

Abstract
Atopic dermatitis (AD) is a disease of immune dysregulation and skin barrier dysfunction with a relapsing, remitting course and has been associated with several different genetic risk variants. Human leukocyte antigens (HLA) represent a highly variable set of genes that code for cell surface protein molecules involved in the antigen-specific immune response, including the regulation or functioning of T-cells, natural killer (NK) cells, and antigen presenting cells. The purpose of this study was to evaluate associations between HLA Class I polymorphisms and the progression of AD over time. We evaluated the associations of AD symptoms and HLA Class I polymorphisms based on high resolution two-field typing in a longitudinal cohort of children with AD (up to 10-years of follow-up). Seven hundred ninety-two children were evaluated every 6 months resulting in 12,752 AD evaluations. Using generalized estimating equations, B*44:02 was found to be associated with AD remission (1.83 [1.35, 2.47]; pcorr=0.0015). The HLA-B residues at position 116 (D-aspartate) and 80 (T-threonine) were associated with remission (1.42 [1.13, 1.76] p=0.003, pcorr= 0.028) and (1.45 [1.17, 1.80] p=0.0008, pcorr=0.0024) respectively. B80T is a killer-cell immunoglobulin-like receptor (KIR) site. Our findings reveal that two axes of immune response (T-cell and NK cell) may influence disease progression. Identifying binding pocket changes in addition to other factors (e.g. allergens) that increase the risk or severity of AD can improve our understanding of the immunologic mechanisms associated with AD and may lead to personalized therapies for improving patient care.
Keywords: Human, Skin, Allergy, MHC, T Cells, Natural Killer Cells
Introduction
Atopic dermatitis (AD) is a chronic inflammatory skin disease that affects about 10% of the US population.(1, 2) At one time, it was thought to primarily affect children and resolve in late childhood, but recently several studies have shown that it affects individuals of all ages.(1, 3, 4) For those with childhood onset, AD can persist into adulthood.(3) Genetic variation has been associated with the risk of having AD as well as with its severity and persistence.(3, 5, 6) The most commonly identified genetic association is with filaggrin (FLG), a skin barrier protein.(5) However, AD is often thought of as an immune mediated illness and has been associated with genes controlling inflammatory processes such as IL-4 and IL-13.(7–9) A recently approved therapy for AD blocks IL-4 and IL-13 signaling.(10)
Human leukocyte antigens (HLA) represent a highly variable set of genes found on chromosome 6 that code for cell surface protein molecules involved in the antigen-specific immune response, including the regulation or functioning of T-cells, natural killer (NK) cells, and antigen presenting cells.(11) Previous studies, including genome-wide association studies (GWAS) and studies using immunophenotyping techniques, have found associations between HLA genes and the risk of AD, as well as other atopic illness such as asthma, seasonal allergies, and food allergies.(12–18) With respect to AD, GWAS of Europeans and Asians have identified a number of single nucleotide polymorphisms (SNPs) within the MHC region to be associated with AD including rs9368677 (between HLA-B and HLA-C)(14), rs2251396 (between HLA-B and MICA)(19), rs6474 (complement region)(19), rs9469099 (TSBP1)(14), rs4713555 and rs28383201 (between DRB1 and DQA1)(13, 20) (21). However, the RSID numbers listed above, merely designate regions of potential relevance to the onset of AD but they do not identify actual HLA genes or alleles, that could be relevant to the onset of AD.
Independently, Natural Killer (NK) cell function has also been shown to be important in the pathogenesis of AD.(22–26) This observation was first noted in 1985.(25) A recent study by Mack et al revealed that the number of circulating NK cells in adults with moderate to severe AD were significantly less than in a healthy control population.(22) NK cell function is dependent on the interaction of HLA class I molecules with killer-cell immunoglobulin-like receptor (KIR) molecules on NK cells. A prior study of a Polish population, demonstrated that KIR2DS1 was associated with a decreased risk of AD.(23, 24) Studying HLA class I regions that interact with KIR may shed light on the innate immune system’s role in the pathophysiology of AD as well as interactions with the adaptive immune system (through T cell engagement).
In this study we focused on HLA class I genes and their polymorphisms that influence critical functionalities of the HLA molecule, such as interactions with peptides or other receptors (T-cell receptor or KIR). Our rationale for focusing on HLAs is based upon past studies evaluating risk of developing AD, as summarized above. We leveraged the availability of up to 10 years of longitudinal data on 792 children with AD(5, 27) to assess whether polymorphisms of HLA class I molecules are associated with clinical course of AD over time. To our knowledge, no previous studies have ever explored the role of HLA class I variation in disease progression of AD.
Methods:
Study subjects were enrolled in the Pediatric Eczema Elective Registry (PEER; www.thepeerprogram.com) which is a United States nationwide cohort of more than 8,000 subjects with pediatric-onset AD. All subjects had a diagnosis of AD confirmed by a physician. The current study represents a sub-cohort of PEER children, those who provided a genetic sample (PEER DNA cohort).(27, 28) In the PEER DNA cohort, both self-described race/ethnicity and ancestry informative markers were used to define ancestry and were previously found to be highly correlated.(27) PEER DNA enrollment occurred between November of 2004 and January 2015. At the time of enrollment, children were two to seventeen years old, had a physician-confirmed diagnosis of AD, and had used pimecrolimus cream for at least six months, but may have used many other therapies.(27) With respect to outcome measures (e.g., the persistence of AD), subjects are followed for up to 10 years and during that time were not required to (and most did not) continue therapy with pimecrolimus.(29) Follow-up is still ongoing.
All subjects who provided DNA or their legal guardians provided written informed consent approved by the University of Pennsylvania’s Institutional Review Board.
Outcome:
The clinical course of AD was determined periodically and defined by whether a child was in remission based on the self-reported outcome of whether or not a child’s skin was AD symptom-free during the previous six-months while the child was not using medication to treat their AD. (27) AD disease activity was based on the survey question: “During the last six months would you say that your child’s skin disease (AD) has shown: complete disease control, good disease control, limited disease control, or uncontrolled disease”. Symptom free was defined as an affirmative response to “complete disease control”. This response has been shown to correlate with other tools used to evaluate symptom control and is likely a marker of long-term disease severity.(30–32) Outcome data collection for PEER is ongoing. For this study, the last date that the outcome was reported for a study subject occurred either at the 10-years of follow-up or up to their last response before March 2020, whichever date occurred first.
HLA Genotyping
DNA was collected using Oragene DNA collection kits (DNA Genotek, Ottawa Canada) as previously reported.(27) The classic Class HLA I genes -A, -B and -C for individuals in the PEER DNA cohort were fully sequenced using one of two next generation sequencing (NGS) approaches. Three hundred fifty-seven subjects were genotyped using the Omixon HLA Holotype kit and 424 subjects were typed using a combination of probes generated by SureDesign and a custom design of HLA probes based on the complete collection of IMGT HLA sequences (version 3.09). Using these NGS methods we report high resolution two-field (HR-2F) typing that was used to determine amino acid variation in HLA Class I molecules.
Analysis:
Analyses were based on HLA amino acid residue variation described by HLA allele HR-2F typing per gene. Our primary analysis plan focused on assessing the association between HLA amino acid residue variation based on the HR-2F HLA genotyping (assuming an additive genetic model) and the likelihood of remission at any given timepoint. We used a generalized estimating equations (GEE) approach for the binary outcome of remission using a logistic model and independence working correlation structure to account for the correlation among repeated measures per participant (outcomes were determined by survey every six months for up to 10 years). GEE belong to a class of semiparametric regression models that measure the average response over the population and are frequently used to evaluate longitudinal data that has repeated outcome measures. The odds ratio measured in this study reflects the odds that a subject with an exposure of interest (e.g., a specific HLA polymorphism) as compared to one who does not have that exposure will be in remission at any given time that the outcome was measured. The primary analysis was of the full cohort. A secondary analysis was conducted based on European or African ancestry in order to confirm the stability of the effect estimates. Odds ratios (OR) for association between variant of interest and AD clinical course (remission (OR >1.0) or persistence (OR ≤ 1.0)) and 95% confidence intervals were estimated from these models.
HR-2F typing was conducted for all HLA-A, HLA-B, and HLA-C variants (results available in the supplement). We used the allelic genotyping to determine protein sequence (residue) variation (IMGT version 3.09). Allelic frequency for all the variants is presented in the supplement as well as the association with our outcome for those with an AF of ≥ 0.05. For residue analysis and based on our HR-2F typing, we used the Simpson reciprocal index (SRI)(33) for each polymorphic residue of the binding cleft in our PEER cohort. The SRI is a metric reflecting the influence particular pocket residues exert in peptide binding.(34) We focused only on those pocket residues with the highest SRI index ( ≥3.0), i.e. the residues that exert the highest influence on peptide binding(33). As a result, positions 9, 97 and 114 of HLA-A, 67 and 116 of HLA-B and positions 156 of HLA-C were evaluated with respect to our outcome, remission or persistence to AD.
In addition, based on the potential role of NK cells in AD, known polymorphic HLA residues that constitute contact points with KIR receptors (and not associated with SRI) were explored. These contact residues are 80 and 83 of HLA-A and HLA-B (i.e., Bw4 KIR binding site), −21T and −21M of HLA-B (areas associated with NK education), and 80 of HLA-C (i.e., C1/C2 KIR binding site) and their associations with respect to AD remission were evaluated using the GEE models described above. (35–39)
Additional models were adjusted for the following baseline variables: sex, analysis of African ancestry race based on ancestral informative markers(27), age of onset of AD, and the presence of asthma and/or seasonal allergies. Missingness was previously evaluated and felt to be completely at random and consistent with GEE assumptions.(9) The Bonferroni procedure was used per gene for multiple testing correction based on the number of amino acid residue variants tested after screening. Hypothesis testing of KIR binding sites were adjusted based on the number of KIR analyses however the association between NK cell function and these specific sites were known a priori.
All statistical analyses were conducted using STATA Version 16.1 (College Station, TX)
Results:
Seven hundred and ninety-two children from the PEER DNA cohort were HLA genotyped using NGS-based technologies. The primary outcome of AD remission/persistence was surveyed 12,752 times in this cohort and on average each child had nine evaluations. At the time of this study, PEER DNA children had been followed for an average of 102 months (sd: 48.36). The 792 children were of African-American ancestry (41.2%), European ancestry (55.4%), and of other ancestries or not reported (3.4%), and 52.6% of the children were female. The average age of onset of AD was reported as 1.92 (sd: 2.66) years with a median age of onset of 0.75 years (IQR: 0.25–2). Overall remission was reported in 16.3% of surveys and on average children reported 13.70 (sd: 23.15) months in remission.
In the PEER DNA cohort, 65 HLA-A, 115 HLA-B and 65 HLA-C unique alleles were identified (supplement Table 1). However, only 5 HLA-A, 4 HLA-B and 7 HLA-C had AF ≥ 0.05 and these alleles were included in the outcome analysis (see supplement Tables 1 and 2). Several alleles appear to be associated with AD remission or persistence but only HLA-B, B*44:02 (Supplement Table 2) was found to be associated with remission after correction (1.83 (1.35, 2.47); pcorr=0.0015).
After filtering HLA Class I isotypes by SRI ≥ 3.0, we analyzed residues 9, 97 and 114 for HLA-A, residues 67 and 116 for HLA-B, and residues 156 for HLA-C (Table 1). The associations between amino acid residue variation and remission are presented in Table 2. Several residue sites are potentially associated with remission, however only a single residue of HLA-B at position 116 (D-aspartate), (1.42 (1.13, 1.76) p=0.003, pcorr= 0.028) was significant after correction (Table 2). Residue 116 of HLA-B is part of pocket F of the HLA Class I molecule. The association of the probability of remission by this variant by ancestry over time is shown in Figure 1. The ORs of association was similar in those of both European or African ancestry (Table 2).
Table 1:
Simpson reciprocal index (SRI) is presented for the full PEER DNA cohort for residues known to occur within HLA Class I binding pockets. Variants within a position are presented by amino acid single letter abbreviation.
| HLA-A | HLA-B | HLA-C | ||||
|---|---|---|---|---|---|---|
| position | SRI | Variants | SRI | Variant | SRI | Variants |
| 9 | 3.0 | F; S; T; Y | 1.8 | D; H; Y | 2.6 | D; F; S; Y |
| 63 | 1.5 | E; N; Q | 2.0 | E; N | 1.0 | E |
| 66 | 1.9 | K; N | 1.17 | I; K; N | 1.3 | K; N |
| 67 | 1.3 | M; V | 3.7 | C; F; M; S; Y | 1.0 | Y |
| 70 | 1.8 | H; Q | 1.7 | K; N; Q; S | 1.0 | Q |
| 74 | 1.6 | D; H; N | 1.8 | D; Y | 1.00 | D |
| 77 | 2.0 | D; N; S | 2.0 | D; N; S | 2.0 | N; S |
| 80 | 1.4 | I; T | 2.2 | I; N; T | 2.0 | K; N |
| 81 | 1.4 | A; L | 1.8 | A; L | 1.0 | L |
| 97 | 3.0 | I; M; R | 2.7 | N; R; S; T; V; W | 1.5 | N; R; W |
| 99 | 1.3 | C; F; Y | 1.0 | S; Y | 1.9 | C; F; S; Y |
| 114 | 3.2 | E; H; Q; R | 2.1 | D; H; N | 1.8 | D; N; V |
| 116 | 2.3 | D; H; Y | 3.7 | D; F; L; S; Y | 2.6 | F; L; S; Y |
| 152 | 2.5 | A; E; R; V; W | 1.8 | E; V | 1.8 | A; E; T; V |
| 156 | 2.3 | L; Q; R; W | 2.2 | D; L; R; W | 3.2 | D; L; Q; R; W |
| 163 | 1.5 | E; R; T | 2.6 | E; L; T | 1.6 | E; L; T |
| 167 | 1.6 | G; W | 1.3 | L; S; W | 1.0 | W |
Table 2: Amino acid residues associations with remission outcome by HLA-A gene.
Amino acids and their letter abbreviation (For example, A97T is HLA-A, position 97, amino acid R (arginine)) are presented with odds ratios and 95% CI estimated by generalized estimating equation (family binomial link logit correlation exchangable). Positions were selected for evaluation based on SRI ≥ 3.0. Odds ratios are presented for the full cohort and by ancestry with 95% confidence intervals and p-values (and when appropriate corrected p-value).
| Position/Variant | Variant Frequency | Full cohort (OR) | European | African |
|---|---|---|---|---|
| A9T | 0.12 (0.10,0.14) | 0.91 (0.67, 1.23) p=0.054 | 0.94 (0.59,1.50) p=0.802 | 1.40 (0.90,2.17) p=0.132 |
| A9Y | 0.18 (0.16,0.20) | 1.03 (0.80, 1.32) p=0.842 | 1.00 (0.73,1.37) p=0.995 | 1.21 (0.76,1.92) p=0.420 |
| A9S | 0.20(0.18,0.22) | 0.88 (0.68, 1.12) p=0.135 | 1.13 (0.82,1.55) p=0.451 | 1.01 (0.66,1.56) p=0.956 |
| A9F | 0.48 (0.45,0.50) | 1.16 (0.96, 1.40) p=0.135 | 1.16 (0.92,1.48) p=0.210 | 0.70 (0.47,1.04) p=0.081 |
| A97M | 0.29 (0.27,0.32) | 1.02 (0.83, 1.25) p=0.852 | 1.11 (0.87,1.42) p=0.393 | 1.20 (0.83,1.75) p=0.337 |
| A97R | 0.31 (0.29,0.34) | 1.26 (1.03, 1.55) p=0.024 | 1.24(0.98,1.58) p=0.077 | 1.03(0.68, 1.55) p=0.894 |
| A97I | 0.35 (0.33,0.38) | 0.84 (0.69, 1.03) p=0.097 | 0.85 (0.67,1.08) p=0.178 | 0.83 (0.57,1.20) p=0.330 |
| A114E | 0.08 (0.07,0.10) | 0.66 (0.45,0.98) p=0.040 | 0.89 (0.44,1.82) p=0.757 | 1.02 (0.61,1.69) p=0.943 |
| A114H | 0.38 (0.35,0.40) | 1.25 (1.03,1.53) p=0.023 | 1.31 (1.03,1.67) p=0.026 | 0.93 (0.64,1.35) p=0.712 |
| A114Q | 0.17 (0.16,0.19) | 1.02 (0.81,1.31) p=0.812 | 0.97 (0.71,1.33) p=0.857 | 1.39 (0.94,2.05) p=0.101 |
| A114R | 0.32 (0.30,0.34) | 0.93 (0.76,1.14) p=0.496 | 0.87 (0.69,1.10) p=0.248 | 0.77 (0.50,1.20) p=0.255 |
| B67C | 0.09 (0.07,0.10) | 1.34 (0.98, 1.82) p=0.062 | 1.05 (0.74,1.48) p=0.781 | 2.09(0.108,4.02)p=0.028 |
| B67F | 0.23 (0.21,0.25) | 0.79 (0.63, 1.00) p=0.051 | 0.77 (0.58,1.01) p=0.059 | 0.93 (0.61,1.42) p=0.732 |
| B67M | 0.06 (0.05,0.07) | 0.94 (0.62, 1.42) p=0.770 | 1.38 (0.81,2.36) p=0.229 | 0.57 (0.26,1.21) p=0.144 |
| B67S | 0.36 (0.33,0.38) | 1.23 (1.02, 1.48) p=0.034 | 1.21(0.98, 1.51) p=0.083 | 1.18(0.82, 1.70) p=0.376 |
| B67Y | 0.14 (0.13,0.16) | 0.98 (0.74, 1.29) p=0.900 | 1.00 (0.70,1.41) p=0.981 | 1.01 (0.63,1.62) p=0.949 |
| B116D | 0.14 (0.12,0.16) | 1.42 (1.13, 1.79) p=0.003 pcorr=0.028 |
1.29(0.99, 1.67) p=0.055 | 1.69(1.02, 2.78) p=0.040 |
| B116S | 0.25 (0.23,0.28) | 0.89 (0.73, 1.09) p=0.259 | 0.97 (0.77,1.23) p=0.820 | 0.91 (0.63,0.32) p=0.629 |
| B116F | 0.08 (0.07,0.09) | 1.07 (0.76, 1.51) p=0.682 | 0.84 (0.57,1.24) p=0.380 | 1.34 (0.62,2.90) p=0.453 |
| B116L | 0.08 (0.07,0.10) | 1.13 (0.82, 1.56) p=0.454 | 1.13 (0.77,1.67) p=0.529 | 1.38 (0.79,2.39) p=0.257 |
| B116Y | 0.34 (0.31,0.36) | 0.91 (0.75, 1.11) p=0.360 | 0.88 (0.70,1.11) p=0.291 | 0.79 (0.53,1.18) p=0.254 |
| C156D | 0.02 (0.02,0.03) | 1.71 (1.02, 2.84) p=0.040 | 2.12 (1.21,3.73) p=0.009 | 0.79 (0.21, 3.03)p=0.731 |
| C156L | 0.39 (0.36,0.41) | 0.87 (0.72,1.06) p=0.172 | 0.89 (0.71,1.12) p=0.327 | 0.66 (0.45,0.97) p=0.038 |
| C156Q | 0.06(0.05,0.08) | 0.65 (0.34,1.25) p=0.199 | 1.25 (0.59,2.62) p=0.559 | 0.30 (0.08,1.19) p=0.087 |
| C156R | 0.28 (0.26,0.30) | 1.05 (0.86,1.29) p=0.632 | 1.05 (0.82,1.36) p=0.663 | 1.10 (0.74,1.62) p=0.636 |
| C156W | 0.15 (0.13,0.17) | 1.19 (0.92,1.53) p=0.175 | 1.13 (0.83,1.53) p=0.448 | 1.38 (0.90,2.12) p=0.136 |
Figure 1:
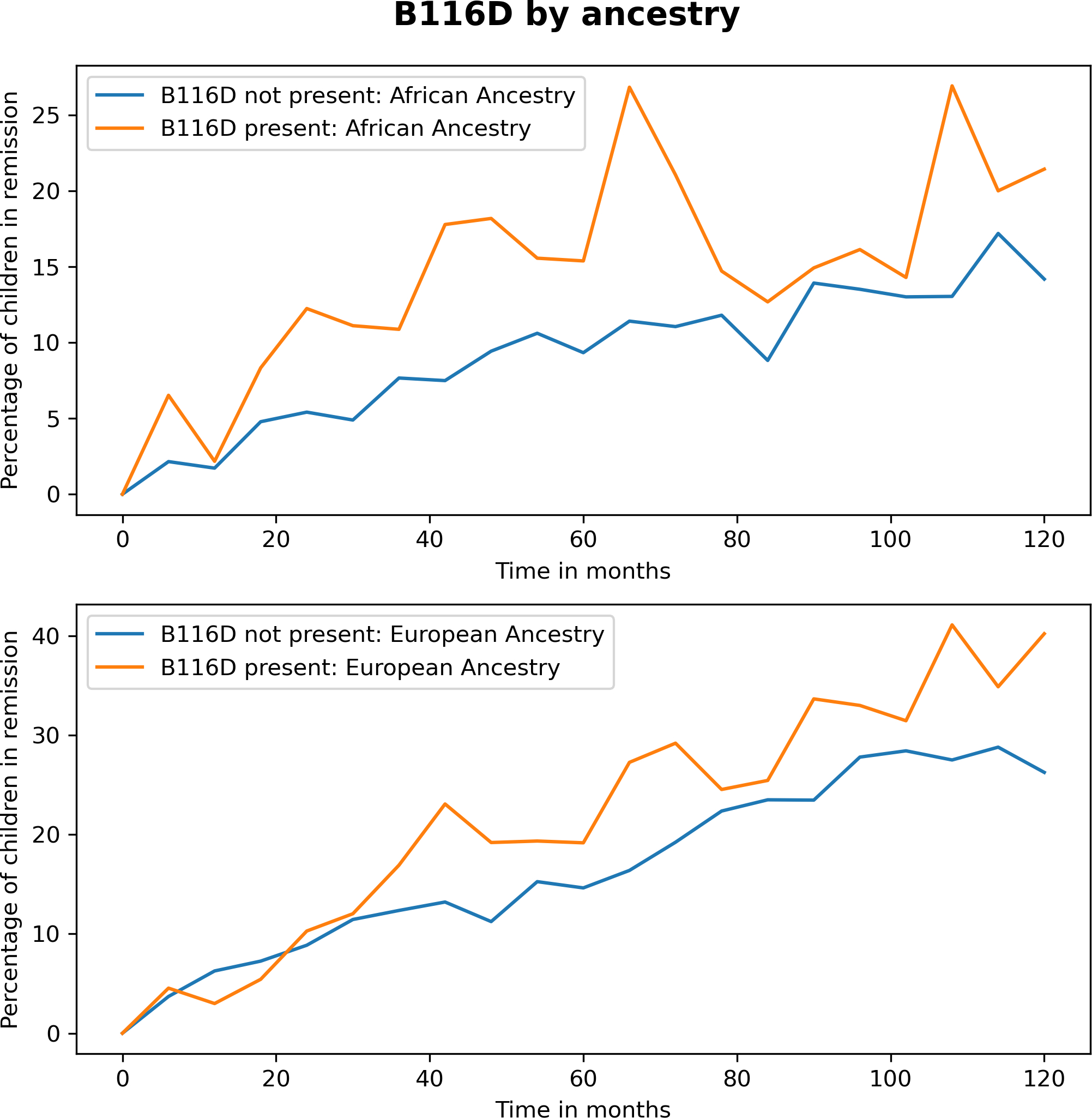
Graph of HLA Class I residue B116D versus other B116 variants over time with respect to the percentage of children symptom and medication free at any evaluation timepoint by ancestry.
Regarding residues that interact with KIR, residues A80 (G/I) and A83 (on HLA-A) were not associated with AD remission (Table 3). On HLA-B, residue B80T (threonine) was associated with remission (1.45 (1.17, 1.80) p=0.0008, pcorr=0.0024), while B80I (isoleucine) was not associated with remission (0.84 (0.65,1.08) p=0.1694). The probability of remission by ancestry due to the presence of B80T over time is presented in Figure 2. The B80T residue is part of pocket F. Residue B83R (arginine) was not associated with remission. HLA-C position 80 with N (asparagine) or K (lysine) was not associated with remission (Table 3). The NK cell education sites of HLA-B, −21M (methionine) (1.12(0.92,1.30) and −21T (0.93(0.76,1.15) were also not associated with remission.
Table 3:
KIR binding sites as represented by amino acid residues association. Frequency with 95% confidence intervals and OR and 95% confidence intervals. Odds ratios are presented for the full cohort and by ancestry with 95% confidence intervals and p-values (and when appropriate corrected p-value).
| Residue | Variant Frequency | Full cohort (OR) | European Ancestry | African Ancestry |
|---|---|---|---|---|
| A80I | 0.34 (0.31,0.36) | 1.03 (0.84,1.26) p=0.762 | 0.78(0.58,1.04) p=0.092 | 0.78 (0.50,1.20) p=0.261 |
| B80T (Bw4) | 0.16(0.14,0.18) | 1.45 (1.17,1.80) p=0.0008399 | 1.34 (0.93, 1.92) p=0.114 | 1.57 (0.95,2.59) p=0.077 |
| B80I (Bw4) | 0.34(0.31, 0.36) | 0.84(0.65, 1.08) p=0.169 | 0.92(0.63,1.34) p=0.657 | 0.66 (0.42,1.03) p=0.066 |
| B83G (Bw4) | 0.53(0.50,0.55) | 0.97 (0.81, 1.17) p=0.772 | 0.82(0.62, 1.10) p=0.185 | 1.25 (0.64,2.92) p=0.411 |
| C80K (C2) | 0.41(0.39,0.44) | 1.01(0.83, 1.22) p=0.932 | 0.88(0.66,1.67) p=0.365 | 1.17 (0.82,1.67) p=0.383 |
| C80N (C1) | 0.48(0.45,0.50) | 1.05 (0.87,1.27) p=0.585 | 1.12 (0.85,1.49) p=0.409 | 0.72 (0.49,1.05) p=0.086 |
Figure 2:
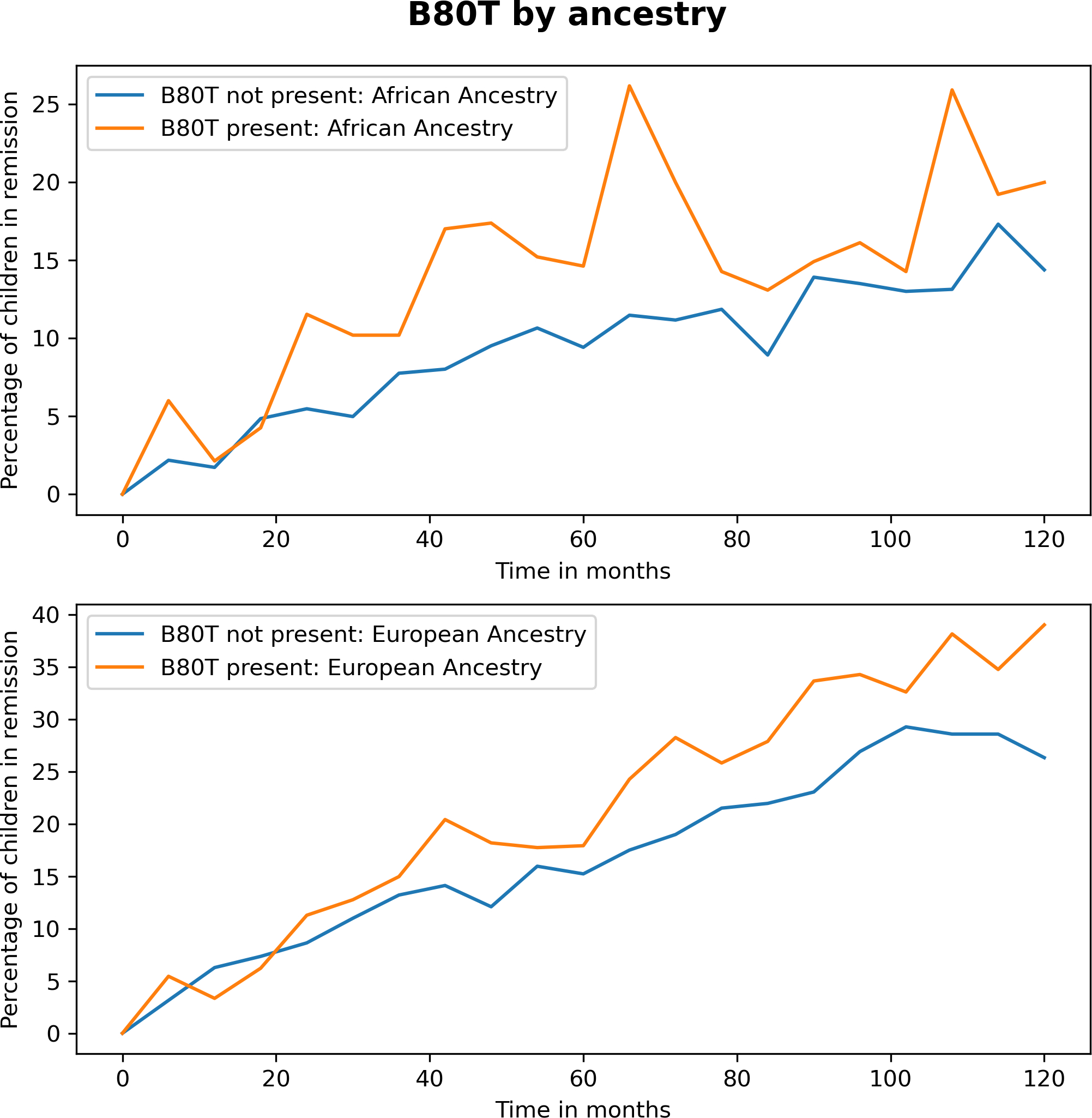
Graph of HLA Class I residue B80T versus other B80 variants over time with respect to the percentage of children symptom and medication free at any evaluation timepoint by ancestry.
As noted, B80 and B116, are both part of pocket F. B80T and B116D are in linkage disequilibrium (LD) (R2 = 0.71), suggesting a possible functional role and that these two residues may have co-evolved. B116D was overall highly associated with Bw4 (B80T or B80I). When B116D did not occur with B80T, B116D occurred with B80I (but never occurred with B80N(Bw6)).
Discussion:
This study is the largest to date assessing the variation of Class I HLA loci of children with AD that uniquely focuses on the clinical course of disease over time. Prior studies have shown that HLAs are associated with the risk of having AD and that there is a role for NK cell activity in the pathogenesis of AD.(12–14, 20, 25, 40) Unlike previous studies, we longitudinally evaluated the clinical course of AD over time. We identified two residues that are both found in HLA Class I binding pocket F. One of the HLA residues is at a location known to be an interaction site for KIR ligand (B80T/I), which is part of Bw4 epitope, and the other HLA molecular variant is at position B116, which is highly likely to be associated with variation in antigen presentation to T-cells. Therefore, the identified associations suggest a role for both the T and NK cells, implying an effect that is T-cell mediated through specific HLA-peptide/TCR interactions and also an NK effect that is HLA/KIR dependent. Based on our findings, we hypothesized that both adaptive and innate immune responses play roles in the clinical course of AD.
B116D and B80T are shared epitopes formed by several HLA-B alleles including B*44:02 (the only allele in our cohort significantly associated with remission that also had an AF >0.05). However, these polymorphisms are also associated with the less common alleles like B*44:03, B*44:27, B*27:02, B*27:05 and B*15:17. By analyzing the amino acid polymorphisms involved in the HLA binding pockets (e.g., composites of many alleles), we were able to include all of the alleles (both rare and common) in our analyses and evaluate their biologically relevant functionality. Our analysis was consistent with the widely accepted notion that HLA disease associations can be defined much more sensitively at the HLA amino acid level than at the classical HLA allelic level.(33, 41)
The B116D and B80T polymorphisms are in LD, so it is not clear whether their functional effects are independent or coordinated. However, it is very likely that two axes of immune response (innate and adaptive) influence longitudinal disease activity (i.e., the clinical course) of AD. B116D can influence interactions with incoming side chains of peptides bound to the HLA binding pockets, but the same B116 residue is also known to influence the optimization of peptide editing/selection by Tapasin, a protein known to influence the presentation of selected of peptides.(42) B116 variation mismatch in bone marrow transplantation has also been shown to be associated with transplant related morbidity like graft versus host disease.(43, 44) Overall, the residue B116D possibly influences more than one function contributing to the formation of the HLA-peptide complex, which may influence the odds of remission from AD symptoms.
There are approximately 15 KIR genes on chromosome 19 and they have important regulatory function for NK cells.(45, 46) Similarly to HLA, the KIR allelic variation results in receptor variation. KIR recognize HLA Class-I molecules and two KIR variants are known to interact with Bw4, KIR3DL1, which inhibits NK cell function, and KIR3DS1, which activates NK cell function.(46) KIR3DL1 and KIR3DS1 are allelic variants of the same gene called KIR3DL1/S1, however 3DL1 variant is most commonly expressed.(35) The association between the HLA-B Bw4 epitope and the KIR receptor variant occurs primarily at amino acid location 80. The Bw4 epitope is represented by HLA-B allelic variation at location 80 represented by amino acids threonine (T) or an isoleucine (I). Threonine is polar and therefore hydrophilic while isoleucine is non-polar and hydrophobic. As a result, biochemically, it is likely that receptor interactions with amino acid T and I differ. In fact, differential KIR interactions by residue 80 (i.e. I or T) have been previously reported.(40, 47) Furthermore, the crystal structure of the receptor/ligand interactions for KIR3DL1, which is centered on location 80, shows more enhanced binding between KIR3DL1 and B80I.(45) This hypothesis of differential Bw4 activity based on the presence of T or I is also substantiated by our observed results. Final confirmation would require KIR genotyping, which was beyond the focus of this study. However, based on our data, future studies evaluating KIR genes and AD are warranted.
In prior studies, we demonstrated that FLG variation, which codes for a skin barrier protein, and TSLP, which codes for a protein initiator of inflammation at barrier surfaces are associated with remission.(7, 9, 27) We now show that HLA Class I proteins found on antigen presenting cells, T-cells, and NK cells are involved in the clinical course of AD thereby creating a supportable and coherent model of genetic variation associated with both immunologic and skin barrier aspects of AD. A simple explanation for the involvement of these genetic variants is that antigen enters through an impaired skin barrier (FLG), is presented by an antigen presenting cell (HLA Class II) resulting in the TSLP activation of T cells and the subsequent production of IgE by plasma cells or the direct interaction of NK cells and HLA Class I KIR ligand. With the inclusion of data from this study, HLA allelic variants may have now been shown to alter the clinical course of AD following this simple pathway. The effects we note in HLA Class I in part could help explain hypothesized “endotypes” of atopic dermatitis base on person-specific varying immunologic dysfunction.(48–51)
In fact, previous studies have shown that polygenic combinations of KIR and HLA Class I receptors are associated with disease outcomes.(45, 46) For example, the protective role of B*53:01 in HIV-1 infection is primarily, because of the KIR binding properties at B80I that cause NK cell activation.(36, 47) However, the B*53:01 HIV-1 protective effect is also related to TCR binding properties.(52) Our finding regarding B116D and B80T may be mechanistically similar with respect to AD disease remission in that HLA disease associations with AD should be evaluated through the perspective that HLAs influence not only antigen presentation to TCR but also NK activation.
As with all epidemiologic studies, this study has limitations. Although this study is the largest study of its kind, we are under-powered to evaluate less common residue variations and for this reason we a priori limited our analyses to residue variants most likely to affect the binding pocket based on SRI. Restricting our analyses to more common variants may limit a fuller understanding of the underlying mechanisms. However, our initial evaluation of allelic variants with frequencies ≥0.05 is arguably clinically meaningful for a common disease that has a prevalence of about 10%. Furthermore, we ultimately evaluated the protein residue altered by the alleles thereby allowing for the inclusion of both common and rarer HLA allelic variants. The PEER DNA study is a subset of the PEER study, a national study that broadly sampled the population of the US. As purposeful sampling techniques were not used for the PEER study and the PEER DNA cohort was composed of volunteers from the original PEER, the sample includes children from across the US and is likely generalizable.
In summary, our findings demonstrate that HLA variation is associated with likelihood of remission of AD over time. Our findings add to the general body of evidence about the genetics of AD and, specifically, the association of HLA Class I residue variation with the likelihood of disease remission. Specifically, in the context of AD disease susceptibility but not disease prognosis, previous studies have shown an associations of HLA variation as well as reports suggesting a role for NK cells in the onset of AD.(12, 20, 24, 25) Further characterization of HLA binding pocket changes and potential allergens that increase the risk or severity of AD can expand our understanding of the immunologic mechanisms associated with AD and hold promise for developing more efficient and personalized therapeutic approaches for AD.
Supplementary Material
Key Points:
Atopic dermatitis (AD) is a chronic waxing and waning illness.
HLA Class I polymorphisms are associated with less severe AD.
AD progression is associated with HLA polymorphisms engaging both T and NK cells
Funding sources:
This work was supported in part by grants from the National Institutes for Health (NIAMS) R01-AR060962 (PI: Margolis) and R01-AR070873 (MPI: Margolis/Monos). The PEER study is funded as the Atopic Dermatitis Registry by Valeant Pharmaceuticals International (PI: Margolis).
Abbreviations:
- AF
Allele frequency
- AD
Atopic dermatitis
- CI
Confidence interval
- DNA
Deoxyribonucleic acid
- FLG
Filaggrin
- GWAS
Genome wide association study
- HR-2F
High resolution two-field typing
- HLA
Human Lymphocyte Antigen
- IMGT
Immunogenetics database
- KIR
Killer-cell immunoglobulin-like receptor
- LD
Linkage disequilibrium
- NMDP
National Marrow Donors Program
- NK
Natural killer cell
- NGS
Next generation sequencing
- OR
Odds Ratio
- p
p-value
- PCR
Polymerize chain reaction
- PEER
Pediatric Eczema Elective Registry
- SNP
Single nucleotide polymorphisms
Footnotes
Potential Conflicts of Interest: David Margolis is or recently has been a consultant for Pfizer, Leo, and Sanofi with respect to studies of atopic dermatitis and serves on an advisory board for the National Eczema Association. Dimitri S. Monos is Chair of the Scientific Advisory Board of Omixon and owns options in Omixon. Dimitri Monos, Deborah Ferriola, and Jamie Duke receive royalties from Omixon.
References
- 1.Chiesa Fuxench ZC, Block JK, Boguniewicz M, Boyle J, Fonacier L, Gelfand JM, Grayson MH, Margolis DJ, Mitchell L, Silverberg JI, Schwartz L, Simpson EL, and Ong PY. 2019. Atopic Dermatitis in America Study: A Cross-Sectional Study Examining the Prevalence and Disease Burden of Atopic Dermatitis in the US Adult Population. J. Invest. Dermatol 139: 583–590. [DOI] [PubMed] [Google Scholar]
- 2.Asher MI, Montefort S, Bjorksten B, Lai CK, Strachan DP, Weiland SK, Williams H, Group IPTS, Asher MI, Montefort S, Bjorksten B, Lai CKW, Strachan DP, Weiland SK, Williams H, and Group IPTS. 2006. Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC Phases One and Three repeat multicountry cross-sectional surveys. Lancet 368: 733–743. [DOI] [PubMed] [Google Scholar]
- 3.Margolis JS, Abuabrar K, Bilker W, Hoffstad O, and Margolis DJ. 2014. Persistance of mild of mild to moderate atopic dermatitis. JAMA Dermatology 150: 593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silverberg JI, Gelfand JM, Margolis DJ, Boguniewicz M, Fonacier L, Grayson MH, Ong PY, Chiesa Fuxench Z, and Simpson EL. 2019. Atopic Dermatitis in US Adults: From Population to Health Care Utilization. J Allergy Clin Immunol Pract 7: 1524–1532.e1522. [DOI] [PubMed] [Google Scholar]
- 5.Brown SJ, and McLean WH. 2012. One remarkable molecule: filaggrin. J. Invest. Dermatol 132: 751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Margolis DJ, Mitra N, Wubbenhorst B, and Nathanson KL. 2019. Filaggrin sequencing and bioinformatics tools. Arch. Dermatol. Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Margolis DJ, Kim B, Apter AJ, Gupta J, Hoffstad O, Papadopoulos M, and Rebbeck TR. 2013. Thymic stromal lymphopoietin variation, filaggrin loss-of-function, and persistence of atopic dermatitis. JAMA Dermatology 150: 254–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang Y, Chang C, and Lu Q. 2016. The Genetics and Epigenetics of Atopic Dermatitis-Filaggrin and Other Polymorphisms. Clin. Rev. Allergy Immunol 51: 315–328. [DOI] [PubMed] [Google Scholar]
- 9.Lou C, Mitra N, Wubbenhorst B, D’Andrea K, Hoffstad O, Kim BS, Yan A, Zaenglein AL, Fuxench ZC, Nathanson KL, and Margolis DJ. 2019. Association between fine mapping thymic stromal lymphopoietin and atopic dermatitis onset and persistence. Ann. Allergy. Asthma. Immunol 123: 595–601.e591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beck LA, Thaci D, Hamilton JD, Graham NM, Bieber T, Rocklin R, Ming JE, Ren H, Kao R, Simpson E, Ardeleanu M, Weinstein SP, Pirozzi G, Guttman-Yassky E, Suarez-Farinas M, Hager MD, Stahl N, Yancopoulos GD, and Radin AR. 2014. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N. Engl. J. Med 371: 130–139. [DOI] [PubMed] [Google Scholar]
- 11.Gough SC, and Simmonds MJ. 2007. The HLA Region and Autoimmune Disease: Associations and Mechanisms of Action. Curr Genomics 8: 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paternoster L, Standl M, Baurecht H, Evans DM, and Weidinger S. 2015. Multi-ethnic genome-wide association study of 21,000 cases and 95,000 controls identifies 11 novel risk loci for atopic dermatitis. Journal of Investigative Dermatology 135: S56–S56. [Google Scholar]
- 13.Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, Curtin JA, Bonnelykke K, Tian C, Takahashi A, Esparza-Gordillo J, Alves AC, Thyssen JP, den Dekker HT, Ferreira MA, Altmaier E, Sleiman PM, Xiao FL, Gonzalez JR, Marenholz I, Kalb B, Pino-Yanes M, Xu CJ, Carstensen L, Groen-Blokhuis MM, Venturini C, Pennell CE, Barton SJ, Levin AM, Curjuric I, Bustamante M, Kreiner-Moller E, Lockett GA, Bacelis J, Bunyavanich S, Myers RA, Matanovic A, Kumar A, Tung JY, Hirota T, Kubo M, McArdle WL, Henderson AJ, Kemp JP, Zheng J, Smith GD, Ruschendorf F, Bauerfeind A, Lee-Kirsch MA, Arnold A, Homuth G, Schmidt CO, Mangold E, Cichon S, Keil T, Rodriguez E, Peters A, Franke A, Lieb W, Novak N, Folster-Holst R, Horikoshi M, Pekkanen J, Sebert S, Husemoen LL, Grarup N, de Jongste JC, Rivadeneira F, Hofman A, Jaddoe VW, Pasmans SG, Elbert NJ, Uitterlinden AG, Marks GB, Thompson PJ, Matheson MC, Robertson CF, Ried JS, Li J, Zuo XB, Zheng XD, Yin XY, Sun LD, McAleer MA, O’Regan GM, Fahy CM, Campbell L, Macek M, Kurek M, Hu D, Eng C, Postma DS, Feenstra B, Geller F, Hottenga JJ, Middeldorp CM, Hysi P, Bataille V, Spector T, Tiesler CM, Thiering E, Pahukasahasram B, Yang JJ, Imboden M, Huntsman S, Vilor-Tejedor N, Relton CL, Myhre R, Nystad W, Custovic A, Weiss ST, Meyers DA, Soderhall C, Melen E, Ober C, Raby BA, Simpson A, Jacobsson B, Holloway JW, Bisgaard H, Sunyer J, Probst-Hensch NM, Williams LK, Godfrey KM, Wang CA, Boomsma DI, Melbye M, Koppelman GH, Jarvis D, McLean WH, Irvine AD, Zhang XJ, Hakonarson H, Gieger C, Burchard EG, Martin NG, Duijts L, Linneberg A, Jarvelin MR, Nothen MM, Lau S, Hubner N, Lee YA, Tamari M, Hinds DA, Glass D, Brown SJ, Heinrich J, Evans DM, and Weidinger S. 2015. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat. Genet 47: 1449–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirota T, Takahashi A, Kubo M, Tsunoda T, Tomita K, Sakashita M, Yamada T, Fujieda S, Tanaka S, Doi S, Miyatake A, Enomoto T, Nishiyama C, Nakano N, Maeda K, Okumura K, Ogawa H, Ikeda S, Noguchi E, Sakamoto T, Hizawa N, Ebe K, Saeki H, Sasaki T, Ebihara T, Amagai M, Takeuchi S, Furue M, Nakamura Y, and Tamari M. 2012. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nature Genetics 44: 1222–1226. [DOI] [PubMed] [Google Scholar]
- 15.Mansur AH, Williams GA, Bishop DT, Markham AF, Lewis S, Britton J, and Morrison JF. 2000. Evidence for a role of HLA DRB1 alleles in the control of IgE levels, strengthened by interacting TCR A/D marker alleles. Clin Exp Allergy 30: 1371–1378. [DOI] [PubMed] [Google Scholar]
- 16.Saeki H, Kuwata S, Nakagawa H, Etoh T, Yanagisawa M, Miyamoto M, Tokunaga K, Juji T, and Shibata Y. 1995. Analysis of diseae-associated amino acid epitopes on HLA class II moleculres in atopic dermatitis. J Allergy Clin Immunol 96: 1061–1068. [DOI] [PubMed] [Google Scholar]
- 17.Park H, Ahn K, Park MH, and Lee SI. 2012. The HLA-DRB1 polymorphism is associated with atopic dermatitis, but not egg allergy in Korean children. Allergy Asthma Immunol Res 4: 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Margolis DJ, Mitra N, Kim B, Gupta J, Hoffstad OJ, Papadopoulos M, Wubbenhorst B, Nathanson KL, Duke JL, Monos DS, and Kamoun M. 2015. Association of HLA-DRB1 genetic variants with the persistence of atopic dermatitis. Human Immunology 76: 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weidinger S, Willis-Owen SA, Kamatani Y, Baurecht H, Morar N, Liang L, Edser P, Street T, Rodriguez E, O’Regan GM, Beattie P, Folster-Holst R, Franke A, Novak N, Fahy CM, Winge MC, Kabesch M, Illig T, Heath S, Soderhall C, Melen E, Pershagen G, Kere J, Bradley M, Lieden A, Nordenskjold M, Harper JI, McLean WH, Brown SJ, Cookson WO, Lathrop GM, Irvine AD, and Moffatt MF. 2013. A genome-wide association study of atopic dermatitis identifies loci with overlapping effects on asthma and psoriasis. Human Molecular Genetics 22: 4841–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paternoster L, Standl M, Chen CM, Ramasamy A, Bonnelykke K, Duijts L, Ferreira MA, Alves AC, Thyssen JP, Albrecht E, Baurecht H, Feenstra B, Sleiman PM, Hysi P, Warrington NM, Curjuric I, Myhre R, Curtin JA, Groen-Blokhuis MM, Kerkhof M, Saaf A, Franke A, Ellinghaus D, Folster-Holst R, Dermitzakis E, Montgomery SB, Prokisch H, Heim K, Hartikainen AL, Pouta A, Pekkanen J, Blakemore AI, Buxton JL, Kaakinen M, Duffy DL, Madden PA, Heath AC, Montgomery GW, Thompson PJ, Matheson MC, Le SP, Australian Asthma Genetics C, St PB, Smith GD, Henderson J, Kemp JP, Timpson NJ, Deloukas P, Ring SM, Wichmann HE, Muller-Nurasyid M, Novak N, Klopp N, Rodriguez E, McArdle W, Linneberg A, Menne T, Nohr EA, Hofman A, Uitterlinden AG, van Duijn CM, Rivadeneira F, de Jongste JC, van der Valk RJ, Wjst M, Jogi R, Geller F, Boyd HA, Murray JC, Kim C, Mentch F, March M, Mangino M, Spector TD, Bataille V, Pennell CE, Holt PG, Sly P, Tiesler CM, Thiering E, Illig T, Imboden M, Nystad W, Simpson A, Hottenga JJ, Postma D, Koppelman GH, Smit HA, Soderhall C, Chawes B, Kreiner-Moller E, Bisgaard H, Melen E, Boomsma DI, Custovic A, Jacobsson B, Probst-Hensch NM, Palmer LJ, Glass D, Hakonarson H, Melbye M, Jarvis DL, Jaddoe VW, Gieger C, C. Genetics of Overweight Young Adults, Strachan DP, Martin NG, Jarvelin MR, Heinrich J, Evans DM, Weidinger S, Genetics EA, and C. Lifecourse Epidemiology. 2012. Meta-analysis of genome-wide association studies identifies three new risk loci for atopic dermatitis. Nature Genetics 44: 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baurecht H, Hotze M, Brand S, Buning C, Cormican P, Corvin A, Ellinghaus D, Ellinghaus E, Esparza-Gordillo J, Folster-Holst R, Franke A, Gieger C, Hubner N, Illig T, Irvine AD, Kabesch M, Lee YA, Lieb W, Marenholz I, McLean WH, Morris DW, Mrowietz U, Nair R, Nothen MM, Novak N, O’Regan GM, Schreiber S, Smith C, Strauch K, Stuart PE, Trembath R, Tsoi LC, Weichenthal M, Barker J, Elder JT, Weidinger S, Cordell HJ, and Brown SJ. 2015. Genome-wide comparative analysis of atopic dermatitis and psoriasis gives insight into opposing genetic mechanisms. Am. J. Hum. Genet 96: 104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mack MR, Brestoff JR, Berrien-Elliott MM, Trier AM, Yang TB, McCullen M, Collins PL, Niu H, Bodet ND, Wagner JA, Park E, Xu AZ, Wang F, Chibnall R, Council ML, Heffington C, Kreisel F, Margolis DJ, Sheinbein D, Lovato P, Vivier E, Cella M, Colonna M, Yokoyama WM, Oltz EM, Fehniger TA, and Kim BS. 2020. Blood natural killer cell deficiency reveals an immunotherapy strategy for atopic dermatitis. Sci. Transl. Med 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kusnierczyk P 2013. Killer cell immunoglobulin-like receptor gene associations with autoimmune and allergic diseases, recurrent spontaneous abortion, and neoplasms. Front. Immunol 4: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niepieklo-Miniewska W, Majorczyk E, Matusiak L, Gendzekhadze K, Nowak I, Narbutt J, Lesiak A, Kuna P, Poninska J, Pietkiewicz-Sworowska A, Samolinski B, Ploski R, Szepietowski JC, Senitzer D, and Kusnierczyk P. 2013. Protective effect of the KIR2DS1 gene in atopic dermatitis. Gene 527: 594–600. [DOI] [PubMed] [Google Scholar]
- 25.Hall TJ, Rycroft R, and Brostoff J. 1985. Decreased natural killer cell activity in atopic eczema. Immunology 56: 337–344. [PMC free article] [PubMed] [Google Scholar]
- 26.von Bubnoff D, Andres E, Hentges F, Bieber T, Michel T, and Zimmer J. 2010. Natural killer cells in atopic and autoimmune diseases of the skin. J Allergy Clin Immunol 125: 60–68. [DOI] [PubMed] [Google Scholar]
- 27.Margolis DJ, Apter AJ, Gupta J, Hoffstad O, Papadopoulos M, Campbell LE, Sandilands A, McLean WH, Rebbeck TR, and Mitra N. 2012. The persistence of atopic dermatitis and filaggrin (FLG) mutations in a US longitudinal cohort. J Allergy Clin Immunol 130: 912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang J, Mitra N, Hoffstad O, and Margolis DJ. 2017. Association of Filaggrin Loss of Function and Thymic Stromal Lymphopoietin Variation With Treatment Use in Pediatric Atopic Dermatitis. JAMA Dermatol 153: 275–281. [DOI] [PubMed] [Google Scholar]
- 29.Margolis DJ, Abuabara K, Hoffstad OJ, Wan J, Raimondo D, and Bilker WB. 2015. Association Between Malignancy and Topical Use of Pimecrolimus. JAMA Dermatol 151: 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang J, Bilker WB, Hoffstad O, and Margolis DJ. 2017. Cross-sectional comparisons of patient-reported disease control, disease severity and symptom frequency in children with atopic dermatitis. Br. J. Dermatol 177: e114–e115. [DOI] [PubMed] [Google Scholar]
- 31.Silverberg JI, Gelfand JM, Margolis DJ, Boguniewicz M, Fonacier L, Grayson MH, Simpson EL, Ong PY, and Chiesa Fuxench ZC. 2018. Patient burden and quality of life in atopic dermatitis in US adults: A population-based cross-sectional study. Ann. Allergy. Asthma. Immunol 121: 340–347. [DOI] [PubMed] [Google Scholar]
- 32.Silverberg JI, Margolis DJ, Boguniewicz M, Fonacier L, Grayson MH, Ong PY, Fuxench ZC, and Simpson EL. 2020. Validation of five patient-reported outcomes for atopic dermatitis severity in adults. Br. J. Dermatol 182: 104–111. [DOI] [PubMed] [Google Scholar]
- 33.van Deutekom HW, and Kesmir C. 2015. Zooming into the binding groove of HLA molecules: which positions and which substitutions change peptide binding most? Immunogenetics 67: 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armstrong RA 2014. When to use the Bonferroni correction. Ophthalmic Physiol. Opt 34: 502–508. [DOI] [PubMed] [Google Scholar]
- 35.Middleton D, and Gonzelez F. 2010. The extensive polymorphism of KIR genes. Immunology 129: 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin MP, Naranbhai V, Shea PR, Qi Y, Ramsuran V, Vince N, Gao X, Thomas R, Brumme ZL, Carlson JM, Wolinsky SM, Goedert JJ, Walker BD, Segal FP, Deeks SG, Haas DW, Migueles SA, Connors M, Michael N, Fellay J, Gostick E, Llewellyn-Lacey S, Price DA, Lafont BA, Pymm P, Saunders PM, Widjaja J, Wong SC, Vivian JP, Rossjohn J, Brooks AG, and Carrington M. 2018. Killer cell immunoglobulin-like receptor 3DL1 variation modifies HLA-B*57 protection against HIV-1. J. Clin. Invest 128: 1903–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saunders PM, Vivian JP, Baschuk N, Beddoe T, Widjaja J, O’Connor GM, Hitchen C, Pymm P, Andrews DM, Gras S, McVicar DW, Rossjohn J, and Brooks AG. 2015. The interaction of KIR3DL1*001 with HLA class I molecules is dependent upon molecular microarchitecture within the Bw4 epitope. J. Immunol 194: 781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saunders PM, Vivian JP, O’Connor GM, Sullivan LC, Pymm P, Rossjohn J, and Brooks AG. 2015. A bird’s eye view of NK cell receptor interactions with their MHC class I ligands. Immunol. Rev 267: 148–166. [DOI] [PubMed] [Google Scholar]
- 39.Horowitz A, Djaoud Z, Nemat-Gorgani N, Blokhuis J, Hilton HG, Beziat V, Malmberg KJ, Norman PJ, Guethlein LA, and Parham P. 2016. Class I HLA haplotypes form two schools that educate NK cells in different ways. Sci Immunol 1: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cella M, Longo A, Ferrara GB, Strominger JL, and Colonna M. 1994. NK3-specific natural killer cells are selectively inhibited by Bw4-positive HLA alleles with isoleucine 80. J Exp Med 180: 1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zerva L, Cizman B, Mehra NK, Alahari SK, Murah R, Zmijewski CM, Kamopun M, and Monos DS. 1996. Arginine at positions 13 or 70–71 in pocket 4 of HLA-DRB1 is associated with susceptibility to tuberculoid leprosy. J Exp Med 183: 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams AP, Peh CA, Purcell AW, McCluskey J, and Elliott T. 2002. Optimization of the MHC class I peptide cargo is dependent on tapasin. Immunity 16: 509–520. [DOI] [PubMed] [Google Scholar]
- 43.Petersdorf EW 2017. In celebration of Ruggero Ceppellini: HLA in transplantation. Hla 89: 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferrara GB, Bacigalupo A, Lamparelli T, Lanino E, Delfino L, Morabito A, Parodi AM, Pera C, Pozzi S, Sormani MP, Bruzzi P, Bordo D, Bolognesi M, Bandini G, Bontadini A, Barbanti M, and Frumento G. 2001. Bone marrow transplantation from unrelated donors: the impact of mismatches with substitutions at position 116 of the human leukocyte antigen class I heavy chain. Blood 98: 3150–3155. [DOI] [PubMed] [Google Scholar]
- 45.Littera R, Piredda G, Argiolas D, Lai S, Congeddu E, Ragatzu P, Melis M, Carta E, Michittu MB, Valentini D, Cappai L, Porcella R, Alba F, Serra M, Loi V, Maddi R, Orrù S, La Nasa G, Caocci G, Cusano R, Arras M, Frongia M, Pani A, and Carcassi C. 2017. KIR and their HLA Class I ligands: Two more pieces towards completing the puzzle of chronic rejection and graft loss in kidney transplantation. PLoS One 12: e0180831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campbell KS, and Purdy AK. 2011. Structure/function of human killer cell immunoglobulin-like receptors: lessons from polymorphisms, evolution, crystal structures and mutations. Immunology 132: 315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin MP, Qi Y, Gao X, Yamada E, Martin JN, Pereyra F, Colombo S, Brown EE, Shupert WL, Phair J, Goedert JJ, Buchbinder S, Kirk GD, Telenti A, Connors M, O’Brien SJ, Walker BD, Parham P, Deeks SG, McVicar DW, and Carrington M. 2007. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat. Genet 39: 733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brunner PM, He H, Pavel AB, Czarnowicki T, Lefferdink R, Erickson T, Canter T, Puar N, Rangel SM, Malik K, Estrada Y, Krueger JG, Guttman-Yassky E, and Paller AS. 2019. The blood proteomic signature of early-onset pediatric atopic dermatitis shows systemic inflammation and is distinct from adult long-standing disease. J Am Acad Dermatol 81: 510–519. [DOI] [PubMed] [Google Scholar]
- 49.Pavel AB, Zhou L, Diaz A, Ungar B, Dan J, He H, Estrada YD, Xu H, Fernandes M, Renert-Yuval Y, Krueger JG, and Guttman-Yassky E. 2020. The proteomic skin profile of moderate-to-severe atopic dermatitis patients shows an inflammatory signature. J Am Acad Dermatol 82: 690–699. [DOI] [PubMed] [Google Scholar]
- 50.Sanyal RD, Pavel AB, Glickman J, Chan TC, Zheng X, Zhang N, Cueto I, Peng X, Estrada Y, Fuentes-Duculan J, Alexis AF, Krueger JG, and Guttman-Yassky E. 2019. Atopic dermatitis in African American patients is TH2/TH22-skewed with TH1/TH17 attenuation. Ann. Allergy. Asthma. Immunol 122: 99–110.e116. [DOI] [PubMed] [Google Scholar]
- 51.Czarnowicki T, He H, Krueger JG, and Guttman-Yassky E. 2019. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol 143: 1–11. [DOI] [PubMed] [Google Scholar]
- 52.Debebe BJ, Boelen L, Lee JC, Sanders EJ, Anzala O, Kamali A, Kaleebu P, Karita E, Kilembe W, Inambao M, Lakhi S, Allen S, Hunter E, Edward VA, Fast PE, Price MA, Gilmour J, Tang J, Thio CL, Astemborski J, Kirk G, Khakoo SI, Donfield SM, Goedert JJ, and Asquith B. 2020. Identifying the immune interactions underlying HLA class I disease associations. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.