

Abstract
Stretch-sensitive Ia afferent monosynaptic connections with motoneurons form the stretch reflex circuit. After nerve transection, Ia afferent synapses and stretch reflexes are permanently lost, even after regeneration and reinnervation of muscle by motor and sensory afferents is completed in the periphery. This loss greatly affects full recovery of motor function. However, after nerve crush, reflex muscle forces during stretch do recover after muscle reinnervation and reportedly exceed 140% baseline values. This difference might be explained by structural preservation after crush of Ia afferent synapses on regenerating motoneurons and decreased presynaptic inhibitory control. We tested these possibilities in rats after crushing the tibial nerve (TN), and using Vesicular GLUtamate Transporter 1 (VGLUT1) and the 65 kDa isoform of glutamic acid-decarboxylase (GAD65) as markers of, respectively, Ia afferent synapses and presynaptic inhibition (P-boutons) on retrogradely labeled motoneurons. We analyzed motoneurons during regeneration (21 days post crush) and after they reinnervate muscle (3 months). The results demonstrate a significant loss of VGLUT1 terminals on dendrites and cell bodies at both 21 days and 3 months post-crush. However, in both cellular compartments, the reductions were small compared to those observed after TN full transection. In addition, we found a significant decrease in the number of GAD65 P-boutons per VGLUT1 terminal and their coverage of VGLUT1 boutons. The results support the hypothesis that better preservation of Ia afferent synapses and a change in presynaptic inhibition could contribute to maintain or even increase the stretch reflex after nerve crush and by difference to nerve transection.
Keywords: glutamic acid decarboxylase, peripheral nerve injury, presynaptic terminals, proprioceptive disorders, rats, reflex, stretch, Wistar, RRID: AB_ 2313637, RRID: AB_887875, RRID: AB_528264, RRID: AB_2315776, RRID: AB_2340411, RRID: AB_ 2340819, RRID: nif_0000_10294, RRID: nif-0000-00314, RRID: nif_0000_00313
1 |. INTRODUCTION
Peripheral nerve injuries may arise from a variety of trauma from nerve crush to complete lacerations, and affect different types of nerves and body regions. Although nerve regeneration occurs in the periphery, overall recovery of motor function is generally disappointing; only 10% of adult patients regain near-normal function after median or ulnar injuries that require surgical repair, although outcomes are much better after milder injuries (Brushart, 2011; Lundborg, 2003). It is thus important to understand the neurobiological mechanisms that impair motor function recovery to different levels after different injuries.
Axon regeneration is fairly inefficient in patients with injuries in large nerves or that occur at long distances from their targets (e.g., a brachial plexus injury). One problem is that the velocity of peripheral axon regeneration is slow (1–3 mm/day) (Sunderland, 1947) and regeneration capacity declines with time (Fu & Gordon, 1995a, 1995b). In addition, pathfinding signals found in early development are not present in the adult and regenerating axons have difficulties finding the correct paths and can be misrouted to wrong muscles or targets (Allodi, Udina, & Navarro, 2012; Brushart & Mesulam, 1980; Brushart, Tarlov, & Mesulam, 1983). Not surprisingly, much experimental attention has focused on mechanisms that can improve the efficiency and specificity of regeneration in the periphery (Chan, Gordon, Zochodne, & Power, 2014; Gordon & English, 2016). However, even when successful peripheral regeneration is achieved after surgical repair, persistent motor deficits remain. For example, full nerve transection and repair of small distal nerves projecting to single hind limb muscles (medial gastrocnemius [MG] nerve or quadriceps nerves) result in rapid and specific muscle reinnervation and full recovery of force within weeks, but stretch reflexes are permanently lost. This occurs even though stretch-sensitive sensory proprioceptors (i.e., Ia afferents) efficiently reinnervate muscle spindle receptors and are capable of encoding and transmitting information about muscle length (Bullinger, Nardelli, Pinter, Alvarez, & Cope, 2011; Cope, Bonasera, & Nichols, 1994; Haftel et al., 2005; Lyle, Prilutsky, Gregor, Abelew, & Nichols, 2016). Deficits in feedback information about muscle length manifest in abnormal interjoint coordination during walking, higher than normal co-contraction of antagonists around single joints and errors in slope walking (Abelew, Miller, Cope, & Nichols, 2000; Maas, Prilutsky, Nichols, & Gregor, 2007; Sabatier, To, Nicolini, & English, 2011). Some of these deficits are compensated by adjustments in the unaffected hind limb joints such that overall limb kinematics and general limb functions are preserved (Chang, Auyang, Scholz, & Nichols, 2009). However, feedback information about muscle length has many important roles during ongoing motor activity including opposing influences from force signals generated by Ib Golgi tendon organs in nearby muscles (Lyle et al., 2016). Their loss implies that tasks involving high forces and/or rapid and large muscle lengthening (steep downslopes) should predictably show significant deficits in performance after nerve regeneration (Abelew et al., 2000; Lyle et al., 2016; Maas et al., 2007).
Deficits in proprioceptive information relayed by stretch-encoding Ia afferents arise because their central synaptic arbors inside the spinal cord are removed from the ventral horn (Alvarez, Bullinger, Titus, Nardelli, & Cope, 2010; Alvarez et al., 2011; Bullinger et al., 2011). This causes the loss of most synaptic collaterals from Ia afferents in lamina IX, and to lesser extent those in lamina VII, greatly denervating motoneurons. Motoneurons lose approximately 60–65% of synapses from Ia afferents on the dendritic arbor and 85–90% on the cell bodies (Rotterman, Nardelli, Cope, & Alvarez, 2014). Functionally, single Ia afferents in normal uninjured animals give input to around 93% of the motor pool, but after injury and complete regeneration in the periphery they only innervate 17% of the pool motoneurons (Bullinger et al., 2011) and remaining synaptic potentials evoked by stretching the muscle are greatly diminished (Bullinger et al., 2011; Haftel et al., 2005). In summary, injured Ia afferent axons can regenerate in the periphery, reinnervate muscle and muscle spindles and become functional (Haftel et al., 2005), however, their central branches do not reestablish the synapses lost in the ventral horn (Alvarez et al., 2010; Bullinger et al., 2011; Rotterman et al., 2014) causing a sensory insufficiency in the stretch reflex and in central processing of muscle length information that alters modulation of motoneuron activity and impairs motor performance.
Interestingly, the stability of the stretch reflex is dependent on the type of nerve injury. In a pioneering study prompted by the types of injuries sustained by British soldiers in World War II, D. Barker and J. Z. Young compared spinal reflexes in rabbits in which peripheral nerves were either fully transected or crushed (Barker & Young, 1947). They found that while stretch reflexes were frequently small or not detectable after full nerve transections, they were fully recovered after nerve crush injuries. A more recent study (Prather et al., 2011), reported supranormal stretch reflexes in cats after recovery from MG nerve crush. Interestingly, Prather et al. (2011) found 70% preservation of stretch-evoked excitatory synaptic potentials (strEPSPs) in MG motoneurons and this contrasts with the large depression that occurs after complete transection of the same nerve (Bullinger et al., 2011; Haftel et al., 2005). These results predict that Ia afferent synapses should be retained to a larger extent after nerve crush compared to nerve transection.
Increased synaptic preservation is certainly a pre-requisite for the results described in Prather et al. (2011), however, additional factors need to be considered to explain why dynamic stretch reflex forces were increased by 145% or greater after nerve crush. This increase in reflex forces can, potentially, be explained by alternative changes in spinal cord circuitry. Presynaptic inhibition of cutaneous sensory afferent mechanoreceptor synapses is reduced in the dorsal horn following peripheral nerve injury, and this is associated with a loss of GABAergic synapses (Castro-Lopes, Tavares, & Coimbra, 1993; Horch & Lisney, 1981; Moore et al., 2002). The effect of crush injury on presynaptic inhibition of ventral proprioceptive Ia afferents is, however, less clear (Enriquez, Jimenez, & Rudomin, 1996; Enriquez-Denton, Manjarrez, & Rudomin, 2004) and has not yet been analyzed at the structural level. Indeed, one potential explanation for the increased stretch-reflex is a loss of presynaptic terminals (P-boutons) (Conradi, 1969). P-boutons on Ia afferents are GABAergic (Maxwell, Christie, Short, & Brown, 1990) and on Vesicular GLUtamate Transporter 1 (VGLUT1) Ia afferent synaptic boutons they can be easily identified by their immunoreactivity to the 65 kDa isoform of glutamic acid decarboxylase (GAD65) (Betley et al., 2009; Hughes et al., 2005).
The goals of the present study are therefore to test whether after crush nerve injury: (a) there is preservation of Ia afferent synapses on regenerating motoneurons using VGLUT1 antibodies as efficient markers to map these synapses on motoneuron dendrites (Alvarez, Villalba, Zerda, & Schneider, 2004; Alvarez et al., 2011; Rotterman et al., 2014); (b) GABAergic presynaptic control of VGLUT1 Ia afferent synaptic boutons is diminished. To accomplish these goals, we analyzed VGLUT1 boutons and their GAD65 P-boutons on retrogradely labeled MG motoneurons after tibial nerve (TN) crush and compared the results to previous data after TN transection (Alvarez et al., 2011). The results confirm that VGLUT1 synapses are better preserved after crush compared to complete nerve transections. Furthermore, a decrease in GAD65 P-bouton coverage of VGLUT1 terminals was also detected suggesting a possible decrease in GABAergic presynaptic control. These differences in structural remodeling of Ia afferent synapses after nerve crush compared to nerve transection contribute to explain the observations on the differential preservation of the stretch reflex in both types of injuries as originally reported by Barker and Young 70 years ago.
2 |. METHODS
All studies were performed on adult female Wistar rats (225–300 g). All animal procedures and nerve surgeries were performed at Wright State University and approved by the institutional laboratory animal use committees of Wright State University and Emory University. Collected tissues were sent to Emory University for processing and analyses.
2.1 |. Nerve injury and injections of retrograde tracers
Adult female Wistar rats were anesthetized with isoflurane until a surgical plane of anesthesia was obtained (induction 4–5%; maintenance 1–3%, both in 100% O2). The TN was exposed at mid-thigh by a midline posterior incision (~1.5 cm) through the skin and underlying connective in the left hind limb. The TN was then crushed with fine #5 forceps for 10 s. After washing with 0.9% sterile saline, the wound was closed in layers and the animals removed from anesthesia. Buprenorphine (subcutaneous, 0.1 mg/kg) was delivered immediately and every 12 hr after surgery prophylactically for 48 hr to alleviate any possible pain and distress. Signs of pain or distress (lethargy, vocalizations, weight loss, absence of grooming) were closely monitored but not observed in any animals. One week prior to the animals being euthanized, the MG muscle of the left leg was exposed and injected with 10–20 μl of 0.1% Alexa 555 conjugated to recombinant cholera toxin subunit b (CTb-555, Invitrogen, ThermoFisher Scientific, Waltham, MA). The total volume was distributed throughout the muscle in four to five 2–5 μl injections. The control group did not undergo any nerve surgeries prior to the CTb-555 injections.
2.2 |. Histology and immunocytochemistry
Control (n = 4) and experimental animals (n = 8) were deeply anesthetized with Euthasol and transcardially perfused with 4% paraformaldehyde in 0.1M phosphate buffer. Experimental animals were sacrificed 21 days or 3 months following nerve crush (n = 4 in each group). These dates were chosen to compare VGLUT1 synaptic coverage on regenerating MG motoneurons at a time point prior to muscle reinnervation (21 days) or well after nerve regeneration and muscle reinnervation is completed (3 months). Spinal cords were collected and post-fixed overnight. Lumbar segments 4 and 5 (L4-L5) were extracted and 50 μm thick sections were obtained in a freezing sliding microtome and processed “free-floating.” After washing the sections in 0.01M phosphate buffer saline (PBS) with 0.3% Triton-x-100 (PBS-Tx), they were blocked in 10% normal donkey serum in PBS-Tx for 1 hr. Sections were then incubated for 24 hr at room temperature in goat polyclonal anticholera toxin b subunit (1:1000, List Biological Laboratories, Campbell, CA; cat#7032A6; RRID: AB_ 2313637), rabbit polyclonal anti-VGLUT1 (1:1000, Synaptic Systems, Goettingen, Germany; cat# 135 303; RRID: AB_887875), and mouse monoclonal anti-GAD65 (1:200, Developmental Studies Hybridoma Bank, Iowa City, Iowa; RRID: AB_528264) all diluted together in PBS-Tx. The immunoreactive sites were revealed with a mixture of anti-goat, anti-rabbit, and anti-mouse IgG secondary antibodies made in donkey (1:100 dilution in PBS-Tx; Jackson ImmunoResearch, West Grove, PA) each tagged to a different fluorochrome (FITC for VGLUT1: RRID: AB_2315776; Cy3 for CTb: RRID: AB_2340411 and Cy5 for GAD65: RRID: AB_ 2340819).
2.3 |. Antibody characterization
The specificity of the rabbit polyclonal VGLUT1 antibody (Synaptic Systems, Goettingen, Germany; cat# 135 303; RRID: AB_887875) was confirmed in VGLUT1 knockout tissue using materials and procedures previously described (Siembab, Gomez-Perez, Rotterman, Shneider, & Alvarez, 2016; Table 1). This VGLUT1 antibody was raised against a Strep-Tag® fusion protein of rat VGLUT1 (aa 456–560). The mouse monoclonal GAD65 antibody used corresponds to the GAD-6 clone that has been amply characterized since it was generated in the mid-1980s (Gottlieb, Chang, & Schwob, 1986). The antibody was generated against GAD partially purified from chicken brains. Clones producing antibodies recognizing GAD were selected thereafter. The antibody produced by clone 6 specifically recognizes an epitope localized between aa 545 and 585 in the lower molecular weight GAD65 isoform (Chang & Gottlieb, 1988; Richter, Shi, & Baekkeskov, 1993) (Developmental Studies Hybridoma Bank, Iowa City, Iowa; RRID: AB_528264). It does not recognize GAD67. Finally, the goat polyclonal antibody against the CTb subunit was raised against recombinant Cholera toxin b subunit (List Biological Laboratories, Campbell, CA; cat#7032A6; RRID AB_ 2313637) and it does not recognize any immunoreactivity sites in spinal cords lacking neural elements that have uptaken CTb. No labeling was seen in the contralateral size that was not injected with CTb.
TABLE 1.
Primary antibody characteristics
| Immunogen | Host (Ab type) | Company | RRID | Specificity | |
|---|---|---|---|---|---|
| VGLUT1 | Strep-Tag® fusion protein of rat VGLUT1 (aa 456–560) | Rabbit (polyclonal) | Synaptic systems | AB_887875 | No reaction in VGLUT1 KO tissue |
| GAD65 (GAD6 clone) | GAD partially purified from chicken brains | Mouse (monoclonal) | Developmental Hybridoma Bank | AB_528264 | Chang and Gottlieb (1988); Richter et al. (1993) |
| Cholera toxin subunit b | Recombinant Cholera toxin B subunit | Goat (polyclonal) | List Biological Laboratories | AB_2313637 | No reaction in tissue lacking CTb |
2.4 |. Confocal imaging and neuron reconstruction
Sections from each animal were imaged first at low magnification (20×, N.A. 0.75) in an Olympus FV1000 confocal microscope. CTb-labeled MG motoneurons with their cell bodies fully contained within the 50 μm-thick section and showing long dendrites were re-imaged at high magnification (60×, N.A. 1.35, oil immersion, 0.5 μm z-steps throughout the tissue thickness). Confocal images were obtained by author TMR and coded such that all quantitative analyses performed by AJS were blinded to the condition of the animal. The images were uploaded into Neurolucida (MicroBrightField, Colchester, VT: RRID: nif_0000_10294) and the whole labeled dendrites traced and reconstructed. The position of each VGLUT1 synapse was “marked” and the GAD65 synapses in contact with marked VGLUT1 synapses were also plotted. VGLUT1 synapses targeting the cell body or dendrites were analyzed separately. Total path length and overall surface of the combined dendrites as well as the surface of the cell body were measured and compared to confirm similar sampling of motoneuron surfaces in all animals (see Table 2). The distribution and density of VGLUT1 synapses was analyzed by obtaining the following two measurements: (a) overall VGLUT1 synaptic densities calculated by dividing the total number of synapses by the total dendritic length (linear), total dendritic surface, or cell body surface; (b) Sholl analysis to examine VGLUT1 distributions in dendrite segments at 50 μm bins of incremental radial distance from the center of the cell body. Linear and surface densities of VGLUT1 contacts within each bin were then calculated. The distribution of GAD65 synapses on VGLUT1 contacts was analyzed also using two separate measurements: (a) the percentage of VGLUT1 contacts receiving contact from GAD65 synapses; (b) the density of contacts from GAD65 synapses was measured by calculating the average ratio of GAD65 to VGLUT1 synapses. We reconstructed and analyzed 40 motoneurons in each of three experimental groups (control, 21 days, 3 months), sampling 10 motoneurons per animal in each group (n = 4 animals per group).
TABLE 2.
Somatic and dendritic surfaces sampled in CTb-labeled MG motoneurons
| Controls (n = 40 motoneurons) | 21 day TN crush (n = 40 motoneurons) | 3 month TN crush (n = 39 motoneurons) | |
|---|---|---|---|
| Somatic surface area (μm2) | 6,150 ± 525 | 6,163 ± 591 | 6,232 ± 525 |
| Dendritic total length (μm) | 802 ± 59 | 837±155 | 774 ± 87 |
| Dendritic total surface (μm2) | 13,068 ± 2,473 | 11,392 ± 3,349 | 10,999 ± 1,371 |
CTb = cholera toxin b; MG = medial gastrocnemius; TN = tibialis nerve.
Values are Mean ± SD. Differences between groups did not reach significance (ANOVA p > .05).
2.5 |. Surface-to-surface analysis
To analyze the percentage of surface contact between both types of boutons, confocal image stacks were loaded into Imaris (Bitplane, Concord, MA; RRID: nif-0000–00314) to obtain 3D surface renderings of VGLUT1 boutons and the GAD65 boutons contacting them (20 VGLUT1 terminals were randomly selected from three animals per condition; all VGLUT1 terminals were pooled together for statistical comparisons). VGLUT1 boutons of similar size were selected to avoid biasing trends on P-bouton coverage dependent on VGLUT1 size. For making surface renderings of the boutons, fluorescent level thresholds were determined based on control VGLUT1 terminals and then applied to the other conditions. Thereafter we used a custom MATLAB script to calculate the surface area of the VGLUT1 terminal contacted by GAD65-IR synapses. For this determination, we defined contact surface between the GAD65 and the VGLUT1 bouton as the region with 0 μm separation in the rendered surfaces.
2.6 |. Statistical analyses
All statistical tests were made using SigmaPlot 12.3 (Systat). We used one-way analysis of variance (ANOVAs) to compare VGLUT1 dendritic linear density, dendritic surface area density, and somatic density between control, 21 day crush, and 3 month crush animals. Post hoc Bonferroni corrected t-tests were used to determine significant differences between experimental groups with respect to control. In all these data sets, normality criteria were fulfilled. Normality, however, failed in data comparing the percentage of VGLUT1 boutons associated with GAD65 P-boutons, the number of P-boutons per VGLUT1 or the coverage of VGLUT1 boutons by P-boutons. In this case, we used Kruskal-Wallis One Way ANOVA followed by post hoc Dunn’s tests versus control. These data sets are therefore displayed as whisker-box plots to better describe the data. Significance was set to p < .05 in all tests.
2.7 |. Figure composition
Figures were composed in CorelDraw (v. 16.0) and graphs were created in SigmaPlot (v. 12.0; Jandel Scientific). Adjustments to images were made only for presentation and publication purposes. All quantification was carried out with unprocessed original images. Images in plates were adjusted for optimal contrast and brightness in Image Pro Plus (v. 5.0; Media Cybernetics, Bethesda, MD; RRID: nif_0000_00313). Some images were sharpened with a “high-gauss” filter. Manipulations were minimal, and the informational content was not altered.
3 |. RESULTS
3.1 |. Technical considerations and data interpretation
Retrogradely labeled MG motoneurons and their dendritic arbors were imaged first at low and then at high magnification (see methods) in single transverse spinal cord sections with the cell body centered in the field of view (Figure 1). Only motoneurons with cell bodies larger than 485 μm2 in cross-sectional area were sampled. This cut-off excludes the smaller γ-motoneurons that do not receive Ia/VGLUT1 inputs (Shneider, Brown, Smith, Pickel, & Alvarez, 2009). Motoneurons with long continuously labeled dendrites were selected for analyses and these were carried out blind to the condition of each motoneuron (see methods). Imaging, sampling, and measurements were chosen to be similar to Alvarez et al. (2011) for optimal comparisons between TN crush (this study) and TN transection (previous study). The extension and size of motoneuron surfaces analyzed on dendrites and cell bodies are similar in both studies (compare Table 2 in this study with Table 2 in Alvarez et al., 2011). However, when compared to full reconstructions of complete motoneuron dendritic trees (Rotterman et al., 2014), the labeling and sampling procedure used here is known to be biased to proximal dendrites with principal trajectories in the transverse plane. This is because imaged dendrite segments are limited to those contained inside the field of view and within the 50 μm deep transverse section. Therefore, dendrites with a preferential rostro-caudal orientation are cut and under sampled (Figure 1c,g,k). Moreover, the longest dendrites extended beyond the field of view and frequently dendrite segments became disconnected because dendrites meander in and out the section. Our preparations contain many labeled dendrites originating in different motoneurons and it is not feasible to follow single dendrites through serial sections. Unfortunately, for the sample sizes used here, it is also not feasible either to generate enough complete full neuron reconstructions of single intracellularly neurobiotin-filled motoneurons imaged through serial sections and with extensive tiling of confocal stacks to sample the whole dendrite territory. Analyses of fully reconstructed motoneurons (Rotterman et al., 2014), however revealed that 50% and 75% of VGLUT1 contacts in uninjured animals are located, respectively, within the first 300 and 550 μm of the dendritic arbor and that the larger loss of VGLUT1 synapses in injured animals occurs within the first 300 μm of dendrite (Rotterman et al., 2014). The large majority (94.7%) of CTb-retrogradely labeled motoneurons analyzed here had dendritic arbors in which the further point was more than 100 μm distal from the cell body dendrite origins. On average, the more distal dendrite location analyzed was 132.8 μm ± 13.6 (±SD) for motoneurons in control animals (n = 4), 131.2 μm ± 17.1, 21 days TN crush and 124.7 μm ± 19.5, 3 months TN nerve crush. Thus, the study is focused on VGLUT1 inputs on the cell body and the more proximal dendritic region.
FIGURE 1.
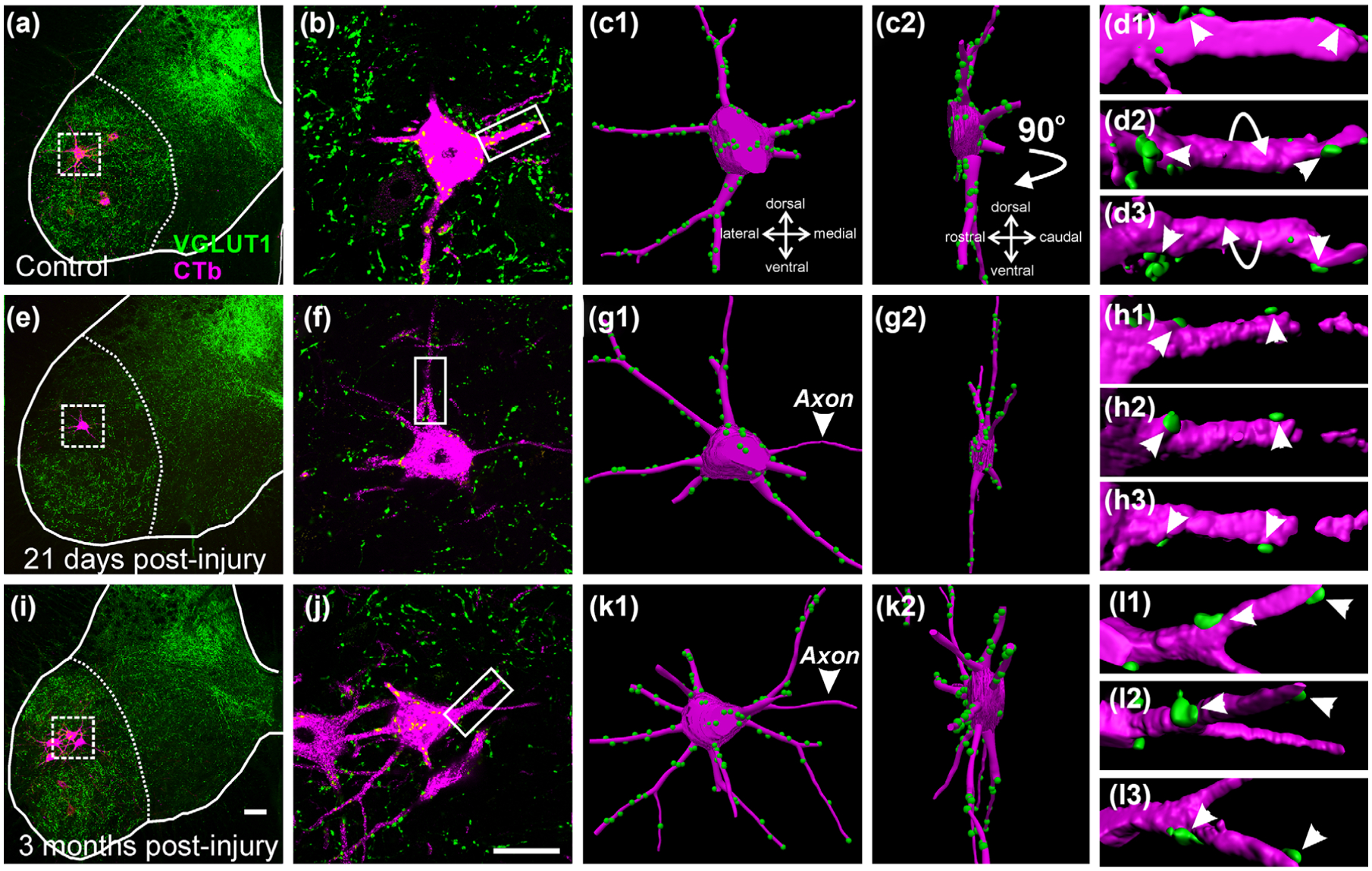
VGLUT1 inputs in control and regenerating motoneurons. (a, e, i) Low magnification confocal images of VGLUT1 (FITC, green) and retrogradely labeled MG motoneurons (magenta neurons in boxes, CTb-555 amplified with Cy3 immunohistochemistry) in control (a), 21 days (e), and 3 months (i) after nerve crush. (b, f, j) High magnification of CTb labeled motoneurons (magenta) and VGLUT1 contacts (green). (c1, g1, k1) Neurolucida 3D reconstructions of the motoneuron cell bodies, dendrites and VGLUT1 contacts. (c2, g2, k2) 90° rotations of motoneuron reconstructions showing in a perpendicular plane the dendritic arbor contained within the 50 μm thick section. (d1–3, h1–3, l1–3) High magnification and surface rendered Imaris 3D reconstructions of VGLUT1 contacts (green) on dendrite segments (magenta) indicated by boxes in (b), (f), and (j). Arrows indicate rotations; Arrowheads indicate the same VGLUT1 contacts in different rotation views demonstrating attachment to the dendrites. The images show that by difference to previous work after a TN transection, TN crush causes limited loss of VGLUT1 synapses difficult to detect in single motoneuron examples. Scale bars: in (i), 100 μm (a and e same magnification); in (j), (k1), and (k2), 50 μm (b, f, c1–2, g1–2, k1–2 same magnification)
CTb retrograde labeling appears increasingly punctate inside finer and more distal dendrites and this characteristic could affect surface estimates which depend on accurate measurement of dendrite diameters at each traced segment interval. We therefore calculated in addition to surface density estimates (number of VGLUT1 contacts per unit of dendrite surface in μm2) the linear dendrite VGLUT1 densities (numbers of VGLUT1 contacts per unit length of dendrite in μm), which are unaffected by possible inaccuracies in dendrite diameter estimates. In summary, the analyses performed assess changes in VGLUT1 synapse densities in the proximal somato-dendritic region and are best compared to Alvarez et al. (2011) using similar methods. Previous studies that analyzed fully reconstructed motoneurons (Rotterman et al., 2014) suggest that these methods focus on the most relevant regions of the motoneuron showing major changes in VGLUT1 density.
3.2 |. Nerve crush causes only a small loss of VGLUT1 contacts on proximal dendrites and cell bodies of regenerating motoneurons
VGLUT1 bouton contact densities on the cell bodies and dendrites were estimated in Neurolucida 3D reconstructions of CTb-labeled motoneurons (Figure 1a–c,e–g,i–k). When necessary, confocal images were imported into Imaris software for confirmation of VGLUT1-IR bouton “contacts” on the surface of CTb-labeled MG motoneurons rendered in three-dimensional space (Figure 1d,h,l). All VGLUT1 bouton “contacts” were considered synapses since previous studies confirmed the presence of bassoon, a presynaptic active zone protein, in the VGLUT1-IR bouton surfaces in contact with labeled motoneuron cell bodies or dendrites (Rotterman et al., 2014). Control motoneurons received on average 0.50 ± 0.04 (±SD) VGLUT1 contacts per 100 μm2 of cell body surface (Figure 2). In both, 21 day and 3 month post-injury groups VGLUT1 contact density was significantly reduced, respectively, by 35.5% and 38.4% from control (One-Way ANOVA, F(2, 9) = 10.909, p = .004; power for α = 0.05: 0.931; post hoc Bonferroni corrected t- tests t = 4.196, p = .07 control versus 3 month and t = 0.179, p = .011 control versus 21 day crush; for these and following comparisons, n = 4 animals per group with each animal data point estimated as average VGLUT1 density calculated from 9 to 10 motoneurons per animal; see Table 3; The number of reconstructed motoneurons augmented accuracy of individual animal estimates and reduced inter-animal variability for statistical comparisons). The magnitude of these depletions is less substantial than the loss reported on MG motoneuron cell bodies after TN transections (69.8% and 57.9% at 6 weeks and 6 months post-injury, respectively; from Alvarez et al., 2011).
FIGURE 2.
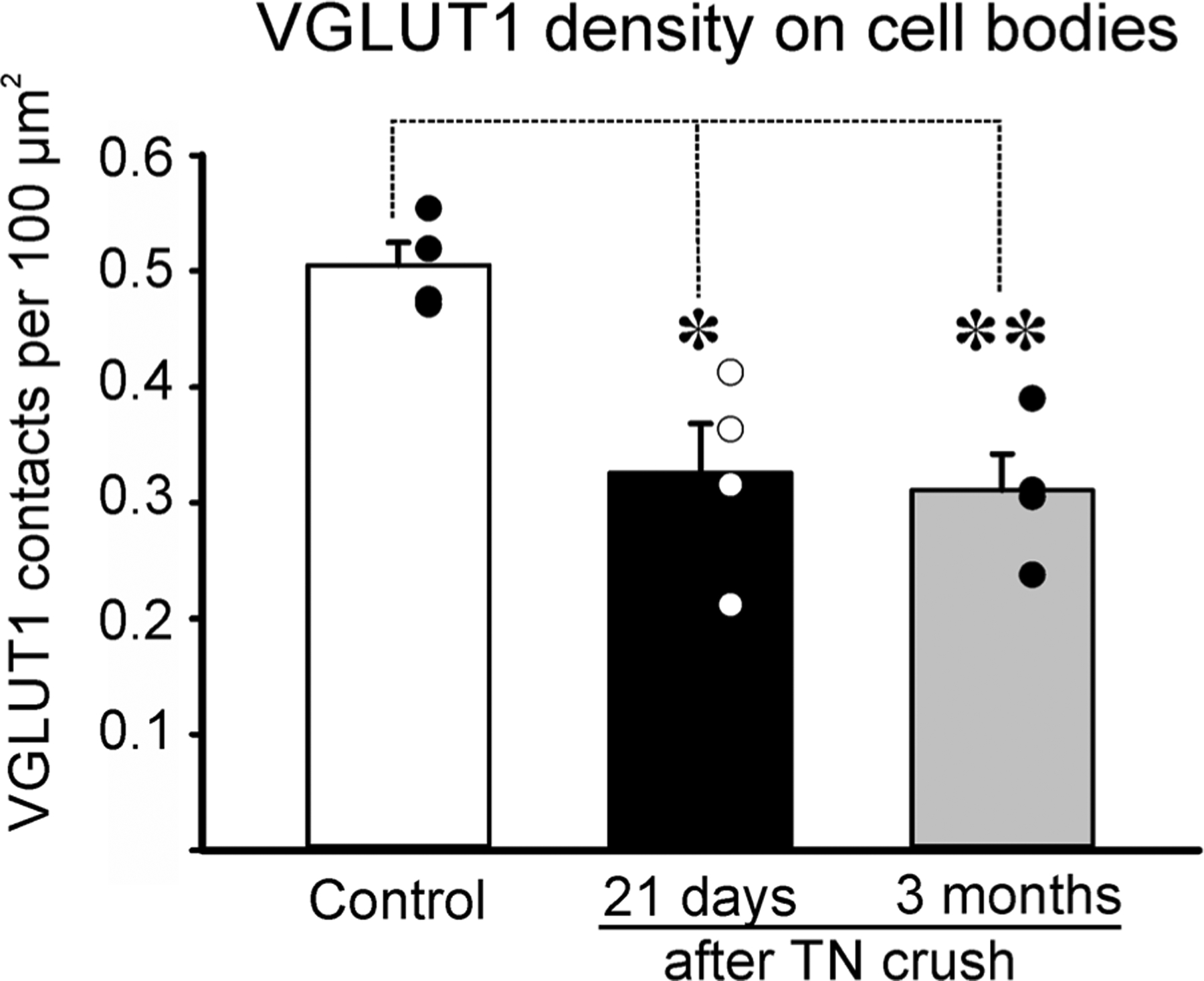
VGLUT1 synaptic density on the surface of control and regenerating motoneuron cell bodies. Averages ± SEM, n = 4 animals analyzed per condition. Each dot represents the average in a different animal obtained from 10 motoneuron cell bodies fully reconstructed per animal. ** p < .01, * p < .05, post hoc Bonferroni t-test comparisons to control
TABLE 3.
VGLUT1 boutons analyzed and percentage covered by GAD65 P-boutons
| Controls (n = 40 motoneurons) (10 in each of four animals) | 21 day TN crush (n = 40 motoneurons) (10 in each of four animals) | 3 month TN crush (n = 39 motoneurons) (9–10 in each of four animals) | |
|---|---|---|---|
| On cell bodies | |||
| Total number | 1,242 | 774 | 737 |
| Average/cell body | 31.1 ± 10.3 | 19.4 ± 7.5 | 18.9 ± 7.5 |
| Average/animal | 310.5 ± 10.1 | 193.5 ± 47.4 | 184.3 ± 31.3 |
| % with P-boutons | 86.9 ± 8.2 | 70.1 ± 17.6 | 76.6 ± 12.2 |
| On dendrites | |||
| Total number | 3,957 | 2,686 | 2,694 |
| Average/motoneuron | 98.9 ± 19.6 | 67.15 ± 19.6 | 69.1 ± 16.3 |
| Average/animal | 989.3 ± 32.6 | 671.5 ± 143.6 | 673.5 ± 113.7 |
| % with P-boutons | 86.9 ± 5.0 | 74.4 ± 13.5 | 80.0 ± 9.2 |
Averages as Mean ± SD. % based on total number of boutons sampled in either dendrites or cell bodies. % not distributed normally; median and distribution properties are graphed in Figure 5a.
VGLUT1 bouton contacts on the dendrites were also reduced to a lesser extent than after TN transection. VGLUT1 dendritic linear density (VGLUT1 density per 100 μm of dendrite segment, Figure 3a) revealed significant depletions after nerve crush compared to control (one-way ANOVA F(2, 9) = 8.885, p = .007; power for α = 0.05: 0.861). Twenty-one days after crush, CTb-labeled motoneuron dendrites showed, on average, a significant 33.3% loss from control (control average = 12.5 ± 0.8 VGLUT1-IR bouton contacts per 100 μm of dendrite length, ±SD, post hoc Bonferroni t-test, t = 3.990, p = .009 compared to control). The 3-month group also exhibited a significant 26.5% depletion from control (post hoc Bonferroni t-test, t = 3.172, p = .034 compared to control). VGLUT1 surface density was also found diminished in both the 21-day (by 21.5%) and 3-month (by 19.5%) groups compared to control values (average surface density in controls = 0.8 ± 0.16 VGLUT1 boutons per 100 μm2 of dendrite surface) (Figure 3b), however, differences in VGLUT1 surface densities did not reach significance (one-way ANOVA, F(2, 9) = 2.389, p = .147; power for α = 0.05: 0.220). Smaller percentage changes and lack of statistical significance in VGLUT1 surface densities compared to linear densities might be due to changes in dendritic surface area caused by either variable levels of thinning of proximal dendrites in regenerating motoneurons or lesser uptake of the retrograde tracer CTb that might not fully fill the dendrite thickness. These factors would, in both cases, increase VGLUT1 surface densities estimates and reduce differences with control. Small differences, if they exist, could not be statistically detected with this number of observations. The power of the performed test (0.22) is below the desired power of 0.8. Nevertheless, the results confirm that changes in VGLUT1 synapses are small after TN crush compared to TN transection (Alvarez et al., 2011)
FIGURE 3.
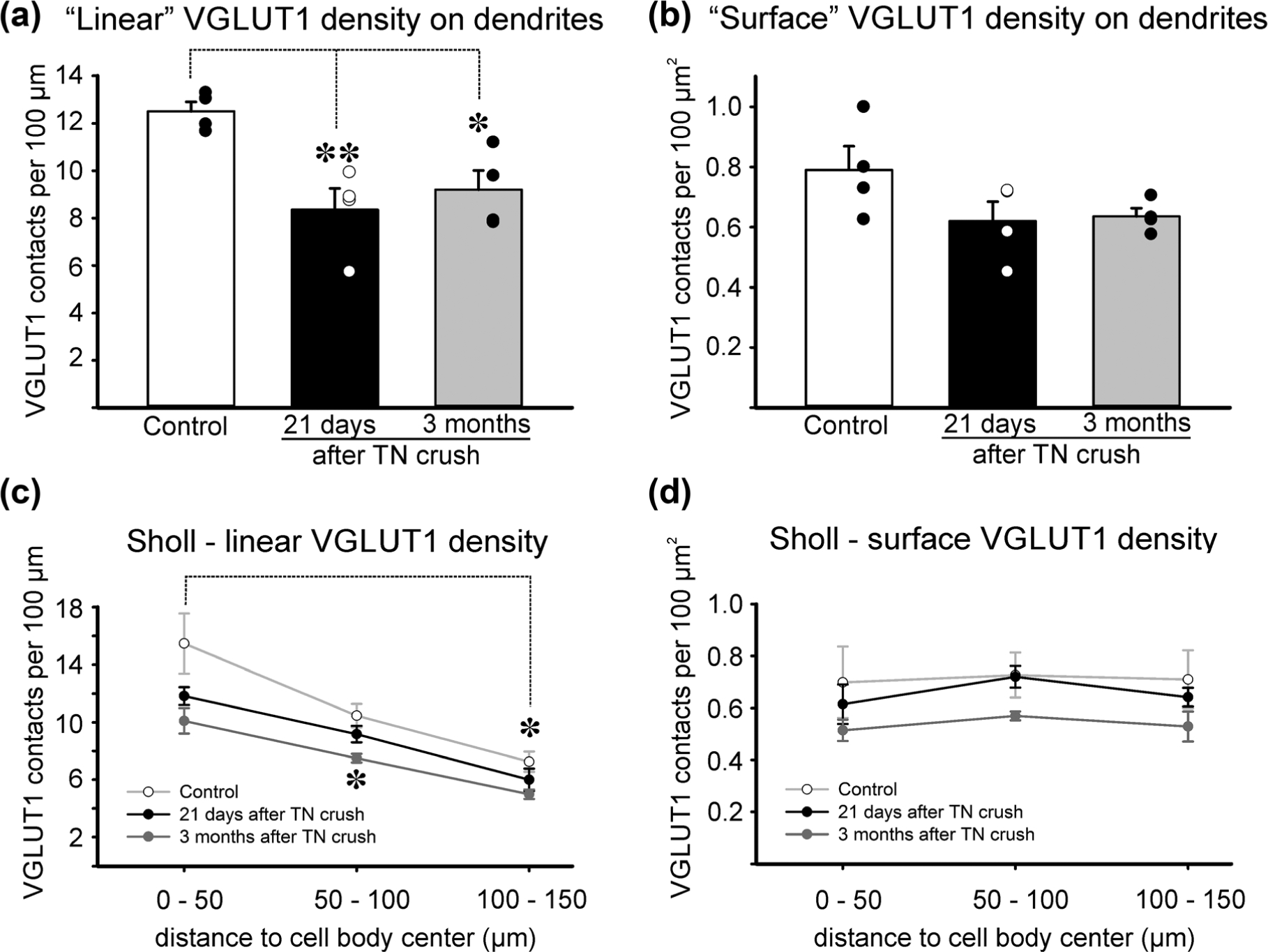
VGLUT1 synaptic densities on control and regenerating motoneuron dendrites. (a, b) Linear and surface densities of VGLUT1 contacts on the traced dendrites (see Table 2 for characteristics of the dendrites sampled). Averages ± SEM, n = 4 animals analyzed per condition. Each dot represents the average in a different animal obtained from dendrite reconstructions of 10 motoneurons per animal. **p < .01, *p < .05, post hoc Bonferroni t-test comparisons to control. A significant depletion was detected in linear, but not in surface density. (c) Density of VGLUT1 contacts per 100 μm distance in dendritic segments localized at different distances from the cell body (x axis). Data points represent averages ± SEM of estimates obtained in each of four animals. The number of VGLUT1 contacts decreased in a proximo-distal gradient (*p < .05, post hoc Bonferroni t-test comparison of dendrite segments located at a distance of 100–150 μm from the cell body center with segments in the first 0–50 μm). (d) Density of VGLUT1 contacts per 100 μm2 of dendrite surface. VGLUT1 surface density is maintained constant in all distance bins and no significant differences were detected between control dendrites and dendrites from regenerating motoneurons (ANOVA, p > .05)
To examine if different dendrite compartments sustained different synaptic loses, we divided the traced dendritic trees in three 50-μm bins of different distance to the cell body center (Figure 3c). As previously described (Alvarez et al., 2011; Rotterman et al., 2014), Sholl analyses in control motoneurons revealed a significant and progressive reduction in VGLUT1 linear density with distance from the cell body (one-way ANOVA, F(2, 9) = 9.306, p = .006; power for α = 0.05: 0.879) while VGLUT1 surface density was maintained constant (one-way ANOVA, F(2, 9) = 0.0151, p = .985; power for α = 0.05: 0.050; in this case, power is too small to detect significant differences in surface VGLUT1 density because there is practically no difference between the three dendritic compartments: 0.70 ± 0.29; 0.73 ± 0.17; 0.71 ± 0.22 VGLUT1 contacts per 100 μm2 ± SD). Surface densities are maintained despite changes in VGLUT1 synapse numbers (linear density) because dendrites taper as they extend from the cell body reducing their surface area. In control motoneurons, decreases in number of VGLUT1 synapses along the dendrite match reductions in available surface thus keeping the synaptic surface density constant. After nerve crush, VGLUT1 density on motoneuron dendrites tended to decrease in all distance bins in both linear and surface density estimates. These ranged from 12 to 35%, but only one data point reached statistical significance compared to control (3 month crush in distance segment of 50–100 μm from the cell body center; post hoc Bonferroni t-test t = 3.566, p = .015). Loss of significance compared to analyses in the full traced dendrites are likely due to the compound effects of the small differences with control and the increased variance caused by segmentation of the traced dendrites into smaller segments. As a result, all comparisons among the same dendritic compartment from animals in the three conditions (control, 21 days and 3 months after TN nerve crush) were underpowered (<0.8), nevertheless the results once again highlight that if any differences could be revealed by increasing the power of statistical analyses, these would be rather small. These results are notably different from TN transection, in which there was a larger and significant loss in both linear and surface VGLUT1 density in the proximal dendrite compartments using similar sampling, measurements, and Sholl analyses (Alvarez et al., 2011).
To investigate whether there was also and change in the size of VGLUT1 immunoreactive punctae, we surface rendered 100 VGLUT1 immunoreactive clusters per condition (25 per animal) and estimated their total volume using Imaris software. In control, animals VGLUT1-immunoreactive clusters had an average volume of 10.6 μm3 ± 0.8 (±SEM; n = 100; median = 8.4) while 21 days after nerve crush their average volume was reduced by 33% to 7.1 μm3 ± 0.8 (n = 100; median = 4.4) and after 3 months, there was a partial recovery to 8.2 μm3 ± 0.7 (n = 100; median = 6.1). Changes with respect to control values were only significant 21 days after crush (one-way ANOVA on Ranks; H(2) = 14.436, p < .001; post hoc Dunn’s test, Q = 3.79, p < .05; in 21 day versus control comparison and Q = 2.12, p > .05 in 3 month versus control comparison). Bouton size is important given the reported relationship between Ia afferent bouton size, active zone size, and other structural parameters indicative of synaptic strength (Pierce & Mendell, 1993). Thus, neither VGLUT1 bouton density nor size is reduced after nerve crush to the levels identified after nerve transection.
3.3 |. Reduction in P-bouton coverage of VGLUT1 boutons following nerve crush
Large preservation of VGLUT1 synapses alone does not fully explain the enhancement of the stretch reflex after nerve crush (Prather et al., 2011). To examine one possible mechanism that may contribute to disinhibition of the Ia-motoneuron synapse, and thus possibly the stretch reflex, we examined the effects of nerve crush on presynaptic GABAergic control of Ia afferents synapses by examining the incidence and density of GAD65 P-boutons on VGLUT1 synapses (Figure 4).
FIGURE 4.
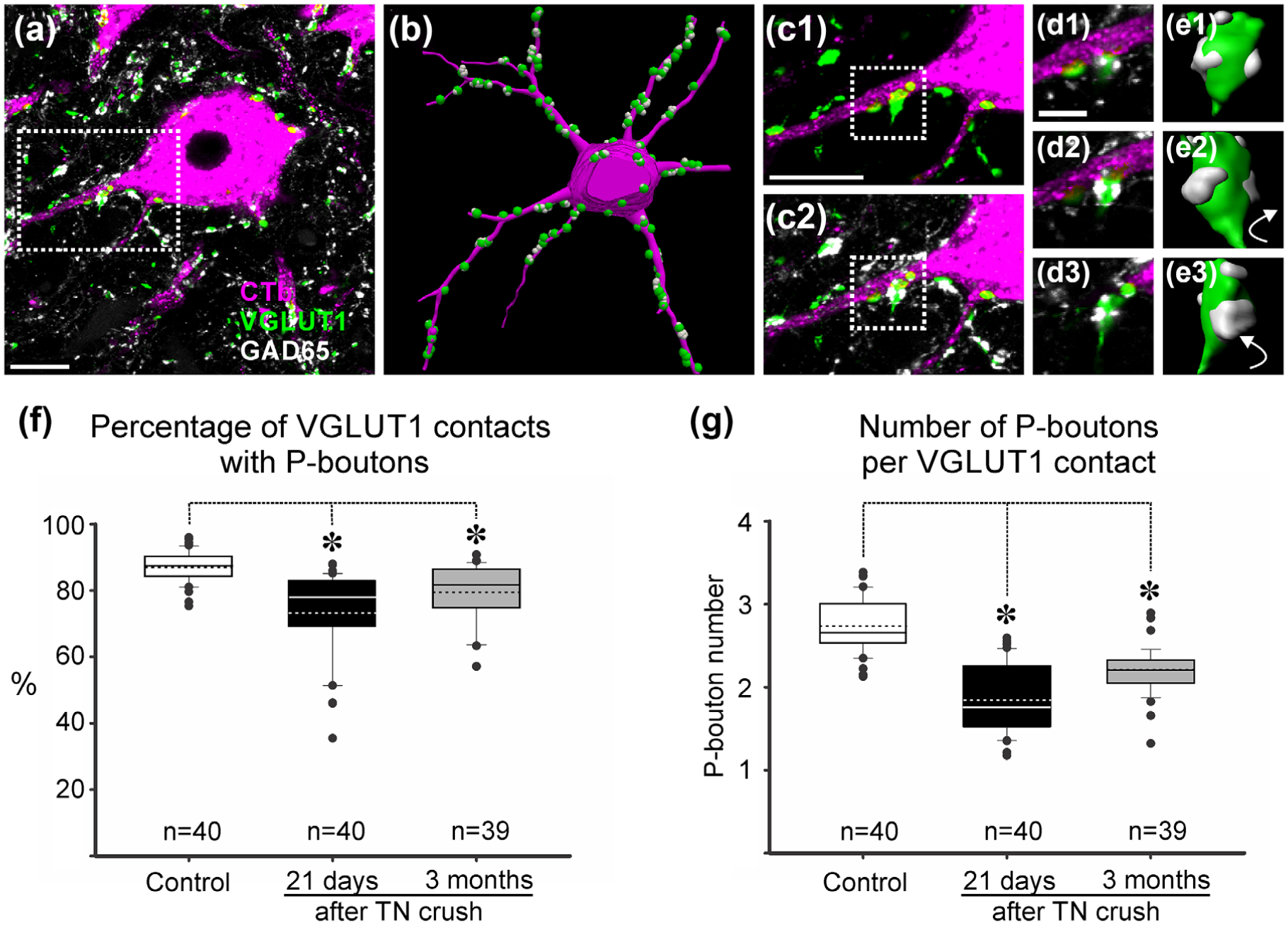
Presynaptic GAD65 P-boutons on VGLUT1 bouton contacts on retrogradely labeled motoneurons. (a) High magnification of a single confocal image plane through the cell body and proximal dendrite of a retrogradely labeled MG motoneurons (magenta, CTb-555 amplified with Cy3 immunohistochemistry) receiving contacts from VGLUT1 terminals (green, FITC) associated GAD65 P-boutons (white, Cy5). (b) Neurolucida 3D reconstruction of motoneuron in (a) with all VGLUT1 contacts plotted on it surfaced and associated GAD65 boutons. (c) High magnification confocal stack projection of dendrite with VGLUT1 contacts (green) (c1) and associated GAD65 P-boutons superimposed (c2). (d) Higher magnification single confocal plane images (d1–d3). (e) Boutons shown in (d) surface reconstructed in Imaris and rotated in the Y plane. (f) Whisker-and-box plots represent the distribution of the percentage of VGLUT1 contacts with P-boutons per motoneuron and in each condition. Continuous lines: median; dashed lines: average; box: 25 and 75% distribution percentiles; whiskers: 5 and 95 percentiles; black circles: outliers beyond the 5–95% distribution; n: number of motoneurons analyzed in each plot. Note skewedness toward lower values after injuries. Asterisks, p < .05 Dunn’s post hoc test. A significant, but small, reduction in the percentage of VGLUT1 contacts associated with P-boutons was detected. (g) Number of P-boutons per VGLUT1 bouton was also diminished. Data presented as whisker-and-box plots as before; Normality test failed. Asterisks, p < .05 Dunn’s post hoc test. The number of P-boutons in VGLUT1 contacts diminished after nerve crush and did not recover. Scale bars: in (a) and (c1), 20 μm; in (d), 5 μm
We examined a total of 3,957 VGLUT1 boutons in contact with dendrites and 1,242 VGLUT1 boutons on the cell bodies of retrogradely labeled control MG motoneurons and these were compared to 2,686 and 774 VGLUT1 boutons on dendrites and cell bodies of MG motoneurons 21 days after TN crush and 2,694 and 737 VGLUT1 boutons, respectively, 3 months post injury (see Table 3 for the characteristics of the sample). The percentage of VGLUT1 boutons receiving presynaptic inhibition in controls (defined by receiving at least one contact from GAD65 P-boutons) was 86.9% on both the dendrites and cell bodies. This estimate is remarkably similar to the 86% of Ia afferent synapses filled with horseradish peroxidase (n = 90) found to receive contacts from P-boutons in an exhaustive serial section electron microscopy analysis (Pierce & Mendell, 1993). Twenty-one days after TN crush, 74.4% and 70.1% of VGLUT1 boutons on, respectively, dendrites and cell bodies of MG motoneurons were associated with P-boutons. These percentages partially recovered 3 months after the injury (80.1 and 76.5%). For statistical analyses, we pooled together VGLUT1 contacts on dendrites and cell bodies in each motoneuron and compared the percentage of VGLUT1 contacts per cell with presynaptic inhibition in control (n = 40 motoneurons), 21 days after crush (n = 40 motoneurons) and 3 months (n = 39 motoneurons). The data was not distributed normally (skew toward lower values was apparent in a number of injured motoneurons; Figure 4f) and therefore a Kruskal-Wallis one way ANOVA was used resulting in significant differences among the three populations (control, 21 days, 3 months; H(2) = 41.158, p < .001). Post hoc Dunn’s comparisons revealed significant differences both at 21 days and 3 months with respect to control (Q = 6.315 [21 days vs. control] and Q = 4.118 [3 months vs. control], both p < .05) (Figure 4f).
In addition, the number of GAD65 boutons per VGLUT1 synapse was significantly decreased. VGLUT1 boutons associated with GAD65 P-boutons and contacting dendrites in the control group were surrounded by an average of 2.8 ± 0.2 (±SD) P-boutons and this was similar for those contacting the cell body (2.7 ± 0.2). Overall VGLUT1 boutons contacting retrogradely labeled motoneurons were associated with 2.7 ± 0.2 P-boutons (Figure 4g). Larger VGLUT1 boutons had more P-boutons than smaller ones but this was not systematically analyzed (see also Pierce and Mendell, 1993). At 21 days after crush, the number of GAD65 P-boutons per VGLUT1 bouton was reduced by 32.8% from the control value. There was some recovery after 3 months such that the estimated reduction at this time point was then only 19% less than control. The reduction in number of P-boutons was significant at both times post-injury (Kruskal-Wallis one ANOVA on ranks, H(2) = 65.508, p < .001; post hoc Dunn’s tests Q = 7.973 [21 days vs. control] and Q = 5.170 [3 months vs. control], both p < .05 in both injured groups vs. control) (Figure 4g).
Given the known relationship between VGLUT1 size and P-bouton number, smaller boutons should be surrounded by fewer P-boutons. Although changes in bouton size were small after nerve crush (see above), we directly tested whether the reduction in P-boutons is independent of changes in VGLUT1 bouton size. For this, we resampled VGLUT1 boutons by selecting only boutons of a typical average size and being careful to be blind to the GAD65 immunofluorescence in the same images (by blanking that channel). Then, we rendered bouton surfaces in Imaris (Figure 5a) and used a MATLAB script to calculate the total surface of the VGLUT1 boutons covered by GAD65 P-boutons (that is the region with 0 μm distance between both rendered surface reconstructions). We analyzed 20 VGLUT1 boutons in each of the three conditions (control, 21 days and 3 months after TN crush) and confirmed that the size of VGLUT1 boutons selected for analysis was rather similar (one-way ANOVA, F(2, 57) = 0.298, p = .744; power for α = 0.050: 0.049) (Figure 5b). The results show statistically significant differences in VGLUT1 surface covered by P-boutons among the three conditions (Kruskal-Wallis one-way ANOVA; H(2) = 9.418, p = 0.009). Control VGLUT1 boutons were covered on 14.3% ± 5.7 (±SD) of their surface in average, while VGLUT1 boutons 21 days and 3 months after TN crush had 9.7% ± 6.9 and 8.6% ± 5.6 of their surface covered by P-boutons (both were statistically different to control VGLUT1 boutons, p < .05, post hoc Dunn’s test; Q = 2.811 comparing 3 months vs. control and Q = 2.472 comparing 21 days vs. control) (Figure 5c). Thus the trend toward recovery of P-bouton numbers observed at 21 days was not confirmed when analyzing the percentage of surface covered in a subsample of VGLUT1 boutons normalized for bouton size.
FIGURE 5.
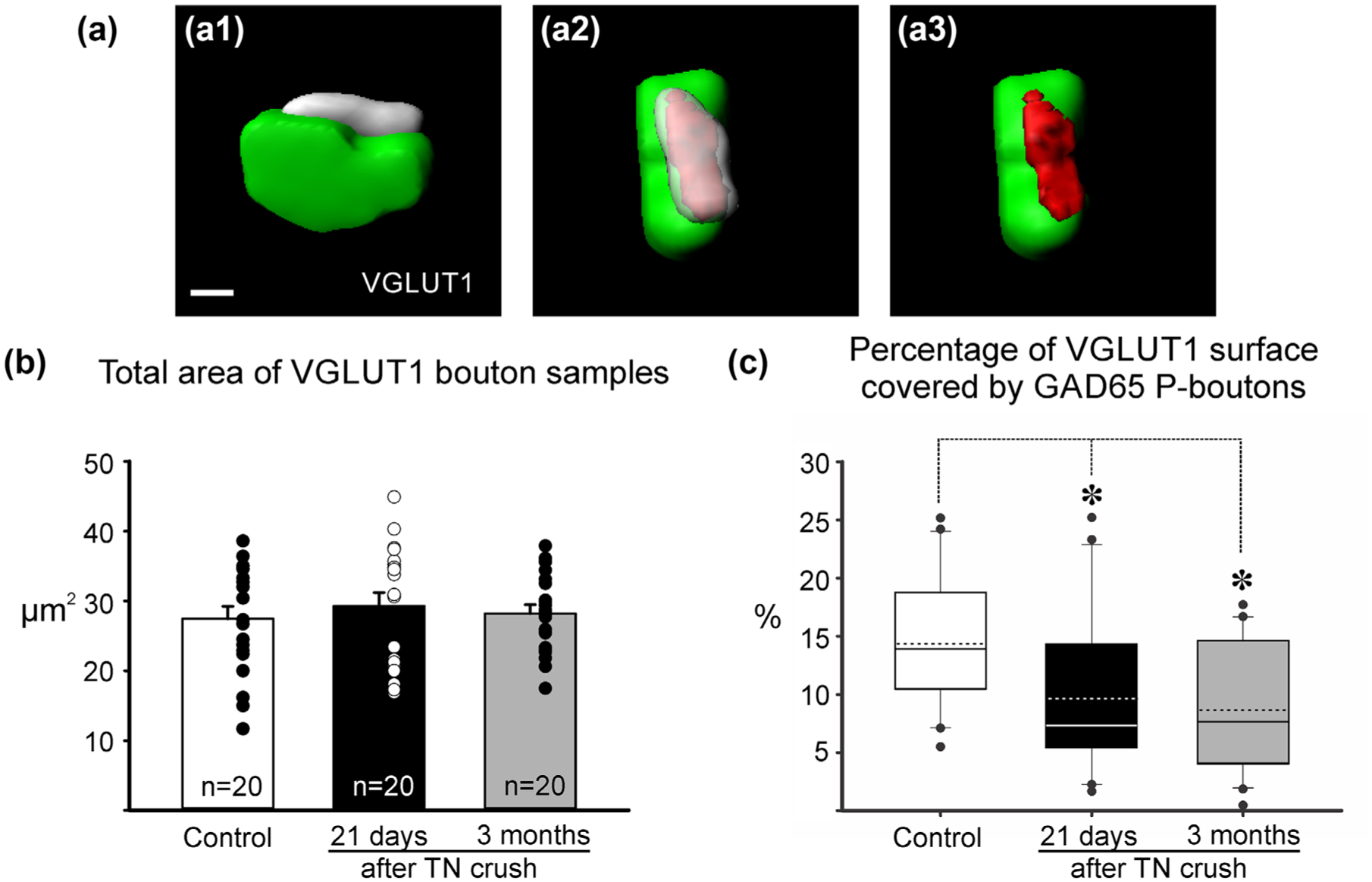
Coverage of VGLUT1 boutons by GAD65 P-boutons. (a) Imaris 3D surface reconstruction of a VGLUT1 bouton and associated GAD65 P-bouton (a1). After rotations to visualize the contact “en face” (a2) a surface of contact is generated. (a3) shows this surface on the VGLUT1 bouton to obtain a percentage coverage on the total VGLUT1 surface covered by the GAD65 P-bouton. (b) VGLUT1 terminals selected for analysis were of similar size. Histograms represent average total surface area and error bars represent standard errors of the mean. Data points are plotted to the side. (c) Percentage of VGLUT1 surface area covered by P-boutons. Data did not distribute normally and is represented as whisker-box plots. Continuous lines: median; dashed lines: average; box: 25 and 75% distribution percentiles; whiskers: 5 and 95 percentiles; black circles: outliers; n: as in (b). There was a statistically significant decrease in surface coverage after crush compared to control (ANOVA p < .001; asterisks: post hoc Dunn’s tests vs. control p < .05)
In summary, slightly fewer VGLUT1 boutons are associated with P-boutons after crush, but VGLUT1 synapses receiving presynaptic control after nerve crush are associated with fewer P-boutons and a significant reduction in VGLUT1 surface coverage. These structural changes in the relations between P-boutons and VGLUT1 synapses could suggest a possible alteration in presynaptic inhibitory control strength.
4 |. DISCUSSION
The major findings of this study indicate that the loss of VGLUT1/Ia afferent input on the soma and dendritic arbor of MG motoneurons following TN crush is less severe than after complete TN transection (Alvarez et al., 2011) and that remaining synapses have reduced coverage by GABAergic P-boutons controlling the strength of synaptic transmission from Ia afferents. Notably, the ~30% reduction in VGLUT1/Ia synapses demonstrated here aligns well with functional data reporting also ~30% loss in the strength of strEPSPs in motoneurons that regenerated and reinnervated muscle after a nerve crush (Prather et al., 2011). However, as discussed below, it is more difficult to relate the partial reduction in P-bouton coverage with the 140% increase in the strength of muscle stretch reflexes.
4.1 |. Differences in stretch reflex synaptic circuit plasticity after different kinds of nerve injury
The milder deficits in VGLUT1/Ia synapses after nerve crush compared to complete nerve transection could be an adaption to the more efficient reinnervation of peripheral targets after nerve crush. Regeneration and recovery from nerve crush injuries is fundamentally different from complete transections because continuity between regenerating axons and endoneurial tubes are better preserved after crush resulting in facilitation of axon growth and greater specificity of target reinnervation (Bodine-Fowler, Meyer, Moskovitz, Abrams, & Botte, 1997; Nguyen, Sanes, & Lichtman, 2002; Valero-Cabre, Tsironis, Skouras, Navarro, & Neiss, 2004). This difference impacts the specificity of connections established in the periphery. It is generally believe that erroneous growth of motor axons to sensory corpuscles or of sensory afferents to neuromuscular junctions is prevented by differential neurotropic and cell-cell adhesion interactions between regenerating axons and Schwann cells lining sensory and motor endoneurial tubes (Brushart, 1993; Hoke et al., 2006; Martini, Schachner, & Brushart, 1994), but there are two other important sources of possible guidance errors. First, motor and sensory axons can enter inappropriate pathways targeting wrong muscles; sometimes the new target might have synergistic actions with the original muscle, but in other situations it might have opposite function. Second, the exact composition of sensory afferents and motor efferents reinnervating muscle spindles might differ from the original innervation.
After full nerve transection, particularly of multifascicular large nerves, regenerating axons enter endoneurial tubes in erroneous fascicles which are then guided to muscles different from the original (Bodine-Fowler et al., 1997; Brushart, 1988; Brushart & Mesulam, 1980; Brushart et al., 1983; Gillespie, Gordon, & Murphy, 1986; Valero-Cabre et al., 2004). Axon misdirection in the periphery disorganizes the spatial order of motor pools inside the spinal cord after nerve transection, but not after nerve crush (Brown & Hardman, 1987; Valero-Cabre et al., 2004). Importantly, the specific connectivity between Ia afferents and motoneurons is dependent on topographic matching the spatial distributions inside the spinal cord of motor pools and terminal synaptic arbors of Ia afferents from different muscles (Surmeli, Akay, Ippolito, Tucker, & Jessell, 2011). Moreover, during regeneration, Ia afferents themselves can also be misrouted in the periphery to novel muscle targets further amplifying the disorganization of Ia inputs to motoneurons centrally. Better preservation after nerve crush of regenerating axons specific targeting in the periphery, implies that functional inputs and modulation of motoneuron activities controlling different muscles and the specificity of muscle stretch information conveyed to spinal neurons might be less disrupted after nerve crush compared to nerve transection, thus requiring less structural synaptic plasticity.
Another important difference between a full nerve transection and nerve crush is the better reinnervation specificity of the muscle spindle apparatus by the original Ia afferents and γ-efferents after nerve crush, as demonstrated in experimental situations in which errors in muscle targeting are prevented (Barker, Scott, & Stacey, 1985; Brown & Butler, 1976; Hyde & Scott, 1983). There is excellent reinnervation of muscle spindles and recovery of normal stretch responses and γ-efferent modulation of spindle responses after crushing nerves, but full transection of the same nerves result in abnormal increases in β-efferent innervation (Brown & Butler, 1976) and in the possibility that Ib afferents are partially responsible for the reinnervation of muscle spindles (Banks & Barker, 1989). Alterations in the source of afferents and efferents innervating muscle spindles would have consequences for both spindle sensitivity modulation during normal motor behaviors in awake animals and also reduce input from stretch-responsive afferents to motoneurons because Ib afferents do not monosynaptically innervate motoneurons in lamina IX.
In summary, central synaptic plasticity in the stretch reflex circuit to overcome unadaptive re-organizations of central motor control circuits resulting from miswired motoneurons, Ia afferents or muscle spindles might be more necessary after nerve transection compared to nerve crush. This suggests that VGLUT1 inputs should be better preserved in regenerating motoneurons after crush, a prediction that was confirmed in this study. Enhancing central synaptic plasticity inside the spinal cord after injuries causing major misrouting of motor axons in the periphery enhances motor recovery, but this is not necessary after nerve crush (Galtrey, Asher, Nothias, & Fawcett, 2007). Our work therefore suggests that endogenous intrinsic signals cause different levels of synaptic plasticity in stretch reflex circuits after different types of injuries. Their exact mechanisms are currently under active investigation. Preliminary data indicate that each type of nerve injury in the periphery induces a different type of neuroinflammatory response inside the spinal cord producing different levels of Ia afferent synapse and axon removal from the ventral horn (Rotterman & Alvarez, 2015). Different inflammatory responses are associated with more or less removal of the pre-terminal Ia axons arborizing in the ventral horn. Ia afferent axons die-back toward lamina V after nerve transection (Alvarez et al., 2011), while much of the Ia axon remains in ventral lamina VII and IX after nerve crush (Rotterman and Alvarez, unpublished observations in VGLUT1-cre mice). Thus, the decrease in VGLUT1 inputs is more extensive and permanent after nerve transection compared to nerve crush.
Functionally, our results suggest that the milder functional deficits in the stretch reflex reported after nerve crush compared to nerve transection (Prather et al., 2011) may be the result of better preservation of the synaptic contacts of Ia afferents onto motoneurons. Interestingly, Prather and colleagues suggested that “roughly 70% of the spindle afferent population recovered function following nerve crush regeneration,” function being measured as the EPSP evoked in MG motoneurons following stretch of the MG muscle (strEPSP). Although there are obvious differences between both studies that include species (cat vs. rat), site of injury (MG nerve vs. TN) and survival (1 year vs. 3 months), it is significant that we found a matching 30% loss of VGLUT1 synapses on the cell bodies and dendrites of MG motoneurons.
4.2 |. Presynaptic inhibition of Ia afferents after nerve crush injuries
Despite small reductions in VGLUT1 synapses and strEPSPs, the stretch reflex is however augmented after recovery from nerve crush (Prather et al., 2011). This suggests that notwithstanding a small reduction in the strength of Ia afferent synapses onto regenerated motoneurons, muscle stretch nevertheless results in increased motor unit firing and/or recruitment. There are several pre- and postsynaptic mechanisms that could be responsible for this response augmentation. These include changes in Renshaw cell mediated recurrent inhibition of motoneurons that is well known to modulate the monosynaptic Ia afferent reflex and alterations in persistent inward currents that amplify Ia afferent synaptic inputs (see Prather et al. 2011 for a more thorough review). While there is evidence of altered recurrent inhibition after nerve transection (Obeidat, Rotterman, Alvarez, & Cope, 2012), it is unknown if a similar reorganization of this circuit happens after nerve crush. On the other hand, possible changes in the synaptic integratory properties of motoneurons after different kinds of nerve injuries are at present unknown. However, the well-established downregulation of presynaptic inhibition in skin afferents after nerve injury (Castro-Lopes et al., 1993; Horch & Lisney, 1981; Moore et al., 2002) prompted us to examine presynaptic inhibition of Ia afferents, which is known to effectively modulate the strength of the Ia-motoneuron synapse and motor unit recruitment by Ia afferent inputs (Rudomin & Schmidt, 1999). The possibility that presynaptic inhibition to Ia afferents is altered after nerve injury was further suggested by a recent study concluding that the formation of presynaptic GABAergic inhibitory terminals (P-boutons) and their strength modulating Ia afferent neurotransmission is regulated by activity-dependent release of glutamate and brain derived neurotrophic factor from Ia synapses (Mende et al., 2016). In uninjured animals, Ia afferents maintain a continuous discharge that is modulated up-or down according to muscle length, therefore, it is conceivable that disconnection of Ia afferents from muscle spindles causes diminished activity that could have dramatic effects on the maintenance of P-boutons on Ia afferent synapses. Indeed, our results suggest a significant change in P-bouton innervation of Ia afferents synapses.
Whether the recorded decrease in P-bouton number correlates with the reported functional impact of nerve crush on stretch-reflexes is more difficult to evaluate. Two previous studies analyzed primary afferent depolarization (PAD, a measurement of presynaptic GABAergic control) in group I afferents (Ia muscle spindle and Ib golgi-tendon organ afferents) after crushing hind limb nerves in the cat and allowing for regeneration and muscle reinnervation to be completed (Enriquez et al., 1996; Enriquez-Denton et al., 2004). Both studies concluded that PAD persisted in group I afferents after nerve regeneration in contrast to previous studies in cutaneous nerves (Horch & Lisney, 1981; Moore et al., 2002) and in agreement with the many VGLUT1 boutons, we found associated with GAD65 P-boutons 21 days and 3 months after crush. However, the electrophysiological studies also reported a change in the organization of PAD to regenerated group I afferents. An extensive body of literature (reviewed in Rudomin and Schmidt, 1999) suggest that Ia, Ib, and type II proprioceptors are specifically modulated by presynaptic interneurons that differ in inputs and modulation. While interneurons presynaptic to Ib afferents are excited by other Ib afferents and also by the reticular formation, the majority of Ia afferents receive PAD from interneurons with convergent excitation from Ia and Ib afferents and that are negatively modulated by the reticular formation. However, after nerve crush, PAD on Ia afferents becomes positively modulated by reticular formation inputs implying a change in P-boutons origins. It could be argued that the small change in VGLUT1 coverage by GAD65 P-boutons could be the result of an initial detachment and thereafter recovery of GAD65 P-boutons that result in both a change in PAD patterns and also a slight decrease in GAD65 P-bouton number and coverage. It is possible that these alterations might be enough to diminish basal presynaptic inhibitory tone and result in increased efficiency of the stretch reflex during natural stimulation (lengthening) of individual muscles like it was done in Prather et al. (2011). However, more work on new animals models are clearly needed to label P-boutons of different origins and confirm these hypotheses.
In summary, the structural changes we report after nerve crush suggest maintenance of VGLUT1 synapses and an alteration of presynaptic inhibition that could contribute to the enhanced neurotransmission between Ia afferents and motoneurons. These two phenomena likely contribute to the better preservation of stretch-evoked EPSPs after nerve crush and may be the supranormal stretch reflex forces. Future experiments should more directly test whether presynaptic disinhibition of Ia afferent synapses is a major contributor to the enhanced stretch reflex observed after nerve crush.
ACKNOWLEDGMENTS
During this study AJS was an undergraduate NET/work fellow supported by the Center for Behavioral Neuroscience in Atlanta. Some of the image analysis software and systems used in this study were made available by the Emory University Integrated Cellular Imaging Microscopy Core. The authors want to thank also Paul Nardelli and Lori Goss (Wright State University) for their help with animal surgeries and Dr. Timothy Cope (Georgia Tech, Atlanta) for his helpful comments in previous versions of the manuscript.
Funding information
NIH-NINDS, Grant/Award Number: P01NS057228; Ruth L. Kirschstein NRSA, Grant/Award Number: F31NS095528; Emory Neurosciences NINDS Core facilities, Grant/Award Number: 5P30NS055077
Footnotes
CONFLICT OF INTEREST STATEMENT
None of the authors have any conflicts of interests regarding the contents of this manuscript.
REFERENCES
- Abelew TA, Miller MD, Cope TC, & Nichols TR (2000). Local loss of proprioception results in disruption of interjoint coordination during locomotion in the cat. Journal of Neurophysiology, 84(5), 2709–2714. [DOI] [PubMed] [Google Scholar]
- Allodi I, Udina E, & Navarro X (2012). Specificity of peripheral nerve regeneration: Interactions at the axon level. Progress in Neurobiology, 98(1), 16–37. [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Bullinger KL, Titus HE, Nardelli P, & Cope TC (2010). Permanent reorganization of Ia afferent synapses on motoneurons after peripheral nerve injuries. Annals of the New York Academy of Sciences, 1198, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez FJ, Titus-Mitchell HE, Bullinger KL, Kraszpulski M, Nardelli P, & Cope TC (2011). Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. I. Loss of VGLUT1/IA synapses on motoneurons. Journal of Neurophysiology, 106(5), 2450–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez FJ, Villalba RM, Zerda R, & Schneider SP (2004). Vesicular glutamate transporters in the spinal cord, with special reference to sensory primary afferent synapses. Journal of Comparative Neurology, 472(3), 257–280. [DOI] [PubMed] [Google Scholar]
- Banks RW, & Barker D (1989). Specificities of afferents reinnervating cat muscle spindles after nerve section. The Journal of Physiology, 408, 345–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker D, Scott JJ, & Stacey MJ (1985). Sensory reinnervation of cat peroneus brevis muscle spindles after nerve crush. Brain Research, 333(1), 131–138. [DOI] [PubMed] [Google Scholar]
- Barker D, & Young JZ (1947). Recovery of stretch reflexes after nerve injury. Lancet, 1(6456), 704–707. [DOI] [PubMed] [Google Scholar]
- Betley JN, Wright CV, Kawaguchi Y, Erdelyi F, Szabo G, Jessell TM, & Kaltschmidt JA (2009). Stringent specificity in the construction of a GABAergic presynaptic inhibitory circuit. Cell, 139(1), 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine-Fowler SC, Meyer RS, Moskovitz A, Abrams R, & Botte MJ (1997). Inaccurate projection of rat soleus motoneurons: A comparison of nerve repair techniques. Muscle & Nerve, 20(1), 29–37. [DOI] [PubMed] [Google Scholar]
- Brown MC, & Butler RG (1976). Regeneration of afferent and efferent fibres to muscle spindles after nerve injury in adults cats. The Journal of Physiology, 260(2), 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, & Hardman VJ (1987). A reassessment of the accuracy of reinnervation by motoneurons following crushing or freezing of the sciatic or lumbar spinal nerves of rats. Brain, 110(Pt. 3), 695–705. [DOI] [PubMed] [Google Scholar]
- Brushart TM (1988). Preferential reinnervation of motor nerves by regenerating motor axons. Journal of Neuroscience, 8(3), 1026–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brushart TM (1993). Motor axons preferentially reinnervate motor pathways. Journal of Neuroscience, 13(6), 2730–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brushart TM (2011). Nerve Repair, Oxford University Press. [Google Scholar]
- Brushart TM, & Mesulam MM (1980). Alteration in connections between muscle and anterior horn motoneurons after peripheral nerve repair. Science, 208(4444), 603–605. [DOI] [PubMed] [Google Scholar]
- Brushart TM, Tarlov EC, & Mesulam MM (1983). Specificity of muscle reinnervation after epineurial and individual fascicular suture of the rat sciatic nerve. The Journal of Hand Surgery, 8(3), 248–253. [DOI] [PubMed] [Google Scholar]
- Bullinger KL, Nardelli P, Pinter MJ, Alvarez FJ, & Cope TC (2011). Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. II. Loss of functional connectivity with motoneurons. Journal of Neurophysiology, 106(5), 2471–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Lopes JM, Tavares I, & Coimbra A (1993). GABA decreases in the spinal cord dorsal horn after peripheral neurectomy. Brain Research, 620(2), 287–291. [DOI] [PubMed] [Google Scholar]
- Chan KM, Gordon T, Zochodne DW, & Power HA (2014). Improving peripheral nerve regeneration: From molecular mechanisms to potential therapeutic targets. Experimental Neurology, 261, 826–835. [DOI] [PubMed] [Google Scholar]
- Chang YC, & Gottlieb DI (1988). Characterization of the proteins purified with monoclonal antibodies to glutamic acid decarboxylase. Journal of Neuroscience, 8(6), 2123–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YH, Auyang AG, Scholz JP, & Nichols TR (2009). Whole limb kinematics are preferentially conserved over individual joint kinematics after peripheral nerve injury. Journal of Experimental Biology, 212(Pt. 21), 3511–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradi S (1969). Ultrastructure of dorsal root boutons on lumbosacral motoneurons of the adult cat, as revealed by dorsal root section. Acta physiologica Scandinavica, Supplementum, 332, 85–115. [PubMed] [Google Scholar]
- Cope TC, Bonasera SJ, & Nichols TR (1994). Reinnervated muscles fail to produce stretch reflexes. Journal of Neurophysiology, 71(2), 817–820. [DOI] [PubMed] [Google Scholar]
- Enriquez M, Jimenez I, & Rudomin P (1996). Changes in PAD patterns of group I muscle afferents after a peripheral nerve crush. Experimental Brain Research, 107(3), 405–420. [DOI] [PubMed] [Google Scholar]
- Enriquez-Denton M, Manjarrez E, & Rudomin P (2004). Persistence of PAD and presynaptic inhibition of muscle spindle afferents after peripheral nerve crush. Brain Research, 1027(1–2), 179–187. [DOI] [PubMed] [Google Scholar]
- Fu SY, & Gordon T (1995a). Contributing factors to poor functional recovery after delayed nerve repair: Prolonged axotomy. Journal of Neuroscience, 15(5 Pt 2), 3876–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu SY, & Gordon T (1995b). Contributing factors to poor functional recovery after delayed nerve repair: Prolonged denervation. Journal of Neuroscience, 15(5 Pt 2), 3886–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtrey CM, Asher RA, Nothias F, & Fawcett JW (2007). Promoting plasticity in the spinal cord with chondroitinase improves functional recovery after peripheral nerve repair. Brain, 130(Pt. 4), 926–939. [DOI] [PubMed] [Google Scholar]
- Gillespie MJ, Gordon T, & Murphy PR (1986). Reinnervation of the lateral gastrocnemius and soleus muscles in the rat by their common nerve. The Journal of Physiology, 372, 485–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon T, & English AW (2016). Strategies to promote peripheral nerve regeneration: Electrical stimulation and/or exercise. European Journal of Neuroscience, 43(3), 336–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb DI, Chang YC, & Schwob JE (1986). Monoclonal antibodies to glutamic acid decarboxylase. Proceedings of the National Academy of Sciences of the United States of America, 83(22), 8808–8812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haftel VK, Bichler EK, Wang QB, Prather JF, Pinter MJ, & Cope TC (2005). Central suppression of regenerated proprioceptive afferents. Journal of Neuroscience, 25(19), 4733–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoke A, Redett R, Hameed H, Jari R, Zhou C, Li ZB, … Brushart TM (2006). Schwann cells express motor and sensory phenotypes that regulate axon regeneration. Journal of Neuroscience, 26(38), 9646–9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horch KW, & Lisney SJ (1981). Changes in primary afferent depolarization of sensory neurones during peripheral nerve regeneration in the cat. The Journal of Physiology, 313, 287–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DI, Mackie M, Nagy GG, Riddell JS, Maxwell DJ, Szabo G, … Todd AJ (2005). P boutons in lamina IX of the rodent spinal cord express high levels of glutamic acid decarboxylase-65 and originate from cells in deep medial dorsal horn. Proceedings of the National Academy of Sciences of the United States of America, 102(25), 9038–9043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde D, & Scott JJ (1983). Responses of cat peroneus brevis muscle spindle afferents during recovery from nerve-crush injury. Journal of Neurophysiology, 50(2), 344–357. [DOI] [PubMed] [Google Scholar]
- Lundborg G (2003). Richard P. Bunge memorial lecture. Nerve injury and repair–A challenge to the plastic brain. Journal of the Peripheral Nervous System, 8(4), 209–226. [DOI] [PubMed] [Google Scholar]
- Lyle MA, Prilutsky BI, Gregor RJ, Abelew TA, & Nichols TR (2016). Self-reinnervated muscles lose autogenic length feedback, but intermuscular feedback can recover functional connectivity. Journal of Neurophysiology, 116(3), 1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas H, Prilutsky BI, Nichols TR, & Gregor RJ (2007). The effects of self-reinnervation of cat medial and lateral gastrocnemius muscles on hindlimb kinematics in slope walking. Experimental Brain Research, 181(2), 377–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R, Schachner M, & Brushart TM (1994). The L2/HNK-1 carbohydrate is preferentially expressed by previously motor axon-associated Schwann cells in reinnervated peripheral nerves. Journal of Neuroscience, 14(11 Pt. 2), 7180–7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell DJ, Christie WM, Short AD, & Brown AG (1990). Direct observations of synapses between GABA-immunoreactive boutons and muscle afferent terminals in lamina VI of the cat’s spinal cord. Brain Research, 530(2), 215–222. [DOI] [PubMed] [Google Scholar]
- Mende M, Fletcher EV, Belluardo JL, Pierce JP, Bommareddy PK, Weinrich JA, … Kaltschmidt JA (2016). Sensory-derived glutamate regulates presynaptic inhibitory terminals in mouse spinal cord. Neuron, 90(6), 1189–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, & Woolf CJ (2002). Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. Journal of Neuroscience, 22(15), 6724–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen QT, Sanes JR, & Lichtman JW (2002). Pre-existing pathways promote precise projection patterns. Nature Neuroscience, 5(9), 861–867. [DOI] [PubMed] [Google Scholar]
- Obeidat AZ, Rotterman TM, Alvarez FJ, & Cope TC (2012). Global changes in recurrent inhibition after peripheral nerve repair. Program No. 789.12. 2012 Neuroscience Meeting Planner. New Orleans, LA: Society for Neuroscience. [Google Scholar]
- Pierce JP, & Mendell LM (1993). Quantitative ultrastructure of Ia boutons in the ventral horn: Scaling and positional relationships. Journal of Neuroscience, 13(11), 4748–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prather JF, Nardelli P, Nakanishi ST, Ross KT, Nichols TR, Pinter MJ, & Cope TC (2011). Recovery of proprioceptive feedback from nerve crush. The Journal of Physiology, 589(Pt. 20), 4935–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter W, Shi Y, & Baekkeskov S (1993). Autoreactive epitopes defined by diabetes-associated human monoclonal antibodies are localized in the middle and C-terminal domains of the smaller form of glutamate decarboxylase. Proceedings of the National Academy of Sciences of the United States of America, 90(7), 2832–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotterman TM, & Alvarez FJ (2015). The severity of the central neuroimmune response following peripheral nerve injury correlates with the amount of proprioceptive IA afferent loss from injured motoneurons. Program No. 607.10. 2015 Neuroscience Meeting Planner. Chicago, IL: Society for Neuroscience. [Google Scholar]
- Rotterman TM, Nardelli P, Cope TC, & Alvarez FJ (2014). Normal distribution of VGLUT1 synapses on spinal motoneuron dendrites and their reorganization after nerve injury. Journal of Neuroscience, 34(10), 3475–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudomin P, & Schmidt RF (1999). Presynaptic inhibition in the vertebrate spinal cord revisited. Experimental Brain Research, 129(1), 1–37. [DOI] [PubMed] [Google Scholar]
- Sabatier MJ, To BN, Nicolini J, & English AW (2011). Effect of axon misdirection on recovery of electromyographic activity and kinematics after peripheral nerve injury. Cells Tissues Organs, 193(5), 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shneider NA, Brown MN, Smith CA, Pickel J, & Alvarez FJ (2009). Gamma motor neurons express distinct genetic markers at birth and require muscle spindle-derived GDNF for postnatal survival. Neural Development, 4, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siembab VC, Gomez-Perez L, Rotterman TM, Shneider NA, & Alvarez FJ (2016). Role of primary afferents in the developmental regulation of motor axon synapse numbers on Renshaw cells. Journal of Comparative Neurology, 524(9), 1892–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunderland S (1947). Rate of regeneration in human peripheral nerves; analysis of the interval between injury and onset of recovery. Archives of Neurology and Psychiatry, 58(3), 251–295. [DOI] [PubMed] [Google Scholar]
- Surmeli G, Akay T, Ippolito GC, Tucker PW, & Jessell TM (2011). Patterns of spinal sensory-motor connectivity prescribed by a dorsoventral positional template. Cell, 147(3), 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero-Cabre A, Tsironis K, Skouras E, Navarro X, & Neiss WF (2004). Peripheral and spinal motor reorganization after nerve injury and repair. Journal of Neurotrauma, 21(1), 95–108. [DOI] [PubMed] [Google Scholar]