

Summary
In vasculitis disorders, inflammation affects blood vessels. Granulomatosis with polyangiitis (GPA) is a chronic systemic vasculitis distinguished by the presence of anti‐proteinase‐3 autoantibodies (anti‐PR3). In this study we analyzed the molecular signature of human umbilical endothelial cells (HUVECs) in response to neutrophil‐derived extracellular vesicles (EVs). EVs were obtained from anti‐PR3‐activated neutrophils, purified and characterized by flow cytometry, nanoparticle tracking and miRNA screening. HUVECs were stimulated with EVs and miRNA/mRNA expression was measured. Cell culture media proteins were identified by antibody microarrays and selected cytokines were measured. Comparison of differentially expressed miRNAs/mRNAs between non‐stimulated and EV‐stimulated HUVECs revealed two regulatory patterns. Significant up‐regulation of 14 mRNA transcripts (including CXCL8, DKK1, IL1RL1, ANGPT‐2, THBS1 and VCAM‐1) was accompanied by 11 miRNAs silencing (including miR‐661, miR‐664a‐3p, miR‐377‐3p, miR‐30d‐5p). Significant down‐regulation was observed for nine mRNA transcripts (including FASLG, CASP8, STAT3, GATA3, IRAK1 and IL6) and accompanied by up‐regulation of 10 miRNAs (including miR‐223‐3p, miR‐142‐3p, miR‐211‐5p). Stimulated HUVECs released IL‐8, Dickkopf‐related protein 1 (DKK‐1), soluble interleukin (IL)‐1 like receptor‐1 (ST2), growth differentiation factor 15 (GDF‐15), angiopoietin‐2, endoglin, thrombospondin‐1 and vascular adhesion molecule‐1 (VCAM‐1). Moreover, transfection of HUVECs with mimics of highly expressed in EVs miR‐223‐3p or miR‐142‐3p, stimulated production of IL‐8, ST2 and endoglin. Cytokines released by HUVECs were also elevated in blood of patients with GPA. The most increased were IL‐8, DKK‐1, ST2, angiopoietin‐2 and IL‐33. In‐vitro stimulation of HUVECs by neutrophil‐derived EVs recapitulates contribution of endothelium in autoimmune vasculitis. Proinflammatory phenotype of released cytokines corresponds with the regulatory network of miRNAs/mRNAs comprising both EVs miRNA and endothelial cell transcripts.
Keywords: autoinflammatory disease, endothelial cells, neutrophils, gene regulation
Endothelial cells can uptake neutrophil‐derived extracellular vesicles and respond to their cargo. Extracellular vesicles were labeled with PKH 67 fuorescent dye.

Introduction
Granulomatosis with polyangiitis (GPA) is a rare autoimmune disease, with the highest prevalence in northern Europeans. Despite unknown aetiology [1], GPA is well characterized clinically by symptoms of upper airways inflammation, renal involvement and presence of circulating autoantibodies against neutrophil serine protease, proteinase‐3 [anti‐PR3 immunoglobulin (Ig)G, anti‐neutrophil cytoplasmic antibodies (ANCA)] [2, 3, 4, 5]. Blood neutrophil is an indispensable component of the immune system; importantly, however, it contributes to the pathophysiology in GPA. In the disease neutrophils are activated by ANCA. The process is initiated by the direct binding of PR3 with the Fab region of the antibody and is assisted by interaction of the Fc region with FcγRs on the neutrophil surface. The consequences of neutrophil activation are degranulation, neutrophil extracellular traps formation, generation of reactive oxygen intermediates and transmigration through the endothelial cell layer, all described in GPA [6, 7, 8]. Cellular exosomes, a subclass of extracellular vesicles (EVs), are released together with ANCA‐mediated neutrophil activation [9]. Population of extracellular particles bound within a phospholipid bilayer is heterogeneous. EVs are divided mainly into exosomes and microparticles [10]. Exosomes are smaller (50–120 nm) and originate from the endocytic compartment, whereas macrovesicles are larger (100–1000 nm) and are produced during the process of plasma membrane budding. During formation of exosomes, invaginations of the endosomal membrane lead to accumulation of intraluminal vesicles, described as endosomal multi‐vesicular bodies. Following fusion with the cell membrane, exosomes are released into the extracellular compartment. As well as a smaller size, exosomes maintain asymmetric distribution of the membrane phospholipids, which is lost in macrovesicles [11]. EVs are important contributors to pathophysiology of several diseases [12, 13, 14]; however, very little is still known about their participation in GPA inflammation. In our previous study we reported on the activation of neutrophils by EVs circulating in plasma of patients with GPA [15]. It was also shown that neutrophil‐derived microparticles activated human umbilical vein endothelial cells (HUVECs), providing a rationale for this study [16]. Thus, some evidence supported a mechanism of vascular inflammation mediated by neutrophil‐derived EVs. In the current study, we focused on miRNA profile of neutrophil‐derived EVs and miRNA/transcripts/proteins interactions in endothelial cells exposed to EVs. To the best of our knowledge, this is the first experimental attempt to analyze molecular mechanisms of endothelial cell activation by neutrophil‐derived EVs.
Materials and methods
Anti‐PR3 IgG preparation, neutrophil isolation and stimulation
Anti‐PR3 IgG antibodies were isolated as the total IgG fraction from pooled sera of GPA patients (n = 10, anti‐PR3 IgG > 200 mU/l; anti‐MPO < 20 mU/l). Total IgG fraction was purified stepwise by ammonium sulphate precipitation and removal of other serum proteins using negative affinity adsorption chromatography (Melon Gel IgG purification kits; Thermo Fisher Scientific, Waltham, MA, USA). Concentration of purified total IgG was determined by immunonephelometry (BN II Nephelometer; Siemens Dade Behring, Marburg, Germany). Specific anti‐PR3 IgG level was assessed by enzyme‐linked immunosorbent assay (ELISA) (anti‐PR3 ELISA kit; Euroimmun Medizinische Labordiagnostika, Lübeck, Germany).
Neutrophil isolation
Neutrophils were isolated from the citrate anti‐coagulated blood of healthy donors (n = 10) by gradient centrifugation (Histopaque‐1077; Sigma‐Aldrich, St Louis, MO, USA) followed by a negative magnetic separation (EasySep human neutrophil enrichment kit; Stemcell Technologies Inc., Vancouver, Canada). Purity of the neutrophil fraction (> 99%) was determined by flow cytometry and viability (> 95%) was tested by trypan blue exclusion staining. Isolated cells were suspended in Hanks’ balanced salts solution (HBSS) with calcium and magnesium containing 2% commercially available exosome‐depleted fetal bovine serum (FBS; Thermo Fisher Scientific).
Preparation of neutrophil‐derived EVs
Neutrophils (4 × 106 cells/ml) of healthy donors were primed with a low concentration of tumour necrosis factor (TNF)‐α (2 ng/ml; Bio‐Techne, Minneapolis, MN, USA) for 20 min and next stimulated with IgG anti‐PR3 antibodies (50 ng/ml of total IgG) for 4 h in HBSS with calcium and magnesium, supplemented with 2% exosome‐depleted FBS. Neutrophil‐derived EVs were purified from the cell medium. First, cellular debris was removed by centrifugation at 1000 g for 10 min. Next, the supernatant was diluted with 0·2 µm filtered PBS (7 ml total volume) and passed through a 0·8 µm syringe filter. EVs were collected by ultracentrifugation in a Sorvall WX 80+ ultracentrifuge equipped with T‐1270 fixed angle rotor (Thermo Fisher Scientific) at 100 000 g for 1·5 h. The pellet was washed with 0·2 µm filtered PBS and centrifuged again. Eventually, the EV pellet was suspended in filtered PBS, aliquoted (10 µl) and stored at −80°C for further experiments.
Neutrophil‐derived EVs surface markers and size distribution
EVs number and size distribution was counted by nanoparticle tracking analysis (NTA) (NanoSight LM10; Malvern Instruments, Malvern, UK). EV preparations were diluted 1 : 1000 with filtered PBS to adjust to the linear range of the apparatus, i.e. below 2–10 × 108/ml. Any presence of anti‐PR3 IgG (ANCA) carry‐over in EVs specimens was excluded by an indirect immunofluorescence method detecting ANCA (ANCA IFA granulocyte Biochip Mosaic Test; Euroimmun Medizinische Labordiagnostika AG, Lübeck, Germany). Analysis of EVs surface markers was performed by flow cytometry, as previously described [17, 18]. In brief, EVs aliquots were diluted in PBS, mixed with a 5 µl suspension of aldehyde/sulphate latex beads (4 μm diameter; Invitrogen, Carlsbad, CA, USA) and incubated at room temperature for 15 min. Next, latex binding sites were blocked with 1 ml of 2% BSA in PBS overnight with rotation at 4°C. Bead‐coupled EV were pelleted by centrifugation at 2700 g for 5 min, washed with 1 ml of 2% BSA in PBS and centrifuged again. The sediment was resuspended in 50 μl of PBS and stained with anti‐CD63 [fluorescein isothiocyanate (FITC), clone: H5C6; BD Biosciences, San Jose, CA, USA] and anti‐CD81 [peridinin chlorophyll (PerCP)‐eFluor 710, clone 1D6‐CD81; Thermo Fisher Scientific] antibodies for 30 min at room temperature.
Endothelial cells culture, stimulation by extracellular vesicles and mimic miRNAs transfection
Human umbilical vein endothelial cells (HUVECs; PromoCell, Heidelberg, Germany) were cultured in full growth medium (endothelial cell growth medium 2; PromoCell) supplemented with exosome‐depleted FBS up to the third passage. Confluent cells were detached, and reseeded on a 24‐well plate (3.0 × 105 cells per well) for 36 h culture to reach 70–80% confluence. Six hours before EVs stimulation, the culture medium was replaced by the full growth medium with reduced FBS concentration (1%, exosome‐depleted). Stimulation of HUVECs with EVs was performed by adding prepared neutrophil‐derived EVs to a final concentration of 1 × 1010 particles/ml. HUVECs were stimulated for 6 h in miRNA/mRNA studies and for 12 h to measure secreted proteins.
HUVEC transfection with selected miRNA mimics was performed using commercially available reagents: mimic miRNAs, Opti‐MEM medium and lipofectamine RNAiMAX (Thermo Fisher Scientific). HUVECs were seeded on a 24‐well plate (3.0 × 105 cells per well) and cultured for 12 h. Next, cells were transfected with selected miRNAs (miRNA mimic negative control 1, mir‐223‐3p mimic, mir‐142‐3p mimic or both mimics miRNAs, 100 pM each) for 24 h. After transfection, the cell culture medium was collected, cells were washed three times with PBS and total RNA was isolated (samples marked as 24 h) or cells were supplemented with the fresh medium and cultured for another day and harvested after similar washes marked as 48‐h cells.
RNA isolation and miRNA/mRNA profiling
Total RNA was isolated from the EV specimens using total exosome RNA and protein isolation kit (Thermo Fisher Scientific) or from HUVECs with RNAZol (Sigma‐Aldrich) combined with a microcolumn purification (total RNA Zol‐Out kit; A&A Biotechnology, Gdynia, Poland). Before each isolation, EV and HUVEC samples were spiked‐in with cel‐39 miRNA (0·64 pM; Invitrogen, Carlsbad, CA, USA) as an internal standard. Reverse transcription was accomplished using the TaqMan MicroRNA reverse transcription kit for miRNA analysis or the high‐capacity cDNA reverse transcription kit for HUVEC transcripts (Thermo Fisher Scientific). Profiling of miRNA was performed using the TaqMan array human MicroRNA card set version 3.0 or the pre‐designed TaqMan assay in experiments involving miRNA mimics. HUVEC mRNAs were analyzed using TaqMan human apoptosis arrays and TaqMan human inflammation panel (TLDA format; Thermo Fisher Scientific). Two transcripts (DKK‐1 and GATA6) were added using a predesigned TaqMan expression primer/probe following bioinformatics analysis and protein profiling experiments (Proteome Profiler Human XL Cytokine Array Kit; Bio‐Techne). Data on the real‐time polymerase chain reaction (PCR) quantification cycle were normalized to cel‐39 and RNU‐44 for miRNA or 18S rRNA and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) for mRNA. Results were calculated using the 2−ΔΔCt (fold change) formula from these endogenous controls [19]. Absolute quantification of the most up‐regulated two miRs was performed using a five‐point calibration curve within the range of 0·064 to 640 pM concentration of cel‐39 (r2 = 0·9987).
EV uptake by endothelial cells
EV aliquots were stained with PKH67 fluorescent tracker dye (PKH67 fluorescent cell linker kit; Sigma‐Aldrich) for 5 min at 37°C. Staining reaction was stopped by dilution with PBS and 1% bovine serum albumin (BSA) and three cycles of wash‐ultrafiltration with Amicon Ultra 0·5 ml centrifugal filters (100 K; EMD Millipore, Burlington, MA, USA). After the last centrifugation, EV concentrates were restituted to their initial volume. EV uptake by HUVECs was assessed at five time‐points: 0·5, 1, 3, 6 and 24 h after incubation at 37°C. Presence of PKH67‐labelled EVs in HUVECs was measured by flow cytometry (BD FACSCanto II; BD Biosciences, San Jose, CA, USA) and confirmed by fluorescence microscopy (Leica Microsystems DMI 3000B, Wetzlar, Germany). To detach cells after stimulation and remove surface‐attached EVs, HUVECs were trypsinized [0·25% (w/v) trypsin, 0·53 mM ethylenediamine tetraacetic acid (EDTA) solution; Lonza, Basel, Switzerland] and then centrifuged at 200 g for 6 min. As a negative control, HUVECs were stimulated on ice with EVs for up to 3 h to exclude passive cell membrane fusion of exosomes. Adherent HUVECs were analyzed by fluorescence microscopy after surface‐attached EVs were removed by three cycles of cold PBS/EDTA solution washes.
Endothelial cells vascular adhesion molecule‐1 (VCAM‐1) expression
Flow cytometry was used to assess VCAM‐1 on HUVECs stimulated with neutrophil‐derived EVs. HUVECs were stimulated by adding EVs at a final concentration of 1·6 × 1010 particles/ml for 12 h. Next, cells were detached using Accutase solution (BD Biosciences) and stained with FITC mouse anti‐human CD106 (clone 51‐10C9; BD Biosciences). Isotype control was included in the analysis.
Cell culture and serum protein measurements
The profile of the proteins secreted by endothelial cells stimulated with EVs was evaluated using the Proteome Profiler Human XL cytokine array kit (Bio‐Techne), according to the manufacturer’s procedure. In brief, 100 µl of HUVEC culture supernatant (control or after EV stimulation) from five different experiments were pooled, diluted (final volume 1·5 ml) and incubated overnight with array membranes spotted with capture antibodies. After washing, membranes were incubated with detection antibodies and then developed with chemiluminescent reagent. Membranes image were collected with C‐DiGit Blot Scanner (Li‐Cor Bioscience, Lincoln, NE, USA). Results from the Proteome Profiler Human XL cytokine array kit were used to select proteins thereafter measured in the HUVEC culture medium following EV stimulation or miRNAs mimic transfection. These proteins were also measured in sera of GPA patients and health controls using xMAP technology Luminex assays (Bio‐Techne) and the Luminex 200 System (Luminex, Austin, TX, USA).
GPA patients and healthy controls
In this observational non‐randomized study we collected serum samples from 55 patients with GPA (26 in the active stage of disease and 29 in remission). Healthy volunteers (n = 31) were matched by age and sex as a control group (for details see Table 1). GPA was diagnosed according to American College of Rheumatology (ACR) 1990 criteria [20]. All GPA patients were positive for IgG anti‐PR3 antibodies and negative for anti‐MPO IgG antibodies. Disease activity was graded using the Birmingham Vasculitis Activity Score (BVAS, version 3), while organ damage was measured using the Vasculitis Damage Index (VDI) scale. Remission was defined as the absence of clinical signs of active disease (BVAS = 0), whereas exacerbation/relapse of the disease was defined as the presence of new or re‐emerging clinical symptoms due to vasculitis (confirmed by the BVAS > 6) requiring intensification of immunosuppressive therapy, according to European League Against Rheumatism (EULAR) recommendations for conducting clinical studies [21]. Patients with co‐existing infection or with kidney failure requiring dialysis were excluded from the study. Basic laboratory tests [complete blood count (CBC), C‐reactive protein (CRP), anti‐PR3 IgG level] were performed in all participants of the study at the time of collection of the peripheral blood. All patients with the active stage of GPA had blood samples collected before onset of high‐dose corticosteroid or immunosuppressive therapy. Written informed consent was obtained from all participants in the study and the study protocol was accepted by the Jagiellonian University Ethic Committee.
Table 1.
Selected characteristics of the study participants
| Active stage of GPA | Remission | Control | |
|---|---|---|---|
| n | 26 | 29 | 31 |
| Age (mean ± s.d.) | 56·1 ± 13·2 | 53·2 ± 13·1 | 52·1 ± 12·3 |
| Gender (F/M) | 14/12 | 16/13 | 16/15 |
| BVAS (min–max) VDI | 6–32 | 0 | – |
| VDI (min–max) | 0–5 | 1–7 | – |
| GC treatment (yes/no) | 15/11 | 25/4 | – |
| GC dose (mg/day) (min–max) | 3–20 | 2–8 | – |
| CYC cumulative dose† | 0–33 | 10–25 | – |
| IgG anti‐PR3 (RU/ml) (min‐max) | 20–200 | < 20–140 | < 20 |
| CRP (mg/ml) | < 5·0–130 | < 5·0–6·9 | < 5·0 |
| Procalcitonin (ng/ml) | < 0·05 | < 0·05 | < 0·05 |
| PBMC (103/µl) | 10·9 ± 3·8* | 8·3 ± 1·9 | 6·2 ± 3·5 |
| PMN (103/µl) | 7·69 ± 4·8* | 5·8 ± 2·5 | 3·9 ± 2·6 |
| PLT (103/µl) | 255 ± 231·6* | 221·6 ± 70·9* | 209·9 ± 33·5 |
| LDH U/l | 550 ± 156 | 506 ± 226 | 360 ± 48 |
BVAS = Birmingham Vasculitis Activity Score; VDI = Vasculitis Damage Index, * P < 0·05 in comparison with controls; GC = glucocorticosteroids; GPA = granulomatosis with polyangiitis; PBMC = peripheral blood mononuclear cells; CRP = C‐reactive protein; PMN = polymorphonuclear cells; PLT = platelet; Ig = immunoglobulin; s.d. = standard deviation. Platelet count results presented as median and interquartile range. †Nine patients in the active‐GPA group were treated with cyclophosphamide (cumulative dose 25 g [9, 32]); all patients with inactive‐GPA previously received cyclophosphamide.
Statistical and bioinformatics analysis
Statistical analysis was performed using GraphPad Prism version 5.0 software (GraphPad Software Inc., San Diego, CA, USA). All comparisons were performed with the Mann–Whitney U‐test or one‐way analysis of variance (anova) with Tukey’s post‐hoc or Kruskal–Wallis tests for non‐parametric distributed variables with Dunn’s post‐hoc test. Descriptive statistics was presented as mean ± standard deviation or median ± interquartile range. Correlations between analyzed factors were calculated by Spearman’s rank method. MiRNA/mRNA expression results were analyzed using Expression Suite Software (Thermo Fisher Scientific). A type I statistical error P < 0·05 was considered significant. Bioinformatics pathway analyses were performed using freely available webtools: DAVID Bioinformatics Resources 6.7, EVEX databases, miRTargetLink Human, miRPath DB 2.0 and miRandola 2017 [22, 23, 24, 25, 26].
Results
Neutrophil‐derived extracellular vesicles characteristics and miRNA profiling
We used NTA analysis to evaluate size distribution and concentration of EVs isolated from the culture medium of healthy donors’ neutrophils stimulated with anti‐PR3 IgG. Average size of the EVs was 110 ± 50 nm and concentration: 2·2 × 1011 ± 7 × 109 particles/ml (Fig. 1a). Both CD81 and CD63 EV markers were expressed (Fig. 1b). Next, using the quantitative (q)PCR method we evaluated the miRNA cargo of EVs. Of 728 miRNAs targets, in all EV samples expression was confirmed for 48 miRNA (Supporting information, Table S1) – with the highest expression of miR‐223‐3p (90·9 pM), miR‐142‐3p (1·8 pM) and miR‐19b‐3p (Fig. 1c).
Fig. 1.
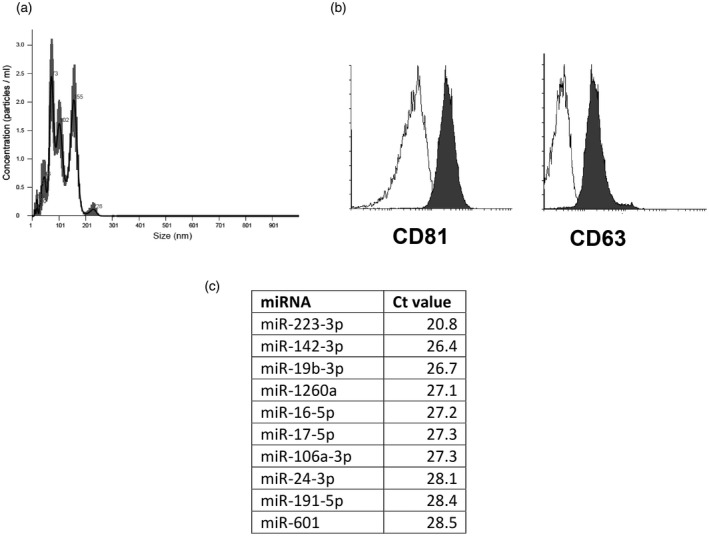
Characteristics of neutrophil‐derived extracellular vesicles (EVs). (a) Representative histogram of a size distribution of extracellular vesicles; (b) expression of extracellular vesicles markers (white = isotype control, dark gray = CD81 or CD63); (c) top 10 miRNAs expressed in neutrophil‐derived EVs.
EVs uptake by endothelial cells
To determine if extracellular vesicles can be taken up by HUVECs we used flow cytometry and fluorescence microscopy. Endothelial cells were cultured in the presence of PKH67‐stained EVs for up to 24 h and analyzed at five time‐points. Increased fluorescence was observed at the first time‐point (0·5 h), then rose steadily to the end of stimulation (24 h, Fig. 2a). The possibility that passive transfer of the PKH67 could produce this result was excluded by comparison of HUVECs cultured in normal conditions (37°C) or on ice (for up to 3 h). Figure 2b shows that HUVECs cultured on ice did not increase fluorescence intensity at any time‐point. Fluorescence microscopy observations (Fig. 2c) were in agreement with the results of the flow cytometry.
Fig. 2.
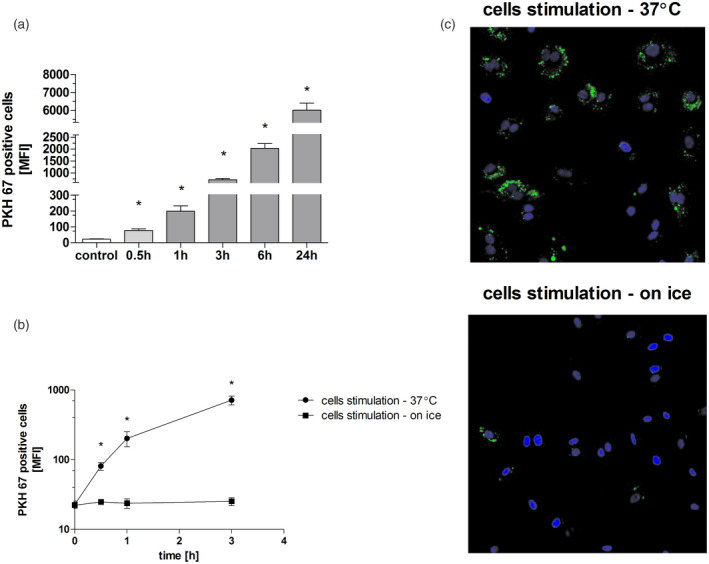
Uptake of neutrophil‐derived extracellular vesicles (EVs) by endothelial cells. (a) Human umbilical endothelial cells (HUVECs) cultured in normal conditions (37°C, 5% CO2), stimulated with PKH67‐stained EVs for up to 24 h and analyzed with the use of flow cytometry. (b) HUVECs cultured in normal conditions (37°C, 5% CO2) or on ice, stimulated with PKH67‐stained EVs for up to 3 h and analyzed with the use of flow cytometry. (c) HUVECs cultured in normal conditions (37°C, 5% CO2) or on ice, stimulated with PKH 67‐stained EVs for 1 h and analyzed with the use of fluorescence microscopy (blue = nuclei stained with Hoechest 33342; green = EVs stained with PKH67 dye). A control of the cells treated with EV not stained with PKH67 was used. In each experiment HUVECs were stimulated with pooled EVs isolated from two different neutrophil donors. Results represent mean ± standard deviation of MnFI of five independent experiments. * P < 0·05 in comparison with control cells, analysis of variance (anova) with Tukey’s post‐hoc test.
Endothelial cell protein evaluation
We used semi‐quantitative protein arrays to analyze the secretome of HUVECs stimulated by neutrophil‐derived EVs. HUVECs were capable to release growth differentiation factor 15 (GDF‐15), Dickkopf‐related protein 1 (DKK‐1), thrombospondin 1(THBS‐1), soluble IL receptor‐like 1 (ST2), endoglin, angiopoietin‐2 (ANGPT‐2) and IL‐8 (Supporting information, Fig. S1). These findings were confirmed by quantitative multiplex immunoassay. All selected proteins were increased in the cell culture medium of HUVECs after EVs stimulation (12 h, all measurements in pg/ml) when compared to non‐stimulated cells: GDF‐15 = 3020 ± 252 versus 1144 ± 155 pg/ml, P < 0·05; DKK‐1 = 2900 ± 219 versus 1741 ± 115 pg/ml, P < 0·05; THBS‐1 = 66·3 ± 16·7 versus 13·9 ± 1·8 pg/ml, P < 0·05; ST2 = 22675 ± 670 versus 12509 ± 1208 pg/ml, P < 0·05; endoglin = 920 ± 162 versus 460 ± 66 pg/ml, P < 0·05; ANGPT‐2 = 29734 ± 1672 versus 14701 ± 1916 pg/ml, P < 0·05; IL‐8 = 1100 ± 301 versus 510 ± 172 pg/ml, P < 0·05 (Fig. 3a). Flow cytometry showed that stimulation of HUVECs with EVs increased expression of VCAM‐1 (Fig. 3b,c).
Fig. 3.
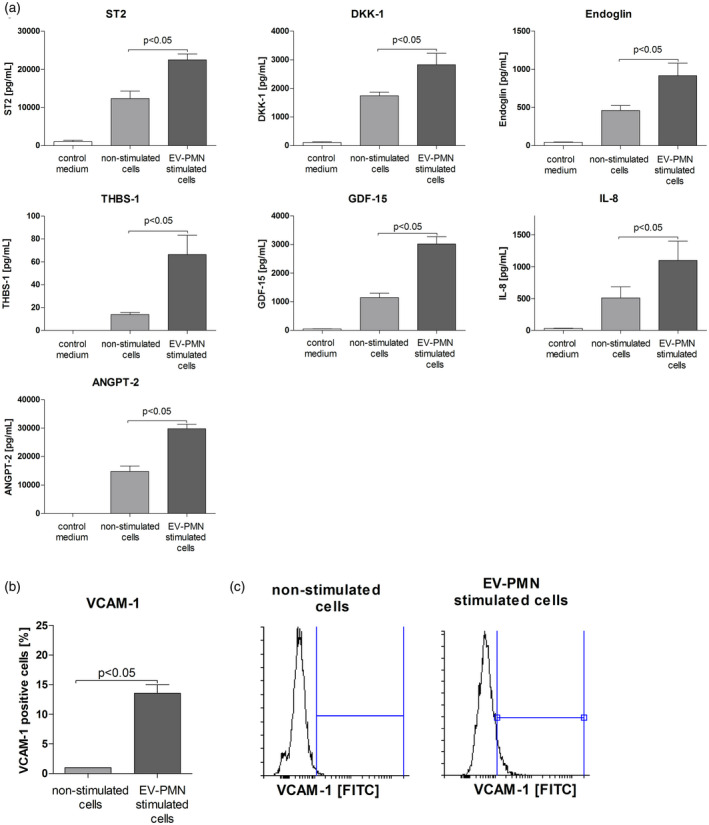
(a) Cell culture supernatant levels of selected proteins released by human umbilical endothelial cells (HUVECs) stimulated with neutrophil‐derived extracellular vesicles (EVs). Control medium = full growth medium supplemented with exosome‐depleted fetal bovine serum (FBS) (1%) and neutrophil‐derived EVs, collected before stimulation. Results for control medium were extrapolated from the standard curve, but were below the detection limit specified by the reagent manufacturer (Bio‐Techne). (b) Surface expression (mean ± standard deviation of MnFI, five independent experiments, Mann–Whitney U‐test) of vascular adhesion molecule‐1 (VCAM‐1) on HUVECs stimulated with EVs. (c) Representative histograms of surface VCAM‐1 expression on HUVECs stimulated with EVs. HUVECs were cultured in full growth medium supplemented with exosomes‐depleted fetal bovine serum (FBS) (1%) and stimulated (6 h) with extracellular vesicles isolated form the culture medium of neutrophils activated by immunoglobulin (Ig)G anti‐proteinase 3 (PR3) antibodies. In each experiment HUVEC cells were stimulated with pooled EVs isolated from two different neutrophil donors.
MiRNA/mRNA profile and regulatory network of EV‐stimulated HUVECs
Of 728 miRNA analyzed in HUVECs stimulated with EVs, we confirmed expression of 450 transcripts. Eleven miRNAs were down‐regulated (fold change < 0·5 in comparison with non‐stimulated cells, P < 0·05): miR‐27a‐3p, miR‐29a‐5p, miR‐30d‐5p, miR‐149‐3p, miR‐361‐3p, miR‐377‐5p, miR‐661, miR‐664a‐3p, miR‐1180‐3p, miR‐1233‐3p and miR‐1275, whereas 10 miRNAs were up‐regulated (fold change > 2 in comparison with non‐stimulated cells, P < 0·05): miR‐15a‐5p, miR‐22‐3p, miR‐101‐3p, miR‐138‐5p, miR‐142‐3p, miR‐223‐3p, miR‐362‐3p, miR‐409‐5p, miR‐484‐3p and miR‐211‐5p, Fig. 4a). Results of mRNA quantification revealed the presence of 512 transcripts of 679 analyzed. Nine mRNAs were down‐regulated (fold change < 0·5 in in comparison with non‐stimulated cells, P < 0·05): STAT3, GATA6, IRAK1, IL6, HRK, CASP8, KLKB1, HTR3A and FASLG, whereas 14 were up‐regulated (fold change > 2 in comparison with non‐stimulated cells, P < 0·05): MyD88, GDF15, THBS1, IL1RL1, DKK1, CXCL8, NALP1, BIRC2, BIRC1, BCL2, VCAM‐1, KLK14 and IL1R2, Fig. 4b).
Fig. 4.
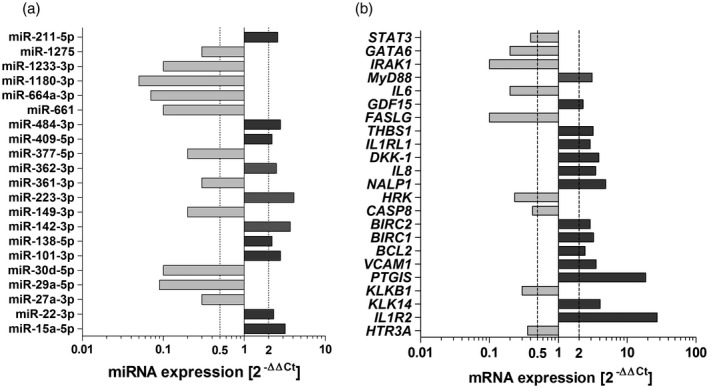
(a) miRNA profile of human umbilical endothelial cells (HUVECs) stimulated with neutrophil‐derived extracellular vesicles (EVs); (b) gene expression in HUVECs stimulated with EVs. HUVECs were cultured in full growth medium supplemented with exosomes‐depleted fetal bovine serum (FBS) (1%) and stimulated (6 h) with extracellular vesicles isolated form the culture medium of neutrophils activated by immunoglobulin (Ig)G anti‐ anti‐proteinase 3 (PR3) antibodies. In each experiment HUVEC cells were stimulated with pooled EVs isolated from two different neutrophil donors. Data represent five independent experiments. MiRNA/mRNA expression was calculated with 2−ΔΔCt formula from glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) and 18SRNA endogenous controls (mRNA) or spiked‐in cel‐39 and RNU‐44 (miRNA) and are presented as median of a fold change (2−ΔΔCt) compared to expression in non‐stimulated HUVECs. Significantly down‐regulated miRNA/mRNA (fold change < 0·5, P < 0·05) are marked light gray and significantly up‐regulated miRNA/mRNA (fold change > 2, P < 0·05) are marked dark gray. Differences between studied groups analyze.
Bioinformatics analyses suggested that stimulation of HUVECs with neutrophil‐derived EVs selectively altered the processes of Toll‐like receptor signalling, nuclear factor kappa B (NF‐κβ) signalling, nucleotide‐binding oligomerization domain (NOD)‐like receptor signalling, apoptosis and cell death, cytokine‐mediated signalling, cell proliferation or inflammatory response (Fig. 5a). Gene ontology terms of differentially expressed genes revealed mutual interactions of studied genes transcripts and miRNAs, aggregated into two regulatory patterns: (1) up‐regulated mRNA/down‐regulated miRNA: DKK1, VCAM‐1, THBS1, GDF‐15, BCL2, CXCL8, ANGPT2, IL1RL1/miR‐377‐3p, miR‐661, miR‐664‐a3p and miR‐30d‐5p and (2) down‐regulated mRNA/up‐regulated miRNA: STAT3, NR3C1, GATA6, IL6, FASLG, IRAK1 , KLKB1/miR‐223‐3p, miR‐211‐5p and miR‐142‐3p (Fig. 5).
Fig. 5.
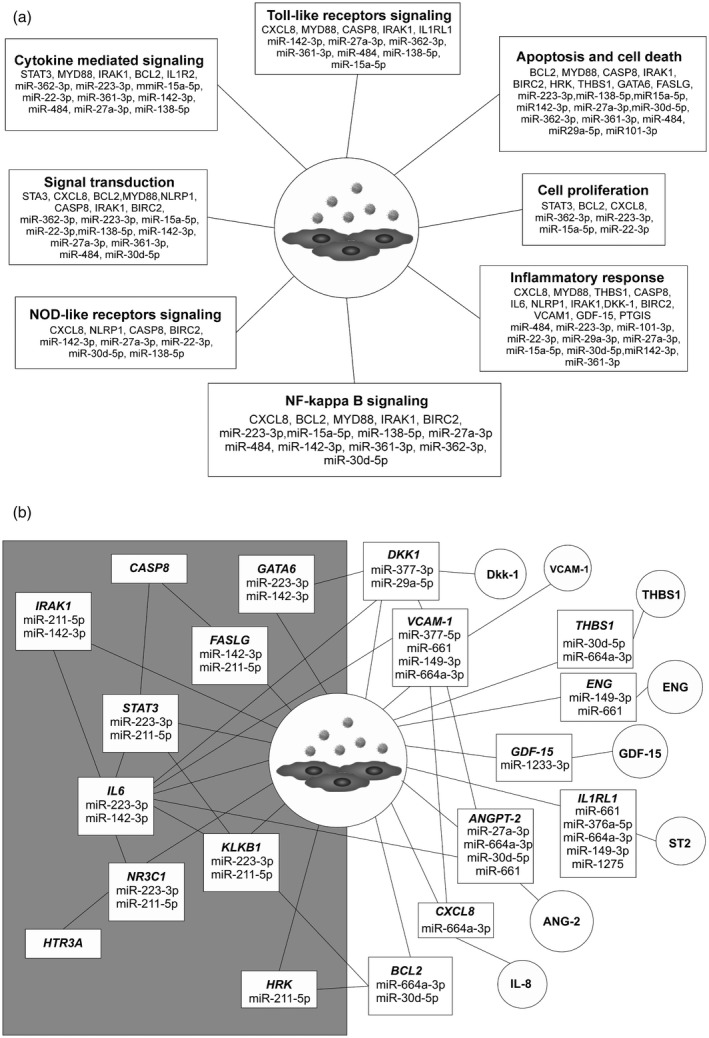
(a) Molecular pathways of human umbilical endothelial cells (HUVECs) stimulated with neutrophil‐derived extracellular vesicles (EVs); (b) miRNA/mRNA/protein network in endothelial cells stimulated with the neutrophil‐derived extracellular vesicles. Circles = proteins released from the cells stimulated with EVs; rectangles = predicted miRNA/mRNA interactions in HUVECs stimulated with EVs. Left side of the panel: down‐regulated mRNA and up‐regulated miRNA; right side of the panel: down‐regulated miRNA and up‐regulated mRNA/proteins. Interaction network is based on the results of miRNA/mRNA/protein profiling of EVs stimulated HUVECs and were constructed using data bases: miRPathDB 2.0, miRTargetLink Human and miRandola 2017 [22, 25, 26].
Transfection of HUVECs with mimic miRNAs
Two miRNAs with the highest expression in EV samples (miR‐223‐3p and miR‐142‐3p) were investigated for their effects on HUVEC release of IL‐8, DKK‐1, THBS‐1, ANGPT‐2, ST2, endoglin and GDF‐15. In the transfection experiments we used commercially available miRNA mimics, where effective cellular entry was documented by a significantly higher expression of miR‐223‐3p and mR‐142‐3p in transfected HUVECs (fold change > 100 versus non‐transfected cells, Supporting information, Fig. S2). Cells transfected with negative control random mimic miRNA did not show any differences in expression of the analyzed miRNAs. Experiments with mimic miRNAs showed their impact upon HUVEC secretome (Fig. 6). The cells transfected with miR‐223‐3p mimic produced IL‐8 (24 h = 998·7 ± 88·2 versus 287 ± 12·3 negative control miRNA mimic and 276 ± 16 pg/ml non‐transfected, P < 0·05; 48 h = 5237·7 ± 771·2 versus 387 ± 80·3 negative control miRNA mimic and 347 ± 46·9 pg/ml non‐transfected, P < 0·05, Fig. 6). However, ST2 and endoglin elevated only on the second day after transfection (ST2, 48 h = 18414·7 ± 621·2 pg/ml versus 14574 ± 305·3 negative control miRNA mimic and 14190 ± 713·9 pg/ml non‐transfected cells; endoglin, 48 h = 757·3 ± 75·2 versus 391 ± 31·3 negative control miRNA mimic and non‐transfected cells 452 ± 52·4 pg/ml, P < 0·05). Analogous transfection of HUVECs with miR‐142‐3p mimic showed similar significant increases of measured proteins: IL‐8, 24 h = 1749·7 ± 204·2 pg/ml; IL‐8, 48 h = 4292·7 ± 199·8 pg/ml; ST2, 48 h = 23601·3 ± 1088·9 pg/ml; endoglin, 24 h = 764 ± 64·4 pg/ml and endoglin, 48 h = 918·7 ± 62·3 pg/ml. There were no differences in DKK‐1, THBS‐1, GDF‐15 and ANGPT‐2 release from HUVECs transfected with mimics or non‐transfected cells.
Fig. 6.

Selected proteins released by human umbilical endothelial cells (HUVECs) transfected with mimic miRNAs. Transfection was performed with miR‐223‐3p mimic (100 pM), miR‐142‐3p mimic (100 pM) or a combination of both miR‐223‐3p and miR‐142‐3p mimics (both 100 pM). As a negative control, commercial miRNA mimic negative control #1 was used (100 pM). The proteins were measured using a custom Luminex assay (Bio‐Techne) after 24 and 48 h post‐transfection in triplicate. Data presented as the median with interquartile range and differences were tested with the use of Kruskal–Wallis non‐parametric analysis of variance (anova) with Dunn’s post‐hoc test.
Clinical blood sample results
In the group of patients with active GPA, 15 patients were newly diagnosed, whereas remaining patients had disease exacerbation. However, no patients with active GPA received immunosuppressive treatment with rituximab, cyclophosphamide or high‐dose corticosteroids before blood sample collection. Laboratory results of circulating blood cell counts showed elevated peripheral blood mononuclear cells (PBMC) and neutrophils in the group of patients with active GPA (P < 0·05 versus healthy controls), while the platelet number was elevated in all GPA patients (Table 1). In all subjects of this study, serum levels of IL‐8, GDF‐15, DKK‐1, THBS‐1, ST2, ANGPT‐2 and IL‐33 (the ligand to ST2) were measured (Fig. 7a). GPA patients had significantly increased levels of all the following cytokines: IL‐8, DKK‐1, ST2, GDF‐15, THBS‐1, ANGPT‐2 and IL‐33. THBS‐1 was increased both in the active phase and in remission (609 ± 38 ng/ml and 643 ± 8·40 ng/ml), compared with thrombospondin in healthy controls (437·5 ± 28 ng/ml, P < 0·05). A similar increase was observed for GDF‐15: 993 ± 121·5 pg/ml in active disease and 863 ± 114 pg/ml in remission versus 301 ± 32 pg/ml in healthy controls. IL‐8, ST2, IL‐33 and DKK‐1 were highest in active GPA (IL‐8 = 4·94 ± 0·9 pg/ml; ST2 = 17173 ± 2596 pg/ml; IL‐33 = 1·8 ± 0·3 pg/ml; DKK‐1 = 632 ± 102 pg/ml) but not in remission (IL‐8 = 1·5 ± 0·24 pg/ml; ST2 = 10 932 ± 796 pg/ml; IL‐33 = 0·9 ± 0·06 pg/ml; DKK‐1 = 406 ± 40 pg/ml) or in healthy controls (IL‐8 = 0·7 ± 0·12 pg/ml; ST2 = 5810 ± 399 pg/ml, IL‐33 = 1·03 ± 0·07 pg/ml, DKK‐1= 301 ± 19 pg/ml, P < 0·05). Moreover, DKK‐1 and ST2 levels correlated with the disease activity score (DKK‐1, σ = 0·73, P < 0·05; ST2, σ = 0·53, P < 0·05, Fig. 7b). However, we did not observe any correlation between measured serum proteins and clinical parameters such as blood PMN and PLT count, corticosteroids dose or VDI. Receiver operator characteristics (ROC) demonstrated the best discrimination between GPA patients versus controls for ST2 (area under ROC = 0.87; 95% confidence interval 0·81–0·94), whereas discrimination based on DKK‐1 was inferior to ST2 (Supporting information, Fig. S3).
Fig. 7.
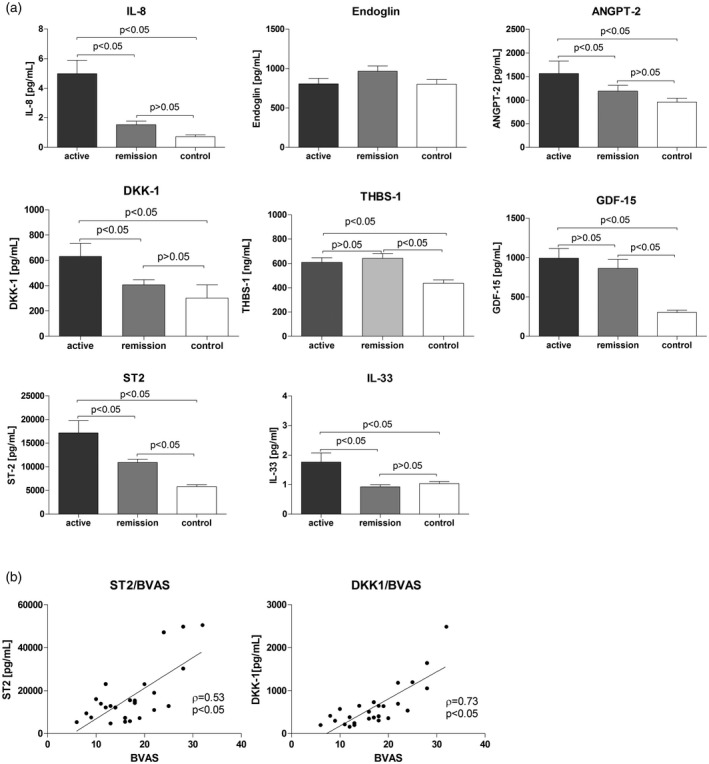
Blood levels of selected proteins in granulomatosis with polyangiitis patients (active GPA n = 26, remission of GPA n = 29) and healthy controls (n = 31). (a) Serum levels of selected proteins in patients, data presented as median with interquartile range, differences tested with the use of Kruskal–Wallis non‐parametric analysis of variance (anova) with Dunn’s post‐hoc test; (b) correlation between serum levels of ST2 or DKK1 and disease activity score (BVAS) in patients with active GPA, Spearman’s rank correlation.
Discussion
Interaction between neutrophils and vascular endothelium was previously studied in GPA [27]. Despite the fact that endothelial cells did not produce PR3 constitutively, Muller et al. [28] showed that anti‐PR3 antibodies could bind to HUVECs and induce E‐selectin expression on their surface. In the current study we wondered if neutrophil‐derived EVs could transfer mediators to explain these findings. There have been relatively few studies published on neutrophil‐derived microparticles. Mesri et al. [16] first showed that overnight incubation of HUVECs with neutrophils microparticles (NeuMPs) resulted in HUVEC activation which indicated that NeuMPs played an important role in vascular inflammation. Hong et al. [9] observed that stimulation of neutrophils with anti‐PR3 IgG antibodies isolated from patients with GPA caused release of NeuMPs, which could activate endothelial cells and stimulate release of IL‐8 and IL‐6. Results presented in our study are in line with these observations. Stimulation of HUVECs with neutrophil‐derived EVs up‐regulated both CXCL8 mRNA and IL‐8 release. Increased IL‐8 release was also observed when cells were transfected with miRNA mimics miR‐223‐3p or miR‐142‐3p, highly expressed in both purified EV specimens and EV‐stimulated HUVECs. Surprisingly, we noticed down‐regulation of IL‐6 expression in HUVECs after stimulation with EVs. Moreover, Pitanga et al. [29] reported that NeuMPs led to endothelial damage, while our observations indicated decreased expression of pro‐apoptotic genes such as CASP8, HRK or FASLG. A possible explanation for these discrepancies could be related to the methodological approach – in both referenced studies [9, 29] microparticles rich with large extracellular vesicles were used, while in our experiments small extracellular vesicles (exosomes) were isolated for stimulation. Several reports indicated that exosomes vary from microvesicles in their protein cargo because of a different biogenesis. Namely, exosomes originate from the endocytic compartment, whereas microparticles derive from budding plasma membranes of the cell. [11, 30]. In our study we did not analyze the protein cargo of EVs; however, it was described that NeuMPs contained MPO, cathepsin G, elastase and other proteins potentially damaging endothelial cells [31]. Small and large extracellular vesicles can also have different miRNA profile [32]. In our study, based on bioinformatics we targeted miRNAs which could interfere with expression of differentially expressed genes: IL‐6, STAT3, NR3C1, KLKB1, CASP8, GATA6 and FASLG in HUVECs. Two of these (miR‐223‐3p and miR‐142‐3p) showed the highest expression in both EVs samples and EV‐stimulated HUVECs. The third miR‐211‐5p was undetectable in EVs but was up‐regulated in stimulated HUVECs. MiR‐223‐3p regulates IL‐6 expression [33] but also modifies biosynthesis of two transcription factors NR3C1, signal transducer and activator of transcription (STAT)‐3 and kallikrein KLKB1 [26]. Moreover, down‐regulation of IL‐6 can suppress expression of NR3C1, STAT3 and KLKB1 [34, 35]. Among predicted targets for miR‐211‐5p are also both transcription factors STAT3 and NR3C1, which regulate mRNA level of FASLG [26]. We did not focus upon any miRNA targeting CASP8, but levels of this pro‐apoptotic gene could be modified by STAT3 and FASLG activities [36, 37].
MiR‐223‐3p and miR‐142‐3p can suppress expression of transcription factor GATA binding protein 6 (GATA6) [26]. This protein is highly expressed in the vascular wall [38, 39, 40]. Zhong et al. reported that GATA6 modulated the WNT/β–catenin pathway by inhibition of DKK‐1 translation [41]. Our results confirmed this observation, as we observed down‐regulation of GATA6 expression and up‐regulation of DKK‐1 in HUVECs mRNA and the secretome. We also observed elevated levels of circulating DKK‐1 in GPA patients. Despite the lack of published data on DKK‐1 levels in serum of patients with vasculitis, this protein was increased in rheumatoid arthritis or systemic lupus erythematosus patients [42, 43]. Moreover, in our study, serum DKK‐1 levels correlated with disease activity score (BVAS) in active GPA patients, indicating that DKK‐1 could be of use as a biomarker of GPA exacerbation. Expression of DKK‐1 is suppressed by IL‐6, at least in synovial fluid of rheumatoid arthritis patients [44]. We also suggest a possible link between miR‐223‐3p and miR‐142‐3p over‐expression and the WNT/β–catenin pathway. The WNT/β–catenin pathway is crucial during embryonic development and in adult homeostasis [45]. It is de‐regulated in several cancers, but its involvement in endothelial function is unclear. Guo et al. [46] and Malhotra et al. [47] reported on interactions of DKK‐1 with another protein over‐expressed in our experiments – VCAM‐1. VCAM‐1 is an essential endothelial sialoglycoprotein mediating leucocyte adhesion and endothelial transmigration [48]. Elevated levels of soluble VCAM‐1 were reported in rheumatoid arthritis [49]. Furthermore, VCAM‐1 is regulated by another vasoactive protein angiopoietin‐2 [50]. Both can be modulated by IL‐6 [51, 52], which was suppressed, and by elevated IL‐8 in our study following EV stimulation [53, 54]. Of note, miR‐661 and miR664a‐3p were suppressed in our study. These miRNAs can silence VCAM‐1 and ANGPT‐2 [26]. ANGPT‐2 is a classical mediator of angiogenesis and inflammation activating the angiopoietin/Tie (tyrosine kinase with Ig and EGF homology domains) signalling pathway [55]. Lomas‐Neira et al. [56] reported that during neutrophil–endothelial interaction, endothelial cells can produce ANGPT‐2. Inhibition of ANGPT‐2 decreases neutrophils influx. In our experiments, a higher secretion of ANGPT‐2 and up‐regulation of ANGPT2 mRNA was present in HUVECs stimulated with neutrophil‐derived EVs. Thus, our results corroborate the published observation [57] reporting elevated levels of circulating ANGPT‐2 in serum of GPA patients with exacerbation of the disease.
Because miR‐661 and miR‐664a‐3p expression was decreased in HUVECs after EV stimulation, this could explain over‐expression of the IL1RL1 transcript encoding ST2. This is a receptor for IL‐33, and ST2/IL‐33 axis is well recognized as a positive regulator of neutrophil chemotaxis [58] and migration [59]. Focusing upon GPA pathophysiology, Van der Windt et al. [60] reported that ST2 can stimulate neutrophils to release neutrophil extracellular traps (NET). In our previous studies we found increased levels of NETs biomarkers (DNA–MPO complexes, mtDNA) in patients with active GPA [61, 62]. Consistently, elevated secretion of ST2 by HUVECs stimulated with EVs or by specific miRNAs mimics, seems to establish a paradigm involving interaction of neutrophils with endothelial cells via EVs in the pathophysiology of GPA. Elevated serum levels of ST2 and IL‐33 in GPA patients observed in this study and reported by other groups [63, 64] along with the positive correlation between ST2 and BVAS, were supportive for validity of our in‐vitro findings.
In conclusion, we designed and accomplished experiments to test for interaction between EVs released from neutrophil by anti‐PR3 IgG antibodies and endothelial cells. By quantitative assessment of miRNA/mRNA/proteins in HUVECs, we found a profound response of the endothelial cell line stimulated with EVs, which recapitulated many elements of GPA pathophysiology. Results from our in‐vitro model were in agreement with clinical measurements of serum cytokines in GPA patients. Apparently, a switch of HUVEC phenotypes following exposure to neutrophil‐derived EVs evokes a miRNA/mRNA profile, suggesting prosurvival, and is permissive for neutrophil interactions properties, marked by a release of several proteins involved in immune cell adhesion, chemotaxis and migration.
There are, however, limitations of this study as a consequence of the experimental approach. A simplified model was used to screen for neutrophil‐derived EV function. In our study we used neutrophils isolated from the healthy donors, whereas we and others have reported that neutrophils from GPA patients are intrinsically different [65, 66], thus their EV cargo could instigate a different response of endothelial cells. Experiments with miRNA transfection at least partly supported EV delivery of miR‐223‐3p and miR‐142‐3p, stimulating endothelial cells to produce IL‐8, ST2 and endoglin. However, the complexity of the miRNA regulatory network remains unexplained, and warrants future studies. Ultimately, the prothrombotic phenotype of endothelial cells was another important feature of GPA pathophysiology, which was not investigated in the current study.
Disclosures
The authors declare no conflicts of interest.
Author contributions
M. Su. performed experiments, analyzed data, discussed result and wrote the draft of the manuscript, S. P. participated in clinical sample collection and performed part of the experiments, J. K. recruited patients and assessed clinical data, M. Sa. discussed the concept of this study and its results and supervised the manuscript.
Supporting information
Fig S1. Screening for proteins secreted from neutrophil‐derived HUVECs into the culture medium.
Fig S2. Real‐time PCR evaluation of the cellular entry of miR‐223‐3p and miR‐142‐3p mimics delivered to HUVECs by transfection.
Fig S3. Biomarker properties of serum DKK‐1 and ST2 in GPA patients.
Table S1. MiRNAs expressed in neutrophil‐derived EVs.
Acknowledgements
This study supported by the National Centre of Science in Poland, grant number: 2016/21/D/NZ6/02123.
Contributor Information
J. Kosałka‐Węgiel, Email: joanna.kosalka@uj.edu.pl.
M. Sanak, Email: marek.sanak@uj.edu.pl.
Data Availability Statement
Raw qRT–PCR data are available from the authors upon request.
References
- 1. Geetha D, Jefferson JA. ANCA‐associated vasculitis: core curriculum 2020. Am J Kidney Dis 2020; 75:124–37. [DOI] [PubMed] [Google Scholar]
- 2. Sinico RA, Radice A. Antineutrophil cytoplasmic antibodies (ANCA) testing: detection methods and clinical application. Clin Exp Rheumatol 2014; 32:112–7. [PubMed] [Google Scholar]
- 3. Osman MS, Tervaert JWC. Anti‐neutrophil cytoplasmic antibodies (ANCA) as disease activity biomarkers in a ‘personalized medicine approach’ in ANCA‐associated vasculitis. Curr Rheumatol Rep 2019; 21:76. [DOI] [PubMed] [Google Scholar]
- 4. Ozaki S. ANCA‐associated vasculitis: diagnostic and therapeutic strategy. Allergol Int 2007; 56:87–96. [DOI] [PubMed] [Google Scholar]
- 5. Kelley JM, Monach PA, Ji C et al. IgA and IgG antineutrophil cytoplasmic antibody engagement of Fc receptor genetic variants influences granulomatosis with polyangiitis. Proc Natl Acad Sci USA 2011; 108:20736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee KH, Kronbichler A, Park DDY et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: a comprehensive review. Autoimmun Rev 2017; 16:1160–73. [DOI] [PubMed] [Google Scholar]
- 7. Surmiak M, Kaczor M, Sanak M. Expression profile of proinflammatory genes in neutrophil‐enriched granulocytes stimulated with native anti‐PR3 autoantibodies. J Physiol Pharmacol 2012; 63:249–56. [PubMed] [Google Scholar]
- 8. Rarok AA, Limburg PC, Kallenberg CGM. Neutrophil‐activating potential of antineutrophil cytoplasm autoantibodies. J Leukoc Biol 2003; 74:3–15. [DOI] [PubMed] [Google Scholar]
- 9. Hong Y, Eleftheriou D, Hussain AAK et al. Anti‐neutrophil cytoplasmic antibodies stimulate release of neutrophil microparticles. J Am Soc Nephrol 2012; 23:49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van der Pol E, Böing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev 2012; 64:676–705. [DOI] [PubMed] [Google Scholar]
- 11. Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 2014; 30:255–89. [DOI] [PubMed] [Google Scholar]
- 12. Becker A, Thakur BK, Weiss JM et al. Extracellular vesicles in cancer: cell‐to‐cell mediators of metastasis. Cancer Cell 2016; 30:836–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 2018; 19:213–28. [DOI] [PubMed] [Google Scholar]
- 14. Oggero S, Austin‐Williams S, Norling LV. The contrasting role of extracellular vesicles in vascular inflammation and tissue repair. Front Pharmacol 2019; 10:1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Surmiak M, Gielicz A, Stojkov D et al. LTB4 and 5‐oxo‐ETE from extracellular vesicles stimulate neutrophils in granulomatosis with polyangiitis. J Lipid Res 2020; 61:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mesri M, Altieri DC. Leukocyte microparticles stimulate endothelial cell cytokine release and tissue factor induction in a JNK1 signaling pathway. J Biol Chem 1999; 274:23111–8. [DOI] [PubMed] [Google Scholar]
- 17. Osteikoetxea X, Balogh A, Szabó‐Taylor K et al. Improved characterization of EV preparations based on protein to lipid ratio and lipid properties. PLOS ONE 2015; 10:e0121184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suárez H, Gámez‐Valero A, Reyes R et al. A bead‐assisted flow cytometry method for the semi‐quantitative analysis of extracellular vesicles. Sci Rep 2017; 7:11271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc 2008; 3:1101–8. [DOI] [PubMed] [Google Scholar]
- 20. Leavitt RY, Fauci AS, Bloch DA et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum 1990; 33:1101–7. [DOI] [PubMed] [Google Scholar]
- 21. Hellmich B, Flossmann O, Gross WL et al. EULAR recommendations for conducting clinical studies and/or clinical trials in systemic vasculitis: focus on anti‐neutrophil cytoplasm antibody‐associated vasculitis. Ann Rheum Dis 2007; 66:605–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Russo F, Di Bella S, Vannini F et al. miRandola 2017: a curated knowledge base of non‐invasive biomarkers. Nucleic Acids Res 2018; 46:D354–D359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 24. Van Landeghem S, Björne J, Wei C‐H et al. Large‐scale event extraction from literature with multi‐level gene normalization. PLOS ONE 2013; 8:e55814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Enright AJ, John B, Gaul U et al. MicroRNA targets in Drosophila. Genome Biol 2003; 5:R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kehl T, Kern F, Backes C et al. miRPathDB 2.0: a novel release of the miRNA Pathway Dictionary Database. Nucleic Acids Res 2020; 48:D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haubitz M, Dhaygude A, Woywodt A. Mechanisms and markers of vascular damage in ANCA‐associated vasculitis. Autoimmunity 2009; 42:605–14. [DOI] [PubMed] [Google Scholar]
- 28. Muller Kobold AC, van Wijk RT, Franssen CF et al. In vitro up‐regulation of E‐selectin and induction of interleukin‐6 in endothelial cells by autoantibodies in Wegener’s granulomatosis and microscopic polyangiitis. Clin Exp Rheumatol 1999; 17:433–40. [PubMed] [Google Scholar]
- 29. Pitanga TN, de França L, Rocha VC et al. Neutrophil‐derived microparticles induce myeloperoxidase‐mediated damage of vascular endothelial cells. BMC Cell Biol 2014; 15:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Akers JC, Gonda D, Kim R, Carter BS, Chen CC. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus‐like vesicles, and apoptotic bodies. J Neurooncol 2013; 113:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dalli J, Montero‐Melendez T, Norling LV et al. Heterogeneity in neutrophil microparticles reveals distinct proteome and functional properties. Mol Cell Proteomics 2013; 12:2205–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen M, Xu R, Rai A et al. Distinct shed microvesicle and exosome microRNA signatures reveal diagnostic markers for colorectal cancer. PLOS ONE 2019; 14:e0210003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu J, Niu P, Zhao Y et al. Impact of miR‐223‐3p and miR‐2909 on inflammatory factors IL‐6, IL‐1ß, and TNF‐α, and the TLR4/TLR2/NF‐κB/STAT3 signaling pathway induced by lipopolysaccharide in human adipose stem cells. PLOS ONE 2019; 14:e0212063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang WL, Yeh HH, Lin CC et al. Signal transducer and activator of transcription 3 activation up‐regulates interleukin‐6 autocrine production: a biochemical and genetic study of established cancer cell lines and clinical isolated human cancer cells. Mol Cancer 2010; 9:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dovio A, Masera RG, Sartori ML, Racca S, Angeli A. Autocrine up‐regulation of glucocorticoid receptors by interleukin‐6 in human osteoblast‐like cells. Calcif Tissue Int 2001; 69:293–8. [DOI] [PubMed] [Google Scholar]
- 36. Fulda S, Debatin KM. IFNγ sensitizes for apoptosis by upregulating caspase‐8 expression through the Stat1 pathway. Oncogene 2002; 21:2295–308. [DOI] [PubMed] [Google Scholar]
- 37. Schlapbach R, Spanaus KS, Malipiero U et al. TGF‐β induces the expression of the FLICE‐inhibitory protein and inhibits Fas‐mediated apoptosis of microglia. Eur J Immunol 2000; 30:3680–8. [DOI] [PubMed] [Google Scholar]
- 38. Yin F, Herring BP. GATA‐6 can act as a positive or negative regulator of smooth muscle‐specific gene expression. J Biol Chem 2005; 280:4745–52. [DOI] [PubMed] [Google Scholar]
- 39. Suzuki E, Evans T, Lowry J et al. The human GATA‐6 gene: structure, chromosomal location, and regulation of expression by tissue‐specific and mitogen‐responsive signals. Genomics 1996; 38:283–90. [DOI] [PubMed] [Google Scholar]
- 40. Zhuang T, Liu J, Chen X et al. Cell‐specific effects of GATA (GATA zinc finger transcription factor family)‐6 in vascular smooth muscle and endothelial cells on vascular injury neointimal formation. Arterioscler Thromb Vasc Biol 2019; 39:888–901. [DOI] [PubMed] [Google Scholar]
- 41. Zhong Y, Wang Z, Fu B et al. GATA6 activates Wnt signaling in pancreatic cancer by negatively regulating the Wnt antagonist Dickkopf‐1. PLOS ONE 2011; 6:e22129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xue J, Yang J, Yang L et al. Dickkopf‐1 Is a biomarker for systemic lupus erythematosus and active lupus nephritis. J Immunol Res 2017; 2017:6861575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seror R, Boudaoud S, Pavy S et al. Increased Dickkopf‐1 in recent‐onset rheumatoid arthritis is a new biomarker of structural severity. Data from the ESPOIR cohort. Sci Rep 2016; 6:18421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yeremenko N, Bisoendial R, Zwerina J et al. TNF and IL‐6 differentially regulate the production of DKK‐1, a master regulator of bone remodelling, by fibroblast‐like synoviocytes. J Transl Med 2010; 8:P43. [Google Scholar]
- 45. MacDonald BT, Tamai K, He X. Wnt/β‐catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17:9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Malhotra S, Kincade PW. Canonical Wnt pathway signaling suppresses VCAM‐1 expression by marrow stromal and hematopoietic cells. Exp Hematol 2009; 37:19–30.18951693 [Google Scholar]
- 47. Guo Y, Mishra A, Howland E et al. Platelet‐derived Wnt antagonist Dickkopf‐1 is implicated in ICAM‐1/VCAM‐1‐mediated neutrophilic acute lung inflammation. Blood 2015; 126:2220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kong DH, Kim YK, Kim MR, Jang JH, Lee S. Emerging roles of vascular cell adhesion molecule‐1 (VCAM‐1) in immunological disorders and cancer. Int J Mol Sci 2018; 19:1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang L, Ding Y, Guo X, Zhao Q. Role and mechanism of vascular cell adhesion molecule‐1 in the development of rheumatoid arthritis. Exp Ther Med 2015; 10:1229–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kümpers P, van Meurs M, David S et al. Time course of angiopoietin‐2 release during experimental human endotoxemia and sepsis. Crit Care 2009;13:R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mofarrahi M, Hussain SNA. Expression and functional roles of Angiopoietin‐2 in skeletal muscles. PLOS ONE 2011; 6:e22882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Oh JW, Van Wagoner NJ, Rose‐John S, Benveniste EN. Role of IL‐6 and the soluble IL‐6 receptor in inhibition of VCAM‐1 gene expression. J Immunol 1998; 161:4992–9. [PubMed] [Google Scholar]
- 53. Manna SK, Ramesh GT. Interleukin‐8 induces nuclear transcription factor‐κB through a TRAF6‐dependent pathway. J Biol Chem 2005; 280:7010–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Grammas P. Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer’s disease. J Neuroinflammation 2011; 8:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Akwii RG, Sajib MS, Zahra FT, Mikelis CM. Role of angiopoietin‐2 in vascular physiology and pathophysiology. Cells 2019; 8:471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lomas‐Neira J, Venet F, Chung CS et al. Neutrophil–endothelial interactions mediate angiopoietin‐2‐associated pulmonary endothelial cell dysfunction in indirect acute lung injury in mice. Am J Respir Cell Mol Biol 2014; 50:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kümpers P, Hellpap J, David S et al. Circulating angiopoietin‐2 is a marker and potential mediator of endothelial cell detachment in ANCA‐associated vasculitis with renal involvement. Nephrol Dial Transplant 2009; 24:1845–50. [DOI] [PubMed] [Google Scholar]
- 58. Watanabe M, Sada M, Nakamoto K et al. Soluble ST2 (sST2)/IL‐33 balance regulates neutrophil chemotaxis release from bronchial epithelial cells. Am J Respir Crit Care Med 2018; 197:A1314. [Google Scholar]
- 59. Artru F, Bou Saleh M, Maggiotto F et al. IL‐33/ST2 pathway regulates neutrophil migration and predicts outcome in patients with severe alcoholic hepatitis. J Hepatol 2020; 72:1052–61. [DOI] [PubMed] [Google Scholar]
- 60. Van Der Windt D, Yazdani HO, Chen H‐W et al. ST2 Stimulation on neutrophils induces neutrophil extracellular trap formation and exacerbates injury after hepatic ischemia/reperfusion. J Am Coll Surg 2016; 223:S79. [Google Scholar]
- 61. Surmiak M, Hubalewska‐Mazgaj M, Wawrzycka‐Adamczyk K et al. Neutrophil‐related and serum biomarkers in granulomatosis with polyangiitis support extracellular traps mechanism of the disease. Clin Exp Rheumatol 2016; 34:S98–104. [PubMed] [Google Scholar]
- 62. Surmiak MP, Hubalewska‐Mazgaj M, Wawrzycka‐Adamczyk K et al. Circulating mitochondrial DNA in serum of patients with granulomatosis with polyangiitis. Clin Exp Immunol 2015; 181:150–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hladinova Z, Hruskova Z, Svobodova B et al. Increased levels of soluble ST2 in patients with active newly diagnosed ANCA‐associated vasculitis. Mediat Inflamm 2015; 2015:603750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hoffmann JC, Patschan D, Dihazi H et al. Cytokine profiling in anti neutrophil cytoplasmic antibody‐associated vasculitis: a cross‐sectional cohort study. Rheumatol Int 2019; 39:1907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Surmiak M, Hubalewska‐Mazgaj M, Wawrzycka‐Adamczyk K, Musiał J, Sanak M. Delayed neutrophil apoptosis in granulomatosis with polyangiitis: dysregulation of neutrophil gene signature and circulating apoptosis‐related proteins. Scand J Rheumatol 2020; 49:57–67. [DOI] [PubMed] [Google Scholar]
- 66. Tang S, Zhang Y, Yin SW et al. Neutrophil extracellular trap formation is associated with autophagy‐related signalling in ANCA‐associated vasculitis. Clin Exp Immunol 2015; 180:408–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Screening for proteins secreted from neutrophil‐derived HUVECs into the culture medium.
Fig S2. Real‐time PCR evaluation of the cellular entry of miR‐223‐3p and miR‐142‐3p mimics delivered to HUVECs by transfection.
Fig S3. Biomarker properties of serum DKK‐1 and ST2 in GPA patients.
Table S1. MiRNAs expressed in neutrophil‐derived EVs.
Data Availability Statement
Raw qRT–PCR data are available from the authors upon request.