

PRESENTATION OF CASE
Dr. Max C. Petersen (Medicine): A 34-year-old woman was evaluated in the diabetes clinic of this hospital for hyperglycemia.
Eleven years before this presentation, the blood glucose level was 126 mg per deciliter (7.0 mmol per liter) on routine laboratory evaluation, which was performed as part of an annual well visit. The patient could not recall whether she had been fasting at the time the test had been performed. One year later, the fasting blood glucose level was 112 mg per deciliter (6.2 mmol per liter; reference range, <100 mg per deciliter [<5.6 mmol per liter]).
Nine years before this presentation, a randomly obtained blood glucose level was 217 mg per deciliter (12.0 mmol per liter), and the patient reported polyuria. At that time, the glycated hemoglobin level was 5.8% (reference range, 4.3 to 5.6); the hemoglobin level was normal. One year later, the glycated hemoglobin level was 5.9%. The height was 165.1 cm, the weight 72.6 kg, and the body-mass index (BMI; the weight in kilograms divided by the square of the height in meters) 26.6. The patient received a diagnosis of prediabetes and was referred to a nutritionist. She made changes to her diet and lost 4.5 kg of body weight over a 6-month period; the glycated hemoglobin level was 5.5%.
Six years before this presentation, the patient became pregnant with her first child. Her prepregnancy BMI was 24.5. At 26 weeks of gestation, the result of a 1-hour oral glucose challenge test (i.e., the blood glucose level obtained 1 hour after the oral administration of a 50-g glucose load in the nonfasting state) was 186 mg per deciliter (10.3 mmol per liter; reference range, <140 mg per deciliter [<7.8 mmol per liter]). She declined a 3-hour oral glucose tolerance test; a presumptive diagnosis of gestational diabetes was made. She was asked to follow a meal plan for gestational diabetes and was treated with insulin during the pregnancy. Serial ultrasound examinations for fetal growth and monitoring were performed. At 34 weeks of gestation, the fetal abdominal circumference was in the 76th percentile for gestational age. Polyhydramnios developed at 37 weeks of gestation. The child was born at 39 weeks 3 days of gestation, weighed 3.9 kg at birth, and had hypoglycemia after birth, which subsequently resolved. Six weeks post partum, the patient’s fasting blood glucose level was 120 mg per deciliter (6.7 mmol per liter), and the result of a 2-hour oral glucose tolerance test (i.e., the blood glucose level obtained 2 hours after the oral administration of a 75-g glucose load in the fasting state) was 131 mg per deciliter (7.3 mmol per liter; reference range, <140 mg per deciliter). Three months post partum, the glycated hemoglobin level was 6.1%. Lifestyle modification for diabetes prevention was recommended.
Four and a half years before this presentation, the patient became pregnant with her second child. Her prepregnancy BMI was 25.1. At 5 weeks of gestation, she had an elevated blood glucose level. Insulin therapy was started at 6 weeks of gestation, and episodes of hypoglycemia occurred during the pregnancy. Serial ultrasound examinations for fetal growth and monitoring were performed. At 28 weeks of gestation, the fetal abdominal circumference was in the 35th percentile for gestational age, and the amniotic fluid level was normal. Labor was induced at 38 weeks of gestation; the child weighed 2.6 kg at birth. Neonatal blood glucose levels were reported as stable after birth. Six weeks post partum, the patient’s fasting blood glucose level was 133 mg per deciliter (7.4 mmol per liter), and the result of a 2-hour oral glucose tolerance test was 236 mg per deciliter (13.1 mmol per liter). The patient received a diagnosis of type 2 diabetes mellitus; lifestyle modification was recommended. Three months post partum, the glycated hemoglobin level was 5.9% and the BMI was 30.0. Over the next 2 years, she followed a low-carbohydrate diet and regular exercise plan and self-monitored the blood glucose level.
Two years before this presentation, the patient became pregnant with her third child. Blood glucose levels were again elevated, and insulin therapy was started early in gestation. She had episodes of hypoglycemia that led to adjustment of her insulin regimen. The child was born at 38 weeks 5 days of gestation, weighed 3.0 kg at birth, and had hypoglycemia that resolved 48 hours after birth. After the birth of her third child, the patient started to receive metformin, which had no effect on the glycated hemoglobin level, despite adjustment of the therapy to the maximal dose.
One year before this presentation, the patient became pregnant with her fourth child. Insulin therapy was again started early in gestation. The patient reported that episodes of hypoglycemia occurred. Polyhydramnios developed. The child was born at 38 weeks 6 days of gestation and weighed 3.5 kg. The patient sought care at the diabetes clinic of this hospital for clarification of her diagnosis.
The patient reported following a low-carbohydrate diet and exercising 5 days per week. There was no fatigue, change in appetite, change in vision, chest pain, shortness of breath, polydipsia, or polyuria. There was no history of anemia, pancreatitis, hirsutism, proximal muscle weakness, easy bruising, headache, sweating, tachycardia, gallstones, or diarrhea. Her menstrual periods were normal. She had not noticed any changes in her facial features or the size of her hands or feet.
The patient had a history of acne and low-back pain. Her only medication was metformin. She had no known medication allergies. She lived with her husband and four children in a suburban community in New England and worked as an administrator. She did not smoke tobacco or use illicit drugs, and she rarely drank alcohol. She identified as non-Hispanic white. Both of her grandmothers had type 2 diabetes mellitus. Her father had hypertension, was overweight, and had received a diagnosis of type 2 diabetes at 50 years of age. Her mother was not overweight and had received a diagnosis of type 2 diabetes at 48 years of age. The patient had two sisters, neither of whom had a history of diabetes or gestational diabetes. There was no family history of hemochromatosis.
On examination, the patient appeared well. The blood pressure was 126/76 mm Hg, and the heart rate 76 beats per minute. The BMI was 25.4. The physical examination was normal. The glycated hemoglobin level was 6.2%.
A diagnostic test was performed.
DIFFERENTIAL DIAGNOSIS
Dr. Miriam S. Udler: I am aware of the diagnosis in this case and participated in the care of this patient. This healthy 34-year-old woman, who had a BMI just above the upper limit of the normal range, presented with a history of hyperglycemia of varying degrees since 24 years of age. When she was not pregnant, she was treated with lifestyle measures as well as metformin therapy for a short period, and she maintained a well-controlled blood glucose level. In thinking about this case, it is helpful to characterize the extent of the hyperglycemia and then to consider its possible causes.
CHARACTERIZING HYPERGLYCEMIA
This patient’s hyperglycemia reached a threshold that was diagnostic of diabetes1 on two occasions: when she was 25 years of age, she had a randomly obtained blood glucose level of 217 mg per deciliter with polyuria (with diabetes defined as a level of ≥200 mg per deciliter [≥11.1 mmol per liter] with symptoms), and when she was 30 years of age, she had on the same encounter a fasting blood glucose level of 133 mg per deciliter (with diabetes defined as a level of ≥126 mg per deciliter) and a result on a 2-hour oral glucose tolerance test of 236 mg per deciliter (with diabetes defined as a level of ≥200 mg per deciliter). On both of these occasions, her glycated hemoglobin level was in the prediabetes range (defined as 5.7 to 6.4%). In establishing the diagnosis of diabetes, the various blood glucose studies and glycated hemoglobin testing may provide discordant information because the tests have different sensitivities for this diagnosis, with glycated hemoglobin testing being the least sensitive.2 Also, there are situations in which the glycated hemoglobin level can be inaccurate; for example, the patient may have recently received a blood transfusion or may have a condition that alters the life span of red cells, such as anemia, hemoglobinopathy, or pregnancy.3 These conditions were not present in this patient at the time that the glycated hemoglobin measurements were obtained. In addition, since the glycated hemoglobin level reflects the average glucose level typically over a 3-month period, discordance with timed blood glucose measurements can occur if there has been a recent change in glycemic control. This patient had long-standing mild hyperglycemia but met criteria for diabetes on the basis of the blood glucose levels noted.
DIABETES
Type 1 and Type 2 Diabetes
Now that we have characterized the patient’s hyperglycemia as meeting criteria for diabetes, it is important to consider the possible types. More than 90% of adults with diabetes have type 2 diabetes, which is due to progressive loss of insulin secretion by beta cells that frequently occurs in the context of insulin resistance. This patient had received a diagnosis of type 2 diabetes; however, some patients with diabetes may be given a diagnosis of type 2 diabetes on the basis of not having features of type 1 diabetes, which is characterized by autoimmune destruction of the pancreatic beta cells that leads to rapid development of insulin dependence, with ketoacidosis often present at diagnosis.
Type 1 diabetes accounts for approximately 6% of all cases of diabetes in adults (≥18 years of age) in the United States,4 and 80% of these cases are diagnosed before the patient is 20 years of age.5 Since this patient’s diabetes was essentially nonprogressive over a period of at least 9 years, she most likely does not have type 1 diabetes. It is therefore not surprising that she had received a diagnosis of type 2 diabetes, but there are several other types of diabetes to consider, particularly since some features of her case do not fit with a typical case of type 2 diabetes, such as her age at diagnosis, the presence of hyperglycemia despite a nearly normal BMI, and the mild and nonprogressive nature of her disease over the course of many years.
Less Common Types of Diabetes
Latent autoimmune diabetes in adults (LADA) is a mild form of autoimmune diabetes that should be considered in this patient. However, there is controversy as to whether LADA truly represents an entity that is distinct from type 1 diabetes.6 Both patients with type 1 diabetes and patients with LADA commonly have elevated levels of diabetes-associated autoantibodies; however, LADA has been defined by an older age at onset (typically >25 years) and slower progression to insulin dependence (over a period of >6 months).7 This patient had not been tested for diabetes-associated autoantibodies. I ordered these tests to help evaluate for LADA, but this was not my leading diagnosis because of her young age at diagnosis and nonprogressive clinical course over a period of at least 9 years.
If the patient’s diabetes had been confined to pregnancy, we might consider gestational diabetes, but she had hyperglycemia outside of pregnancy. Several medications can cause hyperglycemia, including glucocorticoids, atypical antipsychotic agents, cancer immunotherapies, and some antiretroviral therapies and immunosuppressive agents used in transplantation.8 However, this patient was not receiving any of these medications. Another cause of diabetes to consider is destruction of the pancreas due to, for example, cystic fibrosis, a tumor, or pancreatitis, but none of these were present. Secondary endocrine disorders — including excess cortisol production, excess growth hormone production, and pheochromocytoma — were considered to be unlikely in this patient on the basis of the history, review of symptoms, and physical examination.
Monogenic Diabetes
A final category to consider is monogenic diabetes, which is caused by alteration of a single gene. Types of monogenic diabetes include maturity-onset diabetes of the young (MODY), neonatal diabetes, and syndromic forms of diabetes. Monogenic diabetes accounts for 1 to 6% of cases of diabetes in children9 and approximately 0.4% of cases in adults.10 Neonatal diabetes is diagnosed typically within the first 6 months of life; syndromic forms of monogenic diabetes have other abnormal features, including particular organ dysfunction. Neither condition is applicable to this patient.
MODY
MODY is an autosomal dominant condition characterized by primary pancreatic beta-cell dysfunction that causes mild diabetes that is diagnosed during adolescence or early adulthood. As early as 1964, the nomenclature “maturity-onset diabetes of the young” was used to describe cases that resembled adult-onset type 2 diabetes in terms of the slow progression to insulin use (as compared with the rapid progression in type 1 diabetes) but occurred in relatively young patients.11 Several genes cause distinct forms of MODY that have specific disease features that inform treatment, and thus MODY is a clinically important diagnosis. Most forms of MODY cause isolated abnormal glucose levels (in contrast to syndromic monogenic diabetes), a manifestation that has contributed to its frequent misdiagnosis as type 1 or type 2 diabetes.12
Genetic Basis of MODY
Although at least 13 genes have been associated with MODY, 3 genes — GCK, which encodes glucokinase, and HNF1A and HNF4A, which encode hepatocyte nuclear factors 1A and 4A, respectively — account for most cases. MODY associated with GCK (known as GCK-MODY) is characterized by mild, nonprogressive hyperglycemia that is present since birth, whereas the forms of MODY associated with HNF1A and HNF4A (known as HNF1A-MODY and HNF4A-MODY, respectively) are characterized by the development of diabetes, typically in the early teen years or young adulthood, that is initially mild and then progresses such that affected patients may receive insulin before diagnosis.
In patients with GCK-MODY, genetic variants reduce the function of glucokinase, the enzyme in pancreatic beta cells that functions as a glucose sensor and controls the rate of entry of glucose into the glycolytic pathway. As a result, reduced sensitivity to glucose-induced insulin secretion causes asymptomatic mild fasting hyperglycemia, with an upward shift in the normal range of the fasting blood glucose level to 100 to 145 mg per deciliter (5.6 to 8.0 mmol per liter), and also causes an upward shift in postprandial blood glucose levels, but with tight regulation maintained (Fig. 1).13 This mild hyperglycemia is not thought to confer a predisposition to complications of diabetes,14 is largely unaltered by treatment,15 and does not necessitate treatment outside of pregnancy.
Figure 1. Features of GCK-MODY in This Patient.
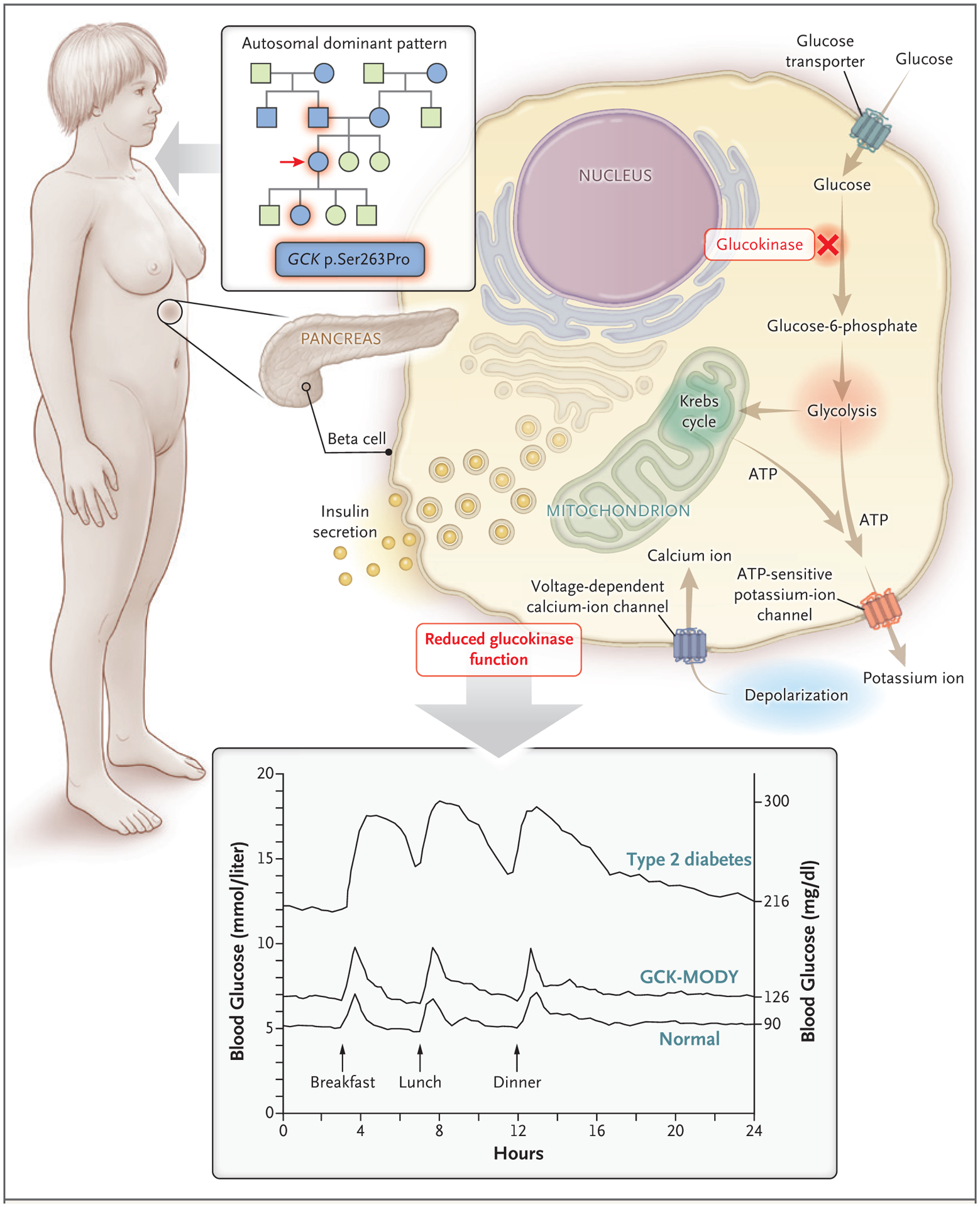
Key features suggesting maturity-onset diabetes of the young (MODY) in this patient were an age of less than 35 years at the diagnosis of diabetes, a strong family history of diabetes with an autosomal dominant pattern of inheritance, and hyperglycemia despite a close-to-normal body-mass index. None of these features is an absolute criterion. MODY is caused by single gene–mediated disruption of pancreatic beta-cell function. In MODY associated with the GCK gene (known as GCK-MODY), disrupted glucokinase function causes a mild upward shift in glucose levels through-out the day and does not necessitate treatment.13 In the pedigree, circles represent female family members, squares male family members, blue family members affected by diabetes, and green unaffected family members. The arrow indicates the patient.
In contrast to GCK-MODY, the disorders HNF1A-MODY and HNF4A-MODY result in progressive hyperglycemia that eventually leads to treatment.16 Initially, there may be a normal fasting glucose level and large spikes in postprandial glucose levels (to >80 mg per deciliter [>4.4 mmol per liter]).17 Patients can often be treated with oral agents and discontinue insulin therapy started before the diagnosis of MODY.18 Of note, patients with HNF1A-MODY or HNF4A-MODY are typically sensitive to treatment with sulfonylureas19 but may also respond to glucagon-like peptide-1 receptor agonists.20
This patient had received a diagnosis of diabetes before 35 years of age, had a family history of diabetes involving multiple generations, and was not obese. These features are suggestive of MODY but do not represent absolute criteria for the condition (Fig. 1).1 Negative testing for diabetes-associated autoantibodies would further increase the likelihood of MODY. There are methods to calculate a patient’s risk of having MODY associated with GCK, HNF1A, or HNF4A.21,22 Using an online calculator (www.diabetesgenes.org/mody-probability-calculator), we estimate that the probability of this patient having MODY is at least 75.5%. Genetic testing would be needed to confirm this diagnosis, and in patients at an increased risk for MODY, multigene panel testing has been shown to be cost-effective.23,24
DR. MIRIAM S. UDLER’S DIAGNOSIS
Maturity-onset diabetes of the young, most likely due to a GCK variant.
DIAGNOSTIC TESTING
Dr. Christina A. Austin-Tse: A diagnostic sequencing test of five genes associated with MODY was performed. One clinically significant variant was identified in the GCK gene (NM_000162.3): a c.787T→C transition resulting in the p.Ser263Pro missense change. Review of the literature and variant databases revealed that this variant had been previously identified in at least three patients with early-onset diabetes and had segregated with disease in at least three affected members of two families (GeneDx: personal communication).25,26 Furthermore, the variant was rare in large population databases (occurring in 1 out of 128,844 European chromosomes in gnomAD27), a feature consistent with a disease-causing role. Although the serine residue at position 263 was not highly conserved, multiple in vitro functional studies have shown that the p.Ser263Pro variant negatively affects the stability of the glucokinase enzyme.26,28–30 As a result, this variant met criteria to be classified as “likely pathogenic.”31 As mentioned previously, a diagnosis of GCK-MODY is consistent with this patient’s clinical features. On subsequent testing of additional family members, the same “likely pathogenic” variant was identified in the patient’s father and second child, both of whom had documented hyperglycemia.
DISCUSSION OF MANAGEMENT
Dr. Udler: In this patient, the diagnosis of GCK-MODY means that it is normal for her blood glucose level to be mildly elevated. She can stop taking metformin because discontinuation is not expected to substantially alter her glycated hemoglobin level15,32 and because she is not at risk for complications of diabetes.14 However, she should continue to maintain a healthy lifestyle. Although patients with GCK-MODY are not typically treated for hyperglycemia outside of pregnancy, they may need to be treated during pregnancy.
It is possible for a patient to have type 1 or type 2 diabetes in addition to MODY, so this patient should be screened for diabetes according to recommendations for the general population (e.g., in the event that she has a risk factor for diabetes, such as obesity).1 Since the mild hyperglycemia associated with GCK-MODY is asymptomatic (and probably unrelated to the polyuria that this patient had described in the past), the development of symptoms of hyperglycemia, such as polyuria, polydipsia, or blurry vision, should prompt additional evaluation. In patients with GCK-MODY, the glycated hemoglobin level is typically below 7.5%,33 so a value rising above that threshold or a sudden large increase in the glycated hemoglobin level could indicate concomitant diabetes from another cause, which would need to be evaluated and treated.
This patient’s family members are at risk for having the same GCK variant, with a 50% chance of offspring inheriting a variant from an affected parent. Since the hyperglycemia associated with GCK-MODY is present from birth, it is necessary to perform genetic testing only in family members with demonstrated hyperglycemia. I offered site-specific genetic testing to the patient’s parents and second child.
Dr. Meridale V. Baggett (Medicine): Dr. Powe, would you tell us how you would treat this patient during pregnancy?
Dr. Camille E. Powe: During the patient’s first pregnancy, routine screening led to a presumptive diagnosis of gestational diabetes, the most common cause of hyperglycemia in pregnancy. Hyperglycemia in pregnancy is associated with adverse pregnancy outcomes,34 and treatment lowers the risk of such outcomes.35,36 Two of the most common complications — fetal overgrowth (which can lead to birth injuries, shoulder dystocia, and an increased risk of cesarean delivery) and neonatal hypoglycemia — are thought to be the result of fetal hyperinsulinemia.37 Maternal glucose is freely transported across the placenta, and excess glucose augments insulin secretion from the fetal pancreas. In fetal life, insulin is a potent growth factor, and neonates who have hyperinsulinemia in utero often continue to secrete excess insulin in the first few days of life. In the treatment of pregnant women with diabetes, we strive for strict blood sugar control (fasting blood glucose level, <95 mg per deciliter [<5.3 mmol per liter]; 2-hour postprandial blood glucose level, <120 mg per deciliter) to decrease the risk of these and other hyperglycemia-associated adverse pregnancy outcomes.38–40
In the third trimester of the patient’s first pregnancy, obstetrical ultrasound examination revealed a fetal abdominal circumference in the 76th percentile for gestational age and polyhydramnios, signs of fetal exposure to maternal hyperglycemia.40–42 Case series involving families with GCK-MODY have shown that the effect of maternal hyperglycemia on the fetus depends on whether the fetus inherits the pathogenic GCK variant.43–48 Fetuses that do not inherit the maternal variant have overgrowth, presumably due to fetal hyperinsulinemia (Fig. 2A). In contrast, fetuses that inherit the variant do not have overgrowth and are born at a weight that is near the average for gestational age, despite maternal hyperglycemia, presumably because the variant results in decreased insulin secretion (Fig. 2B). Fetuses that inherit GCK-MODY from their fathers and have euglycemic mothers appear to be undergrown, most likely because their insulin secretion is lower than normal when they and their mothers are euglycemic (Fig. 2D). Because fetal overgrowth and polyhydramnios occurred during this patient’s first pregnancy and neonatal hypoglycemia developed after the birth, the patient’s first child is probably not affected by GCK-MODY.
Figure 2. Effect of Fetal GCK-MODY on the Relationship between Maternal Blood Glucose Level and Fetal Growth.
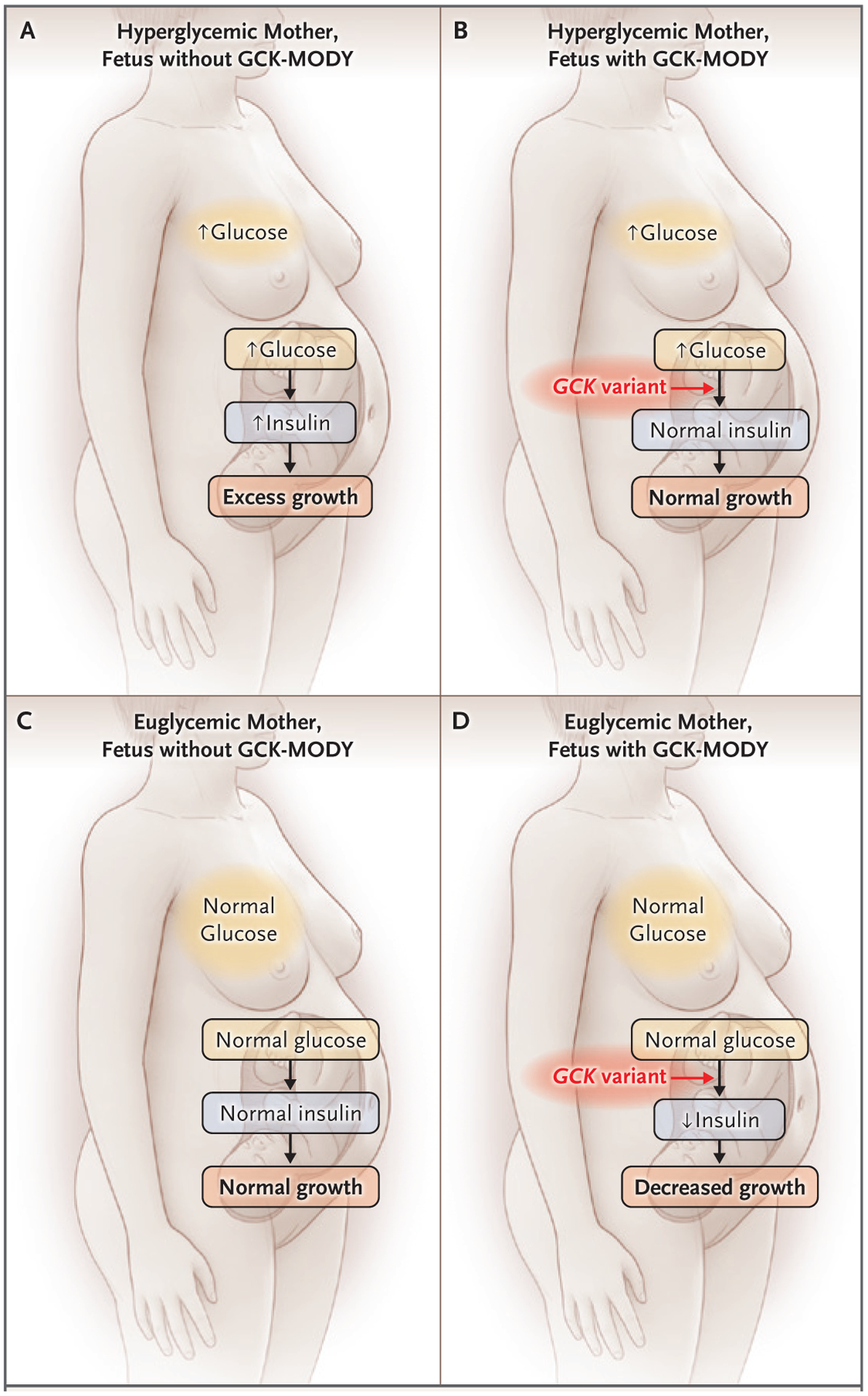
Pathogenic variants that lead to GCK-MODY, when carried by a fetus, change the usual relationship of maternal hyperglycemia to fetal hyperinsulinemia and fetal overgrowth. GCK-MODY–affected fetuses have lower insulin secretion than unaffected fetuses in response to the same maternal blood glucose level. In a hyperglycemic mother carrying a fetus who is unaffected by GCK-MODY, excessive fetal growth is usually apparent (Panel A). Studies involving GCK-MODY–affected hyperglycemic mothers have shown that fetal growth is normal despite maternal hyperglycemia when a fetus has the maternal GCK variant (Panel B). The goal of treatment of maternal hyperglycemia when a fetus is unaffected by GCK-MODY is to establish euglycemia to normalize fetal insulin levels and growth (Panel C); whether this can be accomplished in the case of maternal GCK-MODY is controversial, given the genetically determined elevated maternal glycemic set point. In the context of maternal euglycemia, GCK-MODY–affected fetuses may be at risk for fetal growth restriction (Panel D).
In accordance with standard care for pregnant women with diabetes who do not meet glycemic targets after dietary modification,38,39 the patient was treated with insulin during her pregnancies. In her second pregnancy, treatment was begun early, after hyperglycemia was detected in the first trimester. Because she had not yet received the diagnosis of GCK-MODY during any of her pregnancies, no consideration of this condition was given during her obstetrical treatment. Whether treatment affects the risk of hyperglycemia-associated adverse pregnancy outcomes in pregnant women with known GCK-MODY is controversial, with several case series showing that the birth weight percentile in unaffected neonates remains consistent regardless of whether the mother is treated with insulin.44,45 Evidence suggests that it may be difficult to overcome a genetically determined glycemic set point in patients with GCK-MODY with the use of pharmacotherapy,15,32 and affected patients may have symptoms of hypoglycemia when the blood glucose level is normal because of an enhanced counterregulatory response.49,50 Still, to the extent that it is possible, it would be desirable to safely lower the blood glucose level in a woman with GCK-MODY who is pregnant with an unaffected fetus in order to decrease the risk of fetal overgrowth and other consequences of mildly elevated glucose levels (Fig. 2C).46,47,51 In contrast, there is evidence that lowering the blood glucose level in a pregnant woman with GCK-MODY could lead to fetal growth restriction if the fetus is affected (Fig. 2D).45,52 During this patient’s second pregnancy, she was treated with insulin beginning in the first trimester, and her daughter’s birth weight was near the 16th percentile for gestational age; this outcome is consistent with the daughter’s ultimate diagnosis of GCK-MODY.
Expert opinion suggests that, in pregnant women with GCK-MODY, insulin therapy should be deferred until fetal growth is assessed by means of ultrasound examination beginning in the late second trimester. If there is evidence of fetal overgrowth, the fetus is presumed to be unaffected by GCK-MODY and insulin therapy is initiated.53 After I have counseled women with GCK-MODY on the potential risks and benefits of insulin treatment during pregnancy, I have sometimes used a strategy of treating hyperglycemia from early in pregnancy using modified glycemic targets that are less stringent than the targets typically used during pregnancy. This strategy attempts to balance the risk of growth restriction in an affected fetus (as well as maternal hypoglycemia) with the potential benefit of glucose-lowering therapy for an unaffected fetus.
FOLLOW-UP
Dr. Udler: The patient stopped taking metformin, and subsequent glycated hemoglobin levels remained unchanged, at 6.2%. Her father and 5-year-old daughter (second child) both tested positive for the same GCK variant. Her father had a BMI of 36 and a glycated hemoglobin level of 7.8%, so I counseled him that he most likely had type 2 diabetes in addition to GCK-MODY. He is currently being treated with metformin and lifestyle measures. The patient’s daughter now has a clear diagnosis to explain her hyperglycemia, which will help in preventing misdiagnosis of type 1 diabetes, given her young age, and will be important for the management of any future pregnancies. She will not need any medical follow-up for GCK-MODY until she is considering pregnancy.
FINAL DIAGNOSIS
Maturity-onset diabetes of the young due to a GCK variant.
Acknowledgments
We thank Dr. Andrew Hattersley and Dr. Sarah Bernstein for helpful comments on an earlier draft of the manuscript.
Footnotes
This case was presented at the Medical Case Conference.
No potential conflict of interest relevant to this article was reported.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
REFERENCES
- 1.American Diabetes Association. 2. Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes — 2019. Diabetes Care 2019; 42: Suppl 1: S13–S28. [DOI] [PubMed] [Google Scholar]
- 2.Cowie CC, Rust KF, Byrd-Holt DD, et al. Prevalence of diabetes and high risk for diabetes using A1C criteria in the U.S. population in 1988–2006. Diabetes Care 2010; 33: 562–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radin MS. Pitfalls in hemoglobin A1c measurement: when results may be misleading. J Gen Intern Med 2014; 29: 388–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bullard KM, Cowie CC, Lessem SE, et al. Prevalence of diagnosed diabetes in adults by diabetes type — United States, 2016. MMWR Morb Mortal Wkly Rep 2018; 67: 359–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liese AD, D’Agostino RB Jr, Hamman RF, et al. The burden of diabetes mellitus among US youth: prevalence estimates from the SEARCH for Diabetes in Youth Study. Pediatrics 2006; 118: 1510–8. [DOI] [PubMed] [Google Scholar]
- 6.Pieralice S, Pozzilli P. Latent autoimmune diabetes in adults: a review on clinical implications and management. Diabetes Metab J 2018; 42: 451–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stenström G, Gottsäter A, Bakhtadze E, Berger B, Sundkvist G. Latent autoimmune diabetes in adults: definition, prevalence, beta-cell function, and treatment. Diabetes 2005; 54: Suppl 2: S68–S72. [DOI] [PubMed] [Google Scholar]
- 8.Fathallah N, Slim R, Larif S, Hmouda H, Ben Salem C. Drug-induced hyperglycaemia and diabetes. Drug Saf 2015; 38: 1153–68. [DOI] [PubMed] [Google Scholar]
- 9.Hattersley AT, Greeley SAW, Polak M, et al. ISPAD Clinical Practice Consensus Guidelines 2018: the diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes 2018; 19: Suppl 27: 47–63. [DOI] [PubMed] [Google Scholar]
- 10.Shields BM, Shepherd M, Hudson M, et al. Population-based assessment of a biomarker-based screening pathway to aid diagnosis of monogenic diabetes in young-onset patients. Diabetes Care 2017; 40: 1017–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fajans SS, Bell GI. MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care 2011; 34: 1878–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia 2010; 53: 2504–8. [DOI] [PubMed] [Google Scholar]
- 13.Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med 2001; 345: 971–80. [DOI] [PubMed] [Google Scholar]
- 14.Steele AM, Shields BM, Wensley KJ, Colclough K, Ellard S, Hattersley AT. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA 2014; 311: 279–86. [DOI] [PubMed] [Google Scholar]
- 15.Stride A, Shields B, Gill-Carey O, et al. Cross-sectional and longitudinal studies suggest pharmacological treatment used in patients with glucokinase mutations does not alter glycaemia. Diabetologia 2014; 57: 54–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isomaa B, Henricsson M, Lehto M, et al. Chronic diabetic complications in patients with MODY3 diabetes. Diabetologia 1998; 41: 467–73. [DOI] [PubMed] [Google Scholar]
- 17.Stride A, Vaxillaire M, Tuomi T, et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia 2002; 45: 427–35. [DOI] [PubMed] [Google Scholar]
- 18.Shepherd M, Shields B, Ellard S, Rubio-Cabezas O, Hattersley AT. A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients. Diabet Med 2009; 26: 437–41. [DOI] [PubMed] [Google Scholar]
- 19.Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 2003; 362: 1275–81. [DOI] [PubMed] [Google Scholar]
- 20.Østoft SH, Bagger JI, Hansen T, et al. Glucose-lowering effects and low risk of hypoglycemia in patients with maturity-onset diabetes of the young when treated with a GLP-1 receptor agonist: a double-blind, randomized, crossover trial. Diabetes Care 2014; 37: 1797–805. [DOI] [PubMed] [Google Scholar]
- 21.MODY Probability Calculator. Exeter, United Kingdom: Diabetes Genes, 2019. (https://www.diabetesgenes.org/mody-probability-calculator/).
- 22.Shields BM, McDonald TJ, Ellard S, Campbell MJ, Hyde C, Hattersley AT. The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia 2012; 55: 1265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naylor RN, John PM, Winn AN, et al. Cost-effectiveness of MODY genetic testing: translating genomic advances into practical health applications. Diabetes Care 2014; 37: 202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson SR, Carter HE, Leo P, et al. Cost-effectiveness analysis of routine screening using massively parallel sequencing for maturity-onset diabetes of the young in a pediatric diabetes cohort: reduced health system costs and improved patient quality of life. Diabetes Care 2019; 42: 69–76. [DOI] [PubMed] [Google Scholar]
- 25.Cao H, Shorey S, Robinson J, et al. GCK and HNF1A mutations in Canadian families with maturity onset diabetes of the young (MODY). Hum Mutat 2002; 20: 478–9. [DOI] [PubMed] [Google Scholar]
- 26.Sagen JV, Odili S, Bjørkhaug L, et al. From clinicogenetic studies of maturity-onset diabetes of the young to unraveling complex mechanisms of glucokinase regulation. Diabetes 2006; 55: 1713–22. [DOI] [PubMed] [Google Scholar]
- 27.Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/).
- 28.Zelent B, Odili S, Buettger C, et al. Mutational analysis of allosteric activation and inhibition of glucokinase. Bio-chem J 2011; 440: 203–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fenner D, Odili S, Hong H-K, et al. Generation of N-ethyl-N-nitrosourea (ENU) diabetes models in mice demonstrates genotype-specific action of glucokinase activators. J Biol Chem 2011; 286: 39560–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Negahdar M, Aukrust I, Johansson BB, et al. GCK-MODY diabetes associated with protein misfolding, cellular self-association and degradation. Biochim Bio-phys Acta 2012; 1822: 1705–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shepherd MH, Shields BM, Hudson M, et al. A UK nationwide prospective study of treatment change in MODY: genetic subtype and clinical characteristics predict optimal glycaemic control after discontinuing insulin and metformin. Diabetologia 2018; 61: 2520–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steele AM, Wensley KJ, Ellard S, et al. Use of HbA1c in the identification of patients with hyperglycaemia caused by a glucokinase mutation: observational case control studies. PLoS One 2013; 8(6): e65326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The HAPO Study Cooperative Research Group. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med 2008; 358: 1991–2002. [DOI] [PubMed] [Google Scholar]
- 35.Crowther CA, Hiller JE, Moss JR, McPhee AJ, Jeffries WS, Robinson JS. Effect of treatment of gestational diabetes mellitus on pregnancy outcomes. N Engl J Med 2005; 352: 2477–86. [DOI] [PubMed] [Google Scholar]
- 36.Landon MB, Spong CY, Thom E, et al. A multicenter, randomized trial of treatment for mild gestational diabetes. N Engl J Med 2009; 361: 1339–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pedersen J. Diabetesand pregnancy: blood sugar of newborn infants. Copenhagen: Danish Science Press, 1952: 230. (Ph.D. thesis.) [Google Scholar]
- 38.Committee on Practice Bulletins — Obstetrics. ACOG practice bulletin no. 190: gestational diabetes mellitus. Obstet Gynecol 2018; 131(2): e49–e64. [DOI] [PubMed] [Google Scholar]
- 39.American Diabetes Association. 14. Management of diabetes in pregnancy: Standards of Medical Care in Diabetes — 2019. Diabetes Care 2019; 42: Suppl 1: S165–S172. [DOI] [PubMed] [Google Scholar]
- 40.Committee on Practice Bulletins — Obstetrics. ACOG practice bulletin no. 201: pregestational diabetes mellitus. Obstet Gynecol 2018; 132(6): e228–e248. [DOI] [PubMed] [Google Scholar]
- 41.Bochner CJ, Medearis AL, Williams J III, Castro L, Hobel CJ, Wade ME. Early third-trimester ultrasound screening in gestational diabetes to determine the risk of macrosomia and labor dystocia at term. Am J Obstet Gynecol 1987; 157: 703–8. [DOI] [PubMed] [Google Scholar]
- 42.Buchanan TA, Kjos SL, Montoro MN, et al. Use of fetal ultrasound to select metabolic therapy for pregnancies complicated by mild gestational diabetes. Diabetes Care 1994; 17: 275–83. [DOI] [PubMed] [Google Scholar]
- 43.Hattersley AT, Beards F, Ballantyne E, Appleton M, Harvey R, Ellard S. Mutations in the glucokinase gene of the fetus result in reduced birth weight. Nat Genet 1998; 19: 268–70. [DOI] [PubMed] [Google Scholar]
- 44.Spyer G, Macleod KM, Shepherd M, Ellard S, Hattersley AT. Pregnancy outcome in patients with raised blood glucose due to a heterozygous glucokinase gene mutation. Diabet Med 2009; 26: 14–8. [DOI] [PubMed] [Google Scholar]
- 45.Dickens LT, Letourneau LR, Sanyoura M, Greeley SAW, Philipson LH, Naylor RN. Management and pregnancy outcomes of women with GCK-MODY enrolled in the US Monogenic Diabetes Registry. Acta Diabetol 2019; 56: 405–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bacon S, Schmid J, McCarthy A, et al. The clinical management of hyperglycemia in pregnancy complicated by maturity-onset diabetes of the young. Am J Obstet Gynecol 2015; 213(2): 236.e1–236.e7. [DOI] [PubMed] [Google Scholar]
- 47.Hosokawa Y, Higuchi S, Kawakita R, et al. Pregnancy outcome of Japanese patients with glucokinase-maturity-onset diabetes of the young. J Diabetes Investig 2019; 10: 1586–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bitterman O, Tinto N, Franzese A, et al. Glucokinase deficit and birthweight: does maternal hyperglycemia always meet fetal needs? Acta Diabetol 2018; 55: 1247–50. [DOI] [PubMed] [Google Scholar]
- 49.Chakera AJ, Hurst PS, Spyer G, et al. Molecular reductions in glucokinase activity increase counter-regulatory responses to hypoglycemia in mice and humans with diabetes. Mol Metab 2018;17:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guenat E, Seematter G, Philippe J, Temler E, Jequier E, Tappy L. Counter-regulatory responses to hypoglycemia in patients with glucokinase gene mutations. Diabetes Metab 2000; 26: 377–84. [PubMed] [Google Scholar]
- 51.Fu J, Wang T, Liu J, Wang X, Li M, Xiao X. Birthweight correlates with later metabolic abnormalities in Chinese patients with maturity-onset diabetes of the young type 2. Endocrine 2019; 65: 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spyer G, Hattersley AT, Sykes JE, Sturley RH, MacLeod KM. Influence of maternal and fetal glucokinase mutations in gestational diabetes. Am J Obstet Gynecol 2001; 185: 240–1. [DOI] [PubMed] [Google Scholar]
- 53.Chakera AJ, Steele AM, Gloyn AL, et al. Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care 2015; 38: 1383–92. [DOI] [PubMed] [Google Scholar]