and critical illness polyneuropathy (CIP). CIM is more prevalent than CIP, although problems in the skeletal muscle and peripheral nervous system commonly coincide. 3 , 4 , 5 Of note, both CIM and CIP show a particular high prevalence in septic patients. 6 , 7 Loss of muscle mass due to an imbalance in myofibrillar breakdown and compensatory protein synthesis is the most prominent observation in weak patients, 8 , 9 , 10 which results in a preferential loss of myosin and a shift in muscle fibre type composition. 11 , 12 , 13 Impaired muscle force generation in the critically ill is not merely a consequence of the muscle wasting but is also related to mechanisms that affect the muscles' bioenergetic state and the excitation–contraction coupling. 14 , 15 In addition, neuropathy can express itself as axonal sensory‐motor degeneration in prolonged critically ill patients, 16 , 17 whereas the excitation–contraction coupling might be affected earlier. 14 , 17
Combined research in critically ill patients and animal models has focused on understanding the neuromuscular abnormalities associated with CIM and CIP. Due to the complexity of invasively obtaining patient samples for specialized molecular or functional analyses, human studies have been mostly descriptive, showing the presence of aberrant nerve conduction, axonal degeneration, myosin loss in the skeletal muscle, and loss of structural integrity of the contractile apparatus. 11 , 12 , 13 , 16 , 18 , 19 , 20 Animal studies have further elucidated underlying mechanisms such as neural and muscular channelopathies, altered muscular calcium homeostasis, and disorganized sarcomeres. 21 , 22 , 23 , 24 , 25 Nevertheless, most studies focused on either neural or muscular components of muscle contractions, leaving knowledge on the temporal relationship between neural and muscular components, their interaction, and their contribution to muscle weakness incomplete.
In the current study, we combined functional, structural, and biomechanical analyses of the peripheral nerves and the skeletal muscle to evaluate the neural and muscular contribution to muscle contractions in a validated and clinically relevant mouse model of sepsis‐induced muscle weakness.
Material and methods
Animal study design
The mouse model has been described in detail. 26 In brief, 24‐week‐old male 57BL/6JRj mice (Janvier SAS, Chassal, France) were randomly allocated to ‘sepsis’ or ‘healthy control’ groups. Mice in the sepsis group were anaesthetized with ketamine/xylazine, after which the central jugular vein was catheterized and connected to a swivel device, allowing free movement and continuous fluid perfusion. Sepsis was induced by caecal ligation and puncture (50% ligation and single‐needle through‐and‐through puncture). Mice received intravenous fluid resuscitation post‐op (1:4 mixture of balanced colloids and crystalloids), and subcutaneous antibiotics/analgesia (imipenem/buprenorphine) were given 6 h post‐op and every 12 h until sacrifice. From Day 1 onwards, septic mice received standard mixed parenteral nutrition (Olimel N7E, Baxter, Lessines, Belgium) at 5.8 kcal/day. Mice were excluded if catheter‐related problems occurred. Individually caged healthy mice, pair fed to septic mice, served as controls. Mice were anaesthetized and sacrificed by cardiac puncture on Day 5 (number of included mice: healthy n = 47, sepsis n = 101). Of note, this model mimics many of the complex metabolic, endocrine, and inflammatory changes of human acute and prolonged critical illness. 26 , 27 , 28 After 5 days of sepsis—equivalent to prolonged illness in humans—these mice suffer from severe muscle wasting and weakness (reduced muscle mass, myofibre size, and muscle force). 27 , 28 This carefully chosen time frame for a sepsis‐induced muscle phenotype, together with other measures to mimic clinical care (fluid resuscitation, antibiotic therapy, and administration of parenteral nutrition), largely fits with the recent MQTiPSS guidelines. 29 The Institutional Ethical Committee for Animal Experimentation of the KU Leuven had approved the protocols for these animal studies (internal project numbers P124/2017 and P062/2019).
Electromyography measurements
Electromyography measurements were performed prior to randomization and before sacrifice on anaesthetized mice that were placed on a heating pad. Measurements were performed using 0.4 mm platinum‐coated subdermal needle electrodes (Technomed Europe, Maastricht, the Netherlands) and a Medelec Synergy EMG System (Viasys Healthcare, Conshohocken, PA, USA). Compound muscle action potentials (CMAPs) were acquired by measuring the electrical supramaximal response of the neuromuscular unit at the gastrocnemius muscle and the tail. CMAP latency described the time from nerve signal to muscle stimulus, whereas CMAP amplitude reflected the number of depolarizing myofibres. For the gastrocnemius muscle, the anode and cathode subdermal electrodes were positioned proximal to the sciatic notch, 0.5 cm apart from each other, while the recording electrode was placed at the maximum diameter of the gastrocnemius muscle. A reference electrode was placed at the animal's ankle and the ground on the contralateral side. CMAPs of the tail were recorded along the dorsal caudal tail nerve (DCTN). The stimulating anode and cathode were placed 1 cm from the tip of the tail, separated by 0.5 cm. The recording electrode measured the supramaximal response 3 cm more proximally. The reference electrode was again positioned 0.5 cm closer to the tail base from the recording electrode. The grounding electrode was situated between the cathode and the recording electrode. For both gastrocnemius muscle and tail, stimulation currents were gradually increased until supramaximal responses were reached. All measurements were performed in triplicate and analysed as the averaged delta measurements of Day 5 and Day 0.
In situ muscle force measurements
Anaesthetized mice were placed on a heating pad. The extensor digitorum longus (EDL) muscle was exposed, and the distal tendon was sutured and detached. The deep peroneal nerve was exposed at the back of the knee followed by suturing and detaching it at the proximal end. The animal was then placed on the 809C in situ mouse apparatus (Aurora Scientific, Aurora, ON, Canada), which was heated to 37°C. The paw was fixed to the set‐up, and the distal EDL suture was connected to the lever arm (300C‐LR Dual‐Mode Muscle Lever, Aurora Scientific). The EDL was kept moist with warm saline. Resting length L 0 was set to 25 mN passive force, as determined by pilot data. Supramaximal stimulation intensities were specified for each individual muscle by gradually increasing the stimulation intensity until twitch forces no longer raised. Maximal isometric tetanic forces of the EDL were measured by averaging three consecutive tetanic stimuli (150 Hz stimulation frequency, 200 ms duration, 0.2 ms pulse width, 2 min rest intervals), given via needle electrodes (Aurora Scientific) on the sutured deep peroneal nerve (indirect stimulation) and on the largest width of the EDL muscle (direct stimulation). Data were analysed with analysis software from Aurora Scientific (ASI 611A Dynamic Muscle Analysis v5.300).
Histology
Neuromuscular junction denervation
Snap‐frozen tibialis muscle was sectioned and fixed with 4% paraformaldehyde (PFA). Nerve axons were visualized with antibodies directed against pan‐neuronal marker neurofilament light chain (Alexa‐488 conjugated; Cell Signaling Technology, Danvers, MA, USA; 1:500) and presynaptic marker synaptophysin (Cell Signaling Technology; 1:500). Simultaneous with incubation of the secondary antibody (anti‐rabbit Alexa‐488, 1:1000, Life Technologies, Carlsbad, CA, USA), the neuromuscular endplate was labelled with α‐bungarotoxin (Alexa‐555 conjugated; Life Technologies; 1:500). The percentage of neuromuscular junction innervation (co‐localization of neurofilament light chain/synaptophysin and α‐bungarotoxin labelling) was calculated. Per animal, 177 ± 54 neuromuscular junctions were examined.
Axon analyses
Sciatic nerves were collected in a 0.1 M sodium cacodylate buffer containing 4% PFA and 2.5% glutaraldehyde. Samples were fixed overnight at 4°C and post‐fixed with 2% osmium tetroxide for 2 h at room temperature. After fixation, serial ethanol dehydration was performed. Samples were then immersed in propylene oxide prior to resin impregnation. Fresh agar was prepared (Agar 100 resin 1031, Agar Scientific, Stansted Essex, UK) and gradually impregnated. Resin‐embedded sciatic nerves were prepared for semi‐thin sectioning on a LM Leica EM TRIM2 (Leica Microsystems, Wetzlar, Germany), followed by sectioning at 500 nm using a Leica EM UC7 ultra‐microtome (Leica Microsystems). Myelin was visualized with toluidine blue. The number of axons and their surface area was determined in Fiji using the ‘define particle’ plug‐in (ImageJ NIH, USA). A Fiji macro was created based on the Renyi entropy threshold algorithm. Particles smaller than 75 μm2 or with a circularity outside the range of 0.05–1.00 were excluded. At least 10 images per sciatic nerve per animal were analysed. Myelin sheath abnormalities (in‐/out‐foldings or abnormal myelin thickness) were assessed as the percentage abnormal axons to total axons (1080 ± 320 axons per animal were analysed). Axon g‐ratios (ratio between inner and outer myelin sheath diameter) were calculated in 123 ± 17 axons per animal.
Second harmonic generation microscopy
The EDL muscle was dissected and fixed under slight stretch in phosphate‐buffered saline + 3% PFA. Label‐free multiphoton imaging of the myosin structure of mechanically isolated myofibres was performed on an upright TrimScope II, similar to the previously published system. 30 A U‐AAC condenser lens (OLYMPUS, Tokyo, Japan) was applied to collect transmitted second harmonic generation (SHG) signal. The SHG signal was filtered via a 405/20 nm bandpass filter (Chroma Technology, Olching, Germany), and the signal was detected on non‐descanned photomultiplier tubes (H 7422‐40 LV 5 M, Hamamatsu Photonics, Hamamatsu, Japan). Image stacks of myofibres were acquired with a voxel size of 0.2 × 0.2 × 0.5 μm and a field of view of 100 × 100 μm with a stack depth of typically 50 μm. SHG image analysis: Myofibrillar parallelism was determined by the cosine angle sum (CAS) parameter, as described before with adjustments. 31 The main sarcomere direction vector and local voxel direction vectors were determined in 3D, meaning that the angle φ of each local voxel vector was located in the common plane of and , and not in the XY‐cardinal plane. A 3D fast Fourier transform was performed on the image stacks to obtain from particle analysis in the 3D Fourier space; 9 × 9 × 9 voxel 3D Sobel kernels were convolved with the image stack to obtain the local voxel direction vector for each voxel i. The determination of CAS was performed using MATLAB (MATLAB R2018b, The MathWorks Inc., USA).
Gene expression analyses
Messenger RNA was isolated from the gastrocnemius muscle, and cDNA was quantified in real time as described previously. 27 , 28 Commercial TaqMan® assays (Thermo Fisher Scientific, Waltham, MA, USA) were used to quantify Rn18s (Mm03928990_g1) and Ttn (Mm00621005_m1). Ttn was normalized to Rn18s and expressed as fold change of the mean of controls.
Myosin and actin protein content
Gastrocnemius muscle was homogenized in a buffer containing 20 mM Tris–HCl pH 7.6, 10% glycerol, 1% Nonidet P‐40, 34 μg/mL phenylmethylsulfonyl fluoride, 10 mM Na4P2O7, 100 mM NaF, 10 mM ethylenediaminetetraacetic acid, and protease inhibitors (cOmplete™ EDTA‐free Protease Inhibitor Cocktail, Roche, Basel, Switzerland) with ceramic beads in a Precellys 24 homogenizer (Bertin Instruments, Montigny‐le‐Bretonneux, France). Equal amounts of protein (2 μg) were loaded onto 7.5% polyacrylamide gels (Bio‐Rad Laboratories, Hercules, CA, USA) and separated for 100 min at 120 V. Following electrophoresis, protein bands were visualized using the Silver Stain Plus kit (Bio‐Rad Laboratories). Actin and myosin bands were analysed with the G:BOX Chemi XRQ (Syngene, Bangalore, India) and Syngene software, as described previously. 27 , 28
Single fibre biomechanics
Single fibre biomechanics was assessed using the previously described MyoRobot opto‐biomechatronics system. 32 , 33 Myofibres were mechanically isolated from the EDL muscle and placed in a 1:1 solution of 10 mM dithiothreitol in 99% glycerol and 40 mM HEPES, 20 mM ethyleneglycoltetraacetic acid, 8.82 mM Mg(OH)2, 8 mM Na2ATP, 10 mM sodium creatine phosphate, pH 7 for 5 h at 4°C. Myofibres were stored at −20°C until biomechanical recordings within 4 weeks. The protocols for measuring myofibre biomechanics have been described previously in detail. 32 , 33 Calcium sensitivity of the contractile apparatus: Chemically skinned myofibres were held constant at their resting length L 0 and immersed in wells with sequentially decreasing pCa value to induce isometric contraction. The sequence of wells was composed of different ratios of highly ethyleneglycoltetraacetic acid buffered relaxing and calcium‐rich activating solutions to reflect an increase in free calcium ion concentration. Each pCa immersion allowed 20 s to produce maximum plateau force before proceeding to the next well. Myofibre elasticity: Passive axial compliance was assessed in resting length–tension (RLT) curves by stretching the myofibre to 140% of L 0 at a constantly slow speed of 0.42 μm/s. During the 30 min of experimentation, the sample was kept under relaxing conditions. Sarcomere length and myofibre diameter were derived at the beginning of the experiment. Analysing pCa–force and RLT curve data: For pCa–force recordings, pCa values and normalized force were plotted and fitted to a two‐parameter Hill equation in SigmaPlot (SigmaPlot 13.0, Systat Software Inc.), where c reflects the [Ca50] and (−log10(c)) calculates the pCa50 value and b is the Hill coefficient. The average fit of combined group data was created using the mean value and mean Hill coefficient . For RLT curves, force–length data were separated into sections of 10% stretch of L 0, and a linear fit was applied on each section to compute axial compliance. Normalization to cross‐sectional area with the myofibre diameter allowed conversion of force to stress. From these stress–strain relationships, the Young's modulus, a measure for stiffness and stress development under strain, was calculated for each 10% section as .
Statistics
Normally distributed data were compared with Student's t‐tests, where necessary, after log or (double) square root transformation to obtain a near‐normal distribution (JMP® Pro 14.0, SAS Institute Inc., Cary, NC, USA). Not normally distributed data were analysed with non‐parametric Wilcoxon tests. Two‐sided P‐values ≤0.05 (α‐level of 5%) were considered statistically significant in all analyses. Data are presented as means with standard error of the means or as box plots with median, interquartile ranges, and whiskers representing 10th and 90th percentiles. P‐values are plotted on the figures as *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Results
Five days of sepsis affected electromyography latency and amplitude and caused axonal swelling
To investigate neuromuscular abnormalities, we performed electromyography (EMG) measurements in both the DCTN–tail complex and the sciatic nerve–gastrocnemius muscle complex. Five days of sepsis increased CMAP latency in the tail (Figure 1A), but not in the gastrocnemius muscle (Figure 1B), whereas a reduced CMAP amplitude was present in both the tail and gastrocnemius muscles of septic mice (Figure 1C and 1D). In contrast, septic mice did not require a higher stimulation intensity to achieve supramaximal stimulation than did healthy controls, and this in both the nerve–muscle complexes (Figure 1E and 1F).
Figure 1.
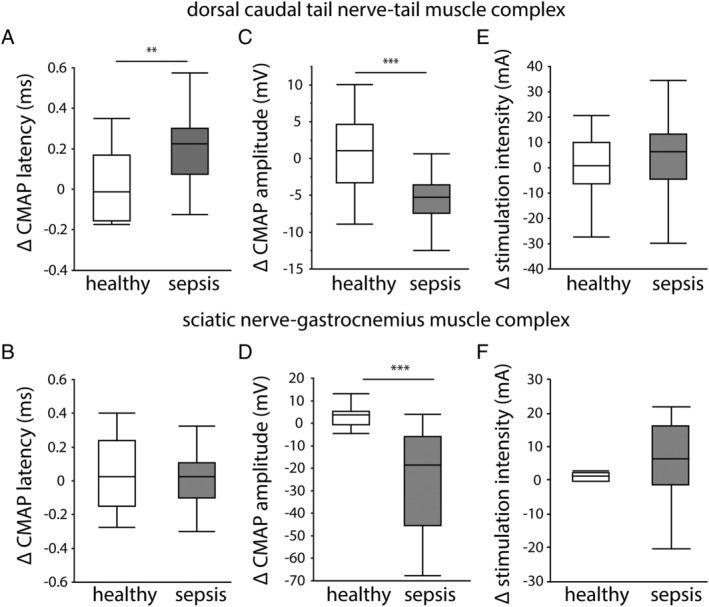
Electromyography (EMG) measurements of the dorsal caudal tail nerve–tail muscle and sciatic nerve–gastrocnemius muscle complexes. In vivo EMG measurements were performed before randomization at Day 0 and before sacrifice at Day 5. Data are presented as the delta of both measurements. (A) Compound muscle action potential (CMAP) latency in the dorsal caudal tail nerve–tail and (B) in the sciatic nerve–gastrocnemius muscle. (C) CMAP amplitude in the dorsal caudal tail nerve–tail and (D) in the sciatic nerve–gastrocnemius muscle. (E) Intensity required to achieve supramaximal stimulation in the dorsal caudal tail nerve–tail and (F) in the sciatic nerve–gastrocnemius muscle. (healthy n = 14, sepsis n = 30) *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Abnormalities in CMAP latency and amplitude commonly suggest neuromuscular junction disorders, demyelination, and axonal degeneration. The number of innervated neuromuscular junctions in the tibialis muscle tended to be lower in septic mice compared with healthy controls (Figure 2A). In the sciatic nerve, the number of axons per 100 μm2 was unaltered by sepsis (Figure 2B). However, a higher percentage of large axons was observed during sepsis (Figure 2C), which corresponded to an overall increased area of sciatic nerve axons in septic mice compared with healthy controls (Figure 2D). Analysis of the axon g‐ratio showed a higher g‐ratio in septic mice compared with healthy controls, irrespective of axon diameter, pointing towards axonal swelling without demyelination (Figure 2E). The percentage of axons with an abnormal myelin sheath (Figure 2F) was similar in septic mice and healthy controls (Figure 2G).
Figure 2.
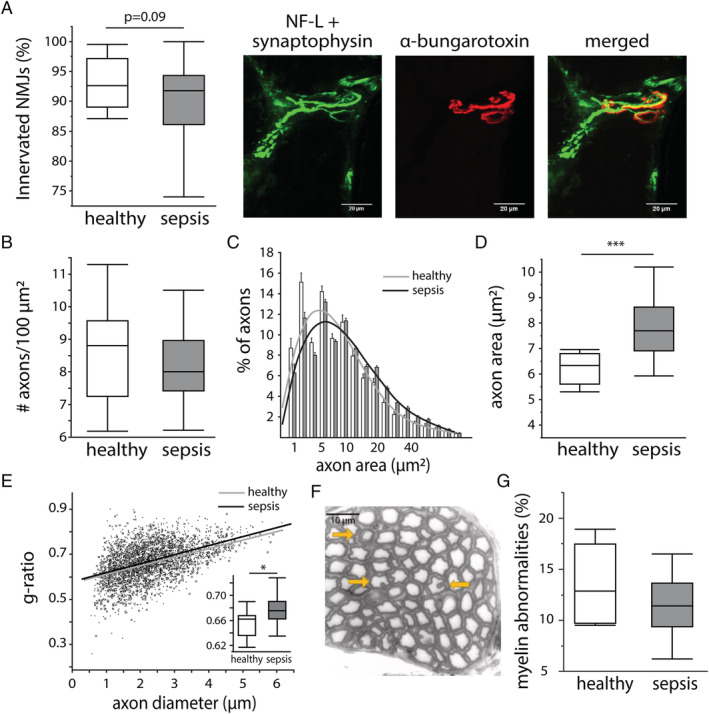
Effect of sepsis on markers of neuromuscular junction (NMJ) denervation, axonal degeneration, and demyelination. (A) Innervation of NMJs, quantified in the tibialis muscle as the percentage of NMJs with co‐localization of neurofilament light chain (NF‐L)/synaptophysin (neural synapses) and α‐bungarotoxin (neuromuscular endplate) staining. Scale bars are 20 μm. (healthy n = 12, sepsis n = 23) (B) The number of axons per 100 μm2, quantified as the median number per animal. (healthy n = 9, sepsis n = 20) (C) Histogram of the axon size and fitted curve. Bars are mean ± standard error of the mean. Grey line = healthy animals, black line = septic animals. (D) Quantification of the median axon size per animal. (healthy n = 9, sepsis n = 20) (E) The g‐ratio in relation to the axon diameter. Grey crosses + fitted grey line = healthy animals, black dots + fitted black line = septic animals. Insert graph shows the median g‐ratio per animal. (healthy n = 8, sepsis n = 20) (F) Myelin abnormalities (abnormal myelin thickness and in‐/out‐foldings) indicated with yellow arrows. Scale bar is 10 μm. (G) Quantification of the percentage of axons with myelin abnormalities. (healthy n = 8, sepsis n = 20) All axon/myelin analyses were performed in toluidine blue stained axons of the sciatic nerve. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Sepsis‐induced effects on the peripheral nerves did not explain reduced muscle force generation
Although EMG measurements can be used to diagnose neuromuscular abnormalities, the CMAP amplitude measurements cannot distinguish problems in peripheral nerves from problems in the skeletal muscle. Therefore, we next compared EDL muscle force generated in response to indirect nerve stimulation with that in response to direct muscle stimulation. After both indirect nerve stimulation and direct muscle stimulation, the absolute maximal force of the EDL muscle was lower in septic mice than in healthy control mice (Figure 3A and 3B). Reductions in muscle force after indirect and direct stimulations were comparable (Figure 3C), suggesting that a dysfunction within the muscle rather than in the nerves explained the reduced muscle force generation.
Figure 3.
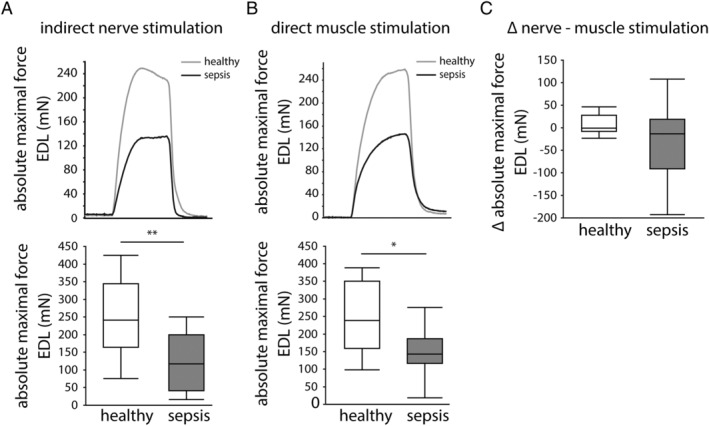
In situ extensor digitorum longus (EDL) muscle force measurements. Representative force traces and quantification of the absolute maximal tetanic force generated by the EDL muscle after (A) indirect stimulation of the deep peroneal nerve and (B) after direct EDL stimulation. Grey line = healthy mice, black line = septic mice. (C) Delta of the absolute maximal tetanic force measurements after indirect nerve and direct muscle stimulations. (healthy n = 8, sepsis n = 26) *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Sepsis did not alter myofibrillar calcium sensitivity nor the muscular myosin‐to‐actin ratio but affected sarcomere and myofibre integrity, which was associated with myofibre force generation
We next assessed the biomechanical and structural properties of the contractile apparatus in isolated myofibres and evaluated sarcomeric proteins on the whole muscle. The myofibrillar calcium sensitivity was evaluated by measuring the produced force at decreasing pCa levels (Figure 4A). Force–pCa curves revealed no differences in pCa50 values and Hill coefficients in septic mice and healthy controls (Figure 4B–4D). Label‐free visualization of myosin filaments (Figure 5A) indicated reduced sarcomere organization with prolonged sepsis, as shown by a lower myofibrillar CAS in septic mice compared with healthy controls (Figure 5B). On the level of the whole muscle, the myosin and actin protein contents were lower in septic mice compared with controls, whereas the myosin‐to‐actin ratio was unaffected by sepsis (Figure 5C and 5D). Passively stretching myofibres to 140% of their resting length L 0 was performed to further assess fibre structural properties (Figure 6A). Myofibres from septic mice were more likely to break during the stretching protocol than those from healthy controls (48% ruptures in healthy controls vs. 73% ruptures in septic mice; P = 0.04; Figure 6B). Myofibre rupturing occurred irrespective of initial sarcomere lengths and at comparable forces (Supporting Information, Figure S1 ), suggesting that baseline parameters were unlikely to have affected rupturing. Ruptured myofibres isolated from septic mice, but not those from healthy controls, showed consistently lower axial compliance and a higher Young's modulus than myofibres that did not rupture, indicating higher myofibrillar stiffness (Figure 6C and 6D). Myofibres that were able to endure 140% stretch of L 0 also behaved differently with sepsis, shown by less passive restoration force at 140% stretch (Figure 6E) and a lower Young's modulus in myofibres from septic mice compared with controls (Figure 6F), whereas compliance was generally not affected (Figure 6G). In myofibres that were used in both the calcium sensitivity measurements and the subsequent stretching protocol, we evaluated the link between myofibre integrity and myofibrillar force generation (active maximal force produced at the lowest pCa step). Subdividing myofibres based on their ability to survive the passive stretching protocol showed that myofibres from septic mice that were unable to be fully stretched produced less maximal force prior to rupturing than those that did not rupture (Figure 6H), an association not observed in myofibres from healthy mice (Figure 6H). On the level of the complete muscle, gene expression of sarcomeric protein titin was reduced in septic mice compared with controls (Figure 6I).
Figure 4.
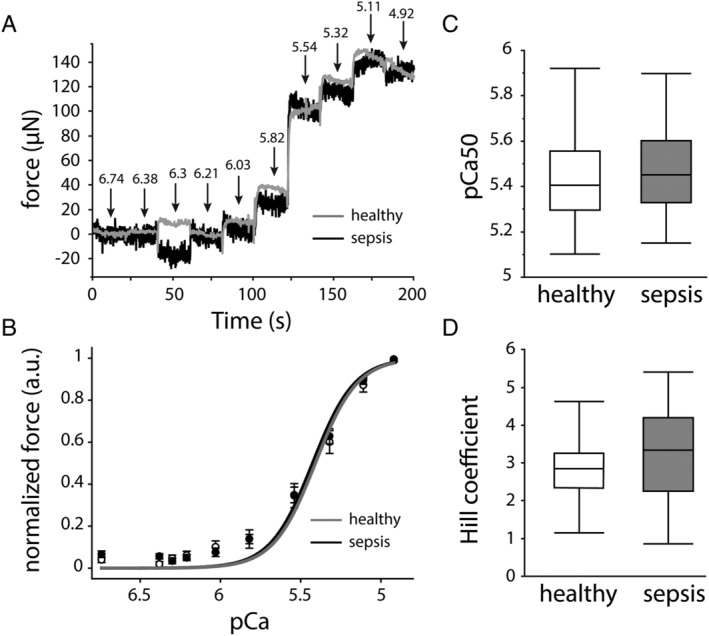
Effect of sepsis on myofibre calcium sensitivity of the contractile apparatus. (A) Representative force traces showing increasing force for each decreasing pCa step. Grey curve = healthy controls, black curve = septic mice. (B) Mean pCa–force group curves with reconstructed Hill fits. Data presented as mean ± standard error of the mean. White circles + grey curve = healthy controls, black circles + black curve = septic mice. (C) Group analyses of pCa50 and (D) Hill coefficients. (healthy n = 23 myofibres from five mice, sepsis n = 45 myofibres from 10 mice) *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Figure 5.
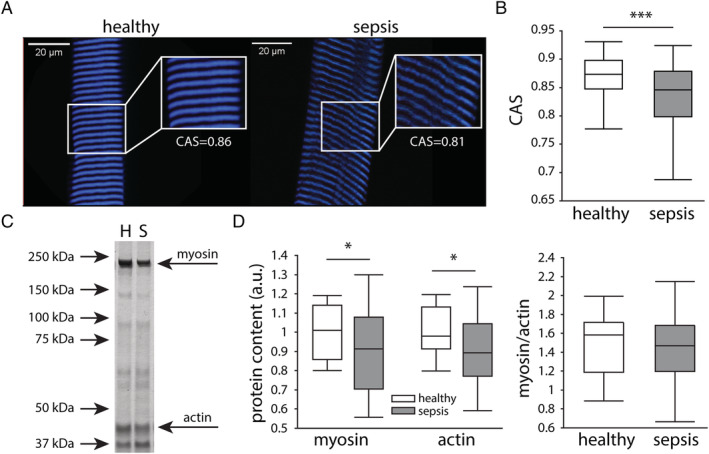
Effect of sepsis on sarcomere organization and muscular myosin/actin content. (A) Sarcomeres were visualized by label‐free multiphoton second harmonic generation (SHG) microscopy. Representative images of SHG measurements in healthy controls and septic mice. Scale bars are 20 μm. (B) Cosine angle sum (CAS) measurements of the SHG signal, indicating the variability in angular myofibrils. (healthy n = 256 from 86 myofibres from six mice, sepsis n = 433 from 145 myofibres from 10 mice) (C) Representative silver staining of myosin and actin in the gastrocnemius muscle. H lane represents a healthy animal, and S a septic one. (D) Muscular myosin and actin protein content as percentage of controls, and myosin‐to‐actin ratio. (healthy n = 15 mice, sepsis n = 30 mice) *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Figure 6.
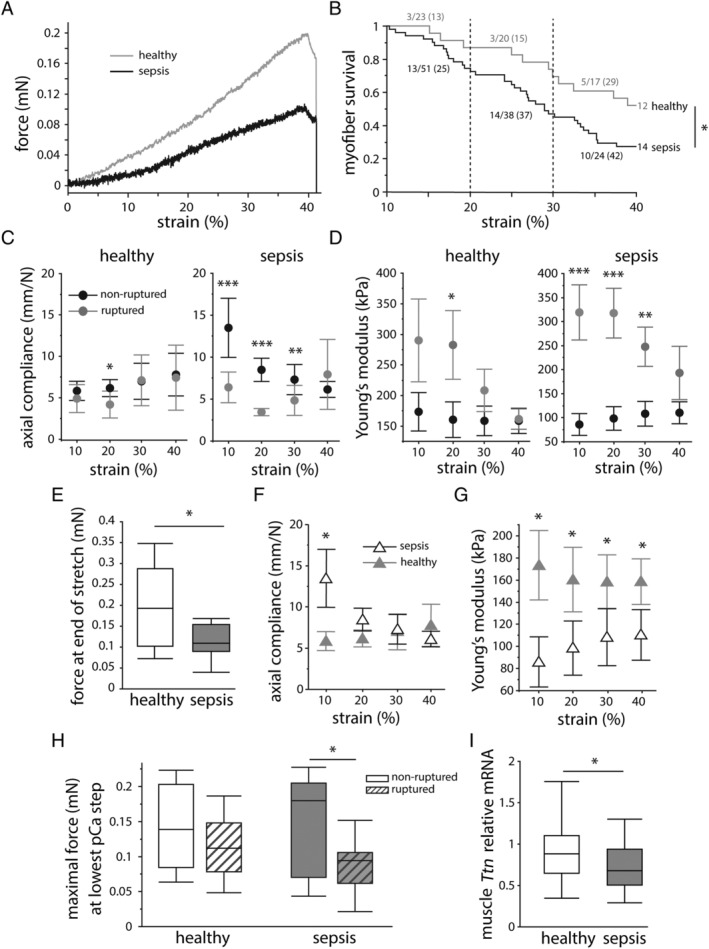
Effect of sepsis on myofibre elasticity. Myofibres were passively stretched to 140% of their resting length (L 0) to assess passive elasticity. (A) Representative recordings of resting length–tension (RLT) curves showing force in relation to percentage of strain. (B) Survival analysis of myofibres subjected to the stretching protocol. Numbers indicate the amount and percentage of ruptured myofibres per 10% strain. (grey line: healthy n = 23 myofibres from four mice, black line: sepsis n = 51 myofibres from 10 mice) Elastic properties of ruptured (grey) and non‐ruptured myofibres (black), evaluated as (C) axial compliance and (D) Young's modulus. (statistic symbols represent difference between ruptured and non‐ruptured myofibres; myofibres from healthy mice: ruptured n = 10, non‐ruptured n = 13; myofibres from septic mice: ruptured n = 37, non‐ruptured n = 14) Properties of myofibres that survived the stretching protocol were assessed as the (E) force at 140% stretch of L 0, (F) axial compliance, and (G) Young's modulus. (statistic symbols represent difference between healthy and septic mice; grey full triangles: healthy n = 13 myofibres, black open triangles: sepsis n = 14 myofibres) Axial compliance and Young's modulus data (C, D, F, G) are shown as mean ± standard error of the mean. (H) Maximal myofibrillar force, produced at the lowest pCa step, shown in relation to myofibre rupturing in the stretching protocol. (healthy mice: ruptured n = 10, non‐ruptured n = 11; septic mice: ruptured n = 22, non‐ruptured n = 13) (I) Relative mRNA expression of sarcomeric protein titin in the whole muscle. (healthy n = 14 mice, sepsis n = 33 mice) *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Discussion
We here investigated key neural and muscular components of muscle contractions in a mouse model of sepsis‐induced muscle weakness. Five days of sepsis caused EMG abnormalities that were most pronounced in the DCTN–tail complex. Signs of axonal swelling were observed in response to sepsis, whereas myelin abnormalities, axonal degeneration, and neuromuscular junction denervation were absent. In situ muscle force measurements showed that skeletal muscle, but not peripheral nerves, predominantly contributed to the muscle weakness. Myofibrillar calcium sensitivity of the contractile apparatus was not affected by sepsis, whereas septic mice did present with disorganized sarcomeres and altered myofibre integrity. This altered integrity appeared related to the myofibrillar force generating capacity. In the whole muscle, sepsis caused a decrease in titin gene expression as well as myosin and actin protein content, but not the myosin‐to‐actin ratio.
After 5 days of sepsis, mice only presented with mild neuropathic changes, including axonal swelling and increased CMAP latency in the DCTN–tail complex, but not the sciatic nerve–gastrocnemius muscle complex. In contrast, signs of neuromuscular junction denervation, myelin abnormalities, or axonal degeneration were absent. Muscle membrane hypoexcitability, 14 , 17 generally linked to sodium channelopathies, 18 , 21 was also not observed. Possibly, neuropathy did not yet develop in the time frame of our study (5 days). Indeed, in CIP, morphological changes are commonly described in long‐term severely ill patients and post‐mortem biopsies, whereas functional changes, such as altered nerve conduction, are thought to arise earlier. 17 , 34 Furthermore, axonal swelling is thought to precede the more severe axonal degeneration, and early neuropathic changes in rodents are most evident in the tail muscle, with other nerve–muscle complexes only being affected when neuropathy becomes more pronounced. 35 , 36 CMAP amplitude was already severely reduced by 5 days of sepsis in both nerve–muscle complexes. This reduction may be the consequence of muscle wasting, rather than of pure neuromuscular abnormalities. Profound muscle wasting that contributes to the muscle weakness has repeatedly been described in caecal ligation and puncture‐induced sepsis. 27 , 28 , 37 In addition, the in situ muscle force measurements did point to myopathy rather than neuropathy as the cause of the muscle weakness. Also, in critically ill patients, electrophysiological signs of CIM have been shown to precede those of CIP. 5
The finding that myofibrillar calcium sensitivity of the contractile apparatus was not affected by 5 days of sepsis was somewhat unexpected, as altered myofibre calcium sensitivity has been observed in a mouse model of critical illness. 23 However, calcium homeostasis was also found to only be transiently affected, with most obvious changes during early sepsis and a normalization after several days. 23 Five days of sepsis did reduce the structural and biomechanical integrity of the myofibres, which was mostly evident as altered myofibre elasticity. Septic mice appeared to have two distinct myofibre subgroups: a large group of less compliant myofibres that ruptured during the quasi‐static stretching protocol and a smaller group of myofibres that did not rupture but also behaved differently to myofibres from healthy controls. This indicates that sepsis alters myofibre elasticity, making them less capable of tolerating strain. Importantly, an association between altered structural integrity and hampered force generation was observed in myofibres from septic mice. Similarly, ultrastructural myofibre abnormalities have been linked to impaired force output in other muscle disorders. 31 , 38 , 39 These changes on the myofibrillar level could also affect the structural integrity and force generation of the complete muscle. Of note, structural changes on the level of the whole muscle have previously been linked to muscle functionality in critically ill patients. 40 , 41 Hence, it could be possible that ultrastructural myofibrillar changes might be related to the observed compromised skeletal muscle bioenergetic status, 15 , 42 although this should be investigated further.
Several skeletal muscle disorders are associated with titin abnormalities, a sarcomeric protein that plays a role in sarcomere organization, passive elastic properties, and active force generation. 43 Combining the biomechanical data and the reduced muscular Ttn gene expression may give an indication that titin might be involved in sepsis‐induced muscle weakness. Indeed, in critically ill patients suffering from CIM, loss of muscle titin has been observed, and titin knockout mice largely exhibit the pathophenotypic muscular changes associated with CIM. 44
While this model of muscle weakness does take known risk factors such as prolonged illness and sepsis into account, some limitations need to be addressed. First, septic mice were not immobilized. Although it is widely known that mice move less when severely ill, the ability of our septic mice to somewhat move around in their cages could have improved their muscle function. Indeed, immobilization is a known risk factor for ICU‐acquired weakness and is thought to greatly contribute to the pathophysiology of CIM. 45 , 46 , 47 , 48 In addition, early mobilization in patients may protect against ICU‐acquired weakness, although results from studies are not univocal. 49 , 50 Second, hyperglycaemia is an important contributor to CIP, and preventing hyperglycaemia reduced the prevalence hereof in critically ill human patients. 20 , 51 Whereas hyperglycaemia is acutely observed in our model, it is not sustained until Day 5 of sepsis, 28 which may have blunted the development of CIP in our model. Third, although we did observe a, probably atrophy‐induced, decrease in muscular actin and myosin content with sepsis, the myosin‐to‐actin ratio remained unchanged. As CIM typically involves the preferential loss of myosin, 11 , 13 the absence of this in our mice may indicate that our model mimics pure sepsis‐induced muscle weakness, rather than the more complex CIM. 14 Lastly, the complex pathophysiology of muscle weakness in critical illness involves several changes that do not all occur simultaneously. Although this 5 day model of sepsis mimics several aspects of the muscle weakness, some factors may require an even more prolonged model combined with immobilization. Indeed, gene expression analysis in a rat model of critical illness has shown down‐regulation of genes involved in the excitation–contraction coupling only beyond Day 5. 52
In conclusion, muscular rather than neural components of muscle contractions were affected after 5 days of sepsis. Although mild effects on neural components were observed, reduced muscle force generation was related to problems in the muscle, rather than the nerves. The main muscular component affected was altered myofibre structural integrity, which was associated with hampered myofibrillar force generation. These findings could point to a structure‐related cytoarchitectural disorder in prolonged sepsis‐induced muscle weakness.
Author contributions
C.G., L.L., G.V.d.B., and O.F. conceived and designed the experiments. C.G., R.W., and S.D. performed the mice studies. C.G., R.W., L.V.H., D.S., M.H., and B.R. developed and optimized experimental methodology/software. C.G., R.W., L.V.H., and B.R. performed experiments and collected data. C.G., L.V.H., D.S., and M.H. analysed experimental data. L.L., G.V.d.B., L.V.D.B., and O.F. provided resources. C.G., S.D., and L.L. managed the research activities. L.L. and G.V.d.B. acquired financial support. C.G. prepared the presentation of the published work. C.G., L.L., G.V.d.B., O.F., L.V.H., D.S., and M.H. contributed to writing the paper. L.L., G.V.d.B., and O.F. supervised the project. All authors reviewed and approved the manuscript. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle. 53
Conflict of interest
The authors have nothing to disclose.
Supporting information
Figure S1. Sarcomere lengths and maximal force of ruptured and non‐ruptured myofibers. Myofibers were passively stretched to 140% of their resting length (L 0) to assess structural integrity. (A) Sarcomere lengths at L 0 of myofibers that did or did not rupture during the stretching protocol. White boxplots = healthy animals, gray boxplots = septic animals. [non‐ruptured myofibers healthy n = 8, sepsis n = 9; ruptured myofibers healthy n = 9, sepsis n = 27] (B) Force at the time of myofiber rupture. [healthy n = 11 myofibers, sepsis n = 37 myofibers] (C) Representative images of measured sarcomere lengths (SL). * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001
Acknowledgements
We thank Marissa Boone, Wouter Vankrunkelsven, Arno Téblick, Thomas Dufour, Sabine Lessig, and Annette Wirth‐Hücking for their technical assistance. G.V.d.B. and L.L., via the KU Leuven, receive long‐term structural research support from the Methusalem Program funded by the Flemish Government (METH14/06) and from the Research Foundation Flanders (G.0C78.17N). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (AdvG 2017‐785809).
Goossens C., Weckx R., Derde S., Van Helleputte L., Schneidereit D., Haug M., Reischl B., Friedrich O., Van Den Bosch L., Van den Berghe G., and Langouche L. (2021) Impact of prolonged sepsis on neural and muscular components of muscle contractions in a mouse model, Journal of Cachexia, Sarcopenia and Muscle, 12, 443–455, 10.1002/jcsm.12668
References
- 1. Fletcher SN, Kennedy DD, Ghosh IR, Misra VP, Kiff K, Coakley JH, et al. Persistent neuromuscular and neurophysiologic abnormalities in long‐term survivors of prolonged critical illness. Crit Care Med 2003;31:1012–1016. [DOI] [PubMed] [Google Scholar]
- 2. Schefold JC, Wollersheim T, Grunow JJ, Luedi MM, Z'Graggen WJ, Weber‐Carstens S. Muscular weakness and muscle wasting in the critically ill. J Cachexia Sarcopenia Muscle 2020;11(6):1399–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bednarik J, Lukas Z, Vondracek P. Critical illness polyneuromyopathy: the electrophysiological components of a complex entity. Intensive Care Med 2003;29:1505–1514. [DOI] [PubMed] [Google Scholar]
- 4. Lefaucheur JP, Nordine T, Rodriguez P, Brochard L. Origin of ICU acquired paresis determined by direct muscle stimulation. J Neurol Neurosurg Psychiatry 2006;77:500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koch S, Spuler S, Deja M, Bierbrauer J, Dimroth A, Behse F, et al. Critical illness myopathy is frequent: accompanying neuropathy protracts ICU discharge. J Neurol Neurosurg Psychiatry 2011;82:287–293. [DOI] [PubMed] [Google Scholar]
- 6. Schefold JC, Bierbrauer J, Weber‐Carstens S. Intensive care unit‐acquired weakness (ICUAW) and muscle wasting in critically ill patients with severe sepsis and septic shock. J Cachexia Sarcopenia Muscle 2010;1:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Khan J, Harrison TB, Rich MM, Moss M. Early development of critical illness myopathy and neuropathy in patients with severe sepsis. Neurology 2006;67:1421–1425. [DOI] [PubMed] [Google Scholar]
- 8. Jespersen JG, Nedergaard A, Reitelseder S, Mikkelsen UR, Dideriksen KJ, Agergaard J, et al. Activated protein synthesis and suppressed protein breakdown signaling in skeletal muscle of critically ill patients. PLoS One 2011;6:e18090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klaude M, Mori M, Tjader I, Gustafsson T, Wernerman J, Rooyackers O. Protein metabolism and gene expression in skeletal muscle of critically ill patients with sepsis. Clin Sci (Lond) 2012;122:133–142. [DOI] [PubMed] [Google Scholar]
- 10. Puthucheary ZA, Rawal J, McPhail M, Connolly B, Ratnayake G, Chan P, et al. Acute skeletal muscle wasting in critical illness. JAMA 2013;310:1591–1600. [DOI] [PubMed] [Google Scholar]
- 11. Derde S, Hermans G, Derese I, Guiza F, Hedstrom Y, Wouters PJ, et al. Muscle atrophy and preferential loss of myosin in prolonged critically ill patients. Crit Care Med 2012;40:79–89. [DOI] [PubMed] [Google Scholar]
- 12. Bierbrauer J, Koch S, Olbricht C, Hamati J, Lodka D, Schneider J, et al. Early type II fiber atrophy in intensive care unit patients with nonexcitable muscle membrane. Crit Care Med 2012;40:647–650. [DOI] [PubMed] [Google Scholar]
- 13. Wollersheim T, Woehlecke J, Krebs M, Hamati J, Lodka D, Luther‐Schroeder A, et al. Dynamics of myosin degradation in intensive care unit‐acquired weakness during severe critical illness. Intensive Care Med 2014;40:528–538. [DOI] [PubMed] [Google Scholar]
- 14. Friedrich O, Reid MB, Van den Berghe G, Vanhorebeek I, Hermans G, Rich MM, et al. The sick and the weak: neuropathies/myopathies in the critically ill. Physiol Rev 2015;95:1025–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Puthucheary ZA, Astin R, McPhail MJW, Saeed S, Pasha Y, Bear DE, et al. Metabolic phenotype of skeletal muscle in early critical illness. Thorax 2018;73:926–935. [DOI] [PubMed] [Google Scholar]
- 16. Latronico N, Fenzi F, Recupero D, Guarneri B, Tomelleri G, Tonin P, et al. Critical illness myopathy and neuropathy. Lancet 1996;347:1579–1582. [DOI] [PubMed] [Google Scholar]
- 17. Latronico N, Friedrich O. Electrophysiological investigations of peripheral nerves and muscles: a method for looking at cell dysfunction in the critically ill patients. Crit Care 2019;23:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koch S, Bierbrauer J, Haas K, Wolter S, Grosskreutz J, Luft FC, et al. Critical illness polyneuropathy in ICU patients is related to reduced motor nerve excitability caused by reduced sodium permeability. Intensive Care Med Exp 2016;4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sander HW, Golden M, Danon MJ. Quadriplegic areflexic ICU illness: selective thick filament loss and normal nerve histology. Muscle Nerve 2002;26:499–505. [DOI] [PubMed] [Google Scholar]
- 20. Hermans G, Wilmer A, Meersseman W, Milants I, Wouters PJ, Bobbaers H, et al. Impact of intensive insulin therapy on neuromuscular complications and ventilator dependency in the medical intensive care unit. Am J Respir Crit Care Med 2007;175:480–489. [DOI] [PubMed] [Google Scholar]
- 21. Novak KR, Nardelli P, Cope TC, Filatov G, Glass JD, Khan J, et al. Inactivation of sodium channels underlies reversible neuropathy during critical illness in rats. J Clin Invest 2009;119:1150–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rossignol B, Gueret G, Pennec JP, Morel J, Giroux‐Metges MA, Talarmin H, et al. Effects of chronic sepsis on the voltage‐gated sodium channel in isolated rat muscle fibers. Crit Care Med 2007;35:351–357. [DOI] [PubMed] [Google Scholar]
- 23. Zink W, Kaess M, Hofer S, Plachky J, Zausig YA, Sinner B, et al. Alterations in intracellular Ca2+‐homeostasis of skeletal muscle fibers during sepsis. Crit Care Med 2008;36:1559–1563. [DOI] [PubMed] [Google Scholar]
- 24. Llano‐Diez M, Cheng AJ, Jonsson W, Ivarsson N, Westerblad H, Sun V, et al. Impaired Ca2+ release contributes to muscle weakness in a rat model of critical illness myopathy. Crit Care 2016;20:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Friedrich O, Yi B, Edwards JN, Reischl B, Wirth‐Hucking A, Buttgereit A, et al. IL‐1α reversibly inhibits skeletal muscle ryanodine receptor. A novel mechanism for critical illness myopathy? Am J Respir Cell Mol Biol 2014;50:1096–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Derde S, Thiessen S, Goossens C, Dufour T, Van den Berghe G, Langouche L. Use of a central venous line for fluids, drugs and nutrient administration in a mouse model of critical illness. JoVE 2017;e55553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goossens C, Marques MB, Derde S, Vander Perre S, Dufour T, Thiessen SE, et al. Premorbid obesity, but not nutrition, prevents critical illness‐induced muscle wasting and weakness. J Cachexia Sarcopenia Muscle 2017;8:89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goossens C, Weckx R, Derde S, Dufour T, Vander Perre S, Pauwels L, et al. Adipose tissue protects against sepsis‐induced muscle weakness in mice: from lipolysis to ketones. Crit Care 2019;23:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Osuchowski MF, Ayala A, Bahrami S, Bauer M, Boros M, Cavaillon JM, et al. Minimum quality threshold in pre‐clinical sepsis studies (MQTiPSS): an international expert consensus initiative for improvement of animal modeling in sepsis. Intensive Care Med Exp 2018;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schneidereit D, Nubler S, Prolss G, Reischl B, Schurmann S, Muller OJ, et al. Optical prediction of single muscle fiber force production using a combined biomechatronics and second harmonic generation imaging approach. Light Sci Appl 2018;7:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Buttgereit A, Weber C, Garbe CS, Friedrich O. From chaos to split‐ups—SHG microscopy reveals a specific remodelling mechanism in ageing dystrophic muscle. J Pathol 2013;229:477–485. [DOI] [PubMed] [Google Scholar]
- 32. Haug M, Meyer C, Reischl B, Prolss G, Nubler S, Schurmann S, et al. MyoRobot 2.0: an advanced biomechatronics platform for automated, environmentally controlled skeletal muscle single fiber biomechanics assessment employing inbuilt real‐time optical imaging. Biosens Bioelectron 2019;138:111284. [DOI] [PubMed] [Google Scholar]
- 33. Haug M, Reischl B, Prolss G, Pollmann C, Buckert T, Keidel C, et al. The MyoRobot: a novel automated biomechatronics system to assess voltage/Ca2+ biosensors and active/passive biomechanics in muscle and biomaterials. Biosens Bioelectron 2018;102:589–599. [DOI] [PubMed] [Google Scholar]
- 34. Zochodne DW, Bolton CF, Wells GA, Gilbert JJ, Hahn AF, Brown JD, et al. Critical illness polyneuropathy. A complication of sepsis and multiple organ failure. Brain 1987;110:819–841. [DOI] [PubMed] [Google Scholar]
- 35. Lauria G, Morbin M, Lombardi R, Borgna M, Mazzoleni G, Sghirlanzoni A, et al. Axonal swellings predict the degeneration of epidermal nerve fibers in painful neuropathies. Neurology 2003;61:631–636. [DOI] [PubMed] [Google Scholar]
- 36. Xia RH, Yosef N, Ubogu EE. Dorsal caudal tail and sciatic motor nerve conduction studies in adult mice: technical aspects and normative data. Muscle Nerve 2010;41:850–856. [DOI] [PubMed] [Google Scholar]
- 37. Hahn A, Kny M, Pablo‐Tortola C, Todiras M, Willenbrock M, Schmidt S, et al. Serum amyloid A1 mediates myotube atrophy via Toll‐like receptors. J Cachexia Sarcopenia Muscle 2020;11:103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Diermeier S, Iberl J, Vetter K, Haug M, Pollmann C, Reischl B, et al. Early signs of architectural and biomechanical failure in isolated myofibers and immortalized myoblasts from desmin‐mutant knock‐in mice. Sci Rep 2017;7:1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Friedrich O, Both M, Weber C, Schurmann S, Teichmann MD, von Wegner F, et al. Microarchitecture is severely compromised but motor protein function is preserved in dystrophic mdx skeletal muscle. Biophys J 2010;98:606–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parry SM, El‐Ansary D, Cartwright MS, Sarwal A, Berney S, Koopman R, et al. Ultrasonography in the intensive care setting can be used to detect changes in the quality and quantity of muscle and is related to muscle strength and function. J Crit Care 2015;30:1151:e9–e14. [DOI] [PubMed] [Google Scholar]
- 41. Puthucheary ZA, Phadke R, Rawal J, McPhail MJ, Sidhu PS, Rowlerson A, et al. Qualitative ultrasound in acute critical illness muscle wasting. Crit Care Med 2015;43:1603–1611. [DOI] [PubMed] [Google Scholar]
- 42. Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002;360:219–223. [DOI] [PubMed] [Google Scholar]
- 43. Ottenheijm CA, Granzier H. Role of titin in skeletal muscle function and disease. Adv Exp Med Biol 2010;682:105–122. [DOI] [PubMed] [Google Scholar]
- 44. Swist S, Unger A, Li Y, Voge A, von Frieling‐Salewsky M, Skarlen A, et al. Maintenance of sarcomeric integrity in adult muscle cells crucially depends on Z‐disc anchored titin. Nat Commun 2020;11: 4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Batt J, dos Santos CC, Cameron JI, Herridge MS. Intensive care unit‐acquired weakness: clinical phenotypes and molecular mechanisms. Am J Respir Crit Care Med 2013;187:238–246. [DOI] [PubMed] [Google Scholar]
- 46. Llano‐Diez M, Renaud G, Andersson M, Marrero HG, Cacciani N, Engquist H, et al. Mechanisms underlying ICU muscle wasting and effects of passive mechanical loading. Crit Care 2012;16: R209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kalamgi RC, Larsson L. Mechanical signaling in the pathophysiology of critical illness myopathy. Front Physiol 2016;7:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ochala J, Gustafson AM, Diez ML, Renaud G, Li M, Aare S, et al. Preferential skeletal muscle myosin loss in response to mechanical silencing in a novel rat intensive care unit model: underlying mechanisms. J Physiol 2011;589:2007–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang L, Hu W, Cai Z, Liu J, Wu J, Deng Y, et al. Early mobilization of critically ill patients in the intensive care unit: a systematic review and meta‐analysis. PLoS One 2019;14:e0223185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wollersheim T, Grunow JJ, Carbon NM, Haas K, Malleike J, Ramme SF, et al. Muscle wasting and function after muscle activation and early protocol‐based physiotherapy: an explorative trial. J Cachexia Sarcopenia Muscle 2019;10:734–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Witt NJ, Zochodne DW, Bolton CF, Grand'Maison F, Wells G, Young GB, et al. Peripheral nerve function in sepsis and multiple organ failure. Chest 1991;99:176–184. [DOI] [PubMed] [Google Scholar]
- 52. Llano‐Diez M, Gustafson AM, Olsson C, Goransson H, Larsson L. Muscle wasting and the temporal gene expression pattern in a novel rat intensive care unit model. BMC Genomics 2011;12:602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019 . J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sarcomere lengths and maximal force of ruptured and non‐ruptured myofibers. Myofibers were passively stretched to 140% of their resting length (L 0) to assess structural integrity. (A) Sarcomere lengths at L 0 of myofibers that did or did not rupture during the stretching protocol. White boxplots = healthy animals, gray boxplots = septic animals. [non‐ruptured myofibers healthy n = 8, sepsis n = 9; ruptured myofibers healthy n = 9, sepsis n = 27] (B) Force at the time of myofiber rupture. [healthy n = 11 myofibers, sepsis n = 37 myofibers] (C) Representative images of measured sarcomere lengths (SL). * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001