

Abstract
Construction of empirical fitness landscapes has transformed our understanding of genotype–phenotype relationships across genes. However, most empirical fitness landscapes have been constrained to the local genotype neighbourhood of a gene primarily due to our limited ability to systematically construct genotypes that differ by a large number of mutations. Although a few methods have been proposed in the literature, these techniques are complex owing to several steps of construction or contain a large number of amplification cycles that increase chances of non-specific mutations. A few other described methods require amplification of the whole vector, thereby increasing the chances of vector backbone mutations that can have unintended consequences for study of fitness landscapes. Thus, this has substantially constrained us from traversing large mutational distances in the genotype network, thereby limiting our understanding of the interactions between multiple mutations and the role these interactions play in evolution of novel phenotypes. In the current work, we present a simple but powerful approach that allows us to systematically and accurately construct gene variants at large mutational distances. Our approach relies on building-up small fragments containing targeted mutations in the first step followed by assembly of these fragments into the complete gene fragment by polymerase chain reaction (PCR). We demonstrate the utility of our approach by constructing variants that differ by up to 11 mutations in a model gene. Our work thus provides an accurate method for construction of multi-mutant variants of genes and therefore will transform the studies of empirical fitness landscapes by enabling exploration of genotypes that are far away from a starting genotype.
Keywords: PCR assembly, genotype, mutagenesis, multi-mutant variants, fitness landscape
Introduction
Empirical fitness landscapes are the key to a better understanding of the principles of genotype–phenotype mapping in biological systems [1–7]. Empirical fitness landscapes have greatly advanced our knowledge of the functional impact of clinically observed mutations on antibiotic resistance genes [8–17], impact of disease mutations on protein stability and aggregation [16, 18–25] and study of splice variants of a gene [26–32]. In addition, fitness landscapes have provided great insights into molecular evolution of proteins [33–35] and RNA molecules [36–42]. However, majority of the empirical fitness landscapes have been limited to the local genotype neighbourhood and to mostly one, two or three mutant variants of a gene [8, 14, 33, 34, 43–51] with the exceptions of small genes such as tRNA genes [39, 41]. Although local neighbourhoods provide important insights into changes in fitness, they fail to capture full evolutionary trajectories occurring over deep evolutionary times. Uncovering the phenotypes of genotypes at large mutational distances can provide unprecedented insights into the interactions between large number of mutations [10, 12, 41, 48, 52–54], their impact on fitness [33, 43, 44, 49, 55–59] and into evolution of novel phenotypes [7, 40, 51, 60, 61].
Construction of fitness landscapes [62] relies on our ability to systematically construct variants of a genotype. There are several approaches that have been employed to construct multi-mutant variants of genes. First, site-directed mutagenesis can be used to introduce targeted mutations in a gene using methods, such as Overlap-extension PCR [63]. However, this method is limited in its ability to introduce more than one or two mutations in a gene [63]. Thus, introduction of a large number of mutations using this technique requires a stepwise introduction of one mutation at a time. In fact, such an approach has been described by Wäneskog et al. [64], where the authors introduced 13 mutations in a gene. However, their method required six PCR steps and some of these steps required many amplification cycles. This made the whole assembly process complex, time-consuming and at the same time increased the chances of introduction of unintended mutations due to the large number of amplification cycles. Similarly, another method by Hejlesen and Füchtbauer [65] utilized prolonged overlap-extension PCR and required 55 amplification cycles. Other studies have tried different approaches [66, 67]; however, they required special primer design with long overlaps between fragments or required special complex PCR steps.
Another set of methods for construction of multi-mutant variants that have been described in the literature are either based on or bear similarity to QuikChange site-directed mutagenesis protocol (Agilent Technologies). These methods utilize single-primer amplification reactions on the whole vector carrying the gene of interest and generate linearized fragments [68–73]. In the next step, the parental template DNA molecules are digested and the linearized plasmid fragments carrying mutations are transformed into bacteria to generate recombinant clones carrying targeted mutations. One of the earliest methods in this regard has been described by Wang and Malcolm [68] where they introduced up to nine mutations in a gene fragment. Several variations of this method have been proposed subsequently [69–73]. Notably, Zeng et al. [73] described one such method and introduced up to 15 mutations in a gene. However, all these methods rely on amplifying the whole vector and thus, risk accumulating mutations in the vector backbone. This can profoundly influence the outcomes of selection experiments that are part of studies on empirical fitness landscapes. In this regard, one study suggested a variation of this method that required amplification of only a part of the plasmid but the process utilized a substantially larger number of cycles [74].
Another recent method for construction of genotype variants utilizes doped oligos [10, 46, 75], which essentially uses a random mutagenesis method [76] with specific probabilities assigned to each type of mutation [54, 77–79]. Although this method has been successfully applied to construct fitness landscapes of protein-coding [46, 75] as well as tRNA genes [39, 78, 79], this method is inherently limited in its capability to construct gene variants with a large number of mutations. Increasing the mutation rate in this method can lead to gene variants that differ by a large number of mutations but in turn will reduce the total number of variants sampled in the local genotype neighbourhood [77]. Further, gene assembly techniques from a large number of oligos can also be employed for construction of gene variants containing a large number of mutations [80–82]. Using these methods, one can combine mutated fragments with wild-type fragments and can get a mutant library with genotypes in the local as well as far-away neighbourhoods. However, these methods require a large number of amplification cycles which increase the likelihood of unwanted mutations in the gene construct.
We hereby describe a simple yet powerful and accurate two-step gene assembly method that enables us to systematically construct genotypes differing by a large number of mutations. Our method utilizes normal amplification primers and low number of amplification cycles, thus ensuring a quick and efficient gene assembly process with extremely low probability of introduction of unintended mutations. We demonstrate the capability of our method by constructing an 11-mutant variant of a model gene. Further, our method can also be adapted to combine wild-type and mutated fragments and can allow construction of gene variants in the immediate genotype neighbourhood as well as at far-away distances. Thus, we believe that our method will substantially boost the capabilities of researchers to include genotypes across large mutational distances in the study of empirical fitness landscapes. This will facilitate developing a deeper understanding of the principles of genotype–phenotype mapping and molecular evolution.
Materials and methods
Template DNA and primers
TEM-1 β-lactamase gene from pUC19 plasmid was chosen as the model gene system for introducing mutations at 11 different amino acid positions. The numbering scheme for residues of TEM-1 gene was obtained from Bush and Jacoby [83].
First PCR amplification
Systematic mutagenesis of TEM-1 gene for 11 different amino acid positions was performed by PCR using Q5 DNA polymerase (New England Biolabs). The reaction was set up as shown in Table 1.
Table 1:
Reaction set-up for first PCR amplification
| Reagent | Amount/reaction (µl) |
|---|---|
| Q5 PCR buffer | 10 |
| 10 mM dNTP | 1 |
| Q5 DNA polymerase | 0.5 |
| 10 µM forward primer | 2.5 |
| 10 µM reverse primer | 2.5 |
| Template | 1 |
| Molecular grade water | 32.5 |
| Total | 50 |
The PCR programme was set as follows : (i) initial denaturation, 30 s at 98°C; (ii) 20 cycles of denaturation for 10 s at 98°C, annealing at 60°C for 30 s, extension at 72°C for 30 s; and (iii) final extension at 72°C for 2 min.
The fragment sizes varied according to the amino acid position. Our desired amino acid positions were 21, 39, 69, 104, 164, 182, 238, 240, 244, 265 and 275 of the TEM-1 protein to introduce mutations as these mutations have been reported from clinical isolates of this gene. More specifically, the mutations L21F, Q39K, M69L, M69I, M69V, E104K, R164S, R164C, R164H, M182T, G238S, E240K, R244H, R244C, R244S, T265M, R275L and R275Q, have been deemed most prevalent across TEM-1 mutants. The PCR was performed according to the procedure briefed above with primers as follows for amplifying individual fragments (Table 2).
Table 2:
Fragments and their sizes
| Serial no. | Fragments generated by primer combinations | Sizes (bp) |
|---|---|---|
| Fragment 1 | Promoter region start to amino acid residue 21 (TEM-1_for+L21F_rev) | 161 |
| Fragment 2 | Residues 21–39 (L21F_for+Q39K_rev) | 81 |
| Fragment 3 | Residues 39–69 (Q39K_for+M69I_rev) | 116 |
| Fragment 4 | Residues 69–104 | 131 |
| (M69I_for+E104K_rev) | ||
| Fragment 5 | Residues 104–164 (E104K_for+R164C/S_rev) | 204 |
| Fragment 6 | Residues 164–182 | 80 |
| (R164C/S_for+M182T_rev) | ||
| Fragment 7 | Residues 182–244 | 209 |
| (M182T_for+Mut238240244_rev) | ||
| Fragment 8 | Residues 244–265 | 101 |
| (Mut238240244_for+T265M_rev) | ||
| Fragment 9 | Residues 265–275 | 61 |
| (T265M_for+R275L/Q_rev) | ||
| Fragment 10 | Residue 275 to end of TEM-1 gene segment (R275L/Q_for+TEM-1_rev) | 61 |
The primer combinations used to generate the fragments are shown inside the parentheses.
Purification and quantification of first PCR products
After the first step of PCR, the quantity and quality of PCR products were checked in 2% Agarose gel. PCR products were then digested with 0.5 µl DpnI (to remove methylated DNA of plasmid template to prevent its interference in the second PCR step) and 2 µl ExoSAP (to hydrolyze excess primers and nucleotides) at 37°C for 1 h. The enzymes were then inactivated at 80°C for 20 min. Next, the fragments were purified by QIAGEN MinElute spin column as per manufacturer’s protocol and were eluted in 15 µl Molecular Grade water. The purified products were checked in 2% agarose gel. The concentrations of purified PCR products were then measured by Qubit Broad Range assay (Invitrogen). Molar mass of each PCR fragment was determined using Sequence Manipulation Suite which calculated the number of moles present per microlitre of solution.
Second PCR for assembly
Equal concentration of each purified fragment (0.5 or 1 pmol) from the first PCR amplification was taken as templates for the next round of reaction. First, a reaction was set up according to Table 1 in a total reaction volume of 90 µl but without adding primers. Thermal cycling conditions were set as follows: 1 cycle at 98°C for 10 s; 10 cycles at 98°C for 10 s, at 55°C for 30 s and at 72°C at 30 s; and a final extension at 72°C for 10 min.
In the next step, the terminal primers (TEM-1_for and TEM-1_rev) were added (5 µl each and total reaction volume of 100 µl). The final amplification programme was done as follows: 1 cycle at 98°C for 2 min; 15 cycles at 98°C for 10 s, at 55°C for 30 s and at 72°C for 30 s; and a final extension at 72°C for 10 min. The final amplified products were checked on 1% agarose gel. In the absence of unspecific bands on gel, the products were purified using QIAGEN PCR purification kit following manufacturer's protocol. In case of unspecific bands visible on gel, band representing assembled gene product was cut from the gel and was purified using QIAGEN Gel extraction kit.
Cloning and Sanger sequencing
The purified or extracted gene product was cloned into the plasmid pUA67 [84] as cloning vector. The vector and insert were digested using high-fidelity restriction enzymes EcoRI and HindIII at 37°C for 16 h followed by inactivation of the enzymes at 80°C for 20 min. The digested vector was dephosphorylated by adding Quick CIP at 37°C for 10 min (and heat inactivating at 80°C for 10 min) to avoid self-ligation. The digested vector and insert were analysed on 1% agarose gel and purified by QIAquick gel extraction kit. The purified products were then ligated using T4 DNA ligase at 16°C for 16 h followed by heat inactivation of the enzyme at 65°C for 10 min. The ligated products were then transformed into chemically competent E. coli DH5α cells using calcium chloride and the transformant colonies were selected on Luria-Bertani (LB) plates supplemented with 100 µg/ml Kanamycin. The colonies were then screened by colony PCR to check for the presence of TEM-1 gene (Fig. 2C). The mutated TEM-1 gene sequence was finally confirmed from the selected colonies by Sanger sequencing.
Figure 2:

Analysis of PCR amplified fragments by agarose gel electrophoresis. (A) Lanes 1 and 12—GeneRuler ULR DNA Ladder (Thermo Scientific). Lanes 2–11—amplified products of each of the 10 fragments carrying the intended mutations after the first PCR (B) Lane1—GeneRuler 1 kb ladder (Thermo Scientific); Lanes 2 and 3—final joined fragments after the second PCR. (C) Lane1—GeneRuler 1 kb ladder; Lanes 2–10—PCR amplified products confirming the presence of the whole construct from transformant E. coli colonies.
Results and discussion
Our method consisted of two PCR amplification steps (Fig. 1). In the first PCR step, we constructed individual fragments containing targeted mutations (Fig. 1). We designed the gene fragments in such a way that primers for amplifying each fragment contained the desired mutations (Fig. 1). Thus, after the first amplification, we obtained gene fragments of variable lengths that contained mutations. In the second step, we assembled these fragments into the whole gene in a single PCR (Fig. 1).
Figure 1:

An outline of the two-step PCR method. The first PCR step yields 10 DNA fragments of various sizes each containing their respective targeted mutations. The primers were designed to contain the mutations and had 12-bp overlap with neighbouring primers to enable fragment assembly in the second and final step of the two-step PCR.
We used the gene TEM-1 beta lactamase for demonstration of the proof-of-concept. We aimed to construct TEM-1 variants containing up to 11 mutations. For TEM-1 gene, we targeted the following amino acid mutations as these have been observed in clinical samples very frequently—L21F, Q39K, M69I, E104K, R164C/S, M182T, G238S, E240K, R244C/S, T265M and R275L/Q. To introduce these mutations, we divided the TEM-1 gene into 10 fragments and designed forward and reverse primers for amplification of each of these fragments. The first fragment contained the TEM-1 promoter region to the L21 amino acid residue. Thus, the reverse primer contained the L21F amino acid mutation. We also ensured that the targeted mutations in the primers were succeeded by at least 12 nucleotides at the 3ʹ-end of the primer to enable efficient PCR amplification (Table 3). The second fragment contained the region starting from L21 residue and ending at Q39 residue. The forward primer contained the L21F mutation and the reverse primer contained Q39K mutation. Similarly, we designed primers for amplifications of other fragments (Table 3). When two mutations were too close to each other for making a fragment by PCR, we constructed these mutations in a single fragment using primers containing both mutations. Further, we designed the primers in such a way that the adjoining fragments had 12-bp overlap for efficient assembly in the next step (Fig. 1 and Table 3).
Table 3:
sequences of primers used with the mutated bases shown in bold
| Primer name | Primer sequence 5′–3′ |
|---|---|
| TEM-1_for | ACGGAATTCCGCGGAACCCCTATTTGTTTATTTTTC |
| TEM-1_rev | ACGAAGCTTCCAATGCTTAATCAGTGAGGCAC |
| L21F_for | GCGGCATTTTGCTTTCCTGTTTTTGCT |
| L21F_rev | AGCAAAAACAGGAAAGCAAAATGCCGC |
| Q39K_for | GATGCTGAAGATAAGTTGGGTGCACGA |
| Q39K_rev | TCGTGCACCCAACTTATCTTCAGCATC |
| M69I_for | CGTTTTCCAATGATCAGCACTTTTAAA |
| M69I_rev | TTTAAAAGTGCTGATCATTGGAAAACG |
| E104K_for | AATGACTTGGTTAAGTACTCACCAGTC |
| E104K_rev | GACTGGTGAGTACTTAACCAAGTCATT |
| R164C/S_for | ACTCGCCTTGATWGTTGGGAACCGGAG |
| R164C/S_rev | CTCCGGTTCCCAACWATCAAGGCGAGT |
| M182T_for | CGTGACACCACGACGCCTGTAGCAATG |
| M182T_rev | CATTGCTACAGGCGTCGTGGTGTCACG |
| Mut238240244_for | AAATCTGGAGCCAGTAAGCGTGGGTCTHGCGGTATCATTGCA |
| Mut238240244_rev | TGCAATGATACCGCDAGACCCACGCTTACTGGCTCCAGATTT |
| T265M_for | GTAGTTATCTACATGACGGGGAGTCAG |
| T265M_rev | CTGACTCCCCGTCATGTAGATAACTAC |
| R275L/Q_for | ACTATGGATGAACDAAATAGACAGATCGCT |
| R275L/Q_rev | AGCGATCTGTCTATTTHGTTCATCCATAGT |
Degenerate bases: W = A or T; D = A or G or T; H = A or C or T, Mut238240244_for and Mut238240244_rev primers denote mutations of three amino acid residues, namely, G238S, E240K, and R244C/S.
We performed the first amplification for 20 cycles using a high-fidelity DNA polymerase (see Materials and methods section). This resulted in 10 gene fragments with sizes of 161, 81, 116, 131, 204, 80, 209, 101, 61 and 61 bp, respectively (Fig. 2A). We then digested these PCR products with DpnI to remove template DNA and with ExoSAP to hydrolyse excess primers and nucleotides. We then column purified the treated DNA fragments and quantified the concentrations of the purified DNA fragments (in mol/µl) by Qubit Broad Range Assay. Next, we mixed 0.5 and 1 pmol of each fragment in a PCR reaction and performed thermal cycling for 10 cycles without addition of any primers. This enabled annealing of overlapping regions of neighbouring fragments and subsequently allowed gap filling. We then added the terminal primers and performed 15 cycles of PCR amplification. This resulted in assembly of the complete gene of size ∼1 kb (Fig. 2B).
To confirm the accuracy of our method, we digested the whole gene fragment with restriction enzymes, ligated with appropriately digested vector and transformed into competent E. coli cells. We confirmed transformation of the gene fragment by colony PCR (Fig. 2C) and verified the introduced mutations in the TEM-1 gene by Sanger sequencing (Fig. 3). We observed introduction of the targeted mutations at specific sites in the TEM-1 gene (Fig. 3).
Figure 3:

Confirmation of mutagenesis by Sanger sequencing. Multiple sequence alignment of the mutated TEM-1 gene sequences from three clones show presence of the desired mutations at the targeted sites.
Further, we observed that two factors are critical for accurate and reproducible reconstruction of mutant variants. The first one is the use of clean and purified PCR product obtained from the first PCR for the second amplification step and secondly, the use of equimolar amounts of products generated by the first PCR in the second assembly step.
We compared different aspects of our method with that of available methods in literature. Our method used relatively small number of amplification cycles and thus had one of the lowest error rates among all methods (Table 4). Methods that used even smaller number of amplification cycles required amplification of the vector along with the gene of interest [68–73] (Table 4). This meant a risk of introducing unwanted mutations in the plasmid backbone which could impact plasmid copy number and antibiotic selection. Changes in plasmid copy number could have unintended critical influence on selection experiments deployed for studies of empirical fitness landscapes. Furthermore, some of these methods generated nicked plasmid as the amplification product which could not be utilized as template in the subsequent amplification cycles [68–70, 85]. This led to use of increased starting template DNA and could lead to formation of hybrid hemi-methylated DNA after amplification that could resist enzymatic digestion [86]. This increased the risk of parental DNA carry-over and contamination with the mutants [69]. Furthermore, some of the published methods required quite complex primer design. For example, Zeng et al. [73] required four primers for each of the mutants that increased the complexity and cost. Some of the other methods required large overlaps that led to long primers again making the process complex and costly.
Table 4:
comparison between the method proposed in this work and the related published methods in terms of the demonstrated capability to introduce multiple mutations in a gene fragment, number of amplification cycles, estimated error rates, risk of plasmid backbone mutations, risk of parental plasmid carry-over and design of oligos
| Methods | This Method | Wang and Malcom, Biotechniques (1999) | Hejlesen and Fuchtbauer, BioTechniques (2020) | Zeng et al., Scientific Reports (2018) | Kuo et al., Biol Proc. Online (2017) | Hallak et al., PLoS ONE (2017) | Kadkhodaei et al., RSC Advances (2016) | Trehan et al., Scientific Reports (2016) | Wäneskog and Bjerling, Analytical Biochemistry (2014) | Edelheit et al., BMC Biotechnology (2009) | Liu and Naismith; BMC Biotechnology (2008) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Maximum no. of mutations introduced | 11 | 9 | 3 | 15 | 2 | 1 | 9 | 2 | 13 | 3 | 2 |
| Amplification product | Gene of Interest | Whole Plasmid (nicked) | Gene of Interest | Whole plasmid | Whole Plasmid | Part of Plasmid | Gene of Interest | Whole Plasmid (nicked) | Gene of Interest | Whole plasmid (nicked) | Whole plasmid (nicked) |
| No. of PCR amplification cycles |
|
|
|
Total: 60 cycles |
14 cycles |
|
|
30 cycles |
|
30 cycles | 12 cycles |
| Estimated error rate (https://pcrfidelityestimator.neb.com/#!/) | 0.7% (for a 1 kb gene) | 0.8–1.5% (for a 3 kb plasmid) | 1.1% (for a 1 kb gene) |
3.5% (for a 3 kb plasmid) |
0.8% (for a 3 kb plasmid) |
1.75% (with starter DNA size of 532 bp) |
0.7–0.9% (for a 1 kb gene) |
1.8% (for a 3 kb plasmid) |
0.9% (for a 1 kb gene) |
1.8% (for a 3 kb plasmid) |
0.7% (for a 3 kb plasmid) |
| Chance of plasmid backbone mutations | No | Yes | No | Yes | Yes | Yes | No | Yes | No | Yes | Yes |
| Size of oligos | 27–42 bp (Overlap 12 bp) | 34–73 bp | 26–33 bp |
25 bp (Overlap 6–10 bp) |
44–46 bp (Overlap 19–22bp) | 22 bp | 50–78 bp (Overlap 50 bp) | 23 bp (Overlap 17 bp) | 31–120 bp | 33–57 bp | 39–51 bp |
| Template amount | 0.5 ng | 50–200 ng | 2–5 ng | 50–500 ng | 10 ng | 0.1 ng | 10–50 ng | 100 ng | 100–250 ng | 500 ng | 10 ng |
| Risk of template carry over | Low | High | Low | Low | Low | Low | Low | Low | Low | High | Low |
| Number and size of fragments | 10; 60–210 bp | N/A | 3; 70–3000 bp | Variable | N/A | N/A | 9; 304–2191 bp | N/A | 13; 63–1800 bp | N/A | N/A |
The method proposed here has among the lowest error rates, has low chance of plasmid backbone mutations and has low risk of parental plasmid carry-over.
Thus, our method provides a balanced approach for the construction of multi-mutant variants in all aspects compared with the published methods. Our method uses simple molecular biology tools and requires only two PCR amplification steps for construction in contrast to many steps adopted by earlier methods [64, 66]. Our method is also robust as it can assemble genes from DNA fragments of sizes ranging from 60 to 210 bp and do not require any complicated primer design or long primers. Further, our method uses a total of 35 cycles across the two steps and hence has low chance of introduction of unintended mutations or indels in the gene fragment. Finally, our method does not require amplification of vector and utilize a very small amount of starting template DNA. This also reduces the chance of parental wild-type DNA contamination with mutants and avoids occurrence of unintended mutations in the plasmid backbone. However, the efficiency of our method for assembling larger DNA fragments remains to be tested.
Taken together, our work describes a powerful tool for construction of genotypes at large mutational distances (Fig. 4). Our method can also be adapted to explore genotypes at any mutational distance from the starting genotype by choosing the number of fragments carrying mutations. In addition, one can also mix wild-type and mutated fragments during the second step of assembly, thus enabling construction of a library containing genotypes at local and far-away neighbourhoods. This can eventually help us to systematically reconstruct the long evolutionary paths of proteins and RNA molecules and can transform our understanding of the principles of molecular evolution.
Figure 4:
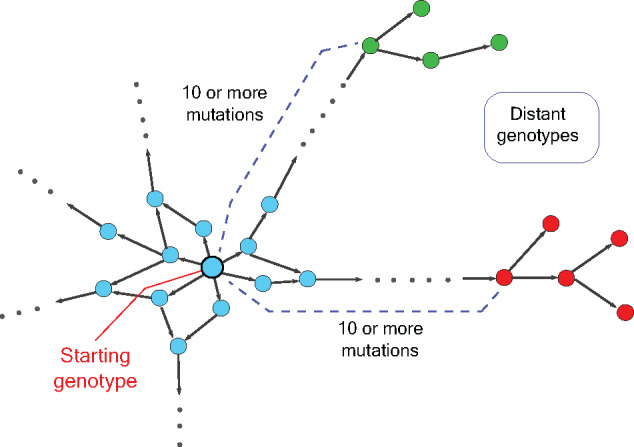
A schematic genotype map encompassing large mutational distances. The network shows nodes representing genotypes with different phenotypes denoted by different colours. Edges connect nodes (genotypes) that can be reached by one mutation. The distant genotypes can be reached after 10 or more mutations from the starting genotype.
Data availability
No new data were generated or analysed in support of this research. The sequencing data generated by Sanger sequencing to confirm the clones are shown in the article.
Funding
Work in the RD lab was funded by an ISIRD grant, IIT Kharagpur and an Early career research (ECR) grant (ECR/2017/002328), Science and Engineering Research Board (SERB), India.
Author contributions
R.D. conceived the study; S.R. and A.A. performed all experiments; S.R., A.A., and R.D. wrote the manuscript. All authors read and approved the manuscript.
Conflict of interest:
The authors declare no conflict of interest.
References
- 1.Poelwijk FJ, Kiviet DJ, Weinreich DM. et al. Empirical fitness landscapes reveal accessible evolutionary paths. Nature 2007;445:383–6. [DOI] [PubMed] [Google Scholar]
- 2.Hayden EJ, Ferrada E, Wagner A.. Cryptic genetic variation promotes rapid evolutionary adaptation in an RNA enzyme. Nature 2011;474:92–5. [DOI] [PubMed] [Google Scholar]
- 3.Crona K, Greene D, Barlow M.. The peaks and geometry of fitness landscapes. J Theor Biol 2013;317:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Visser JAGM, Krug J.. Empirical fitness landscapes and the predictability of evolution. Nat Rev Genet 2014;15:480–90. [DOI] [PubMed] [Google Scholar]
- 5.Mira PM, Meza JC, Nandipati A. et al. Adaptive landscapes of resistance genes change as antibiotic concentrations change. Mol Biol Evol 2015;32:2707–15. [DOI] [PubMed] [Google Scholar]
- 6.Schmiedel JM, Carey LB, Lehner B.. Empirical mean-noise fitness landscapes reveal the fitness impact of gene expression noise. Nat Commun 2019;10:3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng J, Payne JL, Wagner A.. Cryptic genetic variation accelerates evolution by opening access to diverse adaptive peaks. Science 2019;365:347–53. [DOI] [PubMed] [Google Scholar]
- 8.Weinreich DM, Delaney NF, Depristo MA. et al. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 2006;312:111–4. [DOI] [PubMed] [Google Scholar]
- 9.Goulart CP, Mahmudi M, Crona KA. et al. Designing antibiotic cycling strategies by determining and understanding local adaptive landscapes. PLoS One 2013;8:e56040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hietpas RT, Bank C, Jensen JD. et al. Shifting fitness landscapes in response to altered environments. Evolution 2013;67:3512–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chevereau G, Dravecká M, Batur T. et al. Quantifying the determinants of evolutionary dynamics leading to drug resistance. PLoS Biol 2015;13:e1002299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer AC, Toprak E, Baym M. et al. Delayed commitment to evolutionary fate in antibiotic resistance fitness landscapes. Nat Commun 2015;6:7385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodrigues JV, Bershtein S, Li A. et al. Biophysical principles predict fitness landscapes of drug resistance. Proc Natl Acad Sci USA 2016;113:E1470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salverda MLM, Koomen J, Koopmanschap B. et al. Adaptive benefits from small mutation supplies in an antibiotic resistance enzyme. Proc Natl Acad Sci USA 2017;114:12773–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palmer AC, Chait R, Kishony R.. Nonoptimal gene expression creates latent potential for antibiotic resistance. Mol Biol Evol 2018;35:2669–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Lalić J, Baeza-Centurion P. et al. Changes in gene expression predictably shift and switch genetic interactions. Nat Commun 2019;10:3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das SG, Direito SOL, Waclaw B. et al. Predictable properties of fitness landscapes induced by adaptational tradeoffs. Elife 2020;9:e55155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spiller B, Gershenson A, Arnold FH. et al. A structural view of evolutionary divergence. Proc Natl Acad Sci USA 1999;96:12305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimotohno A, Oue S, Yano T. et al. Demonstration of the importance and usefulness of manipulating non-active-site residues in protein design. J Biochem 2001;129:943–8. [DOI] [PubMed] [Google Scholar]
- 20.Bloom JD, Silberg JJ, Wilke CO. et al. Thermodynamic prediction of protein neutrality. Proc Natl Acad Sci USA 2005;102:606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacquier H, Birgy A, Le Nagard H. et al. Capturing the mutational landscape of the beta-lactamase TEM-1. Proc Natl Acad Sci USA 2013;110:13067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Figliuzzi M, Jacquier H, Schug A. et al. Coevolutionary landscape inference and the context-dependence of mutations in beta-lactamase TEM-1. Mol Biol Evol 2016;33:268–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolognesi B, Faure AJ, Seuma M. et al. The mutational landscape of a prion-like domain. Nat Commun 2019;10:4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faber MS, Wrenbeck EE, Azouz LR. et al. Impact of in vivo protein folding probability on local fitness landscapes. Mol Biol Evol 2019;36:2764–77. [DOI] [PubMed] [Google Scholar]
- 25.Bertram J, Masel J.. Evolution rapidly optimizes stability and aggregation in lattice proteins despite pervasive landscape valleys and mazes. Genetics 2020;214:1047–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keren H, Lev-Maor G, Ast G.. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 2010;11:345–55. [DOI] [PubMed] [Google Scholar]
- 27.Ke S, Shang S, Kalachikov SM. et al. Quantitative evaluation of all hexamers as exonic splicing elements. Genome Res 2011;21:1360–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barbosa-Morais NL, Irimia M, Pan Q. et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 2012;338:1587–93. [DOI] [PubMed] [Google Scholar]
- 29.Merkin J, Russell C, Chen P. et al. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 2012;338:1593–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gamazon ER, Stranger BE.. Genomics of alternative splicing: evolution, development and pathophysiology. Hum Genet 2014;133:679–87. [DOI] [PubMed] [Google Scholar]
- 31.Lee Y, Rio DC.. Mechanisms and regulation of alternative pre-mRNA splicing. Annu Rev Biochem 2015;84:291–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Julien P, Miñana B, Baeza-Centurion P. et al. The complete local genotype-phenotype landscape for the alternative splicing of a human exon. Nat Commun 2016;7:11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Firnberg E, Labonte JW, Gray JJ. et al. A comprehensive, high-resolution map of a gene’s fitness landscape. Mol Biol Evol 2014;31:1581–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sarkisyan KS, Bolotin DA, Meer MV. et al. Local fitness landscape of the green fluorescent protein. Nature 2016;533:397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Canale AS, Cote-Hammarlof PA, Flynn JM. et al. Evolutionary mechanisms studied through protein fitness landscapes. Curr Opin Struct Biol 2018;48:141–8. [DOI] [PubMed] [Google Scholar]
- 36.Smith JM.Natural selection and the concept of a protein space. Nature 1970. 7;225:563–4. [DOI] [PubMed] [Google Scholar]
- 37.Macken CA, Perelson AS.. Protein evolution on rugged landscapes. Proc Natl Acad Sci USA 1989;86:6191–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagner A.Robustness and evolvability: a paradox resolved. Proc Biol Sci B 2008;275:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li C, Qian W, Maclean CJ. et al. The fitness landscape of a tRNA gene. Science 2016;352:837–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aguilar-Rodríguez J, Peel L, Stella M. et al. The architecture of an empirical genotype-phenotype map. Evolution 2018;72:1242–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Domingo J, Diss G, Lehner B.. Pairwise and higher-order genetic interactions during the evolution of a tRNA. Nature 2018;558:117–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pressman AD, Liu Z, Janzen E. et al. Mapping a systematic ribozyme fitness landscape reveals a frustrated evolutionary network for self-aminoacylating RNA. J Am Chem Soc 2019;141:6213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bajić D, Vila JCC, Blount ZD. et al. On the deformability of an empirical fitness landscape by microbial evolution. Proc Natl Acad Sci USA 2018;115:11286–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bendixsen DP, Collet J, Østman B. et al. Genotype network intersections promote evolutionary innovation. PLoS Biol 2019;17:e3000300.31136568 [Google Scholar]
- 45.Flynn JM, Rossouw A, Cote-Hammarlof P. et al. Comprehensive fitness maps of Hsp90 show widespread environmental dependence. Elife 2020;9:e53810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hietpas RT, Jensen JD, Bolon DNA.. Experimental illumination of a fitness landscape. Proc Natl Acad Sci USA 2011;108:7896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boucher JI, Cote P, Flynn J. et al. Viewing protein fitness landscapes through a next-gen lens. Genetics 2014;198:461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melamed D, Young DL, Miller CR. et al. Combining natural sequence variation with high throughput mutational data to reveal protein interaction sites. PLoS Genet 2015;11:e1004918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinberg B, Ostermeier M.. Shifting fitness and epistatic landscapes reflect trade-offs along an evolutionary pathway. J Mol Biol 2016;428:2730–43. [DOI] [PubMed] [Google Scholar]
- 50.Aguilar-Rodríguez J, Payne JL, Wagner A.. A thousand empirical adaptive landscapes and their navigability. Nat Ecol Evol 2017;1:45. [DOI] [PubMed] [Google Scholar]
- 51.Starr TN, Thornton JW.. Exploring protein sequence-function landscapes. Nat Biotechnol 2017;35:125–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diss G, Landry C R.. Combining the Dihydrofolate Reductase Protein-Fragment Complementation Assay with Gene Deletions to Establish Genotype-to-Phenotype Maps of Protein Complexes and Interaction Networks. Cold Spring Harb Protoc 2016;11:pdb.prot090035. [DOI] [PubMed] [Google Scholar]
- 53.Filteau M, Vignaud H, Rochette S. et al. Multi-scale perturbations of protein interactomes reveal their mechanisms of regulation, robustness and insights into genotype-phenotype maps. Brief Funct Genomics 2016;15:130–7. [DOI] [PubMed] [Google Scholar]
- 54.Diss G, Lehner B.. The genetic landscape of a physical interaction. Elife 2018;7:e32472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bank C, Hietpas RT, Jensen JD. et al. A systematic survey of an intragenic epistatic landscape. Mol Biol Evol 2015;32:229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bendixsen DP, Østman B, Hayden EJ.. Negative epistasis in experimental RNA fitness landscapes. J Mol Evol 2017;85:159–68. [DOI] [PubMed] [Google Scholar]
- 57.Ferretti L, Weinreich D, Tajima F. et al. Evolutionary constraints in fitness landscapes. Heredity (Edinb) 2018;121:466–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gonzalez CE, Roberts P, Ostermeier M.. Fitness effects of single amino acid insertions and deletions in TEM-1 β-lactamase. J Mol Biol 2019;431:2320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mehlhoff JD, Ostermeier M.. Biological fitness landscapes by deep mutational scanning. Methods Enzymol 2020;643:203–24. [DOI] [PubMed] [Google Scholar]
- 60.Landry CR, Rifkin SA.. Chromatin regulators shape the genotype-phenotype map. Mol Syst Biol 2010;6:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang G, Anderson DW, Baier F. et al. Higher-order epistasis shapes the fitness landscape of a xenobiotic-degrading enzyme. Nat Chem Biol 2019;15:1120–8. [DOI] [PubMed] [Google Scholar]
- 62.Romero PA, Arnold FH.. Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol 2009;10:866–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ho SN, Hunt HD, Horton RM. et al. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989;77:51–9. [DOI] [PubMed] [Google Scholar]
- 64.Wäneskog M, Bjerling P.. Multi-fragment site-directed mutagenic overlap extension polymerase chain reaction as a competitive alternative to the enzymatic assembly method. Anal Biochem 2014;444:32–7. [DOI] [PubMed] [Google Scholar]
- 65.Hejlesen R, Füchtbauer EM.. Multiple site-directed mutagenesis via simple cloning by prolonged overlap extension. Biotechniques 2020;68:345–8. [DOI] [PubMed] [Google Scholar]
- 66.Kadkhodaei S, Memari HR, Abbasiliasi S. et al. Multiple overlap extension PCR (MOE-PCR): an effective technical shortcut to high throughput synthetic biology. RSC Adv 2016;6:66682–94. [Google Scholar]
- 67.Young L, Dong Q.. Two-step total gene synthesis method. Nucleic Acids Res 2004;32:e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang W, Malcolm BA.. Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange(TM) site-directed mutagenesis. Biotechniques 1999;26:680–2. [DOI] [PubMed] [Google Scholar]
- 69.Liu H, Naismith JH.. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 2008;8:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Edelheit O, Hanukoglu A, Hanukoglu I.. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol 2009;9:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Trehan A, Kiełbus M, Czapinski J. et al. REPLACR-mutagenesis, a one-step method for site-directed mutagenesis by recombineering. Sci Rep 2016;6:19121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kuo T-Y, Tsai C-C, Fu H-W.. Enhanced mutant screening in one-step PCR-based multiple site-directed plasmid mutagenesis by introduction of silent restriction sites for structural and functional study of proteins. Biol Proced Online 2017;19:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zeng F, Zhang S, Hao Z. et al. Efficient strategy for introducing large and multiple changes in plasmid DNA. Sci Rep 2018;8:1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hallak LK, Berger K, Kaspar R. et al. Efficient method for site-directed mutagenesis in large plasmids without subcloning. PLoS One 2017;12:e0177788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hietpas R, Roscoe B, Jiang L. et al. Fitness analyses of all possible point mutations for regions of genes in yeast. Nat Protoc 2012;7:1382–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bershtein S, Segal M, Bekerman R. et al. Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein. Nature 2006;444:929–32. [DOI] [PubMed] [Google Scholar]
- 77.Cárcamo E, Roldán-Salgado A, Osuna J. et al. Spiked genes: a method to introduce random point nucleotide mutations evenly throughout an entire gene using a complete set of spiked oligonucleotides for the assembly. ACS Omega 2017;2:3183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li C, Zhang J.. Multi-environment fitness landscapes of a tRNA gene. Nat Ecol Evol 2018;2:1025–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fowler DM, Stephany JJ, Fields S.. Measuring the activity of protein variants on a large scale using deep mutational scanning. Nat Protoc 2014;9:2267–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ge L, Rudolph P.. Simultaneous introduction of multiple mutations using overlap extension PCR. Biotechniques 1997;22:28–30. [DOI] [PubMed] [Google Scholar]
- 81.Gibson DG.Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 2011;498:349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hsieh P-C, Vaisvila R.. Protein engineering: single or multiple site-directed mutagenesis. Methods Mol Biol 2013;978:173–86. [DOI] [PubMed] [Google Scholar]
- 83.Bush K, Jacoby GA.. Updated functional classification of beta-lactamases. Antimicrob Agents Chemother 2010;54:969–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dhar R, Bergmiller T, Wagner A.. Increased gene dosage plays a predominant role in the initial stages of evolution of duplicate TEM-1 beta lactamase genes. Evolution 2014;68:1775–91. [DOI] [PubMed] [Google Scholar]
- 85.Zheng L, Baumann U, Reymond J-L.. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 2004;32:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vovis GF, Lacks S.. Complementary action of restriction enzymes endo R · DpnI and endo R · DpnII on bacteriophage f1 DNA. J Mol Biol 1977;115:525–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analysed in support of this research. The sequencing data generated by Sanger sequencing to confirm the clones are shown in the article.