

Abstract
Objective:
The role of glycolytic enzyme pyruvate kinase M2 (PKM2) in SMC phenotype switching and neointimal hyperplasia is poorly understood. We determined the role of PKM2 in SMC phenotype switching and neointimal hyperplasia.
Approach and Results:
We show that PKM2 is expressed in the SMC-rich neointima of the murine carotid artery and peri-strut areas in bare metal stented human coronary arteries. Platelet-derived growth factor (PDGF-BB) stimulation upregulates the expression of PKM2 in cultured murine and human coronary SMC. To provide conclusive evidence for PKM2 in SMC function, we generated SMC-specific PKM2−/− mutant strain. We report that PKM2 deletion in SMC reduces injury-induced neointimal hyperplasia by inhibiting SMC proliferation and migration, suppressing synthetic phenotype, and reducing aerobic glycolysis associated with decreased ERK, mTOR, and STAT3 signaling. Furthermore, we show that nuclear PKM2 interacts with STAT3 and β-catenin and regulates transcription of MEK5, cyclin D1, GLUT1, and LDHA. Treatment of human coronary SMC with ML265, an activator that induces PKM2 tetramerization and blocks its nuclear translocation, inhibited proliferation, migration, and phenotypic switching. Perivascular application of PKM2 activator reduced neointimal hyperplasia in mice.
Conclusion:
These findings reveal that PKM2 is a key regulator of SMC function in vascular remodeling and implicate PKM2 as a potential target to reduce neointimal hyperplasia.
Keywords: Pyruvate kinase M2, Smooth muscle cell, Phenotype switching, Neointimal hyperplasia
Graphical Abstract
Introduction
Percutaneous coronary intervention (PCI) with balloon angioplasty, often followed by stent implantation, is the most commonly performed procedure for reopening of an atherosclerotic artery. Despite the development of bioresorbable drug-eluting stents, in-stent restenosis limits PCI's success.1 Smooth muscle cells (SMC), the arterial tunica media's primary cell type, have remarkable phenotype plasticity. SMC transition from contractile to synthetic proliferative phenotype plays a significant role in developing in-stent restenosis, neo-atherosclerosis, and other vascular complications.2 Understanding the cellular and molecular mechanisms that regulate SMC plasticity is, therefore, of considerable clinical importance.
SMC undergoes substantial metabolic reprogramming following activation to rapidly provide high energy and meet biosynthetic demands for cell proliferation.3 Increased glycolytic flux is crucial for the bioenergetic shift during SMC proliferation and migration.4 Pyruvate kinase (PK) catalyzes the final step of glycolysis, the transfer of a high-energy phosphate group from phosphoenolpyruvate to ADP, resulting in the formation of ATP and pyruvate. PK exists in four different isoforms (PKM1, PKM2, PKR, and PKL) and is encoded by two distinct genes, PKLR and PKM, in mammals.5 The PKM1 isoform is expressed in most adult tissues with high catabolic demand, such as muscle and brain, whereas PKM2 is primarily expressed in highly proliferative cells such as stem cells and tumor cells characterized by high anabolic demand.6 Unlike other isoforms of PK that function only as tetramers, PKM2 exists in both tetrameric and dimeric forms composed of the same monomers but with different biological activities. While PKM1 and tetrameric PKM2 favor ATP production from oxidative phosphorylation (OXPHOS) through the TCA cycle, dimeric PKM2 regulates aerobic glycolysis.7 Although aerobic glycolysis yields fewer ATP molecules compared to OXPHOS, the ATP generation rate is faster in aerobic glycolysis than OXPHOS.8
PKM2 upregulation has been reported in SMC within murine atherosclerotic plaques and many types of human cancers.9, 10 Dimeric PKM2 is known to facilitate lactate production by regulating aerobic glycolysis.11 When our studies were in progress, another study showed that lactate might promote the SMC synthetic phenotype.12 In addition to its glycolytic activity, several studies suggest that PKM2 has a non-glycolytic activity that may contribute to SMC proliferation. An extracellular signal-regulated kinase (ERK) was shown to phosphorylate PKM2 at Ser 37 and induce its translocation to the nucleus.13 Nuclear dimeric PKM2 can act as a coactivator for transcription factors implicated in the activation of the HIF-1α, STAT3, and β-catenin signaling pathways.14, 15 Despite all these studies, conclusive evidence on the role of PKM2 in SMC biology is lacking. Herein, we generated SMC-specific PKM2−/− mutant strain to provide conclusive evidence for PKM2 in SMC phenotypic switching and neointimal hyperplasia.
Herein, we report that the genetic ablation of PKM2, specifically in murine SMC inhibited cytokine-dependent SMC synthetic phenotype, proliferation, and neointimal hyperplasia in vivo. Mechanistically, we show that nuclear PKM2 interacts with STAT3 and β-catenin and regulates transcription of MEK5, cyclin D1, GLUT1, and LDHA. Treatment of human coronary SMC with ML265, an activator that induces PKM2 tetramerization and blocks its nuclear translocation, inhibited proliferation, migration, and phenotypic switching by downregulating the STAT3 and beta-catenin pathways. Finally, the perivascular application of ML265 strongly reduced neointimal hyperplasia in mice.
Material and Methods
Detailed information on materials and methods is available in the Data Supplement.
Animals
The University of Iowa Animal Care and Use Committee approved all procedures, and studies were performed according to the current Animal Research: Reporting of In Vivo Experiment guidelines (http://www.nc3rs.org/ARRIVE). Details of generating smooth muscle cell-specific PKM2 deficient mice are provided in the Method section in the Data Supplement.
Statistical analysis
The statistical significance was assessed using either unpaired t-test or 1-way or 2-way ANOVA. P < 0.05 was considered significant (Data Supplement for details).
Data Availability
This article adheres to the American Heart Association Journal implementation of the Transparency and Openness Promotion Guidelines. Primary data that support the findings of this study are available from the first author or corresponding author upon request.
Results
PKM2 is expressed in neointimal SMCs and mediates neointimal hyperplasia in mice
To evaluate the potential role of PKM2 in SMC activation and function, we first analyzed the expression pattern of PKM2 in SMCs in the experimental carotid wire-injury model. Immunohistochemistry revealed that mean fluorescence intensity/ cell for PKM2 was higher in the injured carotid artery at 28 days after wire injury when compared to the uninjured control artery (P<0.05, Figure 1A). Next, we examined PKM2 expression in stented human coronary arteries. We focused on samples in which bare-metal stents (BMS) had been implanted for 150 and 361 days (mean duration 286 ± 82 days). Patient coronary artery disease status and stent characteristics are shown in Table 1 in the Data Supplement. Utilizing immunohistochemistry, we found that PKM2 was expressed in the vicinity of SMC-rich neointima and peri-strut areas (Figure 1A in the Data Supplement). PKM2 expression was higher in the neointima as compared to media (Figure 1A in the Data Supplement). In line with these observations, using immunoblotting, we found a time-dependent increase in PKM2 expression in PDGF-BB-stimulated murine aortic SMCs (Figure 1B) and human coronary artery SMCs (HCaSMC) (Figure 1B in the Data Supplement). These observations were confirmed in parallel by immunofluorescence (Figure 1C in the Data Supplement).
Figure 1. PKM2 is expressed in neointimal SMCs and mediates neointimal hyperplasia in mice.

A, The left panels show representative images showing double immunostaining for PKM2 (red) and αSMA (green) in the uninjured and injured carotid artery of wild-type mice harvested after 28 days of injury (n = 6/group). Nuclei are counterstained with Hoechst (blue). Scale bars: 200 μm. The right panel shows the quantification of the PKM2 fluorescence intensity. B, Representative immunoblots and densitometric analysis of PKM2 expression levels in serum-deprived mouse aortic SMCs stimulated with or without PDGF-BB for indicated time points. β-actin was used as a loading control. n = 4/group. C, Immunoblot analysis of PKM2, PKM1, and β-actin in SMC isolated from PKM2fl/fl and PKM2fl/fl-SMMHCCreERT2 mice treated with tamoxifen. #1 and #2 represents samples from two individual mice. D, The left panels show representative photomicrographs of Verhoeff’s van Gieson stained carotid artery sections from PKM2fl/fl and PKM2fl/flSMMHC-CreERT2mice after 28 days of the injury. n = 9/group. Scale bars: 200 μm. The right panels show quantification of intimal area, medial area, and a ratio of intimal to the medial area. Each dot represents a single mouse. E, The left panels show representative BrdU-positive cells (green) counterstained with αSMA (red) and Hoechst (blue). The right panel shows the quantification of percent BrdU-positive cells. Scale bars: 200 μm; n = 6/group. F, The left panels show representative TUNEL-positive cells (green) counterstained with Hoechst (blue). The right panel shows the quantification of TUNEL-positive cells. Scale bars: 200 μm; n = 6/group. Values are represented as mean ± SEM. Statistical analysis: B, One-way ANOVA followed by Bonferroni's post hoc test; A, C, D, E, and F, unpaired Student's t-test. *P < 0.05 versus quiescent.
To confirm a definitive role for PKM2 in SMC biology and neointimal hyperplasia, we generated novel tamoxifen-inducible SMC-specific PKM2-deficient mice (PKM2fl/flSMMHC-CreERT2; Figure 2A in the Data Supplement). Genomic PCR confirmed the presence of the Cre gene in PKM2fl/fl mice (Figure 2B in the Data Supplement). Western blotting showed that treatment with tamoxifen led to ablation of nearly 85% of PKM2 protein in SMC from PKM2fl/flSMMHC-CreERT2 mice when compared to PKM2fl/fl mice (Figure 1C). Bodyweight (not shown) and complete blood count (Table 2 in the Data Supplement) were comparable between PKM2fl/flSMMHC-CreERT2 mice and control PKM2fl/fl mice. Susceptibility to neointimal hyperplasia was studied at 28 days after wire injury in the carotid arteries of male PKM2fl/fl SMMHC-CreERT2 and PKM2fl/fl mice. PKM2fl/fl SMMHC-CreERT2 mice exhibited a decrease in neointimal area as well as neointima/media ratio (P<0.05 versus PKM2fl/fl; Figure 1D). The medial area was comparable between PKM2fl/fl SMMHC-CreERT2 and PKM2fl/fl mice (Figure 1D). Immunostaining revealed that PKM2 expression was minimal and mainly restricted to the luminal and adventitial area in PKM2fl/flSMMHC-CreERT2 mice (Figure 2C in the Data Supplement). Since the decrease in the neointimal area could be due to decreased neointimal cell proliferation or increased apoptosis, we analyzed SMC proliferation and apoptosis in wire-injured carotid artery sections. We found that PKM2 deletion suppressed SMC proliferation but did not affect apoptosis (Figure 1E and 1F).
SMC-derived PKM2 potentiates proliferation, migration, and phenotypic switching
To ablate PKM2, isolated SMC from PKM2fl/fl and PKM2fl/fl SMMHC-CreERT2 mice were treated with 600 nM 4-hydroxytamoxifen for 72 h. Western blotting showed that tamoxifen treatment led to ablation of nearly 85% of PKM2 protein in SMC isolated from PKM2fl/flSMMHC-CreERT2 mice when compared to PKM2fl/fl mice (Figure 2D in the Data Supplement). To obtain further mechanistic insights, we used PDGF-BB-stimulated cultured mouse aortic SMC. PDGF-BB-stimulated aortic SMC from PKM2fl/fl SMMHC-CreERT2 and PKM2fl/fl mice treated with tamoxifen were subjected to BrdU incorporation assays. We found that SMC lacking PKM2 proliferated less when compared to SMC containing PKM2 subjected to PDGF-BB (21.4% ± 1.7% vs. 34.3% ± 3.4%; Figure 2A). PDGF-BB-stimulated aortic SMC from PKM2fl/flSMMHC-CreERT2 mice exhibited a decrease in SMC migration at 12 hours in comparison with control PKM2fl/fl mice (25.7 ± 3.1% vs. 41.1 ± 2.9 %; Figure 2A). SMC modulation from a differentiated "contractile" to a dedifferentiated "synthetic" proliferative phenotype contributes to SMC proliferation and migration. To elucidate the effect of PKM2 deletion on SMC phenotypic switching, aortic SMCs from PKM2fl/fl SMMHC-CreERT2 and PKM2fl/fl mice were immunostained for contractile (SM22α and SM-MHC) and synthetic (vimentin and osteopontin) phenotype markers.16 We found that the expression levels of contractile markers (SM22α and SM-MHC) were higher, whereas synthetic markers (vimentin and osteopontin) were lower in SMC isolated from PKM2fl/flSMMHC-CreERT2 mice as compared with SMC from control PKM2fl/fl mice (Figure 2B). These results were confirmed in parallel in SMC lysates by Western blotting (Figure 2C). To determine whether the observed effect of PKM2 deletion is not limited to PDGF-BB, we stimulated SMC with monocyte chemotactic protein 1 (MCP1), a chemokine produced at the site of vascular injury and has chemotactic and mitogenic activity toward SMC.17, 18 Consistent with PDGF-BB findings, PKM2 deficiency led to a decrease in MCP1-mediated SMC proliferation, migration, and phenotypic switching (Figure 3 in the Data Supplement). Together, these results suggest that SMC-specific PKM2 promotes intimal hyperplasia via potentiating SMC phenotypic switching, proliferation, migration.
Figure 2. SMC-derived PKM2 potentiates proliferation, migration, and phenotypic switching.

Aortic SMCs isolated from PKM2fl/fl and PKM2fl/flSMMHC-CreERT2 mice were treated with 600 nM 4-hydroxytamoxifen for 72 h. For BrdU incorporation assay and SMC phenotypic switching, serum-starved cells were stimulated with PDGF-BB (20 ng/ml) for 24 h. For migration assay, serum-starved cells were stimulated with PDGF-BB (20 ng/ml) for 12 h. A, The left upper panels show representative BrdU-positive cells co-stained with αSMA (green) and Hoechst (blue). Scale bars: 50 μm. The left lower panels show representative phase-contrast images of SMC migration in the scratch assay. The right panel shows the quantification of BrdU-positive cells to the total number of cells and quantification of the migrated area (n = 5/group). Scale bars: 500 μm. B, The left panels show representative immunostaining images for contractile proteins (SM22α, green; and SM-MHC, green) and synthetic proteins (vimentin, red; and osteopontin, red). Scale bars: 50 μm. The right panels show quantification of the fluorescence intensity for SM22α, SM-MHC, vimentin, and osteopontin (n = 5 per group). C, Representative immunoblots and densitometric analysis of SM22α, SM-MHC, vimentin, and osteopontin (n = 4 per group). Values are expressed as mean ± SEM. Statistical analysis: A, two-way ANOVA followed by uncorrected Fisher's LSD test; B and C, unpaired Student’s t-test.
SMC specific PKM2 deletion attenuates ERK, mTOR, and STAT3 signaling
PDGF-BB is known to induce phosphorylation of PKM2 at tyrosine 105 (Y105), which promotes PKM2 nuclear translocation and inhibits PKM2 tetramerization.13 Our results show that PDGF-BB stimulates PKM2 phosphorylation in HCaSMCs. In DSS-crosslinking experiments we found that PDGF-BB stimulation led to a marked increase in the expression of PKM2 dimers and a decrease in PKM2 tetramers as compared to quiescent SMCs (Figure 4 in the Data Supplement). Previous studies in macrophages and cancer cells suggest that PKM2 can activate the ERK/MAPK,19, 20 and mTOR signal pathway21 by phosphorylating STAT3.22 We determined whether specific deletion of PKM2 in SMCs reduces ERK, mTOR, and STAT3 expression levels. PDGF-BB induces two peaks of ERK activation: the first is observed within 15 to 30 minutes, whereas the second is observed between 2 to 6 hours.23 Since we observed increased PKM2 expression at 6 h of PDGF-BB stimulation, we used 6 h time point to determine change in ERK, Akt and mTOR phosphorylation. Using immunoblotting, we found that PDGF-BB stimulated aortic SMCs from PKM2fl/flSMMHC-CreERT2 mice exhibited reduced phospho-ERK1/2 (0.38-fold), phospho-mTOR (0.64-fold), and phospho–STAT3 (0.65-fold) in comparison with control PKM2fl/fl mice (Figure 3A). It is known that PKM2 mediated STAT3 phosphorylation regulates transcription of MEK5 (which plays an important role in cell proliferation), β-catenin transcriptionally induce expression of cyclin D124 and c-Myc dependent expression of glucose transporter 1 (GLUT1) and lactate dehydrogenase A (LDHA).13 Herein, we found that SMC specific PKM2 deletion reduces PDGF-BB induced transcription of MEK5, Cyclin D1, GLUT1, and LDHA (Figure 3B).
Figure 3. SMC specific PKM2 deletion attenuates ERK, mTOR, and STAT3 signaling.

Quiescent SMCs isolated from PKM2fl/fl and PKM2fl/flSMMHC-CreERT2 mice were stimulated with PDGF-BB (20 ng/ml) for 6 h. A, Representative immunoblots and densitometric analysis for p-ERK1/2, total ERK1/2, p-MTOR, total mTOR, p-STAT3, total STAT3, and β-actin (n = 4/group). B, Real-time quantitative PCR analysis of MEK5, LDHA, Cyclin D1, and GLUT1 (n = 5/group). The results are presented as changes in PCR products normalized to GAPDH. C, The bar graph shows lactate levels in cell supernatant (n = 4/group). D, SMCs were seeded in 96 well XF cell culture microplate. OCR and ECAR were measured in response to PDGF-BB (n = 4). Values are expressed as mean ± SEM. Statistical analysis- two-way ANOVA followed by uncorrected Fisher's LSD test. *P < 0.05 vs. PDGF-BB stimulated PKM2fl/fl cells.
A previous study suggested that lactate may promote synthetic phenotype in SMC.12 PKM2 is known to facilitate lactate production by regulating aerobic glycolysis.11 We determined whether PKM2 deletion in SMCs reduces lactate production. We found that PDGF-BB induced lactate production was significantly reduced in PKM2fl/flSMMHC-CreERT2 mice when compared to control PKM2fl/fl mice (Figure 3C). Cells produce ATP via two major energy-producing pathways: glycolysis and oxidative phosphorylation. The glycolytic pathway converts glucose to pyruvate. One fate of the pyruvate is reduction to lactate in the cytosol in an oxygen-independent biochemical reaction resulting in ATP production and net proton production. To maintain the intracellular pH, protons are pumped out of the cell which causes extracellular acidification. Because PKM2 is a glycolytic enzyme, we next determined whether the observed inhibitory effect of PKM2 deletion on SMC proliferation is associated with changes in OXPHOS and aerobic glycolysis. We used the Seahorse extracellular flux analyzer to quantify the oxygen consumption rate (OCR), and extracellular acidification rate (ECAR), which are indirect mitochondrial OXPHOS measures aerobic glycolysis, respectively. Oligomycin, FCCP, and a mix of rotenone A/ Antimycin were serially injected to measure maximal respiration and nonmitochondrial respiration. PKM2 deficiency in SMCs led to a decrease in PDGF-BB-induced glycolytic activity while OXPHOS remained unaltered, indicating that energy was mainly generated from mitochondrial respiration (Figure 3D). Additionally, we found that proton production rate (PPR) and ATP production rate was significantly reduced in SMC isolated from PKM2fl/flSMMHC-CreERT2 mice when compared to control PKM2fl/fl mice. Spare respiratory capacity, which measures the ability of a cell to respond to increased energy demand, was also slightly reduced, albeit not significant (Figure 5 in the Data Supplement). Together, these results suggest that PKM2 regulates ERK, mTOR, and STAT3 signaling during SMC proliferation via both its glycolytic and non-glycolytic activities.
PKM2 activator inhibits STAT3, and β catenin induced transcription of MEK5 and LDHA and suppresses the SMC synthetic phenotype in cultured human coronary artery SMC
In order to examine the functional significance of targeting PKM2 during SMC proliferation, we used ML265, a small molecule activator that induces PKM2 tetramerization.25 We first determined the minimal dose of ML265 that inhibits cell proliferation in a MTT assay. We observed that PDGF-BB-induced proliferation was strongly reduced in SMC pretreated at a dose of 50 μM, but not at lower doses (Figure 6A in the Data Supplement). Similarly, in the BrdU incorporation assay, no significant differences were observed at a dose of 25 μM (Figure 6B in the Data Supplement). Our findings are in line with other studies, where they have used 50-100 μM of PKM2 activator in different cell types.14, 26 Using SMC-specific PKM2-deficient mice, we further determined any off-target effects of ML265 (at 50 μM) in SMC biology. Tamoxifen-treated SMC derived from PKM2fl/flSMMHC-CreERT2 mice were treated with ML265 prior to PDGF-BB stimulation, and SMC proliferation and migration were measured. No inhibitory effects of ML265 on SMC proliferation or migration were observed in SMC lacking PKM2 (Figure 6C in the Data Supplement), suggesting that ML265 at a dose of 50 μM does not exert any off-target effect. Therefore, in all experiments, we used 50 μM ML265 to induce PKM2 tetramerization. Next, we determined whether ML265 inhibits nuclear localization of PKM2 in PDGF-BB-stimulated HCaSMC. We found an increase in PKM2 levels in the nuclear fraction from PDGF-BB-stimulated HCaSMC, which was strongly reduced by ML265 pre-treatment (Figure 4A). No significant difference in PKM2 levels was observed in the cytosolic fraction. Next, we determined the effect of ML265 on SMC proliferation, migration, and phenotypic switching in PDGF-BB-stimulated HCaSMC. In BrdU incorporation and wound-healing assay, a significant decrease in HCaSMC proliferation and migration was observed in ML265 treated groups when compared to vehicle control (Figure 4B). Furthermore, ML265 significantly increased the expression levels of contractile markers, whereas synthetic markers were decreased (Figure 4C). The results were confirmed in parallel in HCaSMC lysates by Western blotting (Figure 4D). To determine if the observed effects also not limited to human coronary SMC, we determined the effect of ML265 on human aortic SMC. We found that similar to HCaSMC, ML265 reduced proliferation and migration, and phenotypic switching of PDGF-BB stimulated human aortic SMC (Figure 7 in the Data Supplement).
Figure 4: PKM2 activator inhibits its nuclear transport and prevents PDGF-BB induced phenotypic switching in human coronary artery SMC.

HCaSMCs were pretreated with ML265 (50 μM) for 1 h and then stimulated with PDGF-BB (20 ng/ml) for 6 h (PKM2 nuclear translocation), 12 h (SMC migration) or 24 h (BrdU incorporation and SMC phenotypic switching). A, Cytosolic and nuclear fractions were isolated. The left panels show representative immunoblot of PKM2, Lamin B1, GAPDH, and β-actin. The right panel shows the densitometric analysis for PKM2/GAPDH in cytosolic extract and PKM2/Lamin B1 in nuclear extract (n = 4/group). B, The top panels show representative BrdU-positive cells co-stained with αSMA (green) and Hoechst (blue). Scale bars: 50 μm. The bottom panels show representative phase-contrast images of SMC migration in the scratch assay. Scale bars: 500 μm. The right panel shows the quantification of BrdU-positive cells to the total number of cells (n = 6/group) and migrated area (n = 6-7/group). C, The left panels show representative immunostaining images for contractile proteins (SM22α, green; and SM-MHC, green) and synthetic proteins (vimentin, red; and osteopontin, red). Scale bars: 100 μm. The right panel shows the quantification of the fluorescence intensity for SM22α, SM-MHC, vimentin, and osteopontin (n = 6/group). D, Representative immunoblots and densitometric analysis of SM22α, SM-MHC, vimentin, and osteopontin (n = 5 /group). Values are mean ± SEM. Statistical analysis- One-way ANOVA followed by Bonferroni’s post hoc test. *P < 0.05 vs. quiescent; #P<0.05 vs. PDGF-BB stimulated cells.
PKM2 translocates to the nucleus (dependent on ERK1/2 activity), where it activates HIF-1α, STAT3, and β-catenin signaling pathways.14, 15 We investigated whether nuclear translocation of PKM2 is ERK and Akt-dependent in PDGF-BB-stimulated HCaSMC. Immunoblotting revealed that inhibition of the ERK1/2 pathway results in a significant decrease in nuclear PKM2 expression, whereas inhibition of Akt signaling showed only a marginal effect (Figure 5A). The absence of GAPDH expression in the immunoblots from the same biological samples suggests that nuclear fraction proteins were not contaminated from the cytoplasmic fraction. PKM2 interacts with several transcription factors to induce gene transcription. Previously it was demonstrated that PKM2 interact and activates STAT3 and this interaction is required to mediate the effects of PKM2 on upregulation of MEK5 transcription.27 In addition to STAT3, PKM2 also interacts with β-catenin and this interaction is required for both proteins to be recruited to the Cyclin D1 promoter and subsequent cyclin D1 expression.28 Based on these observations, we investigated whether PKM2 interacts with STAT3 and β-catenin in PDGF-BB-stimulated HCaSMC. Co-immunoprecipitation showed that both STAT3 and β-catenin interact with PKM2 (Figure 5B). We found that inducing PKM2 tetramerization reduces STAT3 phosphorylation (Figure 5C). To determine if nuclear PKM2 regulates transcription of MEK5 by phosphorylating STAT3, we used chromatin immunoprecipitation (ChIP) assays. We found that PKM2 interacted with the MEK5 promoter and the binding efficiency of PKM2 was significantly increased upon PDGF-BB stimulation. ML265 treatment significantly decreased binding of PKM2 to MEK5 promoter in PDGF-BB stimulated cells but did not showed any effect in quiescent SMCs. (Figure 5D). Furthermore, we found inducing PKM2 tetramerization reduced transcription of MEK5, Cyclin D1, GLUT1, and LDHA in PDGF-BB-stimulated HCaSMC (Figure 5E). We also determined whether inducing PKM2 tetramerization reduces lactate production and thereby may suppress the synthetic phenotype. We found that PDGF-BB stimulation led to a significant increase in lactate production (~2.7 fold) in HCaSMC that was inhibited by ML265 pre-treatment (Figure 5F). Next, we determined whether the inhibitory effect of ML265 on SMC proliferation is associated with changes in OXPHOS and aerobic glycolysis. We found a significant increase in OCR, ECAR, PPR, ATP production rate and spare respiratory capacity in PDGF-BB stimulated HCaSMC when compared to control (Figure 5G and Figure 8 in the Data Supplement). ML265 pre-treatment had little effect on OCR; however, ECAR, PPR, ATP production rate and spare respiratory capacity were significantly reduced in ML265 treated groups when compared to PDGF-BB treated cells (Figure 5G and Figure 8 in the Data Supplement). Mechanistically, these results suggest that SMC-derived PKM2 promotes phenotypic switching, proliferation, and migration via both its glycolytic and non-glycolytic activities.
Figure 5. PKM2 activator inhibits STAT3, and β catenin induced transcription of MEK5 and LDHA in human coronary artery SMC.

A, HCaSMCs were pretreated with Akt inhibitor (LY294002, 10 μM) or ERK pathway inhibitor (U0126, 10 μM) and stimulated with PDGF-BB (20 ng/ml) for 6 h. PKM2, Lamin B1, GAPDH, and β-actin were detected in the nuclear extract by immunoblotting. The left panels show representative immunoblots, and the right panel shows the densitometric analysis for PKM2/Lamin B1 (n = 4/group). B, HCaSMCs were stimulated with PDGF-BB (20 ng/ml) for 6 h, and cell extracts were immunoprecipitated (IP) with PKM2 antibody or control IgG and immunoblotted for PKM2, STAT3, and β-catenin. C, HCaSMCs were pretreated with ML265 (50 μM) and stimulated with PDGF-BB (20 ng/ml) for 6 h. Representative immunoblots and densitometric analysis for p-STAT3, total STAT3, and β-actin (n = 6/group). D, The ChIP assay of the MEK5 promoter in PDGF-BB stimulated HCaSMCs using antibodies against PKM2. ChIP using rabbit Ig as a negative control (n = 6/group). E, Real-time quantitative PCR analysis of MEK5, LDHA, Cyclin D1, and GLUT1 (n = 5/group). The results are presented as changes in PCR products normalized to GAPDH. (F), The bar graph shows lactate levels in cell supernatant (n = 4/group). (G), HCaSMCs were seeded in 96 well XF cell culture microplate. OCR and ECAR were measured in response to PDGF-BB in the presence or absence of ML265 (50 μM) (n =4). Values are mean ± SEM. Statistical analysis: A, C and F, One-way ANOVA followed by Bonferroni’s post hoc test; E, Kruskal-Wallis test (non-parametric One-way ANOVA) followed by uncorrected Dunn’s test; G, two-way ANOVA followed by uncorrected Fisher's LSD test. *P < 0.05 vs. quiescent; #P<0.05 vs. PDGF-BB-stimulated cells.
PKM2 activator inhibits injury-induced neointimal hyperplasia in wild-type mice
We determined the time-dependent nuclear translocation of PKM2 in the experimental carotid wire-injury model. PKM2 nuclear translocation increased significantly (~3.4 fold) at 6 h and remained elevated until 24 h after wire injury when compared to uninjured controls (Figure 6A). No significant change in PKM2 nuclear translocation was observed between 6 and 24 h. Next, we determined the effect of ML265 on PKM2 nuclear translocation in vivo. Pluronic gel with or without ML265 was perivascularly applied to the carotid artery 1 h before wire injury. We found that that ML265 pre-treatment significantly inhibited injury-induced PKM2 nuclear translocation at 6 h after injury (Figure 6B). We next investigated the therapeutic effect of ML265 against neointimal hyperplasia in wild-type mice in the experimental carotid wire-injury model. Verhoeff's/Van Gieson staining revealed that neointima formation at 28 days after injury was significantly reduced in the ML265 treated group as compared to the vehicle-treated control group (Figure 6C). Morphological measurements showed that the ML265 treated group had significantly reduced neointimal area and intima/media area ratio when compared to the control group (Figure 6C). Together, these results demonstrate that inducing PKM2 tetramerization via perivascular delivery of ML265 reduces injury-induced neointimal hyperplasia in mice and, thereby, might be a potential therapeutic intervention.
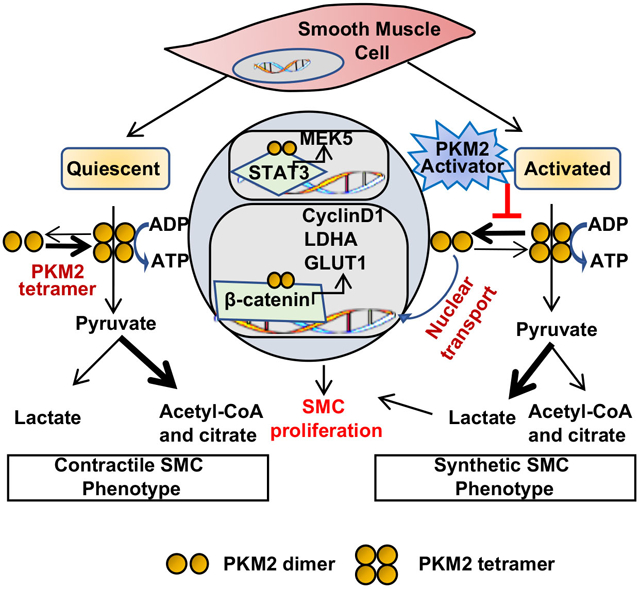
Figure 6. PKM2 activator inhibits injury-induced neointimal hyperplasia in wild-type mice.

A, Wire injury was done in the carotid artery of wild-type mice. Cytosolic and nuclear extracts were prepared at the following time points: 30 min, 60 min, 6 h, and 24 h after wire injury and then subjected to immunoblotting of PKM2, Lamin B1, GAPDH, and β-actin. The lower panel shows the densitometric analysis for PKM2/GAPDH in cytosolic extract and PKM2/Lamin B1 in nuclear extract (n = 4/group). B, Wire injury was done in the carotid artery of wild-type mice pretreated with ML265 (50 μM). Cytosolic and nuclear extracts were prepared 6 h after wire injury and were subjected to Western blotting of PKM2, Lamin B1, GAPDH, and β-actin. The lower panel shows the densitometric analysis for PKM2/GAPDH in cytosolic extract and PKM2/Lamin B1 in nuclear extract (n = 3/group). C, Carotid arteries pretreated with ML265 (50 μM in 30% w/v pluronic gel) were analyzed 28 days after wire injury. The left panel shows representative images of Verhoeff’s van Gieson stained carotid artery sections. Scale bars: 200 μm. The right panels show quantification of intimal area, medial area, and a ratio of intimal to the medial area. (n = 11/group). Each dot represents a single mouse. D, Schematic showing the mechanism by which PKM2 mediates SMC proliferation and neointimal hyperplasia. Values are expressed as mean ± SEM. Statistical analysis: A, B, 1-way ANOVA with Bonferroni’s post hoc test; C, unpaired Student’s t-test. *P < 0.05 vs. uninjured; #P<0.05 vs. wire injury + vehicle.
Discussion
Very little is known about the role of metabolic regulatory mechanisms in SMC phenotypic switching, proliferation, or migration. Herein, utilizing both in vitro and in vivo models, we demonstrate that the glycolytic enzyme PKM2 regulates proliferation, migration, and phenotype switching of SMC and injury-induced neointimal hyperplasia. We believe that these findings are conceptually novel and may have clinical significance for the following reasons: First, we demonstrated the expression of PKM2 in the vicinity of SMC-rich neointima and peri-strut areas in human coronary arteries with bare-metal stents. Second, we generated a novel smooth-specific PKM2-deficient mouse and provide genetic evidence that PKM2 regulates SMC phenotypic switching and mediates injury-induced neointimal hyperplasia via the STAT3 and β-catenin pathways. Third, using an activator that induces PKM2 tetramerization and blocks its nuclear translocation, we showed that targeting PKM2 in HCaSMC strongly reduces SMC proliferation, migration, and phenotypic switching essentially via glycolytic and non-glycolytic signaling pathway. Fourth, as a translational potential, we demonstrated that the perivascular application of PKM2 activator reduces experimental neointimal hyperplasia at 28 days.
When our study was in progress, two studies published this year showed that treating pulmonary artery SMC with PKM2 siRNA reduces proliferation and migration,29 and inhibiting PKM2 activity by inhibitor shikonin reduces oxLDL-induced SMC proliferation and migration in murine SMCs.10 Although the recently published studies suggest a role of PKM2 in SMC proliferation and migration in vitro, in our opinion, the findings reported herein are definitive and novel for the following reasons. None of these published studies has provided genetic evidence or the cell-specific role of PKM2 in SMC biology. Notably, there still remain key gaps in our understanding of the mechanistic role of PKM2 in SMC phenotypic switching and neointimal hyperplasia with therapeutic significance. Because SMC is the prominent cell type that contributes to the bulk of the neointima, and to determine the cell-specific role of PKM2, we generated SMC-specific PKM2 deficient mice. We provided genetic evidence for the first time that SMC-derived PKM2 promotes injury-induced neointimal hyperplasia by potentiating phenotypic switching, proliferation, and migration.
We also investigated the molecular mechanism by which PKM2 promotes SMC proliferation and migration, and thereby neointimal hyperplasia. We demonstrated that nuclear PKM2 interacts with STAT3, promotes phosphorylation of STAT3, and thereby regulates transcription of MEK5 in SMC, which can be inhibited by inducing PKM2 tetramerization. In agreement with our findings, nuclear PKM2 is known to phosphorylate STAT3 to induce expression of MEK5 in the proliferating cells, including cancer cells.24, 27 STAT3 is known to regulate SMC phenotypic switching through interacting with myocardin.30 MEK5 activates ERK5 and is known to play a key role in PDGF-BB-induced SMC migration.31 Additionally, we demonstrated that PKM2 interacts with β-catenin and potentiates the expression of cyclin D1, GLUT1, and LDHA. The upregulation of cyclin D1 and LDHA plays a critical role in cell cycle progression and lactate production in the presence of oxygen.3 Glucose transporters, particularly GLUT1 is abundantly expressed in SMC and regulates glucose transport/glucose metabolism, which in turn mediates SMC proliferation and phenotypic switching.32 Together, these results suggest a cross-talk between nuclear PKM2, STAT3, and β-catenin, which may be an important mechanism contributing to SMC synthetic phenotype, proliferation, and migration.
PKM2 is a glycolytic enzyme. Herein, we found that genetic ablation of PKM2 suppresses SMC synthetic phenotype and was associated with a decrease in lactate production. Although a previous study showed that lactate promotes synthetic phenotype in SMC,12 the authors did not determine the role of PKM2 in phenotypic switching or neointimal hyperplasia. The possibility that PKM2 by regulating lactate production, in addition to its kinase activity, may contribute to SMC synthetic phenotype, proliferation, and migration cannot be ruled out. Why is lactate production increased following SMC activation? PKM2 is a key regulator of aerobic glycolysis (lactate production). It may be an adaptive response to fulfill the high energy demand of activated SMC to change from contractile to the synthetic phenotype. Another possibility is that many intermediates from aerobic glycolysis can be used to synthesize proteins and lipids, which might be required for rapid proliferation. In parallel, we provided evidence that genetic ablation of PKM2 reduces ECAR in SMC; however, to our surprise, OCR remained unchanged. ECAR is the sum of acidification due to lactate and CO2 production. Deletion of PKM2 will completely inhibit the nuclear transport of dimeric/monomeric PKM2 and, thereby, decrease LDHA expression, which in turn will result in reduced lactate production and ECAR. It is intriguing to ask why is ECAR, but not OCR, is reduced by deletion of PKM2. We speculate that PK isomer PKM1 in complete absence of PKM2 favors a constant rate of pyruvate flux into OXPHOS through the TCA cycle. In line with our observations, PKM2 knockdown in pancreatic ductal adenocarcinoma cells or promoting its tetramerization in T cells impairs glycolysis without affecting OXPHOS.26, 33
We found that inducing PKM2 tetramerization by activator ML265 in SMC results in similar phenotype when compared to completely knocking out PKM2 in SMC. We speculate that in proliferating cells, nuclear translocation of dimeric/monomeric PKM2, which acts a kinase, interact with STAT3 and β-catenin and thereby potentiates the transcription of MEK5, Cyclin D1, GLUT1, and LDHA. Inducing PKM2 tetramerization inhibits dimeric/monomeric PKM2 nuclear translocation and hence protein kinase activity, which result in reduced proliferation, migration and phenotypic switching. Similarly, complete PKM2 deficiency shuts down protein kinase activity, thereby reducing proliferation, migration and phenotypic switching. Importantly, PKM2 deficiency does not affect PKM1 expression level and, therefore, total pyruvate kinase activity should be comparable in case of PKM2 deficiency or ML265 treatment. Furthermore, to investigate the translational potential, we demonstrated that inducing PKM2 tetramerization by perivascular delivery of ML265 reduces neointimal hyperplasia in wild-type mice. We adopted the perivascular delivery approach for the following reasons: 1) perivascular delivery prevents the drug from directly entering into the circulation and hence prevents rapid metabolic degradation; 2) local delivery introduces relatively high drug concentrations while bypassing systemic side effects; 3) perivascular delivery could generate a concentration gradient with higher concentrations in the adventitia and lower concentrations in the endothelium and thus have minimal effect on PKM2-mediated endothelial cell function which is critical in determining the success of PCI. In contrast to stenting, perivascular delivery is well suited for open vascular procedures, such as endarterectomy, interposition grafting, or bypass grafting, where antiproliferative agents can be applied directly at the site of operative injury. Notably, the pluronic gel has been widely used as a carrier to deliver viral vectors, inhibitors of the signaling pathway, micro RNAs, and other pharmacological agents to arteries.34-37
A particular strength of our study is that relevance to human coronary artery SMCs and therapeutic significance. We showed that inducing PKM2 tetramerization suppresses the SMC synthetic phenotype in cultured human coronary artery SMC via reducing STAT3 and β catenin induced transcription of MEK5 and LDHA. As a therapeutic significance, in the pre-clinical model, we showed that inducing PKM2 tetramerization by perivascular delivery reduces neointimal hyperplasia at 28 days, the time when peak neointima growth is observed. Despite its strength, our study has limitations. We have used a mouse carotid wire injury model, which partially mimics balloon angioplasty or intraluminal stent placement. Future studies are warranted to confirm the efficacy of targeting PKM2 in large pre-clinical animal models with stent models. In summary, using both pharmacological and genetic approaches, we revealed a critical role for PKM2 in SMC phenotypic switching and neointimal hyperplasia exacerbation. A summary of the proposed mechanism is shown in Figure 6D. We suggest that therapeutically targeting PKM2 may be beneficial in patients to reduce intimal hyperplasia during both open and endovascular procedures.
Supplementary Material
HIGHLIGHTS.
PKM2 regulates SMC phenotypic switching and neointimal hyperplasia in mice.
Inducing PKM2 tetramerization inhibits proliferation, migration, and phenotypic switching of human coronary SMC.
PKM2 regulates SMC phenotype switching via STAT3 and β-catenin pathways.
Perivascular application of PKM2 activator reduces neointimal hyperplasia in mice.
Acknowledgments
Sources of Funding
The A.K.C lab is supported by grants from the National Institutes of Health grant (R35HL139926, R01NS109910 & U01NS113388) and by Established Investigator Award 18EIA33900009 from American Heart Association. The A.V.F lab is supported by grants from CVPath Institute, Leducq Foundation Transatlantic Networks of Excellence Grant (18CVD02) to PlaqOmics Research Network (A.V. Finn)
Non-standard Abbreviations and Acronyms
- PKM
Pyruvate kinase muscle
- PDGF
Platelet-Derived Growth Factor-BB
- SMC
Smooth Muscle Cells
- ERK
Extracellular Signal-Regulated Kinase
- STAT3
Signal Transducer and Activator of Transcription 3
- MEK5
Mitogen/extracellular Signal-Regulated Kinase Kinase-5
- GLUT1
Glucose Transporter 1
- LDHA
Lactate Dehydrogenase A
- DMEM
Dulbecco’s Modified Eagle’s Medium
- BrdU
5-Bromo-2'-Deoxyuridine
- SMMHC
Smooth Muscle Myosin Heavy Chain
- PCI
Percutaneous Coronary Intervention
Footnotes
Disclosures
The authors declare no conflict of interest.
References.
- 1.Shlofmitz E, Iantorno M and Waksman R. Restenosis of Drug-Eluting Stents: A New Classification System Based on Disease Mechanism to Guide Treatment and State-of-the-Art Review. Circ Cardiovasc Interv. 2019;12:e007023. [DOI] [PubMed] [Google Scholar]
- 2.Gomez D and Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim JH, Bae KH, Byun JK, Lee S, Kim JG, Lee IK, Jung GS, Lee YM and Park KG. Lactate dehydrogenase-A is indispensable for vascular smooth muscle cell proliferation and migration. Biochem Biophys Res Commun. 2017;492:41–47. [DOI] [PubMed] [Google Scholar]
- 4.Perez J, Hill BG, Benavides GA, Dranka BP and Darley-Usmar VM. Role of cellular bioenergetics in smooth muscle cell proliferation induced by platelet-derived growth factor. Biochem J. 2010;428:255–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He X, Du S, Lei T, Li X, Liu Y, Wang H, Tong R and Wang Y. PKM2 in carcinogenesis and oncotherapy. Oncotarget. 2017;8:110656–110670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qin S, Yang D, Chen K, Li H, Zhang L, Li Y, Le R, Li X, Gao S and Kang L. Pkm2 can enhance pluripotency in ESCs and promote somatic cell reprogramming to iPSCs. Oncotarget. 2017;8:84276–84284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu S and Le H. Dual roles of PKM2 in cancer metabolism. Acta Biochim Biophys Sin (Shanghai). 2013;45:27–35. [DOI] [PubMed] [Google Scholar]
- 8.Pfeiffer T, Schuster S and Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292:504–7. [DOI] [PubMed] [Google Scholar]
- 9.Hsu MC and Hung WC. Pyruvate kinase M2 fuels multiple aspects of cancer cells: from cellular metabolism, transcriptional regulation to extracellular signaling. Mol Cancer. 2018;17:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao X, Tan F, Cao X, Cao Z, Li B, Shen Z and Tian Y. PKM2-dependent glycolysis promotes the proliferation and migration of vascular smooth muscle cells during atherosclerosis. Acta Biochim Biophys Sin (Shanghai). 2020;52:9–17. [DOI] [PubMed] [Google Scholar]
- 11.Zahra K, Dey T, Ashish, Mishra SP and Pandey U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front Oncol. 2020;10:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang L, Gao L, Nickel T, Yang J, Zhou J, Gilbertsen A, Geng Z, Johnson C, Young B, Henke C, Gourley GR and Zhang J. Lactate Promotes Synthetic Phenotype in Vascular Smooth Muscle Cells. Circ Res. 2017;121:1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC and Lu Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012;14:1295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, Jiang JK, Israelsen WJ, Keane J, Thomas C, Clish C, Vander Heiden M, Xavier RJ and O'Neill LA. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015;21:65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alves-Filho JC and Palsson-McDermott EM. Pyruvate Kinase M2: A Potential Target for Regulating Inflammation. Front Immunol. 2016;7:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain M, Dhanesha N, Doddapattar P, Chorawala MR, Nayak MK, Cornelissen A, Guo L, Finn AV, Lentz SR and Chauhan AK. Smooth muscle cell-specific fibronectin-EDA mediates phenotypic switching and neointimal hyperplasia. J Clin Invest. 2020;130:295–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh NK, Janjanam J and Rao GN. p115 RhoGEF activates the Rac1 GTPase signaling cascade in MCP1 chemokine-induced vascular smooth muscle cell migration and proliferation. J Biol Chem. 2017;292:14080–14091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selzman CH, Miller SA, Zimmerman MA, Gamboni-Robertson F, Harken AH and Banerjee A. Monocyte chemotactic protein-1 directly induces human vascular smooth muscle proliferation. Am J Physiol Heart Circ Physiol. 2002;283:H1455–61. [DOI] [PubMed] [Google Scholar]
- 19.Wang C, Zhang S, Liu J, Tian Y, Ma B, Xu S, Fu Y and Luo Y. Secreted Pyruvate Kinase M2 Promotes Lung Cancer Metastasis through Activating the Integrin Beta1/FAK Signaling Pathway. Cell Rep. 2020;30:1780–1797 e6. [DOI] [PubMed] [Google Scholar]
- 20.Keller KE, Doctor ZM, Dwyer ZW and Lee YS. SAICAR induces protein kinase activity of PKM2 that is necessary for sustained proliferative signaling of cancer cells. Mol Cell. 2014;53:700–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He CL, Bian YY, Xue Y, Liu ZX, Zhou KQ, Yao CF, Lin Y, Zou HF, Luo FX, Qu YY, Zhao JY, Ye ML, Zhao SM and Xu W. Pyruvate Kinase M2 Activates mTORC1 by Phosphorylating AKT1S1. Sci Rep. 2016;6:21524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao X, Wang H, Yang JJ, Chen J, Jie J, Li L, Zhang Y and Liu ZR. Reciprocal regulation of protein kinase and pyruvate kinase activities of pyruvate kinase M2 by growth signals. J Biol Chem. 2013;288:15971–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Millette E, Rauch BH, Defawe O, Kenagy RD, Daum G and Clowes AW. Platelet-derived growth factor-BB-induced human smooth muscle cell proliferation depends on basic FGF release and FGFR-1 activation. Circ Res. 2005;96:172–9. [DOI] [PubMed] [Google Scholar]
- 24.Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK and Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang J, Walsh MJ, Brimacombe KR, Anastasiou D, Yu Y, Israelsen WJ, Hong BS, Tempel W, Dimov S, Veith H, Yang H, Kung C, Yen KE, Dang L, Salituro F, Auld DS, Park HW, Vander Heiden MG, Thomas CJ, Shen M and Boxer MB. ML265: A potent PKM2 activator induces tetramerization and reduces tumor formation and size in a mouse xenograft model Probe Reports from the NIH Molecular Libraries Program Bethesda (MD); 2010. [PubMed] [Google Scholar]
- 26.Angiari S, Runtsch MC, Sutton CE, Palsson-McDermott EM, Kelly B, Rana N, Kane H, Papadopoulou G, Pearce EL, Mills KHG and O'Neill LAJ. Pharmacological Activation of Pyruvate Kinase M2 Inhibits CD4(+) T Cell Pathogenicity and Suppresses Autoimmunity. Cell Metab. 2020;31:391–405 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao X, Wang H, Yang JJ, Liu X and Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K and Lu Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011;480:118–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang A, Yu F, Yu W, Ye P, Liu P, Gu Y, Chen S and Zhang H. Pyruvate kinase M2 activation protects against the proliferation and migration of pulmonary artery smooth muscle cells. Cell Tissue Res. 2020. [DOI] [PubMed] [Google Scholar]
- 30.Liao XH, Wang N, Zhao DW, Zheng DL, Zheng L, Xing WJ, Ma WJ, Bao LY, Dong J and Zhang TC. STAT3 Protein Regulates Vascular Smooth Muscle Cell Phenotypic Switch by Interaction with Myocardin. J Biol Chem. 2015;290:19641–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Izawa Y, Yoshizumi M, Ishizawa K, Fujita Y, Kondo S, Kagami S, Kawazoe K, Tsuchiya K, Tomita S and Tamaki T. Big mitogen-activated protein kinase 1 (BMK1)/extracellular signal regulated kinase 5 (ERK5) is involved in platelet-derived growth factor (PDGF)-induced vascular smooth muscle cell migration. Hypertens Res. 2007;30:1107–17. [DOI] [PubMed] [Google Scholar]
- 32.Pyla R, Poulose N, Jun JY and Segar L. Expression of conventional and novel glucose transporters, GLUT1, -9, -10, and -12, in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2013;304:C574–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yokoyama M, Tanuma N, Shibuya R, Shiroki T, Abue M, Yamamoto K, Miura K, Yamaguchi K, Sato I, Tamai K and Satoh K. Pyruvate kinase type M2 contributes to the development of pancreatic ductal adenocarcinoma by regulating the production of metabolites and reactive oxygen species. Int J Oncol. 2018;52:881–891. [DOI] [PubMed] [Google Scholar]
- 34.Jain M, Singh A, Singh V and Barthwal MK. Involvement of interleukin-1 receptor-associated kinase-1 in vascular smooth muscle cell proliferation and neointimal formation after rat carotid injury. Arterioscler Thromb Vasc Biol. 2015;35:1445–55. [DOI] [PubMed] [Google Scholar]
- 35.Zeng Z, Xia L, Fan X, Ostriker AC, Yarovinsky T, Su M, Zhang Y, Peng X, Xie Y, Pi L, Gu X, Chung SK, Martin KA, Liu R, Hwa J and Tang WH. Platelet-derived miR-223 promotes a phenotypic switch in arterial injury repair. J Clin Invest. 2019;129:1372–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Zheng J, Bai X, Liu B, Liu CJ, Xu Q, Zhu Y, Wang N, Kong W and Wang X. ADAMTS-7 mediates vascular smooth muscle cell migration and neointima formation in balloon-injured rat arteries. Circ Res. 2009;104:688–98. [DOI] [PubMed] [Google Scholar]
- 37.Cai Y, Nagel DJ, Zhou Q, Cygnar KD, Zhao H, Li F, Pi X, Knight PA and Yan C. Role of cAMP-phosphodiesterase 1C signaling in regulating growth factor receptor stability, vascular smooth muscle cell growth, migration, and neointimal hyperplasia. Circ Res. 2015;116:1120–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This article adheres to the American Heart Association Journal implementation of the Transparency and Openness Promotion Guidelines. Primary data that support the findings of this study are available from the first author or corresponding author upon request.