

Abstract
Hepatocyte nuclear factor 4α (HNF4α) is highly enriched in the liver, but its role in the progression of liver steatosis (NAFL) to non-alcoholic steatohepatitis (NASH) has not been elucidated. In this study, we investigated the effect of gain or loss of hepatocyte HNF4α function on the development and progression of non-alcoholic fatty liver disease (NAFLD) in mice. Over-expression of human HNF4α protected against high fat/cholesterol/fructose (HFCF) diet-induced steatohepatitis whereas loss of hepatocyte Hnf4α had opposite effects. HNF4α prevented hepatic triglyceride accumulation by promoting hepatic triglyceride lipolysis, fatty acid oxidation and VLDL secretion. Furthermore, HNF4α suppressed the progression of NAFL to NASH. Over-expression of human HNF4α inhibited HFCF diet-induced steatohepatitis in control mice but not in hepatocyte-specific p53−/− mice. In HFCF diet-fed mice lacking hepatic Hnf4α, recapitulation of hepatic expression of HNF4α targets cholesterol 7α-hydroxylase and sterol 12α-hydroxylase normalized hepatic triglyceride levels and attenuated steatohepatitis.
Conclusions:
The current study indicates that hepatocyte HNF4α protects against diet-induced development and progression of NAFLD by coordinating the regulation of lipolytic, p53 and bile acid signaling pathways. Targeting hepatic HNF4α may be useful for treatment of NASH.
Keywords: HNF4α, P53, bile acid, steatohepatitis, lipolysis, apoptosis
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a spectrum of liver diseases ranging from steatosis (NAFL) to non-alcoholic steatohepatitis (NASH), which may further progress to liver cirrhosis and hepatocarcinoma. Multiple mechanisms are involved in the development and progression of NAFLD. While NAFL may be a protective mechanism to avoid lipotoxicity, NASH is the advanced form of NAFLD that needs medical attention. Several mechanisms, such as insulin resistance, abnormal apoptosis, reactive oxygen species (ROS), lipotoxicity, and inflammation, are believed to contribute to the progression of NAFL to NASH (1–7). In addition, defective lipolysis also plays a role in the pathogenesis of NAFLD (8–10).
Hepatocyte nuclear factor 4α (HNF4α) is a nuclear hormone receptor which is highly conserved between humans and mice. HNF4α is highly expressed in the liver but also found in the intestine, pancreas and kidney (11). HNF4α is constitutively active and usually binds to the promoters of genes to regulate their expression. Gene knockout studies demonstrate that HNF4α is required for the expression of a number of hepatic genes that regulate hepatocyte differentiation, energy metabolism, xenobiotic detoxification, bile acid synthesis, and plasma protein production (12, 13). On a chow diet, liver-specific Hnf4α−/− mice display reduced plasma triglyceride (TG) and cholesterol levels, which are accompanied by reduced microsomal triglyceride transfer protein (MTTP) and ApoB expression (14). Acute loss of hepatic Hnf4α by shRNA also reduces plasma TG and cholesterol levels but increases hepatic TG accumulation (15). In addition, loss of HNF4α results in reduced expression of genes involved in bile acid (BA) synthesis, such as cholesterol 7α-hydroxylase (CYP7A1) and sterol 12α-hydroxylase (CYP8B1) (16), and BA conjugation (17). In NASH patients or obese mice, hepatic HNF4α expression is markedly reduced (18). So far, the role of hepatic HNF4α in the progression of NAFL to NASH has not been investigated. Moreover, the mechanism underlying the regulation of hepatic TG levels by HNF4α has not been fully understood.
The tumor suppressor p53 is a primary stress sensor that controls a variety of biological processes. P53 expression or signaling is increased in the livers of NAFLD patients or experimental NASH (19–21). p53-deficent mice are resistant to methionine- and choline-deficient (MCD) diet-induced apoptosis and steatohepatitis (19). Likewise, inhibition of p53 attenuates high fat diet-induced liver steatosis and injury (22). Consistent with these observations, activation of farnesoid X receptor (FXR) by obeticholic acid (OCA) inhibits diet-induced p53 activation and cell death, and protects against the development of NASH (23). BAs are endogenous ligands for FXR. Interestingly, p53 is shown to inhibit BA synthesis via inducing small heterodimer partner (SHP) (24), a nuclear hormone receptor that is also a target of FXR (25). Nonetheless, it is unclear whether HNF4α regulation of the pathogenesis of NAFLD involves p53 and/or BAs.
In this report, we show that adeno-associated virus 8 (AAV8)-mediated over-expression of human HNF4α (AAV8-ALB-hHNF4α) in hepatocytes protected against high fat/cholesterol/fructose (HFCF) diet-induced hepatosteatosis, inflammation and fibrosis. In contrast, hepatocyte-specific Hnf4α−/− (Hnf4αHep−/−) mice had aggravated steatohepatitis upon fed an HFCF diet. Mechanistically, we show that HNF4α regulated hepatic triglyceride (TG) accumulation by promoting triglyceride hydrolysis (TGH), fatty acid oxidation (FAO) and very low-density lipoprotein (VLDL) secretion. We also show that over-expression of human HNF4α in hepatocytes protected against the development of steatohepatitis in p53fl/fl mice, but not in hepatocyte-specific p53−/− (p53Hep−/−) mice. Lastly, recapitulation of hepatic CYP7A1 and CYP8B1 expression in Hnf4αHep−/− mice normalized hepatic TG levels and alleviated steatohepatitis. Our data suggest that HNF4α regulates the development and progression of NAFLD via coordinating modulation of lipolytic, p53 and BA signaling.
Experimental Procedures
Mice and diets.
C57BL/6J mice (stock # 000664), Fxr−/− mice (stock # 004144), Hnf4αfl/fl mice (stock # 004665) and p53fl/fl mice (stock # 008462) were purchased from the Jackson Laboratories (Bar Harbor, Maine, USA). Hnf4αfl/fl mice and Fxr−/− mice were cross-bred with C57BL/6J mice for at least 10 generations. AAV8-TBG-Null or AAV8-TBG-Cre (produced by Vector Biolabs) was injected to Hnf4αfl/fl mice or p53fl/fl mice to generate control mice (Hnf4αfl/fl mice or p53fl/fl mice), hepatocyte-specific Hnf4α−/− (Hnf4αHep−/−) mice, or hepatocyte-specific p53−/− (p53Hep−/−) mice, respectively. All mice were kept in a temperature- and humidity-controlled room with a 12-h light/12-h dark cycle and free access to water and food. The high fat/cholesterol/fructose (HFCF) diet contained 40% fat/0.2% cholesterol (from TestDiet, AIN-76A), and 4.2% fructose (in drinking water). Mice were fed an HFCF diet for 20 weeks. Unless otherwise stated, male mice were used and were fasted for 5–6 h prior to euthanasia. All the animal experiments were approved by the Institutional Animal Care and Use Committee at Northeast Ohio Medical University (NEOMED).
Adeno-associated viruses (AAVs).
Human HNF4α, CYP7A1, CYP8B1, or p53 coding sequences were cloned into an AAV vector under the control of a mouse albumin promoter to generate AAV-ALB-hHNF4α, AAV-ALB-hCYP7A1, AAV-ALB-hCYP8B1 or AAV-ALB-hP53, respectively. AAV8-TBG-Null (control), AAV8-TBG-Cre, AAV8-ALB-null (control), AAV8-ALB-hHNF4α, AAV8-ALB-hCYP7A1, AAV8-ALB-hCYP8B1 or AAV8-ALB-hP53 was produced using AAV serotype 8 and titrated by Vector BioLabs. Each mouse was i.v. injected with 3 × 1011 genome copies (GC) AAV and then fed a chow diet or an HFCF diet for up to 20 weeks.
OCA treatment.
Obeticholic acid (OCA), a specific FXR agonist, was provided by Intercept Pharmaceuticals, Inc (San Diego, CA). Fxr+/+ or Fxr−/− mice were gavaged with either vehicle (0.5% carboxymethycellulose, Sigma) or OCA (30 mg/kg/day) for 7 days. In a different study, Hnf4αfl/fl mice or Hnf4αHep−/− mice were fed an HFCF diet for 16 weeks. In the last 30 days of the latter study, mice were also gavaged with either vehicle or OCA (20 mg/kg/day).
mRNA and qRT-PCR.
RNA was extracted using Trizol Reagent (Thermo Fisher) and mRNA levels were quantified by quantitative real-time PCR (qRT-PCR) using PowerUP SYBR Green Master Mix (ThermoFisher) on a 7500 Real Time PCR machine (Applied Biosystems). mRNA levels were normalized to 36B4.
Western blot assays.
Western blot assays were performed using whole liver lysates or membrane extracts of the liver samples as described previously (26, 27). Antibodies against HNF4α (cat # sc-374229) or p53 (cat # sc-6243) were purchased from Santa Cruz Biotechnology. Antibodies against CYP7A1 (cat # TA351400) or CYP8B1 (cat # TA313734) were purchased from Origene. Antibodies against SR-BI (cat # NB400–101) or Calnexin (cat # NB100–1965) were purchased from Novus. Antibodies against phospho-Smad2/3 (cat # 8828), total Smad2/3 (cat # 5678), cleaved Caspase 3 (cat # 9661) or total Caspase-3 (cat # 9662) were purchased from Cell Signaling Technology. Antibodies against CES1 (cat # ab45957), CES2 (cat # ab56528), ACAT2 (cat # ab23669) or Tubulin (cat # ab4074) were purchased from Abcam.
Analysis of plasma or hepatic levels of lipids, hydroxyproline, ROS, AST, ALT, β-hydroxybutyrate and apoptosis.
Approximately 100 mg liver tissue was homogenized in methanol and lipids were extracted in chloroform/methanol (2:1 v/v) as described (28). Triglycerides (TG) and cholesterol in the liver and plasma ALT or AST levels were measured using Infinity reagents from Thermo Scientific (Waltham, MA). Hepatic hydroxyproline level was quantified using a kit from Cell BioLabs (Cat # STA675). Hepatic ROS levels were measured using an OxiSelect in Vitro ROS/RNS Assay kit (Cat # STA-347) from Cell Biolabs (San Diego, CA) as described (29). Plasma β-hydroxybutyrate level was determined using a kit from Pointe Scientific (Canton, MI). Apoptosis was determined using a kit from Abcam (Cat # ab206386).
TGH activity assays.
Hepatic microsome proteins were isolated and TGH activity was measured using 3H-triolein as substrate as described (30).
Fatty acid oxidation (FAO).
Primary hepatocytes were isolated and cultured in DMEM in 12-well dishes. FAO was performed using [3H]palmitate as substrate as described (31, 32).
Oil Red O (ORO), H&E or Sirius Red staining.
Liver was fixed in 4% formalin and then embedded in OCT or paraffin. Oil Red O, H&E or Sirius Red staining was performed as descried (32).
Analysis of fatty acid levels and composition.
Hepatic total free fatty acids (FFAs) were measured using a kit from Wako Chemicals USA (Richmond, VA). Hepatic fatty acid composition was analyzed by GC-MS as described (30).
Statistical analysis.
Statistical significance was analyzed using unpaired Student t test or ANOVA (GraphPad Prisim, CA). All values are expressed as mean±SEM. Differences were considered statistically significant at P<0.05.
Results
HNF4α is an essential regulator of hepatic TGH activity and FAO.
Hepatic TG level is controlled by TG synthesis, lipolysis and VLDL secretion. Previous studies have shown that HNF4α regulates hepatic expression of MTTP and ApoB as well as VLDL secretion (14, 15). So far, the role of hepatic HNF4α in lipolysis or FAO has not been examined. Since adenoviruses are known to cause significant liver inflammation, we generated AAV8-ALB-hHNF4α which expressed human HNF4α specifically in hepatocytes. On a chow diet, AAV8-mediated over-expression of human HNF4α reduced hepatic TG and free fatty acid (FFA) levels by ~ 33% (Figure 1A, left and middle panels). Analysis of hepatic fatty acid composition by GC-MS showed that human HNF4α over-expression reduced the levels of C16:0, C18:1, C18:2 and C20:4 fatty acyl-CoA by 50–90% (Figure 1A, right panel). Analysis of hepatic mRNA levels by qRT-PCR showed that human HNF4α induced the expression of genes involved in BA biosynthesis (Cyp7a1, Cyp8b1), lipolysis (Ces1, Ces2), FAO (Pparα, Cpt1, Cpt2, Pdk4), and VLDL secretion (Mttp, Apob) (Figure 1B). There was no change in hepatic mRNA levels of genes involved in de novo lipogenesis (DNL) (Srebp1c, Fasn, Acc1) (Figure 1B) or hepatic levels of total cholesterol (TC), free cholesterol (FC) or cholesteryl esters (CE) (Figure S1A). We have previously shown that CES1 and CES2 have TGH activity, whose over-expression lowers hepatic TG levels and stimulates FAO (30, 32). In line with these findings, over-expression of human HNF4α increased hepatic TGH activity by 76% (Figure 1C, left panel) and FAO by 28% (Figure 1C, right panel), respectively.
Figure 1. HNF4α is an essential regulator of hepatic TGH activity and fatty acid oxidation.
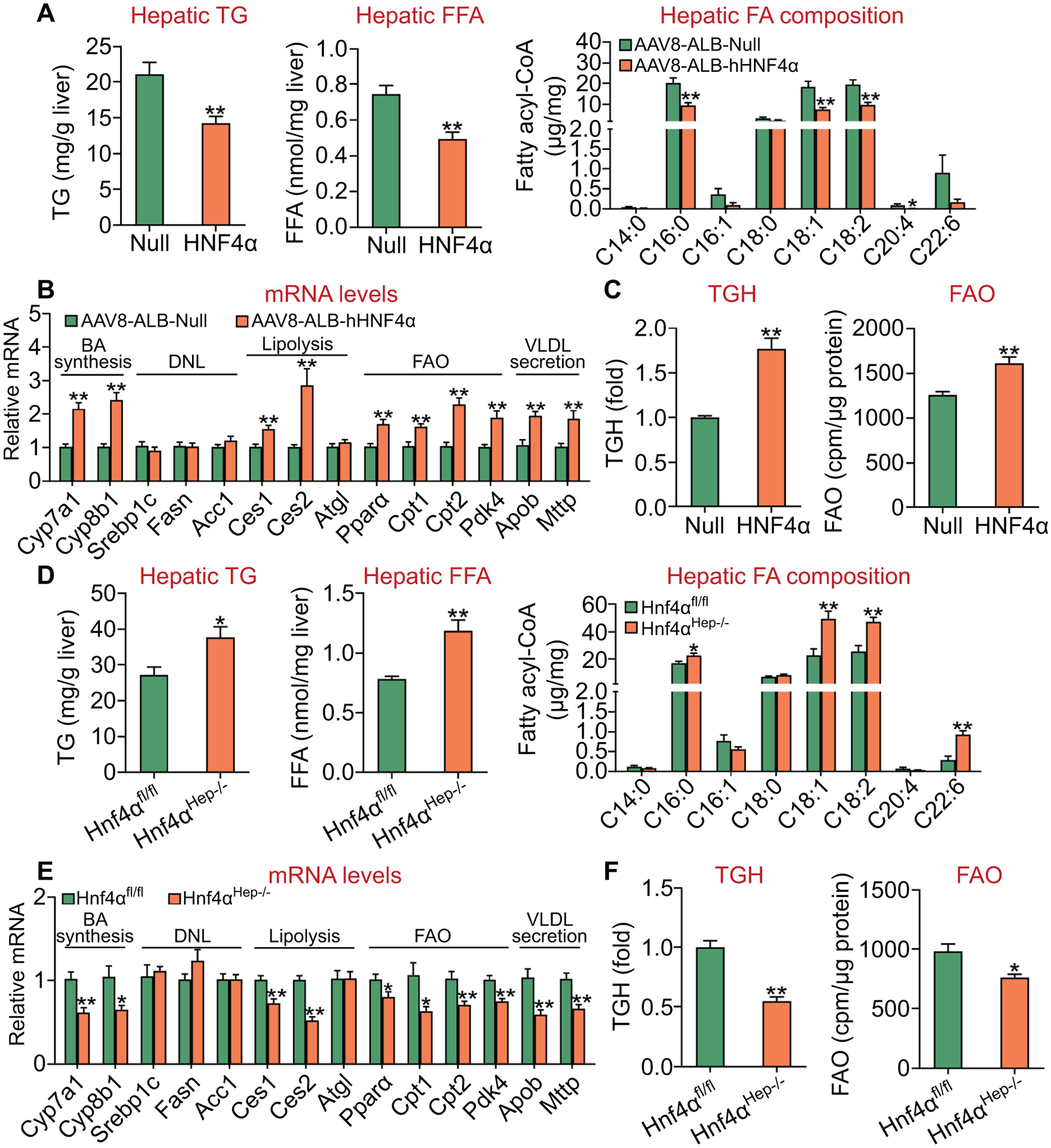
(A-C) C57BL/6J mice were injected i.v. with AAV8-ALB-Null or AAV8-ALB-hHNF4α and then fed a chow diet for 8 weeks (n=8 per group). Hepatic levels of TG and FFA as well as fatty acid composition were determined (A). Hepatic mRNA levels were quantified (B). Hepatic TGH activity (left panel) and FAO in hepatocytes (right panel) were determined (C). (D-F) Hnf4αfl/fl mice and Hnf4αHep−/− mice were fed a chow diet (n=8 per group). Hepatic levels of TG and FFA as well as fatty acid composition were determined (D). Hepatic mRNA levels were quantified (E). Hepatic TGH activity (left panel) and FAO in hepatocytes (right panel) were determined (F). *P<0.05, **P<0.01
In contrast to what was observed in mice over-expressing human HNF4α in the liver, hepatocyte-specific loss of Hnf4α increased hepatic levels of TG by 39% and FFA by 52% (Figure 1D, left and middle panels). Hepatic C16:0, C18:1, C18:2 and C22:6 fatty acyl-CoA levels were increased by 135% to 320% (Figure 1D, right panel). Hnf4αHep−/− mice also had reduced expression of Cyp7a1, Cyp8b1, Ces1, Ces2, Pparα, Cpt1, Cpt2, Pdk4, Mttp or Apob (Figure 1E). There was no change in hepatic expression of genes involved in DNL (Figure 1E) or hepatic levels of TC, FC or CE (Figure S1B). In addition, hepatocyte-specific loss of Hnf4α reduced hepatic TGH activity by 45% (Figure 1F, left panel) and FAO by 22% (Figure 1F, right panel), respectively.
Taken together, our gain- and loss-of-function data demonstrate that HNF4α is an essential regulator of hepatic TGH activity, which may account, at least in part, for the regulation of hepatic TG levels by HNF4α.
Hepatocyte-specific over-expression of human HNF4α is sufficient to protect against HFCF diet-induced NASH.
We have previously shown that hepatic HNF4α expression is markedly reduced in NAFLD patients or high fat diet (HFD)-fed mice (18). So far, it remains to be determined whether gain of hepatocyte HNF4α function is sufficient to protect from diet-induced steatosis or steatohepatitis. To address this question, we injected C57BL/6J mice i.v. with AAV8-ALB-Null or AAV8-ALB-hHNF4α, which were then fed an HFCF diet for 20 weeks. Injection of AAV8-ALB-hHNF4α led to a 5.9–7.5-fold increase in hepatic HNF4α protein level at 8, 16 or 20 weeks (Figure S2A, B). Over-expression of human HNF4α prevented HFCF diet-induced increases in hepatic TG or FFA levels (Figure 2A), and hepatic C16:0, C16:1, C18:1 fatty acyl-CoA levels (Figure 2B). Oil Red O and H & E staining also confirmed a reduction in hepatic neutral lipid accumulation in mice over-expressing HNF4α (Figure 2C). In addition, human HNF4α induced Ces1 and Ces2 mRNA and protein levels, and expression of genes involved in FAO (Pparα, Cpt1, Cpt2, Pdk4) or VLDL secretion (Mttp, Apob) (Figure 2D). Consistent with these data, human HNF4α over-expression increased hepatic TGH activity by 60% (Figure 2E) and FAO by 45% (Figure 2F), respectively, in mice fed an HFCF diet.
Figure 2. Hepatocyte-specific expression of human HNF4α is sufficient to prevent HFCF diet-induced hepatosteatosis.

C57BL/6J mice were injected i.v. with AAV8-ALB-Null or AAV8-ALB-hHNF4α and then fed an HFCF diet for 20 weeks (n=8 per group). (A) Hepatic TG (left panel) and FFA (right panel) levels. (B) Hepatic fatty acid composition was analyzed by GC-MS. (C) Liver sections were stained by Oil Red O (top panel) or H&E (lower panel). (D) Hepatic mRNA levels (left panel) or protein levels (right panel). (E) Hepatic TGH activity. (F) FAO was analyzed using isolated hepatocytes. *P<0.05, **P<0.01
We also investigated the effect of gain of hepatic HNF4α function on the development of NASH. Over-expression of human HNF4α reduced plasma ALT levels by 35%, but had no effect on plasma AST levels (Figure 3A), suggesting that gain of hepatic HNF4α function protects against diet-induced liver injury. Over-expression of human HNF4α significantly inhibited the expression of genes involved in inflammation or fibrogenesis (Tnfα, Il6, Il1β, Mcp1, F4/80, p53, α-Sma, Timp1, Col1α1, Col1α2) (Figure 3B). HNF4α also induced the expression of genes involved in cholesterol catabolism (Cyp7a1, Cyp8b1), HDL uptake (Scarb1), cholesterol efflux (Abcg8), or cholesterol esterification (Acta1, Acat2) with genes involved in cholesterol synthesis or LDL uptake (Hmgcr, Hmgcs, Ldlr) being unchanged (Figure 3B). Consistent with the changes in mRNA levels, hepatic protein levels of HNF4α, CYP7A1, CYP8B1, SR-BI and ACAT2 were increased by ≥2 fold (Figure 3C, left panels). In line with the changes in gene expression, hepatic TC and FC levels were decreased by >38% while hepatic BA levels were increased by 142% (Figure 3C, right panels).
Figure 3. Hepatocyte-specific expression of human HNF4α ameliorates HFCF diet-induced steatohepatitis.
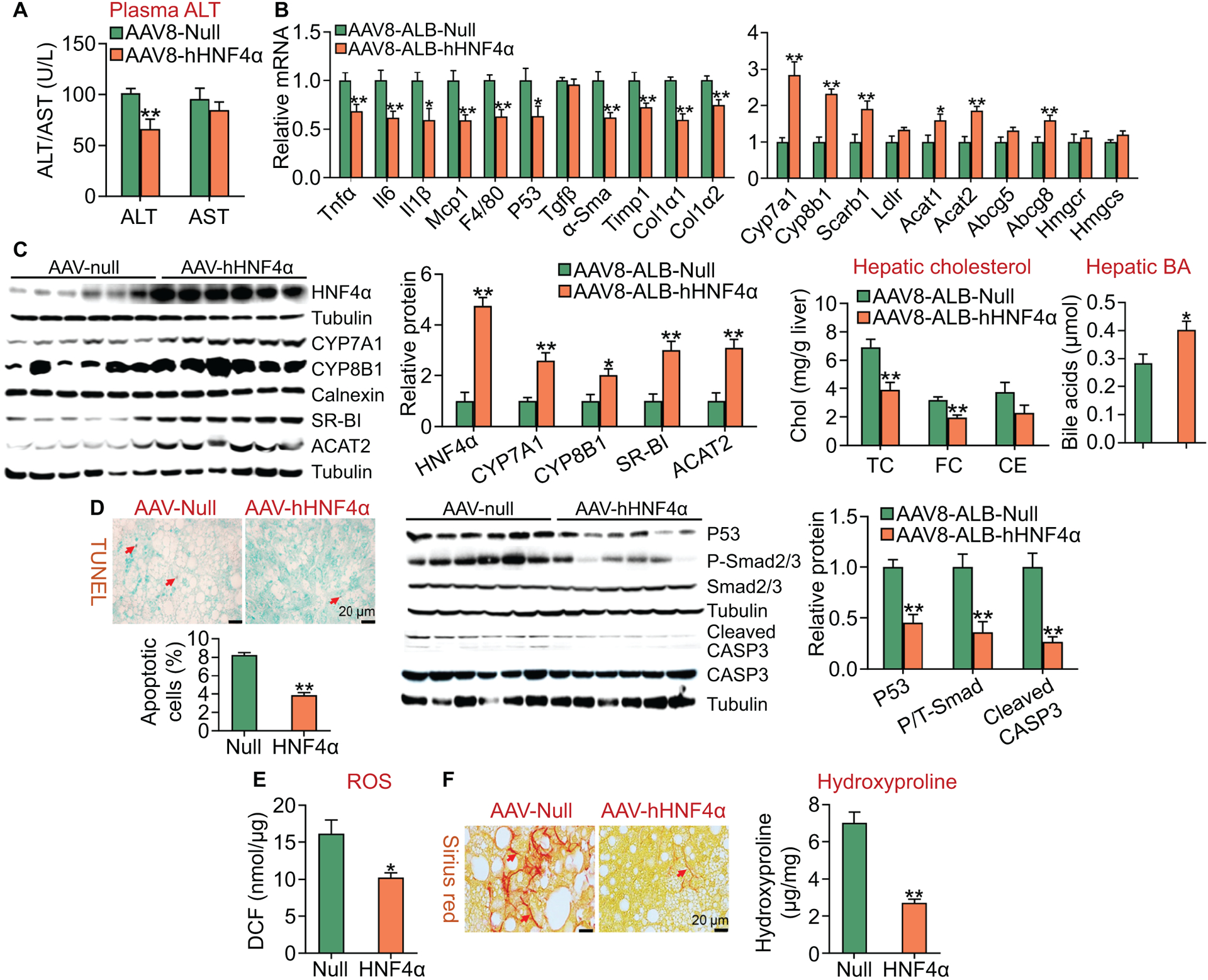
C57BL/6J mice were injected i.v. with AAV8-ALB-Null or AAV8-ALB-hHNF4α and then fed an HFCF diet for 20 weeks (n=8 per group). (A) Plasma ALT and AST levels. (B) Hepatic mRNA levels (left and right panels). (C) Hepatic proteins were analyzed by Western blot assays and then quantified (left panels). Hepatic cholesterol and BA levels were also analyzed (right panels). (D) Hepatic apoptosis was analyzed by TUNEL assays and then quantified (left panel). Hepatic protein levels were analyzed by Western blot assays and then quantified (middle and right panels). P/T-Smad, ratio of phosphorylated Smad2/3 to total Smad2/3. (E) Hepatic reactive oxygen species levels. (F) Hepatic sections were stained by Sirius Red (left panel), and hepatic hydroxyproline levels were quantified (right panel). *P<0.05, **P<0.01
Apoptosis and reactive oxygen species (ROS) play an important role in promoting the progression of NAFL to NASH. Hepatocyte-specific expression of human HNF4α in an HFCF diet-induced NASH model reduced hepatic apoptosis by >50% (Figure 3D, left panel), and hepatic protein levels of p53, phospho-Smad2/3 and cleaved caspase 3 by >50% (Figure 3D, middle and right panels). In addition, human HNF4α also reduced hepatic ROS levels (Figure 3E). Consistent with the reduced apoptosis and ROS levels, human HNF4α over-expression inhibited hepatic fibrogenesis, with hepatic hydroxyproline levels being reduced by >60% (Figure 3F, left and right panels).
Collectively, the data of Figures 2 and 3 demonstrate that hepatocyte-specific over-expression of human HNF4α protects against diet-induced NASH by a mechanism likely involving induction of lipolysis and inhibition of inflammation, apoptosis and ROS production.
Hepatocyte HNF4α is essential for protection against HFCF diet-induced NASH.
To investigate the effect of loss of hepatocyte HNF4α on diet-induced NASH, we fed Hnf4αfl/fl mice and Hnf4αHep−/− mice an HFCF diet for 20 weeks. Loss of hepatic Hnf4α raised hepatic levels of TG and FFA (Figure 4A) as well as C16:0, C16:1 and C18:1 fatty acyl-CoA levels by 32% (P=0.06), 73% and 63%, respectively (Figure 4B). Oil Red O or H & E staining confirmed more neutral lipid accumulation in Hnf4αHep−/− mice (Figure 4C). Analysis of hepatic gene expression showed that hepatic mRNA levels of Ces1, Ces2, Cpt1, Cpt2, Pdk4, Mttp or Apob were significantly reduced (Figure 4D, left panel), and that hepatic CES1 or CES2 protein levels were reduced by ≥ 70% (Figure 4D, right panel). In addition, loss of hepatic Hnf4α reduced hepatic TGH activity by 40% (Figure 4E) and FAO by 24% (Figure 4F).
Figure 4. Hepatocyte HNF4α is required for protection against HFCF diet-induced hepatosteatosis.
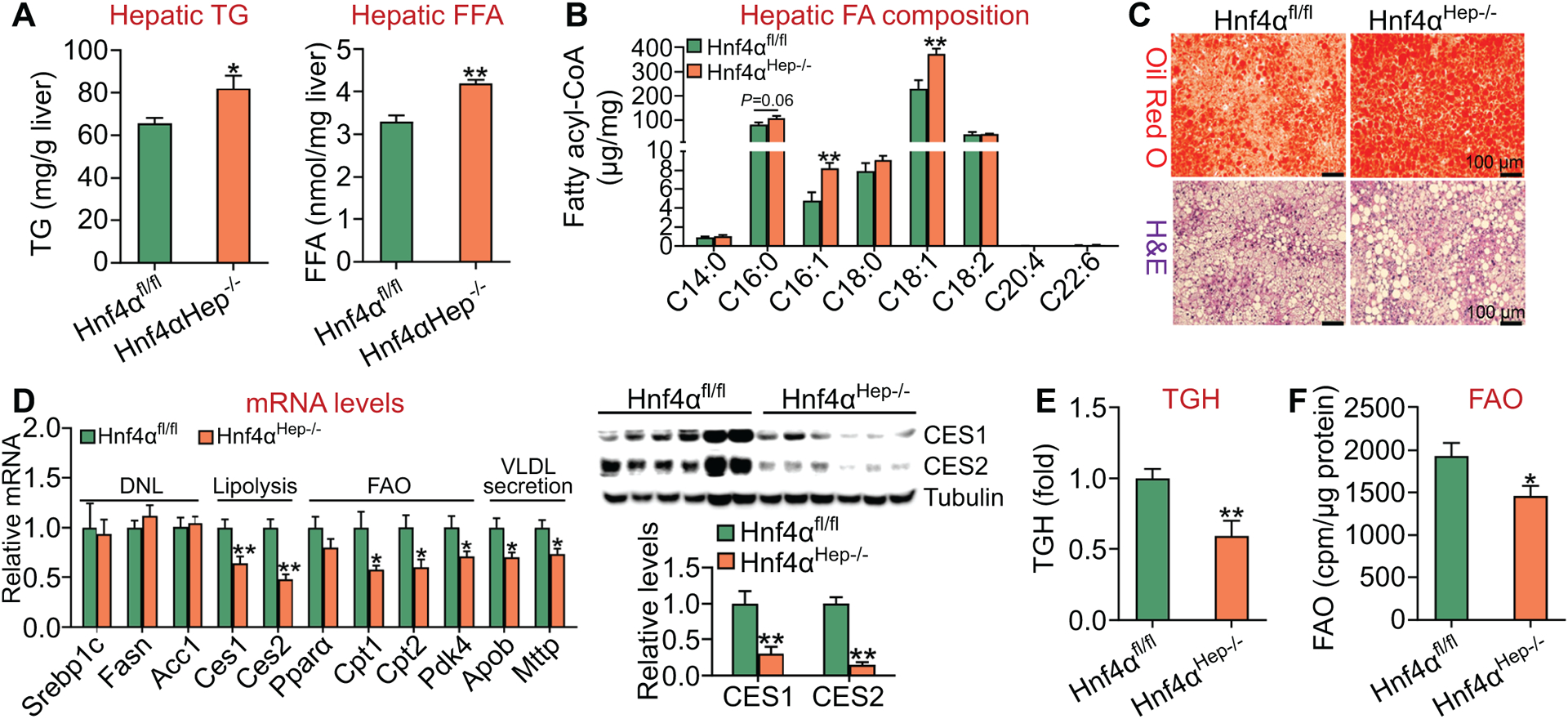
Hnf4αfl/fl mice and Hnf4αHep−/− mice were fed an HFCF diet for 20 weeks (n=8 per group). (A) Hepatic TG (left panel) and FFA (right panel) levels. (B) Hepatic fatty acid composition was analyzed by GC-MS. (C) Liver sections were stained by Oil Red O (top panel) or H&E (lower panel). (D) Hepatic mRNA levels (left panel) or protein levels (right panel). (E) Hepatic TGH activity. (F) FAO was analyzed in hepatocytes. *P<0.05, **P<0.01
Loss of hepatocyte HNF4α also raised plasma ALT levels by 29% (Figure 5A), and induced a number of genes involved in inflammation or fibrogenesis (Tnfα, Il6, Il1β, Mcp1, F4/80, p53, Tgfβ, α-Sma, Timp1, Col1α1, Col1α2) (Figure 5B, left panel), but reduced hepatic expression of genes involved BA/cholesterol metabolism (Cyp7a1, Cyp8b1, Scarb1, Ldlr, Acat2, Abcg5, Abcg8, Hmgcs) (Figure 5B, right panel). In line with the changes in mRNA levels, Hnf4αHep−/− mice had a 45–82% reduction in protein levels of HNF4α, CYP7A1, CYP8B1, SR-BI or ACAT2 (Figure 5C, left panels), and an increase in hepatic TC and FC levels but a decrease in hepatic BA levels (Figure 5C, right panels).
Figure 5. Loss of hepatocyte HNF4α aggravates HFCF diet-induced steatohepatitis.
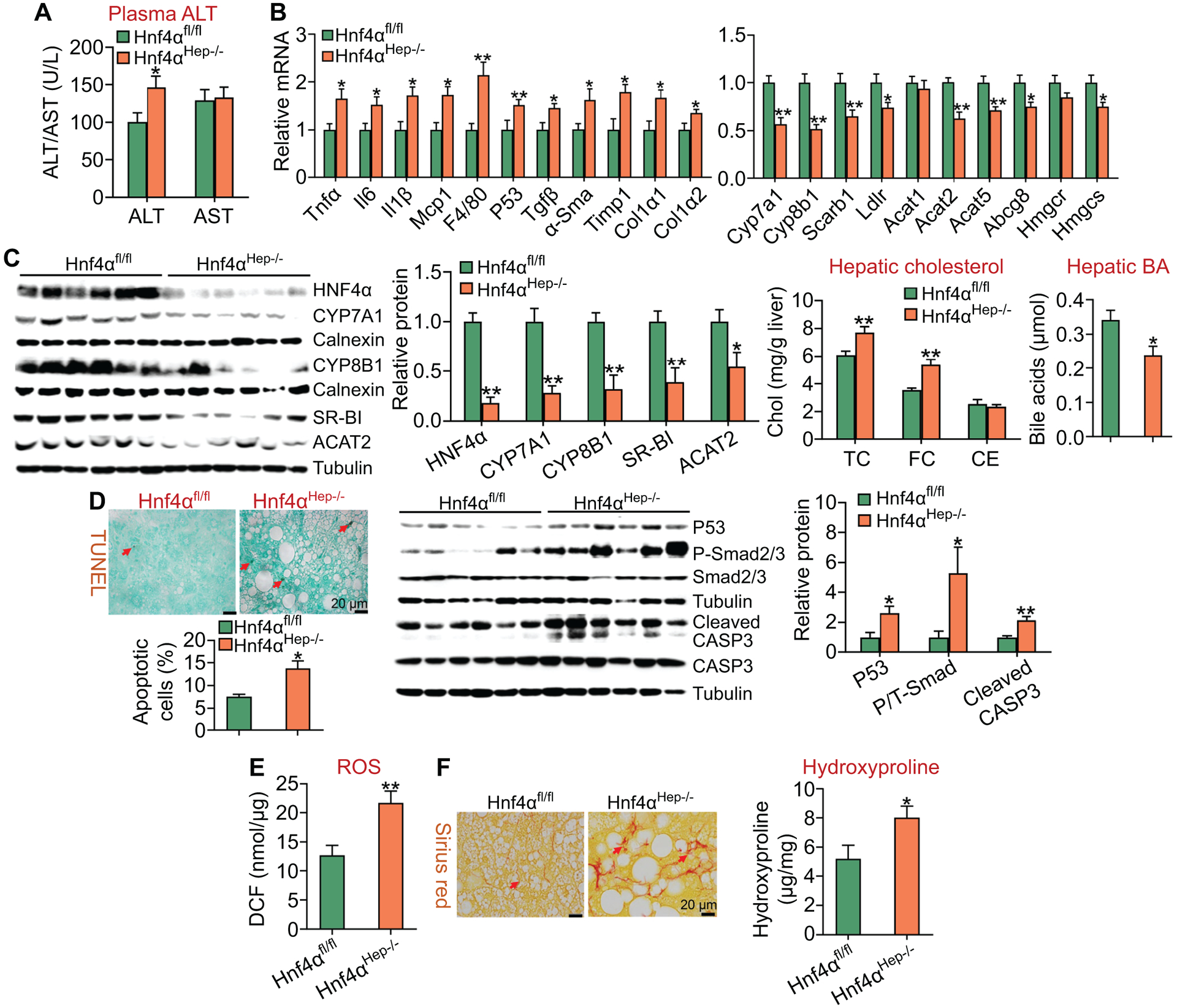
Hnf4αfl/fl mice and Hnf4αHep−/− mice were fed an HFCF diet for 20 weeks (n=8 per group). (A) Plasma ALT and AST levels. (B) Hepatic mRNA levels (left and right panels). (C) Hepatic proteins were analyzed by Western blot assays and then quantified (left panels). Hepatic cholesterol and BA levels were also analyzed (right panels). (D) Hepatic apoptosis was analyzed by TUNEL assays and then quantified (left panel). Hepatic protein levels were analyzed by Western blot assays and then quantified (middle and right panels). (E) Hepatic reactive oxygen species levels. (F) Hepatic sections were stained by Sirius Red (left panel), and hepatic hydroxyproline levels were quantified (right panel). *P<0.05, **P<0.01
In addition, Hnf4αHep−/− mice had an 82% increase in hepatic apoptosis (Figure 5D, left panel), and a >2-fold increase in hepatic protein levels of p53, phopho-Smad2/3 and cleaved caspase 3 (Figure 5D, middle and right panels). Finally, loss of hepatocyte HNF4α also increased hepatic ROS levels by 170% (Figure 5E), and raised hepatic fibrogenesis and hydroxyproline levels by 154% (Figure 5F, left and right panels). Thus, loss of hepatocyte HNF4α aggravates the development of diet-induced NASH.
Human HNF4α protects against diet-induced NASH via inhibition of p53.
P53 is a key regulator of apoptosis and the progression of NAFLD (19). Our data have shown that HNF4α represses hepatic p53 expression by >50% (Figure 3D), suggesting that p53 may play an important role in HNF4α-mediated protection against NASH. To test this hypothesis, we injected p53fl/fl mice and p53Hep−/− mice i.v. with AAV8-ALB-Null or AAV8-ALB-hHNF4α, which were then fed an HFCF diet for 20 weeks. Over-expression of human HNF4α reduced plasma ALT levels in p53fl/fl mice but not in p53Hep−/− mice (Figure 6A). In addition, hepatic mRNA levels of inflammatory genes (Tnfα, Il6, Il1β, Mcp1) (Figure 6B) or fibrogenic genes (Tgfβ, Timp1, α-Sma, Col1α1, Colα2) (Figure 6C) were reduced in p53fl/fl mice but not in p53Hep−/− mice. Consistent with the changes in gene expression, hepatic hydroxyproline levels were reduced in p53fl/fl mice but not in p53Hep−/− mice (Figure 6D, left panel). Interestingly, HNF4α over-expression reduced hepatic TG, FFA and cholesterol levels in both p53fl/fl mice and p53Hep−/− mice (Figure 6D middle and right panels). Over-expression of human HNF4α also inhibited apoptosis (Figure 6E) and reduced the protein levels of phospho-Smad2/3 and cleaved caspase 3 (Figure 6F) in p53fl/fl mice but not in p53Hep−/− mice.
Figure 6. Hepatocyte-specific over-expression of HNF4α attenuates HFCF diet-induced steatohepatitis via inhibition of p53.

p53fl/fl mice and p53Hep−/− mice were injected i.v. with AAV8-ALB-Null or AAV8-ALB-hHNF4α, and then fed an HFCF diet for 20 weeks (n=8 per group). (A) Plasma ALT levels. (B and C) Hepatic mRNA levels. (D) Hepatic levels of hydroxyproline (left panel), TG, FFA and cholesterol (middle and right panels). (E) Hepatic apoptosis was analyzed by TUNEL assays. (F) Hepatic proteins were analyzed by Western blot assays (left panel) and then quantified (right panel). P/T-Smad, ratio of phosphorylated Smad2/3 to total Smad2/3. *P<0.05, **P<0.01
To further investigate whether HNF4α-mediated inhibition of p53 played a role in the development of NAFLD, we generated AAV8-ALB-p53 that over-expressed human p53, which was i.v. injected into C57BL/6J mice together with AAV8-ALB-Null or AAV8-ALB-hHNF4α. The mice were then fed an HFCF diet for 20 weeks. Our data showed that recapitulation of hepatic p53 expression level by 76% of the control mice (Figure S3A) partially restored plasma ALT level (Figure S3B), hepatic TG, cholesterol and hydroxyproline levels (Figure S3C–E), and hepatic fibrogenesis in mice over-expressing HNF4α (Figure S3F). In addition, partial recapitulation of hepatic p53 expression level also partially restored hepatic expression of pro-inflammatory and fibrogenic genes (Tnfα, Il6, Il1β, Mcp1, Timp1, α-Sma, Col1α1, Col1α2) (Figure S4A and S4B), and fully restored apoptosis (Figure S4C and S4D) and phospho-Smad2/3 and cleaved caspase 3 levels in the liver (Figure S4E and S4F).
Taken together, these data demonstrate that gain of hepatic HNF4α function protects against steatohepatitis through, at least in part, inhibition of p53.
Loss of hepatocyte HNF4α aggravates the development of NASH by inhibiting BA signaling.
BAs are endogenous ligands for FXR, whose pharmacological activation inhibits p53 activity and protects against cell death and the development of NASH (23). Since CYP7A1 and CYP8B1 are key enzymes in the BA biosynthetic pathway and are reduced in Hnf4αHep−/− mice, we hypothesized that the reduced BA synthesis/signaling might be responsible, at least in part, for the aggravated steatohepatitis in these mice. To test this hypothesis, we i.v. injected AAV8-ALB-Null or AAV8-ALB-hCYP7A1 plus AAV8-ALB-hCYP8B1 into Hnf4αfl/fl mice or Hnf4αHep−/− mice, which were then fed an HFCF diet for 20 weeks. In Hnf4αHep−/− mice, exogenous expression of human CYP7A1 and CYP8B1 fully normalized hepatic CYP7A1 and CYP8B1 protein expression and hepatic BA levels (Figure 7A), and also partially normalized hepatic p53 protein expression (Figure 7A, left panel; Figure S5). In addition, exogenous expression of human CYP7A1 and CYP8B1 in Hnf4αHep−/− mice also fully normalized hepatic levels of TC, FC (Figure 7B), TG, FFA (Figure 7C) and FAO (Figure 7D), but failed to normalize hepatic TGH activity (Figure 7E). Consistent with these data, hepatic expression of Pparα, Cpt1, Cpt2 or Pdk4, but not Ces1 or Ces2, was normalized in Hnf4αHep−/− mice that expressed human CYP7A1 and CYP8B1 (Figure 7F).
Figure 7. Recapitulation of hepatic CYP7A1 and CYP8B1 expression normalizes hepatic TG levels and FAO in Hnf4αHep−/− mice.
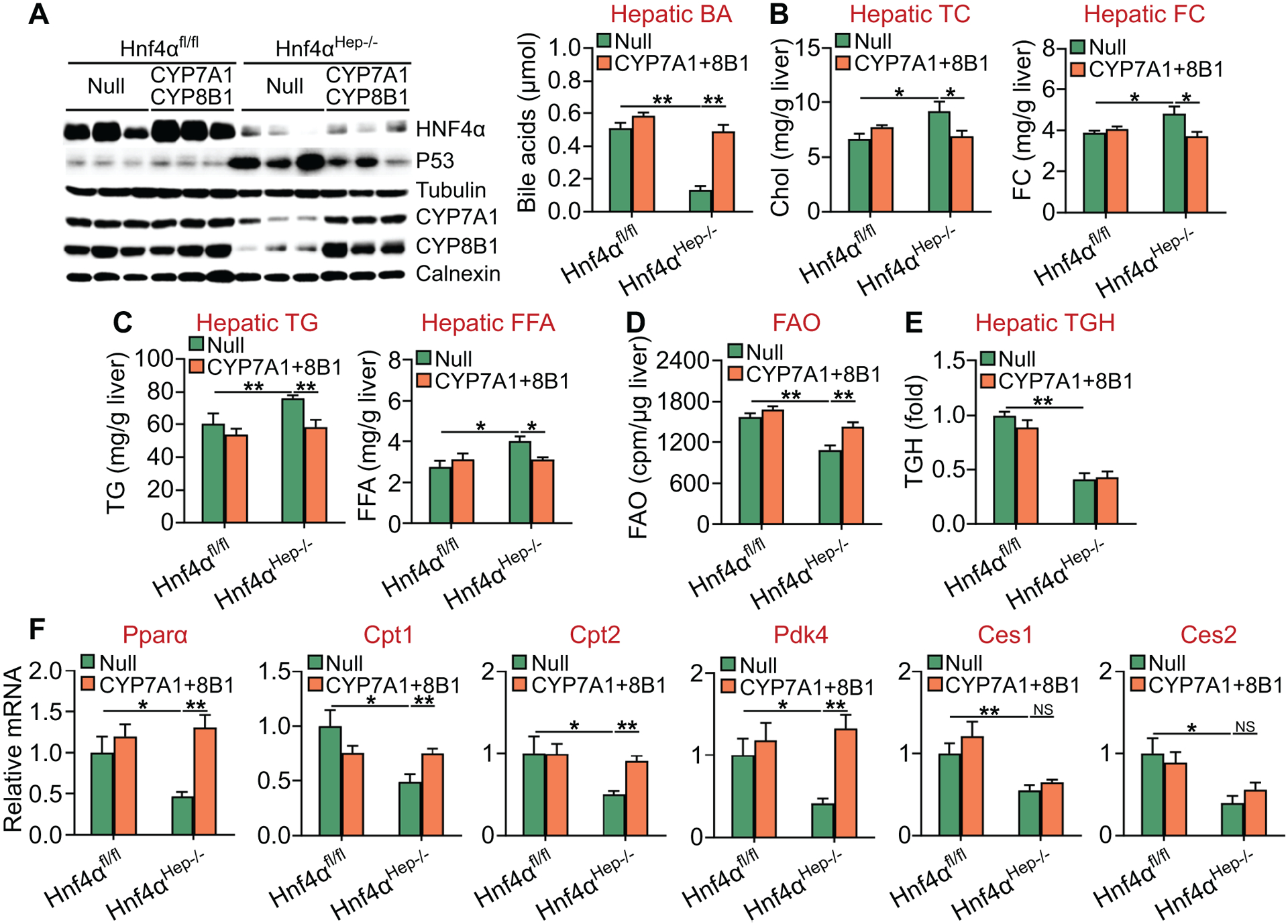
Hnf4αfl/fl mice and Hnf4αHep−/− mice were injected i.v. with 0.5×1011 GC of AAV8-ALB-Null (Null) or AAV8-ALB-hCYP7A1 plus AAV8-ALB-hCYP8B1 (CYP7A1+8B1), and then fed an HFCF diet for 20 weeks (n=8 per group). (A) Hepatic protein levels were analyzed by Western blot assays (left panel) and hepatic BA levels were quantified (right panel). (B) Hepatic TC (left panel) and FC (right panel) levels. (C) Hepatic TG (left panel) and FFA (right panel) levels. (D) FAO was analyzed using isolated hepatocytes. (E) Hepatic TGH activity was analyzed. (F) Hepatic mRNA levels. NS, not significant. *P<0.05, **P<0.01
Furthermore, recapitulation of hepatic CYP7A1 and CYP8B1 expression in Hnf4αHep−/− mice also partially or fully normalized plasma ALT levels (Figure 8A) as well as hepatic levels of hydroxyproline or fibrosis (Figure 8B), apoptosis (Figure 8C), phospho-Smad2/3, cleaved caspase 3 (Figure 8D) and proinflammatory or fibrogenic genes (Tnfα, Il6, Il1β, Mcp1, Timp1, α-Sma, Col1a1, Col1a2) (Figure 8E).
Figure 8. Recapitulation of hepatic CYP7A1 and CYP8B1 expression attenuates diet-induced steatohepatitis in Hnf4αHep−/− mice.
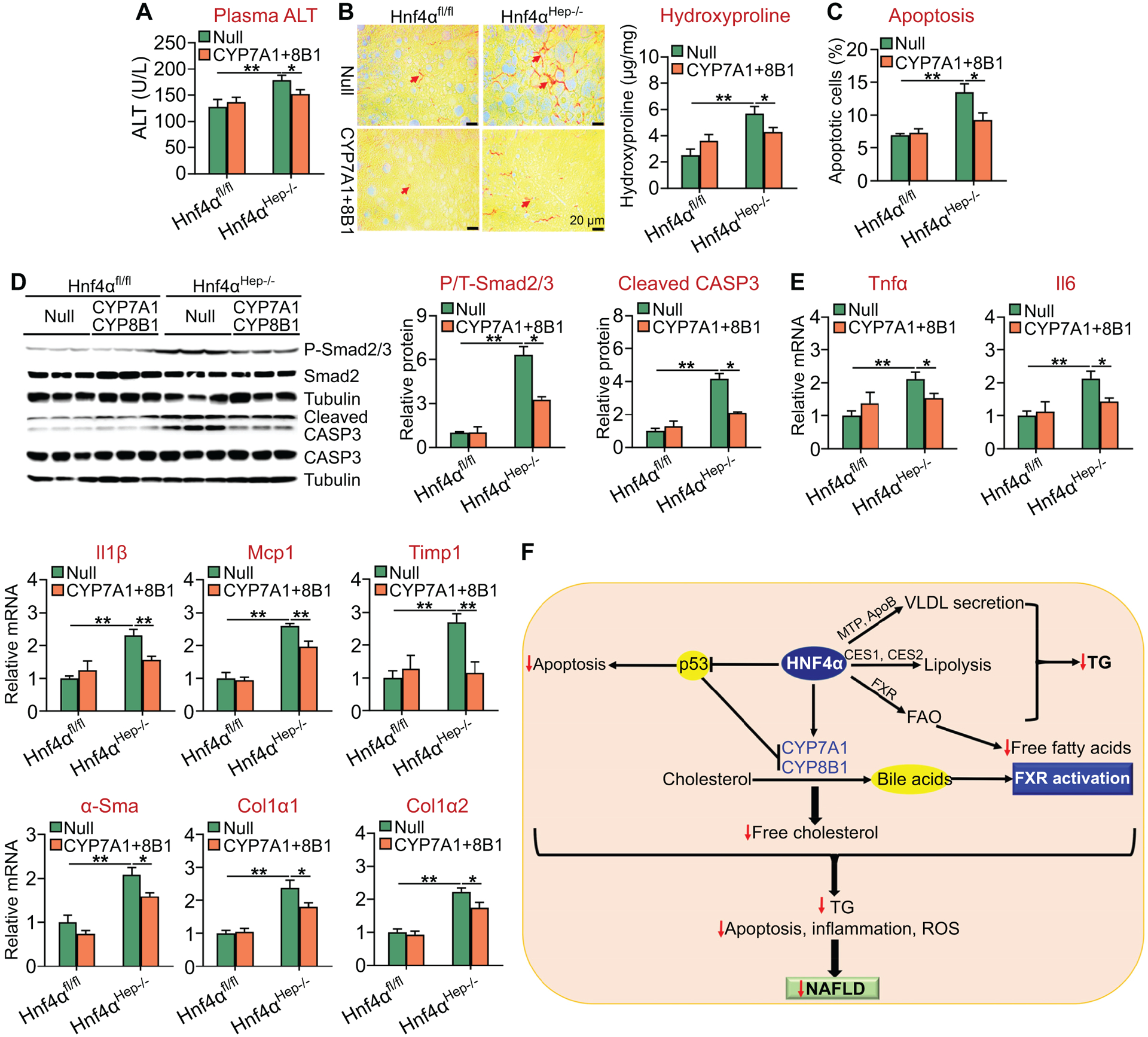
Hnf4αfl/fl mice and Hnf4αHep−/− mice were injected i.v. with 0.5×1011 GC of AAV8-ALB-Null or AAV8-ALB-hCYP7A1 plus AAV8-ALB-hCYP8B1, and then fed an HFCF diet for 20 weeks (n=8 per group). (A) Plasma ALT levels. (B) Liver sections were stained with Sirius Red (left panel) and hepatic hydroxyproline levels were quantified (right panel). (C) Hepatic apoptosis was analyzed by TUNEL assays. (D) Hepatic proteins were analyzed by Western blot assays (left panel) and then quantified (middle and right panels). (E) Hepatic mRNA levels. (F) A model for hepatic HNF4α to regulate the development and progression of NAFLD. HNF4α regulates VLDL secretion, lipolysis, fatty acid oxidation (FAO), bile acid synthesis and p53 expression. As a result, FXR is activated, hepatic free fatty acid, free cholesterol and triglyceride (TG) levels are decreased, and hepatic apoptosis, inflammation, and ROS production are inhibited. These multifacted effects of HNF4α lead to inhibition of development and progression of NAFLD. *P<0.05, **P<0.01
Expression of CYP7A1 and CYP8B1 produces bile acids, which are endogenous ligands for FXR. Indeed, recapitulation of hepatic CYP7A1 and CYP8B1 expression in mice lacking HNF4α induced the expression of FXR target genes, including Shp, bile acid export protein (Bsep), fibroblast growth factor 21 (Fgf21), and organic solute transporter α (Ostα) (25, 33) (Figure S6). Treatment of C57BL/6 mice with either vehicle or the FXR agonist OCA induced hepatic genes involved in FAO, including Pparα, Cpt1, Cpt2 and Pdk4 (Figure S7A). Consistent with these later data, OCA treatment induced FAO in Fxr+/+ hepatocytes, but not in Fxr−/− hepatocytes (Figure S7B). These data may explain why expression of CYP7A1 and CYP8B1 induces FAO.
To investigate whether activation of FXR by a synthetic FXR agonist can mimic the effects of CYP7A1 and CYP8B1 over-expression, we fed Hnf4αfl/fl mice and Hnf4αHep−/− mice an HFCF diet for 20 weeks. In the last 30 days, mice were also gavaged with either vehicle or OCA. Activation of FXR in Hnf4αHep−/− mice reduced plasma ALT level as well as hepatic TG and FFA levels (Figure S8A–C), but induced hepatic mRNA levels of Shp (a known FXR target gene), Pparα, Cpt1, Cpt2 and Pdk4 (Figure S8D). In addition, activation of FXR in Hnf4αHep−/− mice also inhibited liver fibrosis (Figure S9A and S9B), and reduced hepatic mRNA levels of proinflammatory and fibrogenic genes (Tnfα, Il6, Il1β, Mcp1, Timp1, Col1a1, Col1a2) (Figure S9C and S9D). Thus, the data of Figures S6–9 suggest that recapitulation of hepatic CYP7A1 and CYP8B1 expression exert their beneficial effects in mice lacking HNF4α likely by increasing bile acids and subsequent activation of FXR.
Taken together, the data of Figures 7, 8 and S6–9 show that recapitulation of hepatic CYP7A1 and CYP8B1 expression in Hnf4αHep−/− mice partly or fully prevents diet-induced NASH, which can be mimicked by activation of FXR via a synthetic agonist. Thus, disruption of hepatic BA signaling may account, at least in part, for the aggravated NASH in mice lacking hepatocyte Hnf4α.
Discussion
Previous studies have shown that acute or chronic loss of hepatic HNF4α increases hepatic TG levels in chow-fed mice (14, 15), and that hepatic HNF4α expression is markedly reduced in NASH patients and in NASH mouse models (18). However, it remains to be determined whether gain or loss of hepatic HNF4α expression plays a role in Western diet-induced development or progression of NAFLD. In the current study, we provide the first and compelling evidence demonstrating that hepatocyte HNF4α is required and also sufficient to protect against diet-induced development and progression of NASH by integrating lipolytic, p53, and BA signaling (Figure 8F).
The development of NAFLD often initiates from TG accumulation in the liver. The lipolytic pathway is an important mechanism of the development and progression of NAFLD (8–10). Our gain- and loss-of-function studies demonstrate for the first time that HNF4α is a critical regulator of hepatic TGH activity. Importantly, HNF4α also increases FAO, allowing released FFAs to be oxidized so that no toxic FFAs are accumulated in the liver. Our data suggest that the induction of FAO by HNF4α is likely via activation of FXR, as HNF4α promotes activation of FXR and FXR activation induces FAO. We and others have previously shown that HNF4α regulates VLDL secretion (14, 15). Thus, HNF4α regulates hepatic TG levels likely by modulating both TGH and VLDL secretion (Figure 8F).
The mechanism underlying the progression of NAFLD from NAFL to NASH has not been well understood. It is believed that abnormal mitochondrial activity, apoptosis, ROS production, lipotoxicity and inflammation are involved in the progression of NAFLD. P53 is well known for its role in inducing apoptosis and inhibiting tumorigenesis. Mice deficient in p53 are resistant to diet-induced steatohepatitis (19), highlighting a critical role of apoptosis in the progression of NAFLD. Our data show that HNF4α inhibits hepatic p53 expression, and that over-expression of human HNF4α attenuates diet-induced NASH in the control mice but not in mice lacking hepatocyte p53. Importantly, partial recapitulation of hepatic p53 protein expression is sufficient to restore hepatic apoptosis level and prevent HNF4α-mediated amelioration of steatohepatitis. Interestingly, p53 also plays a role in HNF4α-mediated inhibition of pro-inflammatory and fibrogenic gene expression, the TGFβ/Smad pathway, and caspase 3 cleavage. The TGFβ/Smad pathway is known to promote liver fibrosis (34, 35). These data highlight an important role of p53 in the progression of NAFL to NASH. In addition to inhibition of p53 expression, HNF4α also reduces hepatic levels of FC and palmitate, which have been shown to cause lipotoxicity and apoptosis (7, 36–39). The reduction in hepatic FC and palmitate levels may result from increased conversion of free cholesterol to bile acids and increased FAO, respectively. Thus, a reduction in hepatic FC and palmitate levels may also play a role in HNF4α-mediated protection against the development of steatohepatitis (Figure 8F).
Previous studies have shown that activation of FXR inhibits diet-induced p53 signaling and NASH (23). Loss of hepatic HNF4α inhibits both CYP7A1 and CYP8B1 expression, therefore suppressing BA synthesis and FXR signaling. Interestingly, normalization of hepatic CYP7A1 and CYP8B1 expression in mice lacking Hnf4α completely normalizes hepatic TG levels, and also partially attenuates diet-induced p53 expression and steatohepatitis. The reduced hepatic TG levels may partly result from normalized FXR signaling, as activation of FXR is known to lower hepatic TG levels (40). Activation of FXR is also shown to inhibit inflammation (40). Importantly, treatment of mice lacking Hnf4α with a synthetic FXR agonist mimics the effects of CYP7A1 and CYP8B1 normalization. Thus, the impaired bile acid/FXR signaling plays a role in the pathogenesis of NASH in Hnf4αHep−/− mice (Figure 8F). Although both HNF4α and FXR inhibit p53, they also inhibit inflammation and regulate many other pathways. As a result, HNF4α over-expression or FXR activation is not expected to cause tumorigenesis.
In summary, our studies have demonstrated that hepatocyte HNF4α is a key regulator of the development and progression of NASH. HNF4α regulates hepatic TG accumulation by regulating VLDL secretion and lipolysis (Figure 8F). On the other hand, HNF4α also regulates apoptosis, bile acid synthesis and FAO. HNF4α inhibits apoptosis by suppressing p53 expression. HNF4α promotes bile acid production by inducing CYP7A1 and CYP8B1 expression, leading to reduced hepatic free cholesterol levels and activation of FXR. Activation of FXR stimulates FAO and reduces hepatic palmitate levels. The reduced free cholesterol and palmitate levels can reduce lipotoxicity. Thus, HNF4α inhibits the progression of NAFL to NASH likely by suppressing apoptosis, inflammation and ROS production (Figure 8F). Since hepatic HNF4α expression is markedly reduced in NASH patients, targeting hepatic HNF4α may represent a promising approach for treatment of NASH.
Supplementary Material
Financial support:
This work was supported by NIH grants R01HL103227 (Y.Z. and L.Y.), R01DK102619 (Y.Z.), R01HL142086 (Y.Z.) and R01DK118941 (Y.Z.).
Abbreviations:
- AAV
adeno-associated virus
- β-HB
beta-hydroxybutyrate
- BA
bile acid
- CE
cholesteryl ester
- CYP7A1
cholesterol 7α-hydroxylase
- CYP8B1
sterol 12α-hydroxylase
- FC
free cholesterol
- FXR
farnesoid X receptor
- FAO
fatty acid oxidation
- FFA
free fatty acid
- GC
genome copies
- HFCF
high fat/cholesterol/fructose
- HFD
high fat diet
- HNF4α
hepatocyte nuclear factor 4α
- MDA
malondialdehyde
- NAFLD
non-alcoholic fatty liver disease
- NAFL
non-alcoholic simple steatosis
- NASH
non-alcoholic steatohepatitis
- OCA
obeticholic acid
- PPARα
peroxisome proliferator-activated receptor α
- SHP
small heterodimer partner
- SREBP-1c
sterol regulatory element-binding protein 1c
- TG
triglyceride
- TGH
triglyceride hydrolase
- VLDL
very low-density lipoprotein
References
- 1.Edmison J, McCullough AJ. Pathogenesis of non-alcoholic steatohepatitis: human data. Clin Liver Dis 2007;11:75–104, ix. [DOI] [PubMed] [Google Scholar]
- 2.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology 1998;114:842–845. [DOI] [PubMed] [Google Scholar]
- 3.Jou J, Choi SS, Diehl AM. Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin Liver Dis 2008;28:370–379. [DOI] [PubMed] [Google Scholar]
- 4.Lewis JR, Mohanty SR. Nonalcoholic fatty liver disease: a review and update. Dig Dis Sci 2010;55:560–578. [DOI] [PubMed] [Google Scholar]
- 5.Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 2005;129:113–121. [DOI] [PubMed] [Google Scholar]
- 6.Charlton M, Szczepaniak LS, Rasmussen D, Lindor K, Nair KS. Apolipoprotein synthesis in nonalcoholic steatohepatitis. Hepatology 2002;35:898–904. [DOI] [PubMed] [Google Scholar]
- 7.Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol 2018;68:280–295. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs CD, Claudel T, Trauner M. Role of metabolic lipases and lipolytic metabolites in the pathogenesis of NAFLD. Trends Endocrinol Metab 2014;25:576–585. [DOI] [PubMed] [Google Scholar]
- 9.Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am J Physiol Endocrinol Metab 2009;297:E289–296. [DOI] [PubMed] [Google Scholar]
- 10.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sladek FM, Zhong WM, Lai E, Darnell JE Jr. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev 1990;4:2353–2365. [DOI] [PubMed] [Google Scholar]
- 12.Watt AJ, Garrison WD, Duncan SA. HNF4: a central regulator of hepatocyte differentiation and function. Hepatology 2003;37:1249–1253. [DOI] [PubMed] [Google Scholar]
- 13.Parviz F, Matullo C, Garrison WD, Savatski L, Adamson JW, Ning G, Kaestner KH, et al. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet 2003;34:292–296. [DOI] [PubMed] [Google Scholar]
- 14.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol 2001;21:1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin L, Ma H, Ge X, Edwards PA, Zhang Y. Hepatic hepatocyte nuclear factor 4alpha is essential for maintaining triglyceride and cholesterol homeostasis. Arterioscler Thromb Vasc Biol 2011;31:328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoue Y, Yu AM, Yim SH, Ma X, Krausz KW, Inoue J, Xiang CC, et al. Regulation of bile acid biosynthesis by hepatocyte nuclear factor 4alpha. J Lipid Res 2006;47:215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inoue Y, Yu AM, Inoue J, Gonzalez FJ. Hepatocyte nuclear factor 4alpha is a central regulator of bile acid conjugation. J Biol Chem 2004;279:2480–2489. [DOI] [PubMed] [Google Scholar]
- 18.Xu Y, Zalzala M, Xu J, Li Y, Yin L, Zhang Y. A metabolic stress-inducible miR-34a-HNF4alpha pathway regulates lipid and lipoprotein metabolism. Nat Commun 2015;6:7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomita K, Teratani T, Suzuki T, Oshikawa T, Yokoyama H, Shimamura K, Nishiyama K, et al. p53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J Hepatol 2012;57:837–843. [DOI] [PubMed] [Google Scholar]
- 20.Farrell GC, Larter CZ, Hou JY, Zhang RH, Yeh MM, Williams J, dela Pena A, et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J Gastroenterol Hepatol 2009;24:443–452. [DOI] [PubMed] [Google Scholar]
- 21.Panasiuk A, Dzieciol J, Panasiuk B, Prokopowicz D. Expression of p53, Bax and Bcl-2 proteins in hepatocytes in non-alcoholic fatty liver disease. World J Gastroenterol 2006;12:6198–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Derdak Z, Villegas KA, Harb R, Wu AM, Sousa A, Wands JR. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J Hepatol 2013;58:785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goto T, Itoh M, Suganami T, Kanai S, Shirakawa I, Sakai T, Asakawa M, et al. Obeticholic acid protects against hepatocyte death and liver fibrosis in a murine model of nonalcoholic steatohepatitis. Sci Rep 2018;8:8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim DH, Lee JW. Tumor suppressor p53 regulates bile acid homeostasis via small heterodimer partner. Proc Natl Acad Sci U S A 2011;108:12266–12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee FY, Lee H, Hubbert ML, Edwards PA, Zhang Y. FXR, a multipurpose nuclear receptor. Trends Biochem Sci 2006;31:572–580. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Yin L, Hillgartner FB. SREBP-1 integrates the actions of thyroid hormone, insulin, cAMP, and medium-chain fatty acids on ACCalpha transcription in hepatocytes. J Lipid Res 2003;44:356–368. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, Willson TM, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A 2006;103:1006–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 1959;37:911–917. [DOI] [PubMed] [Google Scholar]
- 29.Xu J, Xu Y, Li Y, Jadhav K, You M, Yin L, Zhang Y. Carboxylesterase 1 Is Regulated by Hepatocyte Nuclear Factor 4alpha and Protects Against Alcohol- and MCD diet-induced Liver Injury. Sci Rep 2016;6:24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Zalzala M, Jadhav K, Xu Y, Kasumov T, Yin L, Zhang Y. Carboxylesterase 2 prevents liver steatosis by modulating lipolysis, endoplasmic reticulum stress, and lipogenesis and is regulated by hepatocyte nuclear factor 4 alpha in mice. Hepatology 2016;63:1860–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rune A, Osler ME, Fritz T, Zierath JR. Regulation of skeletal muscle sucrose, non-fermenting 1/AMP-activated protein kinase-related kinase (SNARK) by metabolic stress and diabetes. Diabetologia 2009;52:2182–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu J, Li Y, Chen WD, Xu Y, Yin L, Ge X, Jadhav K, et al. Hepatic carboxylesterase 1 is essential for both normal and farnesoid X receptor-controlled lipid homeostasis. Hepatology 2014;59:1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cyphert HA, Ge X, Kohan AB, Salati LM, Zhang Y, Hillgartner FB. Activation of the farnesoid X receptor induces hepatic expression and secretion of fibroblast growth factor 21. J Biol Chem 2012;287:25123–25138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu F, Liu C, Zhou D, Zhang L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J Histochem Cytochem 2016;64:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci 2002;7:d793–807. [DOI] [PubMed] [Google Scholar]
- 36.Ioannou GN. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol Metab 2016;27:84–95. [DOI] [PubMed] [Google Scholar]
- 37.Arguello G, Balboa E, Arrese M, Zanlungo S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim Biophys Acta 2015;1852:1765–1778. [DOI] [PubMed] [Google Scholar]
- 38.Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, Okada Y, et al. Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014;59:154–169. [DOI] [PubMed] [Google Scholar]
- 39.Van Rooyen DM, Gan LT, Yeh MM, Haigh WG, Larter CZ, Ioannou G, Teoh NC, et al. Pharmacological cholesterol lowering reverses fibrotic NASH in obese, diabetic mice with metabolic syndrome. J Hepatol 2013;59:144–152. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Jadhav K, Zhang Y. Bile acid receptors in non-alcoholic fatty liver disease. Biochem Pharmacol 2013;86:1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.