

Abstract
Many membrane channels, transporters, and receptors utilize a pH gradient or proton coupling to drive functionally relevant conformational transitions. Conventional molecular dynamics simulations employ fixed protonation states, thus neglecting the coupling between protonation and conformational equilibria. Here we describe the membrane-enabled hybrid-solvent continuous constant pH molecular dynamics method for capturing atomic details of proton-coupled conformational dynamics of transmembrane proteins. Example protocols from our recent application studies of proton channels and ion/substrate transporters are discussed.
Keywords: Molecular dynamics, electrostatics, pKa, Proton channels, Secondary active transporters, G-protein-coupled receptors
1. Introduction
1.1. pH-dependent conformational dynamics of membrane proteins
Solution pH plays a critical role in the functions of many transmembrane proteins, including secondary active transporters, proton channels, and G-protein-coupled receptors (GPCRs). For example, pH gradient across the membrane drives the transport of sodium ions through the sodium-proton antiporter NhaA, which is essential for cellular sodium and proton homeostasis in Escherichia coli [54, 60]. Proton channels, such as the voltage-gated proton channel Hv1 [65], conduct protons across the membrane in response to a change in cellular pH [17, 16]. Solution pH also modulates the functions of membrane receptors. For example, the ovarian cancer GPCR1 and the closely related GPR4 regulate cell-mediated responses to acidosis in bone [51].
pH-dependent transmembrane proteins often couple proton sensing with conformational changes [19]. For example, the inward- and outward-facing crystal structures of the sodium-proton antiporter NapA from Thermus thermophilus [46, 14] reveal an elevator-like alternating-access model, although the detailed mechanism of pH-dependent conformational transition remains elusive. In many other cases, however, the conformational change is unknown due to the lack of active-state structure. For example, only inactive crystal structures have been resolved for the aforementioned antiporter NhaA [37, 47] and proton channel Hv1 [68]. Apart from high-resolution structure models, pKa’s of active-site residues are important for mechanistic elucidation, as they inform protonation states at a certain pH, thereby unveiling residues involved in proton binding/release coupled to the conformational transition. However, pKa’s of membrane proteins are extremely challenging if possible at all to experimentally measure. Take NhaA as an example, electrophysiology and biochemical experiments established that it transports two protons in exchange for one sodium above pH 6.5 [67, 61]; however, in the absence of an outward-facing crystal structure and experimental pKa’s, the identities of proton-binding active-site residues remain controversial.
1.2. Continuous constant pH molecular dynamics methods
Constant pH molecular dynamics (pHMD) simulations [9, 72, 12] offer a unique means to describe proton-coupled conformational dynamics and calculate pKa values [72, 76, 3]. Current pHMD methods can be grouped into two classes. In discrete pHMD (DpHMD) methods [5, 55], protonation states of titratable groups are periodically updated using Monte-Carlo sampling following molecular dynamics steps. Rooted in the λ-dynamics method for free energy calculations [45], continuous constant-pH molecular dynamics (CpHMD) methods [48, 41, 73, 18, 74, 13, 34, 4, 35, 26, 27] make use of fictitious λ particles to sample protonation states simultaneously with atomic coordinates. A λ particle carries a mass of a regular heavy atom (e.g., 10 atomic mass units) and the λ coordinate continuously evolves between 0 and 1, where 0 and 1 represent protonated and deprotonated states, respectively. The total Hamiltonian includes the kinetic energy of the fictitious particles and the nonbonded term is modified by linearly scaling electrostatic and van der Waals energies according to λ coordinates. Thus, CpHMD directly couples conformational dynamics of the protein with protonation equilibria of all ionizable sites at a specified pH. Following a CpHMD simulation at a single pH or a set of replica-exchange CpHMD simulations at multiple pH conditions, the trajectories of λ coordinates are extracted, allowing the calculation of pKa’s for all titratable sites.
The first two CpHMD methods [48, 41], implemented in CHARMM molecular dynamics package [6], make use of the generalized Born (GB) implicit-solvent models for both proton titration and conformational dynamics. Since then, several CpHMD methods have been developed and implemented in CHARMM. The hybrid-solvent CpHMD [73] was developed, which takes advantage of accurate description of conformational states in explicit solvent and rapid convergence of protonation state sampling in GB solvent. Additionally, the pH replica-exchange scheme [73] was developed to accelerate convergence of protonation and conformational state sampling. To overcome the limitations of implicit-solvent models, all-atom CpHMDMSλD with force switching was developed that uses explicit solvent to sample both titration and conformational dynamics [26, 27]. To avoid the potential artifacts due to electrostatic cutoff and to further improve accuracy, particle-mesh Ewald (PME) all-atom CpHMD [34] was developed. CpHMD methods have also been implemented in the popular Amber [8] and GROMACS [64] packages, including GBNeck2-CpHMD [35, 28] in Amber and the all-atom CpHMD [18] in GROMACS [64]. Here we focus on the extensively validated [75, 22, 11, 33, 79, 23, 29, 30, 69] pH replica-exchange hybrid-solvent CpHMD method in CHARMM [73], which has been further developed to enable mechanistic studies of membrane proteins [33].
1.3. Membrane-enabled hybrid-solvent CpHMD with pH replica-exchange
In the membrane-enabled hybrid-solvent CpHMD method [33], conformational dynamics of a transmembrane protein is sampled in explicit lipids and water, while protonation states are sampled using the membrane GBSW model [38]. Combined with the pH replica-exchange protocol [73], this method allows detailed description of proton-coupled conformational dynamics of transmembrane proteins (Fig. 1). For example, in the simulation study of NhaA [33], a mechanism emerged that explains the pH-dependent activation of NhaA, reconciling a long standing controversy. According to the calculated pKa values and conformational dynamics [33], Asp164 and Lys300 were identified as proton binding residues, consistent with the recent crystal structures of NhaA [47] and the homologous protein NapA [14]. When NhaA is exposed to the cytoplasmic pH, Asp164 releases the first proton and triggers the hydrophobic gate on the cytoplasm side to open, allowing the entrance of a sodium ion [33]. The sodium ion then binds to three active-site residues, which induces the release of a second proton from Lys300 [33]. The same protocol used to study NhaA has also been applied to investigate the proton-coupled conformational dynamics of the proton channel M2 from influenza virus [11] and the E. coli multi-drug efflux pump AcrB [79]. Most recently, a uniform electric field feature has been added to the membrane-enabled hybrid-solvent CpHMD simulations, allowing us to investigate the activation mechanism of a voltage-gated proton channel Hv1 (Fig. 2).
Fig.1.
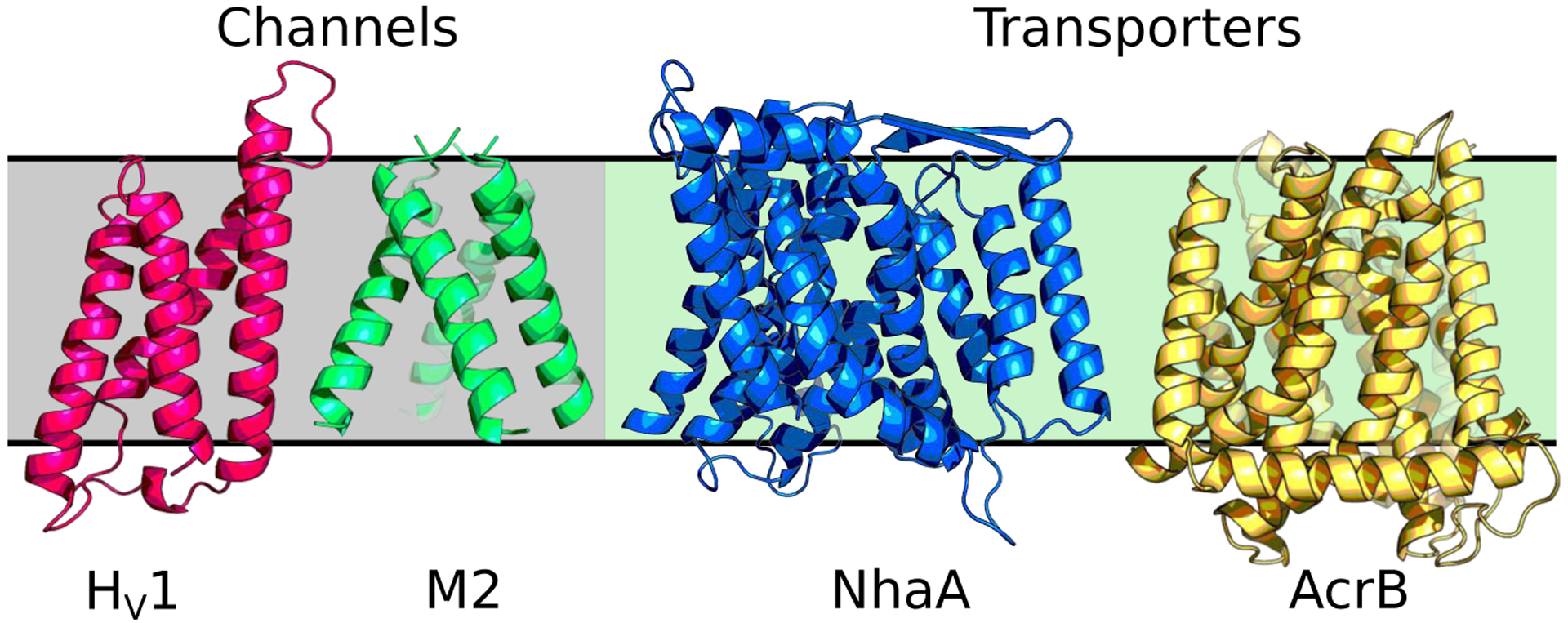
Transmembrane proteins that have been studied by membrane-enabled hybrid-solvent CpHMD simulations with pH replica-exchange [33]: M2 proton channel [11] (PDB: 3LBW [2]), voltage-gated Hv1 proton channel (Henderson and Shen, unpublished data, PDB: 3WKV [68]), sodium-proton antiporter NhaA[33] (PDB: 4AU5 [47]), and multi-drug efflux pump AcrB[79] (PDB: 4DX5 [21]).
Fig.2.
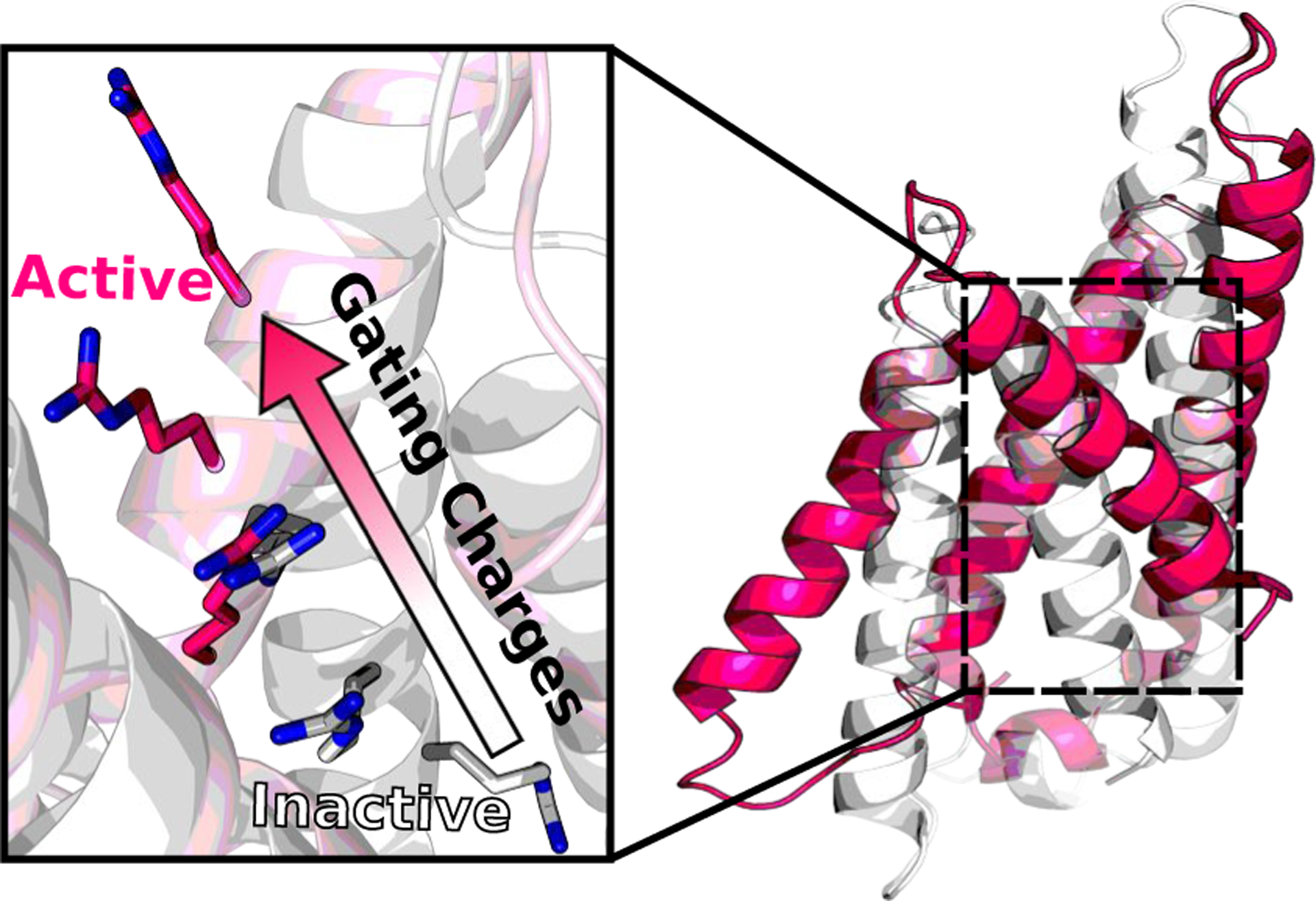
Membrane-enabled hybrid-solvent CpHMD simulations with pH replica-exchange revealed conformational activation of the voltage-gated proton channel Hv1, as indicated by the upward movement of gating charges (Henderson and Shen, unpublished data).
Despite the recent successes of membrane-enabled hybrid-solvent CpHMD simulations (Fig. 1), several important limitations remain. For example, the implicit representation of solvent and bilayer employed in the titration dynamics limits the accuracy of pKa estimation, particularly for buried active-site residues and those interacting with lipids or small-molecule ligands. In general, pKa shifts relative to solution (model) pKa are underestimated for interior residues and overestimated for lipid-interacting ones [33, 11, 79]. Further, explicit interactions with lipids and ions, which may become important for protonation state changes, cannot be accounted for. While PME all-atom CpHMD [34] has the potential to overcome the limitations, our preliminary study of the M2 channel indicated that the calculated pKa’s from all-atom CpHMD have larger systematic errors, likely a result of underpolarization with additive force fields. Thus, the hybrid-solvent CpHMD method remains a method of choice until a solution is found to remedy the force field issue. A future direction is to incorporate polarizable force fields such as the Drude model [49] in PME all-atom CpHMD. Due to the high computational cost of simulations with polarizable force fields, a GPU implementation is desirable.
2. Methods
2.1. Setting up a CpHMD simulation for transmembrane proteins
Here we discuss the protocol of membrane-enabled hybrid-solvent CpHMD simulations in CHARMM [6], which can be divided into three stages (Fig. 3). In Stage 1, the protein/bilayer complex is constructed and undergoes an initial equilibration. In Stage 2, with the protein structure restrained the lipids are equilibrated to the appropriate structural and phase behavior. In Stage 3, protein sidechains are made titratable and CpHMD is invoked to equilibrate the system at a specified pH condition followed by production replica-exchange CpHMD at multiple pH conditions.
Fig.3.
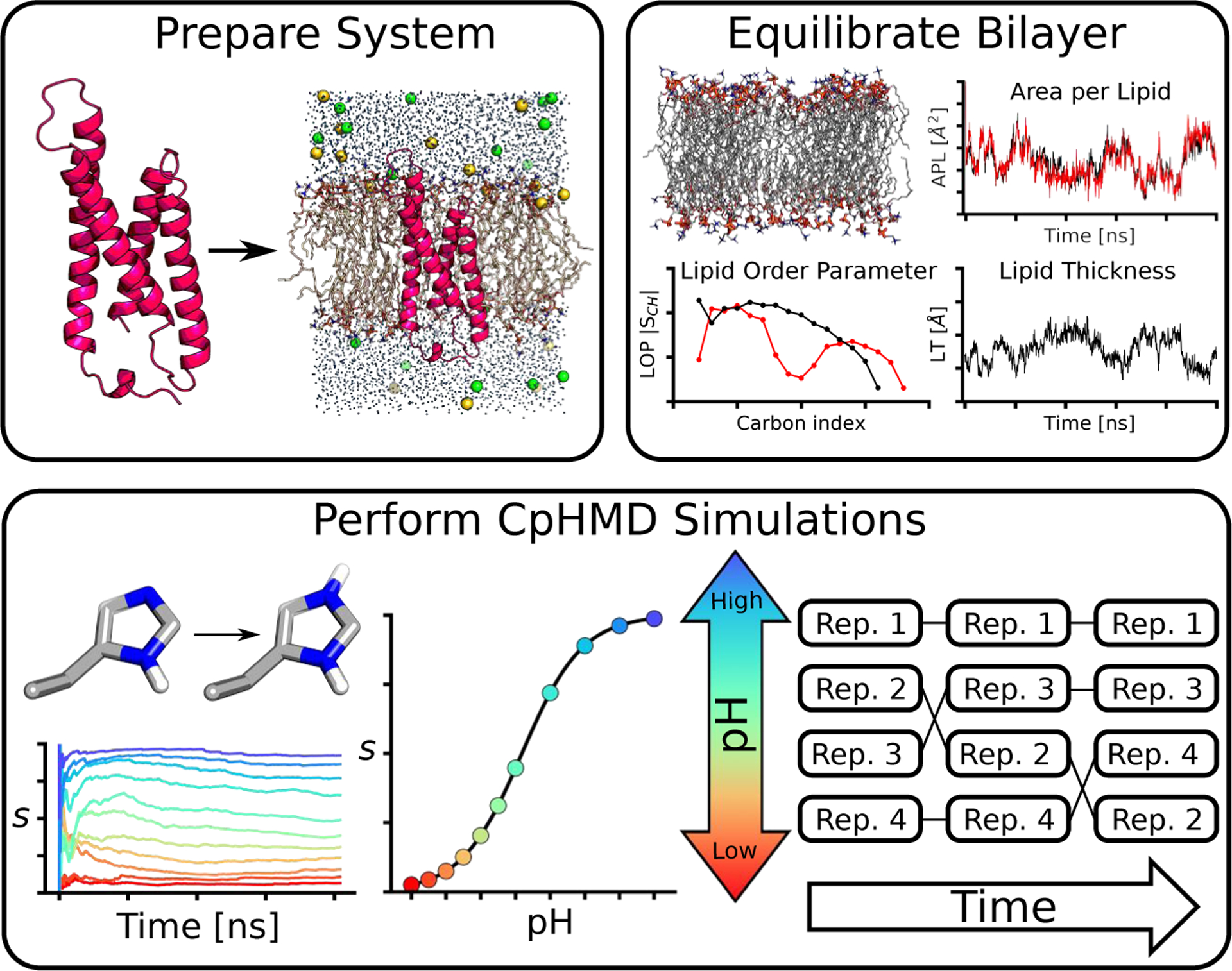
Overview of the three-stage protocol for membrane-enabled hybrid-solvent CpHMD simulations with pH replica-exchange.
Stage 1. Prepare protein/bilayer complex.
First, we retrieve the protein coordinates from the database, Orientations of Proteins in Membranes [50], which provides coordinates of membrane proteins by orienting the structure with respect to the membrane normal using theoretical calculations and comparisons to experimental data [50]. We then use MMTSB toolset [24] or other tools such as SWISS-MODEL [77] to add or replace missing residues, heavy atoms, or mutation sites. The HBUILD facility [7] in CHARMM is used to add missing hydrogens. Note, in this stage, all titratable sites are assumed to be in the “anticipated” protonation states based on the experimental pH condition and hydrogen bond/salt bridge patterns (for His). If no experimental pH can be found, default protonation states corresponding to physiological pH (7.4) should be used: Asp(−)/Glu(−)/Lys(+)/Arg(+)/Cys(0). Unless His is in a salt bridge, HSE or HSD (based on the hydrogen bonding pattern) should be used.
Since amino acid sequences given in a PDB file are often truncated, we suggest capping the terminal groups of each protein chain: for N-terminus use CH3CO (patch ACE), and for C-terminus use NH2 (patch CT2) or NHCH3 (patch CT3). However, if a terminal group interacts with a part of the protein, the free ionized form, and COO−, should be used. Since CpHMD simulation will be conducted at multiple pH in Stage 3, we should consider possible protonation state changes. This consideration is especially relevant for N-amino group, since its solution pKa is 8.5 [59], only one unit above the physiological pH. If N-amino group will be allowed to titrate, make sure the model parameters are included in the PHMD parameter file.
Once the protein structure is complete, a very short minimization in implicit solvent (e.g., with the GBSW model [39]) may be conducted with the steepest descent (SD) and adopted basis Newton-Raphson (ABNR) algorithms to relax unfavorable positions of hydrogens. It is important that during the minimization, protein heavy atoms should be fixed or restrained. For example, two rounds of 50 steps of SD followed by 10 steps of ABNR are performed, first with heavy atoms fixed and then with a harmonic force constant of 5 kcal/mol/Å2. If an extensive number of atoms have been added we also suggest that these sites be energy minimized, but the overall structure should be kept as close as possible to the crystal structure. If crystal waters are resolved in the structure these should be retained.
The rest of Stage 1 follows the CHARMM-GUI protocol [40] for preparing a protein/membrane complex for MD simulations. The protein is placed in a lipid bilayer with water above and below the bilayer. Counterions for neutralizing the system and additional ions to reach the specified ionic strength are added. The assembled protein/membrane complex undergoes a series of initial equilibration steps (Table 1, Stage 1) to relax the lipid positions before subjecting to a lengthy membrane equilibration step in Stage 2.
Table 1.
Example equilibration steps in all three stages
| Step Ensemble | Timestep (fs) | Length (ns) | Restraint force constant (kcal/mol/Å2) | ||||
|---|---|---|---|---|---|---|---|
| Stage 1. Initial equilibration | BBa | SC+XWata | Lipisb | BWatc | |||
| 1 | NVT | 1 | 0.05 | 10.0 | 5.0 | 2.5 | 2.5 |
| 2 | NVT | 2 | 0.05 | 5.0 | 5.0 | 2.5 | 2.5 |
| 3 | NPT | 2 | 0.1 | 5.0 | 5.0 | 1.0 | 1.0 |
| 4 | NPT | 2 | 0.1 | 5.0 | 2.5 | 0.5 | 0.5 |
| 5 | NPT | 2 | 0.1 | 2.5 | 2.5 | 0.1 | 0.1 |
| 6 | NPT | 2 | 0.1 | 2.5 | 2.5 | 0.0 | 0.0 |
| Stage 2. Membrane equilibration | BB + SC | ||||||
| 1 | NVT | 2 | 0.2 | 2.5 | |||
| 2 | NPT | 2 | 100 | 2.5 | |||
| Stage 3. CpHMD equilibration | BB + SC | ||||||
| 1 | NVT | 2 | 0.1 | 1.0 | |||
| 2 | NPT | 2 | 0.1 | 1.0 | |||
| 3 | NPT | 2 | 0.8 | 0.0 | |||
BB and SC refer to protein backbone and side chain heavy atoms, respectively. XWat and BWat refer to crystallographic and bulk water oxygen atoms, respectively.
Lipid heavy atoms are restrained with a planar harmonic potential to ensure that lipid tails are close to the bilayer center (−5 Å < Z < 5 Å), and lipid head groups are close to the bilayer surface (Z = ±19 Å for POPE).
A planar harmonic restraint is applied to the bulk water oxygen atoms to keep them away from the membrane hydrophobic region.
Stage 2. Equilibrate bilayer.
Once a protein/membrane complex is assembled and relaxed, the lipid bilayer needs to be fully equilibrated before protein conformational states can be sampled. Simulation in this stage can be carried out using any scalable simulation engine, such as GROMACS [1], NAMD [63], or OpenMM [20]. Typically, a 0.2-ns restrained MD is performed in the NVT ensemble followed by a 100-ns restrained MD in the NPT ensemble (see Table 1, Stage 2). During the equilibration, protein backbone and side chain heavy atoms are restrained with a harmonic force constant of 1.0 kcal/mol/Å2.
Surface area per lipid, bilayer thickness, and lipid order parameters are quantities commonly used to assess the phase behavior and fine structure of the simulated bilayer [43]. A lipid bilayer is considered equilibrated once all three quantities are converged to experimental values for the specified lipid type or those obtained in the force field validation study [43]. The program VTMC [56] can be used to calculate surface area per lipid; it excludes lipids within 5 Å from the protein, as protein-lipid interactions can artificially decrease the value. Bilayer thickness here refers to the hydrophobic thickness, which is the distance between the average Z-positions of the lipid C2 atoms from the top and bottom leaflets. Lipid order parameters SCH indicate the rigidity of the lipid tails [71] and are defined as
| (1) |
where θ is the angle between the C-H bond of the lipid tail methylene and the membrane normal (z), and the angular brackets denote ensemble average. SCH can be calculated using a Tcl script for VMD [36].
Stage 3. Perform CpHMD titration simulations.
Following equilibration of bilayer, the system can be further equilibrated using membrane-enabled [33] hybrid-solvent CpHMD simulation at a single pH. From the final snap shot of Stage 2, dummy protons need to be added to all Asp, Glu, and His sidechains. For Asp and Glu, dummy protons are placed in the syn position. Note, the dihedral energy barrier to the rotation around the carboxylic bond is raised (to 3 kcal/mol) in the force field [41] to prevent a dummy hydrogen from moving to the less favorable anti position where it becomes unlikely to protonate (“ghost” proton) [55, 41]. For His, a proton is added to either δ or ϵ nitrogen to form a doubly protonated HSP. Addition of dummy hydrogens requires a brief retrained energy minimization, e.g., two rounds of 50 steps of SD followed by 10 steps of ABNR. In the first round all heavy atoms are fixed in positions, and in the second round a harmonic restraint of 5.0 kcal/mol/Å2 is applied to them. Next, three short restrained CpHMD simulations are performed at crystallization or physiological pH (see Table 1, Stage 3). The first and second CpHMD equilibration steps are run for 0.1 ns in the NVT and NPT ensembles, respectively, with protein heavy atoms restrained with a harmonic force constant of 1.0 kcal/mol/Å2. The third unrestrained equilibration step is run in the NPT ensemble for 0.8 ns. To prevent lateral drift of the protein, a cylindrical restraint with a harmonic force constant of 0.1 kcal/mol/Å2 is applied using the MMFP facility in CHARMM [6].
Following equilibration at a single pH, production CpHMD simulations can begin using the pH replica-exchange protocol [73]. First, a simulation pH range is selected by considering the active pH range of the protein or the one that represents experimental or physiological pH conditions. The pH replicas need to be placed close enough to obtain sufficient exchange probabilities (e.g., > 20%), which requires overlap between the potential energy distributions of neighboring replicas. Typically, a 0.5 pH unit increment provides desired exchange probabilities. Each replica is simulated in the NPT ensemble, with exchange attempts occurring every 500 or 1000 MD steps between adjacent replicas. We typically observe an average exchange rate of at least 40%. More replicas at smaller intervals (e.g., 0.25 pH units) may be added to increase the exchange rate. The effectiveness of sampling enhancement can be tracked by monitoring the movement of replicas through pH space as simulation proceeds. At least several replicas should move across the entire pH ladder to achieve enhanced sampling.
Settings in molecular dynamics.
Protein is represented by CHARMM22/CMAP force field [52, 53], lipids and ions are represented by CHARMM36 force field [43], and water is represented by CHARMM modified TIP3P model. van der Waals energies are calculated using a switching function from 10 to 12 Å with a cutoff at 14 Å, consistent with the protocol used in the development of CHARMM36 lipid force field [43]. Simulations are conducted under periodic boundary conditions with the PME method [15] for long-range electrostatic energies and forces. The real space calculation uses a 12-Å cutoff and the reciprocal space calculation uses a 1-Å grid spacing and 6th-order spline interpolation. The nonbond neighbor list is heuristically updated in CHARMM [6] or every 10 MD steps in GROMACS [64].
To allow for a 2-fs integration time step, all bonds involving hydrogens are constrained using SHAKE [66] in CHARMM [6], LINCS [31] in GROMACS [64], or rigidBonds in NAMD [63]. In NPT simulations, temperature is maintained at 310 K by Nóse-Hoover thermostat [58, 32] and pressure is maintained at 1 atm by Langevin piston pressure-coupling algorithm in CHARMM [25] or Parrinello-Rahman barostat [62] in GROMACS.
CpHMD specific settings.
Unless otherwise noted, default CpHMD settings should be used. In membrane-enable hybrid-solvent CpHMD simulations [33], the protein is centered in the lipid bilayer and an exclusion cylinder, e.g., with a radius of 25 Å, is placed along the membrane normal to exclude the low dielectric slab used in the implicit membrane model [38] from influencing the titration of interior residues in the protein. The cylinder should encompass the protein with no or minimal overlap with lipids. A center of mass restraint is applied to the protein with a harmonic force constant of 1.0 kcal/mol/Å2 via the MMFP facility in CHARMM[6]. The thickness of the low-dielectric slab is set to the hydrophobic thickness of the lipid bilayer, defined as the difference between the average z positions of lipid C2 atoms from the top and bottom leaflets, e.g., about 35 Å for POPE. The half of membrane switching length [38] for the dielectric transition region between the low-dielectric slab and bulk water is set to 2.5 Å. The default GBSW atomic input radii [57, 10] are used for protein. For small molecules, the generic atomic radii in the GBSW radii file or van der Waals radii in the force field [44] should be used. Titration (λ) and tautomeric state (x) coordinates are updated every 5 MD steps (default for hybrid-solvent CpHMD), allowing for water and lipid relaxation [73]. Langevin dynamics with a friction coefficient of 5 ps−1 is used (default for CpHMD). The mass of λ and x particles are set to 10 atomic mass units (default for CpHMD).
2.2. pKa calculations
To calculate the pKa of a titratable site, the deprotonated fraction at each pH, S(pH), is obtained and fit to the generalized Henderson-Hasselbalch equation (Hill equation).
| (2) |
where n is the Hill coefficient representing the slope of the transition region in the titration curve. Deviation of n from 1 indicates the extent of cooperative (n > 1) or anticooperative (n < 1) interactions between two or more titrating sites [72]. The deprotonated fraction of a titratable site is calculated as the fraction of time λ samples the deprotonated state,
| (3) |
where deprotonated and protonated states are defined as λ > 0.8 and λ < 0.2, respectively.
For coupled titrating sites, i.e. titration of one site is affected by the protonation state of the other, apparent or macroscopic sequential pKas are of interest [28]. In the statistical mechanics formulation, the two sequential pKa’s, pK1 and pK2, are obtained by fitting the total number of protons bound to the two sites, Nprot, to the coupled titration model below [70, 74].
| (4) |
Alternatively, the total deprotonated fraction of the two sites can be fit to the following uncoupled model [78, 27],
| (5) |
where S1 and S2 are the deprotonated fractions of the two sites. pK1 and pK2 are the two uncoupled pKa’s.
Acknowledgement
The authors acknowledge National Institutes of Health (R01GM098818 and R01GM118772) for funding.
References
- 1.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 [Google Scholar]
- 2.Acharya R, Carnevale V, Fiorin G, Levine BG, Polishchuk AL, Balannik V, Samish I, Lamb RA, Pinto LH, DeGrado WF, Klein ML (2010) Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc. Natl. Acad. Sci. USA 107(34), 15075–15080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexov E, Mehler EL, Baker N, Baptista AM, Huang Y, Milletti F, Nielsen JE, Farrell D, Carstensen T, Olsson MHM, Shen JK, Warwicker J, Williams S, Word JM (2011) Progress in the prediction of pKa values in proteins. Proteins 79, 3260–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arthur EJ, Brooks CL III (2016) Efficient implementation of constant pH molecular dynamics on modern graphics processors. J. Comput. Chem 37, 2171–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baptista AM, Teixeira VH, Soares CM (2002) Constant-pH molecular dynamics using stochastic titration. J. Chem. Phys 117, 4184–4200 [Google Scholar]
- 6.Brooks BR, Brooks CL III, Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brünger AT, Karplus M (1988) Polar hydrogen positions in proteins: Empirical energy placement and neutron diffraction comparison. Proteins 4, 148–156 [DOI] [PubMed] [Google Scholar]
- 8.Case DA, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham T III, Cruzeiro VWD, Darden TA, Duke RE, Ghoreishi D, Gilson MK, Gohlke H, Goetz AW, Greene D, Harris RC, Homeyer N., Huang Y, Izadi S, Kovalenko A, Kurtzman T., Lee TS, LeGrand S, Li P, Lin C, Liu J, Luchko T, Luo R., Mermelstein DJ, Merz KM, Miao Y, Monard G, Nguyen C, Nguyen H, Omelyan I., Onufriev A, Pan F, Qi R, Roe DR, Roitberg A, Sagui C, Schott-Verdugo S, Shen J, Simmerling CL, Smith J, Salomon-Ferrer R, Swails J, Walker RC, Wang J, Wei H, Wolf RM, Wu X, Xiao L, York DM, Kollman PA (2018) AMBER 2018. San Francisco [Google Scholar]
- 9.Chen J, Brooks CL III, Khandogin J (2008) Recent advances in implicit solvent-based methods for biomolecular simulations. Curr. Opin. Struct. Biol 18, 140–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Im W, Brooks CL III (2006) Balancing solvation and intramolecular interactions: toward a consistent generalized Born force field. J. Am. Chem. Soc 128, 3728–3736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen W, Huang Y, Shen J (2016) Conformational activation of a transmembrane proton channel from constant pH molecular dynamics. J. Phys. Chem. Lett 7, 3961–3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen W, Morrow BH, Shi C, Shen JK (2014) Recent development and application of constant pH molecular dynamics. Mol. Simul 40, 830–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen W, Wallace J, Yue Z, Shen J (2013) Introducing titratable water to all-atom molecular dynamics at constant pH. Biophys. J 105, L15–L17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coincon M, Uzdavinys P, Nji E, Dotson DL, Winkelmann I, Abdul-Hussein S, Cameron AD, Beckstein O, Drew D (2016) Crystal structures reveal the molecular basis of ion translocation in sodium/proton antiporters. Nat. Struct. Mol. Biol 23, 248–255 [DOI] [PubMed] [Google Scholar]
- 15.Darden T, York DM, Pedersen L (1993) Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys 98, 10089–10092 [Google Scholar]
- 16.DeCoursey TE (2018) Voltage and pH sensing by the voltage-gated proton channel, Hv1. J. R. Soc. Interface 15, 20180108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeCoursey TE, Hosler J (2014) Philosophy of voltage-gated proton channels. J. R. Soc. Interface 11, 20130799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donnini S, Tegeler F, Groenhof G, Grubmüller H (2011) Constant pH molecular dynamics in explicit solvent with λ-dynamics. J. Chem. Theory Comput 7, 1962–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drew D, Boudker O (2016) Shared molecular mechanisms of membrane transporters. Annu. Rev. Biochem 85, 543–572 [DOI] [PubMed] [Google Scholar]
- 20.Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, Wang LP, Simmonett AC, Harrigan MP, Stern CD, Wiewiora RP, Brooks BR, Pande VS (2017) OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol 13, e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eicher T, Cha H, Seeger MA, Brandstätter L, El-Delik J, Bohnert JA, Kern WV, Verrey F, Grütter MG, Diederichs K, Pos KM (2012) Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc. Natl. Acad. Sci. USA 109, 5687–5692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellis CR, Shen J (2015) pH-dependent population shift regulates BACE1 activity and inhibition. J. Am. Chem. Soc 137, 9543–9546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellis CR, Tsai CC, Hou X, Shen J (2016) Constant pH molecular dynamics reveals pH-modulated binding of two small-molecule BACE1 inhibitors. J. Phys. Chem. Lett 7, 944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feig M, Karanicolas J, Brooks CL III (2004) MMTSB tool set: enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph. Model 22, 377–395 [DOI] [PubMed] [Google Scholar]
- 25.Feller SE, Zhang Y, Pastor RW, Brooks BR (1995) Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys 103, 4613–4621 [Google Scholar]
- 26.Goh GB, Knight JL, Brooks CL III (2012) Constant pH molecular dynamics simulations of nucleic acids in explicit solvent. J. Chem. Theory Comput 8, 36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goh GB, Hulbert BS, Zhou H, Brooks CL III (2014) Constant pH molecular dynamics of proteins in explicit solvent with proton tautomerism. Proteins 82, 1319–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris RC, Shen J (2019) GPU-Accelerated implementation of continuous constant pH molecular dynamics in Amber: pKa predictions with single-pH simulations. J. Chem. Inf. Model 59, 4821–4832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harris RC, Tsai CC, Ellis CR, Shen J (2017) Proton-coupled conformational allostery modulates the inhibitor selectivity for β-secretase. J. Phys. Chem. Lett 8, 4832–4837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henderson JA, Harris RC, Tsai CC, Shen J (2018) How ligand protonation state controls water in protein-ligand binding. J. Phys. Chem. Lett 9, 5440–5444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 18, 1463–1472 [Google Scholar]
- 32.Hoover WG (1985) Canonical dynamics: Equilibration phase-space distributions. Phys. Rev. A 31(3), 1695–1697 [DOI] [PubMed] [Google Scholar]
- 33.Huang Y, Chen W, Dotson DL, Beckstein O, Shen J (2016) Mechanism of pH-dependent activation of the sodium-proton antiporter NhaA. Nat. Commun 7, 12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang Y, Chen W, Wallace JA, Shen J (2016) All-Atom Continuous Constant pH Molecular Dynamics With Particle Mesh Ewald and Titratable Water. J. Chem. Theory Comput 12, 5411–5421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang Y, Harris RC, Shen J (2018) Generalized Born Based Continuous Constant pH Molecular Dynamics in Amber: Implementation, Benchmarking and Analysis. J. Chem. Inf. Model 58, 1372–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 37.Hunte C, Screpanti E, Venturi M, Rimon A, Padan E, Michel H (2005) Structure of a Na+/H+ antiporter and insights into mechanism of action and regulation by pH. Nature 435, 1197–1202 [DOI] [PubMed] [Google Scholar]
- 38.Im W, Feig M, Brooks CL III (2003) An implicit membrane generalized Born theory for the study of structure, stability, and interactions of membrane proteins. Biophys. J 85, 2900–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Im W, Lee MS, Brooks CL III (2003) Generalized Born model with a simple smoothing function. J. Comput. Chem 24, 1691–1702 [DOI] [PubMed] [Google Scholar]
- 40.Jo S, Kim T, Im W (2007) Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2, e880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khandogin J, Brooks CL III (2005) Constant pH molecular dynamics with proton tautomerism. Biophys. J 89, 141–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khandogin J, Brooks CL III (2006) Toward the accurate first-principles prediction of ionization equilibria in proteins. Biochemistry 45, 9363–9373 [DOI] [PubMed] [Google Scholar]
- 43.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr, Pastor RW (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knight JL, Brooks CL III (2011) Surveying implicit solvent models for estimating small molecule absolute hydration free energies. J. Comput. Chem 32, 2909–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kong X, Brooks CL III (1996) λ-dynamics: A new approach to free energy calculations. J. Chem. Phys 105, 2414–2423 [Google Scholar]
- 46.Lee C, Kang HJ, von Ballmoons C, Newstead S, Uzdavinys P, Dotson DL, Iwata S, Beckstein O, Cameron AD, Drew D (2013) A two-domain elevator mechanism for sodium/proton antiport. Nature 501, 573–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee C, Yashiro S, Dotson DL, Uzdavinys P, Lwata S, Sansom MSP, von Ballmoos C, Beckstein O, Drew D, Cameron AD (2014) Crystal structure of the sodium-proton antiporter NhaA dimer and new mechanistic insights. J. Gen. Physiol 144, 529–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee MS, Salsbury FR Jr, Brooks CL III (2004) Constant-pH molecular dynamics using continuous titration coordinates. Proteins 56, 738–752 [DOI] [PubMed] [Google Scholar]
- 49.Lemkul JA, Huang J, Roux B, MacKerell AD Jr (2016) An empirical polarizable force field based on the classical Drude oscillator model: development history and recent applications. Chem. Rev 116, 4983–5013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI (2006) OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625 [DOI] [PubMed] [Google Scholar]
- 51.Ludwig MG, Vanek M, Guerini D, Gasser JA, Jones CE, Junker U, Hofstetter H, Wolf RM, Seuwen K (2003) Proton-sensing G-protein-coupled receptors. Nature 425, 93–98 [DOI] [PubMed] [Google Scholar]
- 52.MacKerell AD Jr, Bashford D, Bellott M, Dunbrack RL Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L., Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T., Nguyen DT, Prodhom B., Reiher WE III, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J., Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 53.MacKerell AD Jr, Feig M, Brooks CL III (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem 25, 1400–1415 [DOI] [PubMed] [Google Scholar]
- 54.Mager T, Rimon A, Padan E, Fendler K (2011) Transport mechanism and pH regulation of the Na+/H+ antiporter NhaA from Escherichia coli. J. Biol. Chem 286, 23570–23581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mongan J, Case DA, McCammon JA (2004) Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem 25, 2038–2048 [DOI] [PubMed] [Google Scholar]
- 56.Mori T, Ogushi F, Sugita Y (2012) Analysis of lipid surface area in protein-membrane systems combining Voronoi tessellation and Monte-Carlo integration methods. J. Comput. Chem 33, 286–293 [DOI] [PubMed] [Google Scholar]
- 57.Nina M, Beglov D, Roux B (1997) Atomic radii for continuum electrostatics calculations based on molecular dynamics free energy simulations. J. Phys. Chem. B 101, 5239–5248 [Google Scholar]
- 58.Nosé S (1984) A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys 52, 255–268 [Google Scholar]
- 59.Nozaki Y, Tanford C (1967) Examination of titration behavior. Methods Enzymol. 11, 715–734 [Google Scholar]
- 60.Padan E (2014) Functional and structural dynamics of NhaA, a prototype for Na+ and H+ antiporters, which are responsible for Na+ and H+ homeostasis in cells. Biochim. Biophys. Acta 1837, 1047–1062 [DOI] [PubMed] [Google Scholar]
- 61.Padan E, Danieli T, Keren Y, Alkoby D, Masrati G, Haliloglu T, Ben-Tal N, Rimon A (2015) NhaA antiporter functions using 10 helices, and an additional 2 contribute to assembly/stability. Proc. Natl. Acad. Sci. USA 112, E5575–E5582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys 52, 7182–7190 [Google Scholar]
- 63.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramsey IS, Moran MM, Chong JA, Clapham DE (2006) A voltage-gated proton-selective channel lacking the pore domain. Nature 440, 1213–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical Integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys 23, 327–341 [Google Scholar]
- 67.Taglicht D, Padan E, Schuldiner S (1991) Overproduction and purification of a functional Na+/H+ antiporter coded by nhaA (ant) from Escherichia coli. J. Biol. Chem 266, 11289–11294 [PubMed] [Google Scholar]
- 68.Takeshita K, Sakata S, Yamashita E, Fujiwara Y, Kawanabe A, Kurokawa T, Okochi Y, Matsuda M, Narita H, Okamura Y, Nakagawa A (2014) X-ray crystal structure of voltage-gated proton channel. Nat. Struct. Mol. Biol 21, 352–357 [DOI] [PubMed] [Google Scholar]
- 69.Tsai CC, Yue Z, Shen J (2019) How electrostatic coupling enables conformational plasticity in a tyrosine kinase. J. Am. Chem. Soc 141, 15092–15101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ullmann GM (2003) Relations between protonation constants and titration curves in polyprotic acids: a critical view. J. Phys. Chem. B 107, 1263–1271 [Google Scholar]
- 71.Vermeer LS, de Groot BL, Réat V, Milon A, Czaplicki J (2007) Acyl chain order parameter profiles in phospholipid bilayers: computation from molecular dynamics simulations and comparison with 2H NMR experiments. Eur. Biophys. J 36, 919–931 [DOI] [PubMed] [Google Scholar]
- 72.Wallace JA, Shen JK (2009) Predicting pKa values with continuous constant pH molecular dynamics. Methods Enzymol. 466, 455–475 [DOI] [PubMed] [Google Scholar]
- 73.Wallace JA, Shen JK (2011) Continuous constant pH molecular dynamics in explicit solvent with pH-based replica exchange. J. Chem. Theory Comput 7, 2617–2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wallace JA, Shen JK (2012) Charge-leveling and proper treatment of long-range electrostatics in all-atom molecular dynamics at constant pH. J. Chem. Phys 137, 184105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wallace JA, Shen JK (2012) Unraveling a trap-and-trigger mechanism in the pH-sensitive self-assembly of spider silk proteins. J. Phys. Chem. Lett 3, 658–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wallace JA, Wang Y, Shi C, Pastoor KJ, Nguyen BL, Xia K, Shen JK (2011) Toward accurate prediction of pKa values for internal protein residues: the importance of conformational relaxation and desolvation energy. Proteins 79, 3364–3373 [DOI] [PubMed] [Google Scholar]
- 77.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TA, Rempfer C, Bordoli L, Lepore R, Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, 296–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Webb H, Tynan-Connolly BM, Lee GM, Farrell D, O’Meara F, Søndergaard CR, Teilum K, Hewage C, McIntosh LP, Nielsen JE (2011) Remeasuring HEWL [DOI] [PubMed]