

Abstract
Flow cytometry is the most widely used method for detecting and quantifying apoptosis in mammalian cells. The multiparametric nature of flow cytometry allows several apoptotic characteristics to be combined in a single sample, making it a powerful tool for analyzing the complex progression of apoptotic death. This chapter provides guidelines for combining single-apoptosis assays such as fluorogenic caspase substrates, annexin V binding, DNA dye exclusion, and covalent viability probes into informative multiparametric assays. This multiparametric approach to analyzing apoptosis provides much more information than single-parameter assays that provide only a percentage apoptotic result, given that multiple early, intermediate, and late apoptotic stages can be observed and quantified simultaneously. While much more informative than single-color assays, these multicolor methods can still be analyzed on relatively simple flow cytometers, making them accessible to many laboratories.
Keywords: Apoptosis, Flow cytometry, Caspase, Fluorogenic caspase substrate, Annexin V, 7-Aminoactinomycin D, Propidium iodide, Pacific Blue, Hoechst dye, Covalent viability probe
1. Introduction
Apoptosis is a critical regulatory process in all tissues, and has been particularly well characterized in the immune system and in tumor cells. Assays based on flow cytometry have been available for over 25 years, and are now by far the most common method for detecting and quantifying apoptosis. They possess many advantages over earlier assays relying on microscopy or cell lysate analysis. They are rapid and quantitative, but most importantly, they analyze cell death in individual cells rather than in bulk populations [1]. The multiparametric nature of flow cytometry also allows the detection of multiple cell death characteristics in a single assay. For example, apoptosis assays that utilize DNA dyes as plasma membrane permeability indicators (such as propidium iodide) can be combined with assays that assess different cellular responses associated with cell death, including mitochondrial membrane potential and annexin V binding to “flipped” phosphatidylserine (PS) [2–5]. Combining multiple measurements for cell death into a single assay has a number of significant advantages. It provides multiple confirmations of apoptotic activity (important in a process that has proven highly variable in character). It also provides a much more comprehensive and multidimensional picture of the entire cell death process, rather than a simple analysis of live versus dead cells. Intrinsic in this second advantage is the ability to combine morphological assays (such as membrane integrity) with assays for biochemical events, such as caspase activation.
Recognition of the critical role of caspases in the death process as both signaling agents and effectors of cell death morphology has led to the development of assays that can measure these important enzymes in situ. Caspase activation represents one of the earliest detectable markers of apoptosis [6]. In most cases, caspase activation precedes degradation in cell permeability, DNA fragmentation, cytoskeletal collapse, and PS “flipping”; caspases are both signal transduction molecules and mediators of these downstream mani-festations of cell death. Combining fluorogenic assays of caspase activation with fluorescence-based assays for later characteristics of cell death (such as PS “flipping” and loss of membrane integrity) can provide a very information-rich view of cell death. It is particularly useful in distinguishing the “early” stages of cell death from later events, allowing better analysis of biochemical events during apoptosis cells prior to the complete collapse of the cell structure [7–11].
Several fluorogenic assays for caspase activity have been described, including the OncoImmunin PhiPhiLux system, the FLICA substrates, and the CellEvent Green or NucView 488 substrates [12–17]. All of these assays involve loading of a cell-permeable fluorogenic caspase substrate into cells following treatment with apoptotic stimuli; all “read out” as an increase in fluorescence in the presence of caspase activity. In general, caspase activity is one of the earliest detectable events in apoptosis, and precedes the later morphological damage usually relied on for cell death assessment.
In this chapter, we describe the combination of the PhiPhiLux, FLICA or CellEvent Green, or NucView 488 substrates caspase substrate system with two simultaneous assays for later stages of cell death, annexin V binding to “flipped” PS residues, and cell membrane integrity using DNA binding dyes or a covalent viability probe [17]. All of the above caspase substrates have characteristics that make them useful for integration with other “live” cell apoptosis assays; they are cell-permeable, and possess varying degrees of caspase specificity. PhiPhiLux and CellEvent Green/NucView 488 are relatively non-fluorescent in the intact state, and become fluorescent upon caspase cleavage. All three substrates are based on fluorescent probes with spectral characteristics similar to commonly used probes like fluorescein and rhodamine; this makes them easy to combine with other fluorescent probes [15–17]. The ability to observe and measure multiple apoptotic phenotypes in a single assay gives a powerful picture of the overall apoptotic process. These assays are applicable to both suspension cells by traditional flow cytometry, and adherent cells using laser scanning cytometry [17]. These assays can take particular advantage of newer flow cytometers with multiple lasers, but are also accessible to older or simpler cytometers with a single 488 nm laser source. Some of these assays require that the cell samples remain “live” and unfixed, requiring prompt analysis. However, others can be fixed with paraformaldehyde following the labeling procedure, allowing analysis at a later time.
2. Materials
The reagents required for multiparametric analysis of apoptosis are described below. To assemble an assay, choose one of the fluorogenic caspase assays, an annexin V conjugate, and either a DNA-binding dye or a covalent viability probe. Caspase assays can be used with DNA-binding dyes or covalent viability probes with no annexin if desired. For cells requiring fixation, FLICA and a covalent viability probe should be used.
2.1. Fluorogenic Caspase Assays
- PhiPhiLux fluorogenic caspase substrates (OncoImmunin, Inc., Gaithersburg, MD) are a series of fluorogenic enzyme substrates that fluoresce upon cleavage of an incorporated consensus domain. The fluorogenic caspase 3/7 substrate (PhiPhiLux G1D2) consists of an 18-amino acid peptide corresponding to the recognition/cleavage sequence from PARP containing the sequence ZVAD, a target for both caspases 3 and 7 [18]. The substrate is homodoubly labeled with one of several fluorophores (in this case, a fluorescein-like molecule) on the opposite sides of the molecule; in this conformation, the fluorochrome molecules are in close physical proximity, and the fluorescence of the resulting complex is largely quenched [16, 19]. After the substrate enters a cell by passive diffusion and is cleaved by caspase 3 or 7, the unquenched fluorescent fragments become somewhat less cell permeable and will remain in the cell for several hours. However, they will eventually diffuse out of the cell [16, 19], requiring flow cytometric analysis soon after labeling.
- The PhiPhiLux nomenclature indicates both its substrate specificity and the conjugated fluorochrome. The first letter refers to the substrate specificity: G refers to caspase 3/7, E to caspase 1, L to caspase 8, J to caspase 6, etc. The first number refers to the conjugated fluorochrome: 1 is a fluorescein-like fluorochrome, 2 is a rhodamine-like molecule, and 6 to the sulforhodamine-like molecule. So G1D2 is specific for caspase 3 with the fluorescein-like probe, and E2D2 is specific for caspase 1 with the rhodamine-like probe. R2D2 is a special case and refers to the Cy5-like molecule, with the caspase indicated beforehand (3-R2D2 for caspase 3, 8-R2D2 for caspase 8). Excitation and emission spectra for all the fluorochrome conjugates (generically referred to as X1D2, X2D2, etc.) are shown in Fig. 1. Special instructions for working with non-fluorescein-based PhiPhiLux reagents are discussed in Note 1.
- PhiPhiLux G1D2 spectrally resembles fluorescein and can be excited with the standard 488 nm argon-ion or solid-state laser found on most flow cytometers. PhiPhiLux G1D2 is spectrally compatible with many conjugates of annexin V, DNA-binding dyes, and covalent viability probes, as described in Subheading 2.5. All methods and data in this chapter employing the PhiPhiLux reagents used the fluorescein-like PhiPhiLux G1D2 specific for caspase 3/7 unless otherwise indicated.
- The PhiPhiLux reagents are roughly 40-fold dimmer in the uncleaved state than following caspase activation.When camptothecin-treated EL-4 lymphoma cells were labeled with PhiPhiLux G1D2 and analyzed by flow cytometry, the apoptotic cells possess 1–3-orders of magnitude higher fluorescence than the viable cells. Note that even truly viable cells with no apoptotic activity labeled with a caspase substrate will have somewhat higher background fluorescence levels than completely unlabeled cells. Primary cell cultures may show somewhat lower levels of caspase activation than cell lines, with subsequent lower levels of substrate fluorescence; however, background fluorescence may be lower with these cells as well (see Note 2).
- The PhiPhiLux reagents are also available with other fluorescent tags, including rhodamine- and sulforhodamine-like fluorochromes, and a proprietary Cy5-like fluorochrome that can be excited with a red laser. Although traditionally used for microscopy, the rhodamine and sulforhodamine substrates can be readily excited using green or yellow lasers, now common on flow cytometers (see Note 1). Unless otherwise indicated, all methods and data in this chapter employing PhiPhiLux labeling use the fluorescein-like PhiPhiLux G1D2 specific for caspase 3/7.
- The PhiPhiLux reagents are commercially provided at concentrations of 5–10 μM in sealed aliquots and can be stored at 4 °C prior to opening; once the ampule is opened, any remaining substrate should be stored at −20 °C. Avoid repeated freezing and thawing. Shelf life at 4 °C is approximately 3–6 months, over 1 year at −20 °C.
- The PhiPhiLux reagents do not covalently bind to caspase molecules, and are therefore not considered caspase inhibitors; theoretically, one caspase enzyme molecule can cleave more than one PhiPhiLux molecule. PhiPhiLux therefore is not immobilized inside the cell. The separated fragments will gradually diffuse out of the cell. For this reason, permeabilization using detergents and fixation using paraformaldehyde is not recommended with PhiPhiLux. This is in contrast to FLICA (described in item 2), which covalently crosslinks to caspase sites and is therefore immobilized in the cell. Detergent treatment and fixation are compatible with FLICA.
- The FLICA reagents (Fluorochrome Linked Inhibitors of Caspase Activity) are the second major group of fluorogenic caspase substrates. The FLICA substrates are composed of a caspase consensus cleave domain, flanked by a fluoromethylketone (FMK) protein reactive group, and a fluorochrome, most commonly fluorescein. They are cell permeable, and are incubated with cells for 30 min to 1 h at 37 °C to load. If a FLICA molecule comes in contact with a caspase via the recognition domain, the FMK group covalently binds to the caspase, and the FLICA molecule is immobilized inside the cell; the caspase enzyme similarly becomes inactivated. Unlike PhiPhiLux, FLICA reagents are always fluorescent. They need to be thoroughly washed out of the cells after labeling to ensure that caspase-negative cells are sufficiently dim to be distinguished from caspase-positives. Caspase-negative cells may be brighter than unlabeled cells due to residual unbound FLICA reagent. Unbound FLICA is removed by multiple centrifugal washings, and the cells are then labeled with additional apoptosis reagents and analyzed. As with PhiPhiLux, caspase-positive cells will be brightly labeled compared to negative cells. FLICA covalently binds to the site of activity via its FMK group. It is therefore immobilized in cells, and is compatible with both subsequent detergent treatment and paraformaldehyde fixation. This is in contrast to PhiPhiLux and CellEvent green/NucView 488 (described in item 3) which are incompatible with fixation.
- The term FLICA is the commercial name for substrates originally developed by Immunochemistry Technologies LLC (Bloomington, MN) and has come to be used generically. Most FLICA reagents are labeled with fluorescein. FLICA substrates sold by Immunochemistry Technologies LLC are designated as FAM-FLICA, referring to the fluorescein conjugation, followed by the caspase name.
- Fluorescein-conjugated FLICA reagents sold by Thermo Fisher Life Technologies (Carlsbad, CA) are termed Vybrant FAM, followed by the caspase designation (i.e., caspase 3/7).
- Sulforhodamine-conjugated FLICA reagents are also available from Immunochemistry Technologies LLC and are designated as SR-FLICA, or by the commercial name Magic Red. Like the PhiPhiLux reagents, these are more commonly used for microscopy than flow cytometry, relying on the green or yellow mercury arc lamp lines for excitation. However, the widespread usage of green and yellow lasers on flow cytometers makes this variant useful for flow cytometry as well. A red fluorochrome variant of FLICA, designated FLICA 660, is also available from Immunochemistry Technologies LLC. This reagent is excited with a red laser, usually a HeNe 633 nm or red laser diode ~640 nm (see Note 1).
- FLICA reagents are available with specificities to multiple caspases. Immunochemistry Technologies LLC manufactures FLICA reagents specific for caspases 1, 2, 3, 6, 7, 8, 9, 10, and 13, as well as a pan or poly caspase version. Thermo Fisher Life Technologies provides fluorescein-conjugated FLICA against caspases 3 and 7 (ZVAD) and a poly caspase version (DEVD). In this chapter, all methods and data show the fluorescein-based caspase 3/7 FLICA reagent unless otherwise noted. The FLICA reagent from both the suppliers is provided as a lyophilized preparation packaged under desiccation. Immediately prior to the assay, solubilize the reagent in dry DMSO, typically at 150× concentration. Use this solution immediately or store it at −20 °C in small aliquots over desiccant for up to 6 months, but avoid freeze/thaw cycles. Dilute the 150× solution in aqueous buffer at a 1:5 ratio to a final 30× concentration. Use this solution within 4 h of preparation and discard any unused reagent. FLICA reagents will rapidly hydrolyze in the aqueous solution, and should not be stored. Add the aqueous FLICA reagent to samples at a final 1× concentration.
- Aminomethylcoumarin (AMC) and other coumarin-based FLICA reagents are available from several suppliers as probes for microplate-based caspase assays. These reagents are generally not recommended for intact cell use; they are not very bright, not very cell permeable, and emit in a region of the spectrum with high autofluorescence.
CellEvent Green (Thermo Fisher Life Technologies) and NucView 488 (Biotium, Hayward, CA) constitute a third group of fluorogenic caspase substrates. These reagents consist of a DNA-binding molecule that is relatively non-fluorescent in the unbound state, but becomes fluorescent upon binding to DNA. The DNA-binding site is inhibited by a structure containing caspase recognition domain peptide. Like PhiPhiLux and FLICA, these reagents can be loaded into cells by passive diffusion. Upon cleave by a caspase molecule, the DNA-binding site is unmasked. The reagent can then diffuse into the nucleus, where it binds to DNA. Caspase-positive cells therefore appear as brightly labeled compared to caspase-negative cells. Currently, these reagents are available only with a fluorescein-like fluorochrome, with excitation at 488 nm and detection in the fluorescein range. They both function by the same mechanism. They are generally available as pre-prepared solutions that can be diluted directly into samples, usually at a final concentration of 1:1000. Some titration may be necessary for some cell types and concentrations. Both are currently available only for the detection of caspases 3 and 7. Thermo Fisher Life Technologies also manufactures a variant termed ReadyProbes that is recommended for slide-based samples and imaging. Like PhiPhiLux, CellEvent Green and NucView 488 do not bind covalently to any cellular components, and should not be fixed with paraformaldehyde.
Fig. 1.
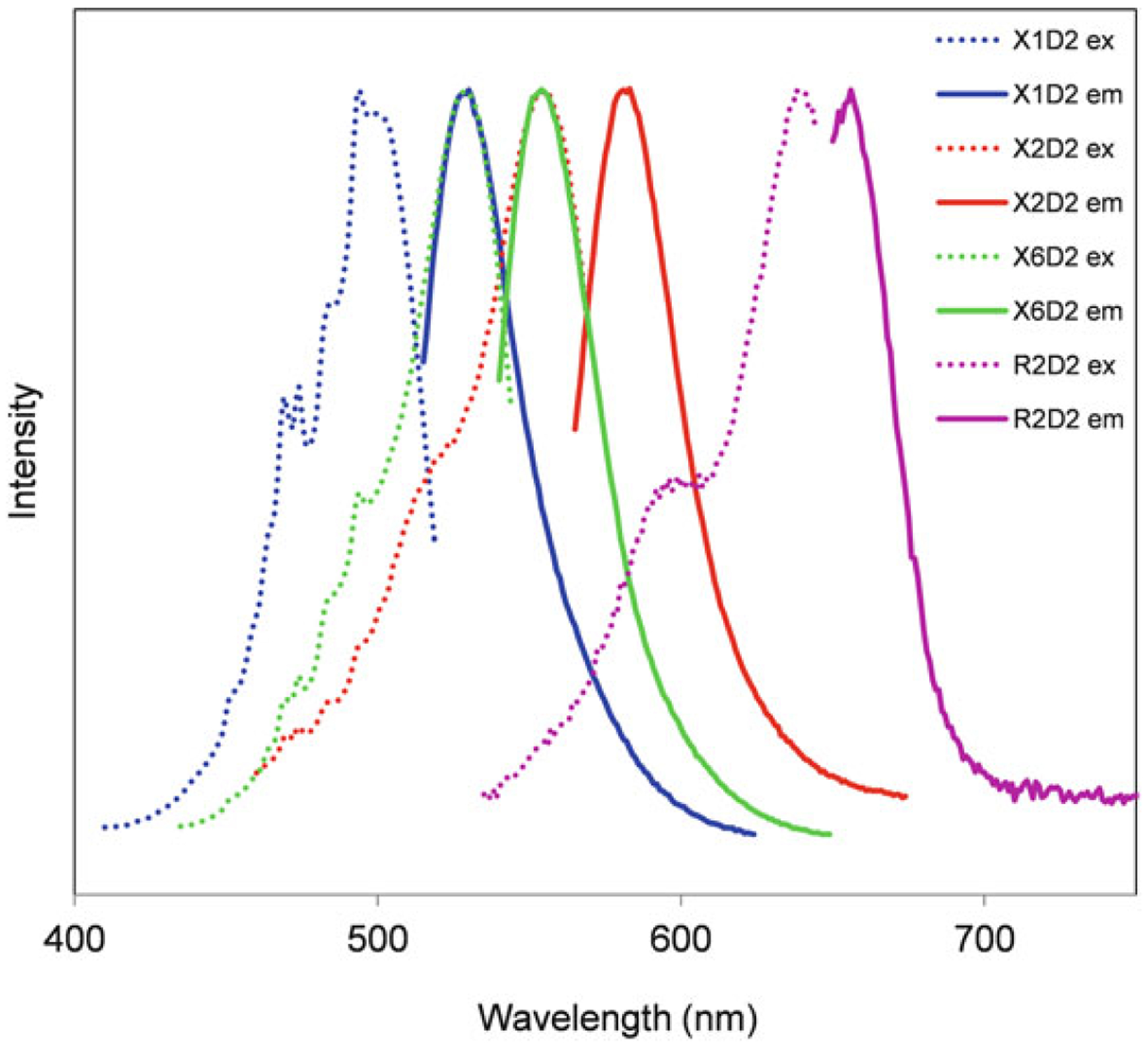
Spectral characteristics of fluorogenic caspase substrates. Excitation (dotted lines) and emission (solid lines) spectra for four caspase substrate conjugates: X1D2 (fluorescein-like, used in most of the data presented in this chapter), X2D2 (rhodamine-like), X6D2 (sulforhodamine-like), and R2D2 (Cy5-like)
2.2. Annexin V
Annexin V binds to apoptotic cells with “flipped” PS residues on their extracellular membrane leaflet, and is a useful and common assay for apoptosis. Annexin V is available conjugated to many fluorochromes, including fluorescein, Alexa Fluor 488, phycoery-thrin (PE), PE-Cy5, allophycocyanin (APC), Cy5, and Alexa Fluor 647, to name a few. It is therefore easy to integrate annexin V labeling into a multicolor protocol. Damaged or necrotic cells with a high degree of membrane permeability can also bind annexin V to their intracellular membrane leaflet, despite their uncertain apoptotic nature (see Note 3); therefore, a DNA-binding dye as a cell permeability indicator should always be incorporated into annexin V-binding assays. In some cell types, annexin V-binding precedes permeability to a DNA dye, allowing the identification of “earlier” apoptotic cells that are annexin V positive and DNA dye negative. Cells that are both annexin V and DNA-binding dye positive may therefore be either advanced apoptotic or necrotic. Annexin V labeling requires buffers with calcium and magnesium cations.
2.3. DNA-Binding Dyes
There are literally dozens of DNA-binding dyes available with potential utility for detecting apoptotic cells. DNA-binding dyes bind stoichiometrically to DNA by a variety of mechanisms, including intercalation and major/minor groove interactions. At high concentrations, many DNA-binding dyes can be used to analyze cell cycle in permeabilized cells; at lower concentrations, they can be added to “live” unfixed cells to assess viability, with the nonviable cells binding the dye and the viable cells excluding it. DNA-binding dyes are one of the oldest viability assays; dyes like propidium iodide are often added during fluorescent immunophenotyping to exclude dead cells from the analysis. Combined with fluorogenic caspase substrates and annexin V, they make excellent labels for identifying late stage apoptotic and necrotic cells. Many DNA-binding dyes are useful for flow cytometry and apoptosis detection. Some are listed here. Keep in mind that all of the DNA-binding dyes described here have somewhat differing cell permeability characteristics. This will affect the ultimate data analysis (see Note 4).
Propidium iodide (PI): An inexpensive and widely available intercalating DNA-binding dye. PI excites at 488 nm and emits in the 570–630 nm range. Dissolve in deionized water at 1 mg/mL and store in the dark at 4 °C for up to 3 months. Use at a final concentration of 1–2 μg/mL.
7-Aminoactinomycin D (7-AAD): An intercalating/groove-binding DNA-binding dye that also excites at 488 nm but emits in the far red, with an emission peak at approximately 670 nm. 7-AAD is a good alternative to PI where a longer wavelength probe is desired. 7-AAD is also somewhat more cell permeable than PI. Dissolve in 95% EtOH at 1 mg/mL and store at −20 °C. 7-AAD is also water-soluble but is not as stable in aqueous solution; such stocks should not be frozen and thawed repeatedly. EtOH solubilized stock solutions are good for 6 months. Use at a final concentration of 5 μg/mL. Diluted stocks should be used within 24 h. A variant of 7-AAD, SYTOX AADvanced™, is available from Thermo Fisher Life Technologies. This version is prepared with its peptide side chains removed, increasing solubility.
Violet-excited DNA-binding dyes: The proliferation of cytometers with multiple lasers has greatly expanded the fluorochromes available for apoptotic analysis. Violet (~405 nm) and red (~640 nm) lasers are now commonly available on flow cytometers. Several red- or violet-excited DNA-binding dyes can substitute for PI or 7-AAD to increase total fluorochrome capability or reduce fluorescence compensation requirements. Hoechst 33258 (several suppliers) is a widely available minor groove DNA-binding dye that is well excited by ultraviolet or violet lasers; it has cell permeability characteristics similar to PI. Prepare Hoechst 33258 as a 1 mg/mL stock in distilled water and store at 4 °C for up to 3 months. Use at a final concentration of 1–5 μg/mL. The DNA dye DAPI (several suppliers) has similar spectral and cell permeability characteristics, and can also be used as a viability dye. Preparation is similar to Hoechst 33258. SYTOX Blue (Thermo Fisher Life Technologies) is another violet-excited DNA dye that can also be used to distinguish dead cells; it is provided as a stock solution at 10 μM and should be diluted at 1:1000 for use. Hoechst 33342 (not to be confused with Hoechst 33258) and DyeCycle Violet (Thermo Fisher Life Technologies) are much more cell permeable than Hoechst 33258 or DAPI; they are not usually used as membrane integrity probes, but can be used to visualize apoptotic chromatin by microscopy.
Red-excited DNA-binding dyes: TO-PRO-3 (Thermo Fisher Life Technologies) is a red-excited DNA dye and is relatively cell-impermeant. It can be used as a membrane integrity dye with red laser equipped cytometers. It is provided as a stock solution and should be diluted at 1:1000 for use. DRAQ7 (Biostatus Ltd. Leicester, UK) is also excited by the red laser and can be used similarly. It is provided as stock solutions at 0.3 and 1 mM and can be used at 0.3–1 μM final concentration. SYTOX Red (Thermo Fisher Life Technologies) is another useful red-excited DNA dye, usually used at 1 μM final concentration.
Cell-permeable DNA dyes: DNA dyes with high cell permeability, including Hoechst 33342, the DyeCycle DNA dyes (Thermo Fisher Life Technologies), and DRAQ5 (Biostatus Ltd), are not recommended for measurement of cell permeability by flow cytometry. Both live and dead cells will take up large amounts of these dyes, making it difficult to distinguish viable from apoptotic cells. However, they can be useful for imaging applications, where the change in chromatin structure can be visualized.
2.4. Covalent Viability Probes
These viability probes are a good alternative to DNA-binding dyes for the assessment of membrane integrity, particularly where fixation is required. They are composed of a fluorochrome coupled to protein reactive group, usually a succinimidyl ester. When added to suspension cells, they bind at higher levels to apoptotic cells and lower levels to viable cells, producing a distribution similar to that seen with DNA-binding dyes. Unlike DNA dyes, however, the binding is covalent; the cells can be subsequently fixed with paraformaldehyde and the labeling preserved. The fluorochromes used in covalent viability probes are also the usual low molecular weight fluorochromes used for cell labeling. Their spectral properties are more similar to dyes like fluorescein and less likely to be spectrally incompatible in multicolor labeling. On the other hand, dyes like PI have extremely broad emission spectra and can occupy a lot of space in a multicolor protocol. The fluorochromes used for covalent viability dyes often show less spectral overlap. Covalent viability probes are available from several manufacturers.
Thermo Fisher Life Technologies makes the Live/Dead dyes in a variety of colors. The dye name (Live/Dead followed by a color) corresponds to the approximate emission wavelength of the dye. Of particular interest are the UV-, violet- and red-excited variants, which can be easily integrated into multicolor apoptosis assays. Live/Dead Fixable Blue is UV-excited and detected through a blue DAPI filter. Live/Dead Fixable Violet, Aqua and Yellow are violet-excited, and emit in the blue, green, and yellow range. Live/Dead Fixable Long Red and Near IR are red-excited, and emit in the long red and near infrared range.
BioLegend (San Diego, CA) manufacturers the Zombie viability probes. They are essentially the same in function as the Live/Dead probes. The color name associated with the dye may correspond to either the excitation or emission wavelength. Zombie UV is UV-excited and detected through a blue DAPI filter. Zombie Violet, Aqua, and Yellow are violet-excited, and emit in the blue, green, and yellow range.
BD Biosciences (San Jose, CA) manufactures the BD Horizon Fixable Viability Stains (FVS), also in a variety of colors. They are labeled according to their emission; so Fixable Viability Stain 450 is violet-excited and emits ~450 nm. Again, the violet- and red-excited variants are of particular interest for flow cytometry.
-
eBiosciences/Affymetrix (San Diego, CA) provides the Fixable Viability Dyes, again in many colors. They are designated by eFluor™ followed by a number roughly corresponding to their emission. So eFluor 450 is violet-excited and emits at ~450 nm.
Note that the dye name may reflect either excitation or emission characteristics. Check Table 1 or the most recent manufacturer’s literature.
Table 1.
Covalent viability probes: names, excitation/emission wavelengths, recommended lasers, and sources
| Covalent viability probe | Excitation max (λ) (nm) | Laser line | Emission max (λ) (nm) |
|---|---|---|---|
| Live/Dead Fixable Bluea | 350 | 355 nm, 375 nm, 405 nm | 450 |
| Live/Dead Fixable Violeta | 416 | 405 nm | 451 |
| Live/Dead Fixable Aquaa | 367 | 355 nm, 375 nm, 405 nm | 526 |
| Live/Dead Fixable Yellowa | 400 | 405 nm | 575 |
| Live/Dead Fixable Greena | 495 | 488 nm | 520 |
| Live/Dead Fixable Reda | 595 | 532 nm, 552 nm, 561 nm, 594 nm | 615 |
| Live/Dead Fixable Far Reda | 650 | 633 nm, 640 nm | 665 |
| Live/Dead Fixable Near IRa | 750 | 633 nm, 640 nm | 775 |
| Zombie UVb | 365 | 355 nm, 375 nm, 405 nm | 452 |
| Zombie Violetb | 376,400 | 355 nm, 375 nm, 405 nm | 420 |
| Zombie Aquab | 380 | 355 nm, 375 nm, 405 nm | 510 |
| Zombie Yellowb | 395 | 405 nm | 572 |
| Zombie Greenb | 487 | 488 nm | 515 |
| Zombie Redb | 600 | 532 nm, 552 nm, 561 nm, 594 nm | 624 |
| Zombie NIRb | 718 | 633 nm, 640 nm | 745 |
| BD Horizon Fixable Viability Stain 450c | 406 | 405 nm | 450 |
| BD Horizon Fixable Viability Stain 510c | 408 | 405 nm | 512 |
| BD Horizon Fixable Viability Stain 520c | 498 | 488 nm | 521 |
| BD Horizon Fixable Viability Stain 570c | 547 | 532 nm, 552 nm, 561 nm | 573 |
| BD Horizon Fixable Viability Stain 620c | 523 | 532 nm, 552 nm, 561 nm | 617 |
| BD Horizon Fixable Viability Stain 660c | 649 | 633 nm, 640 nm | 660 |
| BD Horizon Fixable Viability Stain 700c | 657 | 633 nm, 640 nm | 700 |
| BD Horizon Fixable Viability Stain 780c | 759 | 633 nm, 640 nm | 780 |
| Fixable Viability Dye eFluor 455UVd | 350 | 355 nm, 375 nm | 455 |
| Fixable Viability Dye eFluor 450d | 405 | 405 nm | 450 |
| Fixable Viability Dye eFluor 506d | 405 | 405 nm | 506 |
| Fixable Viability Dye eFluor 520d | 488 | 488 nm | 522 |
| Fixable Viability Dye 3Fluor 660d | 633 | 633 nm, 640 nm | 660 |
| Fixable Viability Dye eFluor 780d | 633 | 633 nm, 640 nm | 780 |
Thermo Fisher Life Technologies (all names are trademarked)
BioLegend (all names are trademarked)
BD Biosciences (all names are trademarked)
eBiosciences/Affymetric (all names are trademarked)
2.5. Combinations of Fluorochromes
The multiparametric assays described in this chapter combine fluorescent labels for three characteristics of cell apoptosis, namely caspase activation, PS “flipping,” and cell permeability. There is considerable flexibility of fluorochrome selection for the investigator depending on the flow cytometric instrumentation available. Three possible combinations are described below, one for analysis on instruments equipped with a single 488 nm laser, a second for instruments equipped with dual 488 nm/red diode or red HeNe lasers, and a third for instruments equipped with blue, red, and violet lasers. While this three-component assay is possible on single-laser instruments, spectral overlap between dyes and required compensation will be minimal on multi-laser cytometers. As with all multicolor flow cytometry experiments, single-color controls to allow calculation of compensation should be prepared and included in the experiment.
2.5.1. Single 488 nm Laser Instruments
These instruments are limited to a single laser, and tend to have only three or four fluorescent detectors. Examples of these include: older flow cytometers from BD Biosciences including the BD FACScan, BD FACSort, or BD FACSCalibur (with no red laser); older flow cytometers from Beckman Coulter (Indianapolis, IN) such as the Coulter Epics XL, or newer cytometers from Beckman Coulter such as the CytoFLEX with the 488 nm only option. At least three fluorescence detectors are needed. The following combination should be used when analysis is limited to this instrument type:
PhiPhiLux G1D2, FLICA, CellEvent Green, or NucView 488: Detect this fluorochrome in the fluorescein or FITC detector on most commercial instruments.
PE-conjugated annexin V: Detect this fluorochome in the PE detector on most instruments. Apply fluorescence compensation to separate the PE signal from the caspase substrate and 7-AAD.
7-AAD: Detect this far-red emitting DNA-binding dye in the far red (or PE-Cy5) detector on most commercial instruments. Unfortunately, there is not a good 488 nm excited long red covalent viability probe to substitute for 7-AAD. Covalent viability probes labeled as red are not suitable here as they are typically excited using a red laser.
2.5.2. Dual 488 nm/Red Laser-Equipped Instruments
Many benchtop flow cytometers are equipped with more than one laser, most commonly a red source (most likely a 635 nm red diode or a 633 nm red HeNe laser). The BD FACSort and BD FACSCalibur usually fall into this category, as do some older and minimally equipped BD LSRIIs (BD Biosciences) and BD FACSArias (BD Biosciences). The BD Accuri C6 (BD Biosciences), FC500 (Beckman Coulter), and Handyem HPC-150 cytometer (du Parc Technologique, Quebec, Canada) are dual laser instruments equipped with blue and red lasers. A red laser allows several red-excited fluorochromes to be incorporated into flow cytometry assays, including APC, Cy5, and Alexa Fluor 647. The DNA dyes TO-PRO-3 and SYTOX Red can also be incorporated if a red laser is available. The following combination is suggested for dual laser instrumentation:
PhiPhiLux G1D2, FLICA, CellEvent Green, or NucView 488: Detect this fluorochrome in the fluorescein detector on most commercial instruments.
APC-conjugated annexin V: Excite this fluorochome with either a red diode or HeNe laser, and detect in the far red range. Little fluorescence compensation is required to separate its signal from PhiPhiLux G1D2 or the DNA-binding dyes described below, making post-acquisition analysis easier. Annexin V conjugates with Cy5 and Alexa Fluor 647 (which are spectrally similar to APC) can be analyzed in the same way.
PI or 7-AAD DNA-binding dyes can be incorporated into a cell death assay with a fluorogenic caspase substrate and APC-annexin V. Detect both in the far red detector, usually with a mid-600 nm bandpass (BP) or longpass (LP) filter on most flow cytometers.
If a red-excited DNA dye (like TO-PRO-3 or SYTOX Red) or a far red covalent viability probe (like Live/Dead Near IR) is used with the red laser, move annexin V to another detector (such as the PE detector).
2.5.3. Triple 488 nm/Red Laser/Violet Laser-Equipped Instruments
Many modern cytometers are equipped with more than two lasers; violet laser diodes (~405 nm) are typically included as a third excitation source. Instruments include the BD LSR II, BD LSR Fortessa (BD Biosciences), Gallios (Beckman Coulter), Navios (Beckman Coulter), CytoFLEX (Beckman Coulter), Stratedigm S1400 (Stratedigm, Inc., Campbell, CA), Partec CyFlow (Partec GmbH, Münster, Germany), MACSQuant (Miltenyi Biotec, San Diego, CA), Guava easyCyte (EMD Millipore, Billerica, MA), and NovoCyte (ACEA Biosciences, San Diego, CA). Violet-excited annexin V conjugates and DNA-binding dyes can be easily incorporated into apoptotic assay combinations. Violet-excited probes do not significantly overlap into other fluorescence channels, making them very useful for multicolor assays. Examples are listed below:
- PhiPhiLux, FLICA, CellEvent Green, or NucView 488: Detect this caspase substrate in the fluorescein detector. Combine it with:
- TO-PRO-3 or SYTOX Red: Either a 488 nm or red-excited DNA-binding dye can be used. PI or 7-AAD could be used here as well, but red-excited DNA dyes will require less compensation.
- Pacific Blue-annexin V: Pacific Blue is a relatively bright violet-excited fluorochrome, and is available in an annexin V conjugate. Pacific Blue does not overlap significantly into other fluorescent channels, and other fluorochromes do not overlap significantly into it, making it very applicable for multiparametric assays.
- Another possible combination still uses the fluorescein detector for the caspase substrate, but uses:
- DAPI, Hoechst 33258, or SYTOX Blue: These DNA-binding dyes use the violet laser for excitation. SYTOX Blue is somewhat more cell permeable than Hoechst 33258 and DAPI, which are roughly equivalent to PI.
- APC-annexin V: A red laser can be used to excite APC-annexin V. This combination uses three lasers to excite three fluorochromes; as a result, virtually no spectral overlap occurs, and almost no fluorescence compensation is required.
Red- or violet-excited covalent viability probes can readily substitute for the DNA dyes mentioned above. Live/Dead Violet, Aqua, and Yellow are all violet-excited and emit in the blue, green, and yellow range. Live/Dead Long Red and Near IR are red-excited and emit in the red and far red region.
2.6. Buffers and Equipment
Wash buffer: Dulbecco’s PBS (containing calcium and magnesium) supplemented with 2% fetal bovine serum. This is used for cell washing after caspase substrate loading and prior to DNA dye addition, and contains FBS to help maintain cell health during the labeling procedure and reduce apoptosis during the assay itself. The inclusion of divalent cations is critical for annexin V binding. Buffers from kits can be substituted as long as calcium and magnesium are present.
Complete medium: RPMI supplemented with 10% FBS.
Dulbecco’s PBS (containing calcium and magnesium).
Fixative: 2% paraformaldehyde in PBS.
Flow cytometer equipped with one, two, or three lasers (see Subheading 2.5 for instrument setup with regard to different combinations of fluorochromes).
3. Methods
In this section, we design a labeling system based on the available flow cytometric instrumentation. A typical labeling protocol will consist of: (a) a fluorogenic caspase substrate (Subheading 3.2), (b) an annexin V conjugate (Subheading 3.3), and (c) either a DNA-binding dye (Subheading 3.4) or a covalent viability dye (Subheading 3.5). Labeling will be done in this order. Any spectrally compatible combination of the following probes can be used for cell samples that do not require fixation. For cell samples requiring fixation with paraformaldehyde, FLICA should be combined with a covalent viability dye, but no annexin V labeling (Subheading 3.6).
3.1. Preparation of Cells
EL-4 cells treated with transcriptional or translational inhibitors such as cycloheximide or actinomycin D, or topoisomerase inhibitors like camptothecin or topotecan were used in the all illustrated data. EL4 cells can be treated with cycloheximide at 50 μg/mL or actinomycin D at 5 μg/mL for 6–8 h to induce apoptosis. Camptothecin induces apoptosis in EL4 cells at 2–5 μM over a longer incubation period of 12–16 h. EL4 cells are easily grown and hardy, and can make a useful positive control for more general use (see Note 2).
Harvest cell lines grown in suspension or cultured primary cells. Count the cells prior to centrifugation using a hemocytometer or other counting devices. Transfer cells to 12 × 75 mm cell culture tubes, and centrifuge at 400 × for 5 min.
Decant supernatant. Maximum removal of the supernatant is critical; the volume of remaining supernatant should be as low as possible to cause minimal dilution of the caspase substrate. Although cells can be washed prior to labeling, performing the assay in the remaining complete medium supernatant will reduce the amount of incidental cell death occurring during the assay. For cells obtained from clinical or other in vivo sources, centrifuge and resuspend in either an enriched buffer like the wash buffer or complete medium prior to use, then centrifuge and decant as described above. Caspase substrate loading can be done in a complete medium.
Each sample should contain 0.5–1 × 106 cells; increasing this number will saturate the detection reagents and reduce caspase and annexin V labeling efficiency. Adherent cells pose special challenges for apoptotic analysis due to the physical trauma and membrane damage that occur with cell dissociation; analysis in the adherent state by laser scanning cytometry is much preferable to flow cytometry under these circumstances (see Note 3).
3.2. Fluorogenic Caspase Substrate Labeling
Instructions are given below for the three main types of fluorogenic caspase substrate. CellEvent Green and NucView 488 instructions are the same. Choose one of the fluorogenic caspase probes below. Since annexin V incubation in Subheading 3.3 will be done with the caspase substrates still present, the total caspase substrate incubation time will be the intervals given below plus 15 min in annexin V. For PhiPhiLux labeling, the fluorescein-like PhiPhiLux G1D2 specific for caspase 3/7 is used as an example. For FLICA labeling, the fluorescein caspase 3/7 variant is used as an example.
3.2.1. PhiPhiPhiLux G1D2 Caspase 3/7 Substrate
Following the wash step in Subheading 3.1, ensure that as much supernatant is removed from the sample tube, to maximize final substrate volume. Tap each tube to resuspend the cell pellet in the remaining supernatant. The supernatant in each tube will be approximately 50 μL in volume (but not to exceed 100 μL).
Add 50 μL of the PhiPhiLux reagent (from 10 μM stock) to each tube and shake gently. The PhiPhiLux reagent should be diluted as little as possible for maximum detection, hence the need for minimal sample supernatant. PhiPhiLux reagent solutions are typically prepared at 10 μM; this will give a final concentration between 3 and 5 μM (in approximately 100–150 μL of total volume).
Incubate the tubes for 30–45 min at 37 °C, in a water bath or an incubator. An incubator may be preferred if CO2 conditions are desired. For both optimal labeling and reasons of economy, the PhiPhiLux reagent can be titered and tested for use between 0.5 and 5 μM. However, this should be done with caution (see Note 5).
Proceed to Subheading 3.3 for annexin V labeling.
3.2.2. FLICA
Prior to beginning the assay, reconstitute and dilute the FLICA substrate according to the manufacturer’s instructions. Generally, this consists of reconstituting the FLICA substrate in DMSO to a 150× stock and diluting at 1:5 in wash buffer to a 30× stock. As a reminder, the DMS reconstituted stock can be frozen in aliquots and stored. The 30× stock should be used promptly, and any excess discarded.
Following the wash step in Subheading 3.1, ensure that as much supernatant is removed from the sample tube, to maximize final substrate volume. Tap each tube to resuspend the cell pellet in the remaining supernatant. The supernatant in each tube will be approximately 50 μL in volume (but not to exceed 100 μL).
Add 300 μL of wash buffer to each tube.
Add 10 μL of the FLICA substrate to each tube.
Incubate the tubes for 30–45 min at 37 °C, in a water bath or an incubator. An incubator may be preferred if CO2 conditions are desired.
Proceed to Subheading 3.3 for annexin V labeling.
3.2.3. CellEvent Green or NucView 488
Both CellEvent Green and NucView are provided as pre-prepared stocks. Warm them to room temperature prior to use.
Following the wash step in Subheading 3.1, ensure that as much supernatant is removed as possible from the sample tube in order to maximize final substrate volume. Tap each tube to resuspend the cell pellet in the remaining supernatant. The supernatant in the tubes will be approximately 50 μL in volume (but not to exceed 100 μL).
Add 1 mL of wash buffer to each tube.
Add 1 μL of either CellEvent Green or NucView 488 to each tube. This amount can be titered up for some cell types.
Incubate the tubes for 15–30 min at 37 °C, in a water bath or an incubator. An incubator may be preferred if CO2 conditions are desired.
Proceed to Subheading 3.3 for annexin V labeling.
3.3. Annexin V Labeling
Cells are then labeled with fluorochrome-conjugated annexin V. Since centrifuge washings are minimized in this method to reduce assay-associated cell death, the cells are not washed following caspase substrate loading but are labeled immediately with fluorochrome-conjugated annexin V. The annexin V incubation period will therefore be part of the caspase substrate incubation period as well. Since most cell culture media and serum supplements contain calcium and magnesium, it is assumed that cation concentrations are sufficient to allow annexin V binding to “flipped” PS residues. However, this should be verified; even brief removal of divalent cations will cause immediate dissociation of the annexin V reagent. Subsequent cell washing is therefore done in the recommended wash buffer containing calcium and magnesium supplemented with FBS (see Note 6). The following steps will work with all of the above caspase substrate labeling methods.
After caspase substrate incubation, remove sample tubes from the incubator or water bath and add the appropriate fluorochrome conjugate of annexin V. Annexin V is generally available in suspension at concentrations ranging from 0.1 to 1 mg/mL. Cell labeling should be carried out at approximately 0.5–5 μg annexin V per sample. Therefore, add 5 μL of a 0.1 mg/mL annexin V solution to PhiPhiLux and FLICA labeled samples (150–300 μL sample volume). For CellEvent Green or NucView 488 labeled samples, add 10 μL of a 0.1 mg/mL annexin V solution (1 mL sample volume). Some titration in advance of the actual experiment might be necessary.
Return sample tubes to the water bath or the incubator and incubate for an additional 15 min.
Add 3 mL of wash buffer to each tube, centrifuge at 400 × for 5 min and decant.
Following the decant step, proceed to Subheading 3.4 for DNA-binding dye labeling or Subheading 3.5 for covalent viability probe labeling. PhiPhiLux, CellEvent Green, and NucView 488 labelings can then proceed directly to the DNA-binding dye or covalent viability probe labeling step. For FLICA labeling, add an additional 3 mL per decanted tube, and centrifuge again at 400 × for 5 min. This additional wash step is critical for FLICA samples, which require removal of unreacted substrate.
3.4. DNA-Binding Dye Labeling
Depending on the instrumentation available, cells can be subsequently labeled with a DNA-binding dye for the assessment of cell permeability in the later stages of apoptosis [6]. Remember that 7-AAD can be used with single-laser instruments. PE-annexin V and PI can be used together on a single-laser instrument, but they are spectrally similar, and compensation of fluorescence may be an issue. 7-AAD is therefore preferable when using PhiPhiLux and PE-annexin V. PI is more readily used with dual-laser instruments (blue-green and red), since annexin V can be detected using the APC detector. TO-PRO-3 and SYTOX Red require a red laser, whereas Hoechst 33258, DAPI, and SYTOX Blue require a violet laser. APC-, Alexa Fluor 647-, and Cy5-conjugated annexin V require a red laser, whereas Pacific Blue-conjugated annexin V requires a violet laser (see Note 4).
Prepare a solution of DNA-binding dye in a complete medium. PI is typically used at 2 μg/mL, 7-AAD at 5 μg/mL, and Hoechst 33258 or DAPI at 2 μg/mL. SYTOX Red and SYTOX Blue are usually supplied at stock solutions of 5 mM, and should be added at 5 μM (1:1000).
Add 0.5 mL of the DNA-binding solution to each decanted sample tube. Maintain samples at room temperature and analyze within 60 min (see Note 7).
3.5. Covalent Viability Probe Labeling
Covalent viability probes make useful indicators for cell permeability and can substitute for the DNA-binding dyes described in Subheading 3.4.
As described in Subheading 2.4, covalent viability probes are typically provided as lyophilized stocks. Add the required amount of DMSO to each vial to reconstitute the reagent. Any reagent left at the end of the assay should be discarded or immediately aliquoted and stored at −20 °C for future use.
Following the wash and decant step after annexin V labeling (Subheading 3.3), add 3 mL of PBS (containing calcium/magnesium but no protein) to each sample tube. Centrifuge at 400 × for 5 min and decant. This wash buffer step must be protein-free to prevent inactivation of the covalent viability probe.
Resuspend the cells in 1 mL of PBS (containing calcium/magnesium but no protein).
Add the covalent viability probe. For most manufacturers, this is a 1:1000 dilution of the DMSO stock.
Incubate at room temperature for 30 min.
Add 3 mL of wash buffer (containing calcium/magnesium and FBS), centrifuge and decant.
Resuspend in wash buffer and analyze within 60 min.
3.6. Fixable Assays Using FLICA and Covalent Viability Probes
All of the above assays utilize annexin V, and are therefore not suitable for paraformaldehyde fixation. In addition, the PhiPhiLux, CellEvent Green, and NucView 488 substrates do not covalently crosslink inside cells, and also do not work well with fixation. If a fixable caspase assay is desirable, FLICA can be combined with a covalent viability probe but without annexin V. This pair can be fixed with paraformaldehyde following labeling.
Prior to beginning the assay, reconstitute and dilute the FLICA substrate according to the manufacturer’s instructions. Generally, this consists of reconstituting the FLICA substrate in DMSO to a 150× stock and diluting at 1:5 in wash buffer to a 30× stock (Subheading 2.1, item 2). As a reminder, the DMSO reconstituted stock can be frozen in aliquots and stored. The 30× stock should be used promptly, and any excess discarded.
Following the wash step in Subheading 3.1, ensure that as much supernatant is removed from the sample tube, to maximize final substrate volume. Tap each tube to resuspend the cell pellet in the remaining supernatant. The supernatant in the tubes will be approximately 50 μL in volume (but not to exceed 100 μL).
Add 300 μL of wash buffer to each sample.
Add 10 μL of the FLICA substrate.
Incubate the tubes for 30–45 min at 37 °C, in a water bath or an incubator. An incubator may be preferred if CO2 conditions are desired.
Add 3 mL of wash buffer to each tube, centrifuge at 400 × for 5 min and decant.
Add 3 mL of PBS, centrifuge at 400 × for 5 min and decant.
Resuspend cells in 1 mL of PBS.
Add the covalent viability probe. For most manufacturers, this is a 1:1000 dilution of the DMSO stock.
Incubate at room temperature for 30 min.
Add 3 mL of wash buffer, centrifuge and decant.
Add fixative. Cells can be stored at 4 °C following fixation until analysis.
3.7. Flow Cytometric Analysis
Unfixed cells should be analyzed as quickly as possible to maximize fluorescent reagent signal and minimize post-assay apoptotic death. The instrument should be set up and ready for sample acquisition immediately upon completion of the assay. PhiPhiLux in particular should be analyzed promptly, as the cleaved substrate will slowly diffuse out of cells. Samples should be kept at room temperature until analysis; storage at 4 °C may reduce dye dissociation, but can itself induce unwanted apoptosis. Single-color controls for the calculation of compensation are highly recommended. Fluorescent detector assignments and analysis issues are described here.
PhiPhiLux, FLICA, CellEvent Green, and NucView 488: These fluorescein-like caspase substrates are detected through the fluorescein detector on most flow cytometers (often with the designation “FITC” or “FL1”) using a 530/30 nm or similar narrow BP filter. The spectral properties of all described caspase substrates are similar to fluorescein, requiring some spectral compensation when used simultaneously with PE or PI (and to a lesser extent with 7-AAD).
PE-conjugated annexin: Like most PE-conjugated reagents, this reagent is detected through the PE detector on most flow cytometers (often with the designation “PE” or “FL2”) using a 575/26 nm or similar BP filter. PE requires some spectral compensation when used with PhiPhiLux G1D2 and 7-AAD.
APC-conjugated annexin: APC is excited with a red laser source and detected through the APC detector on many flow cytometers (sometimes with an “FL4” designation) using a 660/20 nm or similar BP filter. An advantage of APC in multicolor assays is its minimal need for color compensation; there is no significant spectral overlap between caspase substrates, PI or 7-AAD. Cy5 or Alexa Fluor 647 conjugates are spectrally similar to APC, and can be analyzed in the same way.
Pacific Blue-conjugated annexin V: Pacific Blue is analyzed using a violet laser; most instruments so equipped have at least two detectors aligned to this laser source. A 450/50 nm or similar filter is typically used to detect this fluorescent probe. Cascade Blue, Alexa Fluor 405 and Brilliant Violet 421 are spectrally similar to Pacific Blue, and are analyzed in the same way.
PI: This DNA-binding dye is very bright even at low concentrations, and has a broad emission range, requiring compensation when used with any of the fluorogenic caspase substrates. It can be detected in either the PE (575/26 nm filter) or far red detection channel (typically a red 660–690 nm BP or LP filter). The second choice is preferable to reduce spillover into the fluorescein detector. PE and PI can be analyzed together on older single-laser instruments using the traditional PE detector (“FL2” detector, 575/26 nm) for PE detection, and the longer PE-Cy5 detector (“FL3” detector, 675/20 nm BP or 650 nm LP filter) for PI. However, the close proximity of their spectra makes this analysis difficult. Substitution of PI with 7-AAD is preferable. PI is highly charged, and will contaminate instrument tubing, causing unwanted “shedding” of the dye into later samples. After PI use, the instrument should be thoroughly cleaned with 10% bleach or other detergents to remove the dye.
7-AAD: This DNA-binding dye is not as bright as PI and emits in the far red, allowing its detection in the far red channel on most single-laser flow cytometers (PE-Cy5 or “FL3” detector, 675/20 nm BP or 650 nm LP filter). Some compensation will be required when used with the fluorogenic caspase substrates and PE. SYTOX AADvanced is a 7-AAD variant and can be substituted for 7-AAD (see Subheading 2.3, item 2).
Hoechst 33258, DAPI, and SYTOX Blue: Hoechst 33258 is very bright, and can be excited using either an UVor violet laser source. It is detected through a 450/50 nm or similar filter. It will have minimal spectral overlap into other fluorochromes. Do not confuse Hoechst 33258 with 33342; Hoechst 33342 is far more cell permeable, and is less useful as a discriminator of membrane permeability loss. DAPI is also usable as a permeability dye, and has spectral properties similar to Hoechst 33258. SYTOX Blue is also violet-excited and emits in the blue range. Like PI, they are highly charged and will adhere tightly to instrument tubing; the instrument should be cleaned thoroughly with 10% bleach or other detergents after use.
TO-PRO-3, SYTOX Red, and DRAQ5: These DNA-binding dyes are both excited with a red laser source, usually a red laser diode (~640 nm) or less likely a red HeNe laser (633 nm). They emit in the long red range from 650 to 670 nm, and can be measured using filters and detectors set up for APC.
Covalent viability probes: These probes are excited and detected at a variety of wavelengths. See Table 1 for excitation conditions. The color name of the probe may correspond to either the excitation or emission wavelength depending on the manufacturer. For example, Live/Dead Near IR (Thermo Fisher Life Technologies) emits in the near infrared, with excitation in the red; however, Zombie UV (BioLegend) excites with a UV laser, but emits in the blue. It should therefore not be assumed that the dye name refers to one or the other. Consult Table 1 and the manufacturer’s latest information. Covalent viability probes are available that excite with virtually all laser sources available on flow cytometers including ultraviolet, violet, blue-green, green, yellow, and red. They also emit over the entire visible spectrum, and can be selected to integrate with many fluorochrome combinations. As a rule, the violet and red excited variants are the most useful when designing multiparametric assays for flow cytometry. For example, Live/Dead Aqua and Yellow, or Zombie Aqua and Yellow are violet excited probes that emit in the blue and yellow range, and combine well with fluorescein-based caspase substrates and APC or Alexa Fluor 647 annexin V. Similarly, Live/Dead Near IR and Zombie NIR are red-excited probes that emit in the near infrared range, and combine well with FITC and Pacific Blue annexin V. Many compatible combinations are possible.
3.8. Data Acquisition and Analysis
Good gating is critical for meaningful analysis of apoptosis, and a logical gating strategy should be used to identify the various components of the apoptotic process. Typical gating schemes are illustrated.
3.8.1. Scatter Gating
Many cell lines and some primary cells show a dramatic alteration in forward and side scatter measurements late in the onset of apoptosis. Forward and side scatters are approximate indicators of cell size and optical density, respectively, and reflect both the cell volume loss and intracellular breakdown occurring during apoptotic death. This is clear in Fig. 2, where EL4 cells treated with camptothecin show a “scatter viable” cell population with higher forward scatter, and an apoptotic population with reduced forward scatter and increased side scatter. It therefore seems logical to draw a gate around BOTH the scatter viable population AND the apoptotic population, and look at caspase activation, annexin V binding and DNA dye uptake in this total population.
Fig. 2.
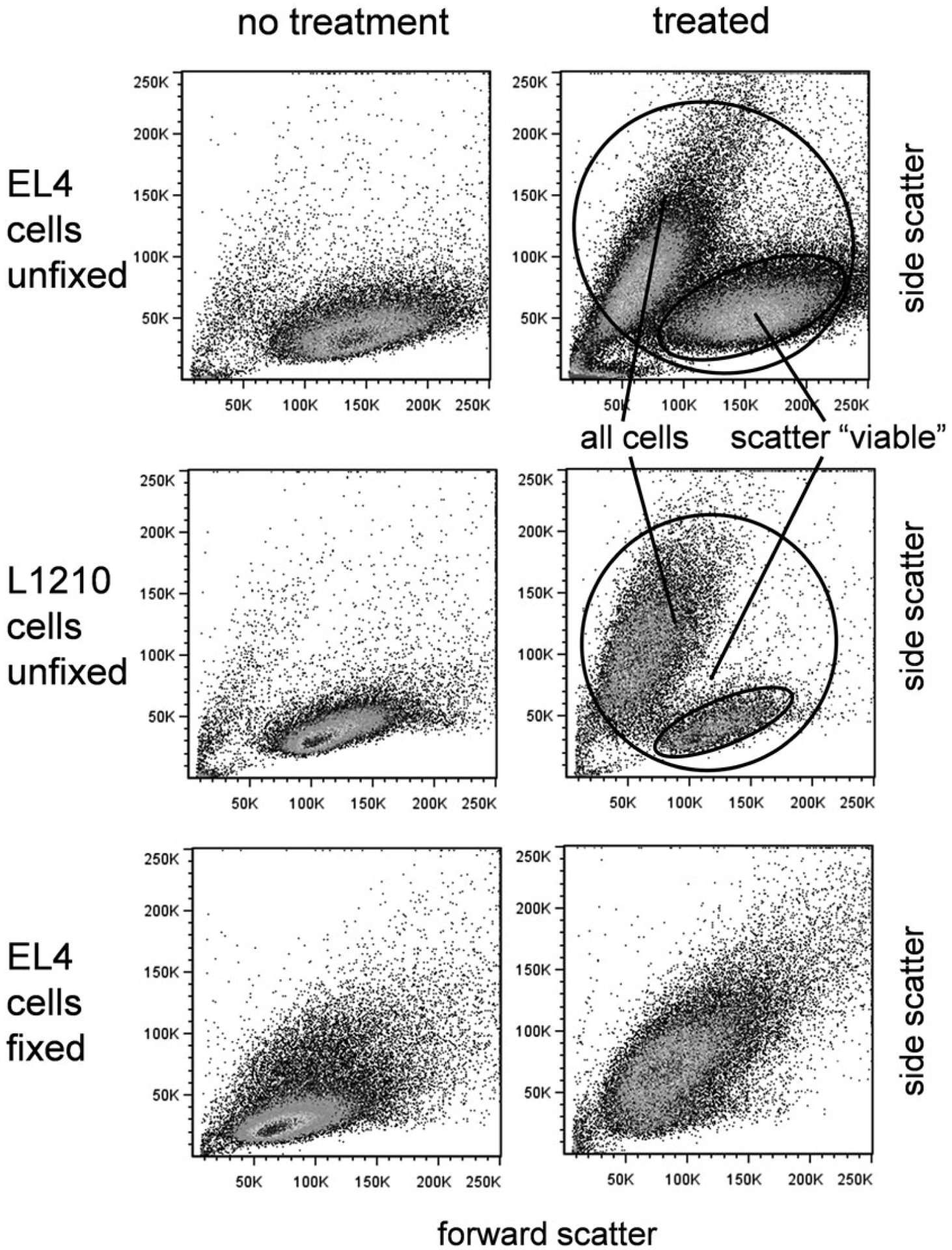
Scatter properties of viable and apoptotic cells. Forward versus side scatter dotplots of unfixed EL4 cells (top row), unfixed L1210 cells (middle row), and fixed EL4 cells (bottom row). (Left column) Untreated cells received no apoptotic agent. (Right column) Treated EL4 cells received camptothecin at 5 μM for 16 h, treated L1210 cells received anti-Fas antibodies (clone Jo2) at 1:1000 final concentration for 12 h
However, the apoptotic population based on scatter is usually at very advanced stage of apoptotic death; the cells are already positive for all markers. The advanced physical perturbation of the cells in this group can also produce positive but highly variable labeling results, interfering with the identification of earlier apoptotic stages. Both viable and apoptotic scatter populations should indeed be gated as the first step in an analysis strategy. However, it is also useful to gate only on the scatter viable cells, and examine early apoptotic markers such as caspase activation only within this group of cells. In many cases, these so-called scatter viable cells are not completely viable; the earliest onset of caspase activation and even annexin V binding can often be observed. This dual approach allows an overall picture of both early and late apoptotic stages, as well as examination of the earliest apoptotic cells. It is therefore recommended that both gating approaches be applied to get a clear picture of the apoptotic process. Examples of gating for both all cells and the scatter viable population are shown in Fig. 2, and in all of the assays.
3.8.2. Annexin V-Binding and DNA-Binding Dye Exclusion
Once the cells are gated for scatter, they should be plotted for annexin V versus DNA dye or covalent viability probe labeling. These events usually occur after caspase activation and are considered “later” markers of apoptosis. Therefore, subpopulations negative or positive for annexin V and DNA dye-binding can be gated for discrimination of “early” and “late” apoptotic cells, and can be subsequently examined for caspase activation. An example of annexin V versus DNA dye labeling for camptothecin-treated EL4 cells is shown in Fig. 3. Scatter viable cells that are both annexin V and DNA dye negative are present, but may show the initiation of caspase activation; this population should be gated. Some cell types may also show either annexin V or DNA dye permeability arising first; these single positive populations can also be gated. Later apoptotic double-positives should be gated as well. These multiple populations can then be displayed for caspase activation.
Fig. 3.
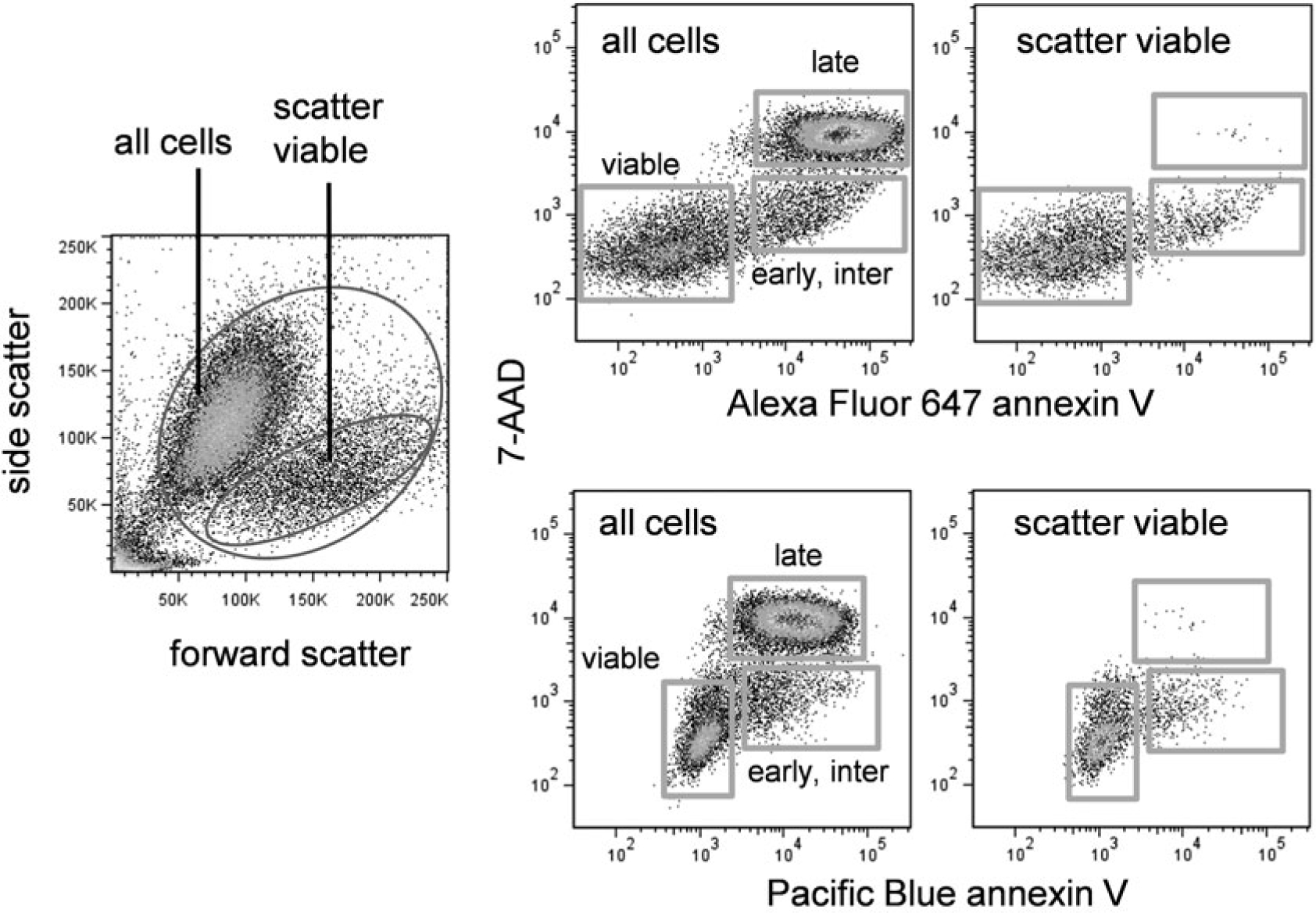
Annexin V and DNA dye labeling in apoptotic cells. EL4 cells were treated with camptothecin at 5 μM for 16 h. Left dotplot shows forward versus side scatter of treated cells. Top right row shows Alexa Fluor 647 annexin V versus 7-AAD labeling for all cells (left plot) or scatter viable cells only (right plot). Bottom right row shows Pacific Blue annexin V versus 7-AAD labeling for all cells (left plot) or scatter viable cells only (right plot)
DNA dyes are not completely interchangeable with regard to exclusion by apoptotic cells (see Note 4). For example, 7-AAD is somewhat more cell permeable than PI and will label an earlier subset of apoptotic cells. This will affect the overall analysis. For example, if 7-AAD-positive cells are excluded from the analysis (in an attempt to quantify very early apoptotic events), this dye’s greater cell permeability will result in a lower apparent number of caspase-positive cells that are DNA dye-negative than if PI were used instead. These differences should be kept in mind when analyzing these early apoptotic subsets.
3.8.3. Annexin V-Binding and Covalent Viability Probes
While functioning by a very different mechanism than DNA-binding dyes, the overall appearance of covalent viability probes will be similar to DNA-binding dyes. Analysis should be similar, identifying and gating on the negatives, single positives (if any) and double positives. The fluorescence distribution of several covalent viability probes is shown in Fig. 4. These probes are usually bright, and viable and apoptotic cells can be easily distinguished.
Fig. 4.
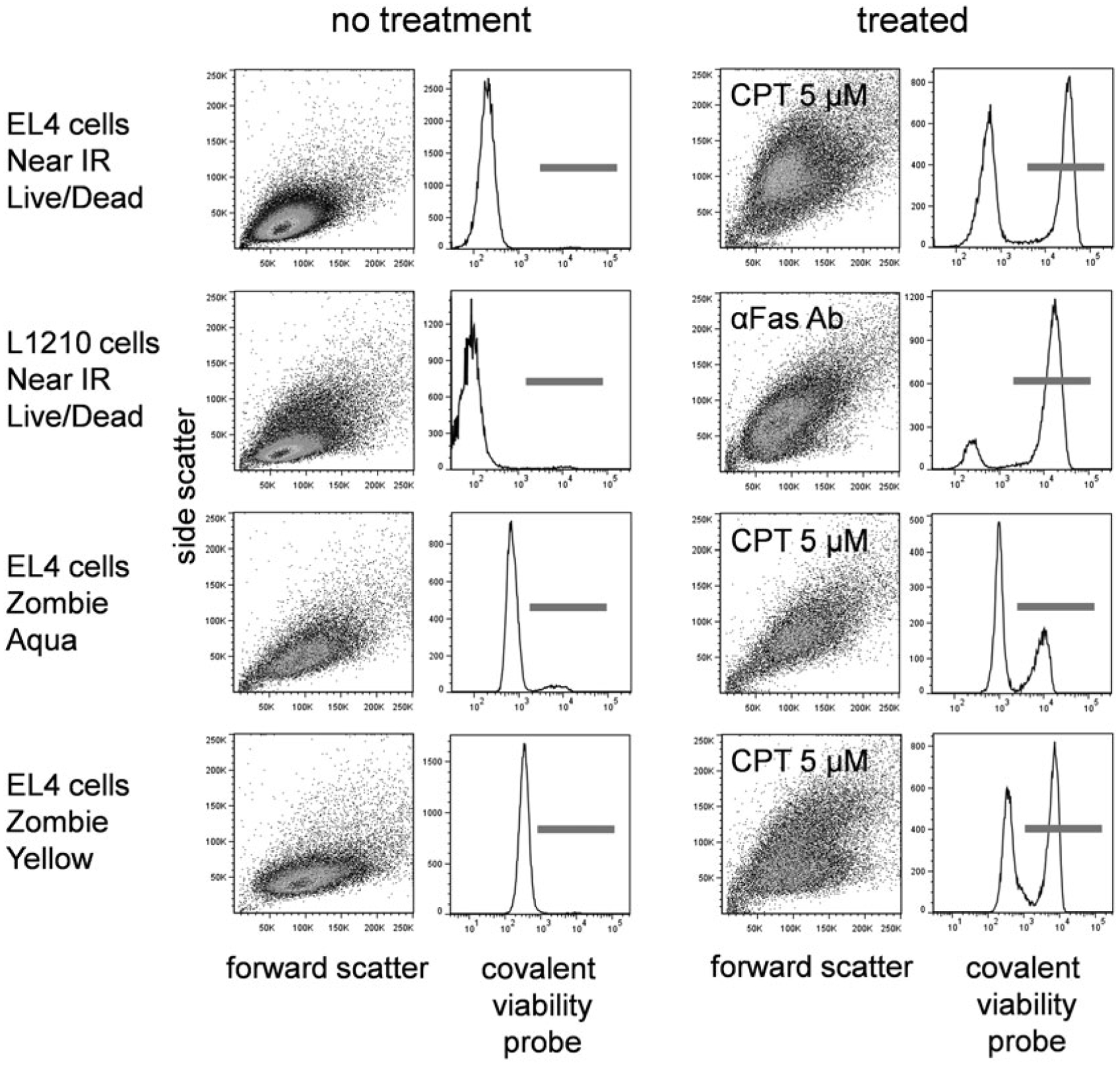
Covalent viability probe labeling. EL4 cells treated with camptothecin at 5 μM for 16 h or L1210 cells treated with anti-Fas antibody (clone Jo2) at 1:1000 final concentration for 12 h were labeled with the indicated covalent viability probe. Forward versus side scatter dotplots and viability probe histograms are shown for untreated (left two columns) and treated (right two columns) cells
3.8.4. Fluorogenic Caspase Substrates
After initial annexin V/viability gating, the cells can be observed for caspase activation. Note that even truly viable cells with no apoptotic activity labeled with a caspase substrate will have somewhat higher background fluorescence levels than completely unlabeled cells. All of the caspase substrates bind to viable cells to some degree. Care should be taken to identify both the viable and apoptotic fraction without using an unlabeled control as a cutoff. This background fluorescence is shown in Fig. 5.
Fig. 5.
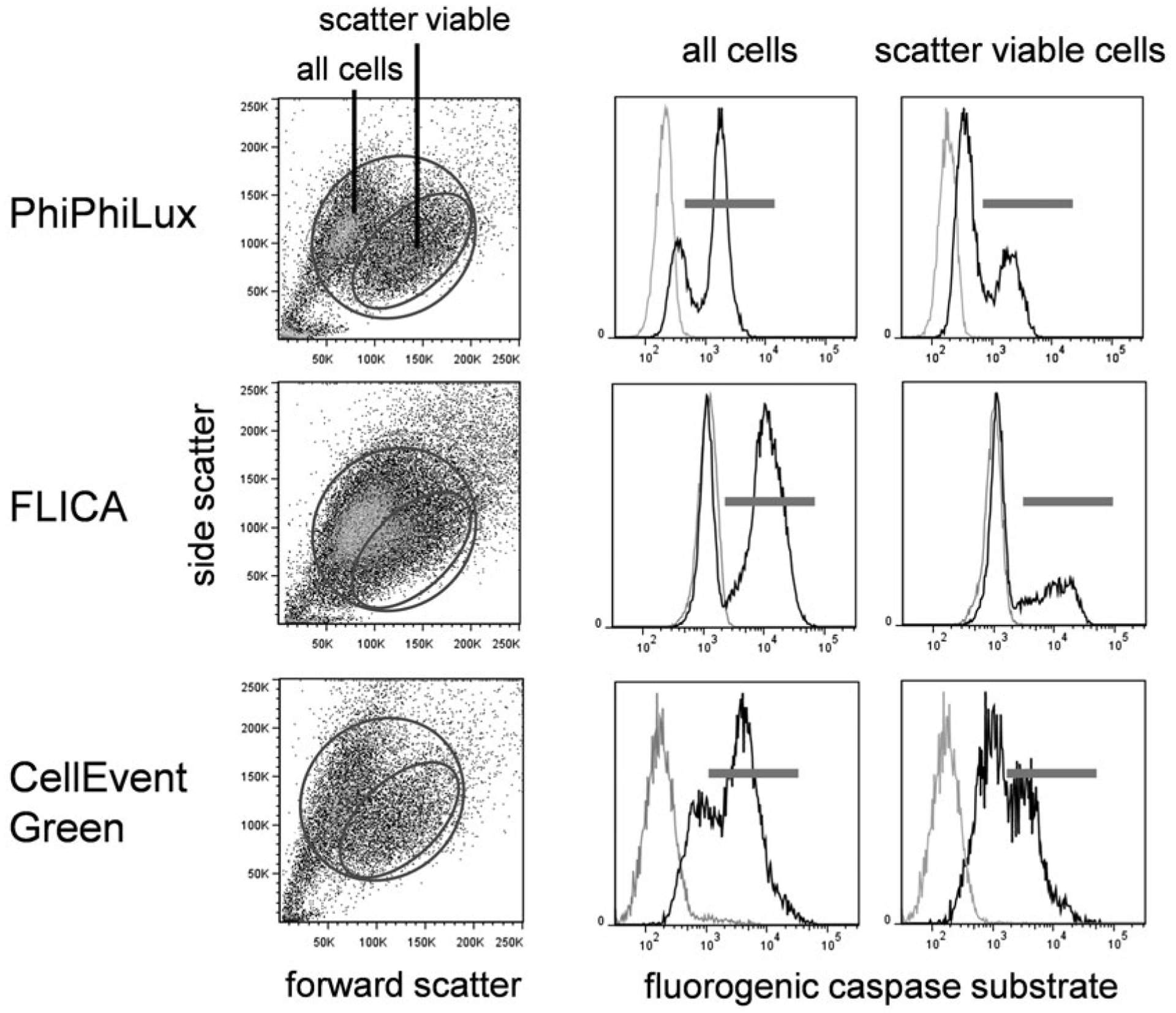
Fluorogenic caspase substrate fluorescence in viable and apoptotic cells versus cellular autofluorescence. EL4 cells treated with camptothecin at 5 μM for 16 h were labeled with PhiPhiLux G1D2 (top row), FLICA (middle row), or CellEvent Green (bottom row). Forward versus side scatter dot plots (left column) with corresponding caspase labeling for all cells (middle column) or scatter viable only (right column). In the fluorogenic caspase histograms, black peaks indicate substrate labeling; gray peaks are unlabeled cells
Once the caspase-negative population is identified, the caspase-positive cells should be clearly distinguishable. As discussed in Subheading 3.8.1, use both a total cell gate and a scatter viable gate to observe the extent of caspase activation. Caspase positive cells are usually present in the scatter viable only population in the presence of an apoptotic inducer.
3.9. Sample Results for Multiparametric Apoptosis Assays
Below are representative data for several combinations of fluorogenic caspase substrate, annexin Vand either a DNA-binding dye or a covalent viability probe. In all of the illustrated results, apoptosis was induced in EL-4 murine thymoma cells by treatment with camptothecin at 10 μM for 12 h. The figures both illustrate expected results for the individual components of the multiparametric cell death assay, and demonstrate how the simultaneous analysis of multiple cell death characteristics in a single assay gives a multidimensional picture of the total apoptotic process.
3.9.1. PhiPhiLux, Annexin V, and DNA-Binding Dye
In Fig. 6, EL4 cells treated with camptothecin were labeled with PhiPhiLux, Pacific Blue annexin V, and 7-AAD. As described in Subheading 3.8.1, cells were visualized for forward versus side scatter and gated to include both scatter viable and apoptotic cells, and scatter viable only. Labeling for all three markers can be seen, with PhiPhiLux-positive cells appearing earlier than the other two markers, and in significant numbers in the scatter viable population. Annexin V binding and 7-AAD permeability appear later.
Fig. 6.
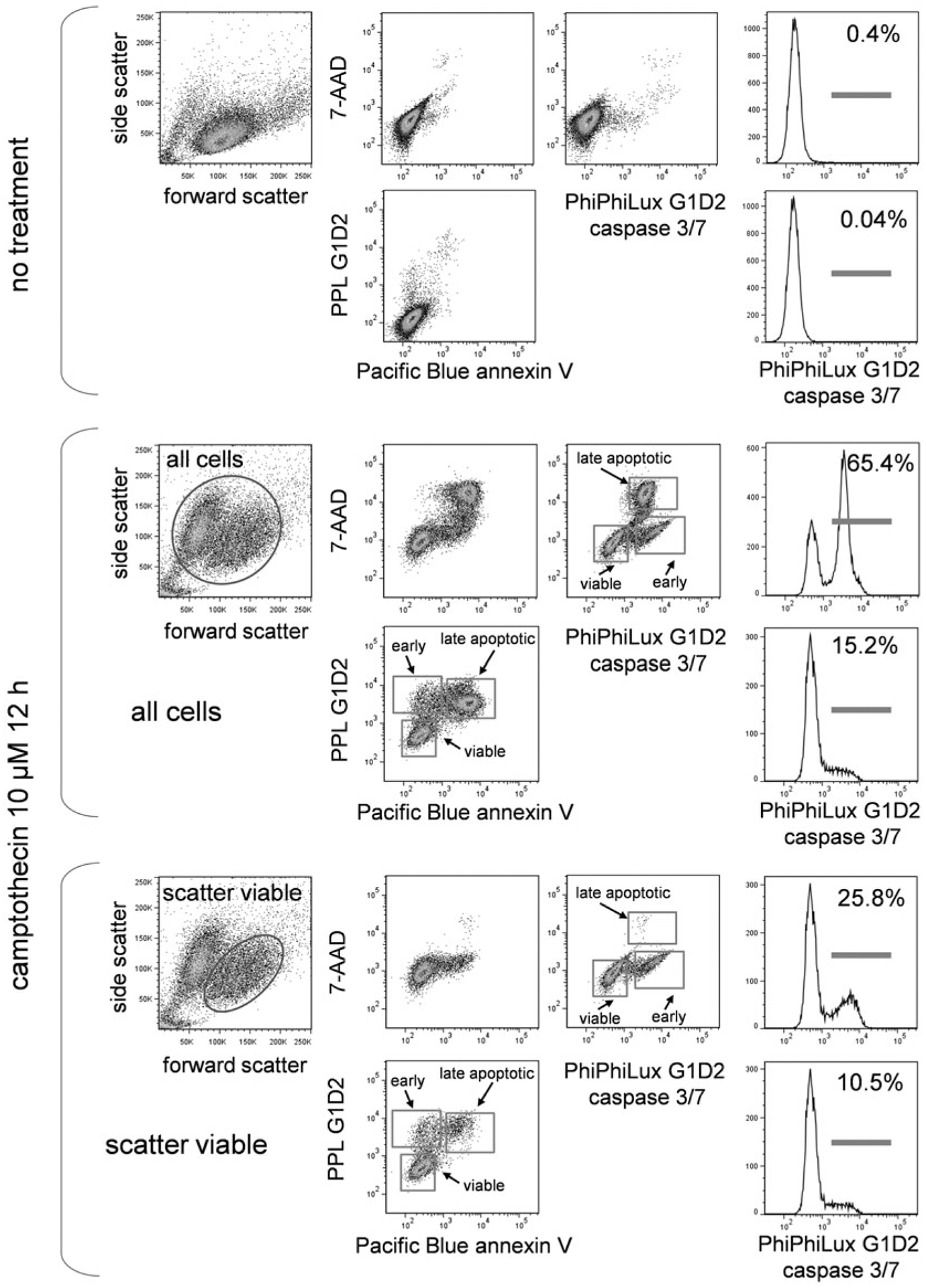
PhiPhiLux, annexin V, and DNA-binding dye labeling. EL4 cells untreated or treated with camptothecin at 5 μM for 16 h were labeled with PhiPhiLux G1D2, Pacific Blue-conjugated annexin V, and the DNA dye 7-AAD. Scatter dot plots and labeling for all three labels in untreated cells (upper group), treated cells gated for all cells (middle group), and treated cells gated for scatter viable cells only (bottom group) are shown. Stages of cell death are indicated
3.9.2. FLICA, Annexin V, and DNA-Binding Dye
In Fig. 7, treated EL4 cells were labeled with FLICA, Pacific Blue annexin V, and 7-AAD. Again, all three markers appear in apoptotic cells, but FLICA labeling precedes the other two and appears in the scatter viable cells as the earliest marker of apoptosis.
Fig. 7.
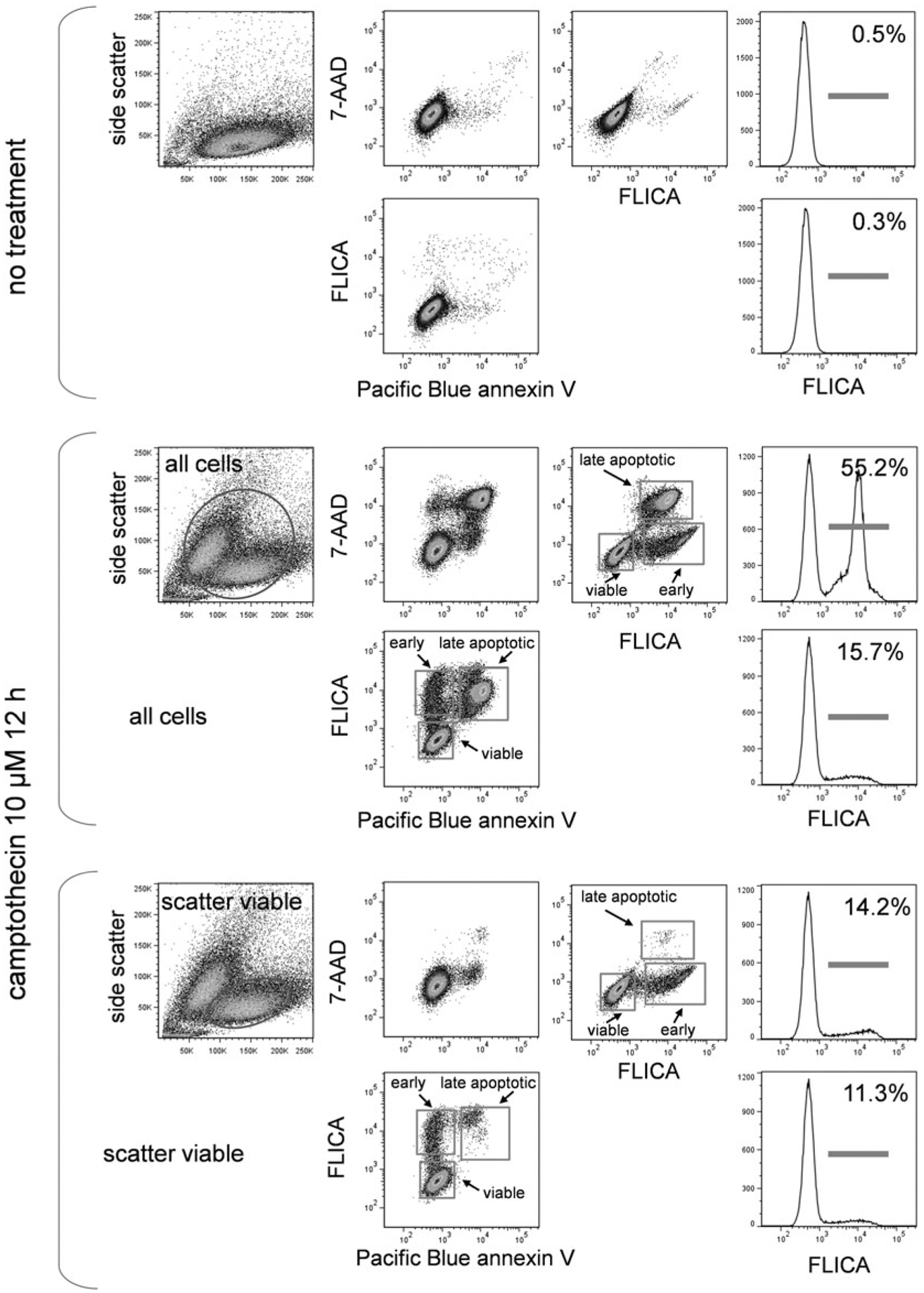
FLICA, annexin V and DNA-binding dye labeling. EL4 cells untreated or treated with camptothecin at 5 μM for 16 h were labeled with FLICA, Pacific Blue conjugated annexin V and the DNA dye 7-AAD. Scatter dot plots and labeling for all three labels in untreated cells (upper group), treated cells gated for all cells (middle group), and treated cells gated for scatter viable cells only (bottom group) are shown. Stages of cell death are indicated
3.9.3. CellEvent Green, Annexin V, and DNA-Binding Dye
In Fig. 8, treated EL4 cells were labeled with CellEvent Green, Pacific Blue annexin V, and 7-AAD. The marker distribution and order of appearance is similar to the PhiPhiLux and FLICA labeling in Figs. 6 and 7.
Fig. 8.
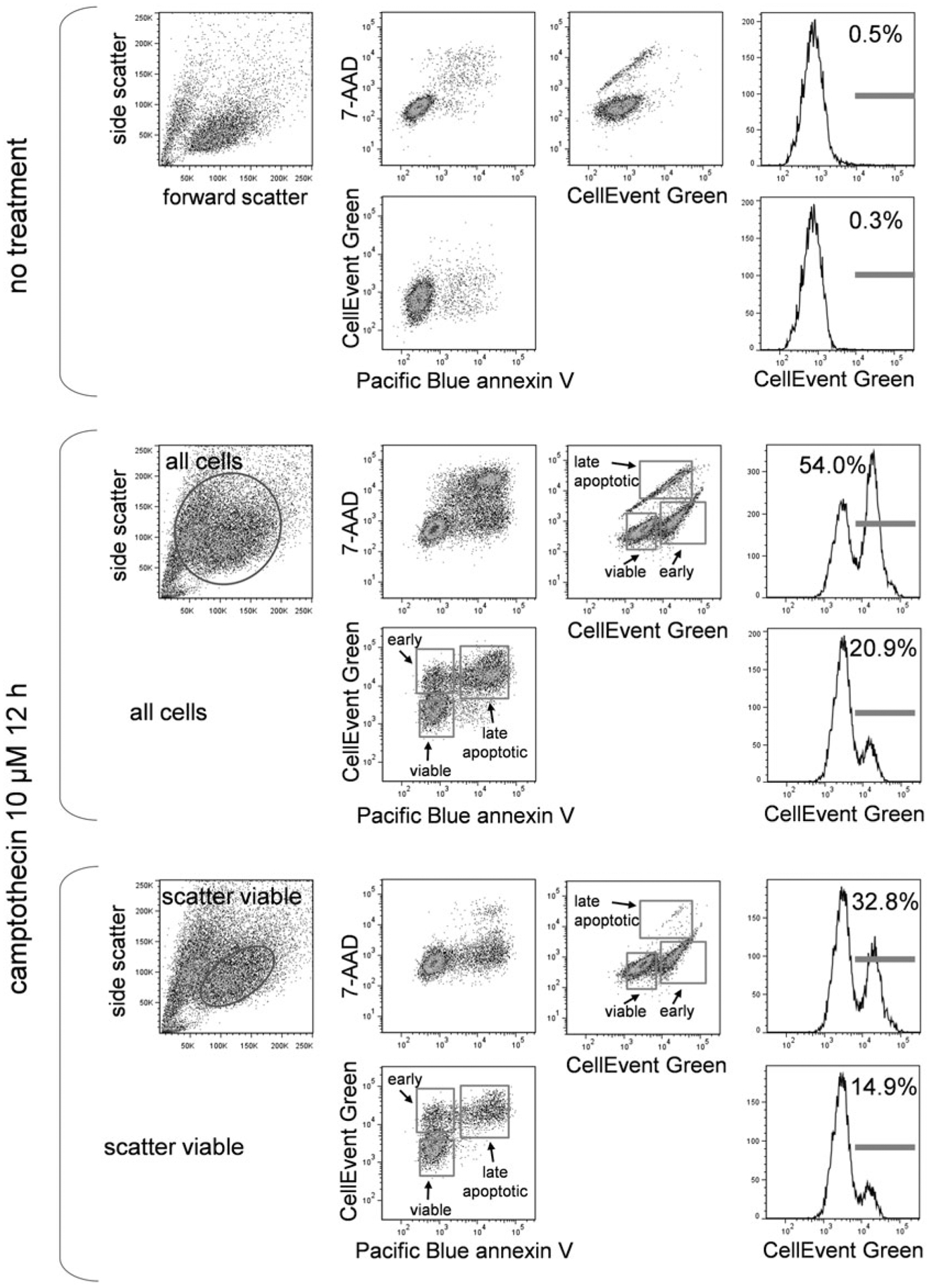
CellEvent Green, annexin V, and DNA-binding dye labeling. EL4 cells untreated or treated with camptothecin at 5 μM for 16 h were labeled with CellEvent Green, Pacific Blue conjugated annexin V and the DNA dye 7-AAD. Scatter dot plots and labeling for all three labels in untreated cells (i), treated cells gated for all cells (middle group), and treated cells gated for scatter viable cells only (bottom group) are shown. Stages of cell death are indicated
3.9.4. FLICA and Covalent Viability Probe
In Fig. 9, treated EL4 cells were labeled with FLICA followed by the covalent viability probe Live/Dead Near IR, and fixed with paraformaldehyde. Again, caspase activation precedes cell permeability, and multiple stages of apoptosis can be identified. This experiment did not include annexin V labeling; had it been included, the cells could not have been fixed prior to analysis.
Fig. 9.
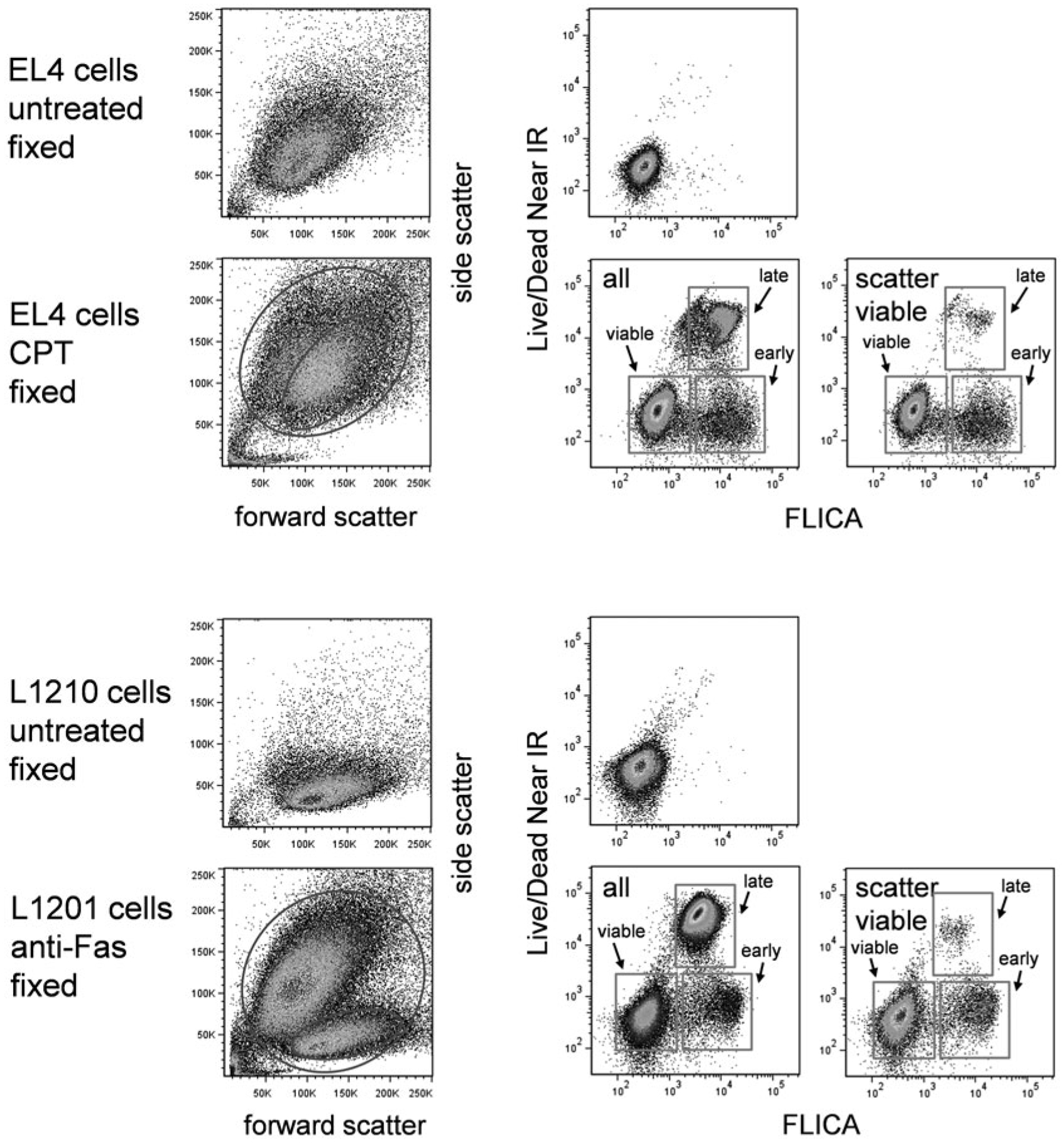
FLICA and covalent viability dye labeling. EL4 cells untreated or treated with camptothecin at 5 μM for 16 h (top two rows) or L1210 cells untreated or treated with anti-Fas antibody (clone Jo2) for 12 h (bottom two rows) were labeled with FLICA and the covalent viability dye Live/Dead Near IR (see Table 1). Scatter dot plots and labeling for both labels in untreated and treated cells are shown. Stages of cell death are indicated
3.10. Conclusion
Apoptosis is a highly variable process involving multiple biochemical pathways; therefore, there are no universal morphological or physiological characteristics that are common to apoptosis in all cell types. Cell death in different cell types (even in physiologically or morphologically similar ones) may present very different phenotypes, and may not necessarily be detectable by the same assays. Multiparametric assays for apoptosis are very amenable to the nature of apoptosis, since the investigator is not limited to one characteristic of cell death. Investigators should also be willing to try other apoptotic assays to fully characterize their particular system.
The protocol described in this chapter is readily amenable to the incorporation of antibody immunophenotyping along with the cell death markers, resulting in a very sophisticated “screening out” of dead cells for the measurement of receptor expression in “viable cells.” A potentially exciting extension of this method would appear to be the phenotyping of early apoptotic cells, positive for caspase expression but negative for later markers. This method should be approached with care; from a cellular standpoint, caspase activation is probably not an “early” event in cell death, and many alterations in the plasma membrane may have occurred by this timepoint, resulting in aberrant antibody binding to cells as is observed in later cell death. Any cell surface marker expression results obtained by such methodology should therefore be interpreted with caution.
4. Notes
Fluorogenic caspase substrates with alternative fluorophores. Fluorogenic caspase substrates coupled to rhodamine-like, sulforhodamine-like, and Cy5-like fluorophores are also available. The spectra of these probes for the PhiPhiLux substrates are shown in Fig. 1. None of these probes excites well at 488 nm; the rhodamine and sulforhodamine probes require a green or yellow laser source (532, 552, or 561 nm laser), and the Cy5 probes require red laser excitation. The green- and yellow-excited probes were originally designed for epifluorescence microscopes, which are usually equipped with mercury arc lamp filters that can provide 546 and 577 nm (green and yellow) excitation light. Many flow cytometers are now equipped with lasers in this range, however, making these probes potentially useful for cytometric analysis. Using the rhodamine and sulforhodamine probes limits DNA dye choices; the spectrum of propidium iodide coincides too well with them, and therefore cannot be used simultaneously. 7-AAD and the red-excited DNA binding dyes should also be avoided. The violet-excited DNA binding dyes including DAPI, Hoechst 33258, or SYTOX Blue should be used instead. The violet-excited covalent viability probes should also be used in this situation (Table 1). All of these longer wavelength probes may give better sensitivity than the fluorescein-like versions; cellular autofluorescence is significantly reduced with green to red excitation (compared to 488 nm blue-green), so overall signal-to-background signal is likely to increase.
Controls. Good “viable” and apoptotic controls are important for apoptotic analysis of apoptosis, and should be used, particularly when a new cell type or apoptotic stimulus is being investigated. Where possible, an untreated negative control and an independent positive control should be included, the latter being induced by an agent other than that under study (such as a cytotoxic drug). The EL4 cells used in this study are a good example of a control system that is easy to maintain and is reliable. Topoisomerase inhibitors including camptothecin and topotecan, transcriptional inhibitors including cycloheximide and translational inhibitors including actinomycin D and puro-mycin are all good inducers, and can induce in other cell lines as well. Samples with both the absence and presence of the caspase reagents are also important to include as controls, since the substrate does possess some low but detectable intrinsic fluorescence in the uncleaved state that can be erroneously interpreted as apoptosis without the appropriate control samples. However, unlabeled cells should not be used as a strict guide for gating on caspase labeled cells; they only allow determination of the increase in background from PhiPhiLux labeling. This background can be seen in Fig. 2.
Multiparametric analysis of apoptosis in adherent cells. Flow cytometric analysis of apoptosis in adherent cell lines poses special challenges, since the removal of cells from their growth substrate may itself induce apoptosis. In addition, cell removal methods (such as trypsinization) can trigger false apoptotic indicators, such as aberrant annexin V binding in the absence of true cell death. By far the best solution to this problem is to utilize a slide-based laser scanning cytometer (LSC) for the analysis of apoptosis in these cell types; this specialized flow cytometer can perform cytometric analysis of cells on a flat surface, allowing minimal disruption during cell preparation [20]. Several apoptosis assays utilizing caspase substrates using laser scanning cytometry have been described [17, 21, 22].
DNA-binding dyes. All DNA-binding dyes do not have identical cell permeability characteristics. Some DNA dyes will gradually cross the plasma membranes of even viable cells, while others are better excluded. These differences can affect the results obtained from the assay. For example, 7-AAD is somewhat more cell permeable than PI, and may give a slightly greater percentage of apoptotic cells when compared directly to PI. Similarly, SYTOX Blue is slightly more cell permeable than Hoechst 33258, and will also identify an earlier set of apoptotic cells. This difference should be kept in mind while designing cell death assays, and may dictate the use of 7-AAD when this property is desired. Highly permeable DNA binding dyes such as Hoechst 33342, the DyeCycle dye series (Thermo Fisher Life Technologies), and DRAQ5 (Biostatus Ltd.) will enter cells and label their chromatin regardless of their viability state. This may limit their usefulness as apoptotic reagents for flow cytometry. They have however been used to identify morphological changes in chromatin during apoptosis by microscopy and laser scanning cytometry.
Caspase substrate specificity and background. While the PhiPhiLux substrates seem reasonably specific for their target caspases, no synthetic substrate is exclusively specific for any particular enzyme. This should be kept in mind for any assay involving specific proteolytic activity. In general, a considerable excess of substrate will encourage low levels of nonspecific cleavage, increasing the non-caspase background of the assay. Titration of the substrate to the lowest concentration able to distinguish activity may be necessary when the specificity of the assay is in doubt.
Annexin V critical parameters. Calcium and magnesium are critical for annexin V binding; even brief removal of divalent cations after the binding reaction will result in rapid dissociation from PS residues. The cells must therefore remain in a calcium/magnesium buffer at all stages up to analysis, including all wash buffers.
Incubation periods and sample storage. All incubation periods and conditions are critical parameters for this assay, as is prompt analysis of samples following the labeling procedure. Insufficient incubation time for the PhiPhiLux substrates will result in poor labeling; prolonged incubation periods will increase the level of nonspecific substrate binding and cleavage, resulting in high background fluorescence and decreased signal-to-noise ratios. In addition, prolonged storage of cells following the removal of the surrounding PhiPhiLux substrate will eventually result in leakage of the cleaved substrate from the cell, despite its reduced cell permeability in the cleaved state. Overly long annexin V incubation periods will also increase the amount of nonspecific binding to cells, making discrimination of “viable” and apoptotic cells more difficult. Although PI (and to a lesser extent 7-AAD) are relatively impermeant to viable cells, prolonged incubation will cause uptake even in healthy cells. If laboratory conditions do not allow prompt analysis of sample, the FLICA assay with covalent viability probe labeling should be considered. Other cell death assays involving fixed cells such as TUNEL assays or immunolabeling of active caspases not described here should also be considered alternatives.
Acknowledgments
The author wishes to acknowledge Veena Kapoor and Nga Hawk of the National Cancer Institute for excellent technical assistance, and Dr. Zbigniew Darzynkiewicz of the New York Medical College for helpful discussion. Jolene Bradford and Gayle Buller at Thermo Fisher Life Technologies (formerly Molecular Probes) provided valuable technical information regarding fluorescent probes. Beverly Packard and Akira Komoriya also provided information regarding the PhiPhiLux probes, technical assistance and useful discussion. This work was supported by intramural research fund provided by the Center for Cancer Research, National Cancer Institute.
References
- 1.Telford WG, King LE, Fraker PJ (1994) Rapid quantitation of apoptosis in pure and hetero-geneous cell populations using flow cytometry. J Immunol Methods 172(1):1–16 [DOI] [PubMed] [Google Scholar]
- 2.Darzynkiewicz Z, Juan G, Li X, Gorczyca W, Murakami T, Traganos F (1997) Cytometry in cell necrobiology: analysis of apoptosis and accidental cell death (necrosis). Cytometry 27(1):1–20 [PubMed] [Google Scholar]
- 3.Del Bino G, Darzynkiewicz Z, Degraef C, Mosselmans R, Fokan D, Galand P (1999) Comparison of methods based on annexin-V binding, DNA content or TUNEL for evaluating cell death in HL-60 and adherent MCF-7 cells. Cell Prolif 32(1):25–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vermes I, Haanen C, Reutelingsperger C (2000) Flow cytometry of apoptotic cell death. J Immunol Methods 243(1–2):167–190 [DOI] [PubMed] [Google Scholar]
- 5.Pozarowski P, Grabarek J, Darzynkiewicz Z (2003) Flow cytometry of apoptosis. Curr Pro-toc Cytom Chapter 7:Unit 7 19. doi: 10.1002/0471142956.cy0719s25 [DOI] [PubMed] [Google Scholar]
- 6.Henkart PA (1996) ICE family proteases: mediators of all apoptotic cell death? Immunity 4(3):195–201 [DOI] [PubMed] [Google Scholar]
- 7.Ormerod MG, Sun XM, Snowden RT, Davies R, Fearnhead H, Cohen GM (1993) Increased membrane permeability of apoptotic thymocytes: a flow cytometric study. Cytometry 14(6):595–602. doi: 10.1002/cyto.990140603 [DOI] [PubMed] [Google Scholar]
- 8.Castedo M, Hirsch T, Susin SA, Zamzami N, Marchetti P, Macho A, Kroemer G (1996) Sequential acquisition of mitochondrial and plasma membrane alterations during early lym-phocyte apoptosis. J Immunol 157(2):512–521 [PubMed] [Google Scholar]
- 9.Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281(5381):1309–1312 [DOI] [PubMed] [Google Scholar]
- 10.Overbeeke R, Steffens-Nakken H, Vermes I, Reutelingsperger C, Haanen C (1998) Early features of apoptosis detected by four different flow cytometry assays. Apoptosis 3(2):115–121 [DOI] [PubMed] [Google Scholar]
- 11.Earnshaw WC, Martins LM, Kaufmann SH (1999) Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem 68:383–424. doi: 10.1146/annurev.biochem.68.1.383 [DOI] [PubMed] [Google Scholar]
- 12.Koester SK, Bolton WE (2001) Cytometry of caspases. Methods Cell Biol 63:487–504 [DOI] [PubMed] [Google Scholar]
- 13.Gorman AM, Hirt UA, Zhivotovsky B, Orrenius S, Ceccatelli S (1999) Application of a fluorometric assay to detect caspase activity in thymus tissue undergoing apoptosis in vivo. J Immunol Methods 226(1–2):43–48 [DOI] [PubMed] [Google Scholar]
- 14.Belloc F, Belaud-Rotureau MA, Lavignolle V, Bascans E, Braz-Pereira E, Durrieu F, Lacombe F (2000) Flow cytometry detection of caspase 3 activation in preapoptotic leukemic cells. Cytometry 40(2):151–160 [DOI] [PubMed] [Google Scholar]
- 15.Bedner E, Smolewski P, Amstad P, Darzynkiewicz Z (2000) Activation of caspases measured in situ by binding of fluorochrome-labeled inhibitors of caspases (FLICA): correlation with DNA fragmentation. Exp Cell Res 259(1):308–313. doi: 10.1006/excr.2000.4955 [DOI] [PubMed] [Google Scholar]
- 16.Komoriya A, Packard BZ, Brown MJ, Wu ML, Henkart PA (2000) Assessment of caspase activities in intact apoptotic thymocytes using cell-permeable fluorogenic caspase substrates. J Exp Med 191(11):1819–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Telford WG, Komoriya A, Packard BZ (2002) Detection of localized caspase activity in early apoptotic cells by laser scanning cytometry. Cytometry 47(2):81–88 [DOI] [PubMed] [Google Scholar]
- 18.Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC (1994) Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371 (6495):346–347. doi: 10.1038/371346a0 [DOI] [PubMed] [Google Scholar]
- 19.Packard BZ, Toptygin DD, Komoriya A, Brand L (1996) Profluorescent protease substrates: intramolecular dimers described by the exciton model. Proc Natl Acad Sci U S A 93(21):11640–11645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamentsky LA, Burger DE, Gershman RJ, Kamentsky LD, Luther E (1997) Slide-based laser scanning cytometry. Acta Cytol 41(1):123–143 [DOI] [PubMed] [Google Scholar]
- 21.Packard BZ, Komoriya A, Brotz TM, Henkart PA (2001) Caspase 8 activity in membrane blebs after anti-Fas ligation. J Immunol 167(9):5061–5066 [DOI] [PubMed] [Google Scholar]
- 22.Smolewski P, Bedner E, Du L, Hsieh TC, Wu JM, Phelps DJ, Darzynkiewicz Z (2001) Detection of caspases activation by fluorochrome-labeled inhibitors: multiparameter analysis by laser scanning cytometry. Cytometry 44(1):73–82 [DOI] [PubMed] [Google Scholar]