

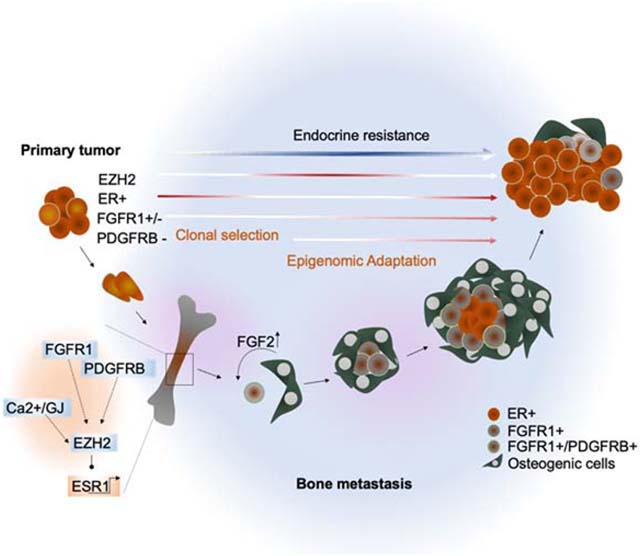
Summary
ER+ breast cancer exhibits a strong bone-tropism in metastasis. How the bone microenvironment (BME) impacts ER signaling and endocrine therapy remains poorly understood. Here, we discover that the osteogenic niche transiently and reversibly reduces ER expression and activities specifically in BMMs, leading to endocrine resistance. As bone micrometastases (BMMs) progress, the ER reduction and endocrine resistance may partially recover in cancer cells away from the osteogenic niche, creating phenotypic heterogeneity in macrometastases. Using multiple approaches including an evolving barcoding strategy, we demonstrated that this process is independent of clonal selection, and represents an EZH2-mediated epigenomic reprogramming. EZH2 drives ER+ BMMs toward a basal and stem-like state. EZH2 inhibition reverses endocrine resistance. These data exemplify how epigenomic adaptation to BME promotes phenotypic plasticity of metastatic seeds, fosters intra-metastatic heterogeneity, and alters therapeutic responses. Our study provides insights into the clinical enigma of ER+ metastatic recurrences despite endocrine therapies.
Keywords: Bone metastasis, Osteogenic cells, bone microenvironment, epigenomic reprogramming, clonal evolution, bone tropism, stemness, endocrine therapies, evolving barcoding, EZH2, FGF2, FGFR, PDGFR, chromatin alteration, endocrine resistance
Graphical Abstract
eTOC Blurb:
Bado et al. demonstrate that the osteogenic niche enhances the phenotypic plasticity of metastatic ER+ breast cancer cells through an EZH2-mediated epigenomic reprogramming. The resulting spatiotemporal heterogeneity of ER expression leads to increased stemness and endocrine resistance. Targeting EZH2 restores endocrine sensitivity and synergizes with standard anti-ER therapies.
INTRODUCTION
Estrogen receptor positive (ER+) breast cancer accounts for over 70% of all breast cancers (BC), and after recurring, causes over 24,000 deaths per year in the US. Adjuvant endocrine therapies target ER and significantly reduce recurrences. However, 20–40% of patients still develop metastases, often after a prolonged latency (Lim et al., 2012; Zhang et al., 2013). Thus, it is imperative to understand how ER+ cancer cells escape endocrine therapies in distant organs and to identify therapies to eliminate these cells.
Bone is the most frequently affected organ by ER+ BC, which is usually luminal-like. Compared to the basal-like subtype, luminal BC exhibits a 2.5-fold increased frequency of bone metastasis (BoM), but a 2.5-fold decreased frequency of lung metastasis (Kennecke et al., 2010; Smid et al., 2008). BoMs of luminal-like BC are usually late-onset, occurring beyond 5 years after surgery. Current tumor-intrinsic biomarkers in primary tumors can predict recurrences within 5 years, but not beyond (Sgroi et al., 2013), suggesting that the capacity of developing late-onset metastasis may not be encoded in cancer cells. We hypothesize that unique interactions between the bone microenvironment (BME) and ER+ disseminated tumor cells (DTCs) may facilitate survival and therapeutic resistance.
Very little is known about how the BME affects ER+ BC cells. There is a paradoxically high discordance rate of ER status between primary tumors and DTCs, suggesting loss of ER in DTCs (Fehm et al., 2008; Jäger et al., 2015). However, most overt BoMs (>85%) remain positive for ER (Hoefnagel et al., 2013), seemingly contradicting the DTC findings. ER+ BoMs can still respond to endocrine therapies, although resistance almost invariably develops.
We have previously developed a series of models and techniques to investigate cancer-bone interaction at a single cell resolution. Herein, we aim to investigate the impact of BME on the ER signaling in ER+ breast cancer cells.
RESULTS
BMMs transiently lose ER expression
To study how ER+ BC cells interact with BME, we identified two patient-derived xenograft models (PDXs), HCI011 and WHIM9, from patients with BoMs (Derose et al., 2011; Li et al., 2013). In immunodeficient mice, orthotopic HCI011 and WHIM9 tumors spontaneously metastasized to bone 5–6 months after surgeries (Figure 1A). Spontaneous BoMs exhibited reduced ER expression in both PDX models (Figure 1B). The BoM size positively correlated with average intensity of nuclear ER by IHC staining (Figure S1A and S1B).
Figure 1: BME induces transient loss of ER expression in ER+ breast cancer cells.
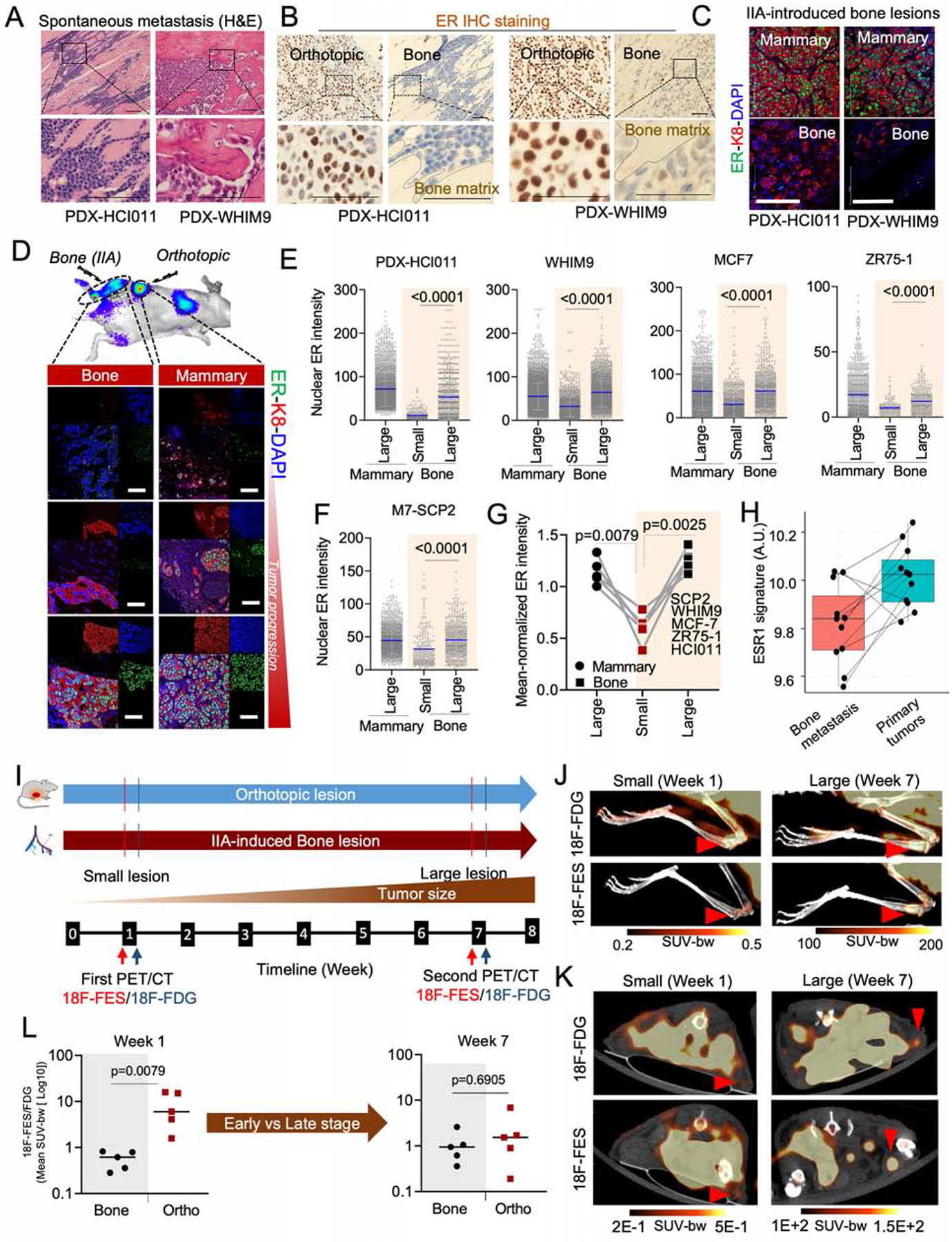
A. H&E staining of spontaneous metastases of HCI011 and WHM9 tumors to spine and hind limb, respectively. Scale bar: 100μm.
B. Human-specific ER IHC staining are shown for spontaneous metastasis of HCI011 and WHIM9, respectively. Scale bar: 50μm.
C. IF staining of ER, keratin 8 (k8), and DAPI in orthotopic (mammary) and IIA-induced BoM models of ER+ PDXs (HCI011 and WHIM9). Scale bars: 100μm.
D. IF images of MCF7 cells following orthotopic and bone transplantation. Changes in ER expression are illustrated at different stages of tumor progression. Scale bars: 50μm.
E. Dot plot depicting nuclear ER intensity of single cells (SCs) in orthotopic and BoM specimens from PDXs (HCI011, WHIM9) and cell lines (MCF7, ZR75-1), following IF staining as illustrated in 1C and 1D (n=3–6 mice per model).
F. Dot plot depicting nuclear ER intensity of SCs in orthotopic and BoM specimens from M7-SCP2. Bone lesions were classified into “small” and “large” as defined in (E); (n=4 mice).
G. Connected scatterplots showing the mean-normalized ER intensity of all cancer models used from Figure 1E to 1F. (n= 5 cell lines).
H. Boxplot depicting changes in ESR1 early signature in matched BoM and primary specimens from BC patients (https://github.com/npriedig/).
I. Scheme describing the PET-CT experiment on MCF7 orthotopic or IIA-induced BoM models. 18F-FES and 18F-FDG imaging were performed 48h apart, at week 1 and 7 post tumor transplantation.
J. PET/CT scans showing the maximum intensity projection (MIP) of 18F-FDG and 18F-FES in bone as described in I. MCF7 BoMs were generated using IIA injection. Red arrows indicate tumor location. Scale: 0.2–0.5 SUV-bw (week 1) and 100–200 SUV-bw (week 7).
K. Axial view of PET/CT scans depicting the uptake of 18F-FDG and 18F-FES in small and large lesions of MCF7 orthotopic tumors. Early time point (Week 1) and late time point (Week 7) were used to depict non palpable orthotopic tumor stage (small < 2mm) and the palpable tumor stage. Red arrows and scale: as in J.
L. Relative quantification of radiolabeled 18F-FES uptake in small and large lesions of orthotopic and BoMs. Each dot represents the mean standard uptake values (mean SUV-bw) of 18F-FES normalized to the mean SUV of 18F-FDG for each mouse. Mann Whitney U-test is used for statistical analysis; n=5 mice per group.
(A–L): P values were computed by two-tailed unpaired Student t-tests unless otherwise noted.
Next, we performed intra-iliac artery (IIA) injection of dissociated PDXs or established ER+ cell lines (MCF-7 and ZR75-1) to introduce experimental BoMs. This approach synchronizes the onset of colonization and enriches BMMs, thereby allowing quantitative examination of bone colonization of relatively indolent cancer cells at different stages (Figure 1C,D) (Yu et al., 2016). As in spontaneous BoMs, a strong correlation was found between ER expression and the size of IIA-induced BoMs (Figure S1C) but not orthotopic tumors (Figure S1D). When we classified BoMs by size, the expression of ER diminished in small lesions compared to mammary tumors, but was restored in large lesions (Figure 1E). Thus, in both spontaneous and experimental BoM models, lesion size seems to be associated with ER expression (Figure 1E–G).
Two possible mechanisms might explain the above association. First, genetically distinct ER-low and ER-high cancer cells may pre-exist, and the former progresses at a slower rate and form small lesions. Second, there may be a transient loss and a subsequent recovery of ER in ER+ cancer cells in BME. To distinguish these, we collected four single cell-derived populations from MCF-7 cells (M7-SCPs). Exome sequencing validated their genetic purity (Figure S1E). The expression of ER in M7-SCPs is still variable from cell to cell, although to a lesser degree compared to parental cells (Figure S1F), suggesting a baseline level of cellular plasticity among ER+ BC cells. Using an M7-SCP of the best clonality (M7-SCP2), we compared the ER expression between BMMs and macrometastases. Like parental MCF7, M7-SCP2 cells exhibited decreased ER expression in small BoMs (Figure 1F, S1G and S1H). Taken together, multiple ER+ models supported that BME induces a loss of ER expression specifically in BMMs (Figure 1G).
The loss of ER was also observed on clinical specimens. In a study comparing patient-matched primary BCs and BoMs, ESR1 was found to be one of the top genes downregulated in BoMs (Priedigkeit et al., 2017). Gene Set Variation Analysis (GSVA) further suggests that there is an even stronger downregulation of acute ER signaling (Figure 1H).
We next monitored longitudinal alteration of ER signaling during BoM progression using positron emission tomography-computed tomography (PET-CT) imaging. A radiolabeled 18F-Fluoroestradiol (18F-FES) PET/CT imaging strategy was adopted (Figure 1I and S1I–K) to measure uptake of estrogen by tumors in parallel with glucose consumption (18F-FDG) (Kurland et al., 2017). We found a significant reduction in estrogen uptake in early lesions of BoM (Figure 1J), compared to mammary tumors (Figure 1K and S1I). The difference was reduced at a later time point (Figure 1J–L), suggesting a reversible process.
An evolving barcode strategy to trace clonal evolution of ER+ BC cells
ER heterogeneity is often observed in the clinic. However, the phylogenetic relationship between ER+ and ER− cancer cells is not defined. We used an evolving barcode system (Kalhor et al., 2017, 2018) to trace the evolution of ER+ cancer cells in BME. This is a variant of the widely used CRISPR-Cas9 system. PAM site is mutated to allow Cas9 to home to the locus encoding guide RNAs (hgRNAs) and introduce mutations, providing an opportunity for multiple parallel lineage tracing.
The evolving barcode system was introduced to MCF-7 cells, and allowed to accumulate mutations before tumor transplantation to the bone by IIA injection (Figure 2A). This created a baseline barcode diversity so that independent clones in the bone can be distinguished. Further evolution after IIA injection helps deduce parent-child relationship between clones. We used laser-captured microdissection (LCM) to isolate cancer cells in different regions of established BoMs (Figure 2B and Figure S2A). DNA purification and PCR-mediated enrichment of hgRNA sequences were performed before sequencing (Figure 2A, 2B, and S2A). The hgRNAs were then used to deduce phylogenetic relationship among cancer cells in different lesions (Figure 2C and S2B). A consecutive section of the bone was used for immunofluorescence (IF) staining of ER so that the nuclear ER expression of each lesion can be superimposed with the barcodes. ER expression was highly variable across different lesions in the femur (Figure 2D and S2C) and tibia (Figure S2D), indicating an increased phenotypic heterogeneity of the BoM.
Figure 2: Tracing ER expression in bone metastases by evolving barcodes and ERE-reporter.
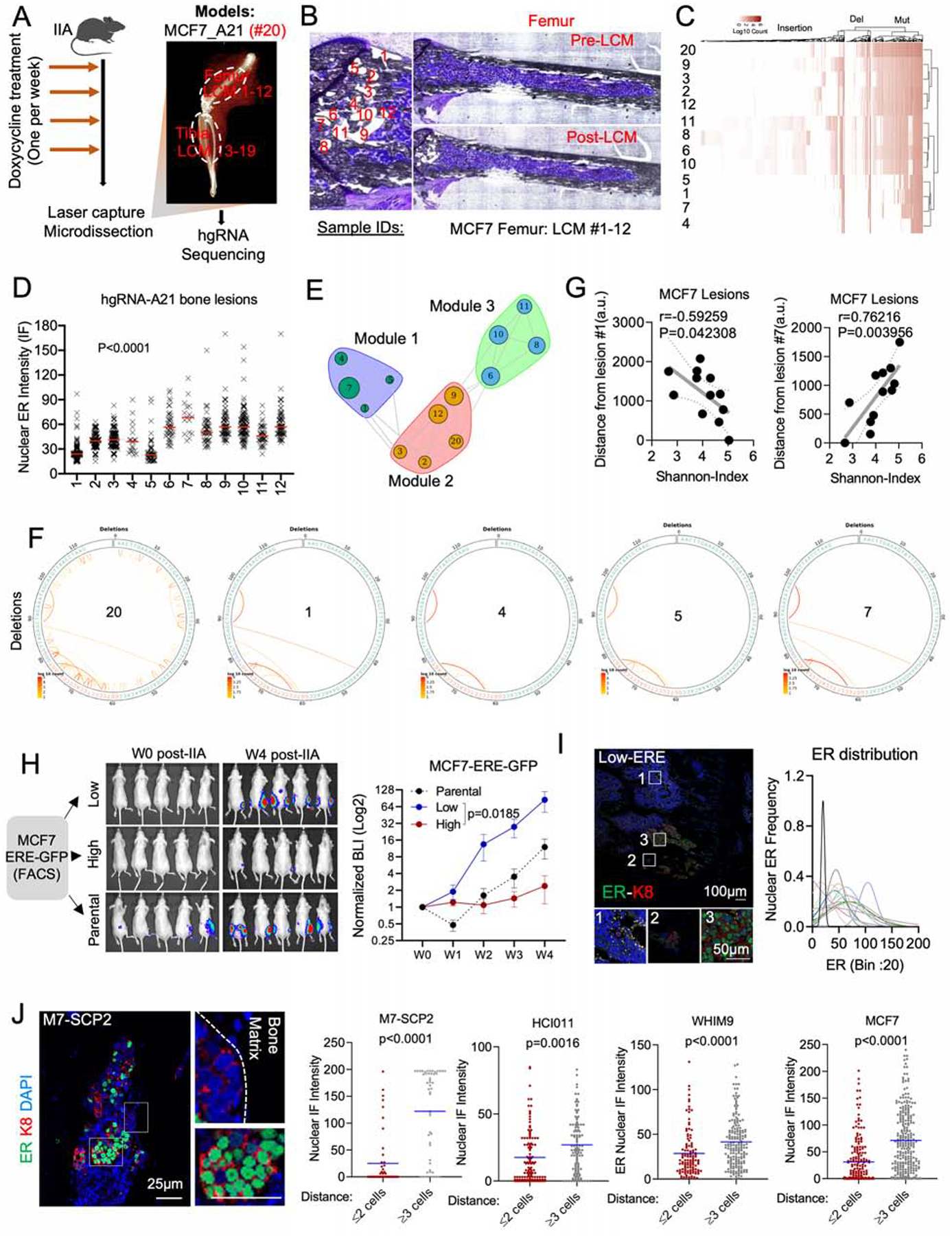
A. Experimental design of BoM lineage tracing using an inducible CRISPR-Cas9 hgRNA evolving barcoding system. iCas9-expressing MCF7 cells were stably transduced with homing guide RNA A21 (hgRNA-A21) before transplantation to bone and induced weekly for four weeks before LCM and targeted sequencing. We collected 19 lesions from femur (#1–12) and tibia (#13–19). Barcoded parental cells were labelled as #20.
B. Exact map of mouse femur before and after LCM of metastatic lesions #1–12.
C. Heatmap showing hierarchical clustering of bone lesions #1–12, based on hgRNA-A21 mutations.
D. IF quantification of SCs nuclear ER in lesions #1–12 (femur). (n=12 lesions); P value: Ordinary one-way ANOVA.
E. Modular organization of lesions #1–12 following a high-dimensional undirected analysis of barcode mutations. Node sizes represent mean intensity of ER expression.
F. Circus plot showing barcode deletions in bone lesions clustered in module 1 (see E). #20: pre-injected cells.
G. Scatter plot showing Pearson correlation (r) between the Shannon index of bone lesions and their relative distance to lesion #1 or #7. P value: two-tailed t-test.
H. IIA-induced BoM from ERE-GFP reporter MCF7 cells. ERE-GFPLow, and ERE-GFPhigh MCF7 cells were sorted based of their GFP expression. Tumor growth was measured by Bioluminescence. (n= 5–8 mice); P value: Two-Way ANOVA.
I. IF staining of ER in BoM derived from ERE-GFPLow MCF7 cells. The Gaussianized ER distribution is based on nuclear intensity of SCs; peak: mean ER expression per lesion.
J. IF staining depicting a spatial distribution of ER based on the location of M7-SCP2 lesions relatively to the bone matrix. Scale bars: 25μm. Dot plots represent nuclear ER expression in cells proximal (≤ 2 cell distance) or distal (≥3 distance) to the bone matrix. P values: two-tailed unpaired Student’s t-tests.
In the metaphyseal area of femur, 12 lesions were grouped into three clusters (Figure S2E). Interestingly, ER expression varies within each cluster, which provides strong evidence against genetic traits as a determinant of ER level in BoM (Figure 2E). Similar observations were found in lesions derived from tibia (Figure S2F). A notable example was the module 1, a cluster formed by lesions No. 1, 4, 5 and 7. The barcodes of these lesions are highly similar (Figure 2F, S2G), indicating close phylogenetic distance. Additionally, these lesions have a distinct spatial distribution with close proximity to the growth plate (Figure S2H). Lesion No. 1 exhibited a low ER expression but the highest Shannon entropy among all 12 lesions, whereas Lesion No. 7 was the opposite. Both ER expression and Shannon entropy of other lesions vary as functions of distance to these two extreme lesions (Figure 2G). Considering that the Shannon entropy is correlated with the “age” of cancer cells (Zhang et al., 2021), Lesion No. 1 is likely to be parental to Regions No. 4, 5, and 7. The fact that their ER level greatly varies, support our hypothesis that ER− lesions may give rise to ER+ lesions as BoMs progresses.
The analysis of all lesions (#1–19) derived from the same hind limb revealed surprising similarities between lesions from femur and tibia (Figure S2E). Despite physical barriers, lesion No. 15 from tibia shares similar mutations with lesions No. 6, 8, 10, and 11 from femur, suggesting a femur-to-tibia seeding (Figure S2E). This metastasis-to-metastasis seeding is further investigated in our accompanied study (Zhang et al., 2021).
ERE-GFPLow cells drive BoM progression and reconstitute ER heterogeneity.
As an independent approach to trace the fate of ER− cancer cells in BoM, we introduced a reporter system, namely GFP expression driven by the estrogen-responsive-elements (ERE-GFP). We performed IIA injection of ERE-GFPhigh and ERE-GFPlow cells, respectively. The bone colonization capacity of ERE-GFPLow cells is over 30-fold higher compared to ERE-GFPhigh cells (Figure 2H and S2I), consistent with the finding that the ER− subset of MCF-7 cells enrich stemness (Fillmore et al., 2010). Interestingly, BoMs established by ERE-GFPLow cells exhibited heterogeneous ER expression, similar to those derived from parental cells (Figure 2I and S2J) and lesion sizes were associated with ER expression (Figure S2K). This experiment provides additional support for the conclusion that ERLow cells may generate ERhigh cells in BME.
BC lesions tend to associate with areas of new bone matrix deposition (Figure S2L). We examined the spatial distribution of ER expression relative to bone matrix. Cancer cells apart from lesion borders were more likely to restore ER expression compared to those at the border (Figure 2J). Thus, ER recovery tends to occur first toward the center of a BoM lesion, leading to the hypothesis that the interactions between metastatic cells and adjacent bone cells drive the transient loss of ER.
Direct interaction with OGs mediates the loss of ER expression
To identify bone cells that causes ER loss in BMMs, we assessed the spatial relationship between ER expression and various bone cells including osteoclasts (RANK+), endothelial cells (CD31+), myofibroblasts/bone stromal cells (αSMA+), and OGs (ALP+). In WHIM9, HCI011 and MCF-7 models, RANK expression exhibited a positive association with nuclear ER intensity at a single cell level (Figure 3A). In contrast, negative associations were observed for endothelial, fibroblasts, and OGs (Figure 3B and 3C). Among these, the correlation of ALP+ OGs is most consistent across different models (Figure 3C), suggesting that OGs may be the cell types driving the loss of ER.
Figure 3: OGs promote the loss of ER expression and reduction of ER activities during early stages of bone colonization.
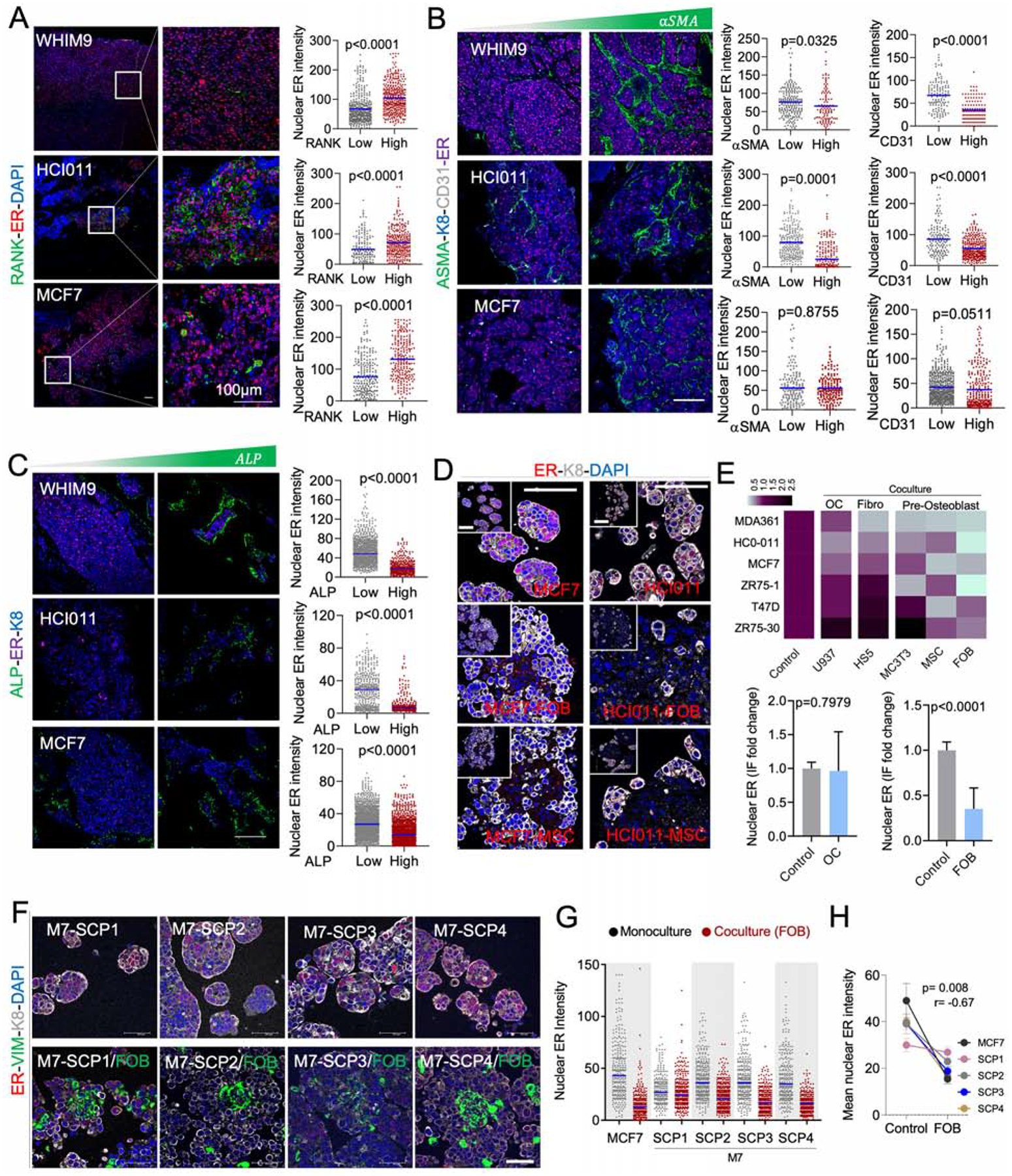
A. IF of BoMs showing ER expression in PDXs (HCI011 and WHIM9) and MCF7 cells, relatively to Receptor activator of nuclear factor-KB (RANK) expression in osteoclasts. Scale bars: 100μm. Dot plots show ER quantification in SCs. (n=3–5 lesions).
B. IF as in A. ER is co-stained with alpha smooth muscle actin (αSMA) and cluster of differentiation 31 (CD31) to depict endothelial cells. Dot plots show ER quantification in SCs. (n=3–5 lesions).
C. IF as in A. ER is co-stained with alkaline phosphatase (ALP). Dot plots show ER quantification in SCs. (n=3–5 lesions).
D. IF images of HCI011-derived primary cells and MCF7 cells in 3D monoculture and co-culture with FOBs and MSCs. Scale bars: 100μm.
E. Heatmap showing the mean intensity of ER in primary cells (HCI011) and BC cell lines (MDA-MB-361, MCF7, ZR75-1, T47D, ZR75-30) in 3D monoculture (control) or co-culture with osteoclast precursors (U937), bone marrow stromal cells (Hs5), mouse pre-osteoblasts (MC3T3), human mesenchymal stem cells (MSC) and human pre-osteoblast (FOB). Histogram shows ER expression in monoculture versus co-culture of multiple cell lines with U937 or FOB cells; (n=3 biological replicates). Error bars: mean +/− SD.
F. IF showing ER expression in M7-SCPs in 3D monoculture or co-culture with FOB cells. Vimentin (VIM). Scale bars: 50μm.
G. IF quantification of ER expression in M7-SCP1 to 4; (n = 3 biological replicates).
H. Graph representing ER expression in cancer cells alone or in co-cultured with FOB cells. Spearman correlation (r); (n = 5 cell lines); Error bars: mean +/− SE
(A–H): P values were computed by two-tailed unpaired Student t-tests unless otherwise noted.
To further study this interaction, we employed a fetal OB line (FOB) and a human MSC line to represent OGs. Luminal-like cancer cells and OGs form heterotypic organoids in 3D suspension co-culture, which recapitulated several aspects of cancer-niche interaction (Wang et al., 2015). Co-staining of ER and keratin 8 (K8, a marker of luminal cells) in 3D co-cultures revealed a loss of ER in MCF-7 cell line and HCI011 PDX-derived organoids (Figure 3D) similar to in vivo BMMs. MSCs and FOB both induced consistent loss of ER expression across multiple models (Figure 3E). In contrast, U937, a human monocytic cell line that is often used to model osteoclast precursors, did not cause the same changes to the ER expression, supporting the specificity of OGs (Figure 3E).
Importantly, M7-SCPs also exhibited the same alterations upon interacting with FOB (Figure 3F and 3G). In M7-SCP2, -SCP3 and -SCP4, the degree of ER downregulation is comparable to parental MCF-7 cells. In contrast, M7-SCP1 exhibited a lesser decrease (Figure 3G and 3H). Interaction with OGs confers growth advantage on cancer cells as we previously showed (Wang et al., 2015, 2018). Similarly to MCF-7 cells (Figure S3A and S3B), M7-SCP1, 2, and 3 also displayed such advantage in 3D co-cultures as compared to mono-cultures. In contrast, the growth of M7-SCP4 was suppressed by FOB (Figure S3C). Thus, different M7-SCPs possess variable capacity of orthotopic tumor-initiation, bone colonization, and FOB-mediated growth promotion and ER downregulation (Figure S3D). This pre-existing heterogeneity supports the importance of clonal selection in metastasis, which has been repeatedly demonstrated in previous studies (Bos et al., 2009; Kang et al., 2003; Minn et al., 2005). However, the BME-induced, adaptive change is less appreciated (Figure 3F–H).
Hyperactive ER activities can lead to ER protein degradation (Nawaz et al., 1999). Therefore, decreased ER expression could paradoxically suggest an enhanced ER signaling. Upon co-culturing with FOB, cancer cells reduced the transcription of ER mRNA (Figure 4A), as well as ER transcription activities as indicated by a luciferase reporter driven by a promoter containing ER-responsive elements (ERE-luciferase) (Figure 4B). The ER target progesterone (PR) was also downregulated in BMMs (Figure 4C). These data indicate that the transient loss of ER is not a reflection of hyperactive ER signaling, but rather the cause of decreased ER signaling.
Figure 4: OGs confer endocrine resistance.
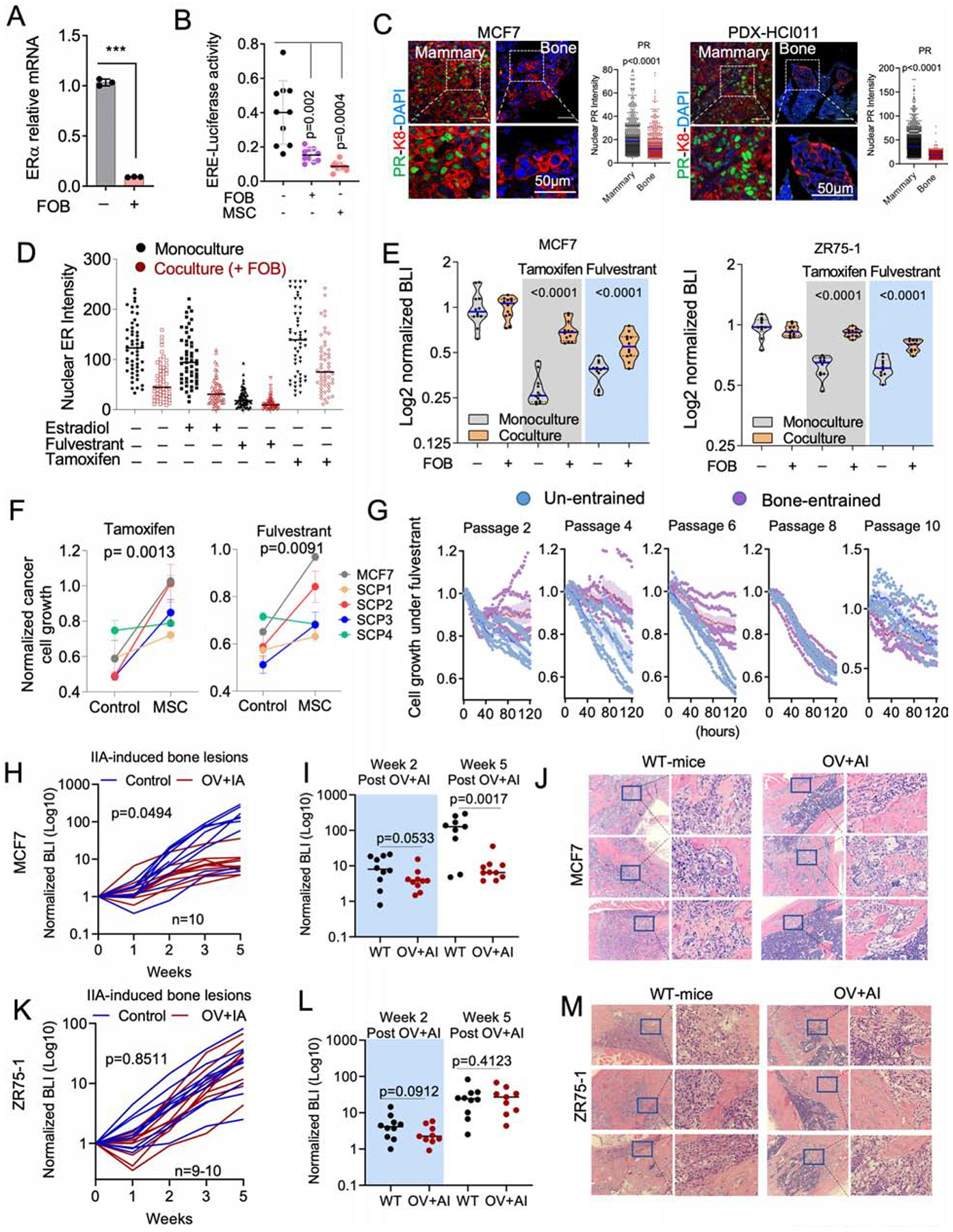
A. Relative mRNA expression of ESR1 in 3D monoculture or co-culture of MCF7 with FOB. Data result from FACS-sorted MCF7. (n=3 technical replicates).
B. Dot plot representing ER transcriptional activity in MCF7 cells expressing pGL2 ERE-luciferase reporter. MCF7 cells were cultured in 3D with or without OGs (FOB and MSC) for 7 days. (n=10 technical replicates); Error bars: mean +/− SD.
C. Confocal images showing the expression of progesterone receptor (PR) in IIA-induced BoM from MCF7 and HCI011. Dot plots show nuclear PR IF intensity. (n=3 lesions).
D. Dot plots depicting ER intensity in 3D mono- and co-culture of MCF7 cells with FOB following 24 hours treatment with 10nM 17β-estradiol, 20nM fulvestrant and 100nM 4-Hydroxytamoxifen (tamoxifen); n=5 fields.
E. Violin plot showing the response of luciferase-labelled MCF7 and ZR75-1 cells to 100nM of 4-Hydroxytamoxifen (Tamoxifen) and 20nM of fulvestrant in 3D mono- or co-culture with OGs (FOB). Bioluminescence was acquired 72 hours post-treatment. (n=12 and 10 technical replicates for and ZR75-1).
F. Graphs representing the proliferation of MCF7 cells and M7-SCP1-4 in monoculture and MSC co-culture following 1 week of treatment with 20nM fulvestrant or 100nM tamoxifen; n= 5 cell lines.
G. Time course experiment depicting growth kinetics of un-entrained and bone-entrained M7-SCP2 cells in vehicle or 20nM fulvestrant conditions; n=6 technical replicates.
H. Growth curve showing response of IIA-induced MCF7 BoMs to estrogen depletion. Ovariectomized (OV) mice were additionally treated with Letrozole (OV+AI), daily. Results are based on BLI. (n=10 mice); P value: Two-Way ANOVA.
I. Dot plot showing BoM growth in wild-type (WT) and OV+AI mice at week 2 and week 5 post tumor transplantation; n=10 mice. P value: two-tailed unpaired Student’s t-test.
J. H&E staining showing MCF7 metastatic lesions in wild-type (WT) and “OV+AI” groups.
K. Growth curve depicting the response of ZR75-1-derived BoM as in H. (n=9–10 mice); P value: Two-Way ANOVA.
L. Dot plot showing statistical growth differences in ZR75-1 as in I.
M. H&E staining of ZR75-1 metastatic lesions as in J.
(A–M): P values were computed by two-tailed unpaired Student t-tests unless otherwise noted.
Interaction with OGs in the BME leads to resistance to endocrine therapies
Downregulation of ER may impact endocrine therapies. We examined the effects of fulvestrant, tamoxifen, and estradiol on ER+ cancer cells with or without co-culture of FOB. The presence of FOB diminished the effects of these agents on ER nuclear localization (Figure 4D) and blunted the anti-proliferative effects of tamoxifen and fulvestrant (Figure 4E). The same results were observed using M7-SCPs (Figure 4F), indicating a process independent of genetic selection. We next examined the reversibility of “bone-entrained” effects by inoculating M7-SCP2 cells into bone via IIA. Cancer cells were retrieved after establishment of BMMs, resulting in “bone-entrained” M7-SCP2 cells (M7-SCP2-Bo) (Figure S4A). M7-SCP2-Bo cells remained resistant to fulvestrant in early passages, but this resistance diminished as cells were expanded in cultures (Figure 4G), suggesting that BME-induced phenotypic shift is not stably inherited.
We next examine responses of ER+ cancer cells to estrogen deprivation in BME (Figure S4B). Combined overiectomy and letrozole treatment could significantly impede orthotopic tumor growth in both MCF-7 and ZR75-1 models (Figure S4C and S4D), but failed to reduce BoM colonization at early time points (Figure 4H–4M). The response to estrogen deprivation was partially recovered in MCF-7 cells after Week 3 (Figure 4H–4J), further supporting that the resistance may be reversible as metastases further progress. However, ZR75-1 bone lesions remained resistant at later time points (Figure 4K–4M). Hence, not all BoMs restore endocrine sensitivity. In ZR75-1, despite the partial reversion of ER expression in macrometastases, PR expression remained repressed (Figure S4E and S4F). Thus, downstream ER signaling may not recover together with ER expression during BoM progression.
Downregulation of ER in BoM is partially mediated by direct cell-cell contact and gap junctions
We previously reported that heterotypic gap junctions between cancer cells and OGs mediate calcium influx to the former and activates calcium signaling (Wang et al., 2018). We asked if the gap junction and calcium signaling may mediate ER downregulation and endocrine resistance. Suppression of gap junction by a peptide, GAP19, or calcium signaling by a small molecule inhibitor, FK506, partially restored ER expression in co-cultures with FOBs (Figure S5A). This effect was small but noticeable, and was further supported by a converse experiment showing that high [Ca2+] in the medium decreased ER expression (Figure S5B). Inhibition of calcium signaling also reduced the grow advantage conferred by FOB (Figure S5C), and enhanced endocrine therapies in bone-in-culture array (BICA) (Figure S5D), which is an ex vivo platform that recapitulate BME and cancer-niche interactions (Wang et al., 2017). Taken together, gap junction and calcium signaling contribute to inhibit ER expression in BMMs.
Unbiased profiling uncovered global phenotypic shift of ER+ cancer cells
We used multiple approaches to discovery additional mechanisms underpinning BME-induced ER downregulation (Figure 5A). Translating ribosome affinity purification followed by RNA-seq (TRAP-seq) was applied to profile transcriptome in cancer cells interacting with OGs in 3D suspension co-cultures without dissociating the two cell types. TRAP-seq revealed that MSCs diminished impact of endocrine perturbations on ER+ cancer cells (Figure S5E). We also validated that GJA1, the gene encoding connexin 43, was upregulated by MSCs in co-cultures and exhibited a strong inverse correlation with ER expression (Figure S5F), further indicating a role of gap junctions in downregulating ER. However, conditioned medium of OGs also causes ER downregulation and endocrine resistance (Figure S5G), indicating paracrine mechanisms.
Figure 5: BME drives a global phenotypic shift involving multiple pathways.
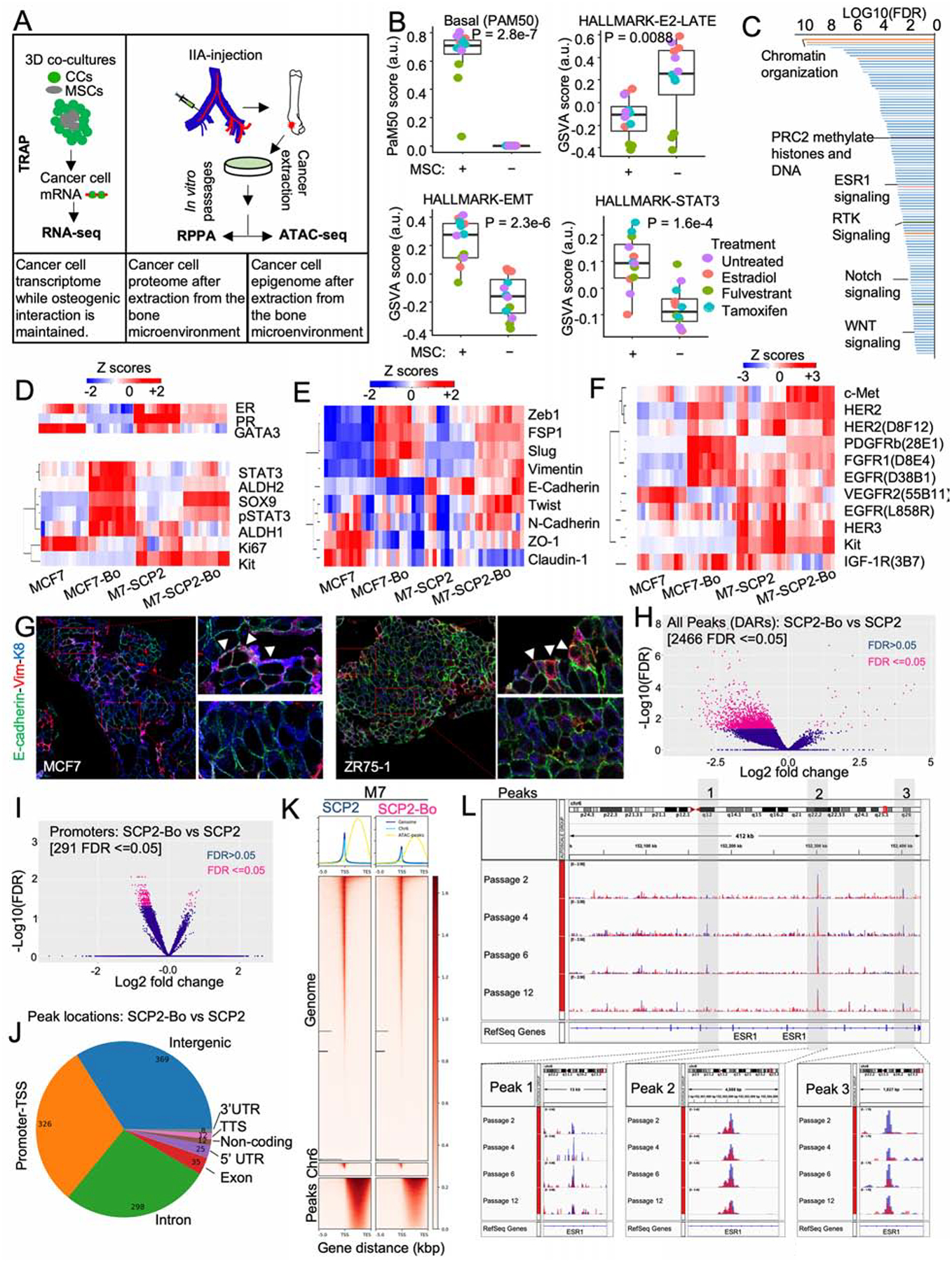
A. Experimental summary diagram to evaluate molecular in cancer cells exposed to BME. TRAP-seq was performed on 3D co-culture of MCF7 and OGs (MSCs), RPPA on un-entrained (MCF7 and M7-SCP2) and bone-entrained cells (MCF7-Bo and M7-SCP2-Bo), and ATAC-seq on un-entrained (M7-SCP2) or bone-entrained (M7-SCP2-Bo) cells.
B. Box plot depicting gene signature alternations in MCF7 monoculture (MSC-) and co-cultures (MSC) from TRAP-seq. Analysis was performed using a non-parametric and unsupervised GSVA (Hänzelmann et al., 2013). Specific colors represent different treatment conditions. (n= 4 biological replicates each with 3 technical replicates); P value: two-tailed unpaired Student’s t-test.
C. Waterfall plot showing the gene ontology analysis of TRAP-seq data from PANTHER classification system, based on false discovery rate (FDR).
D. Heatmap depicting expression changes in luminal and stemness-related markers from RPPA data. Parental cells (MCF7 and M7-SCP2), and bone-entrained BC cells (MCF7-Bo and M7-SCP2-Bo) are compared; n: 4 biological replicates and 3 technical replicates.
E. Heatmap depicting EMT/MET markers from RPPA data as described in D.
F. Heatmap depicting receptor tyrosine kinases from RPPA data as described in D.
G. IF showing vimentin, E-cadherin, and Keratin 8 expression in IIA-induced bone MCF7 and ZR75-1 BoMs.
H. Volcano plot showing epigenetic reprogramming of bone-entrained M7-SCP2 cells based on differentially enriched peaks from ATAC sequencing analysis. 2644 peaks were significantly altered in bone-entrained M7-SCP2 (FDR ≤0.05) and highlighted in pink.
I. Volcano plot based on opened promotors identified by ATAC-seq analysis. FDR < 0.05 is highlighted in pink.
J. Pie chart depicting the genomic distribution of differentially altered peaks between un-entrained and bone-entrained M7-SCP2 cells.
K. Heatmaps and summary plots showing chromatin opening near the transcription start site TSS. 5000 bp before and after TSS are represented.
L. Genomic track showing peak variations in the ESR1 gene of un-entrained (M7-SCP2) and bone-derived (M7-SCP2-Bo) cells in blue and red color, respectively. 3 major peaks are highlighted epigenetic reversibility in M7-SCP2-Bo in vitro.
According to TRAP-seq, over 1,100 genes are significantly increased by MSC co-cultures (FDR < 0.05 and fold change > 2), which indicates a global phenotypic alteration. Using PAM50 signatures, we observed a shift from luminal to basal subtype (Figure 5B). Several pathways changed significantly in MSC-interacting cancer cells, including the decrease of ER signaling and increase of epithelial to mesenchymal transition (EMT) and STAT3 signaling (Figure 5B), all of which indicated dedifferentiation and stem-like activities (Mani et al., 2008; Marotta et al., 2011; Pfefferle et al., 2015). PANTHER classification system identified a number of pathways overrepresented in the altered genes, including several related to epigenomic regulation of gene expression (e.g., PRC2), stemness-related pathways (e.g., WNT and Notch), and receptor tyrosine kinase (RTK) signaling (Figure 5C and S5H). Some of these pathways have been previously implicated in BoM and therapeutic resistance (Andrade et al., 2017; Esposito et al., 2019; Sethi et al., 2011; Zheng et al., 2017). These findings indicate that OGs induce an epigenomic landscape alteration in ER+ BC cells toward more ER-independent and stem-like states.
Reverse phase protein array (RPPA) was used to profile over 236 key proteins and phosphor-proteins in cancer cells that have been extracted from BME. Comparison were made between MCF-7 and MCF-7-Bo and between M7-SCP2 and M7-SCP2-Bo. We identified the proteins and phospho-proteins that are significantly altered (Figure S5I and S5J). The bone-entrained cells exhibited reduced ER signaling (Figure 5D), enhanced stemness (Figure 5D), increased mesenchymal markers (Figure 5E), and increased RTK expression (Figure 5F). The most up-regulated protein in both MCF-7-Bo and M7-SCP2-Bo was PDGFRβ (Figure S5J and S5J). Overall, these changes revealed a global phenotypic shift toward a more dedifferentiated status (Ginestier et al., 2007; Guo et al., 2012; Mani et al., 2008; Tam et al., 2013; Trastuzumab et al., 2013). One notable osteogenic cell-induced change is the acquisition of a hybrid EMT status (Figure 5E). We performed co-IF staining of epithelial markers (E-cadherin and cytokeratin 8) and a mesenchymal marker (Vimentin). A small proportion of double-positive cells were observed, usually located at the border between metastases and bone matrix, where OGs are located (Figure 5G). Thus, the interaction with OGs does not simply cause EMT, but rather induce phenotypic plasticity and confer stemness.
Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) (Buenrostro et al., 2015) was used to evaluate epigenetic changes occurring at the chromatin level of cancer cells extracted from bone. The differentially accessible regions (DAR) between parental and bone-entrained cells were in general decreased in the latter (Figure 5H–5J), indicating a global shift toward heterochromatin formation. This was further supported by a global decrease in chromatin accessibility (Figure 5K). We identified two major peaks around ESR1 gene with reduced chromatin accessible in bone-entrained cells (Figure 5L) (Peaks 1 and 3). The reduction reversed over multiple passages in vitro (Figure 5L and S5K). Overall, these results indicate a reversible epigenetic reprogramming in cancer cells interacting with BME.
FGFR and PDGFR pathways contribute to phenotypic changes in BMMs
Among all pathways altered in BME, PDGFRβ and FGFR1 pathways are of particular interest. PDGFRβ exhibited the highest fold change in both models (Figure S5I and 5J) and was shown to determine the subtype of BC and mediate cancer stem cell activities (Lehmann et al., 2011; Tam et al., 2013). Multiple FGF ligands and receptors were up-regulated in human BoMs compared to matched primary tumors (Priedigkeit et al., 2017). FGF signaling regulates stem cell compartment in ER+ BC (Fillmore et al., 2010). Using a literature-based network analysis platform (https://string-db.org/)(von Mering et al., 2005), we found that FGF2 connects ER, FGFR1 and PDGFRB (Figure 6A), suggesting a pivotal role of FGF2 in ER downregulation and endocrine resistance.
Figure 6: Osteogenic cell-secreted FGFs promote endocrine resistance.
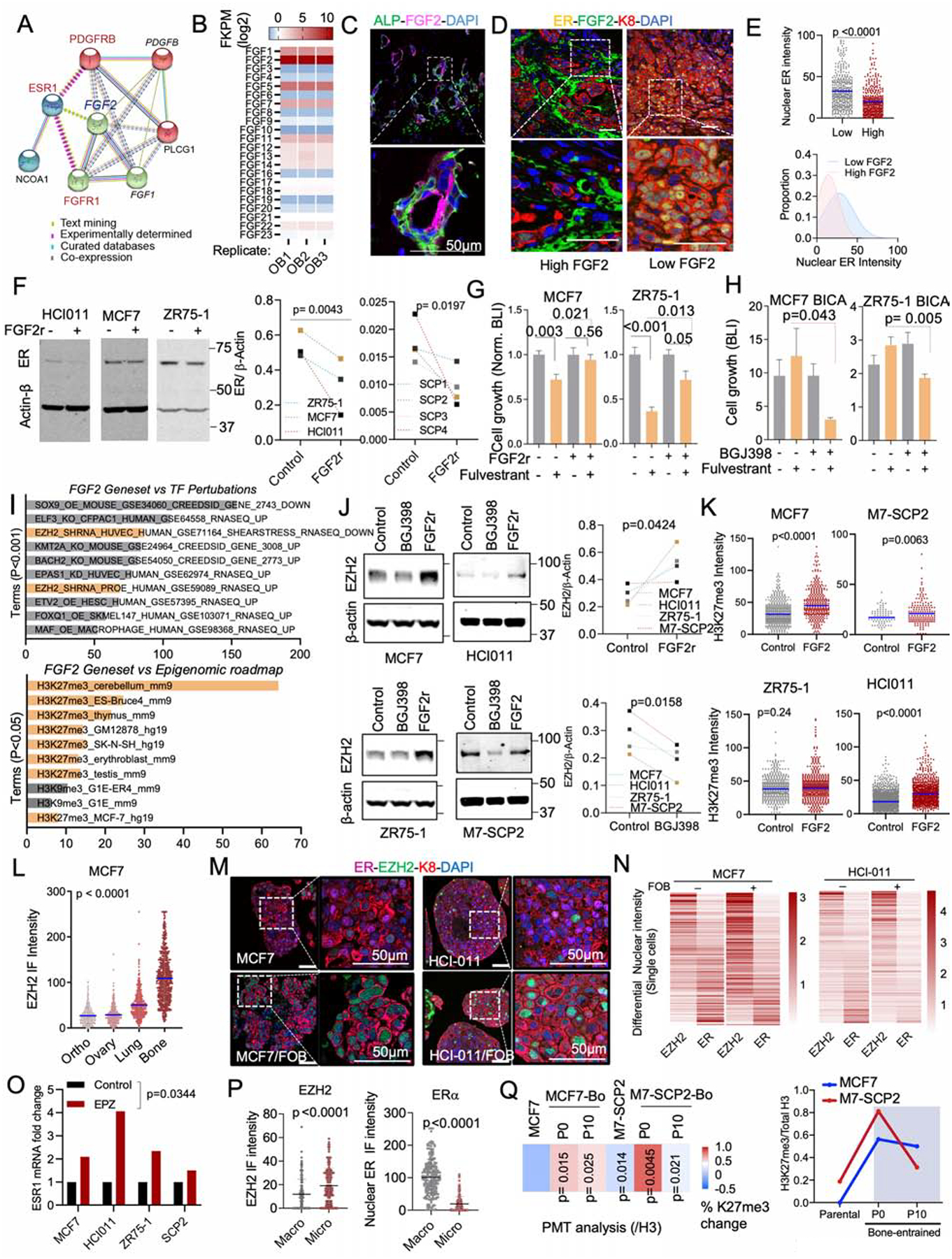
A. Network depicting functional protein association between FGFR1, PDGFRB and ER using the STRING database. Kmeans clustering (k=3) was used to represent 3 major centroids (depicted as red, green, and cyan spheres) and their most closely associated proteins based on unsupervised data mining.
B. Graph showing the expression of all human 22 fibroblast growth factor (FGF) family proteins (FGF1–23) in OGs (FOB) RNA sequencing dataset (n=3 technical triplicates).
C. IF images of Alkaline Phosphatase (ALP) and basic fibroblast growth factor (FGF2/bFGF) in normal bone tissue. Nuclei is shown in blue (DAPI). Scale bar: 50μm
D. IF images showing decreased ER expression in tumors established in FGF2 enriched BME.
E. The scatter dot plot represents ER quantification from tumors according to FGF2 enrichment (Low and High) in adjacent stromal cells (n=3–4 lesions). Mean expression is represented in blue. The Gaussian curve simulates ER distribution based on nuclear intensity. Peaks: mean expression of ER.
F. Immunoblots depicting the effect of recombinant FGF2 (20ng/ml) on ER expression in multiple BC models including PDX HCI011. Cells were treated for 24h. ER expression was summarized as connected dot plot graphs; n = 3 and 4 cell lines. P values: one-tailed unpaired Student t-tests.
G. Histogram showing effect of recombinant FGF2 (20ng/ml) on MCF7 and ZR75-1 cell growth in 3D; n=6 technical replicates.
H. Bone-In-Culture-Array (BICA) assay showing synergistic effects between 2.5μM pan FGFR inhibitor (BGJ398) and 20nM fulvestrant in MCF7 and ZR75-1 models. n=6 technical replicates.
I. Annotated bar plot showing the association of histone modifications with basic FGF (FGF2) gene signatures using the Enrichr platform (https://amp.pharm.mssm.edu/Enrichr/). Processed ChIP-sequencing data was obtained from epigenomic roadmap project (Roadmap Epigenomics Consortium et al., 2015). Histograms represent the association score with FGF2 signaling. Signatures are sorted based on P value ranking. Only p values < 0.05 and <0.01 were shown for the top and bottom panel, respectively.
J. Immunoblotting showing alteration of EZH2 expression in multiple cells following a 24h treatment with 1μM pan FGFR inhibitor (BGJ398) or 20nM FGF2 recombinant (FGF2r). Primary cells generated from HCI011 (ER+ PDX) were cultured in 3D and treated with 1μM pan FGFR inhibitor (BGJ398) or vehicle for 24h. Normalized EZH2 expression (/β-actin) was shown as dot plots. (n=4 cell lines); P value: one-tailed unpaired Student t-test.
K. Dot plots depicting the effect of 20nM recombinant FGF2 on H3k27me3 of multiple ER+ BC 3D models, based on quantified IF images (n=3 technical replicates). P values: One-tailed paired Student t-tests.
L. IF quantification of EZH2 expression in paired metastases and orthotopic tumor. MCF7 cells were transplanted to bone and to mammary gland of nude mice, which led to tumor formation at multiples sites including lung, ovary, bone and mammary gland. Metastatic tissues were harvested for IF quantification and shown as a dot plot graph. (n=3 images); Mean expression of EZH2 is indicated in blue.
M. IF showing co-expression of ER and EZH2 in 3D monocultures and co-cultures (+FOB) models of MCF7 and PDX HCI011. Scale bars: 50μm.
N. Heatmap showing relative expression of nuclear ER and EZH2 in MCF7 and PDX HCI011 SCs as depicted in M.
O. Effect of the EZH2 inhibitor EPZ011989 on ESR1 mRNA expression after 24 hours of treatment; n=4 cell lines.
P. Dot plots showing IF quantification of EZH2 and ER in MCF7 BMMs and macrometastases (n= 3–5 lesions).
Q. Reversibility of epigenetic changes based on post-translational modification (PTM) analysis. The percent changes in H3k27me3 between parental cells (MCF7), M7-SCP2 and bone-derived (MCF7-Bo and M7-SCP2-Bo) after multiple passages in vitro (Passages: P0 and P10). (n=4 biological replicates); P values are relatively to parental cells. The right panel depicts the temporal epigenetic changes (H3k27me3/H3)
(A–Q): P values were computed by two-tailed unpaired Student t-tests unless otherwise noted.
FGF2 is the highest expressed FGF ligands by FOB cells (Figure 6B). IF staining of FGF2 in BMMs revealed a positive correlation with ALP+ OGs (Figure 6C and S6A) and a negative correlation with nuclear intensity of ER (Figure 6D and 6E). Functionally, recombinant FGF2 treatment decreased ER expression in multiple cell lines including M7-SCPs (Figure 6F), and induced resistance to fulvestrant (Figure 6G). Conversely, a potent FGFR inhibitor, BGJ398, reversed fulvestrant resistance of ER+ cancer cells in BICA (Figure 6H).
PDGFRB is highly expressed in the bone-entrained cells at both protein and mRNA levels (Figure S6B). Direct interaction between cancer cells and OGs was required for PDGFRB upregulation (Figure S6C). Among all PDGF ligands, PDGF-DD, but not PDGF-BB or PDGF-CC, significantly promoted the therapeutic resistance (Figure S6D and S6E), while suppressed ER expression (Figure S6F and S6G). Like FGFR, the inhibition of PDGFR signaling by sunitinib partially abolished the cancer-promoting effects of FOB cells in 3D co-cultures (Figure S6H), further supporting the important roles of both FGFR and PDGFR signaling in the BME-induced endocrine resistance.
The complicated impact of BME converges on an EZH2
We next asked how the discovered pathways cooperate to silence ER and increase phenotypic plasticity. In the Epigenomic Roadmap database, FGF2-regulated genes are enriched with trimethylation of H3K27, and sensitive to perturbation of EZH2 (Figure 6I). Consistently, PRC2 methyltransferase activity is enhanced in cancer cells co-cultured with MSCs (Figure 5G). Treatment of recombinant FGF2 increased H3K27me3 and EZH2 (Figure 6J–K and S6I) but did not affect other H3 modifications (Figure S6J). Conversely, treatment of BGJ398 decreased EZH2 expression in 3D cancer-MSC co-cultures (Figure 6J). PDGF-DD yielded similar effects (Figure S6I and S6J). Furthermore, calcium signaling affected EZH2 expression at the RNA level (Figure S6K). Thus, the pathways that downregulate ER seem to converge on the regulation of EZH2.
EZH2 is a reliable marker for cancer stemness (Kim and Roberts, 2016; Zhou et al., 2002). In ER+ cancer cells, the PRC2 target genes were concertedly downregulated, whereas a stemness signature was upregulated, by co-culturing of OGs (Figure S6L–N), validating the connection of EZH2 to cancer stemness. In addition, cancer cell-intrinsic EZH2 expression was specifically increased in BME compared to other organs (Figure 6L).
EZH2 has been shown to silence ER expression in previous studies (Reijm et al., 2011). To validate this, we carried out IF staining in 3D co-cultures and observed inverse correlation between EZH2 and ER expression both in 3D cultures and in bone lesions at a single cell level (Figure 6M and 6N). Inhibition of EZH2 enzymatic activity by an EZH2 inhibitor (EPZ011989) (Campbell et al., 2015) led to restoration of ER expression at the RNA level (Figure 6O). The inverse changes of ER and EZH2 were also observed in vivo between BMMs and macrometastases (Figure 6P). Finally, the BME-induced increase of H3K27me3 was reversible after several passages in vitro – again suggesting a transient impact (Figure 6Q).
EZH2 inhibition induced ER expression in a murine model, which is abolished by OGs in bone lesions.
We sought to validate our findings in immunocompetent hosts. Most murine BC cell lines are ER−, thereby limiting the possibility of syngeneic experiments (Derose et al., 2011). However, some murine models express ER in early-stage tumor progression, e.g., MMTV-PyMT (Lin et al., 2003; Medina et al., 2002). AT3 is a cell line derived from MMTV-PyMT (Guy et al., 1992). We reasoned that it might express ER at some stage, and examined if EZH2 inhibition restores ER expression in AT3. In mammary tumors, the transient treatment of EZH2 inhibitor increased ER expression in a durable fashion and to a level exceeding the threshold defining ER+ tumors (Figure 7A, 7B and S7A). Although the re-expression of ER may not restore downstream estrogen signaling, it recapitulated upstream regulation of ER expression by EZH2. Therefore, we went on to determine how the aberrant expression of ER in AT3 cells might respond to BME. After IIA injection, ER expression in AT3 was lost again in syngeneic mice, and this loss was especially pronounced in regions adjacent to bone matrix (Figure 7C and 7D). Importantly, inducible depletion of osterix-expressing osteoprecursor cells abolished the loss of ER (Figure 7C and 7D). Taken together, these data validated that the OGs suppress ER expression in BME.
Figure 7: EZH2 integrates multiple signals from BME and drives the phenotypic shift of ER+ breast cancer cells.
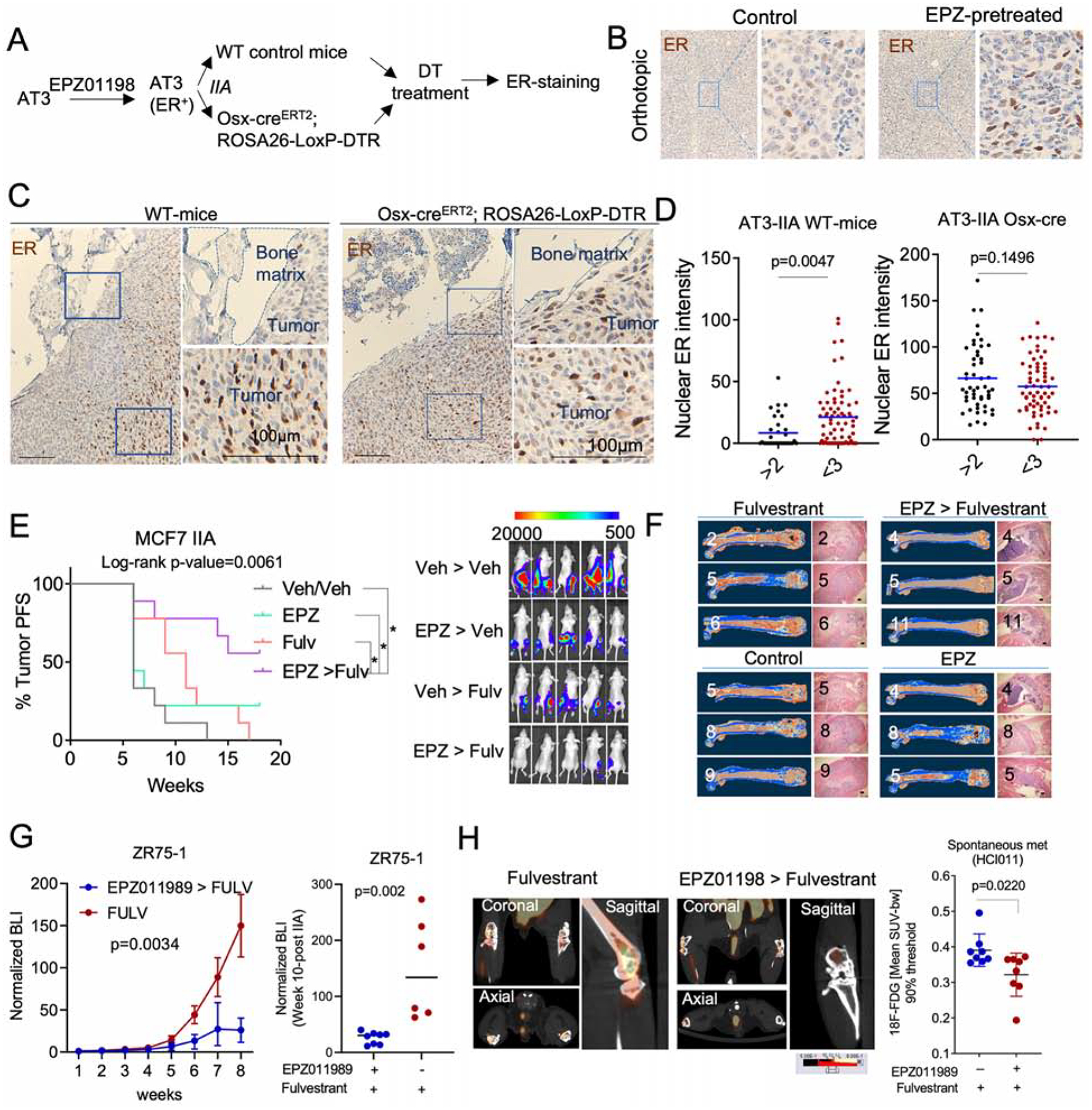
A. Scheme to evaluate ER loss in syngeneic murine models. AT3 cells were pre-treated with EZH2 inhibitor (EPZ011989) for 2 weeks before being transplanted to bone of wild-type or osterix-depleted C57BL/6 mice (Osx-creERT2 ROSA-LoxP-DTR).
B. IHC staining depicting ER expression in orthotopic tumors derived from EZP011989 pretreated AT3 cells.
C. IHC staining of ER in BoM models presented in A.
D. Dot plots showing ER expression in IIA-induced AT3 BoM in control (WT-mice) and osteoprogenitor-depleted (Osx- creERT2 ROSA-LoxP-DTR) mice at SC level.
E. Progression free survival (PFS) curve of IIA-induced BoMs following treatment with the EZH2 inhibitor EPZ011989 or the ER inhibitor fulvestrant in combination or as single agents. BL images show the effect of combination treatment on IIA-induced BoMs. (n=9–10 mice); Log-rank test was used for survival analysis. * p<0.05.
F. microCT and H&E images depicting tumor burden after pre-clinical experiment described in E. All groups revealed BoM formation except for combination treatment group (EPZ > Fulvestrant).
G. Growth curve showing the effect of EPZ011989 pretreatment on the fulvestrant response of endocrine resistant ZR75-1 BoMs. Single agent and combination treatment groups are shown in blue and red, respectively. Multiple ANOVA was used for statistical analysis. BLI of metastatic burden at week 8 was shown as dot plot. (n= 6 and 8 mice for Fulvestrant and combination group, respectively.
H. PET-CT images showing 18F-FDG uptake in spontaneous BoMs (Hind limbs) following single agent (fulvestrant) or combination (EPZ > fulvestrant) treatment. (n=4 mice).
(A-H): P values were computed by two-tailed unpaired Student t-tests unless otherwise noted.
Short-term inhibition of EZH2 restores sensitivity of BMMs to endocrine therapies
Since EZH2 mediates the BME-induced endocrine resistance, we hypothesized that inhibition of EZH2 should reverse this resistance and synergize with endocrine therapies. Using the EZH2 inhibitor EZP011989, we confirmed our hypothesis in vitro using MCF-7 cells. The synergy is especially strong on MCF-7-Bo cells (Figure S7B). Next, a four-arm in vivo experiment was used to ask if combinatory treatment of EPZ011989 and fulvestrant at the microscopic metastasis stage (to mimic adjuvant therapy) could lead to decreased bone colonization. EPZ011989 and fulvestrant had little to modest effects when used as single agents. However, the combined treatment inhibited bone colonization and rendered 50% of mice tumor-free based on bioluminescence signals (Figure 7E), and microCT (Figure 7F, S7C, and S7D). This is a remarkable effect considering that EPZ011989 treatment only last for 3 weeks. EPZ011989 treatment also sensitized ZR75-1 bone lesions to fulvestrant treatment (Figure 7G). Finally, we tested the combinatory treatment on PDX-based spontaneous BoM models using PET imaging. Pretreatment of mice with EPZ011989 inhibited spontaneous metastasis to bone as shown by the reduced 18F-FDG update (Figure 7H, S7E).
DISCUSSION
Phenotypic plasticity has been recognized as a major driving force of normal development, tumor initiation, and tumor progression (Dravis et al., 2018; Gupta et al., 2019; Lambert et al., 2017). The heterogeneity of ER expression in ER+ tumors has long been noticed and may reflect such plasticity. In this study, our data demonstrate that even genetically identical ER+ cancer cells exhibit a substantial level of variation in ER expression. We uncovered that the OGs trigger an adaptive epigenomic change in ER+ metastatic seeds through both paracrine signaling and direct cell-cell contact, and leads to increased phenotypic plasticity and therapeutic resistance. These changes form a transient and reversible effect on cancer cells, including those that are genetically homogeneous, which distinguishes this process from the clonal selection investigated in the past (Bos et al., 2009; Kang et al., 2003; Minn et al., 2005). Indeed, our data suggest a coordinated action between epigenomic adaptation and genetic selection.
Our study identified a number of pathways that are altered in cancer cells by BME. Among these pathways, EZH2-mediated epigenomic reprogramming is a leading candidate for therapeutic intervention. It integrates multiple signals from OGs (e.g., FGF2 and PDGF-DD), and in turn, broadly impacts several downstream pathways related to cancer stemness and metastasis (e.g., WNT and Notch)(Gonzalez et al., 2014; Shi et al., 2007). Moreover, potent and selective EZH2 inhibitors are available and being clinically investigated in other diseases (Italiano et al., 2018), making it relatively easy for future clinical applications. Pharmacological inhibition of EZH2 promotes a global landscape change of histone marks (Huang et al., 2018). Tumors developed resistance to histone demethylase KDM5A/B had increased EZH2 expression (Hinohara et al., 2018). Hence, BME induction of EZH2 in BMMs may trigger an epigenomic disturbance beyond H3K27me3.
The loss of ER expression during BoM appears to be transient. In the advanced stage when the osteolytic vicious cycle starts (Boyce et al., 1999; Kozlow and Guise, 2005; Weilbaecher et al., 2011), ER expression may recover, perhaps driven by the stimulatory effects of other stromal cells recruited to macrometastases. The positive spatial correlation between RANK and ER supports this possibility. However, the BME-conferred endocrine resistance may persist in cells maintaining interactions with OGs suggesting that additional mechanisms may be involved (Eyre et al., 2019). Thus, overt BoMs may be heterogeneous, including a subset whose ER signaling remains repressed, which may be responsible for the rapid development of resistance observed during metastatic treatments (Johnston, 2010).
Although our experiments focused on BoMs, we are not ignoring the fact that other metastases also need to be prevented and cured. Recent genomic analyses revealed frequent metastasis-to-metastasis seeding (Brown et al., 2017; Ullah et al., 2018), suggesting that bone may not be the final destination for cancer cell dissemination. In fact, over two-thirds of bone-only metastases subsequently develop other metastases (Coleman, 2001). The enhancement of stem cell signaling in BME raises the possibility that bone may invigorate disseminated tumor cells for further metastases, and this possibility has recently gained support in our co-submitted manuscript (Zhang et al., 2021). Therefore, investigations on BoMs may have much broader impacts.
Limitations of Study
Our data did not address the question of whether the observed effects are specific to the bone microenvironment. It is possible that in non-bone organs similar mechanisms also lead to increased phenotypic plasticity via other cell types. Our study is also limited by the lack of naturally occurring murine ER+ models that recapitulate endocrine responses and development of resistance, as well as inability to stably tag all PDX models for linage tracing.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contac Dr. Xiang H.-F. Zhang at xiangz@bcm.edu
Material Availability
Plasmids and cell lines generated in the study will be made available upon request.
Data And Code Availability
Datasets were deposited in Gene Expression Omnibus (Edgar, 2002), with the following GEO accession numbers (GSE137245; GSE137270, GSE160566, GSE160582,and GSE161181). The GEO Reference Series connecting all datasets is GSE160583. Barcode analysis pipeline is accessible at: https://github.com/LiuzLab/ER_positive_breast_cancer-manuscirpt.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal studies:
All animal experiments were in compliance with Institutional Animal Care and Use Committee of Baylor College of Medicine. Nude mice [Athymic Nude-Foxn1nu] and SCID/Beige mice [C.B-17/IcrHsd-Prkdc scid Lyst bg-J] were purchased from Envigo. Osx1-GFP-cre/iDTR was generated from Osx1-GFP-cre [B6.Cg-Tg(Sp7-tTA,tetO-EGFP/cre)1Amc/J] and STP-iDTR mice [C57BL/6-Gt(ROSA)26Sortm1(HBEGF) Awai/J] originally obtained from Jackson Laboratory. For all in vivo experiment, 4- to 7-week-old female mice were used.
Patients derived xenografts (PDXs) and Primary cells
ER+ PDX models were kindly provided by Alana L. Welm (HCI011) and Matthew Ellis (WHIM9). All PDXs were maintained in SCID/Beige mice. PDX-HCI011 primary cell line was successfully generated from freshly harvested orthotopic tumors in RPMI medium supplemented with 15–20% FBS, 1X antibiotics Penicillin/Streptomycin and 1X antimycotic Amphotericin (Gibco#15240062). Media was changed every 48 hours.
Cell lines
Human estrogen receptor positive (ER+) breast cancer cell lines MCF7, T47D, MDA-MB-361 and ZR75-30, pre-osteoblast cells hFOB-1.19, mesenchymal stem cells MSC, pre-osteoclast U937, and the mouse pre-osteoblast MC3T3-E1 were purchased from American Type Culture Collection (ATCC). The Human ER+ breast cancer cell line ZR75-1 was kindly provided by Dr. Rachel Schiff. MCF7 single cell derived populations M7-SCP1, M7-SCP2, M7-SCP3, and M7-SCP4 were generated from single clones of parental MCF7 cells. The mouse cell line AT3 was a kind gift of S.I. Abrams at Roswell Park Cancer Institute.
METHOD DETAILS
Ovariectomy and aromatase inhibition:
Mouse ovaries were removed using the previously described surgical procedure (Ström et al., 2012). Briefly, mice were anesthetized with 2% isoflurane, and placed on a temperature-regulated heat pad. The dorsal area covering the lumbar vertebrae was shaved to display a 3×3 cm patch and disinfected. A 1 cm mid incision was performed on the skin and a 0.5 cm incision in the peritoneum allowed access to the ovary. Each ovary was cauterized, removed and peritoneum closed using an absorbable suture (Ethicon Vicryl #J497G). Skin closure was completed using EZ clips (Stoelting # 59027) and mice were monitored for recovery. Paracrine estrogen was blocked with oral administration of 0.5 mg/kg Letrozole as previously described (Brodie et al., 2005).
Intra-Iliac-Artery (IIA) and Mammary Fat Pad (MFP) injections:
Intra-iliac-artery (IIA) injection was performed as previously described (Yu et al., 2016). Briefly, breast cancer cells were trypsinized, pelleted, washed twice with PBS, collected in cold PBS and kept on ice. For established breast cancer cell lines such as MCF7 and ZR75, 5×105 cells were injected into the internal iliac artery to generate bone lesions. For PDX models, 2×105 cells were injected except when specified otherwise. For the syngeneic and aggressive model AT3, 1×105 cells were injected. Mammary fat pad (MFP) injections were performed as previously described (Zhang et al., 2019a). Cells were prepared as for IIA injection. For all xenograft models except spontaneous metastasis from PDX-HCI011 (figure 7), estrogen was provided through drinking water to reduce deleterious side effects. Based on previous studies, 8 μg/ml of 17-β-estradiol was added to mouse water bottles and replaced twice a week (Levin-Allerhand et al., 2003; Welsch et al., 1981). In experiments involving dual IIA and MFP injections as in figure 1D, the same cell number was injected to bone and mammary gland, respectively.
Spontaneous Metastasis from PDX models
To evaluate spontaneous metastases from PDX-HCI011, Foxn1nu mice were orthotopically transplanted with 5×105 cells from freshly dissociated and purified tumor (Mouse Cell Depletion Kit; miltenyibiotec #130-104-694). Estrogen pellets were subcutaneously implanted to increase the tumor take rate. When tumors reached 1×1 cm, a survival surgery was performed to remove both the primary tumors and the remaining estrogen pellets. A three-week treatment with vehicle or EPZ011989 (125 mg/kg; oral gavage; twice daily) was started two weeks after orthotopic tumor removal. Then, 250 mg/kg of fulvestrant was administered weekly via subcutaneous injection to both EPZ011989 and vehicle pretreated groups for 3 consecutive weeks. Mice were monitored for 3 months before 18F-Fluorodeoxyglucose positron emission tomography (PET) and computed tomography (CT) scans were performed.
Drug treatments
In vivo: The selective estrogen receptor degrader (SERD) fulvestrant (Selleckchem #S1191) was solubilized in 5% DMSO and 95% corn oil and administered subcutaneously at 250 mg/kg per mouse, once a week for 2 consecutive weeks. Letrozole was purchased from Selleckchem (#S1235), diluted in 0.5% sodium carboxymethyl cellulose (NaCMC), and administered at 0.5 mg/kg via oral gavage. EPZ-110989 was kindly provided by Epizyme and stock solutions were prepared following the company’s recommendations, using 0.5% sodium carboxymethyl cellulose (NaCMC) and 0.1% Tween-80 as vehicle. A dosage of 125 mg/kg of EPZ-011989 or vehicle was administered twice daily by oral gavage for 3 weeks.
In vitro: Tamoxifen and fulvestrant were used in vitro at a concentration of 100 nM and 20 nM per well, respectively. 1–4 uM EPZ011989 was used for coculture experiments. For calcium signaling studies, we used 1 uM of GAP19 (cat#5353) to inhibit gap junction (CX43), and 1 uM of FK506/Tacrolimus (cat#S5003) to block calcineurin. Similarly, 2.5 μM BGJ398 (selleckchem #S2183) and 10 μM Sunitinib (Selleckchem #S7781) were used to inhibit FGF receptors and PDGF receptor B, respectively.
Immunohistochemistry, Immunofluorescence and immunoblotting:
IHC/IF: Tissues were processed with the help of the Breast Center Pathology Core at Baylor College of Medicine. Immunohistochemistry staining and immunoblotting were performed using antibodies against human ERα (D8H8 and 6F11), Progesterone Receptor (H-190), EZH2 (D2C9), Cytokeratin 8 (TROMA-I), α-Smooth Muscle Actin (D4K9N), Vimentin (D21H3), RANK (64C1385), ALP (ab75635), FGF2 (C2, #sc-74412), CD31 (AF3628), PDGFRβ (28E1), H3K27me3 (C36B11). Western blot: Protein extraction was performed using RIPA buffer as previously described (Rajapaksa et al., 2015). Protein electrophoresis and transfer were performed using the XCell SureLock and the iblot system (Invitrogen), respectively. Immunoblotting was performed using antibodies against Estrogen Receptor α (D8H8), Cytokeratin 19 (BA-17), Red Fluorescent Protein (Rockland-Fisher), β-actin (8H10D10), H3K27me3 (Millipore Cat# 07–449), H3K36me3 (D5A7), H3K9me3 (D4W1U), H4K20me3 (D84D2), H3K4me3 (C42D8), and Histone 3 (D1H2). Images were captured using the Odyssey system (Li-cor).
Image acquisition and quantification:
Images were acquired with the Leica TCS SP5 or the Zeiss LSM 880 with Airyscan FAST Confocal Microscope. A 40x oil objective lens (Immersion oil refractive index n=1.51) was used to capture images for immunofluorescence quantifications except when specified otherwise. We compared only sets of images that were captured under the same microscopic setting. To further reduce technical biases and batch effects, compared samples were processed and stained in parallel. All images were quantified using ImageJ 1.52i. All statistical analyses represent a two-tailed unpaired Student’s t-test except when specified otherwise. Whenever applicable (Figure 1E, 1F, 3A–C, 3E, 3G, 6E, 6K–L, 6P, and 7D), single cells from at least 3 biological replicates were used for statistical analysis. Figure 2J, and 6D are representative of at least 3 independent lesions.
Tumor classification:
Based on size: metastases were classified based on cell number. On average, small lesions were defined as lesions with fewer than 100 cells. The average maximum cell number common among all models was ~150 for macrometastases (large lesions) and ~90 for micrometastases (small lesions) which gives a fold change superior to > 1.5 between the two experimental stages of metastasis. The tumor size in different models (HCI011, WHIM9, MCF7, ZR75-1 and M7-SCP2) of bone metastasis was variable due to differences in tumor aggressiveness. Accordingly, we used a cutoff of median +/− 0.5×S.D. as a more consistent variable to segregate tumors into micrometastasis and macrometastasis. Based on location: cancer cells were classified as proximal if they were directly interacting with the bone matrix or separated from it by less than two cells (≤2 cell distance), or classified as distal if separated from the bone matrix by 3 cells or more (≥3 cell distance).
Recombinant protein and calcium treatments
All experiments involving protein recombinants were performed in low serum media (2% serum). Protein recombinants for FGF2 (#130093838), PDGF-BB (100-14B), PDGF-CC (100-00CC), PDGF-DD (1159-SB-025) were diluted in PBS and used at a concentration of 20 or 100ng/ml. To evaluate endocrine resistance after treatment with FGF2 and PDGF recombinants, cells were starved for 48 hours in 2% charcoal stripped media before a 20 nM fulvestrant treatment. All experiments involving cells growth were performed in 3D culture and bioluminescence was assessed 72 hours post treatment. For western blot, short term treatments were performed for 24 hours and long-term ones for up to 72 hours. Western blot experiments were performed in 2D in most cases, except when specified otherwise. Calcium treatment: 2×106 cells (MCF7 or ZR75-1) were cultured in regular medium for 24 hours. Regular medium was replaced with calcium-free minimum essential medium (S-MEM) and treated with vehicle or 2 mM Calcium chloride for 24 hours. After collecting all cells, we extracted protein lysates to assess the effect of calcium on ER expression by western blot.
Live imaging
For in vivo experiments, all cells were pre-labelled with Luciferase fused to GFP or RFP as previously described (Wang et al., 2015). 5×105 breast cancer cells were injected in bone or mammary fat pad, except when specified otherwise. Tumor growth was monitored using the IVIS Lumina II system. Briefly, mice were anesthetized in an isoflurane chamber (2%) and 100 μl of D-Luciferin was administered through retro-orbital injection to each mouse before image acquisition. For in vitro experiments, 10,000 cells were plated in low attachment 96 well plates to assess cell growth at 72 or 96 hours post-treatment. For conditions demanding estrogen depleted media and starvation, 20,000 cells were cultured per well. Images were acquired after adding 1X concentration of D-luciferin containing media to each well.
Reverse phase protein arrays (RPPA)
MCF7 and M7-SCP2 cell lines were injected to bone using intra-iliac artery injection. After 5 weeks of metastasis formation, bones were collected in aseptic conditions and dissociated to generate bone-entrained MCF7-Bo and M7-SCP2-Bo cell lines. Cells were cultured in DMEM 10% FBS supplemented with 1X antibiotics (penicillin, streptomycin) and antimycotics (amphotericin). Bone-educated cells were FACS-sorted and maintained in 2D culture. Approximately 5×106 non-entrained and bone-entrained MCF7 and M7-SCP2 cells were harvested in freshly prepared RPPA lysis buffer containing protease and phosphatase inhibitors. Protein lysate was cleared twice via centrifugation (14,000 g for 15 min at 4°C). A BCA assay was adopted for protein quantification (ThermoFisher #23225). All samples were diluted in RPPA solution and SDS to a final concentration of 0.5 mg/ml and heated for 8 min at 100°C for protein denaturation. RPPA was performed as previously described (Welte et al., 2016). In brief, samples and control lysates were spotted onto nitrocellulose-coated slides (Grace Bio-labs; array format of 960 lysates/slide or 2880 spots/slide). The automated slide stainer Autolink 48 (Dako) was used to probe 236 antibodies (against total and phospho-proteins) on slides. Control slides were incubated with antibody diluent. A biotinylated secondary antibody was probed by streptavidin-conjugated IRDye680 fluorophore (LI-COR Biosciences) and total protein was detected with Sypro Ruby Protein Blot Stain according to the manufacturer’s instructions (Molecular Probes). All slides were scanned on a GenePix 4400 AL scanner and images were analyzed with GenePix Pro 7.0 (Molecular Devices). Samples were normalized as previously described (Chang et al., 2015). After quality control 233 antibodies remained and were used for subsequent data analysis.
PET/CT Imaging and Analysis
Radiopharmaceuticals and Small-Animal PET-CT: Fluorine-18 labeled fluorodeoxyglucose (18F-FDG), fluoroestradiol (18F-FES), and sodium fluoride (18F-NaF) was purchased from (Cyclotope, Houston, TX). All CT and PET images were acquired using an Inveon scanner (Siemens AG, Knoxville, TN). The mice were injected with 9.25 MBq (250 μCi) of FES and 11.1 MBq (300 μCi) of either 18F-FDG or 18F-NaF radiotracers at any given time. To identify skeletal metastases or measure tumor metabolic activities, 18F-NaF or 18F-FDG were injected intraperitoneally, and to measure estrogen activity 18F-FES was injected intravenously via tail vein. Before 18F-FDG administration, the mice were fasted for approximately 12 hours. PET and CT were performed one hour after injection of radioisotopes. During imaging, a respiratory pad was placed under the abdomen of the animal to monitor respiration (Biovet, Newark, NJ). Mice were anesthetized with isoflurane gas (1–3%) mixed with oxygen at a flow rate of 0.5–1 L/minute, and adjusted accordingly during imaging to maintain normal breathing rates. A CT scan was acquired with the following specifications: 220 acquired projections except for the 18F-NaF imaging which was 360 full scan. Each projection was 290 ms with x-ray tube voltage and current set at 60 kVp and 500 μA, respectively. A 30-minute PET scan was immediately acquired afterward. The PET scans were reconstructed using OSEM3D reconstruction method and registered to the CT scan for attenuation correction. PET Image Analysis: The PET images were quantified using Inveon Research Workspace IRW (IRW, Siemens AG, Knoxville, TN). Using the reconstructed PET scan, bone (hind limbs) and mammary fat pads were manually selected to form regions of interest (ROI) on the PET-CT images. The data was represented as standardized uptake value (SUV) normalized to body weight. For PDX spontaneous metastasis to bone, a 90% SUVmax thresholding was applied to ROI.
microCT imaging and analysis
A microPET/CT scanner (Siemens Medical Solutions USA, Inc; Malvern, PA; USA) was used to acquire microtomography images. Paired murine hindlimb specimens with bone metastases were imaged with a spatial resolution of 20 microns. Images were converted to Dicom format using Inveon software (version 4.2; Siemens) and bone analyses (volume and mineral density) and three-dimensional reconstruction/visualization performed using Skyscan CTAn and CTVox software packages by Bruker (version 1.19 and 2.3.2; Kontich; Belgium).
Mammosphere and coculture assays
5×105 cells were plated in low attachment 6 well plates (Greiner) using regular (10% FBS) of serum free DMEM/F12 media supplemented with 2–3% dextran-coated Charcoal stripped. We used a 1/1 ratio for cocultures between cancer cells and stromal cells except when specified otherwise. Cells were collected after 24, 48 or 72 hours of culture for downstream analyses. For immunofluorescence, cells were fixed with 2% PFA for 24 hours, washed 3 times with PBS, embedded in paraffin, and sectioned for imaging.
Quantitative real-time PCR
Total RNA was extracted using the Direct-zol Zymo according to the manufacturer protocol. Copy DNA was synthesized using the iScript cDNA Synthesis Kit (Biorad). All primers are indicated in Table S1. Real-time PCR was performed on the CFX connect system (Biorad) using PowerUP SYBR Green master mix (ThermoFisher, #A25780) for amplification.
Bulk ATAC sequencing (ATAC-seq) and analysis:
ATAC-seq Assay: ATAC-seq was performed as previously described (Buenrostro et al., 2015). Here, we collected 50,000 cells from parental (M7-SCP2) and bone-entrained (M7-SCP2-Bo) cells at different passages (#2, #4, #6 and #12). DNA transposition was performed on freshly collected cells using the Nextera Tn5 Transposase from Illumina. Purified DNA was stored at −80 °C for each passage before library preparation. All experiments were performed in parallel in both parental and bone-entrained cells.
ATAC-seq Analysis:
Peak Generation Pipeline: Analysis was conducted with a modified version of the Encode Consortium’s ATAC-Seq Pipeline. Adapters were trimmed from input FASTQ files using cutadapt. Alignment was performed using Bowtie2. Samtools and Picard were used for post alignment filtering to remove duplicate, unmapped, and mitochondrial reads. Pseudo-replicates were generated for both individual replicates and pooled replicates by randomly dividing the input into two equal length files. The MACS2 peak caller was utilized to generate peaksets for all true replicates and pseudo-replicates. Peaks with P value < 10−5 were retained for further analysis. Peaks in the Encode DCC consensus blacklist regions were also removed. Then, pairwise comparisons between each pair of biological replicates, the two pseudo-replicates generated from the pooled replicate file, and pseudo-replicates generated from each replicate were conducted. The Encode IDR (irreproducible discovery rate) was used to rank the consistency of each peak region; only peaks with IDR<0.05 were retained. Further analysis was conducted using the overlap IDR thresholded peaks between pooled pseudo-replicates.
Post-peak analysis: A consensus peakset was then generated by merging all the peak regions for the samples of interest using BedTools (Quinlan and Hall, 2010). Promoter regions were retrieved from the UCSC genome browser and were defined as 5000 bases up and 1000 bases down from the TSS. Heatmaps and profile plots for the peak and promoter regions were generated using the DeepTools (Ramírez et al., 2014) utility. The IGV (Thorvaldsdóttir et al., 2013) utility was utilized to generate visualizations for specific gene regions. Further analysis utilized the DiffBind (Stark and Brown, 2011) suite to identify differentially accessible regions (DAR). Both promoter and consensus peak regions were used as peaksets for occupancy and differential binding affinity analysis. DiffBind utilizes DeSeq2 to identify and calculate log fold change and P values for DAR. Contrasts were established between the MCF7 parental and MCF7-Bo samples and the M7-SCP2 and M7-SCP2-Bo samples while controlling for passage number as a confounding factor. DiffBind was also used to generate the PCA and Volcano Plots. DAR generated from Diffbind were then labelled with genes based on the nearest TSS using HOMER (Heinz et al., 2010). HOMER’s findMotifGenome module was also utilized to conduct motif analysis. The input file consisted of regions with lower binding in M7-SCP2-Bo compared to M7-SCP2 with an FDR cutoff of <0.05. A region size of 200 and the masked genome setting were used. All other settings used the default HOMER options.
Whole exome sequencing (WES)
WES library was prepared using the Nextera DNA exome kit (Illumina # 20020616) per manufacturer’s instructions and sequenced on a Novaseq 6000 platform at ~100x depth (paired end 100bp, 50 million reads per sample). FastQC and mulitQc were used for quality control. After adaptor removal using cutadapt and trim galore, reads were aligned to reference genome (hg19) using BWA-MEM. BAM files were filtered, and duplicate reads removed using samtools (Li et al., 2009) and Picard. A normal whole exome sequencing sample was downloaded for 1000-Genomes (ERR031938), aligned to the reference genome (hg19), and downsampled (http://broadinstitute.github.io/picard/). We generated pileups from BAM files using samtools mpileup. Varscan 2 was used to call copy numbers and somatic mutations (Koboldt et al., 2012, 2013). Only variant calls with P value < 10−2 were used for downstream analyses. Data processing was performed on public server (Afgan et al., 2018). To evaluate the heterogeneity and subclonality of all M7-SCP cells, we used the Expands package (http://cran.r-project.org/web/packages/expands). For each sample, a Z-score analysis was performed using the matrix of predicted subpopulation. Dominant subpopulations with a positive Z-score were used to assess tumor heterogeneity represented by pie charts. Tumor purity was estimated based on cellular frequencies of the largest subpopulation as previously described (Andor et al., 2014).
Histone Protein post-Translational Modification (PTM) analysis
Un-entrained (MCF7 and M7-SCP2) cells were either directly purified from bone (Passage 0: P0) or purified from bone and cultured in vitro for 10 passages (P10). Each sample was washed 3 times with PBS and snap frozen pellets were shipped to Active Motif for PTM quantitation (www.activemotif.com). Briefly, a pilot study was performed to determine the optimal histone extraction method for the samples. Using the additional 6 samples, three lysis methods were evaluated: 1) A one-step method wherein histones are acid extracted directly from the frozen cell pellet, 2) A two-step method wherein a sucrose-based hypotonic buffer is used to lyse the cells and histones are acid extracted from isolated nuclei, or 3) A two-step method wherein an IGEPAL-containing hypotonic buffer is used to lyse the cells and histones are acid extracted from isolated nuclei. Acid extraction was performed for two hours at 4 °C, cellular debris was pelleted, and lysate aliquots were frozen in a methanol-dry ice bath and stored at −80 °C until testing. Histone yields for the three methods were evaluated using the Histone H3 Total bead and a two-fold five-point dilution series of the samples. The IGEPAL-containing hypotonic lysis buffer method gave the highest yield and was selected for use with the experimental samples. Lysate Preparation: Histones were extracted from the experimental samples using the method described above. Cellular debris was pelleted, and lysate aliquots were frozen in a methanol-dry ice bath and stored at −80°C until testing. Next, relative histone H3 concentrations in the samples were determined using the H3 Total bead. Multiplex assays were performed with the beads of interest using sample volumes normalized for histone H3 concentration. Assay protocol: 1) Beads were added to wells in 25 μl Assay Buffer supplemented with Inhibitor Cocktails (ABIC) for proteases, phosphatases and HDACs. 2) Samples as a four-point 1.4 dilution series were added to wells in 25 μl ABIC in duplicate and incubated for 1 hour at room temperature. 3) Three 100 μl washes with 1X Wash Buffer (PBS containing 0.05% Tween-20) were performed using plate magnet to retain beads. 4) 50 μl biotinylated Histone H3 antibody diluted 1:500 in Assay Buffer was added for the high abundance PTM multiplex assay for 1 hour with agitation. 50 μl biotinylated Histone H3 antibody diluted 1:250 in Assay Buffer was added for the low abundance PTM multiplex assay for 1 hour with agitation. 5) Washes were performed as above. 6) 50 μl of SAPE diluted 1:100 in Assay Buffer was added to each well and incubated for 30 min with agitation. 7) Beads were collected on a plate magnet and the SAPE solution discarded. 8) The assay plate was removed from the plate magnet and beads resuspended in 100 μl 1X Wash Buffer and read on the Luminex LX100 Instrument. Data analysis: Histone H3 Total matched data sets were used to determine PTM/H3 ratios, the PTM percent change relative to each other and Student t-test P values.
Translating Ribosome Affinity Purification (TRAP) sequencing (TRAP-seq)
TRAP assay was adopted from previous studies (Heiman et al., 2014). Here, we performed all experiments in 3D. We stably labeled MCF7 cells with GFP-RPL10a plasmids kindly provided by Dr. William Pu from Harvard. Cells were sorted using FACSAria II to enrich for GFP-positive cells. GFP-RPL10a-expressing MCF7 cells were maintained in 2% charcoal stripped medium for 48 hours from which 1 million cells were seeded in 100 mm low attachment plates (Corning, cat #05-539-101) either alone or in coculture with human mesenchymal stem cells (MSCs). These cultures were incubated overnight and treated with 10 nM 17β-estradiol, fulvestrant or 100 nM Tamoxifen for 24h hours. Cells were collected for TRAP sequencing. Library was prepared using illumina Nextera XT Kit and paired-end sequencing was performed on a Nextseq 550 System. All sequencing experiments were performed at the Genomic and RNA Profiling core (GARP) at Baylor College of Medicine.
Tracing Metastasis Expansion in Bone using CRISPR-Cas9/hgRNA System
CRISPR-Cas9 barcoding: The hgRNA A21 vector was previously characterized and published (Kalhor et al., 2017). MCF7 cells were stably infected with Lenti-iCas9-neo (Addgene #85400) and hgRNA-A21 (Addgene #100570) using Neomycin/puromycin antibiotic selection before intra-iliac artery injection (IIA). To activate cas9 expression, mice were administered 2 mg/kg of doxycycline via intraperitoneal injection at day 1 post-IIA. Doxycycline treatment was repeated once a week for 3 more weeks (see experimental design Figure 1A).
Laser capture micro-dissection (LCM) and barcode sequencing: Tumor bearing limbs (femur and tibia) were isolated, embedded in Tissue-Tek O.C.T., snap-frozen in liquid nitrogen, and stored at −80°C until sectioning. 10 μm cryosections of each bone were generated using Leica CM3050S cryotome equipped with a low-profile microtome blade. The chamber temperature was set at −26°C. Sectioning was facilitated with the CryoJane Tape Transfer System and then placed on the PET membrane slides (MMI, Prod. No. 50103). Sections were fixed in ethanol and stained with DAPI and ArcturusTM HistoGeneTM solution (Applied Biosystems) according to the instruction manual. Microdissection was performed using the Leica LMD7000 instrument. DNA was purified from each LCM-derived lesion using the Quick-DNA/RNA Microprep Plus Kit from Zymo (D7005). The evolving barcode library was generated as previously described (Kalhor et al., 2017). A paired-end sequencing was performed using the Hiseq 4000 system.
Bioinformatic Analysis of the evolving barcode system: The R1 sequences were aligned and annotated using the TraceQC package (https://github.com/LiuzLab/TraceQC). First, the CRISPR barcode sequences were aligned to the hgRNA_A21 reference construct using the following score system: match +2, mismatch −2, gap opening −6, gap extension −0.1. After annotating the aligned sequences, the adapters were trimmed off and sequences with low alignment scores (<200) were filtered out. Sequences with less than 10 count were subsequently filtered out. TraceQC extracted mutation events from the sequence into 4 attributes: 1) the mutation type (insertion, deletion or point mutation), 2) the starting position of mutation, 3) the length of mutation, and 4) the altered sequence. We combined the mutation events for all the samples into a mutation count matrix and normalized samples using the read count per million (RPM) approach. The hierarchical clustering based on mutations revealed 3 major modules. Within each module, the mutation count matrix was binarized into whether each mutation event exists in the samples or not: TRUE (mutation present) or FALSE (mutation absent). Then, we used maximum parsimony to establish the lineage relations within each module. To build the cell lineage network, we performed graphic LASSO using the Huge package. First, the mutation count matrix was Gaussianized using non-paranormal transformation provided by Huge package. Then, the graphic LASSO was applied to the Gaussianized mutation count matrix. We selected lambda = 0.52 to make the graph have the maximum sparsity while remaining fully connected. Next, we applied a random walk-based community detection algorithm to detect the 3 modules in the graph. The algorithm is provided by the iGraph package. Detailed analysis pipeline is accessible at: https://github.com/LiuzLab/ER_positive_breast_cancer-manuscirpt.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis was conducted in GraphPad Prism (v8.0.1) and R (version 3.4 R). Specific statistical approaches are indicated for each figure in the figure legend. For PET imaging, Mann Whitney U-test was used for statistical analysis. Pearson correlation was used for correlation studies.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Estrogen Receptor alpha (D8H8) | Cell Signaling Technology | 8644S |
| EZH2 (D2C9) | Cell Signaling Technology | 5246S |
| Red Fluorescent Protein | Rockland-Fisher | 600-401-379 |
| Beta-actin (8H10D10) | Cell Signaling Technology | 3700S |
| Cytokeratin 19 (BA-17) | Genetex | GTX27755 |
| FGF2 (C-2) | Santa Cruz | sc-74412 |
| Anti-FGF-2/basic FGF (neutralizing), clone bFM-1 | EMD Millipore | Cat# 05–117 |
| Human PDGF R beta Antibody | R&D Systems-Fisher | Cat# AF385-SP |
| Fgf Receptor 1 (D8E4) XP | Cell Signaling Technology | 9740S |
| Human Vimentin (VIM-V9-L-CE) | Leica | VIM-V9-L-CE |
| Vimentin (D21H3) | Cell Signaling Technology | 5741S |
| Gata 3 (D13C9) | Cell Signaling Technology | 5852S |
| PDGF Receptor β (28E1) Rabbit mAb | Cell Signaling Technology | 3169S |
| Estrogen Receptor alpha Monoclonal (6F11) | Thermo Fisher Scientific | MA1–80216 |
| GFP (Htz-GFP19C8) | Sloan-kettering | N/A |
| GFP (Htz- GFP19F7) | Sloan-kettering | N/A |
| Cytokeratin 8 (k8) | DSHB | TROMA-I-s |
| Donkey Anti-Rat IgG 594 | Jackson ImmunoResearch | Cat# 712-546-153 |
| Donkey Anti-Mouse IgG 647 | Jackson ImmunoResearch | Cat# 715-605-151 |
| Goat Anti-Rabbit IgG 488 | Cat# 111-585-144 | |
| Tri-methyl-Histone H3 (Lys27) Antibody | EMD Millipore | Cat# 07–449 |
| Tri-methyl-Histone H3 (Lys27) Antibody | Cell Signaling Technology | Cat# C36B11 |
| Donkey Anti-Rabbit IgG 488 | Jackson ImmunoResearch | Cat# 711-545-152 |
| PR Antibody (H-190) | Santa Cruz Biotechnology | Sc-7208 |
| Total Histone 3 | Biolegend | Cat# MMS-5230 |
| Acetyl-Histone H3 (Lys27) (D5E4) XP | Cell Signaling Technology | 8173S |
| Alkaline Phosphatase | Abcam | Cat# ab108337 |
| Tri-Methyl-Histone H3 (Lys4) (C42D8) | Cell Signaling Technology | 9751S |
| Tri-Methyl-Histone H4 (Lys20) (D84D2) | Cell Signaling Technology | 5737S |
| Tri-Methyl-Histone H3 (Lys36) (D5A7) XP | Cell Signaling Technology | 4909S |
| Tri-Methyl-Histone H3 (Lys9) (D4W1U) | Cell Signaling Technology | 13969S |
| Bacterial and Virus Strains | ||
| XL1-Blue | Agilent | Cat# 200236 |
| Stbl3 | Thermo Fisher Scientific | C737303 |
| Biological Samples | ||
| PDX HCI011 | Gift from Dr. Alana L. Welm’s Laboratory, University of Utah | N/A |
| PDX WHIM9 | Gift from Dr. Matthew Ellis’s Laboratory, Baylor College of Medicine | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human FGF-2 | Miltenyi Biotec | Cat# 130093838 |
| Human PDGF-BB | PeproTech-Fisher | Cat# 100-14B |
| Human PDGF-CC | PeproTech-Fisher | Cat# 100-00CC |
| Human PDGF-DD | R&D Systems | Cat# 1159-SB-025 |
| β-Estradiol | Sigma-Aldrich | Cat# E2758 |
| Streptavidin MyOne T1 Dynabeads | ||
| Biotinylated protein L | ||
| Cycloheximide, ultrapure, 98% | Alfa Aesar™-Fisher | Cat# J6690103 |
| 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC) | 1,2-dihexanoyl-sn-glycero-3-phosphocholine | Cat# 850305P-500 mg |
| Fulvestrant | Selleck Chemicals | Cat# S1191 |
| 4-Hydroxytamoxifen (4-OHT) | Sigma-Aldrich | Cat# H6278–10MG |
| EPZ-011989-9 | Gift from Epizyme | N/A |
| Anti-Fibroblast MicroBeads, human | Miltenyi Biotec | Cat# 130-050-601 |
| BGJ398 (NVP-BGJ398) | Selleck Chemicals | Cat# S2183 |
| FK506 (Tacrolimis) | ApexBio Technology LLC | Cat# B2143 |
| Carbenoxolone disodium (CBX) | Tocris | Cat# 3096 |
| Sunitinib | Selleckchem-Fisher | S7781 |
| D-Luciferase | Goldbio | Cat# LUCNA-1G |
| Critical Commercial Assays | ||
| Nextera XT DNA Library Prep Kit | Illumina | Cat# FC-131–1096 |
| Nextseq 500/500 high output v2 Kit | Illumina | Cat# FC-404–2002 |
| CellTrace™ Far Red Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34572 |
| iScript™ Reverse Transcription Supermix | Bio-rad | Cat# 1708841 |
| Lipofectamine RNAiMAX Transfection Reagent | Thermo Fisher Scientific | Cat#13778030 |
| iBlot® Transfer Stack, nitrocellulose, mini | Thermo Fisher Scientific | Cat# IB301032 |
| NuPAGE™ Novex™ 4–12% Bis-Tris Protein Gels, 1.5 mm, 15-wel | Thermo Fisher Scientific | Cat# NP0336BOX |
| Deposited Data | ||
| SuperSeries for all sequencing datasets | This Paper | GSE160583 |
| MCF7-MSC TRAP-sequencing dataset | This Paper | GSE137270 |
| Human osteoblast dataset | This Paper | GSE137245 |
| MCF7 bone metastasis evolving barcode dataset | This Paper | GSE160566 |
| Bulk-ATAC-seq SCP2 dataset | This Paper | GSE160582 |
| Whole exome sequencing (WES) | This Paper | GSE161181 |
| Experimental Models: Cell Lines | ||
| Human breast cancer MCF7 | ATCC | Cat# HTB-22, RRID:CVCL_0031 |
| Human breast cancer T47D | ATCC | Cat# HTB-133, RRID:CVCL_0553 |
| Human breast cancer ZR75-1 | Gift from Dr. Rachel Schiff’s Lab, Baylor College of Medicine | Cat# CRL-1500, RRID:CVCL_0588 |
| Human breast cancer ZR75-30 | ATCC | Cat# CRL-1504, RRID:CVCL_1661 |
| Human breast cancer MDA-MB-361 | ATCC | Cat# HTB-27, RRID:CVCL_0620 |
| Human pre-osteoblast FOB-1.19 | ATCC | Cat# CRL-11372, RRID:CVCL_3708 |
| Human mesenchymal stem cell MSC | Gift from Dr. Max Wicha’s Laboratory, University of Michigan | N/A |
| Human Bone Marrow | Lonza | Cat# 2M-125C |
| Human pre-osteoclast U937 | ATCC | Cat# CRL-1593, RRID:CVCL_0007 |
| Mouse pre-osteoblast MC3T3-E1 | ATCC | Cat# CRL-2953, RRID:CVCL_0409 |
| Human bone marrow fibroblast Hs5 | ATCC | Cat# CRL-11882, RRID:CVCL_3720 |
| Human primary cells HCI011 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Athymic Nude mice [Athymic Nude-Foxn1 nu] | Envigo | N/A |
| Scid/Beige mice [C.B-17/IcrHsd-Prkdc scid Lyst bg-J] | Envigo | N/A |
| Osx1-GFP-cre/iDTR | This paper | N/A |
| Oligonucleotides | ||
| Silencer™ Negative Control | Thermo Fisher Scientific | AM4611 |
| EZH2 siRNA (human) - ID: s4918 | Thermo Fisher Scientific | 4392420 |
| EZH2 siRNA (human)- ID: s4916 | Thermo Fisher Scientific | 4392420 |
| Oligos for qPCR | This paper (Supplementary table 1) | N/A |
| Recombinant DNA | ||
| 3X ERE TATA luciferase | A gift from Dr. Donald McDonnell to Addgene, Duke University | Plasmid #11354, RRID_Addgene_11354 |
| GFP-RPL10 | Dr. William T. Pu’ Laboratory, Harvard Stem Cell Institute | NA |
| pMD2.G | A gift from Didier Trono to Addgene, EPFL | Plasmid# 12259, RRID_Addgene_12259 |
| psPAX2 | A gift from Didier Trono to Addgene, EPFL | plasmid #12260, RRID_Addgene_12260 |
| Pwpt-Fluc/GFP | Wang et al. 2015 | N/A |
| Pwpt-Fluc/RFP | Wang et al. 2015 | N/A |
| ERE-pWPXL/GFP | This paper (modified from Plasmid # 12257; gift from Didier Trono to addgene) EPFL | N/A |
| hgRNA-A21_pLKO-Puro | A gift from George Church, Harvard (Addgene plasmid # 10057) | Plasmid # 100570. RRID:Addgene 100570 |
| Lenti-iCas9-neo | A gift from Qin Yan, Yale (Addgene plasmid # 85400) | Plasmid # 85400 RRID:Addgene_85400 |
| Software and Algorithms | ||
| Expanding Ploidy and Allele-Frequency on Nested Subpopulations | Stanford University | https://CRAN.R-project.org/package=expands |
| Evolving barcode analysis | This Paper | https://github.com/LiuzLab/ER_positive_breast_cancer-manuscirpt |
| TraceQC | Baylor College of Medicine | https://github.com/LiuzLab/TraceQC/ |
| Rstudio | Rstudio | http://www.rstudio.com/ |
| Galaxy project | Penn State, Johns Hopkins University, Oregon Health & Science University, and the Galaxy Community | http://galaxyproject.org/ |
| ImageJ | National Institute of Health | https://fiji.sc/ |
| Flow Jo V10.0 | FlowJo, LLC | https://www.flowjo.com |
| LAS AF Lite | Leica Microsystems | RRID:SCR_013673 |
| Living Image 4.5.2 | PerkinElmer | https://www.perkinelmer.com/lab-products-and-services/resources/in-vivo-imaging-software-downloads.html |
| Inveon Research Workplace (IRW) | SIEMENS | https://www.siemens-healthineers.com/en-us/molecular-imaging/preclinical-imaging/inveon-workplace/ |
| Graphpad Prism | GraphPad Software, Inc. | https://www.graphpad.com/scientific-software/prism/ |
| Combenefit | Cancer Research UK Cambridge Institute | http://sourceforge.net/projects/combenefit/ |
Highlights:
ER+ breast cancer cells transiently reduce ER expression in bone micrometastases
Interaction with osteogenic cells causes reversible ER loss and endocrine resistance
Epigenomic mechanisms lead to ER reduction, cancer stemness and cellular plasticity
EZH2 mediates the impact of bone and is a target to alleviate endocrine resistance
Acknowledgements
We thank the Breast Center Pathology Core, the Antibody-based Proteomics Core (CPRIT Core Facility Award (RP170005) and P30 Cancer Center Support Grant (NCI-CA125123)), the Small Animal Imaging Facility (SAIF) at Texas Children Hospital, and the Genomic and RNA Profiling core (GARP) for their technical support. We also thank Epizyme (Cambridge, MA) for providing the EZH2 inhibitor EPZ-011989. We express our gratitude to Drs. Rosen Jeffrey, Michael Lewis, and Xi Chen, for advice and technical support. This project was supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the CPRIT Core Facility Support Award (CPRIT-RP180672), the NIH (CA125123 and RR024574) and the assistance of Joel M. Sederstrom. X.H.-F.Z. is supported by US Department of Defense DAMD W81XWH-16-1-0073 (Era of Hope Scholarship), NCI CA183878, NCI CA251950, DAMD W81XWH-20-1-0375, Breast Cancer Research Foundation, and McNair Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests
REFERENCES:
- Afgan E, Baker D, Batut B, Van Den Beek M, Bouvier D, Ech M, Chilton J, Clements D, Coraor N, Grüning BA, et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andor N, Harness JV, Müller S, Mewes HW, and Petritsch C (2014). Expands: Expanding ploidy and allele frequency on nested subpopulations. Bioinformatics, 30, 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade K, Fornetti J, Zhao L, Miller SC, Randall RL, Anderson N, Waltz SE, McHale M, and Welm AL (2017). RON kinase: A target for treatment of cancer-induced bone destruction and osteoporosis. Sci. Transl. Med 9, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PD, Zhang XH-F, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, et al. (2009). Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce BF, Yoneda T, and Guise TA (1999). Factors regulating the growth of metastatic cancer in bone. Endocr. Relat. Cancer 6, 333–347. [DOI] [PubMed] [Google Scholar]
- Brodie A, Jelovac D, Macedo L, Sabnis G, Tilghman S, Goloubeva O, Osborne K, Santen R, Johnston S, Yee D, et al. (2005). Therapeutic observations in MCF-7 aromatase xenografts. In Clinical Cancer Research, 11, 884s–888s [PubMed] [Google Scholar]
- Brown D, Smeets D, Székely B, Larsimont D, Marcell Szász A, Adnet P-Y, Rothé F, Rouas G, Nagy ZI, Faragó Z, et al. (2017). Phylogenetic analysis of metastatic progression in breast cancer using somatic mutations and copy number aberrations. Nat. Commun 8, 14944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 5, 21.29.1–21.29.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, Raimondi A, Klaus CR, Rioux N, Yokoi A, et al. (2015). EPZ011989, A potent, orally-available EZH2 inhibitor with robust in vivo activity. ACS Med. Chem. Lett 6, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Zhang M, Rajapakshe K, Coarfa C, Edwards D, Huang S, and Rosen JM (2015). Mammary Stem Cells and Tumor-Initiating Cells Are More Resistant to Apoptosis and Exhibit Increased DNA Repair Activity in Response to DNA Damage. Stem Cell Reports 5, 378–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RE (2001). Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev 27, 165–176. [DOI] [PubMed] [Google Scholar]
- Derose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MTW, Factor R, Matsen C, Milash BA, Nelson E, et al. (2011). Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med 17, 1514–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravis C, Chung CY, Lytle NK, Herrera-Valdez J, Luna G, Trejo CL, Reya T, and Wahl GM (2018). Epigenetic and Transcriptomic Profiling of Mammary Gland Development and Tumor Models Disclose Regulators of Cell State Plasticity. Cancer Cell. 34, 466–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito M, Mondal N, Greco TM, Wei Y, Spadazzi C, Lin SC, Zheng H, Cheung C, Magnani JL, Lin SH, et al. (2019). Bone vascular niche E-selectin induces mesenchymal–epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol 21, 627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre R, Alférez DG, Santiago-Gómez A, Spence K, McConnell JC, Hart C, Simões BM, Lefley D, Tulotta C, Storer J, et al. (2019). Microenvironmental IL1β promotes breast cancer metastatic colonisation in the bone via activation of Wnt signalling. Nat. Commun 10, 5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehm T, Krawczyk N, Solomayer EF, Becker-Pergola G, Dürr-Störzer S, Neubauer H, Seeger H, Staebler A, Wallwiener D, and Becker S (2008). ERalpha-status of disseminated tumour cells in bone marrow of primary breast cancer patients. Breast Cancer Res. 10, R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, and Kuperwasser C (2010). Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc. Natl. Acad. Sci. U. S. A 107, 21737–21742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, et al. (2007). ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell. 1, 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez ME, Moore HM, Li X, Toy KA, Huang W, Sabel MS, Kidwell KM, and Kleer CG (2014). EZH2 expands breast stem cells through activation of NOTCH1 signaling. Proc. Natl. Acad. Sci 111, 3098–30103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zürrer-Härdi U, Bell G, et al. (2012). Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148, 1015–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PB, Pastushenko I, Skibinski A, Blanpain C, and Kuperwasser C (2019). Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 24, 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CT, Cardiff RD, and Muller WJ (1992). Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell. Biol 12, 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S, Castelo R, and Guinney J (2013). GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Kulicke R, Fenster RJ, Greengard P, and Heintz N (2014). Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP). Nat. Protoc 9, 1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefnagel LDC, Van Der Groep P, Van De Vijver MJ, Boers JE, Wesseling P, Wesseling J, Van Der Wall E, and Van Diest PJ (2013). Discordance in ERα, PR and HER2 receptor status across different distant breast cancer metastases within the same patient. Ann. Oncol 24, 3017–3023. [DOI] [PubMed] [Google Scholar]
- Huang X, Yan J, Zhang M, Wang Y, Chen Y, Fu X, Wei R, Zheng X. ling, Liu Z, Zhang X, et al. (2018). Targeting Epigenetic Crosstalk as a Therapeutic Strategy for EZH2-Aberrant Solid Tumors. Cell 175, 186–199.e19. [DOI] [PubMed] [Google Scholar]
- Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, Coindre JM, Blakemore SJ, Clawson A, Suttle B, et al. (2018). Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 19, 649–659. [DOI] [PubMed] [Google Scholar]
- Jäger BAS, Finkenzeller C, Bock C, Majunke L, Jueckstock JK, Andergassen U, Neugebauer JK, Pestka A, Friedl TWP, Jeschke U, et al. (2015). Estrogen receptor and HER2 status on disseminated tumor cells and primary tumor in patients with early breast cancer. Transl. Oncol 8, 509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SRD (2010). New strategies in estrogen receptor-positive breast cancer. Clin. Cancer Res 16, 1979–1987. [DOI] [PubMed] [Google Scholar]
- Kalhor R, Mali P, and Church GM (2017). Rapidly evolving homing CRISPR barcodes. Nat. Methods 14, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalhor R, Kalhor K, Mejia L, Leeper K, Graveline A, Mali P, and Church GM (2018). Developmental barcoding of whole mouse via homing CRISPR. Science 361, eaat9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, and Massague J (2003). A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549. [DOI] [PubMed] [Google Scholar]
- Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, Nielsen TO, and Gelmon K (2010). Metastatic behavior of breast cancer subtypes. J. Clin. Oncol 28, 3271–3277. [DOI] [PubMed] [Google Scholar]
- Kim KH, and Roberts CWM (2016). Targeting EZH2 in cancer. Nat. Med 22, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Larson DE, and Wilson RK (2013). Using varscan 2 for germline variant calling and somatic mutation detection. Curr. Protoc. Bioinforma 44, 15.4.1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlow W, and Guise TA (2005). Breast cancer metastasis to bone: mechanisms of osteolysis and implications for therapy. J. Mammary Gland Biol. Neoplasia 10, 169–180. [DOI] [PubMed] [Google Scholar]
- Kurland BF, Peterson LM, Lee JH, Schubert EK, Currin ER, Link JM, Krohn KA, Mankoff DA, and Linden HM (2017). Estrogen receptor binding (18F-FES PET) and glycolytic activity (18F-FDG PET) predict progression-free survival on endocrine therapy in patients with ER+ breast cancer. Clin. Cancer Res 23, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AW, Pattabiraman DR, and Weinberg RA (2017). Emerging Biological Principles of Metastasis. Cell. 168, 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, and Pietenpol JA (2011). Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest 121, 2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin-Allerhand JA, Sokol K, and Smith JD (2003). Safe and Effective Method for Chronic 17β-Estradiol Administration to Mice. Contemp. Top. Lab. Anim. Sci 42, 33–35. [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, He X, Liu S, Hoog J, Lu C, et al. (2013). Endocrine-Therapy-Resistant ESR1 Variants Revealed by Genomic Characterization of Breast-Cancer-Derived Xenografts. Cell Rep. 4, 1116–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim E, Metzger-Filho O, and Winer EP (2012). The natural history of hormone receptor-positive breast cancer. Oncology (Williston Park) 26, 688–694. [PubMed] [Google Scholar]
- Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, and Pollard JW (2003). Progression to Malignancy in the Polyoma Middle T Oncoprotein Mouse Breast Cancer Model Provides a Reliable Model for Human Diseases. Am. J. Pathol 163, 2113–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. (2008). The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta LLC, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, et al. (2011). The JAK2/STAT3 signaling pathway is required for growth of CD44 +CD24− stem cell-like breast cancer cells in human tumors. J. Clin. Invest 121, 2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina D, Kittrell FS, Shepard A, Stephens LC, Jiang C, Lu J, Allred DC, McCarthy M, and Ullrich RL (2002). Biological and genetic properties of the p53 null preneoplastic mammary epithelium. FASEB J. 16, 881–883. [DOI] [PubMed] [Google Scholar]
- Von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, Jouffre N, Huynen MA, and Bork P (2005). STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 33, D433–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, and Massagué J (2005). Genes that mediate breast cancer metastasis to lung. Nature. 436, 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawaz Z, Lonard DM, Dennis AP, Smith CL, and O’Malley BW (1999). Proteasome-dependent degradation of the human estrogen receptor. Proc. Natl. Acad. Sci. U. S. A 96, 1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferle AD, Spike BT, Wahl GM, and Perou CM (2015). Luminal progenitor and fetal mammary stem cell expression features predict breast tumor response to neoadjuvant chemotherapy. Breast Cancer Res. Treat 149, 425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priedigkeit N, Watters RJ, Lucas PC, Basudan A, Bhargava R, Horne W, Kolls JK, Fang Z, Rosenzweig MQ, Brufsky AM, et al. (2017). Exome-capture RNA sequencing of decade-old breast cancers and matched decalcified bone metastases. JCI Insight 2, e95703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapaksa G, Nikolos F, Bado I, Clarke R, Gustafsson J, and Thomas C (2015). ERβ decreases breast cancer cell survival by regulating the IRE1/XBP-1 pathway. Oncogene 34, 4130–4141. [DOI] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). DeepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijm EA, Jansen MPHM, Ruigrok-Ritstier K, Van Staveren IL, Look MP, Van Gelder MEM, Sieuwerts AM, Sleijfer S, Foekens JA, and Berns EMJJ (2011). Decreased expression of EZH2 is associated with upregulation of ER and favorable outcome to tamoxifen in advanced breast cancer. Breast Cancer Res. Treat 125, 387–394. [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi N, Dai X, Winter CG, and Kang Y (2011). Tumor-Derived Jagged1 Promotes Osteolytic Bone Metastasis of Breast Cancer by Engaging Notch Signaling in Bone Cells. Cancer Cell 19, 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgroi DC, Sestak I, Cuzick J, Zhang Y, Schnabel CA, Schroeder B, Erlander MG, Dunbier A, Sidhu K, Lopez-Knowles E, et al. (2013). Prediction of late distant recurrence in patients with oestrogen-receptor-positive breast cancer: A prospective comparison of the breast-cancer index (BCI) assay, 21-gene recurrence score, and IHC4 in the TransATAC study population. Lancet Oncol. 14, 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu H, Sun L, Zhang Y, Chen Y, Li R, et al. (2007). Integration of Estrogen and Wnt Signaling Circuits by the Polycomb Group Protein EZH2 in Breast Cancer Cells. Mol. Cell. Biol 27, 5105–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smid M, Wang Y, Zhang Y, Sieuwerts AM, Yu J, Klijn JG, Foekens JA, and Martens JW (2008). Subtypes of breast cancer show preferential site of relapse. Cancer Res. 68, 3108–3114. [DOI] [PubMed] [Google Scholar]
- Stark R, and Brown G (2011). DiffBind : differential binding analysis of ChIP-Seq peak data. Bioconductor. DOI: 10.18129/B9.bioc.DiffBind. [DOI] [Google Scholar]
- Ström JO, Theodorsson A, Ingberg E, Isaksson IM, and Theodorsson E (2012). Ovariectomy and 17β-estradiol replacement in rats and mice: A visual demonstration. J. Vis. Exp 64, E4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, Wu ZJ, Krall JA, Bierie B, Guo W, et al. (2013). Protein Kinase C α Is a Central Signaling Node and Therapeutic Target for Breast Cancer Stem Cells. Cancer Cell 24, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform 14, 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trastuzumab A, Ithimakin S, Day KC, Malik F, Zen Q, Dawsey SJ, Bersano-Begey TF, Quraishi AA, Ignatoski KW, Daignault S, et al. (2013). HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: Implications for efficacy of. Cancer Res. 73, 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullah I, Karthik GM, Alkodsi A, Kjällquist U, Stålhammar G, Lövrot J, Martinez NF, Lagergren J, Hautaniemi S, Hartman J, et al. (2018). Evolutionary history of metastatic breast cancer reveals minimal seeding from axillary lymph nodes. J. Clin. Invest 128, 1355–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yu C, Gao X, Welte T, Muscarella AM, Tian L, Zhao H, Zhao Z, Du S, Tao J, et al. (2015). The Osteogenic Niche Promotes Early-Stage Bone Colonization of Disseminated Breast Cancer Cells. Cancer Cell 27, 193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Tian L, Goldstein A, Liu J, Lo H-C, Sheng K, Welte T, Wong STC, Gugala Z, Stossi F, et al. (2017). Bone-in-culture array as a platform to model early-stage bone metastases and discover anti-metastasis therapies. Nat. Commun 8, 15045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Tian L, Liu J, Wong STC, Gugala Z, and Zhang XH (2018). The Osteogenic Niche Is a Calcium Reservoir of Bone Micrometastases and Confers Unexpected Article The Osteogenic Niche Is a Calcium Reservoir of Bone Micrometastases and Confers Unexpected Therapeutic Vulnerability. Cancer Cell 34, 823–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilbaecher KN, Guise TA, and McCauley LK (2011). Cancer to bone: a fatal attraction. Nat. Rev. Cancer 11, 411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsch CW, Swim EL, Jean McManus M, White AC, and McGrath CM (1981). Estrogen induced growth of human breast cancer cells (MCF-7) in athymic nude mice is enhanced by secretions from a transplantable pituitary tumor. Cancer Lett. 14, 309–316. [DOI] [PubMed] [Google Scholar]
- Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, Holdman XB, Herschkowitz JI, Pond A, Xie G, et al. (2016). Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat. Cell Biol 18, 632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Wang H, Muscarella A, Goldstein A, Zeng H-C, Bae Y, Lee BHI, and Zhang XH-F (2016). Intra-iliac Artery Injection for Efficient and Selective Modeling of Microscopic Bone Metastasis. J. Vis. Exp 115, 53982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang GL, Zhang Y, Cao KX, and Wang XM (2019a). Orthotopic injection of breast cancer cells into the mice mammary fat pad. J. Vis. Exp 2019. [DOI] [PubMed] [Google Scholar]
- Zhang W, Bado IL, Hu J Wan Y-W, Wu L, Wang H,Gao Y, Jeong H-H, and Zhang XH-F (2021). The bone microenvironment invigorates metastatic seeds for further dissemination. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XHF, Giuliano M, Trivedi MV, Schiff R, and Kent Osborne C (2013). Metastasis dormancy in estrogen receptor-positive breast cancer. Clin. Cancer Res 19, 6389–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Bae Y, Kasimir-Bauer S, Tang R, Chen J, Ren G, Yuan M, Esposito M, Li W, Wei Y, et al. (2017). Therapeutic Antibody Targeting Tumor- and Osteoblastic Niche-Derived Jagged1 Sensitizes Bone Metastasis to Chemotherapy. Cancer Cell 32, 731–747.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Barrette TR, Kumar-Sinha C, Ghoshk D, Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. (2002). The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 388–390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets were deposited in Gene Expression Omnibus (Edgar, 2002), with the following GEO accession numbers (GSE137245; GSE137270, GSE160566, GSE160582,and GSE161181). The GEO Reference Series connecting all datasets is GSE160583. Barcode analysis pipeline is accessible at: https://github.com/LiuzLab/ER_positive_breast_cancer-manuscirpt.