

Abstract
As one of the leading causes of morbidity and mortality worldwide, diabetes affects an estimated 422 million adults, and it is expected to continue expanding such that by 2050, 30% of the U.S. population will become diabetic within their lifetime. Out of the estimated 422 million people currently afflicted with diabetes worldwide, about 5% have type 1 diabetes (T1D), while the remaining ~95% of diabetics have type 2 diabetes (T2D). Type 1 diabetes results from the autoimmune-mediated destruction of functional β-cell mass, whereas T2D results from combinatorial defects in functional β-cell mass plus peripheral glucose uptake. Both types of diabetes are now believed to be preceded by β-cell dysfunction. T2D is increasingly associated with numerous reports of deficiencies in the exocytosis proteins that regulate insulin release from β-cells, specifically the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins. SNARE protein’s functionality is further regulated by a variety of accessory factors such as Sec1/Munc18 (SM), double C2-domain proteins (DOC2), and additional interacting proteins at the cell surface that influence the fidelity of insulin release. As new evidence emerges about the detailed mechanisms of exocytosis, new questions and controversies have come to light. This emerging information is also contributing to dialogue in the islet biology field focused on how to correct the defects in insulin exocytosis. Herein we present a balanced review of the role of exocytosis proteins in T2D, with thoughts on novel strategies to protect functional β-cell mass.
Keywords: glucose-stimulated insulin secretion, islet beta cell, SNARE proteins, insulin granule, SM proteins
Introduction to islet cell function and the deficiencies associated with type 2 diabetes
Glucose-stimulated insulin secretion
Glucose-stimulated insulin secretion (GSIS) is the release of insulin from pancreatic β-cells in response to circulating glucose. First-phase GSIS occurs first as a transient insulin spike, which lasts ~10 min. Beginning when insulin release is ~2–5-fold above the basal secretion level, a second phase of GSIS occurs, and this second phase can be sustained for hours in the presence of stimulatory extracellular glucose [1,2]. A long second phase can ensure the assimilation of nutrients that continue to be absorbed from the intestine after a meal. Although first-phase GSIS garners attention because of its sharp rise to a high-amplitude peak, the first phase is relatively brief (~10 min) and the area under the curve (a measure of total insulin release) for the first phase is often far less than that for the second phase, because second-phase GSIS can continue for hours.
Early models of insulin exocytosis mechanisms were formulated from electron micrographs showing some ISGs within 100–200 nm of the plasma membrane (PM) and others located more distantly from the PM. Based on these observations and analogous images from neurons, it was presumed that insulin secretory granules (ISGs) would approach and congregate at the inner surface of the PM, and the membrane-proximal ISGs were thus called predocked ISGs. The first phase of GSIS was presumed to be wholly derived from these predocked ISGs, known as the readily releasable pool (RRP) of ISGs [3–5]. After the depletion of the RRP, more docked ISGs must be primed and/or more ISGs must be recruited from the cell interior to sustain the second phase of GSIS; these are referred to as newcomer ISGs (reviewed in Ref. [6]).
β-cell defects in type 2 diabetes
The importance of islet β-cells in type 2 diabetes (T2D) was established more than two decades ago when dysfunctional insulin release was observed in humans with T2D (reviewed in Ref. [7]). Using in vivo hyperglycemic clamp analysis, the researchers detected a dramatic decrease in the first phase of GSIS in people with impaired glucose tolerance who were not yet diabetic [8]. Despite the foundational impact of this work, there was another effect of T2D that went largely unrecognized—an order-of-magnitude loss of second-phase GSIS. The y-axis in the graph differed by an order of magnitude for the T2D compared with that for the nondiabetic participant graph [9], which may have caused the oversight. The substantial loss of second-phase GSIS can be recapitulated ex vivo using perifused human T2D islets [10]. The results are graphed on the same y-axis scale in Fig. 1 to depict the loss of amplitude in both the first and second phase of GSIS.
Fig. 1. Type 2 diabetic (T2D) human islets show deficient first- and second-phase glucose-stimulated insulin release (GSIS).
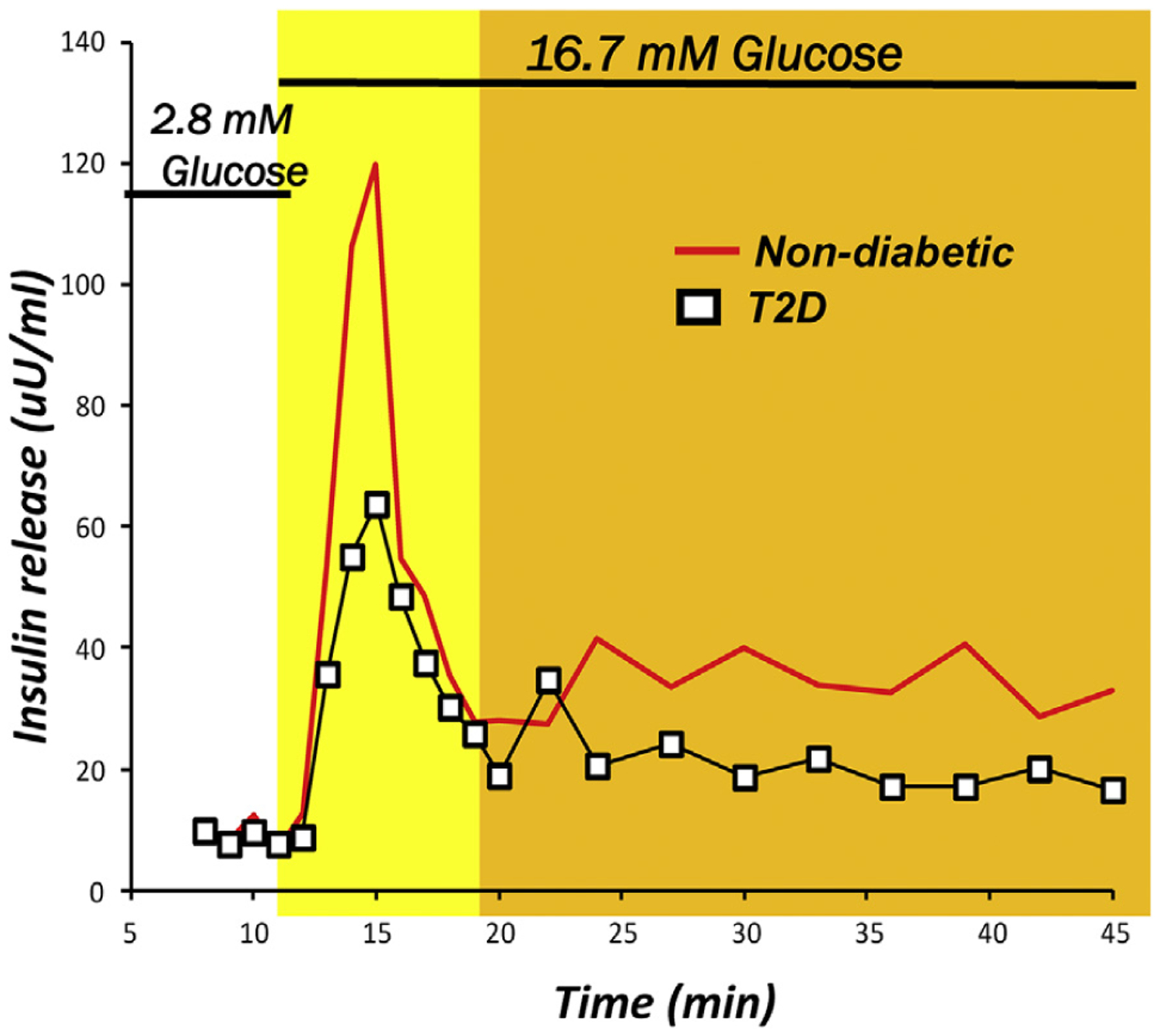
Biphasic insulin release in response to a glucose stimulus measured in isolated human islets from age, gender, and ethnicity matched human donors who are nondiabetic (red line) or T2D (black line). First-phase release begins within 2 min and lasts for ~10 min (yellow region), whereas second-phase release can last as long as several hours, if the glucose stimulus persists (orange region). Note that first phase does not return to baseline when the second phase begins. Adapted from Ref. [10], with permission from JCEM.
In this review, we provide an updated view of the β-cell exocytotic machinery. Importantly, we have focused on recent insights into deficiencies in the exocytotic machinery during diabetes and the major controversies that have arisen. New concepts derived from this active area of research have led us to propose exciting new avenues of treatment, which will also be discussed.
Mechanisms of biphasic glucose-stimulated insulin secretion
Stimulus-secretion coupling pathway
Insulin is released into the circulation when the pancreatic islet β-cells sense increases in extracellular glucose that are sufficient to elicit signaling via the stimulus-secretion coupling pathway (Fig. 2). This pathway begins when extracellular glucose enters the β-cell through cell surface glucose transporters (GLUT2 in rodents [11], GLUT1 in humans [12]) and is rapidly metabolized, thereby increasing the ATP/ADP ratio. PM-localized ATP-sensitive potassium channels (KATP) respond to this ratio by closing, which causes PM depolarization to induce the opening of PM voltage-dependent calcium channels [13] and allow calcium influx into the β-cells. The increased intracellular concentration of calcium leads to ISG-PM fusion to form the fusion pore and release insulin from predocked and primed ISGs, thus creating the insulin spike associated with first-phase GSIS [4,14–17]. The next portion of the stimulus-secretion coupling pathway, which delivers the newcomer ISGs to the PM for exocytosis, requires ISG recruitment from the cell interior, ISG tethering and docking on the PM, and priming to prepare the ISGs to become fusion-competent and releasable (reviewed in Refs. [6,18]).
Fig. 2. Basic steps in insulin granule exocytosis.
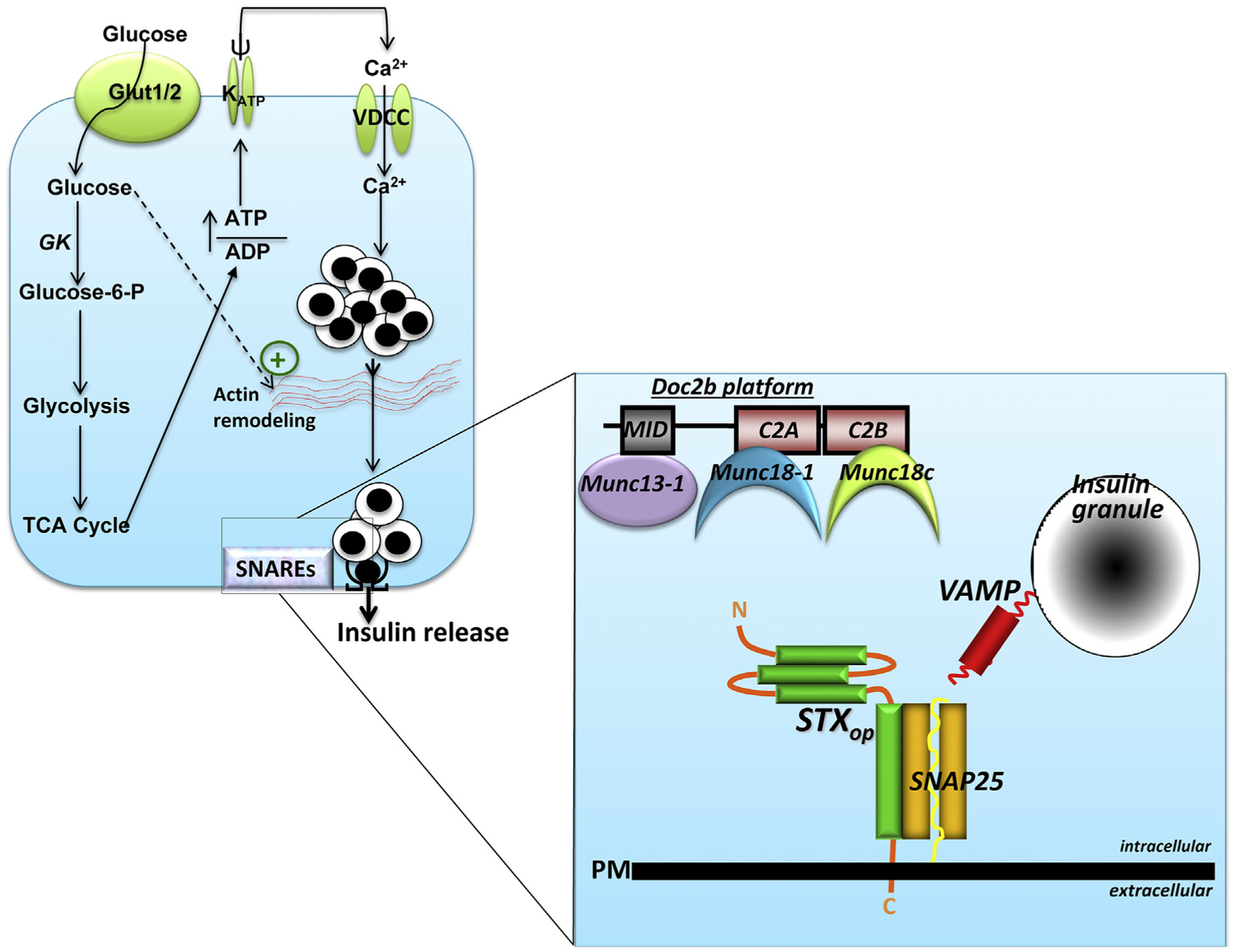
β-cells sense elevated circulating glucose via the glucose transporter (Glut1 in humans, Glut2 in rodents) at the plasma membrane (PM). Glucose is rapidly metabolized, which increases the ATP/ADP ratio, triggering KATP channel closure, PM depolarization (ψ), and opening of the voltage-dependent calcium channels (VDCC) to increase intracellular calcium. Glucose also promotes actin remodeling to facilitate the steady flow of insulin granules to the SNAREs at the PM. The details of the SNAREs are shown in an enlargement at the right, where the three SNAREs (STXop, open form of syntaxin; SNAP25, and VAMP) are assembling to form the heterotrimeric SNARE complex. After SNARE complex formation, the ISGs fuse with the PM and the insulin cargo is released into the extracellular space. The SNARE complex assembly process is regulated by accessory proteins that can attach to a scaffold protein, DOC2B. The DOC2B scaffold acts as a floating platform so that when the Munc18 proteins release from their cognate STX partners, they can associate with the nearby Doc2b platform. Munc18–1 binds to Doc2b′s C2A domain, and Munc18c binds to Doc2b′s C2B domain; the priming protein Munc13–1 can associate with Doc2b via its Munc13-interacting domain (MID).
SNARE proteins and accessory proteins
These sequential steps of exocytosis are mediated by highly conserved proteins called soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) on the ISG (VAMP) and PM (syntaxin [STX] and SNAP25) and many SNARE-associated proteins that assist in the assembly of the three SNARE proteins to form the primed SNARE complex. ISG exocytosis is thought to closely mimic neuronal synaptic vesicle fusion [19,20]. Here, synaptic vesicles, as well as ISGs, are now recognized as being associated with the PM in a hemi-fused state, predocked and waiting for calcium release to complete vesicle-PM fusion [19,20]. Most, but not all, of these predocked ISGs become primed, and upon stimulation, calcium influx from voltage-gated calcium channels promotes ISG-PM fusion (reviewed in Refs. [21,22]). In healthy β-cells, distinct SNARE complexes typically regulate the fusion of predocked and newcomer ISGs. The three SNARE proteins that mediate fusion of predocked ISGs are primarily VAMP2, STX1A, and SNAP25, which form a “primed” or fusion-ready complex after activation by Munc18–1 [19–22]. Fusion of newcomer ISGs is mediated by distinct SM and SNARE proteins consisting of Munc18b activation of a STX3-SNAP25-VAMP8 complex [23–26] and also Munc18c activation of a STX4-SNAP25 complex [27,28], when the latter binds VAMP8 (discussed further).
Many accessory proteins then regulate the assembly of these SNARE proteins into a primed SNARE complex. Sec1/Munc18-like (SM) proteins, particularly neuronal Munc18–1 (also called Munc18a), bind STX1A in a dormant “closed” conformation (STXcl) [29]. Munc18–1 then converts STX1A into an activated “open” conformation (STXop) that can associate with SNAP25 and VAMP2 in a 1:1:1 stoichiometry [19,20,30]. This priming process is assisted by signaling pathway components such as diacylglycerol-activated Munc13–1 [31,32] and cAMP/PKA-activated RIM (RIM2 in β-cells) and Epac2 [33,34]. Because fusion is a calcium-sensitive process, the SNARE complex requires calcium sensors, which are primarily the synaptotagmins (synaptotagmin-7 in β-cells) [35]. Another calcium sensor in β-cells is DOC2B, which contains calcium-sensing C2 domains. In β-cells, DOC2B binds to Munc18–1 and Munc18c via its C2A and C2B domains, respectively [36–38] (Fig. 2), and can influence both first- and second-phase GSIS [39,40].
Although the SNARE complex can spontaneously carry out PM fusion in vitro, albeit slowly, this spontaneous PM fusion does not occur in vivo. In regulated secretory cells, there are “clamps,” regulated by the complexins (complexin-2 in β-cells) [41] and tomosyns (tomosyn-2 in β-cells) [42], which prevent spontaneous ISG fusion, although a recent report relegated complexins to have a relatively minor role in the clamping action [43]. Furthermore, multiple protein conformational changes must be well orchestrated, including the activation of calcium sensors and release of the clamps. These changes allow complete formation of the SNARE complex, presumed to entail “zippering up” from a hemi-fused state as observed by many in vitro, to culminate in mixing of the phospholipid bilayers of the vesicle/granule and PM to form the fusion pore. Zippering and phospholipid bilayer mixing has been observed in vitro, and in non-islet cells and organelles [44–47] and awaits to be tested in β-cells.
Many of the aforementioned studies that defined the stepwise assembly of these fusion proteins have been based on biochemical data from cell homo-genates, which had predicted evolving sequential models that had served as working hypotheses. These models have been more recently tested using high spatiotemporal live-cell imaging analysis of their molecular interactions in the physiologically relevant timescale and have led to modifications of the original models. Recent work using high-resolution total internal fluorescence (TIRF) microscopy has begun to reveal and redefine the details underlying the coordinated dance of these exocytotic proteins. For instance, it was shown that Munc18–1 binds STX1A molecules to form a cluster of 50–70 STX1A molecules on the PM, which serve as docking sites to recruit SNAP25. These proteins form the acceptor complex, which can then sequentially recruit Munc13–1, synaptotagmin, and finally VAMP2 [48–50]. A more critical assessment in the β-cell showed a different sequence in how these proteins come to form clusters on the PM to then bind calcium channels [50], with the surprising finding that Munc13–1 could directly bind and serve to recruit calcium channels to the ISG [51]. Using two-photon microscopy, it was suggested that the cognate SNAREs may not have been preassembled (as still believed by most) and instead undergo assembly slowly during stimulation and that the different states of assembly would have profound effects on the state of fusion readiness of the SNARE complex [52]. Finally, because these clusters of SNARE complex proteins form at focal sites on the PM, polarized secretion can occur in the morphologically nonpolarized β-cells [50]. In fact, we and others have noted that repeated ISG recruitment and fusion occur at these polarized sites, especially when these SNARE clusters also include calcium channels [51,53,54].
Defective exocytosis in diabetes and therapeutic insights
Given the complexity of ISG exocytosis, it is therefore easy to imagine why any misstep in this mechanism would either reduce ISG fusion efficiency or block fusion completely. This is the case in T2D β-cells, wherein the reduced levels of certain exocytosis proteins [10,55–61] impair the exocytosis of predocked ISGs. This exocytosis deficiency has been long known to account for the disappearance of first-phase GSIS [3]. Conversely, rescuing the deficiencies in key exocytotic proteins could restore biphasic GSIS, a strategy that could be developed to treat human diabetic islets [10,22].
For type 1 diabetes (T1D), if it was possible to stably enrich the expression of key exocytotic proteins in human islets, then it is conceivable that a smaller number of islets would be needed for transplantation, which would greatly expand this limited therapeutic resource to a larger number of T1D patients. However, which protein or proteins should be enriched? Genetic STX1A overexpression paradoxically reduces β-cell exocytosis [62]. On the other hand, in T2D islets, STX4 overexpression rescues the deficiency in STX1A and restores GSIS [10]. The inhibitory effects of overexpressed STX1A are at least in part attributed to its inhibitory effects on calcium channels, resulting in reduced calcium influx [62]; one explanation for this outcome was that the overexpressed STX1A stabilizes calcium channel inactivation [63]. However, an alternate explanation for these inhibitory effects involves the necessity for stoichiometric pairing of the three SM proteins and their cognate SNARE complexes, as well as complex interactions of these SM/SNARE complexes and associated proteins with the calcium channel [51].
In T2D islets, the levels of some SM/SNARE and accessory proteins are reduced, whereas some remain relatively normal [10,26]. Nonstoichiometric pairing of SNAREs leads to the formation of incomplete SNARE complexes that are inhibitory and compete with the complete SNARE complexes. Overexpression of an SM protein, such as Munc18–1, can lead to an excess of SM/closed conformation STX complexes [64]. Similarly, overexpression of Munc18c is thought to sequester STX4 in its inactive closed conformation and prevent the formation of fusion-competent SNARE complexes; indeed, islets from transgenic mice overexpressing Munc18c showed impaired GSIS [65]. On the other hand, overexpression of Munc18b rescued deficient GSIS in islets from T2D human and T2D rats [26], likely because Munc18b could recruit and promote the assembly of deficient SNARE proteins into functional SNARE complexes [26]. Therefore, it seems that a complex mechanism governs the impact of nonstoichiometric SNARE complex interactions on β-cell function and the role of stoichiometry must be considered when predicting the outcome of candidate therapeutic interventions.
Key questions
Question 1. Do clinical parameters and/or genetics contribute to the variable expression/functionality of exocytosis proteins?
Numerous reports correlate deficiencies in SNARE and regulatory proteins to T2D and obesity in human subjects [10,57,66]. Although there is general agreement that the levels of the SM protein Munc18–1 and SNARE proteins STX1A and VAMP2 are reduced in T2D islets, there is a wide degree of variation in the reported deficiencies. STX4 is reduced in T2D islets, and reduced STX4 is associated with the T2D that co-occurs with psoriasis [67]. Macrophage-derived nitrosative stress induces phosphorylation of STX4 at serine residue 78, which induces proteosomal degradation of STX4 [68]. The prevailing hypothesis is that deficiencies in SNAREs are likely caused by diabetes and that the differences in SNARE protein loss might be attributed to differences in the disease states. In fact, glucotoxicity and lipotoxicity, both of which are associated with T2D, accentuate the loss on these SNARE proteins, but with variable effects, and resolution of the glucolipotoxicity could restore the levels of some of these deficient SNARE proteins that contributed to the improvement in insulin secretion [59,69]. It remains to be seen whether the variability in SNARE protein loss will be explained by stratification of SNARE levels by clinical parameters such as BMI, adiposity, and serum analyte levels.
Furthermore, genetic polymorphisms may impact SNARE protein function in T2D. For instance, polymorphisms in STX1A have been linked to impaired glucose metabolism, particularly in insulin release from β-cells, in overweight and obese humans [70], and to the age of T2D onset and degree of insulin requirement in T2D individuals [71]. High-impact variants for STX4 that are strongly associated with elevated BMI are reported (Type 2 Diabetes Knowledge Portal, type2diabetesgenetics.org). It will be important to retrospectively evaluate whether these polymorphisms are overrepresented in the populations of islet donors with low loss of STX1A and STX4 but poor functionality, now that rapid and inexpensive genetic testing is commonplace.
Question 2. What is the purpose of having alternative SNARE complexes in β-cells?
Whereas a single set of SM (Munc18–1) and cognate SNARE proteins (STX1A, VAMP2, SNAP25) would theoretically suffice to mediate biphasic GSIS, empirical studies of depletion of Munc18c [72] and STX1A [73] have been shown to cause first-phase- and second-phase selective reduction of GSIS, respectively. Findings such as these have led to the concept that alternative SNARE-SM protein partnerships can form functionally distinct fusion complexes to regulate first- and second-phase GSIS (reviewed in Ref. [6]). According to this alternative partnership concept, STX4 is activated by Munc18c to bind VAMP2 and SNAP25, which forms a SNARE complex that mediates predocked ISG fusion in first-phase GSIS [28,72]. This process of mediating predocked ISG fusion is functionally redundant to the activation of STX1A by Munc18–1 to form SNARE complexes with SNAP25 and VAMP2. Munc18b-linked STX3-based SNARE complexes as well as Munc18c-linked STX4-based SNARE complexes are the primary mediators of newcomer ISG fusion (reviewed in Refs. [6,23,74]). One would therefore assume that these SM and SNARE protein interactions have high fidelity, to support the preferential formation of SNARE complexes that interact with only predocked or newcomer ISGs. However, there appears to be an element of promiscuity in the formation of SNARE complexes, a concept derived from in vitro protein-binding studies [75]. We speculate that there may be a need for functional redundancy under pathophy-siological conditions.
This concept of promiscuous binding of non-cognate SNAREs sufficient to form SNAREpins that bring two membranes in proximity (i.e., PM and vesicle/granule) and “zipper up” to effect fusion had been proposed earlier based upon reconstituted fusion assays [76]. These in vitro fusion assays indicated that while fusion mediated by non-cognate SNARE complexes can be demonstrated, their kinetic actions may not be fast enough to be meaningful under physiological conditions. To do so would require not only the subcellular compartmental specificity of the v- and t-SNAREs [77], but also a host of cognate regulatory factors (Munc18, Munc13, synaptotagmin, complexin) to increase fusion efficiency, as discussed earlier. This hypothesis, however, requires vigorous testing in a genuine biological system that was previously thought not to be possible (therefore not supporting a physiologic role of non-cognate SNAREs) because a fuller complement of catalytic factors would have to be present. As discussed earlier, the β-cell contains the full complement of SNAREpins, catalytic and restraining factors to modify fusion efficiency, whereby the promiscuity of fusion by non-cognate SNAREs could occur if faced by a deficiency of cognate SNAREs and catalytic factors caused by T2D. We speculate that if the content of catalytic factors remained sufficient to drive fusion of the non-cognate SNAREpins, this may have the capacity to effect, albeit somewhat inefficient, secretion in the T2D β-cell.
Promiscuous SM/SNARE complex formation was supported by early in vitro binding assays showing that STX1–3 binds to Munc18–1 and Munc18b [78–80], whereas only STX4 binds to Munc18c with high affinity [81,82]. Furthermore, studies in vivo and in human islet β-cells have shown that Munc18c prefers to bind STX4 in a closed conformation, and Munc18–1 prefers the closed conformation of STX1A (Fig. 3A). However, newer in vitro assays have revealed that Munc18c and Munc18–1 will bind with lower affinity to STX1A and STX3, respectively [75], indicating that promiscuous partnership can occur (Fig. 3B). In response to diabetogenic stress-induced depletion of SNARE and SM proteins in islet β-cells, alternative SNARE complexes may permit some ISG docking and fusion, but with a reduced rate and/or extent. In support of this model, adding Munc18b to human T2D islets harboring reduced levels of Munc18–1 restored GSIS, via its ability to bind to STX3 (and residual STX1A [26]). Overexpression of STX4 binds to Munc18c to compensate for deficiencies in Munc18–1 and STX1A (Fig. 3C). Interestingly, in β-cells replete in STX1A and STX4, the overexpression of Munc18–1 boosted GSIS, and this increase in insulin secretion required STX4 [55]. Therefore, it is possible that promiscuity occurs in any cells wherein the normal stoichiometry of SM and SNARE proteins is disturbed, and not just in disease states. Another possibility is that the overexpressed Munc18–1 bound and sequestered the recently identified inhibitory STX, STX2 [83], thereby increasing insulin release. Knowing now that promiscuous SM and SNARE interactions occur (discussed further), and that STX2 is inhibitory for GSIS (discussed next), this boosting effect of Munc18–1 will require re-evaluation.
Fig. 3. Fidelity and promiscuity in β-cell SNARE pairing.
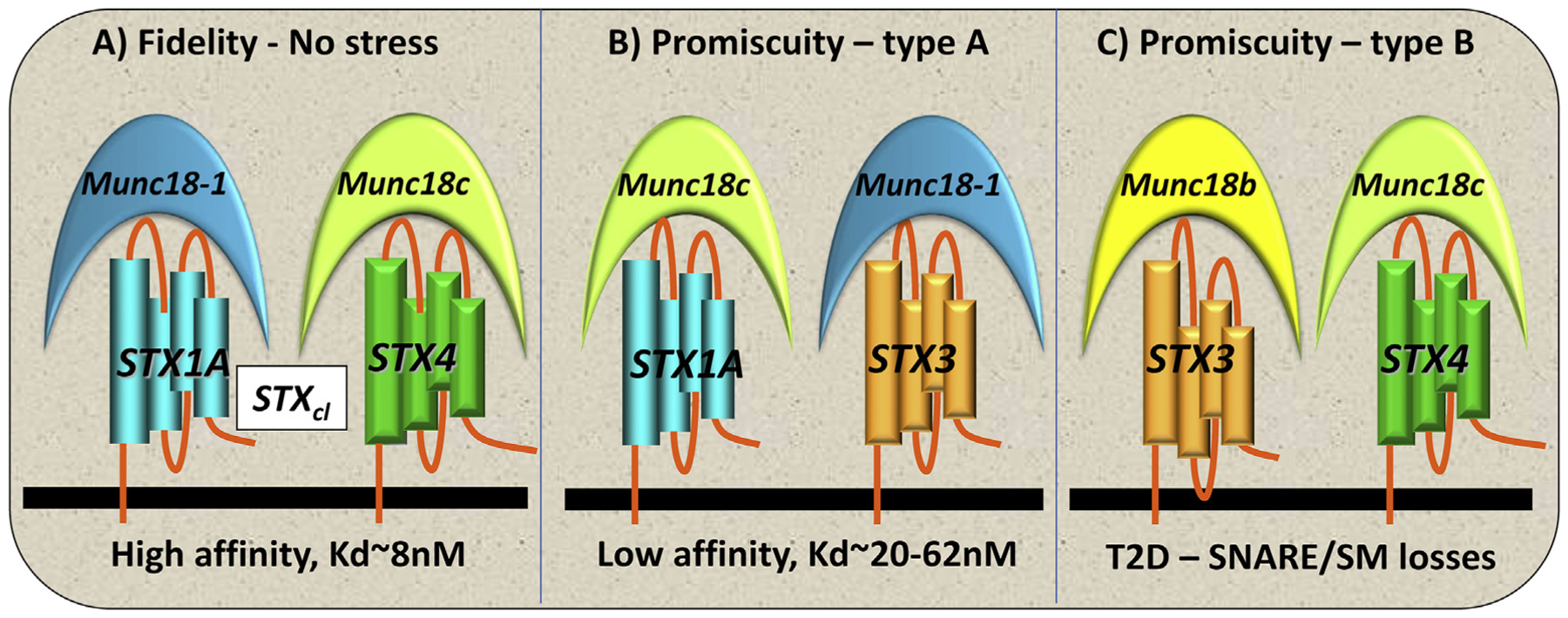
(A) Fidelity occurs in healthy β-cells, with STX and Munc18 isoforms pairing according to highest affinity partnerships (Kd ~8 nM). (B) Promiscuity type A occurs under conditions of stress where lower-affinity partnerships are formed (Kd ~20–62 nM) and GSIS function is reduced. (C) Promiscuity type B occurs in the absence or greatly reduced amount of a primary SNARE (e.g., STX1A) or Munc18 isoform (e.g., Munc18–1) in T2D islets, which can be overcome by the overexpression of Munc18b or STX4, resulting in functionally redundant rescue of those deficient exocytotic proteins.
Inhibitory SNAREs
STX2 can bind all three SM proteins at high affinity, which is similar to the affinity of the SM proteins for their primary STX partners [75]. At first glance, one would assume STX2 to have a redundant functional role to the proexocytotic proteins STX1A, STX3, and STX4. On the contrary, STX2 was reported to play an unexpected inhibitory role by competing with STX1A and STX3 for their SNARE partners, thus blocking fusion of both predocked and newcomer ISGs [83]. SNAP23, also thought to be functionally redundant to SNAP25, likewise competes with SNAP25 for SNARE complexes, thereby suppressing ISG fusion and GSIS [84]. It therefore appears that these “inhibitory” SNAREs may modulate and fine-tune GSIS in response to ever-changing demand. This insight has therapeutic implications: blocking these inhibitory proteins could improve GSIS. In support of this therapeutic potential, blocking SNAP23 action with a small molecule drug was shown to increase GSIS in vivo [84]. The therapeutic potential of blocking STX2 remains to be evaluated.
Promiscuous alternate SM/SNARE complexes for predocked and newcomer ISGs
STX4 and Munc18c were shown to also mediate second-phase GSIS [28,72], which is commonly attributed to newcomer ISG fusion. Indeed, shRNA knockdown of Munc18c [85] or STX4 [86] in human β-cells reduced both predocked and newcomer ISG fusions, thereby reducing both first- and second-phase GSIS. Furthermore, genetic deletion of STX1A in mouse β–cells depleted not only predocked ISGs but also newcomer ISGs, albeit to a lesser extent [87]. These unexpected effects of STX4 and STX1A on newcomer ISG fusion have been attributed to their promiscuous binding of VAMP8 [24,85,86] (as alternative to binding to VAMP2, their primary partner that is known to mediate predocked ISG fusion). We therefore postulated that the identity of the VAMP (VAMP2 or VAMP8) [24] may be a stronger determinant of whether an ISG behaves as a predocked or newcomer ISG compared with the identities of the STX and SM proteins. However, cognate SMs and STXs likely induce preferential assembly with a favored VAMP partner. For example, Munc18b activates STX3 to mediate newcomer ISG fusion via VAMP8, but can also activate STX1A to mediate predocked ISG fusion via VAMP2 [26]. Taken together, it is the promiscuity in SM/STX partnering, and also the subsequent assembly with the VAMPs, that promotes different SNARE complexes and mediates the ISG fusions underlying first- and second-phase GSIS.
When the SNARE field started, it was dogma that distinct SM/SNARE complexes would be formed with high affinity and fidelity to mediate very specific fusion events [19]. In the normal state with normal levels of SM/SNARE (and accessory) proteins exhibiting preferential affinity for their partners, high fidelity is the norm. So, is this promiscuity discussed earlier experimentally contrived or can it become a biological necessity? STX4 overexpression in human T2D islets can rescue the disruptions to both phases of GSIS [10]. Munc18b overexpression in T2D human islets and T2D Goto-Kakizaki rats also rescued biphasic GSIS in vitro and, remarkably, in vivo [26]. It therefore appears that in severely SNARE-deficient T2D β–cells, SM proteins (Munc18b or Munc18c for deficient Munc18–1), STXs (STX4 for deficient STX1A), and VAMPs (VAMP8 for deficient VAMP2) can replace the deficient fusion proteins. This promiscuous binding can generate sufficient fusion-competent SM/SNARE complexes to at least partially rescue biphasic GSIS. Hence, when the dance of exocytosis is missing few dancers, the dance can still go on because they can simply switch partners. However, this dance of promiscuous SM/SMARE partners with lower affinity for each other may not be perfect and may increase the variability of fusion efficiency. Such variable fusion efficiency could introduce considerable heterogeneity into GSIS, which would depend on the disease status of T2D. In fact, different states of T2D induce dynamic effects on SM/SNARE protein levels. This also has strong therapeutic implications because the most versatile SNARE proteins, the dancers that can dance with the most partners (e.g., Munc18b, STX4, VAMP8), could be selected for therapeutic development to restore biphasic GSIS in T2D islets [10,26].
While a single set of SM (Munc18–1) and cognate SNARE proteins (STX1A, VAMP2, SNAP25) could theoretically suffice to explain biphasic GSIS, the earlier discussion on the numerous reports supporting the role of promiscuous pairing of SM proteins and SNARE complexes raises the corollary concept of functional redundancy. Functional redundancy serves a teleological purpose, particularly for the β–cell that is vulnerable to disease (diabetes) and exposed to physiological insults (glucolipotoxicity) that induce transient or chronic secretory deficiency states [88,89]. Inefficient fusion, overseen by “less-cognate” SNARE interactions with various enabling catalysts (Munc18, Munc13, synaptotagmins), can play an important role under pathological conditions, effectively replacing the deficient SM and SNARE proteins to provide some level of GSIS. It is important to note that SNARE complex assembly is rapidly followed by disassembly and SNARE recycling for subsequent rounds of docking and fusion [90]. However, we speculate that under pathological conditions, where levels of some SNARE proteins may be compromised, resultant from genetic or epigenetic/environmental exposures, the reassembly may not be as faithful and efficient, and alternate SNARE complexes may be utilized.
Question 3. Are there other types of ISG fusion that significantly contribute to GSIS?
Therapeutic potential of increasing newcomer ISG recruitment and fusion
While much work had focused on the role of predocked ISG priming and fusion in first-phase GSIS, it now has been proposed that increasing newcomer ISG recruitment and fusion could compensate for the loss of first-phase GSIS in T2D. In T2D β-cells, the peak of the first phase is flattened to a similar level as the second phase. This residual release in the first phase is attributed to newcomer ISGs. Although the SM and SNARE proteins that mediate predocked ISG fusion are depleted in T2D, the levels of the SM and SNARE proteins mediating newcomer ISG exocytosis are relatively intact in the T2D islet [26]. In support of the therapeutic potential of increasing newcomer ISG recruitment, adenoviral overexpression of Munc18b in T2D human β-cells increased newcomer ISG fusion in both phases of GSIS. In vivo administration of the Munc18b adenovirus to T2D Goto-Kakizaki rats improved glucose homeostasis for a sustained period (4 months); remarkably, this improvement surpassed the therapeutic efficacy of conventional daily treatment with a sulfonylurea [26].
While many groups, including ours, have made similar observations of the existence and exocytotic kinetics of newcomer ISG fusion [6,18,23–26,91], others have not consistently observed such events [50]. We and others have defined newcomer ISGs to be the ISGs that are not predocked. Therefore, these ISGs are not seen as fluorescent hotspots at the PM by TIRF microscopy, and they appear de novo from the darkness deeper within the cytosol. One reason for the discrepancy may be technical, because it is possible for the fluorescence on newcomer ISGs to be bleached during TIRF microscopy imaging. Another possible explanation is that some groups might consider unprimed predocked ISGs to be newcomer ISGs. Primed predocked ISGs could also lose their primed status as part of as yet undefined “aging” process (discussed further) [92,93], and perhaps this depriming is accelerated in T2D. The latter two possibilities beg the following question: do ISGs, including unprimed and deprimed ISGs, undergo recruitment from the cytosol to the PM and undergo rapid priming and fusion?
Do newcomer ISGs undergo more rapid priming?
Based on TIRF experiments, newcomer ISGs are reported have a short residence time at the PM before fusion, suggesting that they can undergo rapid priming; this may be because these ISGs have a higher calcium sensitivity than predocked ISG for fusion [94]. Although the set of SM and SNARE proteins required for newcomer ISGs is often distinct from those proteins involved in predocked ISGs, both complexes share similar priming (Munc13–1) and calcium sensor (synaptotagmin-7) proteins [35,95]; the newcomer ISGs might even be more sensitive to these priming protein and calcium sensor in the predocked ISGs. More work using higher spatiotemporal resolution TIRF imaging, as was done for the predocked ISG exocytotic machinery [50], needs to be performed to decipher the sequential interactions of components of the newcomer exocytotic machinery and explain the more rapid priming.
Are newcomer and predocked ISGs different populations of ISGs?
While it might seem that newcomer and predocked ISGs exist as distinguishable entities, this is likely not the case. For example, all ISGs arrive at the PM as some type of “newcomer” that requires some type of priming [52]. Under basal conditions, such as the fasted state, there are already ISGs visibly predocked at the PM, although prior to this predocking, they were newcomer ISGs. It remains in dispute whether there is a preference for the predocked ISG to undergo a more complete SNARE complex assembly, because this predocked ISG SNARE complex can also undergo a loss of priming. If so, it could be speculated that to ensure “fusion-readiness,” it would be preferable for SNARE proteins to be only weakly and partially assembled that then undergo rapid assembly proceeding to fusion during stimulation [52]. Indeed, some predocked ISGs are well known to lose fusion competence, described by some as “aged” ISGs [92,93]; this process of loss of priming and fusion competence is accelerated in T2D. In this case, the SNARE machineries intended to facilitate the oncoming newcomers can undergo a new cycle of assembly to attain (or reattain) the primed status and fusion competence for the ISGs and produce an intact second-phase GSIS. It is intriguing to speculate whether the newcomer SNARE complex in the unprimed or deprimed predocked ISGs might be capable of being (re) activated to restore fusion competence.
How important is the contribution of compound and sequential ISG-ISG fusion to GSIS?
Multiple SGs homotypically fused (i.e., SG-SG fusion) that then fuse with the PM with the intent to release massive amounts of SG content is termed or compound exocytosis, which was initially presumed to be insignificant in β-cells (only 2–3% of total exocytosis events), has been demonstrated by several groups to account for as much as 20% with the inclusion of an amplifying stimulus such as GLP-1 [96], cholinergic stimulation [97], or overexpression of Munc18b [98] (and perhaps STX3 [25] or SNAP25 [99] as well). As it turns out, the SM and SNARE protein isoforms that mediate newcomer ISG fusion also mediate compound fusion [24,25,98,99]. Some of these studies were convincingly shown by high-quality two-photon microscopy or electron microscopy [25,98,99]. The compound fusion that occurs in β-cells is not like that classically described in mast cells. Compound fusion in a mast cell is complete and almost immediate, and it involves massive multi-SG fusion to release its contents (i.e., histamine) that would induce an allergic or anaphylactic reaction [100,101]. In β-cells, compound fusion occurs in a slow, stepwise manner, reaching a state where five ISGs fuse together in response to cholinergic stimulation [97], and up to eight ISGs fuse in cells overexpressing Munc18b [98].
With very fine slicing and 3D-reconstructed electron microscopy images, sequentially fused ISGs were observed to have only small fusion pores connecting them; all these compound-fused ISGs were situated within two ISG diameters (~200–300 nm) from the PM [98], presumably allowing exposure to maximum calcium influx through the voltage-dependent calcium channels. Furthermore, some of the sequentially fused ISGs were observed to be undergoing fusion with both the PM and the lead ISG [98]. The ability of Munc18b to rescue GSIS in T2D β-cells (human and Goto-Kakizaki rat) [26] may be due to increased compound fusion that increased the efficiency of insulin release, particularly if there were fewer fusion sites on the PM. One issue with the interpretation of compound fusion is that the small fusion pores among the fused ISGs may not be large enough for the large crystallized insulin core to traverse; the hexameric insulin crystal was shown to be limiting for release by exocytosis mechanisms such as “kiss-and-run” and would require full ISG fusion with the PM [102]. It is, however, possible that some of the insulin cargo is in a partially dissolved state, which could exit though the small pores [103]. These small “kiss-and-run” fusion pores are large enough to empty small ISG cargo, such as ATP and neurotransmitters (e.g., GABA, serotonin), which have paracrine actions [14]. Although more work on this topic is warranted, technical challenges with electron microscopy or two-photon microscopy will need to be surmounted in order to more clearly assess the fusion machinery involved in compound ISG fusion [98].
Interactions between SNAREs and other cellular components
SNARE interactions with ion channels
The SNARE proteins STX1A and SNAP25 bind L- and R-type voltage-dependent calcium channels to form excitosomes [104,105], which tether ISGs to PM sites where maximum calcium influx will occur, in order to reach the high calcium concentration required to produce ISG fusion [51,105]. In rodent β-cells, L-type (Cav1.2, Cav1.3) and R-type (Cav2.3) calcium channels mediate first- and second-phase GSIS, respectively [106,107]. ISGs recruited to sit on the L-type calcium channels constitute the RRP that is the first to be released, which is absent in human T2D β-cells and postulated to account for the loss of first-phase GSIS [51]. Interestingly, chronic exposure of β-cells to palmitate, which mimics lipotoxicity in T2D, can cause dissociation of ISGs from the calcium channels, reducing both first- and second-phase GSIS [51,108]. Binding of these calcium channels (L-type vs R-type) to distinct SNARE proteins (STX1A vs. STX3) may determine the mechanism for how the predocked and newcomer ISGs are recruited to fuse with the PM [53]. The priming protein Munc13–1 was recently reported to bind and recruit calcium channels to the ISGs, which would facilitate the recruitment of ISGs for fusion [51]. Loss of Munc13–1, which occurs in T2D β-cells [57], disrupted the clustering of the calcium channels and reduced the size of the RRP [51].
In addition to mediating calcium channel functionality, SNARE proteins can also bind to and modulate voltage-gated potassium channels (Kv2.1) and ATP-sensitive potassium channels (KATP in β-cells to regulate membrane excitability, which controls calcium channel opening [109]. However, the binding of SNARE proteins to Kv2.1 could also regulate ISG exocytosis in a manner independent of electrical activity [110]. Similar to the calcium channels [51], Kv2.1 channels also form clusters on the PM. Through binding of STX1A and STX3 at distinct domains on the Kv2.1 C-terminus, they have been postulated to act as reservoirs for docked and newcomer ISGs, respectively [111,112]. Of note, ISG fusion occurs adjacent to the Kv2.1 clusters [111,113], presumably where the calcium channel clusters are located. Nonetheless, it appears that Kv2.1 clusters at least partly account for the compartmentalized pattern of ISG fusion sites [113]. In fact, loss of Kv2.1, which has been observed in T2D β-cells [111–113], contributes to the loss of this spatial architecture of ISG fusion, with fewer fusion sites on the PM. Restoration of Kv2.1 expression in T2D cells was sufficient to restore the pattern of spatial organization and the number of ISG fusion sites [112,113]. These new insights into insulin ISG loading into a novel Kv2.1 cluster reserve pool or onto calcium channel clusters to form a larger primed releasable pool suggest that these SNARE–ion channel interactions could be targeted therapeutically to increase capacity of the reserve and releasable pools.
SNARE interactions with the cytoskeleton
Overexpression of STX1A selectively in mouse β-cells inhibits GSIS [62], but overexpression of STX4, also selectively in mouse β-cells, enhances GSIS [28,114] (Fig. 4). This result implies that there are important functional differences between these t-SNARE proteins. Amino acid alignment of STX1A and STX4 shows only 45% sequence similarity, supporting the concept that STX4 partners with different intracellular factors than STX1A. One such factor that partners with a subset of STX proteins is filamentous actin (F-actin). Through its unique N-terminal α-spectrin-like domain, STX4 is the only SNARE protein capable of directly binding to F-actin [115–118]. Moreover, in β-cells, STX4 can associate with the actin binding-and-severing protein gelsolin [117], which is consistent with a model wherein actin depolymerization is involved in ISG exocytosis from β-cells [119–121]. Actin polymerization/depolymerization is proposed to coordinate the timing of STX4 activation with the arrival of incoming granules/vesicles. If true, STX4’s interaction with the actin cytoskeleton may create an excitosome [122,123]. Overexpression of STX4 may support more excitosomes, thereby supplying the β-cell with more docking sites for exocytosis. DOC2B overexpression may also boost exocytosis via promoting maximal STX4 and/or STX1A activation potential by acting as a platform for Munc18 proteins [36] when the STX proteins are in their open conformation (STXop, Fig. 2).
Fig. 4. Functional and dysfunctional responses to disruptions in stoichiometry.
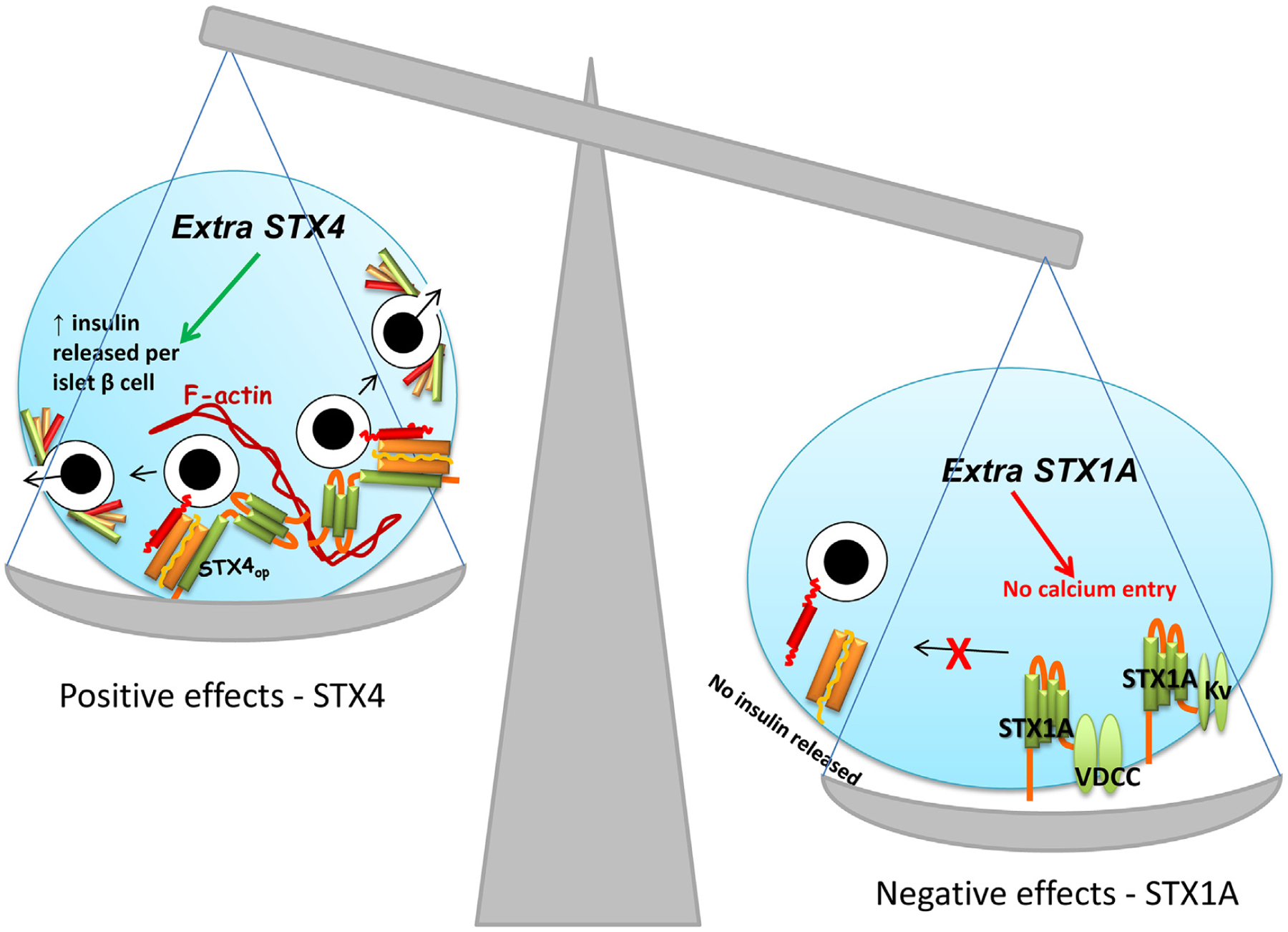
(A) An overabundance of STX4 in the β-cell increases the amplitude of both phases of GSIS and enhances glucose homeostasis in vivo. STX4 binds directly to F-actin and indirectly via association with the actin binding protein gelsolin. Additional units of STX4 increase the abundance of STX4-based SNARE complexes. (B) An overabundance of STX1A decreases VDCC and Kv channel activity/gating, linked to STX1A’s ability to bind to the channels and reduce function, which in vivo decreases GSIS and disrupts glucose homeostasis.
The SNARE accessory protein DOC2B can bind to the microtubule protein KLC1 in skeletal muscle cells [124] and to Tctex-1, another microtubule binding factor in neuronal cells [125,126]. While this localizes DOC2B to the microtubule machinery, the precise role of DOC2B in ISG trafficking along microtubules is unclear. Microtubules in β-cells are largely considered to serve as tracks to shuttle ISGs out to the cell surface to the SNARE machinery for subsequent docking, fusion, and insulin release, although a recent study suggests that the role of microtubules in GSIS may be more complex [127]. Future studies will be required to understand the role of DOC2B, and perhaps other SNAREs and SNARE accessory factors, as microtubule-associated proteins in ISG exocytosis.
Exciting perspectives for potential future Directions
It is intriguing that some SNARE proteins involved in GSIS are deficient in islets from T2D humans and rodent models [10,55–59], but the degrees of reduction were not uniform in these reports. This may in part be due to the stages of T2D that were not uniform, along with the small number of human T2D samples, as well as the exposure levels to glucolipotoxicity as discussed earlier [59,66,69]. T2D comprises a spectrum of pathophysiologies that have as their common element the inability of the insulin-producing beta cell to manage the glycemic load, and a recent finding that this T2D spectrum could be subdivided into five general clusters provides the potential opportunity for a more personalized treatment approach to yield enhanced effectiveness in a timelier manner [128]. Stratification of T2D human islets by these clustered parameters, both retrospectively and prospectively, will provide valuable information to discern if, or if not, certain SNARE deficiencies associate with particular clusters.
Does this suggest that there exists a common mechanism regulating SNARE protein levels? Possibly. For instance, it is possible that for many of the SNARE proteins (but not all), their expression is downregulated due to their common susceptibility to increased degradation or reduced expression. However, some SNARE proteins, namely STX2 and STX3 [26] (and SNAP23, unpublished observations), are not decreased in T2D β-cells, suggesting that they may be more stable or resistant to degradation. Therefore, STX3 could be an alternative way to correct the STX4 deficiency in second-phase GSIS; and normal levels of SNAP23 and STX2 would be detrimental in T2D β-cells because they act as inhibitory SNAREs.
The concept that T2D activates a common SNARE protein degradation pathway remains largely untested, despite agreement that SNARE proteins can be deficient in T2D islets. One candidate degradation mechanism is S-nitrosylation. However, although STX4 undergoes S-nitrosylation in response to proinflammatory cytokine stress in β-cells [129], STX1A does not undergo this posttranslational modification in β-cells [129], suggesting that S-nitrosylation is not a common pathway of degradation of all STX proteins. Human islet RNAseq datasets and congenic rodent databases show deficits in SNARE transcripts associated with diabetes [130,131], suggesting that reduced mRNA expression is a possible common pathway. However, no SNAREs have been reported as T2D susceptibility genes per se, and the SNAREs found to be deficient in T2D are scattered throughout the genome, not clustered in disease-affiliated loci. Recently we noted that STX4 mRNA (and protein) was decreased in nondiabetic human islets following exposure to proinflammatory cytokines [132]. This suggests that diabetogenic stimuli can impact SNARE gene transcription and/or mRNA stabilities. Consistent with this concept, SNAREs are targets of miRNAs [133,134] and lncRNAs [135,136], suggesting that epigenetics and mRNA dynamics will need to be investigated to understand how T2D reduces SNARE expression.
Recent studies show that enhancing SNARE protein levels in human islet β-cells protects against proinflammatory cytokine-induced apoptosis [132,137]. Furthermore, overexpression of the SNARE regulatory protein DOC2B conferred protection against the endoplasmic reticulum stressor thapsigargin [137]. STX4 overexpression prevented the cytokine-induced escalation of chemokine ligands CXCL9 and CXCL10, and this was linked to STX4 interference in NF-kB signaling and transactivation of the chemokine ligand genes [132]. However, the enrichment and depletion of SNARE proteins are not based upon a common pathway. For example, overexpression of STX1A solely in β-cells interferes with calcium channel gating due to interaction of STX1A with the channel [62]. This indicates that those SNAREs that confer benefit when overexpressed are doing so at the protein level; i.e. by providing benefit via indirect signaling pathways (e.g., STX4 suppressing NF-κB translocation), or perhaps via direct protein–protein interactions yet to be elucidated. These findings mark only the beginning of our understanding of how SNAREs support β-cell mass, but at the very least provide indications that SNAREs may provide the means to enhance islet function and survival and may provide novel therapeutic strategies for diabetes.
Acknowledgments
We thank Dr. Raja Veluthakal for assistance with the artwork in Fig. 2. This work was supported in part by grants from the NIH (DK067912 and DK112917) and JDRF (2-SRA-2015-138-S-B) to D.C.T.; and grants from the Canadian Institute for Health Research PJT-159741 and MOP 86544 to H.Y.G. Editing assistance was provided by Nancy Linford, PhD.
References
- [1].Curry DL, Bennett LL, Grodsky GM, Dynamics of insulin secretion by the perfused rat pancreas, Endocrinology 83 (1968) 572–584. [DOI] [PubMed] [Google Scholar]
- [2].Porte D Jr., Pupo AA Insulin responses to glucose: evidence for a two pool system in man, J. Clin. Investig 48 (1969) 2309–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Daniel S, Noda M, Straub SG, Sharp GW, Komatsu M, Schermerhorn T, et al. , Identification of the docked granule pool responsible for the first phase of glucose-stimulated insulin secretion, Diabetes 48 (1999) 1686–1690. [DOI] [PubMed] [Google Scholar]
- [4].Barg S, Eliasson L, Renstrom E, Rorsman P, A subset of 50 secretory granules in close contact with L-type Ca(2+) channels accounts for first-phase insulin secretion in mouse beta-cells, Diabetes 51 (2002) S74–S82. [DOI] [PubMed] [Google Scholar]
- [5].Olofsson CS, Gopel SO, Barg S, Galvanovskis J, Ma X, Salehi A, et al. , Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells, Pflüg. Arch 444 (2002) 43–51. [DOI] [PubMed] [Google Scholar]
- [6].Gaisano HY, Here come the newcomer granules, better late than never, Trends Endocrinol. Metab 25 (2014) 381–388. [DOI] [PubMed] [Google Scholar]
- [7].Kahn SE, Porte D Jr., Islet dysfunction in non-insulin-dependent diabetes mellitus, Am. J. Med 85 (1988) 4–8. [DOI] [PubMed] [Google Scholar]
- [8].van Haeften TW, Pimenta W, Mitrakou A, Korytkowski M, Jenssen T, Yki-Jarvinen H, et al. , Relative contributions of beta-cell function and tissue insulin sensitivity to fasting and postglucose-load glycemia, Metabolism 49 (2000) 1318–1325. [DOI] [PubMed] [Google Scholar]
- [9].Nesher R, Cerasi E, Modeling phasic insulin release: immediate and time-dependent effects of glucose, Diabetes 51 (2002) S53–S59. [DOI] [PubMed] [Google Scholar]
- [10].Oh E, Stull ND, Mirmira RG, Thurmond DC, Syntaxin 4 up-regulation increases efficiency of insulin release in pancreatic islets from humans with and without type 2 diabetes mellitus, J. Clin. Endocrinol. Metab 99 (2014) E866–E870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Thorens B, Gerard N, Deriaz N, GLUT2 surface expression and intracellular transport via the constitutive pathway in pancreatic beta cells and insulinoma: evidence for a block in trans-Golgi network exit by brefeldin A, JCB (J. Cell Biol.) 123 (1993) 1687–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McCulloch LJ, van de Bunt M, Braun M, Frayn KN, Clark A, Gloyn AL, GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: implications for understanding genetic association signals at this locus, Mol. Genet. Metab 104 (2011) 648–653. [DOI] [PubMed] [Google Scholar]
- [13].Ashcroft FM, Harrison DE, Ashcroft SJ, Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells, Nature 312 (1984) 446–448. [DOI] [PubMed] [Google Scholar]
- [14].MacDonald PE, Braun M, Galvanovskis J, Rorsman P, Release of small transmitters through kiss-and-run fusion pores in rat pancreatic beta cells, Cell Metabol. 4 (2006) 283–290. [DOI] [PubMed] [Google Scholar]
- [15].Cook DL, Hales CN, Intracellular ATP directly blocks K+ channels in pancreatic B-cells, Nature 311 (1984) 271–273. [DOI] [PubMed] [Google Scholar]
- [16].Rorsman P, Ashcroft FM, Trube G, Single Ca channel currents in mouse pancreatic B-cells, Pflüg. Arch 412 (1988) 597–603. [DOI] [PubMed] [Google Scholar]
- [17].Satin LS, Cook DL, Voltage-gated Ca2+ current in pancreatic B-cells, Pflüg. Arch 404 (1985) 385–387. [DOI] [PubMed] [Google Scholar]
- [18].Seino S, Shibasaki T, Minami K, Dynamics of insulin secretion and the clinical implications for obesity and diabetes, J. Clin. Investig 121 (2011) 2118–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sudhof TC, Rothman JE, Membrane fusion: grappling with SNARE and SM proteins, Science 323 (2009) 474–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pfeffer SR, A prize for membrane magic, Cell 155 (2013) 1203–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kwan EP, Gaisano HY, New insights into the molecular mechanisms of priming of insulin exocytosis, Diabetes Obes. Metab 9 (Suppl 2) (2007) 99–108. [DOI] [PubMed] [Google Scholar]
- [22].Aslamy A, Thurmond DC, Exocytosis proteins as novel targets for diabetes prevention and/or remediation? Am. J. Physiol. Regul. Integr. Comp. Physiol 312 (2017) R739–R752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gaisano HY, Recent new insights into the role of SNARE and associated proteins in insulin granule exocytosis, Diabetes Obes. Metab 19 (Suppl 1) (2017) 115–123. [DOI] [PubMed] [Google Scholar]
- [24].Zhu D, Zhang Y, Lam PP, Dolai S, Liu Y, Cai EP, et al. , Dual role of VAMP8 in regulating insulin exocytosis and islet beta cell growth, Cell Metabol. 16 (2012) 238–249. [DOI] [PubMed] [Google Scholar]
- [25].Zhu D, Koo E, Kwan E, Kang Y, Park S, Xie H, et al. , Syntaxin-3 regulates newcomer insulin granule exocytosis and compound fusion in pancreatic beta cells, Diabetologia 56 (2013) 359–369. [DOI] [PubMed] [Google Scholar]
- [26].Qin T, Liang T, Zhu D, Kang Y, Xie L, Dolai S, et al. , Munc18b increases insulin granule fusion, restoring deficient insulin secretion in type-2 diabetes human and gotokakizaki rat islets with improvement in glucose homeostasis, EBioMedicine 16 (2017) 262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Oh E, Spurlin BA, Pessin JE, Thurmond DC, Munc18c heterozygous knockout mice display increased susceptibility for severe glucose intolerance, Diabetes 54 (2005) 638–647. [DOI] [PubMed] [Google Scholar]
- [28].Spurlin BA, Thurmond DC, Syntaxin 4 facilitates biphasic glucose-stimulated insulin secretion from pancreatic beta cells, Mol. Endocrinol 20 (2006) 183–193. [DOI] [PubMed] [Google Scholar]
- [29].Misura KM, Scheller RH, Weis WI, Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex Nature 404 (2000) 355–362. [DOI] [PubMed] [Google Scholar]
- [30].Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Sudhof TC, et al. , A conformational switch in syntaxin during exocytosis: role of munc18, EMBO J. 18 (1999) 4372–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kang L, He Z, Xu P, Fan J, Betz A, Brose N, et al. , Munc13–1 is required for the sustained release of insulin from pancreatic beta cells, Cell Metabol. 3 (2006) 463–468. Epub 2006 May 11. [DOI] [PubMed] [Google Scholar]
- [32].Sheu L, Pasyk EA, Ji J, Huang X, Gao X, Varoqueaux F, et al. , Regulation of insulin exocytosis by Munc13–1, J. Biol. Chem 278 (2003) 27556–27563. Epub 2003 May 13. [DOI] [PubMed] [Google Scholar]
- [33].Kwan EP, Xie L, Sheu L, Ohtsuka T, Gaisano HY, Interaction between Munc13–1 and RIM is critical for glucagon-like peptide-1 mediated rescue of exocytotic defects in Munc13–1 deficient pancreatic beta-cells, Diabetes 56 (2007) 2579–2588. Epub 007 Jul 16. [DOI] [PubMed] [Google Scholar]
- [34].Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, et al. , cAMP-GEFII is a direct target of cAMP in regulated exocytosis, Nat. Cell Biol 2 (2000) 805–811. [DOI] [PubMed] [Google Scholar]
- [35].Dolai S, Xie L, Zhu D, Liang T, Qin T, Xie H, et al. , Synaptotagmin-7 functions to replenish insulin granules for exocytosis in human islet beta-cells, Diabetes 65 (2016) 1962–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ramalingam L, Lu J, Hudmon A, Thurmond DC, Doc2b serves as a scaffolding platform for concurrent binding of multiple Munc18 isoforms in pancreatic islet beta-cells, Biochem. J 464 (2014) 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Verhage M, de Vries KJ, Roshol H, Burbach JP, Gispen WH, Sudhof TC, DOC2 proteins in rat brain: complementary distribution and proposed function as vesicular adapter proteins in early stages of secretion, Neuron 18 (1997) 453–461. [DOI] [PubMed] [Google Scholar]
- [38].Ke B, Oh E, Thurmond DC, Doc2beta is a novel Munc18c-interacting partner and positive effector of syntaxin 4-mediated exocytosis, J. Biol. Chem 282 (2007) 21786–21797. [DOI] [PubMed] [Google Scholar]
- [39].Ramalingam L, Oh E, Yoder SM, Brozinick JT, Kalwat MA, Groffen AJ, et al. , Doc2b is a key effector of insulin secretion and skeletal muscle insulin sensitivity, Diabetes 61 (2012) 2424–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ramalingam L, Oh E, Thurmond DC, Doc2b enrichment enhances glucose homeostasis in mice via potentiation of insulin secretion and peripheral insulin sensitivity, Diabetologia 57 (2014) 1476–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Abderrahmani A, Niederhauser G, Plaisance V, Roehrich ME, Lenain V, Coppola T, et al. , Complexin I regulates glucose-induced secretion in pancreatic beta-cells, J. Cell Sci 117 (2004) 2239–2247. [DOI] [PubMed] [Google Scholar]
- [42].Bhatnagar S, Oler AT, Rabaglia ME, Stapleton DS, Schueler KL, Truchan NA, et al. , Positional cloning of a type 2 diabetes quantitative trait locus; tomosyn-2, a negative regulator of insulin secretion, PLoS Genet. 7 (2011), e1002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Courtney NA, Bao H, Briguglio JS, Chapman ER, Synaptotagmin 1 clamps synaptic vesicle fusion in mammalian neurons independent of complexin, Nat. Commun 10 (2019) 4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lakomek NA, Yavuz H, Jahn R, Perez-Lara A, Structural dynamics and transient lipid binding of synaptobrevin-2 tune SNARE assembly and membrane fusion, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 8699–8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yavuz H, Kattan I, Hernandez JM, Hofnagel O, Witkowska A, Raunser S, et al. , Arrest of trans-SNARE zippering uncovers loosely and tightly docked intermediates in membrane fusion, J. Biol. Chem 293 (2018) 8645–8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Melia TJ, Weber T, McNew JA, Fisher LE, Johnston RJ, Parlati F, et al. , Regulation of membrane fusion by the membrane-proximal coil of the t-SNARE during zippering of SNAREpins, J. Cell Biol 158 (2002) 929–940. Epub 2002 Sep. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Matos MF, Mukherjee K, Chen X, Rizo J, Sudhof TC, Evidence for SNARE zippering during Ca2+-triggered exocytosis in PC12 cells, Neuropharmacology 45 (2003) 777–786. [DOI] [PubMed] [Google Scholar]
- [48].Knowles MK, Barg S, Wan L, Midorikawa M, Chen X, Almers W, Single secretory granules of live cells recruit syntaxin-1 and synaptosomal associated protein 25 (SNAP-25) in large copy numbers, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 20810–20815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].de Wit H, Walter AM, Milosevic I, Gulyas-Kovacs A, Riedel D, Sorensen JB, et al. , Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes, Cell 138 (2009) 935–946. [DOI] [PubMed] [Google Scholar]
- [50].Gandasi NR, Barg S, Contact-induced clustering of syntaxin and munc18 docks secretory granules at the exocytosis site, Nat. Commun 5 (2014) 3914. [DOI] [PubMed] [Google Scholar]
- [51].Gandasi NR, Yin P, Riz M, Chibalina MV, Cortese G, Lund PE, et al. , Ca2+ channel clustering with insulin-containing granules is disturbed in type 2 diabetes, J. Clin. Investig 127 (2017) 2353–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Takahashi N, Sawada W, Noguchi J, Watanabe S, Ucar H, Hayashi-Takagi A, et al. , Two-photon fluorescence lifetime imaging of primed SNARE complexes in presynaptic terminals and beta cells, Nat. Commun 6 (2015) 8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Xie L, Dolai S, Kang Y, Liang T, Xie H, Qin T, et al. , Syntaxin-3 binds and regulates both R- and L-type calcium channels in insulin-secreting INS-1 832/13 cells, PLoS One 11 (2016), e0147862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Low JT, Zavortink M, Mitchell JM, Gan WJ, Do OH, Schwiening CJ, et al. , Insulin secretion from beta cells in intact mouse islets is targeted towards the vasculature, Diabetologia 57 (2014) 1655–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Oh E, Kalwat MA, Kim MJ, Verhage M, Thurmond DC, Munc18–1 regulates first-phase insulin release by promoting granule docking to multiple syntaxin isoforms, J. Biol. Chem 287 (2012) 25821–25833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Nagamatsu S, Nakamichi Y, Yamamura C, Matsushima S, Watanabe T, Ozawa S, et al. , Decreased expression of t-SNARE, syntaxin 1, and SNAP-25 in pancreatic beta-cells is involved in impaired insulin secretion from diabetic GK rat islets: restoration of decreased t-SNARE proteins improves impaired insulin secretion, Diabetes 48 (1999) 2367–2373. [DOI] [PubMed] [Google Scholar]
- [57].Ostenson CG, Gaisano H, Sheu L, Tibell A, Bartfai T, Impaired gene and protein expression of exocytotic soluble N-ethylmaleimide attachment protein receptor complex proteins in pancreatic islets of type 2 diabetic patients, Diabetes 55 (2006) 435–440. [DOI] [PubMed] [Google Scholar]
- [58].Chan CB, MacPhail RM, Sheu L, Wheeler MB, Gaisano HY, Beta-cell hypertrophy in fa/fa rats is associated with basal glucose hypersensitivity and reduced SNARE protein expression, Diabetes 48 (1999) 997–1005. [DOI] [PubMed] [Google Scholar]
- [59].Gaisano HY, Ostenson CG, Sheu L, Wheeler MB, Efendic S, Abnormal expression of pancreatic islet exocytotic soluble N-ethylmaleimide-sensitive factor attachment protein receptors in Goto-Kakizaki rats is partially restored by phlorizin treatment and accentuated by high glucose treatment, Endocrinology 143 (2002) 4218–4226. [DOI] [PubMed] [Google Scholar]
- [60].Andersson SA, Olsson AH, Esguerra JL, Heimann E, Ladenvall C, Edlund A, et al. , Reduced insulin secretion correlates with decreased expression of exocytotic genes in pancreatic islets from patients with type 2 diabetes, Mol. Cell. Endocrinol 364 (2012) 36–45. [DOI] [PubMed] [Google Scholar]
- [61].Waanders LF, Chwalek K, Monetti M, Kumar C, Lammert E, Mann M, Quantitative proteomic analysis of single pancreatic islets, Proc. Natl. Acad. Sci. U. S. A 106 (2009) 18902–18907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lam PP, Leung YM, Sheu L, Ellis J, Tsushima RG, Osborne LR, et al. , Transgenic mouse overexpressing syntaxin-1A as a diabetes model, Diabetes 54 (2005) 2744–2754. [DOI] [PubMed] [Google Scholar]
- [63].Bezprozvanny I, Scheller RH, Tsien RW, Functional impact of syntaxin on gating of N-type and Q-type calcium channels, Nature 378 (1995) 623–626. [DOI] [PubMed] [Google Scholar]
- [64].Zhang W, Efanov A, Yang SN, Fried G, Kolare S, Brown H, et al. , Munc-18 associates with syntaxin and serves as a negative regulator of exocytosis in the pancreatic beta -cell, J. Biol. Chem 275 (2000) 41521–41527. [DOI] [PubMed] [Google Scholar]
- [65].Spurlin BA, Thomas RM, Nevins AK, Kim HJ, Kim YJ, Noh HL, et al. , Insulin resistance in tetracycline-repressible Munc18c transgenic mice, Diabetes 52 (2003) 1910–1917. [DOI] [PubMed] [Google Scholar]
- [66].Ostenson CG, Efendic S, Islet gene expression and function in type 2 diabetes; studies in the Goto-Kakizaki rat and humans, Diabetes Obes. Metab 9 (Suppl 2) (2007) 180–186. [DOI] [PubMed] [Google Scholar]
- [67].AlFadhli S, Al-Zufairi AAM, Nizam R, AlSaffar HA, Al-Mutairi N, De-regulation of diabetic regulatory genes in psoriasis: deciphering the unsolved riddle, Gene 593 (2016) 110–116. [DOI] [PubMed] [Google Scholar]
- [68].Perrotta C, Cervia D, Di Renzo I, Moscheni C, Bassi MT, Campana L, et al. , Nitric oxide generated by tumor-associated macrophages is responsible for cancer resistance to cisplatin and correlated with syntaxin 4 and acid sphingomyelinase inhibition, Front. Immunol 9 (2018) 1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ostenson CG, Chen J, Sheu L, Gaisano HY, Effects of palmitate on insulin secretion and exocytotic proteins in islets of diabetic Goto-Kakizaki rats, Pancreas 34 (2007) 359–363. [DOI] [PubMed] [Google Scholar]
- [70].Romeo S, Sentinelli F, Cavallo MG, Leonetti F, Fallarino M, Mariotti S, et al. , Search for genetic variants of the SYNTAXIN 1A (STX1A) gene: the −352 A>T variant in the STX1A promoter associates with impaired glucose metabolism in an Italian obese population, Int. J. Obes 32 (2008) 413–420. Epub 2007 Oct 2. [DOI] [PubMed] [Google Scholar]
- [71].Tsunoda K, Sanke T, Nakagawa T, Furuta H, Nanjo K, Single nucleotide polymorphism (D68D, T to C) in the syntaxin 1A gene correlates to age at onset and insulin requirement in Type II diabetic patients, Diabetologia 44 (2001) 2092–2097. [DOI] [PubMed] [Google Scholar]
- [72].Oh E, Thurmond DC, Munc18c depletion selectively impairs the sustained phase of insulin release, Diabetes 58 (2009) 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ohara-Imaizumi M, Fujiwara T, Nakamichi Y, Okamura T, Akimoto Y, Kawai J, et al. , Imaging analysis reveals mechanistic differences between first- and second-phase insulin exocytosis, J. Cell Biol 177 (2007) 695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Jewell JL, Oh E, Thurmond DC, Exocytosis mechanisms underlying insulin release and glucose uptake: conserved roles for Munc18c and syntaxin 4, Am. J. Physiol. Regul. Integr. Comp. Physiol 298 (2010) R517–R531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Morey C, Kienle CN, Klopper TH, Burkhardt P, Fasshauer D, Evidence for a conserved inhibitory binding mode between the membrane fusion assembly factors Munc18 and syntaxin in animals, J. Biol. Chem 292 (2017) 20449–20460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ, Selective activation of cognate SNAREpins by Sec1/Munc18 proteins, Cell 128 (2007) 183–195. [DOI] [PubMed] [Google Scholar]
- [77].McNew JA, Parlati F, Fukuda R, Johnston RJ, Paz K, Paumet F, et al. , Compartmental specificity of cellular membrane fusion encoded in SNARE proteins, Nature 407 (2000) 153–159. [DOI] [PubMed] [Google Scholar]
- [78].Riento K, Jantti J, Jansson S, Hielm S, Lehtonen E, Ehnholm C, et al. , A sec1-related vesicle-transport protein that is expressed predominantly in epithelial cells, Eur. J. Biochem 239 (1996) 638–646. [DOI] [PubMed] [Google Scholar]
- [79].Riento K, Kauppi M, Keranen S, Olkkonen VM, Munc18–2, a functional partner of syntaxin 3, controls apical membrane trafficking in epithelial cells, J. Biol. Chem 275 (2000) 13476–13483. [DOI] [PubMed] [Google Scholar]
- [80].Hata Y, Slaughter CA, Sudhof TC, Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin, Nature 366 (1993) 347–351. [DOI] [PubMed] [Google Scholar]
- [81].Tellam JT, Macaulay SL, McIntosh S, Hewish DR, Ward CW, James DE, Characterization of Munc-18c and syntaxin-4 in 3T3-L1 adipocytes. Putative role in insulin-dependent movement of GLUT-4, J. Biol. Chem 272 (1997) 6179–6186. [DOI] [PubMed] [Google Scholar]
- [82].Thurmond DC, Ceresa BP, Okada S, Elmendorf JS, Coker K, Pessin JE, Regulation of insulin-stimulated GLUT4 translocation by munc18c in 3T3L1 adipocytes, J. Biol. Chem 273 (1998) 33876–33883. [DOI] [PubMed] [Google Scholar]
- [83].Zhu D, Xie L, Kang Y, Dolai S, Bondo Hansen J, Qin T, et al. , Syntaxin 2 acts as inhibitory SNARE for insulin granule exocytosis, Diabetes 66 (2017) 948–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kunii M, Ohara-Imaizumi M, Takahashi N, Kobayashi M, Kawakami R, Kondoh Y, et al. , Opposing roles for SNAP23 in secretion in exocrine and endocrine pancreatic cells, J. Cell Biol 215 (2016) 121–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zhu D, Xie L, Karimian N, Liang T, Kang Y, Huang YC, et al. , Munc18c mediates exocytosis of pre-docked and newcomer insulin granules underlying biphasic glucose stimulated insulin secretion in human pancreatic beta-cells, Mol Metab 4 (2015) 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Xie L, Zhu D, Dolai S, Liang T, Qin T, Kang Y, et al. , Syntaxin-4 mediates exocytosis of pre-docked and newcomer insulin granules underlying biphasic glucose-stimulated insulin secretion in human pancreatic beta cells, Diabetologia 58 (2015) 1250–1259. [DOI] [PubMed] [Google Scholar]
- [87].Liang T, Qin T, Xie L, Dolai S, Zhu D, Prentice KJ, et al. , New roles of syntaxin-1A in insulin granule exocytosis and replenishment, J. Biol. Chem 292 (2017) 2203–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Lytrivi M, Anne-Laure C, Poitout V, Cnop M, Recent insights into mechanisms of beta-cell lipo- and glucolipotoxicity in type 2 diabetes, J. Mol. Biol 432 (2019) 1514–1534, 10.1016/j.jmb.2019.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Fontes G, Zarrouki B, Hagman DK, Latour MG, Semache M, Roskens V, et al. , Glucolipotoxicity age-dependently impairs beta cell function in rats despite a marked increase in beta cell mass, Diabetologia 53 (2010) 2369–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Littleton JT, Chapman ER, Kreber R, Garment MB, Carlson SD, Ganetzky B, Temperature-sensitive paralytic mutations demonstrate that synaptic exocytosis requires SNARE complex assembly and disassembly, Neuron 21 (1998) 401–413. [DOI] [PubMed] [Google Scholar]
- [91].Ohama T, Hori M, Momotani E, Elorza M, Gerthoffer WT, Ozaki H, IL-1beta inhibits intestinal smooth muscle proliferation in an organ culture system: involvement of COX-2 and iNOS induction in muscularis resident macrophages, Am. J. Physiol. Gastrointest. Liver Physiol 292 (2007) G1315–G1322. [DOI] [PubMed] [Google Scholar]
- [92].Duncan RR, Greaves J, Wiegand UK, Matskevich I, Bodammer G, Apps DK, et al. , Functional and spatial segregation of secretory vesicle pools according to vesicle age, Nature 422 (2003) 176–180. [DOI] [PubMed] [Google Scholar]
- [93].Ivanova A, Kalaidzidis Y, Dirkx R, Sarov M, Gerlach M, Schroth-Diez B, et al. , Age-dependent labelingand imaging of insulin secretory granules, Diabetes 62 (2013) 3687–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Pedersen MG, Sherman A, Newcomer insulin secretory granules as a highly calcium-sensitive pool, Proc. Natl. Acad. Sci. U. S. A 106 (2009) 7432–7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Xie L, Zhu D, Gaisano HY, Role of mammalian homologue of Caenorhabditis elegans unc-13–1 (Munc13–1) in the recruitment of newcomer insulin granules in both first and second phases of glucose-stimulated insulin secretion in mouse islets, Diabetologia 55 (2012) 2693–2702. [DOI] [PubMed] [Google Scholar]
- [96].Kwan EP, Gaisano HY, Glucagon-like peptide 1 regulates sequential and compound exocytosis in pancreatic islet beta-cells, Diabetes 54 (2005) 2734–2743. [DOI] [PubMed] [Google Scholar]
- [97].Hoppa MB, Jones E, Karanauskaite J, Ramracheya R, Braun M, Collins SC, et al. , Multivesicular exocytosis in rat pancreatic beta cells, Diabetologia 55 (2012) 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lam PP, Ohno M, Dolai S, He Y, Qin T, Liang T, et al. , Munc18b is a major mediator of insulin exocytosis in rat pancreatic beta-cells, Diabetes 62 (2013) 2416–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Takahashi N, Hatakeyama H, Okado H, Miwa A, Kishimoto T, Kojima T, et al. , Sequential exocytosis of insulin granules is associated with redistribution of SNAP25, J. Cell Biol 165 (2004) 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Klein O, Sagi-Eisenberg R, Anaphylactic degranulation of mast cells: focus on compound exocytosis, J. Immunol. Res 2019 (2019) 9542656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Alvarez de Toledo G, Fernandez JM, Compound versus multigranular exocytosis in peritoneal mast cells, J. Gen. Physiol 95 (1990) 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ma L, Bindokas VP, Kuznetsov A, Rhodes C, Hays L, Edwardson JM, et al. , Direct imaging shows that insulin granule exocytosis occurs by complete vesicle fusion, Proc. Natl. Acad. Sci. U. S. A 101 (2004) 9266–9271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Michael DJ, Ritzel RA, Haataja L, Chow RH, Pancreatic beta-cells secrete insulin in fast- and slow-release forms, Diabetes 55 (2006) 600–607. [DOI] [PubMed] [Google Scholar]
- [104].Cohen R, Atlas D, R-type voltage-gated Ca(2+) channel interacts with synaptic proteins and recruits synaptotagmin to the plasma membrane of Xenopus oocytes, Neuroscience 128 (2004) 831–841. [DOI] [PubMed] [Google Scholar]
- [105].Wiser O, Trus M, Hernandez A, Renstrom E, Barg S, Rorsman P, et al. , The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the exocytotic machinery, Proc. Natl. Acad. Sci. U. S. A 96 (1999) 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Schulla V, Renstrom E, Feil R, Feil S, Franklin I, Gjinovci A, et al. , Impaired insulin secretion and glucose tolerance in beta cell-selective Ca(v)1.2 Ca2+ channel null mice, EMBO J. 22 (2003) 3844–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Jing X, Li DQ, Olofsson CS, Salehi A, Surve VV, Caballero J, et al. , CaV2.3 calcium channels control second-phase insulin release, J. Clin. Investig 115 (2005) 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Hoppa MB, Collins S, Ramracheya R, Hodson L, Amisten S, Zhang Q, et al. , Chronic palmitate exposure inhibits insulin secretion by dissociation of Ca(2+) channels from secretory granules, Cell Metabol. 10 (2009) 455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Leung YM, Kwan EP, Ng B, Kang Y, Gaisano HY, SNAREing voltage-gated K+ and ATP-sensitive K+ channels: tuning beta-cell excitability with syntaxin-1A and other exocytotic proteins, Endocr. Rev 28 (2007) 653–663. [DOI] [PubMed] [Google Scholar]
- [110].Dai XQ, Manning Fox JE, Chikvashvili D, Casimir M, Plummer G, Hajmrle C, et al. , The voltage-dependent potassium channel subunit Kv2.1 regulates insulin secretion from rodent and human islets independently of its electrical function, Diabetologia 55 (2012) 1709–1720. [DOI] [PubMed] [Google Scholar]
- [111].Fu J, Dai X, Plummer G, Suzuki K, Bautista A, Githaka JM, et al. , Kv2.1 clustering contributes to insulin exocytosis and rescues human beta-cell dysfunction, Diabetes 66 (2017) 1890–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Greitzer-Antes D, Xie L, Qin T, Xie H, Zhu D, Dolai S, et al. , Kv2.1 clusters on beta-cell plasma membrane act as reservoirs that replenish pools of newcomer insulin granule through their interaction with syntaxin-3, J. Biol. Chem 293 (2018) 6893–6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Fu J, Githaka JM, Dai X, Plummer G, Suzuki K, Spigelman AF, et al. , A glucose-dependent spatial patterning of exocytosis in human beta-cells is disrupted in type 2 diabetes, JCI Insight 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Spurlin BA, Park SY, Nevins AK, Kim JK, Thurmond DC, Syntaxin 4 transgenic mice exhibit enhanced insulin-mediated glucose uptake in skeletal muscle, Diabetes 53 (2004) 2223–2231. [DOI] [PubMed] [Google Scholar]
- [115].Band AM, Ali H, Vartiainen MK, Welti S, Lappalainen P, Olkkonen VM, et al. , Endogenous plasma membrane t-SNARE syntaxin 4 is present in rab11 positive endosomal membranes and associates with cortical actin cytoskeleton, FEBS Lett. 531 (2002) 513–519. [DOI] [PubMed] [Google Scholar]
- [116].Jewell JL, Luo W, Oh E, Wang Z, Thurmond DC, Filamentous actin regulates insulin exocytosis through direct interaction with Syntaxin 4, J. Biol. Chem 283 (2008) 10716–10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Kalwat MA, Wiseman DA, Luo W, Wang Z, Thurmond DC, Gelsolin associates with the N-terminus of syntaxin 4 to regulate insulin granule exocytosis, Mol. Endocrinol 26 (2012) 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Woronowicz K, Dilks JR, Rozenvayn N, Dowal L, Blair PS, Peters CG, et al. , The platelet actin cytoskeleton associates with SNAREs and participates in alpha-granule secretion, Biochemistry 49 (2010) 4533–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Howell SL, Tyhurst M, Interaction between insulin-storage granules and F-actin in vitro, Biochem. J 178 (1979) 367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Kalwat MA, Yoder SM, Wang Z, Thurmond DC, A p21-activated kinase (PAK1) signaling cascade coordinately regulates F-actin remodeling and insulin granule exocytosis in pancreatic beta cells, Biochem. Pharmacol 85 (2013) 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Orci L, Gabbay KH, Malaisse WJ, Pancreatic beta-cell web: its possible role in insulin secretion, Science 175 (1972) 1128–1130. [DOI] [PubMed] [Google Scholar]
- [122].Suckow AT, Zhang C, Egodage S, Comoletti D, Taylor P, Miller MT, et al. , Transcellular neuroligin-2 interactions enhance insulin secretion and are integral to pancreatic beta cell function, J. Biol. Chem 287 (2012) 19816–19826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Zhang C, Caldwell TA, Mirbolooki MR, Duong D, Park EJ, Chi NW, et al. , Extracellular CADM1 interactions influence insulin secretion by rat and human islet beta-cells and promote clustering of syntaxin-1, Am. J. Physiol. Endocrinol. Metab 310 (2016) E874–E885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Zhang J, Oh E, Merz KE, Aslamy A, Veluthakal R, Salunkhe VA, et al. , DOC2B promotes insulin sensitivity in mice via a novel KLC1-dependent mechanism in skeletal muscle, Diabetologia 62 (2019) 845–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Nagano F, Orita S, Sasaki T, Naito A, Sakaguchi G, Maeda M, et al. , Interaction of Doc2 with tctex-1, a light chain of cytoplasmic dynein. Implication in dynein-dependent vesicle transport, J. Biol. Chem 273 (1998) 30065–30068. [DOI] [PubMed] [Google Scholar]
- [126].Williams JC, Xie H, Hendrickson WA, Crystal structure of dynein light chain TcTex-1, J. Biol. Chem 280 (2005) 21981–21986. [DOI] [PubMed] [Google Scholar]
- [127].Zhu X, Hu R, Brissova M, Stein RW, Powers AC, Gu G, et al. , Microtubules negatively regulate insulin secretion in pancreatic beta cells, Dev. Cell 34 (2015) 656–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Ahlqvist E, Storm P, Karajamaki A, Martinell M, Dorkhan M, Carlsson A, et al. , Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables, Lancet Diabetes Endocrinol. 6 (2018) 361–369. [DOI] [PubMed] [Google Scholar]
- [129].Wiseman DA, Kalwat MA, Thurmond DC, Stimulus-induced S-nitrosylation of syntaxin 4 impacts insulin granule exocytosis, J. Biol. Chem 286 (2011) 16344–16354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Lawlor N, George J, Bolisetty M, Kursawe R, Sun L, Sivakamasundari V, et al. , Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes, Genome Res. 27 (2017) 208–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Keller MP, Choi Y, Wang P, Davis DB, Rabaglia ME, Oler AT, et al. , A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility, Genome Res. 18 (2008) 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Oh E, Ahn M, Afelik S, Becker TC, Roep BO, Thurmond DC, Syntaxin 4 expression in pancreatic beta-cells promotes and protects functional beta-cell mass, Diabetes 67 (2018) 2626–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Latreille M, Hausser J, Stutzer I, Zhang Q, Hastoy B, Gargani S, et al. , MicroRNA-7a regulates pancreatic beta cell function, J. Clin. Investig 124 (2014) 2722–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Lovis P, Gattesco S, Regazzi R, Regulation of the expression of components of the exocytotic machinery of insulin-secreting cells by microRNAs, Biol. Chem 389 (2008) 305–312. [DOI] [PubMed] [Google Scholar]
- [135].Fadista J, Vikman P, Laakso EO, Mollet IG, Esguerra JL, Taneera J, et al. , Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism, Proc. Natl. Acad. Sci. U. S. A 111 (2014) 13924–13929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Esguerra JL, Eliasson L, Functional implications of long non-coding RNAs in the pancreatic islets of Langerhans, Front. Genet 5 (2014) 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Aslamy A, Oh E, Olson EM, Zhang J, Ahn M, Moin ASM, et al. , Doc2b protects beta-cells against inflammatory damage and enhances function, Diabetes 67 (2018) 1332–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]