

Abstract
Successful pregnancy relies on maternal immunologic tolerance mechanisms limit maladaptive immune responses against the semi-allogeneic fetus and placenta and support fetal growth. Preeclampsia is a common disorder of pregnancy that affects 4–10% of pregnancies and is a leading cause of maternal and neonatal morbidity and mortality. Preeclampsia clinically manifests as maternal hypertension, proteinuria, and progressive multi-organ injury likely triggered by hypoxic injury to the placenta, resulting in local and systemic anti-angiogenic and inflammatory factor production. Despite the steady rising rates of preeclampsia in the United States, effective treatment options are limited to delivery, which improves maternal status often at the cost of prematurity in the newborn. Preeclampsia also increases the lifelong risk of cardiovascular disease for both mother and infant. Thus, identifying new therapeutic targets is a high priority area to improve maternal, fetal, and infant health outcomes. Immune abnormalities in the placenta and in the maternal circulation have been reported to precede the clinical onset of disease. In particular, excessive systemic and placental complement activation and impaired adaptive T cell tolerance with Th1/Th2/Th17/Treg imbalance has been reported in humans and in animal models of preeclampsia. In this review, we focus on the evidence for the immune origins of preeclampsia, discuss the promise of immune modulating therapy for prevention or treatment, and highlight key areas for future research.
Keywords: Preeclampsia, complement, maternal-fetal tolerance, regulatory T cells, pregnancy, Th17, immune modulation
INTRODUCTION
The human placenta is a fetal-derived organ that interfaces directly with the maternal uterine decidua—a vascular-immunologic tissue interface that nourishes and facilitates peaceful coexistence of the semi-allogenic fetus.1,2 The placental extravillous trophoblast cells invade directly into maternal uterine vessels to facilitate efficient nutrient exchange; thus, the maternal immune system experiences direct exposure to foreign fetal antigens locally in the decidua. Systemic shedding of placental and fetal material occurs throughout the pregnancy—in fact, this chimerism is exploited clinically in non-invasive prenatal screening methods, which detect placental DNA in maternal circulation for assessing risk for fetal Trisomy 21 and other common fetal chromosomal disorders.3–6 Active immune tolerance mechanisms in pregnancy are critical to prevent fetal rejection, which may manifest as early pregnancy loss, preterm birth, fetal growth restriction, or preeclampsia (PE).
PE is a heterogeneous obstetric disorder that is clinically defined by new-onset maternal hypertension (systolic blood pressure ≥140 mmHg and/or diastolic blood pressure ≥90 mmHg) after 20 weeks’ gestational age that is usually accompanied by new-onset proteinuria (>300mg in 24 hours).7 PE complicates 4–10% of pregnancies and is a leading cause of maternal and fetal/neonatal mortality.8,9 The only definitive treatment for PE is delivery of the placenta and fetus, often accompanied by the neonatal morbidity and mortality of prematurity. Clinically, PE is a systemic syndrome varying by gestational age at onset and severity. Some patients with PE can be asymptomatic and perhaps not diagnosed until delivery, while others may develop early-onset preeclampsia with rapid progression to multi-organ dysfunction, including neurologic symptoms (headache, seizure, stroke), hematologic symptoms (thrombocytopenia, disseminated intravascular coagulation), acute renal failure, hepatic (subcapsular hematoma or rupture) symptoms, or HELLP (hemolysis, elevated liver enzymes, low platelets) syndrome. The clinical manifestations are heterogeneous, suggesting that the biologic underpinnings also are likely heterogenous. Biologically, PE is associated with systemic vascular response and immune activation in reaction to placental syncytiotrophoblast stress and ischemia induced by a combination of angiogenic, immunologic, genetic, or maternal/environmental factors.9 Risk factors for PE, such as nulliparity, change in paternity from prior pregnancies, donor gamete in vitro fertilization, baseline maternal autoimmune disorders, and multifetal gestation, all support impaired maternal immune tolerance to paternal antigens as a potential etiology.
In this review, we cover the mechanisms of adaptive and innate immune mechanisms that support healthy pregnancy, evidence for local and systemic immune dysregulation in pregnancy pathology (with a focus on PE), as well as current and future therapeutic strategies that target immune mechanisms of PE.
1. Immune mechanisms of healthy pregnancy and preeclampsia
1.1. The complement system in healthy pregnancy
The complement system is part of the innate immune system and actuates potent inflammatory responses, direct cellular damage through the membrane attack complex, and targets pathogens for phagocytosis. The complement system is comprised of circulating plasma proteins and cell-bound components primarily produced by the liver that react in a proteolytic cascade when triggered through either the classical, alternative, or lectin pathways (Fig 1).10 The classical pathway refers to antigen-antibody immune complex-mediated activation, whereas the alternative pathway refers to spontaneous and continuous amplification of the system in response to tissue damage. Finally, the lectin pathway refers to reaction triggered by the presence carbohydrates in the cell walls of microorganisms.11 Each of these mechanisms results in activation of the C3 convertase, which releases C3b. C3b combines with the C3 convertase to cleave C5 and allows C5b to combines with C6, C7, C8, and a C9 components to create the terminal membrane attack complex (MAC) that forms a lytic pore in its target. C3a and C5a cleavage products are potent pro-inflammatory mediators. The complement system is crucial to mediate defense against pathogens, but excessive activation also is associated with clinical disease such as hemolytic uremic syndrome, a close imitator of PE and HELLP syndrome.
Figure 1. Local and systemic complement activation in preeclampsia and healthy pregnancy and complement-modulating medications.
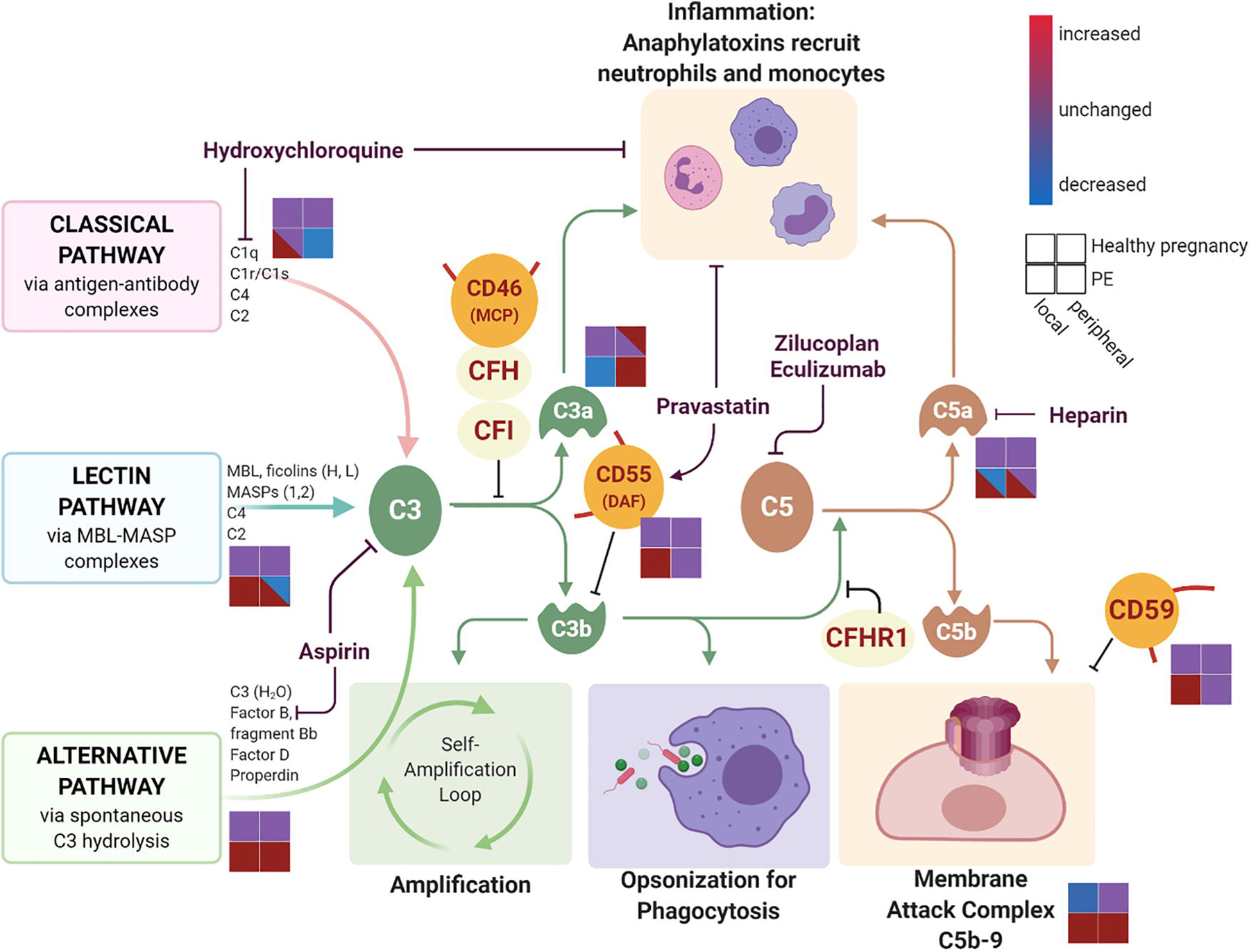
The complement cascade is initiated through the classical, lectin, and alternative pathways. Complement activation results in inflammation primarily mediated through anaphylatoxins C3a and C5a, pathogen opsonization for phagocytosis via C3b, and cell lysis through the terminal membrane attack complex formation. Endogenous soluble regulators of complement activation (CFH, CFI, CFHR1) and membrane-bound regulators (MCP, DAF, and CD59) work through inhibition of the complement pathway at different steps. Various pharmacologic agents also target the complement activation at different steps. Schematic for the level of complement component activation is represented for healthy pregnancy (upper two squares) and in preeclampsia (PE, lower two squares) and separated by local decidua complement activation (left squares) and peripheral complement activation (right squares). Complement component levels are increased (red), decreased (blue), or unchanged (purple) depending on the disease state and location in pregnancy. Created with BioRender.com
During pregnancy, the complement system is upregulated to ensure protection of the fetal-placental unit from pathogens.11 Systemic complement activation in pregnancy results in deposition of degradation products in the syncytiotrophoblasts during normal pregnancy to protect against potential infections.12,13 Local complement production by human syncytiotrophoblasts and uterine decidua endothelial cells also contributes to pathogen protection.14,15 Pregnant C1q-deficient mice develop features of placental insufficiency and PE, supporting a role for complement deposition in trophoblast migration and spinal artery remodeling critical for normal placentation.11 However, the evidence is more variable in humans, with one study demonstrating a correlation between C1q deposition in syncytiotrophoblast and healthy placentation16 while others found no changes C1q syncytiotrophoblast in healthy placentas compared to PE.17,18 Cross-sectional studies have demonstrated that healthy pregnant patients exhibit higher levels of complement degradation products compared to non-pregnant patients, supporting the need for enhanced complement activation during healthy pregnancy.10
Increased complement activation is important to protect against pathogens and likely for healthy placentation; however, too much complement activity can result in recognition and rejection of fetal-derived tissues. Soluble complement inhibitory proteins complement factor H (CFH), complement factor I (CFI), and CFH-related 1 (CFHR1) protect against aberrant complement activation.19 Membrane-bound complement inhibitory proteins decay-accelerating factor (DAF also known as CD55), membrane cofactor protein (MCP, also known as CD46), and CD59 are expressed directly by syncytiotrophoblasts and prevent excessive complement activation and damage to the placenta.11,20
1.2. The complement system and adverse pregnancy outcomes
Excessive complement activation or imbalance in the activation and inhibitory pathways is associated with adverse pregnancy outcomes (summarized in Table 1). Systemic elevation in complement activation products from the classic and lectin pathway such as C3a, C5a, and MAC are predictive of or present in excess during clinical presentation of PE.19,21–23 In one study, systemic complement activation products such as C4d, C3a, soluble C5b9 were significantly higher in pregnant patients with PE compared to both non-pregnant and healthy pregnant patients.24 Elevated levels of complement fragment Bb, a marker of alternative complement pathway activation in systemic circulation also precedes onset of clinical PE.25 These results implicate early activation and terminal pathway activation in the development of PE. Significantly higher expression of C4d and MAC present on the syncytiotrophoblasts membranes in PE also was identified by immunofluorescence staining, suggesting that local complement activation in PE can potentially lead directly to placental damage and ischemia.26 Furthermore, in cell culture, complement activation on syncytialized human trophoblasts induced upregulation of soluble fms-like tyrosine kinase receptor 1 (sFLT1), an anti-angiogenic factor that has been linked with the pathogenesis of the maternal syndrome.27 In animal models, complement activation and in particular C5a has been shown to be required intermediary event in the pathogenesis of placental injury by inducing dysregulation of angiogenic factors required for normal placental development.13 Furthermore, treatment with various complement inhibitors (soluble complement receptor 1, C3a receptor antagonist, or C5a receptor antagonist) resulted in decreased blood pressure in the RUPP (reduced utero-placental perfusion pressure) rat model of PE.18 Taken together with human studies that demonstrate a robust correlation of C4d with placental sFLT1 expression,26 placental complement activation could be a major trigger for placental damage and release of anti-angiogenic factors in PE.
Table 1:
Overview of local and peripheral immune pathogenesis in preeclampsia
| Immune Pathway | Maternal-fetal Interface | Peripheral Blood |
|---|---|---|
| Complement cascade | ||
| Classical pathway | C1q deposition in chorionic villi, placental blood vessel endothelia90–93 | Lower levels of C1q93 |
| Lectin pathway | Higher C4d, ficolins H, L deposition in syncytiotrophoblast90,94–96 | Lower levels of C4, Ficolins H, L93,96–98 Higher levels of C4d24,99 |
| Alternative pathway | C3 deposition in decidua tissue, villous endothelial cells91,100–102 | Higher levels of fragment Bb103 |
| C3a and C5a (anaphylatoxins) | Lower C3a receptor and conflicting results of higher and lower C5a expression in preeclampsia18 | Majority of studies with higher C3a18,19 Reports of both higher19 and similar C5a93 |
| Terminal MAC (C5b-9) | Increased MAC deposition in stroma and syncytiotrophoblast26,92 | Most demonstrate higher MAC19,22 |
| Complement regulatory proteins | DAF and CD59 upregulated90 | |
|
T helper bias | ||
| Th1 | Increased TNFα49,50 | Increased TNFα49,50 |
| Th2 | Reduction in IL-449,50 | Reduction in IL-449,50 |
| Regulatory T cells (Treg) | Reduced FoxP3 and IL-10 expression in first trimester104 and at delivery105,106,107 Decreased Treg clonal expansion47,108 |
Decrease in Treg proportion105,109 and suppressive capacity44 Lower activation of memory Treg35 |
| Th17 | Increased proportion of CD4 T cells (Collier, unpublished) | Upregulation in Th1749 |
DAF= decay accelerating factor
MAC= membrane attack complex
Treg= regulatory T cells
Systemic lupus erythematosus and antiphospholipid antibody syndrome are immune-mediated disorders associated with dysregulated complement activity, and they confer much higher risk of obstetric complications, including preterm PE and fetal growth restriction—another manifestation of placental disease.28 In a large cohort study of pregnant patients with lupus and/or antiphospholipid antibody syndrome, early trimester elevation in soluble MAC and alternative pathway products was strongly associated with adverse pregnancy outcomes, including PE.29 Genetic sequencing of the complement regulatory proteins in the patients with lupus who developed PE revealed that 18% had heterozygous mutations in CFI, MCP, and CFH genes, further supporting the critical importance of complement regulation in the pathogenesis of PE. Genetic variants were found in a cohort of women with PE without underlying autoimmune disease, strengthening the link between complement dysregulation and PE.30
Fetal rejection phenotypes have been observed in chronic histiocytic intervillositis, a placental finding of mononuclear cell deciduitis found in association with PE, fetal growth restriction, and pregnancy loss.31 Chronic histiocytic intervillositis has been associated with M2-polarized placental macrophages and Hoffbauer cells that express complement receptor 4, providing further evidence of complement activation as a triggering event for fetal rejection.32
1.3. Adaptive T cell tolerance in healthy pregnancy
Historically, the paradigm of healthy pregnancy was thought to be driven by a helper T cell (Th) bias, with Th2 favoring maternal tolerance to the allogeneic fetus compared to pro-inflammatory Th1. This dichotomy does not fully capture the dynamic Th repertoire of human T cell biology; thus, the paradigm has been expanded to include pro-inflammatory Th17 cells and regulatory T cell (Treg) subtypes.33 (Fig 2). Th1 cells secrete TNFα and IFNγ and are upregulated in early pregnancy to support early trophoblast invasion into the uterine spiral arteries. After placentation, there is a shift from Th1 to Th2 immunity (characterized by IL-4, IL-10 production), supporting a more anti-inflammatory state until parturition. Th17 cells are another class of pro-inflammatory helper T cells present at the maternal-fetal interface. Th17 cells are critical in launching pro-inflammatory IL-17A anti-bacterial and anti-fungal responses and have been pathologically implicated in autoimmune disorders such as lupus, psoriasis, and multiple sclerosis.34 Th2 cells are able to suppress Th17 activation through IL-10 secretion and suppress Th1 activation through IL-4 production. In addition to Th1, Th2, and Th17, the fourth, and arguably most critical, players in the maintenance of maternal T cell tolerance to fetal and placental antigens are the suppressive regulatory T cells (Treg).35,36 The canonical natural Treg are produced centrally in the thymus and are characterized by high FoxP3 expression. Treg are increased in normal pregnancy systemically37 and present locally in human decidua. More recently, multiple Treg subtypes matching the description of induced type 1 regulatory (Tr1) cells38 have been described in first-trimester and term human decidua with the capacity to suppress effector T cell proliferation and suppressive IL-10 cytokine production.39 The necessity of Treg suppression for healthy pregnancy development is illustrated in mouse models in which rejection of antigen distinct male fetuses manifests as pregnancy loss or decrease in male birthweight occurs after Treg depletion.40,41
Figure 2. T helper cell changes locally and peripherally in healthy pregnancy and preeclampsia.
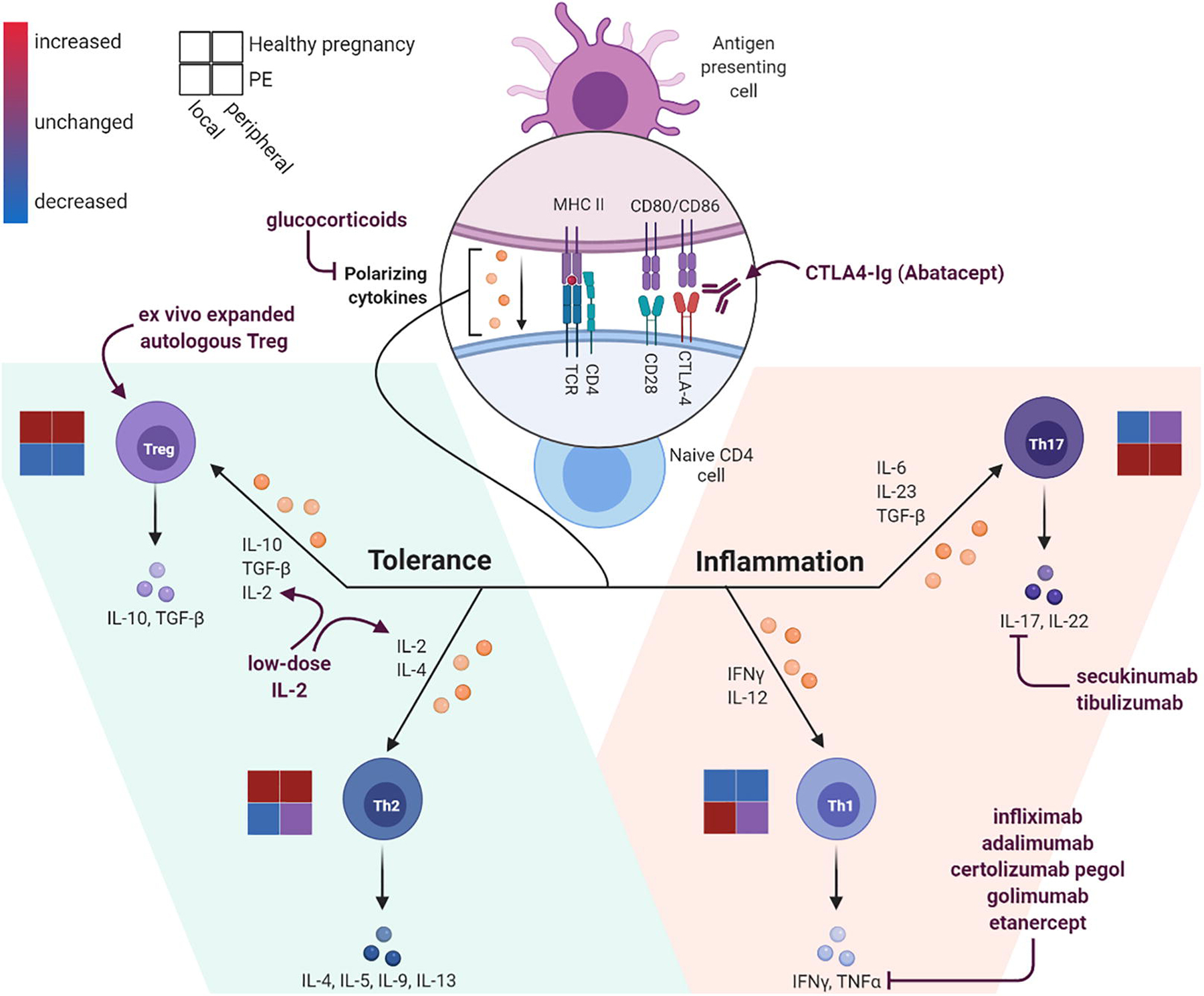
When naïve CD4 T cells interface with cognate antigen-MHC complexes, the presence of co-stimulatory (CD28) and co-inhibitory (CTLA-4) activation along with type of polarizing cytokines present will determine the helper T cell (Th) subset differentiation. Regulatory T cells (Treg) and Th2 polarization favors a more suppressive, immune tolerant state (left); whereas Th1 in and Th17 polarization favors a more pro-inflammatory state (right). Various immune modulating medications have been developed to promote differentiation into Treg and Th2 subsets and to inhibit inflammatory Th1 and Th17 differentiation or inhibit resultant cytokines. Schematic for the level of Th pathway activation is represented for healthy pregnancy (upper squares) and in preeclampsia (PE, lower squares) and separated by local decidua T cell activation (left squares) and peripheral T cell activation (right squares). Levels of Th subset activation are increased (red), decreased (blue), or unchanged (purple) depending on the disease state and location in pregnancy. Created with BioRender.com
Normal human pregnancy is associated with an increase in FoxP3-expressing Treg.37 Multiple findings support that defective placentation in PE may be a manifestation of an immunologic event resulting in a break in maternal-fetal immune tolerance (Table 1). In a study in Iran, the GG genotype of the rs4824747 single nucleotide polymorphism in the promoter region of the FoxP3 gene was more frequent in women with PE compared with controls.42 However, this finding was not reproduced in a similar study in the US.43 Nevertheless, there does appear to be a relationship between Treg function and Th imbalance in PE. Treg are significantly reduced in maternal blood in PE and HELLP syndrome, with associated defects in effector T cell suppressive function.44,45 Hu et al. have proposed that low maternal serum acetate (known to contribute to Treg cell generation) is associated with development of PE and this maternal metabolic derangement is transferred to the offspring resulting in associated prolonged impaired fetal and childhood thymic Treg development.46 Recently, the T cell antigen receptor (TCR) clonal expansion of decidua Treg has been assessed using human samples from the first trimester, third trimester healthy pregnancy, miscarriage, and in PE. In healthy pregnancy, the decidua Treg TCR repertoire becomes more constrained from first to third trimester. In contrast to healthy controls, PE is associated with reduced clonal expansion at delivery.47 This suggests that the skewed TCR repertoire in decidua Treg is important in maintaining immune tolerance. The antigenic targets of clonal Treg at the maternal-fetal interface have not been identified in humans.
1.4. Impaired T cell tolerance in preeclampsia
Similar to the Th cytokine imbalances seen in autoimmunity, PE is associated with an imbalance in both systemic and local pro-inflammatory Th1 and Th17 cytokines (TNFα and IL-17) and a decrease in suppressive Treg and Th2 cytokines (IL-10 and IL-4).48–50 The predominance of IL-17-expressing CD4 T cells in human intervillous blood samples in PE compared with healthy controls at delivery (Collier, unpublished) further supports a pro-inflammatory, fetal rejection phenotype of PE. The RUPP rat model of PE, which utilizes uterine artery ligation to mimic clinical symptoms of PE, depends on the presence of Th17 cells for the clinical phenotype.51 Another animal model in which vasopressin is infused into mice to induce preeclampsia results in increased Th1 and Th17 cells and reduced Treg and Th2 cytokine production, providing a link from vasculopathy to Th1/Th2/Treg/Th17 imbalance in the pathogenesis of PE.52
1.5. Intracellular complement activation in T cells
Thus far, we have reviewed the role of complement and T cell dysregulation as separate pathogenic mechanisms for PE. However, the two are connected. There is an intracellular pathway of complement activation in T cells known as the “complosome.”53,54 CD4 T cells can produce C3 and C5 within intracellular T cell lysosomes. With engagement of the TCR by cognate antigen, C3 is released from the lysosome and cleaved into C3b by lysosomal cathepsin L, resulting in further T cell activation through complement receptors on the T cells’ surface. In addition, intracellular C5 is cleaved into C5a, resulting in intracellular signaling and Th1 T cell differentiation (marked by IFNγ release and canonical transcription factor, Tbet expression).12 Intracellular complement activation has been implicated in intestinal damage during ischemia/reperfusion and may represent an explanatory mechanism linking placenta ischemia to complement activation and Th1/Th17 bias.
2. Current and future therapeutic strategies for preeclampsia
2.1. Pharmacologic approaches currently used for preeclampsia prevention
Many pharmacologic interventions with anti-inflammatory and immune modulating properties are either being used to prevent PE or are under investigation for the treatment of PE ((Fig. 1 and Fig. 2). The most evidence-based preventive treatment currently in use is low-dose aspirin, administered at 81–150 mg daily by mouth, typically beginning prior to 16 weeks’ gestational age.55–57 Aspirin is an anti-pyretic and anti-platelet agent that inhibits prostaglandin thromboxane A2 and is thought to improve trophoblastic invasion into uterine spiral arteries and reduce C3 and factor B expression in the syncytiotrophoblasts. This intervention is recommended for patients with one strong risk factor for PE or more than one moderate risk factor and can reduce the risk of preterm PE. PE. Heparin is another anticoagulant typically given with aspirin to women with antiphospholipid antibody syndrome, which confers particularly high risk for early pregnancy loss and PE. Heparin and low-molecular weight heparin also likely improve uterine artery blood flow and work on immune dysregulation by reducing trophoblast C5a production.58–60 It is recommended that patients with systemic lupus erythematosus continue hydroxychloroquine in pregnancy for both treatment of autoimmune disease (to reduce the risk of flares and achieve remission in pregnancy) and to reduce the risk of PE.61 Hydroxychloroquine is an anti-malarial agent and works through several mechanisms as an anti-inflammatory, antioxidant, and antithrombotic agent and to impair complement-dependent antibody opsonization. This results in impaired antigen processing machinery in antigen-presenting cells, resulting in reduced T cell activation.
2.2. Pharmacologic approaches under investigation for preeclampsia
Other anti-inflammatory and immune modulating therapies currently under investigation to prevent and treat PE and its complications include pravastatin and eculizumab. Pravastatin is a statin that blocks cholesterol synthesis and may improve placental perfusion in PE due to the acute atherosclerotic changes seen in the uterine arteries in PE. In mouse models, pravastatin appears to reduce complement activation through upregulation of complement inhibitor DAF and reduction of C5a activation in the cervix.62 In an open-label, non-randomized trial including 21 patients with antiphospholipid antibody syndrome who developed PE despite use of aspirin and heparin, the addition of pravastatin 20 mg daily resulted in improved placental blood flow, improved clinical features of PE, and pregnancy prolongation when compared with aspirin and heparin only.63 This provides preliminary data to support the potential use of pravastatin for PE treatment. A phase 3 randomized clinical trial evaluating the use of pravastatin versus placebo in secondary prevention of PE in patients with a history of preterm PE requiring delivery prior to 34 weeks (NCT03944512) is underway. Eculizumab is a monoclonal antibody inhibiting C5 that is FDA-approved for use in pregnant patients with paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Eculizumab was used to treat HELLP syndrome in a case report resulting in improved laboratory parameters and pregnancy prolongation (19 days),64 and a phase 1 open-label trial recently opened for enrollment of patients with HELLP syndrome between 23–28 weeks’ gestation (NCT04103489). Zilucoplan is a small molecule inhibitor of C5a that is currently being used to treat myasthenia gravis and also may have potential for treatment of HELLP or PE.
2.3. Immune modulation strategies
IL-17 is a key mediator of inflammation and antibacterial responses and has been implicated in impaired tolerance in PE and other clinical disease states such as autoimmunity, contact dermatitis, and transplant rejection. Secukinumab is a monoclonal antibody against IL-17 that has been used to alter the Th imbalance in contact dermatitis (NCT02778711), psoriasis, and discoid lupus erythematosus (NCT03866317). Tibulizumab (LY3090106) is a dual antagonist tetravalent antibody against IL-17 and BAFF (B cell activating factor) marketed for Sjogrens disorder (NCT04563195) and also may prove a useful strategy for reducing inflammation that may be either causal or a downstream mediator in PE. Other biologic agents (TNF-α blockade) have been used with success to reduce inflammation in inflammatory bowel disease and have been safely used in pregnancy. These may prove useful in reducing the Th1 imbalance observed in PE.
Inflammation mediated through innate immune complement activation and adaptive T cell polarization are features of PE and carry many features that parallel pathogenesis in autoimmunity and transplant rejection. Given the Treg dysfunction in PE, there is a potential for introducing therapy that might support autologous Treg function as a preventive or therapeutic modality. A recent review by Robertson et al. outlined a three-part recommendation to improve Treg function in pregnancy.65 The first is to allow adequate prior priming to male partner alloantigens to establish induced Treg to acquire antigen experience and memory. This may include advice on preconception planning and new diagnostics to assess for established Treg memory in the reproductive tract. The second recommendation is to implement strategies to increase endogenous Treg cell numbers or function through lifestyle interventions, such as sunlight, exercise, or pharmacologic reduction in IL-6, which reduces the STAT5 signaling important for Treg activity. The final strategy is to trial cell therapy that involves ex vivo generation or expansion of autologous polyclonal Treg for infusion. Autologous Treg therapy currently is being investigated in clinical trials for autoimmune diabetes, graft versus host disease, and renal transplants (NCT01210664, NCT02772679, NCT02519816, NCT03284242).66 In diabetes, T cell receptor activity and specific cell therapy against relative antigens has been shown to improve Treg cell recruitment.67 Low-dose IL-2 infusions also have been demonstrated to support Treg homeostasis in patients with newly diagnosed autoimmune type 1 diabetes and may be a useful, safe strategy for improving endogenous Treg numbers and function.68
2.4. Immune checkpoint molecules
Immune checkpoint molecules are co-stimulatory receptors found on the surface of T cells and other immune cells, which bind co-stimulatory molecules (e.g. CD80 and CD86) on the surface of antigen-presenting cells. (Fig 2). In response to ligand binding, immune checkpoint molecules such as CTLA-4, TIM-3, and PD-1 can transduce inhibitory intracellular signals or induce anergy or T cell exhaustion.69 Treg often rely on signaling through CTLA-4 to induce indoleamine 2,3 dioxygenase expression by antigen-presenting cells. Use of CTLA4-Ig (Abatacept) for immune checkpoint inhibition has been demonstrated to increase duration of endogenous insulin production by a median of 9.6 months in newly diagnosed patients with autoimmune type 1 diabetes (NCT00505375).70 This effect size could prove promising for delayed progression of PE in the timeframe of weeks to months needed to drastically reduce perinatal morbidity/mortality from prematurity.
3. Unanswered questions and future research directions
3.1. Phenotyping of preeclampsia syndromes and longitudinal sampling
Not all PE is likely to be immune-mediated. Current work aims to better separate PE syndromes into distinct clusters by using a combination of clinical data and placental histologic and transcriptomic profiles71,72 to create more homogenous designations. While this has many important research applications (limits confounding of clinically similar groups with different molecular fingerprints) and potential clinical applications (ability to design etiology-dependent treatments), this strategy is still limited by the inability to phenotype prior to delivery. Longitudinal cohorts that can tie biomarkers from preconception, interconception, and during pregnancy prior to the onset of PE to differentiate PE syndromes will be important to improve prediction and to develop a personalized medicine approach to treatment. For instance, it is very likely that term preeclampsia is a different pathophysiology and would therefore necessitate a different treatment strategy than preterm PE.1
We have emphasized the importance of evaluation of local immunology at the maternal-fetal interface in pregnancy. Decidua tissue typically is sampled at delivery when the clinical syndrome has already manifested or sampled from pregnancy termination specimens, which prohibits knowledge of clinical outcomes in later pregnancy. Sampling the decidua from ongoing pregnancies is associated with risk of pregnancy loss and thus is not done for research purposes only. Decidua sampling of ongoing pregnancies occurs clinically during first-trimester chorionic villus sampling (CVS) for prenatal genetic testing. The maternal decidua is a bystander contaminating tissue in this scenario, where the target tissue for clinical testing is the chorionic villi that share fetal genetics. Preconception endometrial sampling and discarded first-trimester maternal decidua from CVS procedures are human samples of convenience that have been used previously to link antecedent pathobiology to healthy and adverse pregnancy outcomes.73 Although some patients choose to undergo the risks of elective CVS for prenatal testing, the population undergoing CVS tends to be biased towards higher-risk pregnancies (suspected fetal aneuploidy, multifetal gestations). Close collaboration between gynecologic and obstetric health care providers with access to samples across the reproductive care and reproductive immunologists will be critical to optimizing clinical and biologic data capture from these limited human specimens.
3.2. Multi-omics approaches for exploring the immune landscape of human decidua
Future preclinical studies are needed to better understand the key epigenetic and genetic variants, as well as transcripts regulating decidua immune cells during healthy and pathologic pregnancy. Technologies combining single-cell transcriptomics with antigen receptor sequencing and cell surface proteomic profiling have been utilized to explore the landscape of the murine and human decidua.75–77 These single-cell techniques will be useful for further defining the differential expression, antigen-specificity, and distinct decidua immune cell subtype associated with clinical “decidualopathies” such as preeclampsia, preterm labor, and fetal growth restriction. The uterine natural killer cell (uNK) is abundant and well characterized in decidualization physiology and may play a role in the abnormal placentation observed in PE.82–84 However, cross-talk between uNK cells and the adaptive immune system remains unexplored.
Furthermore, although reduced clonal expansion of T cells expressing the same T cell receptor is associated with PE, we have not identified the antigenic target.47 High-dimensional MHC-peptide and linear peptide arrays have been utilized to identify antigen targets for T cells and antibodies in HIV,78,79 autoimmunity,80 and cancer.81 Using an array of alloantigens and placental antigens we may be able to use such technologies to identify the exact antigenic target for PE or chronic histiocytic intervillositis. Cross-disciplinary collaboration between bioinformaticists, immunologists, and clinicians is necessary to make progress in discovering the key antigenic and cellular targets in PE.74
3.3. Animal models and reverse translational research
One limitation to developing targeted therapies for PE is the lack of valid animal models for use in preclinical studies.85 Multi-omics data from decidua immune cells may identify crucial transcriptional targets that can be evaluated using a reverse translational approach, whereby disease pathogenesis is validated through creation of new animal models. Targeted deletion of placental targets using CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 genomic editing presents a unique method for investigating the decidua leukocyte-trophoblast interactions and clinical consequences in animal models.1
3.4. Future clinical trials
Future clinical trials need to be designed for pregnancy extrapolating from other transplant and autoimmune disease treatments. Trials evaluating efficacy of autologous T cell therapy,86 immune checkpoint inhibitors,69,87,88 or biologics already used in autoimmune disease have the potential to be safely applied to treatment of decidualopathies in pregnancy. The use of tissue-specific liposomes for delivery of immune modulators to the uterus have been investigated to treat preterm labor and may provide a valid vehicle for specific modulation of immune dysfunction occurring specifically at the maternal-fetal interface.89
4. CONCLUSIONS
Preeclampsia is a common disease in pregnancy and a leading cause of maternal and perinatal morbidity and mortality. Healthy pregnancy depends on the coordination of the innate and adaptive maternal immune system that must balance protection of the fetus against pathogens and tolerance of the allogeneic fetus and placenta. PE arises from insufficient immune priming against paternal antigens and an elevated inflammatory load, which is supported by known clinical risk factors for developing PE, including nulliparity, multifetal gestation, in vitro fertilization, prolonged interpregnancy interval, and presence of underlying maternal autoimmune or cardiometabolic disorders. Excessive complement activation and pro-inflammatory Th cell bias are strongly coupled with the development of PE. Consistent disease phenotyping, longitudinal sampling, cross-disciplinary collaboration, and a multi-omics approach to investigating PE will lead to earlier diagnosis and allow for targeted immune tolerance-promoting strategies for treatment and prevention. Available therapies are limited; however, with improved understanding of the immunologic mechanisms of PE, we may be able to leverage existing immune modulating strategies from other fields.
ACKNOWLEDGEMENTS
AYC received support from the grant K12HD000849, awarded to the Reproductive Scientist Development Program by the Eunice Kennedy Shriver National Institute of Child Health & Human Development and Burroughs Wellcome Fund.
List of abbreviations:
- Bb
fragment of complement factor B
- C1q
complement component 1q
- C3
complement component 3
- C3a
complement component 3a
- C3b
complement component 3b
- C4
complement component 4
- C4d
complement component 4d
- C5
complement component 5
- C5a
complement component 5a
- C5b
complement component 5b
- C5b-9
terminal complement complex or membrane attack complex
- CD28
cluster of differentiation 28, co-stimulatory receptor for B7 ligands
- CD4
cluster of differentiation 4 marker of helper T cells
- CD46
cluster of differentiation 46, also known as membrane cofactor protein (MCP)
- CD55
cluster of differentiation 55, also known as decay-accelerating factor
- CD59
cluster of differentiation 59, also known as protectin
- CD80
cluster of differentiation 80, also known as B7–1, a surface ligand on antigen-presenting cells
- CD86
cluster of differentiation 86, also known as B7–2 a surface ligand on antigen-presenting cells
- CFH
complement factor H
- CFHR1
complement factor H related 1
- CFI
complement factor I
- CTLA-4
cytotoxic T-lymphocyte associated protein 4, an immune checkpoint molecule that down regulated immune responses; homology with co-stimulatory receptor CD28
- DAF
decay-accelerating factor, a complement regulatory protein, also known as CD55
- FOXP3
forkhead box P3 or scurfin
- HELLP
hemolysis, elevated liver enzymes, low platelet count
- IFNγ
interferon gamma
- IL-10
interleukin 10
- MAC
membrane attack complex
- MASPs
mannose associated serine proteases
- MBL
mannose binding lectin
- MHC
peptide major histocompatibility complex
- MCP
membrane cofactor protein or CD46
- PD-1
programmed cell death protein 1, an immune checkpoint molecule
- PE
preeclampsia
- Protectin
complement regulatory protein, also known as CD59
- TCR
T cell receptor
- TGF-β
Transforming growth factor beta
- Th1
T helper type 1
- Th17
T helper cell 17
- Th2
T helper type 2
- TIM-3
T cell immunolglobulin and mucin-domain containing-3, an immune checkpoint molecule
- TNFα
tumor necrosis factor alpha
- Tr1
Type 1 regulatory cells
- Treg
regulatory T cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest Statement
Dr. Karumanchi is co-listed as co-inventors on patents related to preeclampsia biomarkers that are held at Beth Israel Deaconess Medical Center. He has financial interest in Aggamin LLC and also reports serving as a consultant to Roche Diagnostics and Thermofisher. Dr. Karumanchi has received research funding from Siemens and Thermofisher. Other authors report no conflicts.
REFERENCES
- 1.PrabhuDas M et al. Immune mechanisms at the maternal-fetal interface: perspectives and challenges vol. 16 328–334 (Nature Publishing Group, 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ander SE, Diamond MS & Coyne CB Immune responses at the maternal-fetal interface. Sci. Immunol 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khosrotehrani K & Bianchi DW Multi-lineage potential of fetal cells in maternal tissue: A legacy in reverse. J. Cell Sci (2005) doi: 10.1242/jcs.02332. [DOI] [PubMed] [Google Scholar]
- 4.Bianchi DW et al. Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obs. Gynecol 119, 890–901 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Lo YM et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2, 61ra91 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Norton ME et al. Cell-free DNA analysis for noninvasive examination of trisomy. N Engl J Med 372, 1589–1597 (2015). [DOI] [PubMed] [Google Scholar]
- 7.ACOG Practice Bulletin No. 202: Gestational Hypertension and Preeclampsia. Obstet. Gynecol (2019) doi: 10.1097/AOG.0000000000003018. [DOI] [PubMed] [Google Scholar]
- 8.Collier ARY & Molina RL Maternal mortality in the united states: Updates on trends, causes, and solutions. Neoreviews (2019) doi: 10.1542/neo.20-10-e561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rana S, Lemoine E, Granger J & Karumanchi SA Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res 124, 1094–1112 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Richani K et al. Normal pregnancy is characterized by systemic activation of the complement system. J. Matern. Neonatal Med 17, 239–245 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denny KJ, Woodruff TM, Taylor SM & Callaway LK Complement in Pregnancy: A Delicate Balance. American Journal of Reproductive Immunology vol. 69 3–11 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Girardi G, Lingo JJ, Fleming SD & Regal JF Essential Role of Complement in Pregnancy: From Implantation to Parturition and Beyond. Frontiers in Immunology vol. 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Girardi G, Yarilin D, Thurman JM, Holers VM & Salmon JE Complement activation induces dysregulation of angiogenic factors and causes fetal rejection and growth restriction. J. Exp. Med 203, 2165–2175 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bulla R et al. Complement production by trophoblast cells at the feto-maternal interface. J. Reprod. Immunol (2009) doi: 10.1016/j.jri.2009.06.124. [DOI] [PubMed] [Google Scholar]
- 15.Bulla R et al. Decidual endothelial cells express surface-bound C1q as a molecular bridge between endovascular trophoblast and decidual endothelium. Mol. Immunol 45, 2629–2640 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lokki AI et al. Complement Activation and Regulation in Preeclamptic Placenta. Front. Immunol 5, 312 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buurma A et al. Preeclampsia is characterized by placental complement dysregulation. Hypertension 60, 1332–1337 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Pierik E et al. Dysregulation of Complement Activation and Placental Dysfunction: A Potential Target to Treat Preeclampsia? Front. Immunol 10, 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burwick RM & Feinberg BB Complement activation and regulation in preeclampsia and hemolysis, elevated liver enzymes, and low platelet count syndrome. American Journal of Obstetrics and Gynecology (2020) doi: 10.1016/j.ajog.2020.09.038. [DOI] [PubMed] [Google Scholar]
- 20.Girardi G, Bulla R, Salmon JE & Tedesco F The complement system in the pathophysiology of pregnancy. Molecular Immunology vol. 43 68–77 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Regal JF, Gilbert JS & Burwick RM The complement system and adverse pregnancy outcomes. Molecular Immunology (2015) doi: 10.1016/j.molimm.2015.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burwick RM et al. Terminal Complement Activation in Preeclampsia. Obstet. Gynecol (2018) doi: 10.1097/AOG.0000000000002980. [DOI] [PubMed] [Google Scholar]
- 23.C.M., V. et al. Improved diagnosis of preeclampsia with severe features and end organ injury using complement activation measurement in urine and plasma. Am. J. Obstet. Gynecol (2018). [Google Scholar]
- 24.Derzsy Z, Prohászka Z, Rigó J, Füst G & Molvarec A Activation of the complement system in normal pregnancy and preeclampsia. Mol. Immunol 47, 1500–1506 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Lynch AM et al. The Relationship of Longitudinal Levels of Complement Bb During Pregnancy with Preeclampsia. Am. J. Reprod. Immunol 75, 104–111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yonekura Collier A-R et al. Placental sFLT1 is associated with complement activation and syncytiotrophoblast damage in preeclampsia. Hypertens. pregnancy 38, 193–199 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banadakoppa M, Balakrishnan M & Yallampalli C Upregulation and release of soluble fms-like tyrosine kinase receptor 1 mediated by complement activation in human syncytiotrophoblast cells. Am. J. Reprod. Immunol 80, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shamonki JM, Salmon JE, Hyjek E & Baergen RN Excessive complement activation is associated with placental injury in patients with antiphospholipid antibodies. Am J Obs. Gynecol 196, 167 e1–5 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim MY et al. Complement activation predicts adverse pregnancy outcome in patients with systemic lupus erythematosus and/or antiphospholipid antibodies. Ann Rheum Dis 77, 549–555 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salmon JE et al. Mutations in complement regulatory proteins predispose to preeclampsia: a genetic analysis of the PROMISSE cohort. PLoS Med 8, e1001013 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen A & Roberts DJ Placental pathologic lesions with a significant recurrence risk – what not to miss! APMIS (2018) doi: 10.1111/apm.12796. [DOI] [PubMed] [Google Scholar]
- 32.Hussein K et al. Complement receptor-associated CD163 + /CD18 + /CD11c + /CD206 - /CD209 - expression profile in chronic histiocytic intervillositis of the placenta. Placenta (2019) doi: 10.1016/j.placenta.2019.02.007. [DOI] [PubMed] [Google Scholar]
- 33.Wang W, Sung N, Gilman-Sachs A & Kwak-Kim J T Helper (Th) Cell Profiles in Pregnancy and Recurrent Pregnancy Losses: Th1/Th2/Th9/Th17/Th22/Tfh Cells. Frontiers in Immunology vol. 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Littman DR & Rudensky AY Th17 and Regulatory T Cells in Mediating and Restraining Inflammation. Cell vol. 140 845–858 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Kieffer TEC, Scherjon SA, Faas MM & Prins JR Lower activation of CD4+ memory T cells in preeclampsia compared to healthy pregnancies persists postpartum. J. Reprod. Immunol 136, (2019). [DOI] [PubMed] [Google Scholar]
- 36.Aluvihare VR, Kallikourdis M & Betz AG Regulatory T cells mediate maternal tolerance to the fetus. Nat Immunol 5, 266–271 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Somerset DA, Zheng Y, Kilby MD, Sansom DM & Drayson MT Normal human pregnancy is associated with an elevation in the immune suppressive CD25+ CD4+ regulatory T-cell subset. Immunology 112, 38–43 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gagliani N et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med 19, 739–746 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Salvany-Celades M et al. Three Types of Functional Regulatory T Cells Control T Cell Responses at the Human Maternal-Fetal Interface. Cell Rep 27, 2537–2547.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Kahn DA & Baltimore D Pregnancy induces a fetal antigen-specific maternal T regulatory cell response that contributes to tolerance. Proc Natl Acad Sci U S A 107, 9299–9304 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aluvihare VR, Kallikourdis M & Betz AG Tolerance, suppression and the fetal allograft. J Mol Med 83, 88–96 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Norouzian M, Rahimzadeh M, Rajaee M, Arabpour F & Naderi N FoxP3 gene promoter polymorphism affects susceptibility to preeclampsia. Hum. Immunol 77, 1232–1238 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Metz TD, Nelson LM, Stoddard GJ & Silver RM FOXP3 gene polymorphisms in preeclampsia. in American Journal of Obstetrics and Gynecology vol. 206 165.e1–165.e6 (Mosby Inc., 2012). [DOI] [PubMed] [Google Scholar]
- 44.Steinborn A et al. Pregnancy-associated diseases are characterized by the composition of the systemic regulatory T cell (T reg) pool with distinct subsets of T regs. Clin. Exp. Immunol 167, 84–98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sasaki Y et al. Proportion of peripheral blood and decidual CD4(+) CD25(bright) regulatory T cells in pre-eclampsia. Clin Exp Immunol 149, 139–145 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu M et al. Decreased maternal serum acetate and impaired fetal thymic and regulatory T cell development in preeclampsia. Nat. Commun 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsuda S et al. Clonally Expanded Decidual Effector Regulatory T Cells Increase in Late Gestation of Normal Pregnancy, but Not in Preeclampsia, in Humans. Front. Immunol 9, 1934 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toldi G, Rigó J, Stenczer B, Vásárhelyi B & Molvarec A Increased prevalence of IL-17-producing peripheral blood lymphocytes in pre-eclampsia. Am. J. Reprod. Immunol (2011) doi: 10.1111/j.1600-0897.2011.00987.x. [DOI] [PubMed] [Google Scholar]
- 49.LaMarca B et al. Identifying immune mechanisms mediating the hypertension during preeclampsia. American Journal of Physiology - Regulatory Integrative and Comparative Physiology vol. 311 R1–R9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Darmochwal-Kolarz D et al. The predominance of Th17 lymphocytes and decreased number and function of Treg cells in preeclampsia. J. Reprod. Immunol (2012) doi: 10.1016/j.jri.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 51.Harmon A et al. IL-10 supplementation increases Tregs and decreases hypertension in the RUPP rat model of preeclampsia. Hypertens. Pregnancy 34, 291–306 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scroggins SM et al. Elevated vasopressin in pregnant mice induces T-helper subset alterations consistent with human preeclampsia. Clin. Sci. (Lond) 132, 419–436 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arbore G et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science (80-. ) 352, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lajoie S & Wills-Karp M New Twist on an Ancient Innate Immune Pathway. Immunity vol. 39 1000–1002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Committee on Obstetric Practice Society for Maternal-Fetal Medicine. Low-Dose Aspirin Use During Pregnancy. Obstet. Gynecol (2018).
- 56.Rolnik DL et al. Aspirin versus Placebo in Pregnancies at High Risk for Preterm Preeclampsia. N. Engl. J. Med (2017) doi: 10.1056/nejmoa1704559. [DOI] [PubMed] [Google Scholar]
- 57.Roberge S, Bujold E & Nicolaides KH Aspirin for the prevention of preterm and term preeclampsia: systematic review and metaanalysis. American Journal of Obstetrics and Gynecology vol. 218 287–293.e1 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Pierik E et al. Dysregulation of Complement Activation and Placental Dysfunction: A Potential Target to Treat Preeclampsia? Frontiers in Immunology vol. 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roberge S et al. Prevention of pre-eclampsia by low-molecular-weight heparin in addition to aspirin: a meta-analysis. Ultrasound Obstet. Gynecol 47, 548–553 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Wang X & Gao H Prevention of preeclampsia in high-risk patients with low-molecular-weight heparin: a meta-analysis. J. Matern. Neonatal Med 33, 2202–2208 (2020). [DOI] [PubMed] [Google Scholar]
- 61.Seo MR et al. Hydroxychloroquine treatment during pregnancy in lupus patients is associated with lower risk of preeclampsia. Lupus vol. 28 722–730 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Garrett N et al. Pravastatin therapy during preeclampsia prevents long-term adverse health effects in mice. JCI insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lefkou E et al. Pravastatin improves pregnancy outcomes in obstetric antiphospholipid syndrome refractory to antithrombotic therapy. J. Clin. Invest 126, 2933–2940 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burwick RM & Feinberg BB Eculizumab for the treatment of preeclampsia/HELLP syndrome. Placenta 34, 201–203 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Robertson SA et al. Therapeutic potential of regulatory T cells in preeclampsia-opportunities and challenges. Frontiers in Immunology vol. 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McDonald-Hyman C, Turka LA & Blazar BR Advances and challenges in immunotherapy for solid organ and hematopoietic stem cell transplantation. Science Translational Medicine vol. 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Allan SE et al. CD4+ T-regulatory cells: Toward therapy for human diseases. Immunological Reviews vol. 223 391–421 (2008). [DOI] [PubMed] [Google Scholar]
- 68.Hartemann A et al. Low-dose interleukin 2 in patients with type 1 diabetes: A phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 1, 295–305 (2013). [DOI] [PubMed] [Google Scholar]
- 69.Miko E, Meggyes M, Doba K, Barakonyi A & Szereday L Immune checkpoint molecules in reproductive immunology. Frontiers in Immunology vol. 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Orban T et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled trial. Lancet 378, 412–419 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leavey K et al. Unsupervised Placental Gene Expression Profiling Identifies Clinically Relevant Subclasses of Human Preeclampsia. Hypertens. (Dallas, Tex. 1979) 68, 137–47 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Leavey K, Grynspan D & Cox BJ Both ‘canonical’ and ‘immunological’ preeclampsia subtypes demonstrate changes in placental immune cell composition. Placenta 83, 53–56 (2019). [DOI] [PubMed] [Google Scholar]
- 73.Prins JRR et al. Altered expression of immune-associated genes in first-trimester human decidua of pregnancies later complicated with hypertension or foetal growth restriction. Placenta 33, 453–455 (2012). [DOI] [PubMed] [Google Scholar]
- 74.Maric-Bilkan C et al. Research Recommendations From the National Institutes of Health Workshop on Predicting, Preventing, and Treating Preeclampsia. Hypertension 73, 757–766 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li Y et al. Decidual-placental immune landscape during syngeneic murine pregnancy. Front. Immunol (2018) doi: 10.3389/fimmu.2018.02087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vazquez J et al. Transcriptional and Functional Programming of Decidual Innate Lymphoid Cells. Front. Immunol 10, 3065 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vento-Tormo R et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563, 347–353 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stephenson KE et al. Antibody responses after analytic treatment interruption in human immunodeficiency virus-1-infected individuals on early initiated antiretroviral therapy. Open Forum Infect. Dis 3, 1–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stephenson KE et al. Quantification of the epitope diversity of HIV-1-specific binding antibodies by peptide microarrays for global HIV-1 vaccine development. J. Immunol. Methods 416, 105–123 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hecker M et al. Computational analysis of high-density peptide microarray data with application from systemic sclerosis to multiple sclerosis. Autoimmunity Reviews (2012) doi: 10.1016/j.autrev.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 81.Soen Y, Chen DS, Kraft DL, Davis MM & Brown PO Detection and characterization of cellular immune responses using peptide-MHC microarrays. PLoS Biol (2003) doi: 10.1371/journal.pbio.0000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiong S et al. Maternal uterine NK cell-activating receptor KIR2DS1 enhances placentation. J. Clin. Invest 123, 4264–4272 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moffett A & Colucci F Uterine NK cells: Active regulators at the maternal-fetal interface. Journal of Clinical Investigation vol. 124 1872–1879 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hiby SE et al. Maternal activating KIRs protect against human reproductive failure mediated by fetal HLA-C2. J. Clin. Invest 120, 4102–4110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maric-Bilkan C et al. Research Recommendations From the National Institutes of Health Workshop on Predicting, Preventing, and Treating Preeclampsia. Hypertension 73, 757–766 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bluestone JA, Trotta E & Xu D The therapeutic potential of regulatory T cells for the treatment of autoimmune disease. Expert Opinion on Therapeutic Targets vol. 19 1091–1103 (2015). [DOI] [PubMed] [Google Scholar]
- 87.Zhao Y et al. Immune checkpoint molecules on T cell subsets of pregnancies with preeclampsia and gestational diabetes mellitus. J. Reprod. Immunol 142, (2020). [DOI] [PubMed] [Google Scholar]
- 88.Sharma P & Allison JP The future of immune checkpoint therapy. Science (80-. ) 348, 56–61 (2015). [DOI] [PubMed] [Google Scholar]
- 89.Paul JW et al. Drug delivery to the human and mouse uterus using immunoliposomes targeted to the oxytocin receptor. in American Journal of Obstetrics and Gynecology vol. 216 283.e1–283.e14 (Mosby Inc., 2017). [DOI] [PubMed] [Google Scholar]
- 90.Buurma A et al. Preeclampsia is characterized by placental complement dysregulation. Hypertension 60, 1332–1337 (2012). [DOI] [PubMed] [Google Scholar]
- 91.Sinha D, Wells M & Page Faulk W Immunological studies of human placentae: Complement components in pre-eclamptic chorionic villi. Clin. Exp. Immunol 56, 175–17584 (1984). [PMC free article] [PubMed] [Google Scholar]
- 92.Lokki AI et al. Complement activation and regulation in preeclamptic placenta. Front Immunol 5, 312 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Agostinis C et al. Complement component C1q as potential diagnostic but not predictive marker of preeclampsia. Am. J. Reprod. Immunol 76, 475–481 (2016). [DOI] [PubMed] [Google Scholar]
- 94.Kim EN et al. Placental C4d deposition is a feature of defective placentation: Observations in cases of preeclampsia and miscarriage. Virchows Arch 466, 717–725 (2015). [DOI] [PubMed] [Google Scholar]
- 95.Lashley LEELO et al. Preeclampsia in autologous and oocyte donation pregnancy: Is there a different pathophysiology? J. Reprod. Immunol 109, 17–23 (2015). [DOI] [PubMed] [Google Scholar]
- 96.Wang CC et al. Innate Immune Response by Ficolin Binding in Apoptotic Placenta Is Associated with the Clinical Syndrome of Preeclampsia. Clin. Chem 53, 42–52 (2007). [DOI] [PubMed] [Google Scholar]
- 97.Kestlerová A et al. Immunological and biochemical markers in preeclampsia. J. Reprod. Immunol 96, 90–94 (2012). [DOI] [PubMed] [Google Scholar]
- 98.Sarween N et al. Humoral immunity in late-onset Pre-eclampsia and linkage with angiogenic and inflammatory markers. Am. J. Reprod. Immunol 80, e13041 (2018). [DOI] [PubMed] [Google Scholar]
- 99.Halmos A et al. Circulating ficolin-2 and ficolin-3 in normal pregnancy and pre-eclampsia. Clin. Exp. Immunol 169, 49–56 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.TEDESCO F et al. Immunohistochemical detection of terminal complement complex and S protein in normal and pre-eclamptic placentae. Clin. Exp. Immunol 80, 236–240 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hering L et al. Trophoblasts reduce the vascular smooth muscle cell proatherogenic response. in Hypertension vol. 51 554–559 (Lippincott Williams & Wilkins, 2008). [DOI] [PubMed] [Google Scholar]
- 102.Wang W et al. Autoantibody-mediated complement c3a receptor activation contributes to the pathogenesis of preeclampsia. Hypertension 60, 712–721 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hoffman MC, Rumer KK, Kramer A, Lynch AM & Winn VD Maternal and Fetal Alternative Complement Pathway Activation in Early Severe Preeclampsia. Am. J. Reprod. Immunol 71, 55–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kieffer TEC et al. Lower FOXP3 mRNA Expression in First-Trimester Decidual Tissue from Uncomplicated Term Pregnancies with a Male Fetus. J. Immunol. Res 2018, 1950879 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sasaki Y et al. Proportion of peripheral blood and decidual CD4 + CD25 bright regulatory T cells in pre-eclampsia. Clin. Exp. Immunol 149, 139–145 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Saito S, Sakai M, Sasaki Y, Nakashima A & Shiozaki A Inadequate tolerance induction may induce pre-eclampsia. Journal of Reproductive Immunology vol. 76 30–39 (2007). [DOI] [PubMed] [Google Scholar]
- 107.Hennessy A, Pilmore HL, Simmons LA & Painter DM A deficiency of placental IL-10 in preeclampsia. J. Immunol (1999). [PubMed] [Google Scholar]
- 108.Tsuda S, Nakashima A, Shima T & Saito S New paradigm in the role of regulatory T cells during pregnancy. Frontiers in Immunology (2019) doi: 10.3389/fimmu.2019.00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Prins JR et al. Preeclampsia is associated with lower percentages of regulatory T cells in maternal blood. Hypertens. Pregnancy 28, 300–311 (2009). [DOI] [PubMed] [Google Scholar]