

Abstract
We have recently shown the strong negative impact of multiple myeloma (MM)-bone marrow mesenchymal stromal cell (BMMSC) interactions to several immunotherapeutic strategies including conventional T cells, chimeric antigen receptor (CAR) T cells, and daratumumab-redirected NK cells. This BMMSC-mediated immune resistance via the upregulation of antiapoptotic proteins in MM cells was mainly observed for moderately cytotoxic modalities. Here, we set out to assess the hypothesis that this distinct mode of immune evasion can be overcome by improving the overall efficacy of immune effector cells. Using an in vitro model, we aimed to improve the cytotoxic potential of KHYG-1 NK cells toward MM cells by the introduction of a CD38-specific CAR and a DR5-specific, optimized TRAIL-variant. Similar to what have been observed for T cells and moderately lytic CAR T cells, the cytolytic efficacy of unmodified KHYG-1 cells as well as of conventional, DR5-agonistic antibodies were strongly reduced in the presence of BMMSCs. Consistent with our earlier findings, the BMMSCs protected MM cells against KHYG-1 and DR5-agonistic antibodies by inducing resistance mechanisms that were largely abrogated by the small molecule FL118, an inhibitor of multiple antiapoptotic proteins including Survivin, Mcl-1, and XIAP. Importantly, the BMMSC-mediated immune resistance was also significantly diminished by engineering KHYG-1 cells to express the CD38-CAR or the TRAIL-variant. These results emphasize the critical effects of microenvironment-mediated immune resistance on the efficacy of immunotherapy and underscores that this mode of immune escape can be tackled by inhibition of key antiapoptotic molecules or by increasing the overall efficacy of immune killer cells.
Introduction
Multiple myeloma (MM) is a plasma cell malignancy that mainly resides in the bone marrow, where it establishes tight communications with the surrounding cells, resulting in tumor growth, survival, and resistance toward conventional drug therapies. In addition to these well-documented effects, we have recently shown that the cellular interactions between MM cells and bone marrow mesenchymal stromal cells (BMMSCs) can have a significant negative influence on the efficacy of several immunotherapeutic strategies, including conventional cytotoxic T cells (CTLs), chimeric antigen receptor (CAR)-transduced T cells, and daratumumab-redirected NK cells.1–3 We have demonstrated that this specific mode of immune escape can be readily abrogated by small molecules YM155 and FL118, which have strong inhibitory effects on multiple antiapoptotic proteins including Survivin, Mcl-1, and XIAP.1–5 Unlike several well-known immunosuppressive mechanisms, this BMMSC-mediated immune escape represents resistance mechanisms in MM cells that protect them against the cytotoxic machinery of immune cells. In addition, we have recently discovered a strong inverse correlation between the ability of BMMSCs to protect MM cells against CAR T cells and the overall lytic capacity of these immune effector cells, which led to the hypothesis that strategies that improve the intrinsic cytotoxic machinery or the avidity of immune effector cells may overcome the BMMSC-mediated immune resistance.1
To broaden our understanding of this concept, we now tested the impact of BMMSCs on the efficacy of the NK cell line KHYG-1. The efficacy of these NK cells to kill MM cells is moderate but can be significantly improved by the expression of a CD38-CAR, resulting in increased avidity toward CD38+ MM cells (manuscript under review). In addition, we evaluated the potential to improve the cytotoxic activity of KHYG-1 NK cells. To this end, we transduced KHYG-1 cells with a specific TRAIL variant (TRAILv), which was engineered to selectively bind DR5 with greater affinity compared with wild-type TRAIL and avoids binding to decoy receptors and osteoprotegerin.6
Thus, to evaluate our hypotheses, we tested BMMSC-mediated protection in MM cell lines and in patient-derived MM cells against native KHYG-1 cells and against KHYG-1 cells that express either the CD38-CAR or the TRAILv, in earlier established, compartment-specific model systems. In line with our previous studies, we also examined whether the protective effects of BMMSCs could be abrogated with the small proapoptotic molecule FL118.
Materials and methods
Bone marrow mononuclear cells
Bone marrow mononuclear cells from BM aspirates of MM patients were isolated by Ficoll-Hypaque density-gradient centrifugation and cryopreserved in liquid nitrogen until use. All patient material has been obtained after informed consent according to the code of conduct for medical research developed by The Council of the Federation of Medical Scientific Societies (FEDERA, https://www.federa.org/codes-conduct).
Bone marrow-derived mesenchymal stromal cells
Bone marrow-derived mesenchymal stromal cells were isolated from diagnostic BM aspirates from MM patients and used to culture BMMSCs as previously described.7 To minimalize interindividual variation, all assays were performed with a single batch that consisted a pool of BMMSCs derived from 12 MM patients at the earliest possible passage (passage 3).
MM cell lines
Luciferase (LUC)-transduced MM cell lines MM1.s (p53 wild-type) and RPMI-8226 (p53 mutant)5 were cultured in RPMI 1640 (Life Technologies), supplemented with 10% HyClone Fetal Clone I serum (GE Healthcare Life Sciences) and antibiotics (penicillin 10.000 U/mL and streptomycin 10.000 µg/mL; Invitrogen). Authenticity of the cell lines were verified by STR profiling (GenePrint 10 System, Promega).
KHYG-1 NK cells and genetic engineering
KHYG-1 NK cells were cultured in RPMI 1640, supplemented with 10% HyClone Fetal Clone I serum, antibiotics, and 300U/mL recombinant human IL-2 (R&D Systems). Retroviral constructs containing an optimized affinity CD38-CAR (A4 scFv) in a second generation design (CD3ζ and CD28 costimulation)8 or the DR5-specific TNF-related apoptosis-inducing ligand (TRAIL)-variant (D269H/E195R)6 were used to engineer KHYG-1 cells with standard retroviral transduction technology as previously described.8
Reagents
DR5-agonistic antibodies Drozitumab and TRA-8 (both from Absolute Antibody) were stored at 4°C. FL118 was dissolved in dimethyl sulfoxide (DMSO), aliquoted, and stored at –20°C.
BLI-based compartment-specific MM cell lysis assays
BMMSCs were plated in white opaque, 96-well flat-bottom plates in DMEM (Life Technologies) supplemented with 10% FBS (Sigma) and antibiotics, at 2.4 × 104 cells/well. After 24 hours, LUC-transduced MM cell lines were added at 1.2 × 104 cells/well to BMMSC-seeded or medium-only wells for 24 hours. When DR5-agonistic antibodies were used to induce MM cell kill, they were supplemented with freshly isolated peripheral blood mononuclear cells (PBMCs) at a PBMC to target ratio of 40 to 1 to induce FcγR-mediated clustering of DR5.9 When KHYG-1 NK cells were used as effector cells, they were added at increasing effector to target (E:T) ratios. Where indicated, FL118 was added alone or together with antibodies or effector cells. After 24 hours of incubation, the LUC substrate beetle luciferin (125 µg/ml; Promega) was added and incubated for 20 minutes. Then, the survival of MM cells was determined by bioluminescence imaging (BLI). Percentage MM cell lysis was determined using the following formula: 1—BLI signal in treated wells/mean BLI signal in untreated wells ×100%.
Flow cytometry-based ex vivo MM cell lysis assays
BMMSCs were plated in 96-well flat-bottom plates at 2 × 104 cells/well. After 24 hours, cryopreserved bone marrow mononuclear cells derived from MM patients, containing 3%–74% MM cells, were added to BMMSC-seeded or medium-only wells. After 16–24 hours, 10 µg/mL drozitumab and 50 nM FL118 were added and incubated for 24 hours. After addition of flow-count fluorospheres (Beckman Coulter), cells were harvested using Stem-Pro Accutase (Life Technologies), blocked with human immunoglobulin (100 µg/mL, Sanquin), and stained for CD38, CD138, CD45, (Beckman Coulter), DR5 (Biolegend), and live/dead cell marker (LIVE/DEAD Fixable Near-IR; Life Technologies) to determine absolute numbers of viable CD138+ CD38+ CD45dim MM cells using flow cytometry. The percentage of patient MM cell lysis was determined using the following formula: 1—absolute number of viable MM cells in treated wells/mean absolute number of viable MM cells in untreated wells ×100%.
Statistics
Comparisons between variables were performed using 2-tailed paired or unpaired Student’s t-test using Prism software (Graphpad Software Inc., v.7). The Chou-Talalay method was used to quantify KHYG-1-FL118 combinatorial effects with Combination Index (CI) values of <1 indicating synergy, of 1 indicating additive effects, and of >1 indicating antagonism.10 When drozitumab was used, that did not reach increasing MM cell lysis values with increasing concentrations, combinatorial effects with FL118 were estimated with a BLISS model, in which expected lysis values from combinatorial treatments were calculated using the following formula: (% lysis with drozitumab + % lysis with FL118) – % lysis with drozitumab × % lysis with FL118.11–13 The null hypothesis of “additive effects” was rejected if the observed values were significantly different than the expected values. Where indicated, the E:T ratio of KHYG-1 cells required to reach half maximal MM cell lysis was determined by nonlinear regression of lysis values obtained with increasing E:T ratios. Comparisons between multiple groups were then performed using one-way ANOVA. P values below 0.05 were considered significant.
Results
BMMSC-mediated resistance against NK cells and DR5-agonistic antibodies
We started the investigation by testing the efficacy of native KHYG-1 NK cells to kill MM1.s and RPMI-8226 MM cells in absence versus presence of BMMSCs. In accordance with previous studies, we found that the KHYG-1 NK cell-mediated lysis of both MM cell lines was significantly reduced in the presence of BMMSCs, across increasing E:T ratios (Figure 1A).
Figure 1.
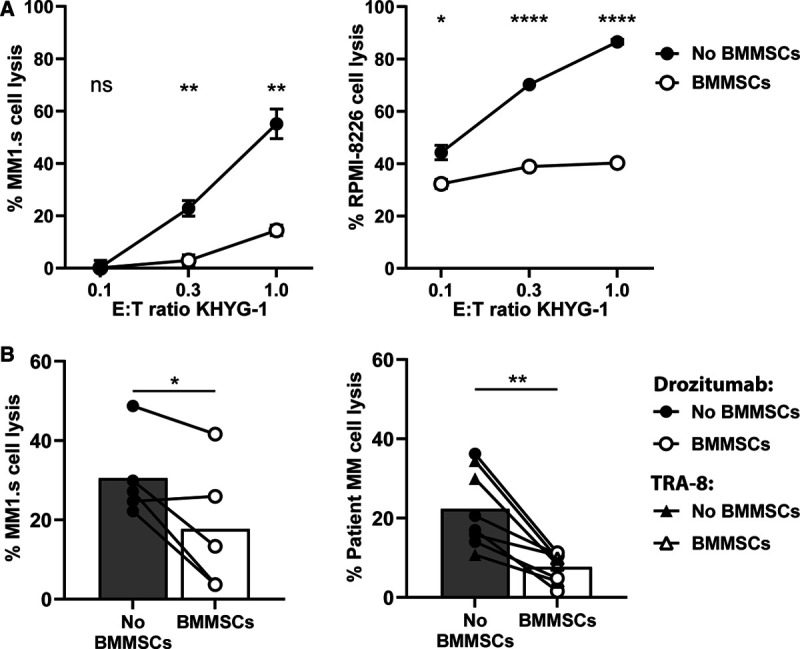
BMMSC-mediated resistance against KHYG-1 NK cells and DR5-agonistic antibodies. (A) LUC-transduced MM1.s and RPMI-8226 cell lines were cultured in the absence or presence of BMMSCs and treated with serial E:T ratios of KHYG-1 NK cells. MM cell survival was determined by BLI, 24 hours after treatment. Results are representative of 3 independent assays. Error bars represent means with SEM of triplicate cultures. (B) The MM1.s cell line (left) and BMMNC samples from 5 MM patients (right) were cultured in the absence or presence of BMMSCs. MM1.s was treated for 24 hours with 1 µg/mL drozitumab and PBMCs from healthy donors (n = 5). BMMNC samples were treated with 10 µg/mL drozitumab (circles) or TRA-8 (triangles) for 24 hours. Viable MM1.s cells were enumerated by BLI and viable CD138+ CD38+ CD45dim patient MM cells were enumerated by flow cytometry. Bars represent the median values. The statistical differences between the culture conditions without versus with BMMSCs were calculated using unpaired (A) or paired (B) t-tests (*P < 0.05; **P < 0.001; ****P < 0.0001). ns = not significant; BMMNC = bone marrow mononuclear cells; BMMSC = bone marrow mesenchymal stromal cell; LUC = Luciferase; MM = multiple myeloma; PBMC = peripheral blood mononuclear cell.
Since we aimed at improving the cytotoxic capacity of KHYG-1 cells through engineering a DR5-specific TRAILv, we next investigated if BMMSCs could protect MM cells from DR5-mediated induction of apoptosis. To this end, we used 2 DR5-agonistic antibodies, the human monoclonal antibody drozitumab and the murine monoclonal antibody TRA-8, for which the humanized versions have previously been tested in clinical trials.14–17 Indeed, the efficacy of these 2 DR5-agonistic antibodies to kill MM1.s cells as well as patient-derived MM cells was significantly reduced in presence of BMMSCs (Figure 1B). These protective effects of the BMMSCs were irrespective of DR5 expression levels on patient MM cells (Supplemental Digital Figure 1; http://links.lww.com/HS/A151).
BMMSC-mediated protection is effectively abrogated by FL118, an inhibitor of antiapoptotic proteins Survivin, Mcl-1, and XIAP
In a subsequent series of experiments, we tested whether BMMSC-induced resistance against KHYG-1 NK cells and DR5-agonistic antibodies could be modulated with the small molecule FL118, at doses that showed no toxic effects on BMMSCs or NK cells.1,4,5 Despite careful dosing, the combination of FL118 with KHYG-1 NK cells was moderately antagonistic in the absence of BMMSCs, indicating that there was still some inhibitory effect of FL118 on KHYG-1 NK cells at the tested concentrations (Figure 2A and B). Importantly however, in the presence of BMMSCs, FL118 showed a clear synergism with KHYG-1 NK cells and effectively abrogated BMMSC-induced protection of MM1.s cells (Figure 2A) and reduced the protection of RPMI-8226 cells (Figure 2B) against KHYG-1 NK cell-mediated lysis. FL118 also completely abrogated BMMSC-mediated protection against DR5-agonistic antibody drozitumab in MM1.s cells as well as in patient-derived MM cells in a synergistic fashion (Figure 2C and D) and irrespective of the level of DR5 expression on MM cells (Supplemental Digital Figure 1; http://links.lww.com/HS/A151). Altogether, these results demonstrated that BMMSC-induced protection against the cytotoxic attack of KHYG-1 NK cells and DR5-agonistic antibodies could be corrected by modulation of key anti-apoptotic molecules in MM cells, despite some inhibitory effects of FL118 on NK cells, similar to what we have previously observed for conventional CTLs, CAR T cell and NK cell-mediated antibody-dependent cellular cytotoxicity (ADCC).1
Figure 2.
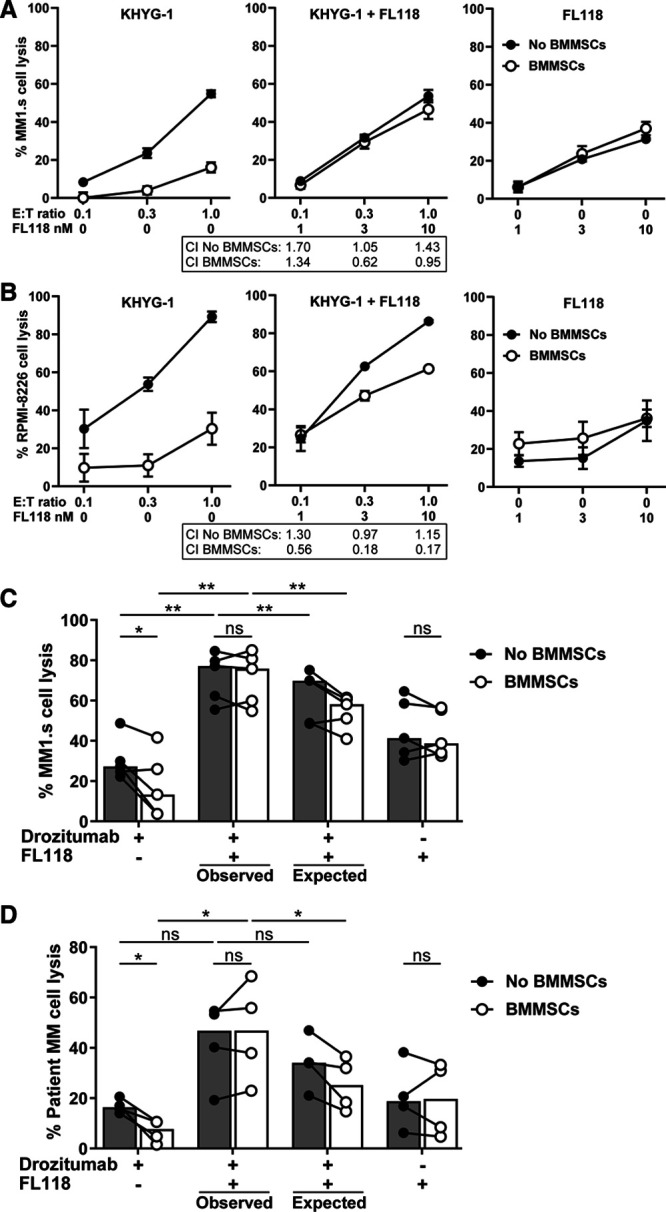
Abrogation of BMMSC-mediated immune resistance in combination with FL118 treatment. The LUC-transduced MM1.s (A) and RPMI-8226 (B) cell lines were cultured in the absence or presence of BMMSCs and treated with serial E:T ratios of KHYG-1 NK cells and serial concentrations of FL118 as shown, for 24 hours. MM cell survival was determined by BLI. Results are representative of 2 independent assays. Error bars represent means with SEM of triplicate cultures. CI values for the treatment of KHYG-1 NK cells with FL118 were quantified with the Chou-Talalay method. (C) The MM1.s cell line was cultured in the absence or presence of BMMSCs and treated with 1 µg/mL drozitumab and 5 nM FL118 for 24 hours. PBMCs from healthy donors (n = 5) were added to all treatment groups. MM cell survival was determined by BLI. (D) BMMNC samples from 4 MM patients were cultured in the absence or presence of BMMSCs and treated with 10 µg/mL drozitumab and 50 nM FL118 for 24 hours. Patient MM cell survival was determined by flow cytometry. Bars represent the median values. The observed lysis levels with combination treatment were compared with the expected lysis levels, as is described in the Materials and methods section. The statistical differences between the indicated groups were calculated using paired t-tests (*P < 0.05; **P < 0.001). BLI = bioluminescence imaging; BMMNC = bone marrow mononuclear cells; CI = Combination Index; LUC = Luciferase; MM = multiple myeloma; ns = not significant; PBMC = peripheral blood mononuclear cell.
Increasing the efficacy of NK cells can abrogate BMMSC-mediated immune resistance
After showing the important influence of BMMSCs on KHYG-1 NK cell and DR5 antibody-mediated lysis of MM cells, we investigated whether the BMMSC-mediated immune resistance against KHYG-1 NK cells could be overcome by improving its avidity toward MM cells through CD38-CAR expression or by increasing its cytotoxic activity through TRAILv expression. Expression of the CD38-CAR on KHYG-1 NK cells, in comparison to MOCK control KHYG-1 NK cells, increased the lysis of MM1.s and RPMI-8226 MM cells (Figure 3A and B). While the BMMSC-mediated lysis inhibition was gradually diminished by increasing the E:T ratio, more importantly, the BMMSC-mediated protection of MM1.s as well as RPMI-8226 cells against KHYG-1 NK cells was significantly reduced after CD38-CAR transduction of KHYG-1 NK cells in all E:T ratios, except the lowest ratio in MM1.s cells, probably due to limited physical interaction between NK cells and MM cells, which caused very low levels of MM cell lysis (Figure 3C). Similarly, expression of the TRAILv on KHYG-1 NK cells enabled the NK cells to kill the MM cells with greater efficacy (Figure 3A and B). Importantly, TRAILv expression on KHYG-1 NK cells reduced the BMMSC-mediated protection of MM cells against the KHYG-1 NK cells (Figure 3C). Combination of CD38-CAR and TRAILv did not further reduce the BMMSC-mediated immune resistance (Figure 3A-C). Together, these results supported our hypothesis that improvement of the overall avidity and cytotoxic machinery of NK cells toward MM cells can overcome BMMSC-induced immune resistance.
Figure 3.
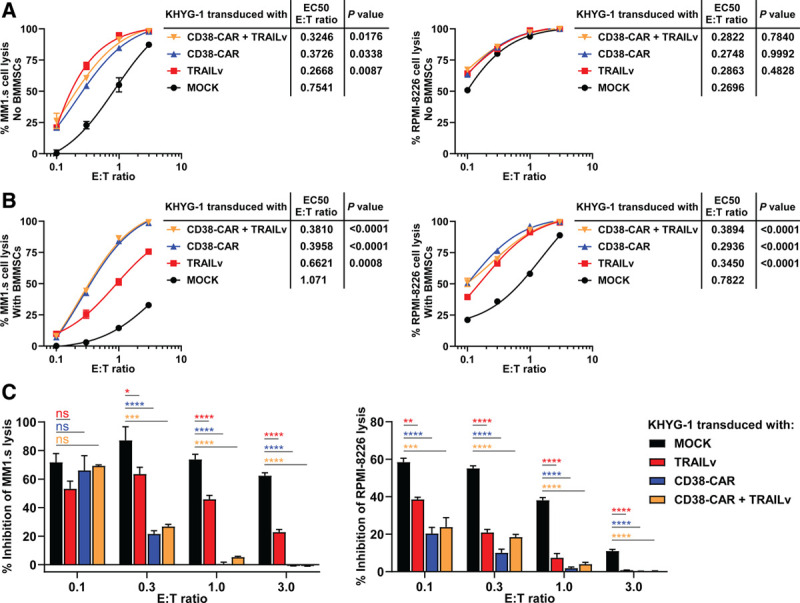
BMMSC-mediated resistance against KHYG-1 NK cells is significantly reduced by the incorporation of CD38-specific CAR or DR5-specific TRAILv. LUC-transduced MM1.s and RPMI-8226 cells were cultured in the absence (A) or presence (B) of BMMSCs and treated with serial E:T ratios of KHYG-1 cells that expressed MOCK control, CD38-CAR and TRAILv. MM cell survival was determined by BLI, 24 hours after treatment. The half maximal effective E:T ratios (EC50) of modulated KHYG-1 NK cells were compared with EC50 values of MOCK control using one-way ANOVA. (C) Results represent the percentages of inhibition of MM cell lysis (or MM cell survival) in the presence of BMMSCs. Percentages of inhibition were compared with MOCK control using one-way ANOVA (*P < 0.05; **P < 0.001; ***P < 0.0005; ****P < 0.0001). Results are representative of 2 independent assays. Error bars represent means with SEM of triplicate cultures. BLI = bioluminescence imaging; BMMNC = bone marrow mononuclear cells; BMMSC = bone marrow mesenchymal stromal cell; CAR = chimeric antigen receptor; CI = Combination Index; LUC = Luciferase; MM = multiple myeloma; ns = not significant; PBMC = peripheral blood mononuclear cell.
Discussion
The main aim of this study was to further evaluate the hypothesis that BMMSC-mediated immune resistance could be overcome by improving the target-binding and the cytotoxic machinery of immune effector cells. Using the model NK cell line KHYG-1, we demonstrated that the incorporation of a CD38-specific CAR or a DR5-specific TRAILv into KHYG-1 cells increases its cytotoxic capability against MM cells and significantly reduces BMMSC-mediated protection. These results are in agreement with our previous findings and emphasize the importance of microenvironment-mediated immune resistance as a potentially important obstacle for the efficacy of immunotherapy.
Relevant in this context is the protective effect of BMMSCs not only from KHYG-1 NK cells but also from DR5-agonistic antibodies, which showed poor therapeutic response rates in earlier clinical trials.14,16–19 In fact, the proposed resistance mechanisms against these antibodies include the increased expression of several anti-apoptotic proteins like c-FLIP, Bcl-2 and Inhibitor of Apoptosis Proteins (IAPs),20,21 most of which are strongly upregulated through interaction of MM cells with BMMSCs.22 Preclinical evaluation of those antibodies never included BMMSCs. Thus, our results emphasize the importance of including BMMSCs in preclinical evaluation of novel therapeutic strategies, especially if the strategies mediate MM cell killing by caspase-mediated apoptosis induction via the classical intrinsic- and extrinsic- apoptotic pathways. Furthermore, the capacity of FL118, a small molecule inhibitor of multiple anti-apoptotic proteins,4,5 to abrogate the BMMSC-mediated immune resistance against NK cells, even though it had a modest negative influence on the effector function of KHYG-1 NK cells, further underscore the importance of anti-apoptotic signaling in this type of immune escape and suggest that immunotherapy could benefit from effective inhibition of such antiapoptotic molecules. Thus, our results warrant further research towards the feasibility of such strategies in the clinical setting.
Finally, the other important message of our study is the possibility to overcome the BMMSC-mediated immune evasion by strategies that increase the overall killing efficacy of immune killer cells. First of all, our preclinical study showed that increasing the KHYG-1 NK cell to MM cell ratio, resulting in improved MM cell lysis, reduced BMMSC-mediated protection, as was observed for MOCK KHYG-1 NK cells in the RPMI-8226 cell line (Figure 3C right panel). But, as we have also shown, a more clinical applicable strategy is the equipment of NK cells or T cells with tumor-reactive CARs. In principle, CAR activity itself can also be improved by the inclusion of strong co-stimulatory domains or high affinity single chain variable fragment (scFv) sequences.8,23 In addition, dual-targeted CARs can enhance the strength of CAR T cell-target cell interactions24 and may hereby avoid BMMSC-mediated resistance against classical single antigen-targeted CARs. Next to the CAR technology, the cytotoxic activity of the effector cells can be improved by increasing the cytotoxic potential of immune killer cells. We have shown that this objective can be achieved by ectopic expression of a DR5 specific TRAIL variant on the NK cell surface.25 Of note, the potency of the TRAILv to overcome BMMSC-mediated resistance seemed more effective in the RPMI-8226 cell line, which had higher DR5 levels in comparison to the MM1.s cell line (Figure 3C and Supplemental Digital Figure 2; http://links.lww.com/HS/A151). This again suggests the importance of an effective induction of immune cell killer mechanisms to overcome BMMSC-mediated resistance. Nonetheless, we observed a substantial difference between the two MM cell lines already for MOCK-transduced NK cells, which was not neutralized by the TRAILv or CAR (Figure 3). Thus, it seems that the intrinsic resistance of MM cells towards the cytotoxic machinery of NK cells may also have an important and independent impact on therapy resistance, which deserves further investigation.
Expression of a DR5-specific TRAILv on effector cells may in addition have potential to modulate immune suppressive elements within the tumor microenvironment, such as regulatory T cells and myeloid-derived suppressor cells, which are known to upregulate DR5 in cancer patients.26,27 Alternative to TRAIL molecules, effector cells may be equipped with Granzyme A, that induces apoptosis via caspase independent mechanisms,22,28,29 to increase their cytotoxic potential. Further research is necessary to investigate the feasibility and potency of these strategies to overcome BMMSC-mediated immune resistance.
Overall, the results presented in the current study demonstrate the value of different strategies to tackle microenvironment-mediated immune escape, such as inhibition of key antiapoptotic proteins and improvement of the overall efficacy of immune effector cells.
Acknowledgments
The authors would like to thank the Company of Canget BioTekpharma LLC (www.canget-biotek.com), Buffalo, NY, USA, for providing FL118.
Disclosures
FL: The anticancer drug FL118 will be further developed in Canget BioTekpharma LLC (www.canget-biotek.com), a Roswell Park Comprehensive Cancer Center-spinoff company. FL is one of the initial investors in Canget for the development of FL118. RWJG has received Research support from Takeda. SZ has received research support from Janssen Pharmaceuticals and Takeda, and serves in advisory boards for Janssen Pharmaceuticals, Celgene, BMS, Amgen, Takeda, Sanofi and Oncopeptides. NWCJvdD has received research support from Janssen Pharmaceuticals, AMGEN, Celgene, Novartis, and BMS, and serves in advisory boards for Janssen Pharmaceuticals, AMGEN, Celgene, BMS, Takeda, Roche, Novartis, Bayer, and Servier. MOD has received research support from Janssen Pharmaceuticals, Celgene and BMS, serves consultancy for Abbvie, and is director (equity ownership) of ONK Therapeutics. TM has received research support from Takeda, Genmab, Janssen, Novartis and ONK Therapeutics. For the remaining authors, no relevant conflicts of interest were declared.
Sources of funding
This study was partly supported by research fundings from the Dutch Cancer Society (VU2014-6567) and ONK Therapeutics.
Supplementary Material
Footnotes
Current address for A. Stikvoort: Karolinska Institutet, Solna, Sweden.
Supplemental digital content is available for this article.
References
- 1.Holthof LC, van der Schans JJ, Katsarou A, et al. Bone marrow mesenchymal stromal cells can render multiple myeloma cells resistant to cytotoxic machinery of CAR T cells through inhibition of apoptosis. Clin Cancer Res. 2021In press [DOI] [PubMed] [Google Scholar]
- 2.de Haart SJ, van de Donk NW, Minnema MC, et al. Accessory cells of the microenvironment protect multiple myeloma from T-cell cytotoxicity through cell adhesion-mediated immune resistance. Clin Cancer Res. 2013; 19:5591–5601 [DOI] [PubMed] [Google Scholar]
- 3.de Haart SJ, Holthof L, Noort WA, et al. Sepantronium bromide (YM155) improves daratumumab-mediated cellular lysis of multiple myeloma cells by abrogation of bone marrow stromal cell-induced resistance. Haematologica. 2016; 101:e339–e342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ling X, Cao S, Cheng Q, et al. A novel small molecule FL118 that selectively inhibits survivin, Mcl-1, XIAP and cIAP2 in a p53-independent manner, shows superior antitumor activity. PLoS One. 2012; 7:e45571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holthof LC, van der Horst HJ, van Hal-van Veen SE, et al. Preclinical evidence for an effective therapeutic activity of FL118, a novel survivin inhibitor, in patients with relapsed/refractory multiple myeloma. Haematologica. 2020; 105:e80–e83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Sloot AM, Tur V, Szegezdi E, et al. Designed tumor necrosis factor-related apoptosis-inducing ligand variants initiating apoptosis exclusively via the DR5 receptor. Proc Natl Acad Sci U S A. 2006; 103:8634–8639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prins HJ, Rozemuller H, Vonk-Griffioen S, et al. Bone-forming capacity of mesenchymal stromal cells when cultured in the presence of human platelet lysate as substitute for fetal bovine serum. Tissue Eng Part A. 2009; 15:3741–3751 [DOI] [PubMed] [Google Scholar]
- 8.Drent E, Poels R, Ruiter R, et al. Combined CD28 and 4-1BB costimulation potentiates affinity-tuned chimeric antigen receptor-engineered T cells. Clin Cancer Res. 2019; 25:4014–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson NS, Yang B, Yang A, et al. An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell. 2011; 19:101–113 [DOI] [PubMed] [Google Scholar]
- 10.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006; 58:621–681 [DOI] [PubMed] [Google Scholar]
- 11.Nijhof IS, Groen RW, Noort WA, et al. Preclinical evidence for the therapeutic potential of CD38-targeted immuno-chemotherapy in multiple myeloma patients refractory to lenalidomide and bortezomib. Clin Cancer Res. 2015; 21:2802–2810 [DOI] [PubMed] [Google Scholar]
- 12.van der Veer MS, de Weers M, van Kessel B, et al. Towards effective immunotherapy of myeloma: enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica. 2011; 96:284–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev. 1995; 47:331–385 [PubMed] [Google Scholar]
- 14.Reck M, Krzakowski M, Chmielowska E, et al. A randomized, double-blind, placebo-controlled phase 2 study of tigatuzumab (CS-1008) in combination with carboplatin/paclitaxel in patients with chemotherapy-naïve metastatic/unresectable non-small cell lung cancer. Lung Cancer. 2013; 82:441–448 [DOI] [PubMed] [Google Scholar]
- 15.Rocha Lima CM, Bayraktar S, Flores AM, et al. Phase Ib study of drozitumab combined with first-line mFOLFOX6 plus bevacizumab in patients with metastatic colorectal cancer. Cancer Invest. 2012; 30:727–731 [DOI] [PubMed] [Google Scholar]
- 16.Camidge DR, Herbst RS, Gordon MS, et al. A phase I safety and pharmacokinetic study of the death receptor 5 agonistic antibody PRO95780 in patients with advanced malignancies. Clin Cancer Res. 2010; 16:1256–1263 [DOI] [PubMed] [Google Scholar]
- 17.Cheng AL, Kang YK, He AR, et al. Safety and efficacy of tigatuzumab plus sorafenib as first-line therapy in subjects with advanced hepatocellular carcinoma: a phase 2 randomized study. J Hepatol. 2015; 63:896–904 [DOI] [PubMed] [Google Scholar]
- 18.Forero A, Bendell JC, Kumar P, et al. First-in-human study of the antibody DR5 agonist DS-8273a in patients with advanced solid tumors. Invest New Drugs. 2017; 35:298–306 [DOI] [PubMed] [Google Scholar]
- 19.Fuchs CS, Fakih M, Schwartzberg L, et al. TRAIL receptor agonist conatumumab with modified FOLFOX6 plus bevacizumab for first-line treatment of metastatic colorectal cancer: a randomized phase 1b/2 trial. Cancer. 2013; 119:4290–4298 [DOI] [PubMed] [Google Scholar]
- 20.Micheau O, Shirley S, Dufour F. Death receptors as targets in cancer. Br J Pharmacol. 2013; 169:1723–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fulda S, Meyer E, Debatin KM. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene. 2002; 21:2283–2294 [DOI] [PubMed] [Google Scholar]
- 22.Holthof LC, Mutis T. Challenges for immunotherapy in multiple myeloma: bone marrow microenvironment-mediated immune suppression and immune resistance. Cancers (Basel). 2020; 12:988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drent E, Themeli M, Poels R, et al. A rational strategy for reducing on-target off-tumor effects of CD38-chimeric antigen receptors by affinity optimization. Mol Ther. 2017; 25:1946–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Larrea CF, Staehr M, Lopez AV, et al. Defining an optimal dual-targeted CAR T-cell therapy approach simultaneously targeting BCMA and GPRC5D to prevent BCMA escape-driven relapse in multiple myeloma. Blood Cancer Discov. 2020; 1:146–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheard MA, Asgharzadeh S, Liu Y, et al. Membrane-bound TRAIL supplements natural killer cell cytotoxicity against neuroblastoma cells. J Immunother. 2013; 36:319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dominguez GA, Condamine T, Mony S, et al. Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clin Cancer Res. 2017; 23:2942–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diao Z, Shi J, Zhu J, et al. TRAIL suppresses tumor growth in mice by inducing tumor-infiltrating CD4(+)CD25 (+) Treg apoptosis. Cancer Immunol Immunother. 2013; 62:653–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lieberman J. Granzyme A activates another way to die. Immunol Rev. 2010; 235:93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan Z, Beresford PJ, Oh DY, et al. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell. 2003; 112:659–672 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.