

Summary
The formicamycins are promising antibiotics first identified in Streptomyces formicae KY5, which produces the compounds at low levels. Here, we show that by understanding the regulation of the for biosynthetic gene cluster (BGC), we can rewire the BGC to increase production levels. The for BGC consists of 24 genes expressed on nine transcripts. The MarR regulator ForJ represses expression of seven transcripts encoding the major biosynthetic genes as well as the ForGF two-component system that initiates biosynthesis. We show that overexpression of forGF in a ΔforJ background increases formicamycin production 10-fold compared with the wild-type. De-repression, by deleting forJ, also switches on biosynthesis in liquid culture and induces the production of additional, previously unreported formicamycin congeners. Furthermore, combining de-repression with mutations in biosynthetic genes leads to biosynthesis of additional bioactive precursors.
Keywords: natural products, Streptomyces, antibiotics, regulation, biosynthesis, formicamycins, fasamycins, two-component system
Graphical abstract
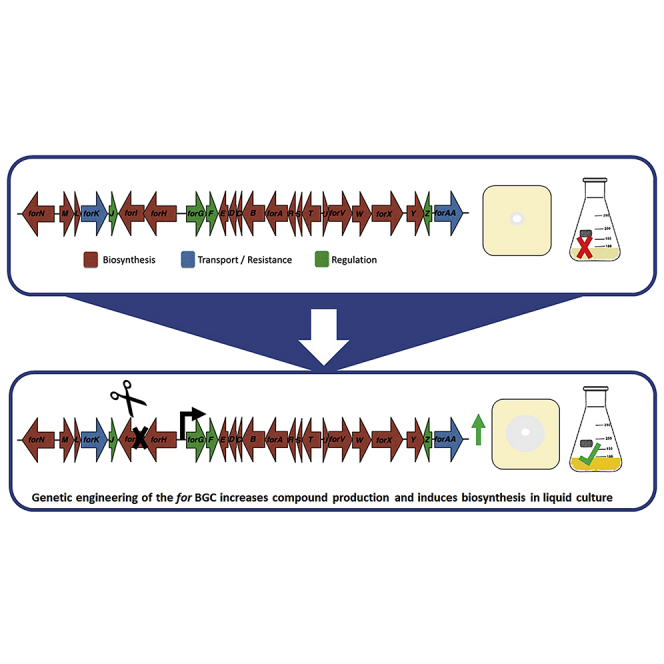
Highlights
-
•
Formicamycin biosynthesis requires 24 genes expressed on nine transcripts
-
•
Deleting the MarR regulator ForJ increases formicamycin biosynthesis
-
•
De-repressing formicamycin biosynthesis induces production in liquid culture
-
•
Re-wiring regulation and biosynthesis results in the production of new congeners
Formicamycins are promising antibiotics with activity against drug-resistant pathogens. Here, Devine et al. show formicamycin biosynthesis is repressed by a MarR-family regulator and activated by a two-component system. Using CRISPR/Cas9 genetic engineering, Devine et al. have re-wired formicamycin biosynthesis to increase titers and enable further development of these promising compounds.
Introduction
Almost half of all known antibiotics are derived from the specialized metabolites of filamentous actinomycetes, particularly Streptomyces species, many of which were discovered during the golden age of antibiotic discovery that occurred between 1940 and 1960 (Kämpfer et al., 2014). Since this time, few new classes of antibiotics have been introduced into the clinic, and increasing problems of antimicrobial resistance pose a significant threat to modern medicine. Many synthetic antibiotics have failed to progress through clinical trials, so interest has returned to natural products (Hutchings et al., 2019). Streptomyces are primarily known as soil bacteria; however, their ability to produce antibiotics makes them competitive in a wide range of environments. By searching under-explored environments and ecosystems, new species can be isolated that may produce useful specialized metabolites (Devine et al., 2017). Furthermore, advances in genomic techniques has revealed that bacterial and fungal species encode many more biosynthetic gene clusters (BGCs) than previously thought, with only around 10% being expressed under laboratory conditions, meaning many more specialized metabolites remain to be discovered from their cryptic BGCs. Isolating bacteria from under-explored environmental niches and mining their genomes for novel BGCs is therefore a promising route toward antibiotic development (Genilloud, 2019). Following this hypothesis, we previously isolated a number of actinomycetes from the domatia of the African fungus-growing plant-ant Tetraponera penzigi, including Streptomyces formicae KY5 (Seipke et al., 2013). The genome of S. formicae encodes numerous specialized metabolite BGCs, including a type 2 polyketide synthase (PKS) BGC that is responsible for the biosynthesis of formicamycins (Holmes et al., 2018). These antibiotics are potent inhibitors of vancomycin-resistant enterococci (VRE) and methicillin-resistant Staphylococcus aureus (MRSA), with no resistance observed in vitro (Qin et al., 2017).
Previous work on the formicamycin (for) BGC has shown that the encoded biosynthetic pathway makes two distinct families of compounds in addition to the formicamycins. The fasamycins are biosynthetic precursors of the formicamycins that also exhibit antibacterial activity; they have been isolated from a number of actinomycete strains and given the alternative names accramycins, naphthacemycins, and streptovertimycins (Feng et al., 2012; Maglangit et al., 2019; Huo et al., 2020; Yang et al., 2020). In addition to the fasamycins, the formicapyridines are shunt metabolites produced when the cyclization stage of the biosynthetic pathway is derailed (Qin et al., 2017, 2019). Conversion of fasamycin precursors into formicamycins involves a unique two-step ring-expansion, ring-contraction pathway that proceeds via a Baeyer-Villigerase-derived lactone intermediate that undergoes a unique reduction Favorskii-like rearrangement (Qin et al., 2020) (Figure 1). In this work, we aimed to understand how S. formicae KY5 regulates the production of formicamycins with the view to refactoring the BGC to produce increased titers of these potentially valuable antibiotics. We show that the for BGC consists of 24 genes expressed on nine transcripts and is controlled by the combined actions of three cluster-situated regulators (CSRs). The MarR-family transcriptional regulator ForJ represses the expression of the majority of the biosynthetic genes, while the two-component system (TCS) ForGF is required to activate formicamycin biosynthesis. A third CSR, the MarR-family regulator ForZ, appears to autorepress its own expression and activate expression of the putative, divergent MFS transporter gene forAA. Deletion of the forGF operon abolished the production of fasamycins and formicamycins in the wild-type strain while deleting forJ increased formicamycin titers approximately 5-fold. Introducing a second copy of forGF into the ΔforJ mutant increased production of formicamycins to approximately 10 times the wild-type levels. De-repression of the BGC by deleting forJ also induced the production of fasamycins and formicamycins in liquid culture, including previously unreported congeners.
Figure 1.

Formicamycin biosynthesis requires 24 genes expressed on nine transcripts
The minimal for BGC contains 24 genes required for formicamycin biosynthesis; red = biosynthetic genes, blue = transporters, green = regulatory genes. Cappable RNA sequencing identified 10 transcription start sites in the for BGC, nine of which are in intergenic regions that likely represent promoter regions for the biosynthetic genes. Formicamycin biosynthesis occurs by the formation of fasamycins through the action of the PKS and associated gene products, including methyltransferases (MTase) and a halogenase. ForX-catalyzed hydroxylation and ring expansion leads to a lactone intermediate that undergoes a reductive ring contraction catalyzed by the flavin-dependent oxidoreductase ForY to yield the formicamycin backbone.
Results
Formicamycin biosynthesis requires 24 genes expressed on nine transcripts
We previously showed that Cas9-mediated deletion of 46 kbp of DNA encompassing the for BGC in S. formicae abolished formicamycin biosynthesis. Production was restored in the S. formicae Δfor strain by introducing the phage-derived artificial chromosome (ePAC) pESAC13-215-G, which carries the entire for BGC plus 40–80 kbp of DNA on each side, thus proving this genomic region encodes biosynthesis of these molecules (Qin et al., 2017). Chromatin immunoprecipitation (ChIP) sequencing and cappable RNA sequencing cc (see below) suggested 24 genes form this cluster, and we confirmed this by deleting genes on each side of this 24-gene cluster contained on pESAC13-215-G and demonstrating their ability to induce formicamycin biosynthesis in the S. formicae Δfor strain. To elucidate the transcriptional organization of the for BGC we mapped the transcription start sites (TSSs) using cappable RNA sequencing (accession number E-MTAB-7975) and identified 10 TSSs, nine of which are in intergenic regions. We thus conclude that the 24 genes required for formicamycin biosynthesis and export are expressed as nine transcripts comprising forN, forMLK, forJ, forHI, forGF, forTSRABCD, forUVWXY, forZ, and forAA. The other TSS is located within the forV coding region and likely maintains the expression levels of forWXY. The forX and forY gene products are required for the unique ring conversion that changes a fasamycin precursor into a formicamycin (Qin et al., 2020) (Figure 1).
Formicamycin biosynthesis is repressed by the MarR-family regulator ForJ and activated by the TCS ForGF
The for BGC encodes three CSRs that were predicted to regulate the expression of the transcripts required for formicamycin biosynthesis and export. There are two putative MarR-family transcriptional regulators encoded by forJ and forZ and a TCS encoded by forGF. To determine the roles of these CSRs, we made CRISPR/Cas9-mediated deletions in each of their coding genes and measured formicamycin biosynthesis in the resulting mutants. Formicamycin and fasamycin biosynthesis were abolished in the ΔforGF mutant, whereas ectopic expression of an additional copy of forGF from the native promoter led to an almost doubling (1.9-fold increase) in formicamycin biosynthesis when grown in solid culture. The loss of production caused by deletion of forGF was rescued by ectopic expression of forGF. In contrast, formicamycin production increased 5-fold in the ΔforJ mutant compared with the wild-type, with the levels of fasamycin precursors increasing more than 28-fold. This suggests that the conversion of a fasamycin to a formicamycin is the limiting step in this biosynthetic pathway (the absolute titers of fasamycins are generally lower than those of the formicamycins). Overall, the combined production of fasamycin and formicamycin metabolites increased 6.7-fold in the ΔforJ mutant grown in solid culture. Introduction of forJ cloned downstream of either the putative forJ promoter or the constitutive ermE∗ promoter failed to complement the ΔforJ mutant for reasons we cannot explain, but overexpression of forJ under control of the ermE∗ promoter in the wild-type strain significantly reduced fasamycin and formicamycin biosynthesis. Deletion of forZ reduced formicamycin biosynthesis to approximately 60% of the wild-type levels (~70% of total metabolites). Taken together, these results show that formicamycin biosynthesis is activated by ForGF and repressed by ForJ, while ForZ may be involved in activation of formicamycin biosynthesis but is not absolutely required and most likely regulates the divergent transporter gene forAA. We then deleted both forJ and forGF in combination and this resulted in a mutant that over-produced formicamycins and accumulated fasamycins, even though loss of forGF was expected to result in biosynthesis being abolished. Furthermore, as noted above, the ectopic expression of a second copy of forJ under a native promoter is enough to almost abolish biosynthesis in the wild-type strain, even in the presence of the activating TCS. These combined data suggest that forJ sits at the top of the for BGC regulatory network as the effects of manipulating the other CSRs are overcome by its activity (Figure 2, Table 1).
Figure 2.

Manipulation of BGC-situated regulators affects formicamycin biosynthesis during solid culture
Deletion of forJ results in overproduction of formicamycins and accumulation of the fasamycin precursors. Deletion of forGF abolishes fasamycin and formicamycin production. Deletion of forJ combined with a second copy of forGF results in 10-fold higher formicamycin production than the wild-type strain. Deletion of forJ can also be combined with mutations in biosynthetic machinery to generate strains that accumulate precursors and intermediates. Deletion of forZ results in a reduction of formicamycin biosynthesis to around 60% of the wild-type strain on solid agar. Manipulation of forJ is enough to overcome any other regulatory mutation. Error bars represent SD across experimental replicates. Values are mean ± SD; Wild-type, n = 16; mutants, n = 3.
Table 1.
Fasamycin and formicamycin production by engineered S. formicae strains on solid soya flour mannitol agar and in liquid soya flour mannitol
| Strain | Fasamycins titer (μM) |
Formicamycins titer (μM) |
Combined titer (μM) |
Fasamycins titer (μM) |
Formicamycins titer (μM) |
Combined titer (μM) |
|---|---|---|---|---|---|---|
| Solid | Liquid | |||||
| Wild-type | 6.5 ± 9.6 | 81.3 ± 21.6 | 87.9 ± 74.2 | 0 | 0 | 0 |
| Wild-type + forJ | 0 | 24.8 ± 4.7 | 24.8 ± 4.7 | 0 | 0 | 0 |
| ΔforJ | 186.2 ± 22.2 | 406.5 ± 42.1 | 592.7 ± 69.4 | 6 ± 0.2 | 624.5 ± 29.4 | 630.5.4 ± 29.6 |
| ΔforJ + forJa | 170.8 ± 15.4 | 558.8 ± 53.5 | 729.6 ± 74.8 | 2.7 ± 0.6 | 657.3 ± 40.7 | 660 ± 41.3 |
| ΔforJ + forJb | 78.0 ± 60.2 | 784.0 ± 18.9 | 862.0 ± 36.6 | 210 ± 36.0 | 423.1 ± 72.4 | 633.1 ± 138.1 |
| Wild-type + forGF | 8.6 ± 0.02 | 155.7 ± 18.7 | 164.3 ± 23.7 | Not tested | not tested | not tested |
| ΔforGF | 0 | 0 | 0 | not tested | not tested | not tested |
| ΔforGF + forGF | 13.1 ± 6.7 | 153.7 ± 73.6 | 165.7 ± 86.1 | not tested | not tested | not tested |
| ΔforJ + forGF | 56.7 ± 52.9 | 814.2 ± 139.8 | 873.4 ± 195.9 | 275.4 ± 11.6 | 759.8 ± 193.8 | 1035.2 ± 212.6 |
| ΔforJ ΔforGF | 76.3 ± 36.8 | 648.1 ± 112.7 | 724.5 ± 147.8 | 242.9 ± 21.6 | 355.3 ± 82.1 | 598.1 ± 85.7 |
| ΔforJ ΔforZ | 38.6 ± 31.2 | 388.5 ± 24.8 | 427.1 ± 31.6 | 516.48 ± 297.4 | 409.1 ± 95.4 | 925.6 ± 341.2 |
| ΔforZ | 11.1 ± 10.6 | 49.8 ± 3.7 | 60.9 ± 19.7 | not tested | not tested | not tested |
| ΔforZ + forZ | 13.6 ± 15.8 | 21.0 ± 15.7 | 31.8 ± 38 | not tested | not tested | not tested |
| ΔforV | 72.8 ± 60.3 | 0 | 72.8 ± 60.3 | 1.3 ± 1.8 | 0 | 1.3 ± 1.8 |
| ΔforJΔforV | 587.5 ± 268.1 | 0 | 587.5 ± 268.1 | 79.4 ± 8.1 | 0 | 79.4 ± 8.1 |
| ΔforX | 42.3 ± 22.6 | 0 | 42.3 ± 22.6 | 2.54 ± 4.6 | 0 | 2.54 ± 4.6 |
| ΔforJΔforX | 782.6 ± 147.8 | 0 | 782.6 ± 147.8 | 274.3 ± 325.4 | 0 | 274.3 ± 325.4 |
Values are mean ± SD; wild-type, n = 16; mutants, n = 3.
forJ under control of the native promoter.
forJ under control of the ermE∗ promoter.
To determine how these CSRs control the expression of the for biosynthetic genes, we generated 3x-FLAG-tagged fusion constructs to complement the deletion mutants and used ChIP sequencing to identify where these CSRs bind across the S. formicae chromosome (accession number E-MTAB-8006). The results show that ForF binds to a single site in the for BGC at the promoter region between the forGF operon and the divergent forHI operon, and therefore likely autoregulates and controls expression of forHI. ForZ binds to a single site between the divergent forZ and forAA genes and likely acts as a typical MarR-family regulator by controlling expression of itself and the MFS transporter gene forAA. ForJ, the apparent master CSR, binds to multiple locations across the BGC. There are binding sites between the divergent forN and forMLK operons, in the intergenic regions between the divergent forHI and forGF operons, and between the forTSRABCDE and forUVWXY operons that encode the majority of the core biosynthetic machinery. These data are consistent with the observation that ForJ represses the biosynthesis of fasamycins and formicamycins by repressing the transcription of most of the biosynthetic genes. ForJ also binds to its own coding region, presumably to autorepress via a roadblock mechanism, as well as binding within the coding region of forE at the end of the long forTSRABCDE transcript. We predict that its likely function here is to act as a roadblock to prevent the RNA polymerase transcribing the forTSRABCDE operon from running into RNA polymerase transcribing forGF since this transcript is required for activation of the for BGC (Figure 3). Only three significant enrichments occurred outside the for BGC: ForF binds upstream of KY5_0375, which encodes a putative NLP/P60 family protein, while ForJ binds upstream of both KY5_3182, which encodes a putative MoxR-type ATPase, and KY5_5812, which encodes a hypothetical protein. The significance of these binding events is not known, and they were not considered further in this study.
Figure 3.

The for CSRs bind to multiple promoter regions within the for BGC
ForJ binds to multiple locations across the for BGC to regulate the expression of the majority of the genes required for formicamycin biosynthesis. ForGF binds to a single promoter within the for BGC to regulate itself and the divergent forHI transcript. ForZ binds a single promoter between itself and the divergent transporter gene forAA and is not predicted to directly regulate biosynthesis of the formicamycins.
To determine the roles of ForGF, ForJ, and ForZ in regulating for BGC expression, we compared the mRNA levels of all for transcripts in the ΔforJ, ΔforGF, and ΔforZ mutants with those in the wild-type strain, with the exception of forN as no suitable primers for qRT-PCR could be found within this relatively short transcript (Figure 4). The results show that levels of all transcripts encoding core biosynthetic machinery were higher in the ΔforJ mutant compared with the wild-type strain, consistent with the hypothesis that binding of ForJ represses the expression of these transcripts. Since the forJ transcript is missing from the ΔforJ mutant, we made a transcriptional fusion between the forJ promoter and gusA (encoding β-glucuronidase [GUS]) and found that GUS activity was 2-fold higher in ΔforJ relative to the wild-type, suggesting ForJ is autorepressed (Table S1). In contrast, levels of the core biosynthetic transcripts were greatly reduced in the ΔforGF mutant compared with the wild-type strain, which is consistent with the fact that this mutant does not make fasamycins or formicamycins. Activity levels of the forG and forH promoters was also reduced in the ΔforGF mutant, suggesting ForGF auto-activates. Interestingly, levels of the forJ transcript were slightly increased in the forGF mutant, suggesting levels of repression from forJ are higher in the absence of activation by forGF. In the ΔforZ mutant, levels of the forAA transcript were decreased more than 3-fold, suggesting that ForZ is required to activate the production of this putative transporter. Levels of some of the transcripts encoding biosynthetic machinery were also reduced in the forZ mutant compared with the wild-type strain, suggesting that forZ may play an indirect role in activating formicamycin biosynthesis (Figure 4). Interestingly, the activity of the forZ promoter was increased in the ΔforZ mutant, suggesting that ForZ may be an example of a dual activator-repressor MarR regulator that activates expression of the divergent transporter while repressing its own transcription. Together these results show that ForJ represses the majority of the core biosynthetic genes by binding to multiple regions across the for BGC. We also propose that ForGF is required to activate the divergent forGF and forHI promoters. The forHI genes encode subunits of the acetyl-CoA carboxylase, which converts acetyl-CoA into malonyl-CoA, the substrate of the for PKS, and are essential for the initiation of formicamycin biosynthesis. We hypothesize that expression of the remaining biosynthetic genes is repressed by ForJ in order to ensure biosynthesis only begins when sufficient levels of malonyl-CoA for formicamycin biosynthesis have been achieved. The results also indicate that ForZ auto-represses while activating the transporter gene forAA to ensure that compounds do not accumulate intracellularly once biosynthesis has been initiated.
Figure 4.

Deletion of forJ results in increased transcription of for genes and deletion of forGF decreases expression
In accordance with observed titers, deletion of forJ results in an increase in the expression of all other for cluster transcripts. Deletion of forGF results in a decrease in expression of all biosynthetic transcripts but an increase in the expression of the repressor gene forJ, thereby inhibiting biosynthesis. In the forZ deletion mutant, expression of the resistance transporter forAA is decreased, suggesting forZ is required to activate its transcription. There is a small decrease in the levels of other transcripts in the forZ mutant, suggesting this regulator may also indirectly influence expression of these genes without binding to their promoters. Error bars represent SD across three biological and two technical experimental replicates.
BGC de-repression results in the production of additional formicamycins and induces biosynthesis in liquid culture
Wild-type S. formicae does not produce fasamycins or formicamycins during liquid culture and production levels on solid agar are low. This limits the scope for further investigation of the antibiotic potential of these compounds since they cannot easily be purified on a large scale. As noted, the ForGF TCS is essential for biosynthesis in the wild-type, while deletion of forJ leads to 5-fold higher production of formicamycins (6.7-fold increase in combined fasamycin and formicamycins) on solid medium. We therefore expressed a second copy of the forGF operon in the ΔforJ strain and found the resulting strain makes 10-fold higher levels of formicamycins on solid culture compared with the wild-type (Figure 2, Table 1). Further analysis of the ΔforJ and ΔforJ + forFG strains gave the surprising result that deletion of forJ induced production of the formicamycins in liquid medium. The ΔforJ mutant produced 1.5-fold more formicamycins when grown in liquid culture compared with on solid culture, although with significantly reduced levels of accumulated fasamycins; this equates to approximately the same overall production level of both sets of metabolites combined, and to an overall 7.1-fold increase in total productivity versus the wild-type strain grown on solid culture. The ΔforJ + forGF mutant was even more impressive, producing 1.6-fold the total levels of metabolites compared with the ΔforJ mutant in liquid culture and 11.8-fold more total metabolites (9.3-fold more formicamycins) than the wild-type strain grown in solid culture. By enabling production of the formicamycins during liquid culture, these mutants will facilitate fermenter-scale production to accelerate their further investigation. Deletion of forJ also induced the production of two additional formicamycin congeners (formicamycins S and R), each carrying five chlorine atoms (Figure 5) (Devine et al., 2020). Previously we had observed a maximum of four chlorination events for any congener. Both molecules exhibited potent antibacterial activity against both methicillin-sensitive S. aureus (MSSA) and MRSA (Table 2).
Figure 5.

Fasamycin and formicamycin congeners isolated from de-repressed for cluster mutants
Deletion of forJ results in accumulation of all previously identified congeners from the for BGC in addition to the production of two additional formicamycin congeners (R and S), which exhibit a unique C4 chlorination (highlighted in blue). De-repressing the for BGC by deleting forJ in the S. formicae ΔforX mutant results in the production of six additional fasamycin congeners (L to Q), which have different chlorination and methylation patterns (highlighted in red) compared with fasamycins C–E produced by the wild-type and S. formicae ΔforX strains. All these congeners displayed potent antibacterial activity against MRSA.
Table 2.
Minimal inhibitory concentration of fasamycin and formicamycin congeners against S. aureus ATCC BAA-1717 (MRSA) and ATCC 6538P (MSSA)
| Minimum inhibitory concentration (μg/mL) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fasamycin |
Formicamycin |
||||||||||
| C | E | L | M | N | O | P | Q | J | R | S | |
| MRSA | 16 | 4 | 2 | 4 | 2 | 2 | 4 | 4 | 4 | 2 | 2 |
| MSSA | 16 | 4 | 2 | 4 | 4 | 4 | 2 | 4 | 4 | 2 | 2 |
Combining mutations in regulatory and biosynthetic genes results in the accumulation of additional fasamycin congeners
Our observations suggested that combining de-repression (deletion of forJ) with deletion of key biosynthetic genes would lead to strains that accumulate elevated levels of pathway intermediates and, potentially, more congeners. We previously showed that the halogenase ForV performs a gatekeeper function, with chlorination of fasamycin intermediates controlling the ability of downstream enzymes to utilize these molecules as substrates and their conversion into the formicamycin skeleton (Qin et al., 2017). We thus constructed an S. formicae ΔforJΔforV strain and found that it accumulates fasamycin C exclusively at a titer similar to that of the combined fasamycins and formicamycins produced by the ΔforJ strain in solid culture; this corresponds to a titer 90-fold higher than the total fasamycins produced by the wild-type strain (Table 1, Figure 2). This provides a powerful route for selectively producing non-halogenated fasamycin congeners, and the lack of formicamycin biosynthesis by this strain is consistent with the proposed gatekeeper function of ForV.
In a similar vein, we next made a strain lacking forJ and forX, which encodes the flavin-dependent monooxygenase ForX, the enzyme responsible for the first ring-expansion step involved in converting fasamycin precursors into formicamycins (Qin et al., 2020). The resulting ΔforJΔforX strain did not make formicamycins but instead accumulated chlorinated fasamycin congeners to approximately 120 times the level of the wild-type strain (8.9-fold increase in total metabolites) when grown in solid culture (Table 1, Figure 2). Moreover, the congener profile of this strain changed considerably and six additional fasamycin congeners (L-Q) were isolated (Figure 5) (Devine et al., 2020). These include molecules carrying up to four chlorine atoms, whereas a maximum of two had previously been observed for fasamycins isolated from the wild-type strain. These congeners all displayed potent antibacterial activity against both MSSA and MRSA (Table 2).
The structures of these fasamycin and formicamycin congeners were assigned using high-resolution liquid chromatography-mass spectrometry and 2D NMR based on our published data (Qin et al., 2017). The substituent variations in chlorination and O-methylations were determined from the 2D heteronuclear single quantum coherence spectroscopy (HSQC) and 1H-1H correlation spectroscopy (NOESY) data. Supplementary chemistry data are available here: https://doi.org/10.6084/m9.figshare.13222070.v1.
Discussion
Using targeted gene deletions and cappable RNA sequencing, we have shown that 24 genes are required for formicamycin biosynthesis and export. These are expressed as nine transcriptional units and are under the control of three CSRs: ForJ and ForZ are MarR-type regulators, while ForGF is a TCS. The latter is responsible for activating expression of these transcripts by binding to a single divergent promoter in the for BGC and is essential for formicamycin biosynthesis in the wild-type strain but not in a ΔforJ deletion mutant. TCSs are one of the major ways in which bacteria sense changes in their environment, and the large genomes of Streptomyces species generally encode for high numbers of TCSs, which enable them to survive in dynamic and sometimes extreme environments (Hutchings et al., 2004). Due to the availability of genome sequencing data and protocols for genetic manipulation, the majority of characterized TCSs are from model organisms such as Streptomyces coelicolor and Streptomyces venezuelae (Rodríguez et al., 2013). Of the TCSs studied in these model species, several have been shown to control secondary metabolism directly, while the majority coordinate secondary metabolism and morphological development either via global regulation (e.g., PhoPR, MtrA/B) or by interacting with CSRs (e.g., AfsQ1) (McLean et al., 2019). In contrast, ForGF is a rare example of a cluster-situated TCS that specifically activates transcription of the for biosynthetic genes. Another example of a cluster-situated TCS is cinKR, which, when deleted, abolishes biosynthesis of the lanthipeptide antibiotic cinnamycin much like deletion of forGF in S. formicae (O'Rourke et al., 2017). However, it should be noted that CinKR is responsible for the activation of a resistance transporter that is absolutely required for cinnamycin biosynthesis, rather than directly activating biosynthetic genes as ForGF appears to. Cluster-situated TCSs that activate biosynthesis in this way represent a promising target for overexpression to activate BGCs that may be cryptic or expressed at low levels, like the for BGC. Indeed, ectopic expression of an extra copy of forGF resulted in a significant increase (2-fold) in the level of formicamycin production compared with the wild-type strain in solid culture.
MarR-family regulators such as ForJ usually repress transcription of their target genes by binding to DNA sequences within promoter regions (Grove, 2017). We have shown that ForJ is the major for cluster repressor and deletion of forJ leads to increased production of multiple pathway products and the induction of biosynthesis during liquid culture of S. formicae. Deletion of CSRs is a known method of inducing biosynthesis from cryptic BGCs (Aigle and Corre, 2012); however, to our knowledge, MarR regulators that repress entire biosynthetic pathways in this way are relatively rare. MarR regulators typically bind to intergenic regions to autoregulate their own gene expression and the divergently transcribed gene (Wilkinson and Grove, 2006). Often, the genes under their control are involved in the control of export of secondary metabolites (Perera and Grove, 2010). ForZ appears to function in this way: through binding to the forZ-forAA intergenic region it autoregulates and controls expression of the divergent forAA gene, encoding a putative resistance transporter. Interestingly, ForZ appears to activate transcription of forAA rather than repressing it, which is unusual, although not unique. ForZ is not required for formicamycin biosynthesis and ChIP sequencing data show that it specifically binds to only a single site within the for BGC. However, our data indicate that ForZ may indirectly activate the expression of some for BGC transcripts to increase formicamycin biosynthesis.
Based on these data, we propose a model in which ForG senses an (unknown) environmental change and phosphorylates ForF, which then activates expression of forHI, which encode subunits of the acetyl-CoA carboxylase that converts acetyl-CoA to the polyketide precursor malonyl-CoA. MarR-family regulators are also known to bind small molecule ligands, and often these ligands are products of the pathways within which they are encoded (Perera and Grove, 2010). We therefore hypothesize that production of malonyl-CoA above a certain threshold level either directly or indirectly induces ForJ and leads to de-repression of the for BGC. This results in the expression of the seven transcripts under the regulation of ForJ, which contain the biosynthetic machinery required for formicamycin biosynthesis. To prevent these compounds accumulating intracellularly, we suggest that ForZ activates transcription of forAA to export formicamycins. Where MarR regulators have been shown to regulate an efflux pump, they generally bind and respond to the molecule required for export (Grove, 2013). An example is OtrR, encoded within the oxytetracycline BGC of Streptomyces rimosus, which controls the expression of the divergent transporter gene otrB in response to the presence of oxytetracycline and biosynthetic pathway intermediates (Pickens and Tang, 2010; Mak et al., 2014). It is possible that ForZ is sensitive to fasamycin and/or formicamycin levels and activates expression of forAA to prevent toxic accumulation while ensuring expression of other for transcripts remains low until this resistance mechanism is activated. This will be investigated in future studies.
The formicamycins represent promising candidates for investigation as a new structural class of antibiotics due to their potent bioactivity against drug-resistant pathogens and their high barrier to the development of resistance (Qin et al., 2017). Until now, further investigation into the formicamycins has been hindered by their low production levels in S. formicae and the fact they were only produced in solid culture. In this work, we show that de-repression of the for BGC results in not only increased titers but also the induction of biosynthesis during liquid culture. This is significant because the majority of industrial antibiotic production is performed in liquid cultures (Manteca and Yagüe, 2018). Furthermore, using our knowledge of for regulation and biosynthesis, we created a series of targeted mutants by combining mutations in genes encoding the biosynthetic machinery with pathway de-repression. This led to S. formicae strains that produce high titers of specific metabolites in both solid and liquid culture. This work will greatly accelerate further investigations into these exciting molecules and demonstrates the importance of studying both the biosynthesis and regulation of BGCs encoding specialized metabolites with antibiotic potential.
Significance
Antimicrobial resistance poses a major threat to public health, therefore the discovery and development of new antibiotics is vital. The formicamycins are promising antibiotics with potent activity against drug-resistant pathogens like MRSA. Further development of the formicamycins has so far been limited by the fact that they are only produced in low levels during solid culture. In this study, CRISPR/Cas9 genome editing was used to rewire formicamycin biosynthesis in the native host to increase titers and induce production during liquid culture. As well as leading to the discovery of additional formicamycin congeners and biosynthetic intermediates, this work will accelerate further investigations into these clinically relevant molecules and demonstrates the power of synthetic biology for refactoring antibiotic biosynthesis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial strains | ||
| Streptomcyes formicae wild-type | Lab stock | N/A |
| Streptomcyes formicae mutants | This work | For details, see Table S3 |
| Staphylococcus aureus (MSSA) | ATCC | ATCC 6538P |
| Staphylococcus aureus (MRSA) | ATCC | ATCC BAA-1717 |
| Chemicals, peptides, and recombinant proteins | ||
| PCRBIO Taq DNA Polymerase Mix, Red | PCR Biosystems | PB10.13-02 |
| Q5 High-fidelity DNA polymerase | NEB | M0491S |
| Gibson assembly master mix | NEB | E2611S |
| ANTI-FLAG M2 Affinity Gel | Sigma | A2220 |
| Luna Universal qPCR Master Mix | NEB | M3003S |
| Critical commercial assays | ||
| Plasmid prep mini kit | Qiagen | 12123 |
| Gel purification kit (QIAquick) | Qiagen | 28506 |
| RNAeasy mini kit | Qiagen | 74104 |
| LunaScript RT SuperMix | NEB | ES010S |
| Deposited data | ||
| Cappable RNA-Seq data | This work | E-MTAB-7975 |
| ChIP-Seq data | This work | E-MTAB-8006 |
| Oligonucleotides | ||
| For details of oligonucleotides generated and used, see Table S5 | This work | N/A |
| Recombinant DNA | ||
| pCRISPomyces-2 for CRISPR/Cas deletions | (Cobb, Wang and Zhao, 2015) | N/A |
| pMF96 for GUS assay | (Feeney et al., 2017) | N/A |
| pMS82 for integration of DNA into S. formicae | (Gregory, Till and Smith, 2003) | N/A |
| pESAC-13 215-G ePAC containing the formicamycin BGC | BioS&T and (Qin et al., 2017) | N/A |
| For details of other plasmids used or generated in this study, see Table S4 | This work | N/A |
| Software and algorithms | ||
| Integrated Genome Browser | (Nowlan, Norris and Loraine, 2016) | https://www.bioviz.org/download.html |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
Resource availability
Lead contact
Further requests for information should be addressed to Matthew I. Hutchings at matt.hutchings@jic.ac.uk.
Materials availability
Requests for materials should be made via the lead contact.
Data and code availability
Sequencing data are available at EMBL-EBI (accession numbers E-MTAB-7975 and E-MTAB-8006). Other data and code are available on request via the lead contact.
Experimental model and subject details
For details of strains used and generated in this study see Table S3. Generally, Streptomyces strains were grown at 30°C and other organisms at 37°C, with shaking at 200 rpm for liquid cultures, unless otherwise stated (Table S3). Streptomyces spores were harvested from confluent lawns streaked from single colonies using a sterile cotton bud and stored in 1.5 ml 20% glycerol at -80°C. Glycerol stocks were made by resuspending overnight culture in 50:50 LB and glycerol (final concentration 20%). All plasmids and ePACs used in this study are described in Table S4 and all primers in Table S5. Standard DNA sequencing was conducted by Eurofins Genomics using the Mix2Seq kit (Ebersberg, Germany).
Method details
Chemicals and reagents were laboratory standard grade or above and purchased from Sigma Aldrich (UK) or Thermo Fisher Scientific (UK) unless otherwise stated. All media and solutions were made using deionised water (dH2O) except where stated otherwise (Table S2).
Standard molecular techniques
Genomic DNA and ePACs were isolated by resuspending 1 ml overnight culture in 100 μl solution I (50 mM Tris/HCl, pH 8; 10 mM EDTA). Alkaline lysis was performed by adding 200 μl solution II (200 mM NaOH; 1% SDS) followed by 150 μl solution III (3M potassium acetate, pH 5.5). The supernatant was extracted in 400 μl phenol:chloroform:isoamyl alcohol and the upper phase mixed with 600 μl 2-propanol and incubated on ice for precipitation of the DNA. The DNA was pelleted and washed with 200 μl 70% ethanol, air dried and resuspended in sterile dH2O. Plasmid DNA was prepared using the QIAprep Spin Miniprep kit (QIAGEN) according to the manufacturer’s instructions. All DNA samples were quantified using the Nanodrop 2000 UV-Vis Spectrophotometer and the Qubit assay using the Qubit® fluorimeter 2.0.
PCRs were conducted using either the PCRBIO Taq DNA Polymerase (PCR Biosystems) for diagnostic reactions or the Q5 High-fidelity DNA polymerase for amplification of fragments required for cloning. Both were used according to the manufacturer’s recommendations with a final concentration of 100 nM primers. PCR products were analysed using gel electrophoresis using 1% agarose gels in TBE buffer (90 mM Tris HCl, 90 mM Boric Acid, 2 mM EDTA) with 2 μg/ml ethidium bromide and visualised by UV-light. When required, bands of interest were excised from the gel and the DNA recovered using the QiaQuick Gel Extraction Kit (QIAGEN) according to the manufacturer’s instructions.
For golden gate assembly, 100 ng purified backbone was incubated with 0.3 μl insert, 2 μl T4 ligase buffer (NEB), 1 μl T4 ligase (NEB) and 1 μl of the relevant restriction enzyme in a total volume of 20 μl made up in dH2O. Reactions were incubated under the following conditions: 10 cycles of 10 minutes at 37°C and 10 minutes at 16°C, followed by 5 minutes at 50°C and 20 minutes at 65°C. Plasmids were digested in 50-100 μl total volumes with restriction enzymes and their appropriate buffer (ether Roche or NEB) in accordance with the manufacturers recommendations (typically 1 μg of DNA was digested with 1 unit of enzyme for 1 hour at 37°C). Restriction enzymes were heat inactivated at 65°C for 10 minutes and 2 μl shrimp alkaline phosphatase was added to single-enzyme digestions to prevent re-ligation. Ligations were carried out using T4 DNA Ligase according to the manufacturer’s recommendations with a standard ratio of 1:3 plasmid:insert. Multiple DNA fragments were assembled into digested vector backbones using Gibson assembly by designing overlaps of between 18 and 24 nucleotides. Gel extracted DNA fragments were incubated in a ratio of 1:3 of plasmid to insert (1:5 for inserts smaller than 300 nucleotides) in the presence of Gibson Assembly master mix (NEB) at 50°C for 1 hour.
Plasmids were transformed into E. coli using heat shock (30 seconds at 42°C) for chemically competent cells or electroporation in a BioRad electroporator (200 Ω, 25 μF and 2.5 kV). ePACs were moved into E. coli using tri-parental mating by incubating 20 μl of each strain in the centre of an LB agar plate at 37°C and then re-streaking the spot onto selective agar for the desired plasmid combination. Plasmids were conjugated into S. formicae via the non-methylating E. coli ET12567/pUZ8002 as described previously using between 10 and 200 μl of spores depending on the application (more spores for conjugation of pCRISPomyces-2) (MacNeil et al., 1992; Keiser et al., 2000; Gust et al., 2004).
To analyse protein content, whole cell lysates were incubated at 100°C for 10 minutes in 50 μl SDS loading buffer (950 μl Bio-Rad® Laemmli buffer, 50 μl β-mercaptoethanol) and analysed by gel electrophoresis on a standard resolving gel of 16% (w/v) acrylamide:Bis-Acrylamide 37.5:1 (Fisher BioReagents). To confirm expression of tagged proteins, proteins were analysed by western blot by transferring to a nitrocellulose membrane (Pall Corporation) in a Trans-Blot transfer cell (10 V, 1 hour). After blocking in 5% (w/v) fat-free skimmed milk powder in TBST (50 mM Tris Cl pH 7.5, 150 mM NaCl, 1% Tween) overnight at room temperature, 20 ml HRP-conjugated anti-FLAG antibody diluted 1 in 20000 in TBST was added to the membrane for 1 hour. The membrane was washed 3 times for 10 minutes in TBST before developing in a 50:50 mix of 100 mM Tris pH 8.5, 100 μl luminal, 45 μl coumaric acid and 100 mM Tris pH 8.5, 6 μl 30% hydrogen peroxide for imaging using the ECL setting on a SYNGENE G:Box.
Editing pESAC-13 to define the borders of the for BGC
Targeted mutagenesis of pESAC-13 was conducted by ReDirect as described previously (Gust et al., 2004) except that the oriT was removed from the apramycin cassette to avoid undesired recombination events with the ePAC. The truncated cassette was PCR amplified, gel purified and electroporated into E. coli BW25113 pIJ1790 transformed (by tri-parental mating) with pESAC-13 215-G (containing the for BGC, supplied by BioS&T, Quebec(Qin et al., 2017)) using the transfer plasmid pR9604 (Piffaretti, Arini and Frey, 1988; Datsenko and Wanner, 2000). Primer overhangs targeting the edges of the formicamycin cluster were designed so that genes at either the left-hand or right-hand edge were deleted. The PCR-confirmed, edited ePACs were isolated and conjugated into S. formicae Δfor and the resulting mutants were grown under formicamycin producing conditions (as above). The metabolites were extracted as described below and then analysed by chromatography over a Phenomenex Gemini reversed-phase column (C18, 100 Å, 150×2.1 mm) using an Agilent 1100 series HPLC system and eluting with the following gradient method: 0–2 min 50% B; 2–16 min 100% B; 16–18 min 100% B; 18–18.1 min 100–50% B; 18.1–20 min 50% B; flowrate 1 mL min−1; injection volume 10 μL; mobile phase A: water+0.1% formic acid; mobile phase B: methanol. UV absorbance was monitored at 250, 285, 360 and 415 nm.
Cappable RNA sequencing
Samples for RNA-sequencing were crushed in liquid nitrogen using a sterile pestle and mortar on dry ice and resuspended by vortexing in 1 mL RLT Buffer (Qiagen) supplemented with β-mercaptoethanol (10 μl in every 1 ml buffer). Following lysis in a QIA-shredder column (Qiagen) the sample was extracted in 700 μl acidic phenol-chloroform and the upper phase mixed with 0.5 volumes 96% ethanol. RNA was purified using the RNeasy Mini spin column (Qiagen) according to the manufacturers protocol, including on column DNase treatment. Following elution, the Turbo-DNase kit was used according to the manufacturers protocol and a further Qiagen RNeasey mini clean-up was conducted before samples were flash frozen in liquid nitrogen for storage at -80°C. Once quantified by Nanodrop and gel electrophoresis, RNA was sent to Vertis Biotechnologie (Freising, Germany) for sequencing and analysed by capillary electrophoresis to map transcription start sites.
Generating S. formicae mutants
Gene deletions were made using the pCRISPomyces-2 system as described previously (Cobb, Wang and Zhao, 2015; Qin et al., 2017, 2019). Approximately 20 bp protospacers were designed so that the last 15 nucleotides, including the PAM (NGG) were unique in the genome. These were annealed and assembled into the sgRNA using a Golden Gate reaction with BbsI and pCRISPomyces-2. The resulting plasmid was then digested with XbaI to allow for the insertion of the repair template (usually approximately 1 Kb from either side of the deletion) using Gibson assembly. Once confirmed by PCR and sequencing, the final vector was conjugated into S. formicae via the non-methylating E. coli ET12567/pUZ8002. The deletion was confirmed using PCR to amplify the region across the repair template and into the surrounding genomic DNA. Once confirmed, loss of the editing plasmid was encouraged by repeated re-streaking on plates lacking the antibiotic selection and incubating at 37°C.
Complementation of gene deletions were achieved by fusing the gene to either a native promoter in pMS82(Gregory, Till and Smith, 2003) by Gibson assembly or by ligating the digested gene product downstream of the constitutive ermE∗ promoter in pIJ10257(Hong et al., 2005) and transferring the plasmid into the relevant mutant by conjugation.
ChIP-sequencing
For sampling, spores were inoculated onto cellophane discs and grown for 2, 3 or 4 days. These time points were chosen as day 5 is the earliest formicamycins have been observed in the culture extract and expression of regulator genes would be predicted to happen before biosynthesis. Day 2 was the earliest point that enough biomass could be harvested. Expression of the genes of interest was also confirmed using RT-PCR using the OneStep RT-PCR Kit (Qiagen) before the experiment was conducted. At sampling, discs were removed and the mycelium submerged in 10 ml 1% (v/v) formaldehyde for 20 minutes, followed by 10 ml 0.5 M glycine for 5 minutes. After washing the discs in 25 ml ice-cold PBS (pH 7.4), the samples were frozen at -80°C for storage and a small aliquot retained for western blot analysis to confirm expression of the tagged proteins in each sample.
For chromatin immunoprecipitation, cell pellets were resuspended in 2 ml lysis buffer (10 mM Tris-HCl pH 8.0, 50 mM NaCl, 10 mg/ml lysozyme, EDTA-free protease inhibitor) and incubated at 37°C for 30 minutes. To fragment the DNA, 1 ml IP buffer (100 mM Tris-HCl pH 8.0, 500 mM NaCl, 1% v/v Triton-X, EDTA-free protease inhibitor) was added and samples sonicated 20 times at 50 Hz for 10 seconds per cycle, ensuring cool-down on ice for at least 2 minutes between pulses. A small sample of this crude lysate was extracted with phenol:chloroform, treated with RNaseA and analysed by gel electrophoresis to confirm DNA fragments were within the desired size range for sequencing. The remaining crude lydate was cleared by centrifugation and incubated with 40 μl prepared Anti-FLAG M2 beads (Sigma-Aldridge) with rotation at 4°C overnight. To elute the bound DNA, the samples were then incubated in 100 μl elution buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 15 SDS) overnight at 65°C. Another 50 μl elution buffer was then added and the samples incubated for a further 5 minutes at 65°C before DNA from the total 150 μl eluate was purified by adding proteinase K and incubating at 55°C for 1.5 hours. DNA was extracted in 150 μl phenol-chloroform and purified on a QIAquick column (Qiagen) and eluted in 50 μl EB buffer (10 mM Tris-HCl pH 8.5) for quantification and sequencing by Genewiz (Leipzig, Germany) using the Illumina HiSeq platform. Data analysis was conducted as described previously (Bush et al., 2016; Munnoch et al., 2016; Som et al., 2017). Briefly, reads were aligned to the genome of S. formicae and Samtools v1.8 were used to sort and index BAM files produced by Bowtie2 and compute the depth of reads at every nucleotide position. Perl scripts were used to calculate local enrichment in 30 nucleotide windows along the entire genome. Assuming normal distribution, adjusted p-values were calculated and regions p < 0.00005 were selected and visualised in Integrated Genome Browser.
qRT-PCR
RNA samples were confirmed to be DNA-free by conducting test PCRs on the 16S rRNA gene and converted to cDNA using the LunaScript RT SuperMix Kit (NEB) according to the manufacturer’s instructions. Primers for each transcript were optimised using serial dilutions of ePAC template DNA and checked for specificity by melt-curve analysis and gel electrophoresis. Reactions were run in biological triplicate and technical duplicate using the Luna Universal qPCR Master Mix according to the manufacturer’s guidelines in a total volume of 20 μl with approximately 100 ng template cDNA and a final concentration of 0.25 μM of each primer. ΔCT values were normalised to the 16S rRNA gene.
GUS assay
Strains were generated using pMF96 plasmids containing the gusA vector under to control of each relevant promoter and a pMF23 plasmid as a negative control (Feeney et al., 2017) (Table S3). GUS plasmids were conjugated into to S. formicae WT, ΔforJ, ΔforGF and ΔforZ strains via ET12567/pUZ8002. Three biological replicates of each new GUS strain were then confirmed by PCR.
GUS strains were grown on SFM agar with a cellophane disk for 4 days before mycelium was harvested by scraping with a sterile metal spatula and resuspended in 1 mL dilution buffer (50 mM phosphate buffer, 0.1 % Triton X-100 [v/v], 5mM DTT). Samples were sonicated in 3 rounds of 8-10 seconds at 80 Hz before centrifugation at 13,000 rpm, 4°C for 10 minutes. A total of 95 μL of each sample supernatant was added to a 96 well plate and freeze-thawed at -80°C and 37°C, respectively. As strains were grown on agar plates rather than in liquid culture, the protein concentration of lysates were quantified by measuring absorbance at 280 nm rather than measuring cell density at 600 nm. The hydrolysis of PNPG by β-glucuronidase produces galactose and chromophoric 4-nitrophenol (PNP) with a peak absorbance at 420 nm. The intensity of this peak is dependent upon the quantity of enzyme present which correlates with transcription at the inserted promoter. PNPG was added to each well (final concentration 3 μg/mL) to initiate the reaction. Samples were loaded into a plate reader at 37°C for optimum enzyme activity and quantified at 415 nm (due to some interference with fasamycin peak absorbance at 419 nm) and 550 nm every 5 minutes for 40 minutes, inclusive. Softmax® Pro7 used to extract raw data which was used calculate Miller units/ mg protein (1 unit = 1000∗[Abs415–{1.75∗Abs550}]/{t∗v∗Abs280}]). Each promoter’s transcriptional activity was tested in both biological and technical triplicate and mean Miller Units/mg protein calculated. All statistical analysis was performed using IBM SPSS™ Statistics 23.
Fasamycin and formicamycin congener analysis
Solid culture
S. formicae WT (n = 16) and mutant strains (n = 3) were grown on soya flour mannitol (SFM) agar at 30°C for 10 days. Equal size agar plugs (1 cm3) were taken in triplicate from each plate and shaken with ethyl acetate (1 mL) for 1 hour before being centrifuged at 3100 rpm for 5 minutes. Ethyl acetate (300 μl) was transferred to a clean tube and solvent was removed under reduced pressure. The resulting extract was dissolved in methanol (200 μL) before being analysed by HPLC (Agilent 1290 UHPLC). To verify peak identity a representative set of samples were analysed by LCMS (Shimadzu IT-ToF LCMS platform). Chromatography was undertaken for both HPLC and LCMS analysis using the following method: Phenomenex Gemini NX C18 column (150 × 4.6 mm); mobile phase A: water + 0.1% formic acid; mobile phase B: methanol. Elution gradient: 0–2 min, 50% B; 2–16 min, 50–100% B; 16–18 min, 100% B; 18–18.1 min, 100–50% B; 18.1–20 min, 50% B; flow rate 1 mL min−1; injection volume 10 μL.
Liquid culture
S. formicae WT or mutant strains (n = 3) were grown in liquid TSB (10 mL) for 2 days. 100 μL of 2-day liquid culture was aliquoted into 10 mL of SFM liquid media into sterile 50 mL falcon tubes with sterile bungs. Strains were incubated at 30°C with shaking at 250 rpm. After 10 days, an aliquot (1 mL) of culture was removed from each sample in triplicate and shaken with ethyl acetate (1 mL) for 1 hour and then centrifuged at 3100 rpm for 5 minutes. Ethyl acetate extract (300 μl) was transferred to a clean tube and solvent was removed under reduced pressure. The resulting extract was dissolved in methanol (200 μl) before being analysed by HPLC and LCMS as described before.
Titre determination
Titres of fasamycin and formicamycins were determined by comparing peak areas from the above HPLC analysis to those of standard calibration curves and correcting for the concentration that occurred during the extraction process. Calibration curves were determined using standard solutions of fasamycin E (10, 20, 50, 80 and 200 μM) and formicamycin I (10, 20, 50, 100, 200 and 400 μM) (Figure S1). The content of fasamycin E and formicamycin I were determined by UV absorption at 418 nm and 285 nm respectively. Each standard solution was measured three times.
Scale up fermentation of S. formicae ΔforJ and ΔforJX
A seed culture was prepared by inoculating the fresh mycelium from an MS agar plate into TSB liquid medium (50 mL) and incubating for 24 h at 30°C with shaking at 250 rpm. The seed culture was then used to inoculate TSB (3 L; 6 × 2.5 L flasks, 1:500 inoculation). The cultures were incubated for seven days under the same condition as above after which the whole culture broth was extracted twice with equal amount of ethyl acetate. The organic phase was separated, and the solvent removed by evaporation under reduced pressure. The resulting material was dissolved in methanol and first analysed by LCMS and then subjected to semi-prep purification. In this way six additional fasamycin congeners (fasamycins L to Q) were isolated from the ΔforJX mutant, and two additional formicamycin congeners (formicamycins R and S) from the ΔforJ mutant. The structures of these compounds were determined using HRLCMS and 2D NMR as described in the Supplementary section.
Analytical LCMS method
For LCMS following analytical LCMS method was used: Phenomenex Kinetex C18 column (100 Å~ 2.1 mm, 100 Å); mobile phase A: water +0.1% formic acid; mobile phase B: acetonitrile. Elution gradient: 0–1 min, 20% B; 1–8 min, 20–100% B; 8–11 min, 100% B; 11–11.1 min, 100–20% B; 11.1–12 min, 20% B; flow rate 0.6 mL min−1; injection volume 10 μL.
Semi-prep HPLC method for isolation of formicamycins R & S
Chromatography was achieved over a Phenomenex Gemini-NX semi-prep reversed-phase column (C18, 110 Å, 150 Å~ 10 mm) using an Agilent 1100 series HPLC system and eluting with the following gradient method: 0–2 min 60% B; 2–18 min 60–100% B; 18–23 min 100% B; 23–23.1 min 100–60% B; 23.1–26 min 60% B; flowrate 3.5 mL min−1; injection volume 20 μL; mobile phase A: water +0.1% formic acid; mobile phase B: acetonitrile. The UV absorbance was monitored at 250 and 285 nm.
Semi-prep HPLC method for isolation of fasamycins L to Q
Chromatography was achieved over a Phenomenex Gemini-NX semi-prep reversed-phase column (C18, 110 Å, 150 Å~ 10 mm) using an Agilent 1100 series HPLC system and eluting with the following gradient method: 0–2 min 70% B; 2–18 min 70–100% B; 18–20 min 100% B; 20–20.1 min 100–70% B; 20.1–24 min 70% B; flowrate 3.5 mL min−1; injection volume 100 μL; mobile phase A: water + 0.1% formic acid; mobile phase B: acetonitrile. UV absorbance was monitored at 250, 285, 360 & 415 nm.
Bioassays
Resazurin assays were performed to determine minimum inhibitory concentrations. A stock solution of compound was prepared in DMSO and diluted in LB or TSB media to give a concentration range of 256 μg/ml to 1 μg/ml containing 5% DMSO. Positive control (PC) for preparations in LB and TSB was apramycin at 50 μg/ml. Negative control (NC) and media control (MC) contained media (LB or TSB) and DMSO at 5%. Methods were followed as detailed in Heine et al (Heine et al., 2018). In brief, cultures were grown to confluence overnight and then diluted 1/100 in fresh media and grown to 0.4 OD600nm, diluted to match a 0.5 McFarlands standard and further diluted 1/100. 5 μl of culture was then aliquoted into each well of the 96 well plate excluding the MC well. Plates were grown at 37°C before being inoculated with 5 μl resazurin dye (6.75 mg / ml, Sigma Aldrich). Colorimetric results were observed 4 hours after inoculation. MSSA (ATCC 6538P) and MRSA (ATCC BAA-1717) indicator strains were obtained from the American Type Culture Collection.
Quantification and statistical analysis
In general, data were analysed in Excel and figures plotted in R using ggplot2. Details of replicates and data analysis for each experiment can be found in the figure legends.
Acknowledgments
This work was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) via BBSRC Responsive Mode Grants (BB/S00811X/1 and BB/S009000/1) to M.I.H. and B.W. (R.D. and C.A.), and via Institute Strategic Program Project BBS/E/J/000PR9790 to the John Innes Center (Z.Q.). H.M. and K.N. were supported by BBSRC PhD studentships (BBSRC Doctoral Training Program grant BB/M011216/1), and Norwich Research Park BBSRC Postdoctoral Training Program Studentship (BB/M011216/1) (H.M. and K.N.). We thank our colleague Dr Tung Le (John Innes Center) for useful discussions.
Author contributions
R.D., H.M., Z.Q., C.A., K.N., B.W., and M.I.H. designed the research. R.D., H.M., K.N., B.W., and M.I.H. wrote the paper and all authors commented. R.D., H.M., K.N., and C.A. performed the molecular genetics experiments, H.M. and Z.Q. performed the chemical experiments. G.C. conducted the bioinformatics analysis.
Declaration of interests
The authors declare no competing interests.
Published: January 12, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.chembiol.2020.12.011.
Contributor Information
Rebecca Devine, Email: rebecca.devine@jic.ac.uk.
Barrie Wilkinson, Email: barrie.wilkinson@jic.ac.uk.
Matthew I. Hutchings, Email: matt.hutchings@jic.ac.uk.
Supplemental information
References
- Aigle B., Corre C. Waking up Streptomyces secondary metabolism by constitutive expression of activators or genetic disruption of repressors. Meth. Enzymol. 2012;517:343–366. doi: 10.1016/B978-0-12-404634-4.00017-6. [DOI] [PubMed] [Google Scholar]
- Bush M., Chandra G., Bibb M.J., Findlay K.C., Buttner M.J. Genome-wide chromatin immunoprecipitation sequencing analysis shows that WhiB is a transcription factor that cocontrols its regulon with WhiA to initiate developmental cell division in Streptomyces. mBio. 2016;7:516–523. doi: 10.1128/mBio.00523-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb R., Wang Y., Zhao H. High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth. Biol. 2015;4:723–728. doi: 10.1021/sb500351f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine R., McDonald H., Qin H., Arnold C., Noble K., Chandra G., Wilkinson B., Hutchings M.I. Structure determination of new fasamycin and formicamycin congeners. 2020. https://figshare.com/articles/dataset/Structure_determination_of_new_fasamycin_and_formicamycin_congeners/12951974
- Devine R., Hutchings M., Holmes N. Future directions for the discovery of antibiotics from actinomycete bacteria. Emerging Top. Life Sci. 2017;1:1–12. doi: 10.1042/ETLS20160014. [DOI] [PubMed] [Google Scholar]
- Feeney M.A., Chandra G., Findlay K.C., Paget M.S.B., Buttner M.J. Translational control of the Sigr directed oxidative stress response in Streptomyces via IF3-mediated repression of a noncanonical GTC start codon. mBio. 2017;8:e00815-17. doi: 10.1128/mBio.00815-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z., Chakraborty D., Dewell S.B., Reddy B.V., Brady S.F. Environmental DNA-encoded antibiotics fasamycins A and B inhibit FabF in type II fatty acid biosynthesis. J. Am. Chem. Soc. 2012;134:2981–2987. doi: 10.1021/ja207662w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genilloud O. Natural products discovery and potential for new antibiotics. Curr. Opin. Microbiol. 2019;51:81–87. doi: 10.1016/j.mib.2019.10.012. [DOI] [PubMed] [Google Scholar]
- Gregory M.A., Till R., Smith M.C. Integration site for Streptomyces phage ϕBT1 and development of site-specific integrating vectors. J. Bacteriol. 2003;185:5320–5323. doi: 10.1128/JB.185.17.5320-5323.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove A. MarR family transcription factors. Curr. Biol. 2013;23:R142–R143. doi: 10.1016/j.cub.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Grove A. Regulation of metabolic pathways by MarR family transcription factors. Comput. Struct. Biotechnol. J. 2017;15:366–371. doi: 10.1016/j.csbj.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gust B., Chandra G., Jakimowicz D., Yuqing T., Bruton C.J., Chater K.F. λ red-mediated genetic manipulation of antibiotic-producing Streptomyces. Adv. Appl. Microbiol. 2004;54:107–128. doi: 10.1016/S0065-2164(04)54004-2. [DOI] [PubMed] [Google Scholar]
- Heine D., Holmes N.A., Worsley S.F., Santos A.C.A., Innocent T.M., Scherlach K., Patrick E.H., Yu D.W., Murrell J.C., Vieria P.C. Chemical warfare between leafcutter ant symbionts and a co-evolved pathogen. Nat. Commun. 2018;9:2208–2219. doi: 10.1038/s41467-018-04520-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes N., Devine R., Qin Z., Seipke R.F., Wilkinson B., Hutchings M.I. Complete genome sequence of Streptomyces formicae KY5, the formicamycin producer. J. Biotechnol. 2018;265:116–118. doi: 10.1016/j.jbiotec.2017.11.011. [DOI] [PubMed] [Google Scholar]
- Hong H.J., Hutchings M.I., Hill L.M., Buttner M.J. The role of the novel fem protein VanK in vancomycin resistance in Streptomyces coelicolor. J. Biol. Chem. 2005;280:13055–13061. doi: 10.1074/jbc.M413801200. [DOI] [PubMed] [Google Scholar]
- Huo C., Zheng Z., Xu Y., Ding Y., Zheng H., Mu Y., Niu Y., Gao J., Lu X. Naphthacemycins from a Streptomyces sp. as protein-tyrosine phosphatase inhibitors. J. Nat. Prod. 2020;83:1394–1399. doi: 10.1021/acs.jnatprod.9b00417. [DOI] [PubMed] [Google Scholar]
- Hutchings M.I., Hoskisson P.A., Chandra G., Buttner M.J. Sensing and responding to diverse extracellular signals? Analysis of the sensor kinases and response regulators of Streptomyces coelicolor A3(2) Microbiology (Reading, Engl) 2004;150:2795–2806. doi: 10.1099/mic.0.27181-0. [DOI] [PubMed] [Google Scholar]
- Hutchings M.I., Truman A.W., Wilkinson B. Antibiotics: past, present and future. Curr. Opin. Microbiol. 2019;51:72–80. doi: 10.1016/j.mib.2019.10.008. [DOI] [PubMed] [Google Scholar]
- Kämpfer P., Glaeser S., Parkes L., van Keulen G., Dyson P. The family Streptomycetaceae. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., Thompson F., editors. The Prokaryotes – Actinobacteria. Springer Berlin Heidelberg; 2014. pp. 889–1010. [Google Scholar]
- Keiser T., Bibb M.J., Buttner M.J., Chater K.F., Hopwood D.A. The John Innes Foundation; 2000. Practical Streptomyces Genetics. [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2'. Nat. Methods. 2012;9 doi: 10.1038/nmeth.1923. pp357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNeil D.J., Gewain K.M., Ruby C.L., Dezeny G., Gibbons P.H., MacNeil T. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene. 1992;111:61–68. doi: 10.1016/0378-1119(92)90603-m. [DOI] [PubMed] [Google Scholar]
- Maglangit F., Fang Q., Leman V., Soldatou S., Ebel R., Kyeremeh K., Deng H. Accramycin A, a new aromatic polyketide, from the soil bacterium, Streptomyces sp. MA37. Molecules. 2019;24:3384. doi: 10.3390/molecules24183384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak S., Xu Y., Nodwell J.R. The expression of antibiotic resistance genes in antibiotic-producing bacteria. Mol. Microbiol. 2014;93:391–402. doi: 10.1111/mmi.12689. [DOI] [PubMed] [Google Scholar]
- Manteca Á., Yagüe P. Streptomyces differentiation in liquid cultures as a trigger of secondary metabolism. Antibiotics. 2018;7:41–54. doi: 10.3390/antibiotics7020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean T.C., Lo R., Tschowri N., Hoskisson P.A., Al Bassam M.M., Hutchings M.I., Som N.F. Sensing and responding to diverse extracellular signals: an updated analysis of the sensor kinases and response regulators of Streptomyces species. Microbiol. Soc. 2019;165:929–952. doi: 10.1099/mic.0.000817. [DOI] [PubMed] [Google Scholar]
- Munnoch J.T., Martinez M.T., Svistunenko D.A., Crack J.C., Le Brun N.E., Hutchings M.I. Characterization of a putative NsrR homologue in Streptomyces venezuelae reveals a new member of the Rrf2 superfamily. Sci. Rep. 2016;6:31597. doi: 10.1038/srep31597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowlan H.F., Norris D.C., Loraine A.E. Integrated genome browser:visual analytics platform for genomics. Bioinformatics. 2016;32:2089–2095. doi: 10.1093/bioinformatics/btw069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke S., Widdick D., Bibb M. A novel mechanism of immunity controls the onset of cinnamycin biosynthesis in Streptomyces cinnamoneus DSM 40646. J. Ind. Microbiol. Biotechnol. 2017;44:563–572. doi: 10.1007/s10295-016-1869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera I.C., Grove A. Molecular mechanisms of ligand-mediated attenuation of DNA binding by MarR family transcriptional regulators. J. Mol. Cell. Biol. 2010;2:243–254. doi: 10.1093/jmcb/mjq021. [DOI] [PubMed] [Google Scholar]
- Pickens L.B., Tang Y. Oxytetracycline biosynthesis. J. Biol. Chem. 2010;285:27509–27515. doi: 10.1074/jbc.R110.130419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piffaretti J., Arini A., Frey J. pUB307 mobilizes resistance plasmids from Escherichia coli into Neisseria gonorrhoeae. Mol. Gen. Genet. 1988;212:215–218. doi: 10.1007/BF00334687. [DOI] [PubMed] [Google Scholar]
- Qin Z., Munnoch J.T., Devine R., Holmes N.A., Seipke R.F., Wilkinson K.A., Wilkinson B., Hutchings M.I. ‘Formicamycins, antibacterial polyketides produced by Streptomyces formicae isolated from African Tetraponera plant-ants †’. Chem. Sci. 2017;8:3218–3227. doi: 10.1039/c6sc04265a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z., Devine R., Hutchings M.I., Wilkinson B. A role for antibiotic biosynthesis monooxygenase domain proteins in fidelity control during aromatic polyketide biosynthesis. Nat. Commun. 2019;10:3611. doi: 10.1038/s41467-019-11538-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z., Devine R., Booth T.J., Farrar E.H.E., Grayson M.N., Hutchings M.I., Wilkinson B. Formicamycin biosynthesis involves a unique reductive ring contraction. Chem. Sci. 2020;11:8125–8131. doi: 10.1039/d0sc01712d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez H., Rico S., Díaz M., Santamaría R.I. Two-component systems in Streptomyces: key regulators of antibiotic complex pathways. Microb. Cell Fact. 2013;12:127–137. doi: 10.1186/1475-2859-12-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seipke R.F., Barke J., Heavens D., Yu D.W., Hutchings M.I. Analysis of the bacterial communities associated with two ant-plant symbioses. MicrobiologyOpen. 2013;2:276–283. doi: 10.1002/mbo3.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Som N.F., Heine D., Holmes N., Munnoch J.T., Chandra G., Seipke R.F., Hoskisson P.A., Wilkinson B., Hutchings M.I. The conserved actinobacterial two-component system MtrAB coordinates chloramphenicol production with sporulation in Streptomyces venezuelae NRRL B-65442. Frontiers Microbiol. 2017;8:1145. doi: 10.3389/fmicb.2017.01145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S.P., Grove A. Ligand-responsive transcriptional regulation by members of the MarR family of Winged Helix proteins. Curr. Issues Mol. Biol. 2006;8:51–62. [PubMed] [Google Scholar]
- Yang L., Li X., Wu P., Xue J., Xu L., Li H., Wei X. ‘Streptovertimycins A–H, new fasamycin-type antibiotics produced by a soil-derived Streptomyces morookaense strain’. J. Antibiot. (Tokyo) 2020;73:283–289. doi: 10.1038/s41429-020-0277-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data are available at EMBL-EBI (accession numbers E-MTAB-7975 and E-MTAB-8006). Other data and code are available on request via the lead contact.