

Abstract
Severe acute respiratory syndrome (SARS) is an emerging infectious disease caused by a novel human coronavirus. Viral maturation requires a main protease (3CLpro) to cleave the virus-encoded polyproteins. We report here that the 3CLpro containing additional N- and/or C-terminal segments of the polyprotein sequences undergoes autoprocessing and yields the mature protease in vitro. The dimeric three-dimensional structure of the C145A mutant protease shows that the active site of one protomer binds with the C-terminal six amino acids of the protomer from another asymmetric unit, mimicking the product-bound form and suggesting a possible mechanism for maturation. The P1 pocket of the active site binds the Gln side chain specifically, and the P2 and P4 sites are clustered together to accommodate large hydrophobic side chains. The tagged C145A mutant protein served as a substrate for the wild-type protease, and the N terminus was first digested (55-fold faster) at the Gln-1-Ser1 site followed by the C-terminal cleavage at the Gln306-Gly307 site. Analytical ultracentrifuge of the quaternary structures of the tagged and mature proteases reveals the remarkably tighter dimer formation for the mature enzyme (Kd = 0.35 nm) than for the mutant (C145A) containing 10 extra N-terminal (Kd = 17.2 nm) or C-terminal amino acids (Kd = 5.6 nm). The data indicate that immature 3CLpro can form dimer enabling it to undergo autoprocessing to yield the mature enzyme, which further serves as a seed for facilitated maturation. Taken together, this study provides insights into the maturation process of the SARS 3CLpro from the polyprotein and design of new structure-based inhibitors.
Severe acute respiratory syndrome (SARS)1 is a severe febrile respiratory illness caused by a newly identified coronavirus, SARS-associated coronavirus (SARS-CoV) (1, 2, 3, 4). In the period from February to June, 2003, SARS rapidly spread from its likely origin in southern China to 32 countries in the world. SARS-CoV belongs to a coronaviridae family that includes porcine transmissible gastroenteritis virus (TGEV), human coronavirus (HCoV) 229E, mouse hepatitis virus, bovine coronavirus, and infectious bronchitis virus (5, 6, 7). These coronaviruses are large, enveloped, positive single-stranded RNA viruses (27-31 kb) that cause respiratory and enteric diseases in humans and other animals. The SARS-CoV genome comprises about 29,700 nucleotides and encodes two overlapping polyproteins, pp1a (486 kDa) and pp1ab (790 kDa) that mediate all the functions required for viral replication and transcription (5, 7). The functional polypeptides are released from the polyproteins by extensive proteolytic processing. This is primarily achieved by the 34.6-kDa main protease (Mpro) which is frequently called 3C-like protease (3CLpro), because its substrate specificity is similar to those of picornavirus 3C proteases (8, 9). SARS-CoV 3CLpro cleaves the polyproteins at eleven sites involving a conserved Gln at the P1 position and a small amino acid (Ser, Ala, or Gly) at the P1′ position, a process initiated by enzyme's own autolytic cleavage (autoprocessing) (10).
Several crystal structures of coronavirus 3CLpro (apo form or with suicide inhibitors) reported from TGEV, HCoV 229E, and SARS-CoV (11, 12, 13) revealed a common feature in 3CLpro: two chymotrypsin-like β-domains (residues 1-184) and one α-helical dimerization domain (residues 201-303). The active site of SARS-CoV 3CLpro is located in the center of the cleft between domains I and II and includes a catalytic dyad consisting of His41 and Cys145. Domain III was proposed to mediate dimer formation, because the C-terminal helical domain III (residues 201-306) alone formed a tight dimer (14). However, the electron densities of the C-terminal residues (301-306) of SARS-CoV-3CLpro could not be detected in the previous structure (13).
Here, we determined the crystal structures of the wild-type and the C145A mutant 3CLpro. Unlike the previous three-dimensional structure of the wild-type protease (13), the new crystal structure of C145A shows clearly visible C-terminal residues that are intercalated into the neighboring protomer creating a product-bound structure that may resemble intermediates during autoprocessing. Autoprocessing has been known to be an essential step for viral maturation, but its detailed molecular mechanism is still hypothetical. To further understand the maturation process, we constructed a wild-type SARS-CoV 3CLpro with 10 additional amino acids that are part of the polyprotein sequence at the N and/or C termini (termed 10aa-WT, WT-10aa, and 10aa-WT-10aa), respectively. We also used analytical ultracentrifugation (AUC) to determine their quaternary structures. In addition, thioredoxin (Trx) and glutathione S-transferase (GST) tags were appended to observe their autoprocessing by SDS-PAGE analysis. The inactive mutant C145A with the tags (Trx-10aa-C145A-10aa-GST) served as a substrate for facilitated processing by the wild-type protease.
Because 3CLpro is responsible for polyprotein maturation, it is a potential target for anti-SARS drug development. We had previously used a fluorescence-based assay to characterize the protease and identified some C2 symmetry peptidomimetic compounds and metal-conjugated compounds as inhibitors of SARS-CoV 3CLpro (15, 16, 17). Other small molecules targeting SARS-CoV 3CLpro were identified from several compound libraries, including bifunctional aryl boronic acids (18), a quinolinecarboxylate derivative (19), a thiophenecarboxylate (20), and phthalhydrazide-substituted ketoglutamine analogues (21). These are all active site inhibitors. The new structures presented here suggest the possibility of drug design targeting protease dimerization during maturation. Overall, our study provides insights to substrate recognition, protein maturation, and drug discovery for SARS 3CLpro.
EXPERIMENTAL PROCEDURES
Materials—Fluorogenic peptide substrate Dabcyl-KTSAVLQSG-FRKME-Edans was prepared as previously described (16). The plasmid mini-prep kit, DNA gel extraction kit, and Ni-NTA resin were purchased from Qiagen. FXa and the protein expression kit (including the pET32Xa/LIC vector and competent JM109 and BL21 cells) were obtained from Novagen. The pGEX vector was obtained from Amersham Biosciences. All commercial buffers and reagents were of the highest grade.
Expression and Purification of SARS-CoV 3CLpro—Different constructs (wild-type and C145A mutant, with and without extra amino acids in their N and C termini) of the SARS proteases were cloned in pET32Xa/LIC (with N-terminal Trx, His tag, and FXa site), pET28 (with C-terminal His tag), or pGEX (with N-terminal GST tag). The previously cloned gene encoding the wild-type SRAS-CoV 3CLpro was used as a template, and the mutant forward primer (5′-CCTTAATGGATCAGCTGGTAGTGTTGGT-3′) for C145A was used in a polymerase chain reaction (PCR) to create the mutant gene. For constructing 10aa-C145A (C145A mutant containing 10 extra amino acids QTSITSAVLQ derived from the natural pp1a polyprotein attached to the N terminus), the forward primer (5′-GGTATTGAGGGTCGCCAGACATCAATCACTTCTGCTGTTCTGCAGAGTGGTTTTAGGAAAATGGCA-3′) was used; for C145A-10aa (10 extra amino acids GKFKKIVKGT derived from the natural pp1a polyprotein attached to its C terminus), the backward primer (5′-AGAGGAGAGTTAGAGCCTTAAGTGCCCTTAACAATTTTCTTGAACTTACCTTGGAAGGTAACACCAGAGCA-3′) was used; and for 10aa-C145A-10aa, the above forward and backward primers were used. The 5′-GGTATTGAGGGTCGCAGTGGTTTTAGG-3′ part of the forward primer and the 5′-AGAGGAGAGTTAGAGCCTTATTGGAAGGTAACACC-3′ part of the reverse primer 5′ are for cloning into the pET32Xa/LIC vector. These primers were also used to prepare the wild-type SARS protease containing extra N- and C-terminal amino acids (10aa-WT, WT-10aa, and 10aa-WT-10aa). In a PCR reaction, thirty cycles of PCR were performed using a thermocycler (Applied Biosystems) with the melting temperature at 95 °C for 2 min, annealing temperature at 42 °C for 1 min, and polymerization temperature at 68 °C for 1 min. The PCR product was subjected to electrophoresis on 1.2% agarose gel in TAE buffer (40 mm Tris-acetate, 5 mm EDTA, pH 8.0), and then the gel was stained with ethidium bromide. The band with the correct size was excised, and the DNA was recovered using a DNA elution kit. The construct was ligated to the pET-32Xa/LIC vector by incubation for 1 h at 22 °C. For preparation of the N-terminal Trx-tagged and C-terminal GST-tagged protease (Trx-10aa-C145A-10aa-GST), the GST gene in the pGEX 6P-1 vector (Amersham Biosciences) amplified with the primers (forward primer 5′-GGTAAGTTCAAGAAAATTGTTAAGGGCACTATGTCCCCTATACTAGGTTA-T-3′ and reverse primer 5′-AGAGGAGAGTTAGAGCCTCAATCCGATTTTGGAGGATGGT-3′) was used as a template. This template, containing the GST portion, and the second template of 10aa-C145A-10aa were used with the forward primer of 5′-GGTATTGAGGGTCGCCAGACATCAATCACTTCTGCTGTTCTGCAGAGTGGTTTTAGGAAAATGGCA-3′ and the backward primer of 5′-AGAGGAGAGTTAGAGCCTCAATCCGATTTTGGAGGATGGT-3′ to clone into pET32Xa/LIC vector.
The recombinant protease plasmid was then used to transform Escherichia coli JM109 competent cells that were streaked on a Luria-Bertani (LB) agar plate containing 100 μg/ml ampicillin. Ampicillin-resistant colonies were selected from the agar plate and grown in 5 ml of LB culture containing 100 μg/ml ampicillin overnight at 37 °C. The entire SARS protease gene of the plasmid obtained from the overnight culture was sequenced. The correct construct was subsequently transformed into E. coli BL21 for protein expression. The protein purification followed our reported procedure using Ni-NTA column chromatography (16). To prepare the protease without tags (10aa-C145A and C145A-10aa) for AUC studies, the N-terminal Trx and His tags were removed by FXa protease digestion, and the mixture was loaded onto another Ni-NTA column to recover the highly purified untagged protein. To purify the Trx-10aa-C145A-10aa-GST to be used as the substrate for facilitated processing, both Ni-NTA and GST columns were used. For protein crystallization, SARS-CoV 3CLpro wild type and mutant C145A clones were incorporated in pGEX-6p-1 plasmid DNA (Amersham Biosciences) with a Factor Xa cutting site immediately before the N-terminal Ser1 of the target gene. GST-tagged protein was purified using a GST column and after the tag cleavage by FXa, the mixture was loaded onto a HiTrap™ 16/10 QFF column (Amersham Biosciences) and eluted with the buffer (20 mm Tris-HCl, pH 8.0, 1 mm EDTA, and 1 m NaCl). The flow-through fractions containing 3CLpro were pooled and concentrated for growing crystals.
Protein Crystallization—Wild-type 3CLpro was stored in a buffer containing 10 mm Tris-HCl (pH 7.5), 1 mm dithiothreitol, and 1 mm
EDTA. The protein was crystallized using the sitting drop diffusion method by mixing 2 μl of the wild-type 3CLpro protein solution (10 mg/ml) with 2 μl of the reservoir solution (1.0 m sodium malonate, pH 7.0, and 4% isopropanol) onto a sitting drop post, equilibrated with 500 μl of the reservoir solution. The crystallization proceeded at 25 °C in the dark for 7 days.
Purified C145A was concentrated by Amicon ultrafiltration and desalted using a HiPrep 26/10 column and a buffer containing 10 mm Tris-HCl (pH 7.5) and 1 mm EDTA. The protein was crystallized using the sitting drop diffusion method by mixing 2 μl of the C145A solution (18 mg/ml) with 2 μl of the reservoir solution (0.1 m Bicine, pH 9.0, and 20% polyethylene glycol 6000) and 1 μl of additive (10 mm cobalt sulfate) onto a sitting drop post, equilibrated with 500 μl of the reservoir solution. Crystallization was carried out at 25 °C in the dark for 3 days.
Data Collection and Processing—Crystals for data collection were rinsed with the reservoir buffer and cryo-cooled in liquid nitrogen. The preliminary x-ray analysis was performed with an in-house Micro-Max002 x-ray generator with a Rigaku R-Axis IV++ image plate system. High resolution data for wild type and C145A crystals were collected at BL17B2 beamline in National Synchrotron Radiation Research Center (NSRRC) (Taiwan) and Taiwan beamline BL12B2 in SPring-8 (Japan), respectively. Data were processed and integrated by using the program HKL2000 (22).
Structure Determination, Refinement, and Model Building—The wild-type structure was determined by molecular replacement using one monomer of HCoV 229E Mpro (PDB ID code 1P9S) (12) as the starting model. Cross-rotation function and translation function searches were performed with the program CCP4 (23). The geometric adjustments were made with XtalView (24) under the guidance of (2Fo - Fc) sum difference maps. The Crystallography and NMR System (CNS) program (25) was used for structure refinement, including stimulated annealing procedure, positional, and B-factor refinements.
Autoprocessing Experiments—Six constructs were individually expressed in E. coli. The 5-ml overnight culture of a single transformant was used to inoculate 500 ml of fresh LB medium containing 100 μg/ml ampicillin. The cells were grown to A 600 = 0.6 and induced with 1 mm isopropyl-β-thiogalactopyranoside. After 4-5 h, the cells were harvested by centrifugation at 7,000 × g for 15 min. The enzyme purification was conducted at 4 °C. The 2-liter cell culture was collected to yield ∼20 g of cell paste, which was suspended in 80 ml of lysis buffer containing 12 mm Tris-HCl, pH 7.5, 120 mm NaCl, and 0.1 mm EDTA in the presence of 1 mm dithiothreitol plus 7.5 mm β-mercaptoethanol. A French-press (AIM-AMINCO®, Spectronic Instruments) was used to disrupt the cells at 12,000 p.s.i. The lysis solution was centrifuged, and the debris was discarded. The cell-free extract was loaded onto a Ni-NTA column, which was pre-equilibrated with the lysis buffer. After washing the column exhaustively with the lysis buffer plus 5 mm imidazole, the column was eluted with the lysis buffer plus 300 mm imidazole. The eluant was concentrated to ∼1 mg/ml for SDS-PAGE analysis to check the existence of the protease (the tag-cleaved protease by autoprocessing cannot be obtained after Ni-NTA column chromatography).
Kinetics of Maturation Assayed by SDS-PAGE—The processing reactions at the N and C termini of the tagged C145A mutant were followed by SDS-PAGE analysis to monitor the degradation of the substrate and the formation of the products with time. The reaction mixture containing Trx-10aa-C145A-10aa-GST (5 μm) and the active protease (0.5 μm) was incubated at 25 °C for 10, 100, 500, 1000, or 1500 min in 20 mm Bis-Tris (pH 7.0), and the reaction was terminated by 100 μm ZnCl2 (a known SARS 3CLpro inhibitor) and subject to SDS-PAGE analysis, which allowed the resolution of substrate and products.
Analytical Ultracentrifuge Experiments—Wild-type and C145A mutant SARS-CoV 3CLpro with extra N- or C-terminal amino acids at a concentration of 1 mg/ml (∼14.3 μm of dimer) were used for the AUC analysis of AUC with the buffer 12 mm Tris-HCl, pH 7.5, 120 mm NaCl, 0.1 mm EDTA, 1 mm dithiothreitol, and 7.5 mm β-mercaptoethanol. Sedimentation coefficients (s values) were estimated by a Beckman-Coulter XL-A analytical ultracentrifuge with an An60 Ti rotor as described before (26). Sedimentation velocity analysis was performed at 40,000 rpm at 25 °C with standard double sector aluminum centerpieces. The UV absorption of the cells was scanned every 5 min for 4 h. Data were analyzed with the SedFit program (version 8.7). The Sednterp program (version 1.07) was used to obtain solvent density, viscosity, and Stokes radius (R s). The Sedphat program (version 1.5b) was used to obtain the dimer-monomer equilibrium dissociation constant of all proteins tested.
RESULTS
Overall Structure of the SARS-CoV Wild-Type 3CLpro—Data collection and refinement statistics for the wild-type and C145A 3CLpro are summarized in Table I . The wild-type crystal belongs to the P21212 space group with one monomer in an asymmetric unit so that the two monomers in the dimer are symmetric (Fig. 1A ). Like the previous 3CLpro structures from HCoV 229E, TGEV, and SARS-CoV (11, 12, 13), our SARS-CoV 3CLpro structure is composed of three domains, including a chymotrypsin-like fold for domain I (residue 1-101) and antiparallel β-barrel for domain II (residue 102-184). The active site is located between domains I and II. The six C-terminal residues in the wild-type protease are flexible (shown below) in contrast to those of the C145A structure. The sequences surrounding the N- and C-terminal cutting sites of 3CLpro in different coronaviruses are shown in Supplemental Fig. 1S.
Table I.
Data collection and refinement statistics for wild-type and C145A 3CLpro
| Wild type | C145A | |
|---|---|---|
| Data collection statistics | ||
| Beamline | BL17B2, NSRRC | BL12B2, SPring-8 |
| Wavelength (Å) | 1.12710 | 1.00000 |
| Space group | P21212 | C2 |
| Unit cell dimensions (Å) | 107.2, 45.1, 54.0 | 125.7, 80.2, 63.9, β = 92.5° |
| Resolution range (Å)a | 30−2.8 (2.9−2.8) | 50−2.8 (2.9−2.8) |
| Total observations | 45,430 | 60,899 |
| Unique observations | 6,711 | 17,504 |
| Completeness (%) | 97.9 (100) | 99.7 (99.7) |
| Rmerge (%)b | 3.8 (7.6) | 8.1 (37.1) |
| I/σ(I) | 40.3 (25.4) | 16.8 (3.7) |
| Refinement statistics | ||
| Reflections | 6,608 (F > 2σ) | 13,891 (F > 2σ) |
| Rcryst/Rfree (%)c | 24.15/28.82 | 20.13/27.45 |
| Room mean square deviation from ideal geometry | ||
| Bonds (Å) | 0.007 | 0.007 |
| Angles (°) | 1.40 | 1.44 |
| No. of non-hydrogen atoms/average B factor, Å2 | ||
| Protein/main chain | 1,204/53.8 | 2,448/37.3 |
| Protein/side chain | 1,129/55.6 | 2,292/38.7 |
| Water |
141/40.1 |
201/28.5 |
Values in parentheses refer to the highest resolution bin.
, where Ihkl is the integrated intensity of a given reflection.
, where Fo and Fc are observed and calculated structure factors.
Fig. 1.

Crystal structures of the wild-type and C145 mutant 3CLpro. In A and B, the overall three-dimensional structures of wild-type and C145A mutant 3CLpro are shown as ribbons (protomer A is green and protomer B is blue), and the “product” in the active site cleft between domain I and II is shown in yellow. In C, the dimer structure is composed of protomer A (shown with a solid tube in green) and protomer B (shown with charge potentials). The C terminus of protomer B′ (shown in cyan) in another asymmetric unit is intercalated into the active site of protomer B. D, an enlarged view of C near the active site, showing the C-terminal amino acids of protomer B′ as well as the N-terminal amino acids of protomer A in the neighborhood of the active site of protomer B.
Overall Structure of SARS-CoV C145A 3CLpro—The 3CLpro (C145A) crystal belongs to the C2 space group with one dimer in the asymmetric unit, so the two monomers in a dimer are not identical (Fig. 1B). The two protomers of the dimeric C145A, denoted “A” and “B,” are oriented perpendicular to each other, and each protomer contains three domains as in the wild type. A novel finding here is that the active site of protomer B is intercalated with the C-terminal residues 301-306 of protomer B′ (shown as a cyan ribbon in Fig. 1C and as a cyan stick in Fig. 1D) from the dimer in another asymmetric unit. The N-terminal residues of protomer A (shown as a green ribbon in Fig. 1C and as a green stick in Fig. 1D) are located near the active site of protomer B. This structure reveals the way in which the product is bound in the active site during the maturation process, and the six amino acids at the C terminus of protomer B′ represent the P6 to P1 sites of the autoprocessed product. The inter- and intra-interactions of SARS 3CLpro dimer are shown in Table II .
Table II.
Inter- and intra-interactions of SARS 3CLpro dimers
Atom pairs in the interacting amino acid residues with contact distances <3.5 Å are listed.
| Residue 1 | Atom 1 | Residue 2 | Atom 2 | Distance |
|---|---|---|---|---|
| Å | ||||
| Interactions of N- terminus from protomer A | ||||
| A Ser1 | Oγ[ρ] | B Glu166 | Oϵ2 | 2.56 |
| A Ser1 | N | B Glu166 | Oϵ1 | 2.48 |
| A Ser1 | N | B His172 | N | 2.83 |
| A Gly2 | O | B Ser139 | Oγ[ρ] | 2.90 |
| A Lys5 | N | A Glu290 | Oϵ1 | 2.53 |
| A Ala7 | N | B Val125 | O | 3.01 |
| A Ala7 | O | B Val125 | N | 2.74 |
| Interactions of N- terminus from protomer B | ||||
| B Ser1 | N | A Glu166 | Oϵ1 | 2.95 |
| B Ser1 | N | A His172 | N | 3.16 |
| B Ala7 | N | A Val125 | O | 3.04 |
| B Ala7 | O | A Val125 | N | 2.80 |
| Interactions of C- terminus from protomer A | ||||
| A Ser301 | N | A Val297 | N | 2.62 |
| A Val303 | O | B Ser123 | Oγ[ρ] | 3.53 |
| A Gln306 |
Oϵ1 |
A Lys12 |
Nζ[ρ] |
3.19 |
Autoprocessing of Tagged SARS-CoV 3CLpro during Lysate Preparation—To further understand autoprocessing, we generated the six 3CLpro constructs listed in Fig. 2A . During the cell lysate preparation of Trx-10aa-WT (construct 1; here Trx refers to Trx-6×His-FXa site), the protein underwent autoprocessing to yield mature SARS protease by self-cleaving the 10 amino acids preceding the N terminus of mature 3CLpro. The processed protease failed to bind to the Ni-NTA column, because the His tag was removed by autoprocessing. The SDS-PAGE (Fig. 2B) shows no band at ∼50 kDa (Trx plus 3CLpro) after Ni-NTA column chromatography for this construct (lane 1). The same result was observed for construct 3, which contained cleavage sites in both N and C termini (lane 3, Fig. 2B), because it was also autoprocessed. In contrast, construct 2 without the cleavage site in the N terminus was retained in the Ni-NTA column (lane 2, Fig. 2B). Constructs 4, 5, and 6 of mutant C145A all yielded a purified band in lanes 4-6 on SDS-PAGE (Fig. 2B), because the C145A mutation prevented autoprocessing.
Fig. 2.
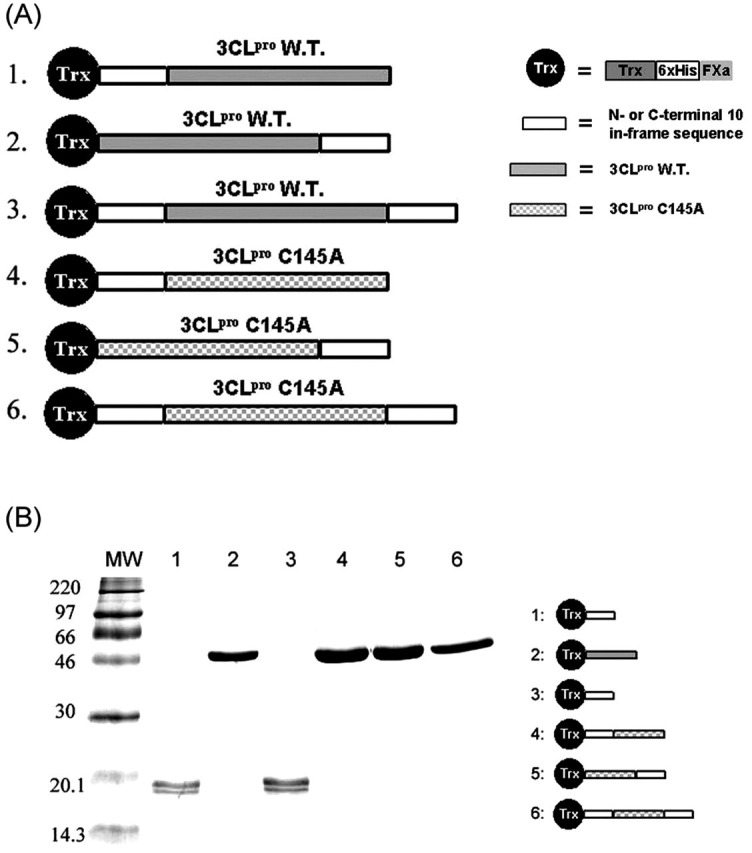
SDS-PAGE analysis of the maturation of SARS-CoV recombinant proteases. In A, the six constructs of the recombinant protease are listed, including the wild-type or C145A mutant enzyme containing N- and/or C-terminal 10 additional amino acids and the N-terminal Trx tag. In B, MW represents the molecular weight markers. Lanes 1 and 3 represent 300 mm imidazole eluant from Ni-NTA column of constructs 1 and 3, respectively, where the protease disappeared, because it underwent autoprocessing and lost the tags. Lanes 2, 4, 5, and 6 represent the protease of constructs 2, 4, 5, and 6, respectively, eluted by 300 mm imidazole from the Ni-NTA column. They either lack the N-terminal cleavage sequence or contain C145A mutant, so they were retained in the Ni-NTA column before elution.
Facilitated Processing—We prepared the inactive protein Trx-10aa-C145A-10aa-GST (5 μm) with tags to examine its processing by using 0.5 μm active 3CLpro. The processed products can be easily resolved from the substrate on SDS-PAGE. As shown in Fig. 3 , the N-terminal Trx tag was cleaved first, followed by cleavage of the C-terminal GST. The bands were assigned to the fragments shown in the right panel. The band (∼34 kDa) corresponding to the added 3CLpro appeared in every lane except in the substrate-only lane, which contained no enzyme. The time course data in Fig. 3B show that the initial rate for the formation of Trx tag (N-terminal cleavage) and GST tag (C-terminal cleavage) is 0.22 and 0.004 μm/min, respectively. Thus, the N terminus is processed much faster (55-fold) than the C-terminal end.
Fig. 3.

Facilitated processing of Trx-10aa-C145A-10aa-GST by the active 3CL protease.A, the inactive C145A (to prevent autoactivation) containing N- and C-terminal tags was prepared as a substrate for active 3CLpro. The substrate protein (lane S) was treated with 1/10 of wild-type 3CLpro (lane E), and the products were monitored with SDS-PAGE analysis after specified periods of incubation time shown on the top of the figure. According to the SDS-PAGE data, the N-terminal tag was digested followed by the C-terminal tag cleavage, as indicated by the product fragments on the right panel. B, the time course of formation of Trx tag (▴) and GST tag (□) from facilitated processing.
AUC Analysis of Wild-type and Mutant Proteases—We then utilized AUC to examine the quaternary structures of 10aa-C145A, C145A-10aa, and the wild-type 3CLpro and compare their dimer Kd values. The AUC data for the wild-type SARS protease as shown in Fig. 4 indicate that the determined molecular weight is that of a dimer and the dimeric wild-type protein has a Kd of 0.35 nm. In contrast, 10aa-C145A and C145A-10aa, which contain N- or C-terminal 10 extra amino acids, shows 49- and 16-fold larger Kd values (17.2 nm and 5.6 nm), respectively. Even with only 10 extra amino acids in the N- or C terminus, dimer formation is inhibited with the N-terminal having a larger impact. The sub-micromolar Kd value of the tagged protease indicates that immature 3CLpro can form a small amount of dimer enabling it to undergo autoprocessing to yield the mature enzyme, which further serves as a seed for facilitated maturation.
Fig. 4.
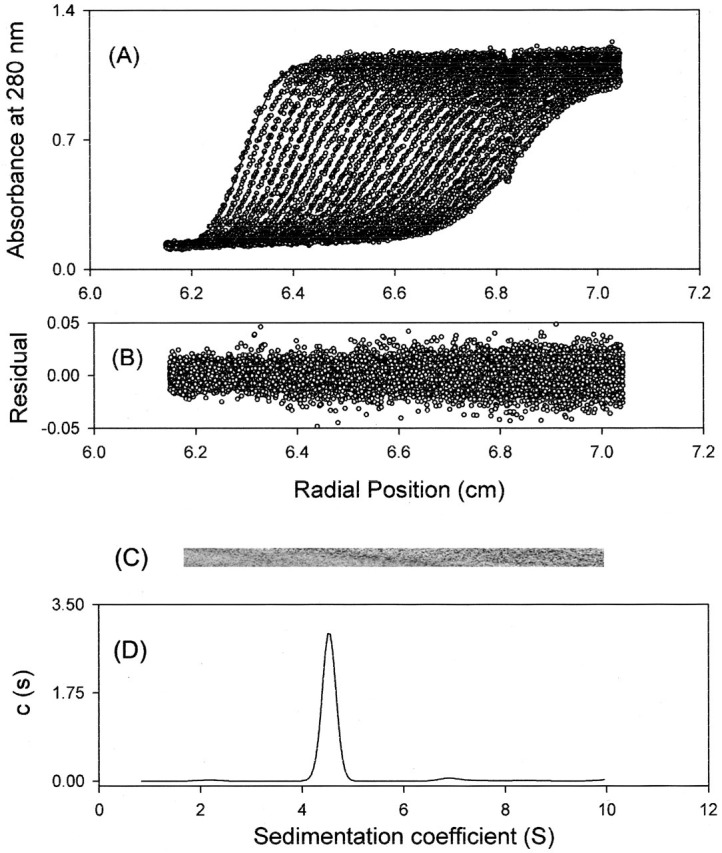
AUC experiments of wild-type SARS 3CLpro. Shown here is an example of using AUC to measure the Kd of the wild-type protease dimer-monomer equilibrium. The A280 absorbance of the protein as a function of radius and time was recorded to calculate the sedimentation coefficient and the Kd value A, circles represent the experimental data, and the lines are the computer-generated results from fitting the data to the Lamm equation with the SedFit program. B, fitting residuals plotted as a function of radial position. C, grayscale of the residual bitmap. The randomly distributed residuals and bitmap show the quality of the data fitting. D, continuous sedimentation coefficient of wild-type 3CLpro derived from the data shown in A.
Processing Intermediate-like, Product-bound C145A Structure—The P1-P6 peptide from protomer B′ shown in the omit map occupies the active site of C145A protomer B (Fig. 5A ). The detailed molecular contacts of the peptide with the active site amino acids are shown in Fig. 5B. In the S1 site, the side-chain Oϵ1 of Gln306 (P1) forms a hydrogen bond with side chain Nϵ2 of His163. The side-chain Nϵ2 of P1-Gln donates an H-bond to the side-chain carbonyl of Glu166. Moreover, the oxygen anion at the free carboxylate end of P1-Gln forms H-bonds with the backbone N atoms of Gly143. Carbon atoms of P1-Gln interact with His41 and Ala145 by hydrophobic interactions. If Ala145 is replaced by Cys using computer modeling (shown in blue) to generate the active form, the Sγ atom of Cys145 will interact with the P1 carboxyl group (Fig. 5B).
Fig. 5.
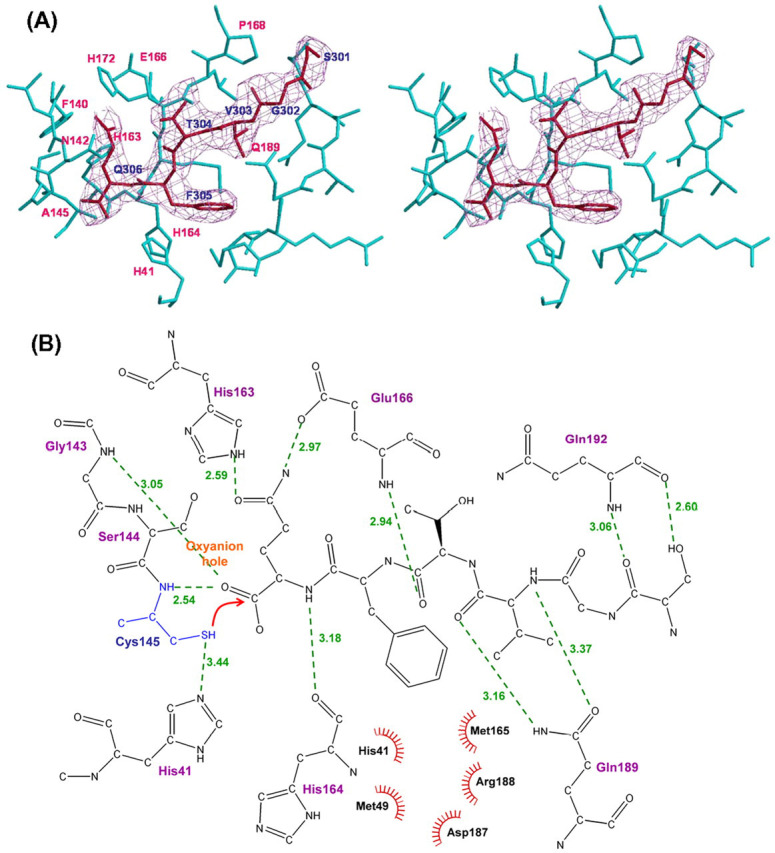
Molecular interactions of the active site residues of protomer B with the C-terminal residues of protomer B′.A, stereo view of the electron density map of the C-terminal region (red stick) of protomer B′ bound in the S pockets (cyan stick) of protomer B. B, details of the molecular interactions between the active site S1-S6 pockets of protomer B and the C-terminal residues of protomer B′. H-bonds are shown as green broken lines.
Residues 140-145 and 163-166 form the “outer wall” of the S1 site. The S2 site of C145A is formed by the main-chain atoms of Val186, Asp187, Arg188, and Gln189 as well as the side-chain atoms of His41, Met49, and Met165, suggesting that the P2 site prefers a bulky side chain such as Val, Leu, or Phe. The N atom in the main chain of P2-Phe interacts with the O atom of His164, and the side chain interacts with Met49, Met165, Asp187, and Arg188 through hydrophobic contacts. Residues 186-188 line the S2 subsite with some of their main-chain atoms. The side chain of P3-Thr is oriented toward bulk solvent. The O atom of the Thr accepts a H-bond (2.9 Å) from the N atom of Glu166. Residues Met165, Leu167, Ser189, Thr190, and Gln192 surround the S4 subsite, which also favors a hydrophobic side chain. The O atom of P4-Val accepts a 3.1-Å H-bond from the Nϵ2 atom of Gln189 and the N atom of the Val donates a H-bond to the Oϵ1 atom of Gln189. The side chain of P4-Val interacts with Met165 and Gln189 via hydrophobic interactions. The S5 subsite is composed of main-chain atoms of Thr190, Ala191, and Gln192. P5-Gly is not in contact with the protease. The S6 site is almost positioned at the outer area of the protein. However, the O atom and Oγ of P6-Ser still interact with the backbone N and O atoms of Gln192.
Comparison of 3CLpro Structures—The conformations of the apo form (protomer A, blue) and the product-bound form (protomer B, cyan) of the C145A active sites are compared in Fig. 6A . We also superimposed the previously solved crystal structure of chain A of SARS 3CLpro complexed with a substrate-like chloromethyl ketone irreversible inhibitor (coded 1UK4), in which the bound inhibitor and water molecules were removed (shown in green), and chain B without inhibitor bound (shown in gold). The crystal structures of 3CLpro of HCoV 229E (1P9S, crimson) and TGEV (1P9U, with inhibitor; pink) were also superimposed. For SARS 3CLpro, the positions of the active-site residues of C145A protomer B (product-bound form) and those of 1UK4 chain A (inhibitor-bound form) are similar. However, C145A protomer A (free form) has slightly different conformation, in which Thr190 is displaced from the active site so that the pocket hole is larger than the product-bound form. In comparison with the active site pocket of 3CLpro of other coronaviruses, the size is TGEV > SARS-CoV > HCoV-229E.
Fig. 6.

Superposition of 3CLpro active sites and inhibitors.A, superimposition of the active site of five 3CLpro protease structures: cyan and blue, protomer A and B of C145A; light green and gold, protomer A and B of the wild type, respectively; crimson, HCoV 229E 3CLpro (1P9S); and pink, TGEV 3CLpro (1P9U). B, superimposition of the six C-terminal residues of SARS 3CLpro (SGVTFQ) (cyan), the inhibitor of TGEV 3CLpro (1P9U, pink), the inhibitor of TGEV 3CLpro (VNSTLQ) at the active site of SARS 3CLpro (1UK4, gold), and the inhibitor of Rhinovirus 3CLpro, AG7088 at the active site (1CQQ, green).
So far, five 3CLpro inhibitor/product-bound crystal structures have been reported. As shown in Fig. 6B, the structure of the six C-terminal residues QFTVGS of SARS 3CLpro (shown in cyan) superimposes well on that of the inhibitor QLTSNV (shown in pink) of TGEV 3CLpro reported previously (12) except for P5 and P6 and inhibitor QLTSNV (shown in gold) in the active site of 1UK4 protomer B beyond the P1 site. Otherwise, AG7088, an effective inhibitor of Rhinovirus 3C protease (1CQQ) (27), has been suggested to be a potential inhibitor for SARS-CoV 3CLpro (12). Therefore we superimposed AG7088 (shown in green), which spans the P1-P2-P3 sites, with the six C-terminal residues of protomer B′. However, the alignment is poor (Fig. 6B). In our enzymatic assay, AG7088 at the maximal tested concentration (100 μm) did not inhibit SARS-CoV 3CLpro (data not shown).
DISCUSSION
Herein, we report clear electron densities for the C-terminal six amino acids of SARS 3CLpro protomer B′, which insert into the active site of the nearby symmetry-related protomer B. The product-bound structure provides the first evidence of an intermediate during SARS viral protease maturation. SARS 3CLpro has its N and C termini both close to the active site, providing clear guidance for designing a novel inhibitor to block the viral maturation before the mature dimer is formed. In a dimer, the N-terminal amino acids (also called the N-finger) have many specific interactions with domains II and III of the parent monomer and domain III of the other monomer. From the crystal structure of TGEV Mpro, it had been suggested that, after in trans autocleavage, the N terminus of one monomer slides over the active site of the other monomer and adopts a position at the edge of the active site (12). That report also hypothesized that the replication complex could be anchored to the membrane in an uncleaved form, and later, when the precursor proteins accumulate to high concentrations at particular region, the 3CL protease could release itself by intermolecular cleavage, thereby triggering the trans-processing reaction. Our structural data enable the maturation process to be better understood. It is likely that the main protease forms a dimer after autocleavage that immediately enables the catalytic site to act on other cleavage sites in the polyprotein.
Our AUC data show that wild-type SARS 3CLpro displays a remarkably small dimer Kd and that the extra amino acids in the N or C terminus cause a significant increase in the Kd value. This is consistent with the previous AUC study that the full-length SARS 3CLpro containing an un-natural C-terminal hexa-His tag that yielded a Kd = 89 nm at pH 7.6 (28). Therefore, the wild-type enzyme containing the N- or C-terminal tags probably still forms a small amount of the active dimer, which undergoes autoactivation during cell lysate preparation (see Fig. 7 as explained below). This does not happen for the inactive mutant C145A protease with extra amino acids of the N and C termini. The kinetics of autoprocessing determined from SDS-PAGE shows that the N-terminal cleavage occurs before the C-terminal cleavage. This is consistent with observations that a short peptide with the N-terminal cleavage sequence is a better substrate than one with the C-terminal cleavage sites determined using an high-performance liquid chromatography assay (10). After autoprocessing, the newly formed active dimeric SARS main protease can serve as a seed for further chain reaction to activate the premature protease and the other proteins. In vitro autoactivation had also been observed for a number of proteases such as caspase and cathepsin K (29, 30).
Fig. 7.

Proposed scheme of SARS 3CLpro maturation. Two polyproteins are shown in pink and cyan, each with three domains (I, II, and III). The maturation processing is composed of Step 1: polyprotein (cyan) approaches a second polyprotein (pink) and inserts its N terminus into the active site to be cleaved; Step 2: the N terminus of the uncleaved polyprotein (pink) then inserts its N terminus into the active site for processing; Step 3: after N-terminal processing, the polyprotein with the N terminus flips over to its new position from the active site to form a premature dimer; Step 4: the C terminus of the partially digested polyprotein in the premature dimer is inserted into the active site of another immature dimer to be cleaved and finally the mature dimer is formed.
A neighboring protomer can catalyze the autoprocessing, as indicated by the crystal structure of C145A in a processing intermediate-like product-bound form (Fig. 1), and the processing rate at the N terminus is faster than that at the C terminus (Fig. 3). Moreover, at the mature 3CLpro dimer the N terminus of one protomer is close to the active site near domain II of the other protomer (Fig. 1D). These findings allow us to develop a hypothesis of SARS 3CLpro maturation. As illustrated in Fig. 7, with three domains in the protease represented as I, II, and III, and with domain I at the N terminus and domain III at the C terminus, the maturation processing can be divided into four steps. Step 1: The N-terminal tail of one polyprotein (cyan) approaches the active site of the other polyprotein (pink) to be cleaved. Because the monomeric protease is inactive (10), the intra-chain processing for maturation is impossible. Step 2: The N-terminal tail of the polyprotein (pink) is cleaved by the partially cleaved polyprotein (cyan). Step 3: After N-terminal cleavage, the polyprotein (cyan) can flip over to its position in the immature dimer. A shift of ∼10.3 Å of the N terminus is estimated from the distance between the bound P1 carboxylate in the C145A structure and the N terminus of the mature wild type, as shown in Fig. 1D. Step 4: The C-terminal tail cleavage then follows by inserting the C-terminal tail into the neighboring immature dimer (resembling the product-bound form of C145A), and then the mature dimer is formed after cleavage.
The crystal structure of C145A in a product-bound form presented here not only suggests the mechanism for maturation, but also guided the structure-based protease inhibitor design. AG7088 did not inhibit SARS protease because the structural features of the active site pocket of SARS 3CLpro differ from those of Rhinovirus 3CLpro. Thr142 of Rhinovirus 3CLpro is absent from SARS 3CLpro, so that there cannot be effective hydrogen bonding with the lactam moiety at P1 site. Because both of the N and C termini are close to the active site of the SARS-CoV 3CLpro according to the new structure of C145A in a product-bound form, inhibitors targeting the dimer interface may block the protease maturation. In fact, several drugs targeting the viral protease interface were in different phases of clinical trials (31). Our new structure of the C145A mutant thus provides the template for design of the inhibitors to block the maturation process, which is ongoing in our laboratories.
Footnotes
The abbreviations used are: SARS, severe acute respiratory syndrome; SARS-CoV, SARS-coronavirus; TGEV, transmissible gastroenteritis virus; 3CLpro, 3C-like protease; Mpro, main protease; Trx, thioredoxin; GST, glutathione S-transferase; Dabcyl, 4-(4-dimethylaminophenylazo)benzoic acid; Edans, 5-[(2-aminoethyl)amino]naphthalene-1-sulfonic acid; FXa, factor Xa; Ni-NTA, nickel nitrilotriacetic acid; AUC, analytical ultracentrifuge; IC50, 50% inhibitory concentration; Bicine, N,N-bis(2-hydroxyethyl)glycine; Bis-Tris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; HCoV, human coronavirus; aa, amino acid(s).
Supplementary Material
References
- 1.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Fouchier R.A., Kuiken T., Schutten M., van Amerongen G., van Doornum G.J., van den Hoogen B.G., Peiris M., Lim W., Stohr K., Osterhaus A.D. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 6.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 7.Ruan Y.J., Wei C.L., Ee A.L., Vega V.B., Thoreau H., Su S.T., Chia J.M., Ng P., Chiu K.P., Lim L., Zhang T., Peng C.K., Lin E.O., Lee N.M., Yee S.L., Ng L.F., Chee R.E., Stanton L.W., Long P.M., Liu E.T. Lancet. 2003;361:1779–1785. doi: 10.1016/S0140-6736(03)13414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorbalenya A.E., Donchenko A.P., Blinov V.M., Koonin E.V. FEBS Lett. 1989;243:103–114. doi: 10.1016/0014-5793(89)80109-7. [DOI] [PubMed] [Google Scholar]
- 9.Hegyi A., Ziebuhr J. J. Gen. Virol. 2002;83:595–599. doi: 10.1099/0022-1317-83-3-595. [DOI] [PubMed] [Google Scholar]
- 10.Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J., Lai L. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 13.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi J., Wei Z., Song J. J. Biol. Chem. 2004;279:24765–24773. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu J.T., Kuo C.J., Hsieh H.P., Wang Y.C., Huang K.K., Lin C.P., Huang P.F., Chen X., Liang P.H. FEBS Lett. 2004;574:116–120. doi: 10.1016/j.febslet.2004.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuo C.J., Chi Y.H., Hsu J.T., Liang P.H. Biochem. Biophys. Res. Commun. 2004;318:862–867. doi: 10.1016/j.bbrc.2004.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu C.Y., Jan J.T., Ma S.H., Kuo C.J., Juan H.F., Cheng Y.S., Hsu H.H., Huang H.C., Wu D., Brik A., Liang F.S., Liu R.S., Fang J.M., Chen S.T., Liang P.H., Wong C.H. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Biochemistry. 2004;43:4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 19.Kao R.Y., Tsui W.H., Lee T.S., Tanner J.A., Watt R.M., Huang J.D., Hu L., Chen G., Chen Z., Zhang L., He T., Chan K.H., Tse H., To A.P., Ng L.W., Wong B.C., Tsoi H.W., Yang D., Ho D.D., Yuen K.Y. Chem. Biol. 2004;11:1293–1299. doi: 10.1016/j.chembiol.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanchard J.E., Elowe N.H., Huitema C., Fortin P.D., Cechetto J.D., Eltis L.D., Brown E.D. Chem. Biol. 2004;11:1445–1453. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jain R.P., Pettersson H.I., Zhang J., Aull K.D., Fortin P.D., Huitema C., Eltis L.D., Parrish J.C., James M.N., Wishart D.S., Vederas J.C. J. Med. Chem. 2004;47:6113–6116. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- 22.Otwinowski Z., Minor W. In: Processing of X-ray Diffraction Data Collected in Oscillation Mode. Sweet R.M., editor. Academic; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 23.Collaborative Computational Project, Number 4 Acta Crystallogr D Biol. Crystallogr. 1994;50:760–763. [Google Scholar]
- 24.McRee D.E. J. Struct. Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 25.Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., Read R.J., Rice L.M., Simonson T., Warren G.L. Acta Crystallogr D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 26.Chang H.C., Chang G.G. J. Biol. Chem. 2003;278:23996–24002. doi: 10.1074/jbc.M213242200. [DOI] [PubMed] [Google Scholar]
- 27.Matthews D.A., Dragovich P.S., Webber S.E., Fuhrman S.A., Patick A.K., Zalman L.S., Hendrickson T.F., Love R.A., Prins T.J., Marakovits J.T., Zhou R., Tikhe J., Ford C.E., Meador J.W., Ferre R.A., Brown E.L., Binford S.L., Brothers M.A., DeLisle D.M., Worland S.T. Proc. Natl. Acad. Sci. U. S. A. 1999;96:11000–11007. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chou C.Y., Chang H.C., Hsu W.C., Lin T.Z., Lin C.H., Chang G.G. Biochemistry. 2004;43:14958–14970. doi: 10.1021/bi0490237. [DOI] [PubMed] [Google Scholar]
- 29.McQueney M.S., Amegadzie B.Y., D'Alessio K., Hanning C.R., McLaughlin M.M., McNulty D., Carr S.A., Ijames C., Kurdyla J., Jones C.S. J. Biol. Chem. 1997;272:13955–13960. doi: 10.1074/jbc.272.21.13955. [DOI] [PubMed] [Google Scholar]
- 30.Rotonda J., Nicholson D.W., Fazil K.M., Gallant M., Gareau Y., Labelle M., Peterson E.P., Rasper D.M., Ruel R., Vaillancourt J.P., Thornberry N.A., Becker J.W. Nat. Struct. Biol. 1996;3:619–625. doi: 10.1038/nsb0796-619. [DOI] [PubMed] [Google Scholar]
- 31.De Clercq E. Nat. Rev. Drug. Discov. 2002;1:13–25. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.