

Abstract
The immune system is tasked with identifying malignant cells to eliminate or prevent cancer spread. This involves a complex orchestration of many immune cell types that together recognize different aspects of tumor transformation and growth. In response, tumors have developed mechanisms to circumvent immune attack. Type I interferons (IFN-Is) are a class of proinflammatory cytokines produced in response to viruses and other environmental stressors. IFN-Is are also emerging as essential drivers of antitumor immunity, potently stimulating the ability of immune cells to eliminate tumor cells. However, a more complicated role for IFN-Is has arisen, as prolonged stimulation can promote feedback inhibitory mechanisms that contribute to immune exhaustion and other deleterious effects that directly or indirectly permit cancer cells to escape immune clearance. We review the fundamental and opposing functions of IFN-Is that modulate tumor growth and impact immune function and ultimately how these functions can be harnessed for the design of new cancer therapies.
Keywords: type I interferon, IFN-I, cancer immunology, hallmarks of cancer, immunotherapy, IFN-stimulated gene, ISG
INTRODUCTION
Twenty years ago, Hanahan & Weinberg (1) defined the hallmarks of cancer as the patho-physiological events required for tumor growth and survival. The hallmarks of cancer included unchecked proliferation, cellular immortalization, angiogenesis, tissue invasion, and metastasis. In the decades since, the hallmarks have been updated to include emerging concepts as our appreciation for the complexity of cancer pathogenesis grows. Genomic instability, metabolic perturbation, inflammation, and immune evasion are all crucial factors now understood to contribute to rapid tumor growth and metastasis. Importantly, a new understanding has emerged of how the immune system interacts within the tumor microenvironment (TME) and with cancer cells themselves, in large part due to the success of immune-targeted therapies to treat many types of cancer (2–5). In fact, it is becoming evident that the immune system plays an overarching influence on all the diverse hallmarks of cancer, and underlying these immune, tissue, and cancer effects are type I interferons (IFN-Is).
In humans, IFN-Is are a family of proteins consisting of 13 isoforms of IFN-α, IFN-β, IFN-ω, and IFN-ε. IFN-Is are essential for antiviral immunity, and in the decades since their discovery, their fundamental role in antitumor immunity has been realized. Despite the numerous isoforms of IFN-Is, they all signal through the IFN-α/β receptor (IFNAR). IFNAR consists of the heterodimeric subunits IFNAR1 and IFNAR2, and their dimerization upon binding IFN-I leads to phosphorylation of JAK1 and TYK2, which activate STAT1/STAT2 heterodimers (Figure 1). Phosphorylated STAT1/STAT2 forms a complex with the interferon regulatory factor 9 (IRF9), termed interferon-stimulated gene factor 3 (ISGF3) (6). ISGF3 translocates to the nucleus, binding to IFN signaling response elements to induce hundreds of IFN-stimulated genes (ISGs) that then affect the diverse outcomes of IFN-Is, including pathogen resistance and immune activation. In addition to canonical STAT1/STAT2 signaling, IFN-Is can activate signaling by STAT3–6 and by STAT-independent signaling pathways, including the c-Jun N-terminal kinase (JNK), extracellular signal regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK), and mammalian target of rapamycin (mTOR) pathways. Differential signaling from IFN-I through these various pathways ultimately leads to the diverse and seemingly opposing immunologic outcomes of IFN-I signaling. Thus, in addition to their widely recognized antipathogen and proinflammatory functions, IFN-I-induced genes regulate all aspects of the immune response, including antigen presentation, cellular proliferation, protein translation, cellular apoptosis, metabolism, and in some cases, immunosuppression.
Figure 1.
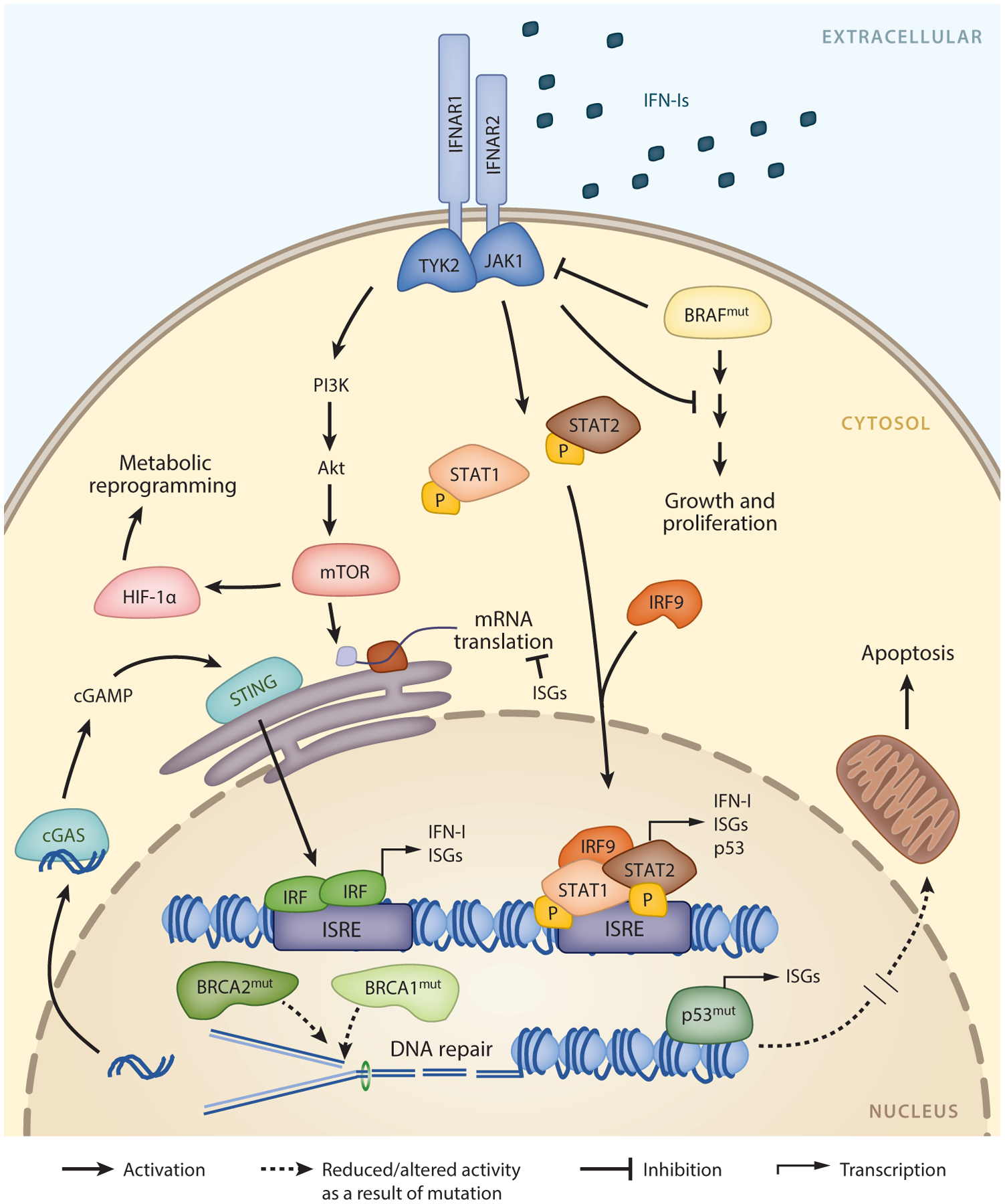
IFN-I signaling in cancer. IFN-Is signal through the IFNAR, which is composed of IFNAR1 and IFNAR2 subunits. Activation of IFNAR triggers a signaling cascade through JAK-STAT, leading to the formation of the ISGF3 complex containing STAT1, STAT2, and IRF9. ISGF3 binds to ISREs, which induces the expression of hundreds of ISGs and other key genes such as TP53. Additional pathways downstream of IFNAR include PI3K/Akt signaling, resulting in the activation of mTOR and leading to HIF-1α stabilization and subsequent metabolic reprogramming. Tumor driver mutations in molecules such as BRAF and p53 can attenuate IFN-I signaling while promoting tumor growth and survival. Gain-of-function BRAF mutation leads to downregulation of IFNAR, and mutations in p53 can blunt the production of ISGs. Dysregulation of the DNA damage response and repair mechanisms, for example, through mutation of BRCA1 or BRCA2, leads to the accumulation of mutations and genomic instability. Resulting cytosolic DNA extruded in response to this replication stress is sensed by cGAS, which activates STING, ultimately initiating and amplifying IFN-I and ISG production. Abbreviations: IFN-I, type I interferon; IFNAR, IFN-α/β receptor; ISG, IFN-stimulated gene; ISRE, IFN-stimulated response element.
Because of the fundamental functions of IFN-Is in antiviral immunity, IFNAR is expressed on all nucleated cells. Therefore, every cell can potentially respond to IFN-Is, although certain cell types are more sensitive than others, and the impact of IFN-Is is highly dependent on cell type (6, 7). Furthermore, although all IFN-Is bind to the same receptor, different isoforms have differing binding affinities and have been attributed to unique functions of IFN-Is (8, 9). Consistent with their many roles, IFN-Is are induced by several different pathways, including DNA/RNA sensing through Toll-like receptors (TLRs), cGAS-STING, and other pattern and danger recognition receptors. Pattern and danger-associated molecular patterns such as viral DNA/RNA are recognized by endosomal TLR3, TLR7/8, and TLR9 or by cytosolic sensors MDA5, RIG-I, IFI-16, and cGAS (6). Endosomal TLRs activate MyD88 signaling, leading to phosphorylation of IRF7, a transcription factor that amplifies IFN-I and ISG production. The cytosolic nucleotide sensors MAVS (through MDA5 or RIG-I) and STING (through cGAS, IFI16, or DAI) stimulate IFN-I production by subsequent phosphorylation and activation of IRF3 (6). Once secreted, IFN-Is can act in a paracrine or autocrine fashion, amplifying the IFN-I signaling pathways (6).
Although generally considered to exert proinflammatory functions, or to switch from an inflammatory to a suppressive state with the onset of chronic disease, IFN-Is simultaneously activate the expression of both proinflammatory and anti-inflammatory ISGs to drive both activating and suppressive programs. The observed balance of these effects can change in a context-dependent manner, for example, as occurs in the transition from acute to chronic disease states, such as chronic infections and cancer (10, 11). In acute scenarios, the benefits of IFN-Is appear to outweigh the negative consequences, although suppressive ISGs are also rapidly expressed, as exemplified by the early expression of the negative regulator programmed death ligand-1 (PD-L1) in acute infections as part of a negative feedback loop to temper T cell receptor signaling and control inflammation (7, 12). During chronic infection and cancer, a new homeostasis is reached such that the negative effects of IFN-Is can become more dominant, initiating a cycle wherein incomplete clearance of the threat prolongs inflammation, which sustains the expression of IFN-I-driven negative regulators (11, 13). However, even in these chronic states, IFNs continue to support proinflammatory functions, as evidenced by the rapid increase in chronic viral replication when IFNAR signaling is transiently blocked during chronic viral infection (11, 14). Although like most processes regulated by IFN-Is, this is not always the case, because blockade of IFNAR signaling in a humanized mouse model of HIV infection led to a rapid decrease in HIV titers (15). Similarly, in cancer IFN-Is drive a state of cellular dormancy, whereby tumor cells are not completely eliminated but rather maintained at a low burden (16, 17). Ultimately, the factors determining the outcome of IFN-Is are complex, depending not only on isoform but also on timing, cell type, and context of the inflammatory milieu. Adding to this complexity, type II (IFN-γ) and type III (IFN-λ) IFNs can also activate expression of overlapping subsets of ISGs upon binding their respective receptors (IFN-γR and IL-10R2/IFN-λR1), and it can often be difficult to resolve type I, type II, and type III IFN gene expression signatures. Trying to understand the dynamics of IFN-I responses is currently a major push in the field and, as discussed below, could lead to new cancer therapies that enhance the antitumor and proinflammatory functions of IFNs while minimizing their suppressive ones. Herein, we review how IFN-Is simultaneously protect against and drive tumor formation to contribute to both anticancer immunity and immunosuppression. We discuss the central role of IFN-Is as mediators of all the hallmarks of cancer and how newly developing concepts in IFN-I research are being translated to the clinic for the future of cancer therapy.
IMMUNE EVASION
Tumor-Intrinsic Type I Interferon Activation
One of the most important emerging hallmarks of cancer is the ability of tumor cells to evade the immune system. Cancer cells are adept at escaping detection by immune cells that are constantly scanning their environment for signs of danger and transformed cells to be eliminated (Figure 2). These cancer cells leverage IFN-I responses to hide in plain sight, skewing immune cells toward a dysfunctional or immunosuppressive phenotype. By contrast, the IFN-Is they induce are also critical for the initial events that stimulate antitumor immunity (10, 18, 19), thus leading to a complex system whereby both inhibition of IFN-Is and stimulation of IFN-Is can contribute to disease progression.
Figure 2.
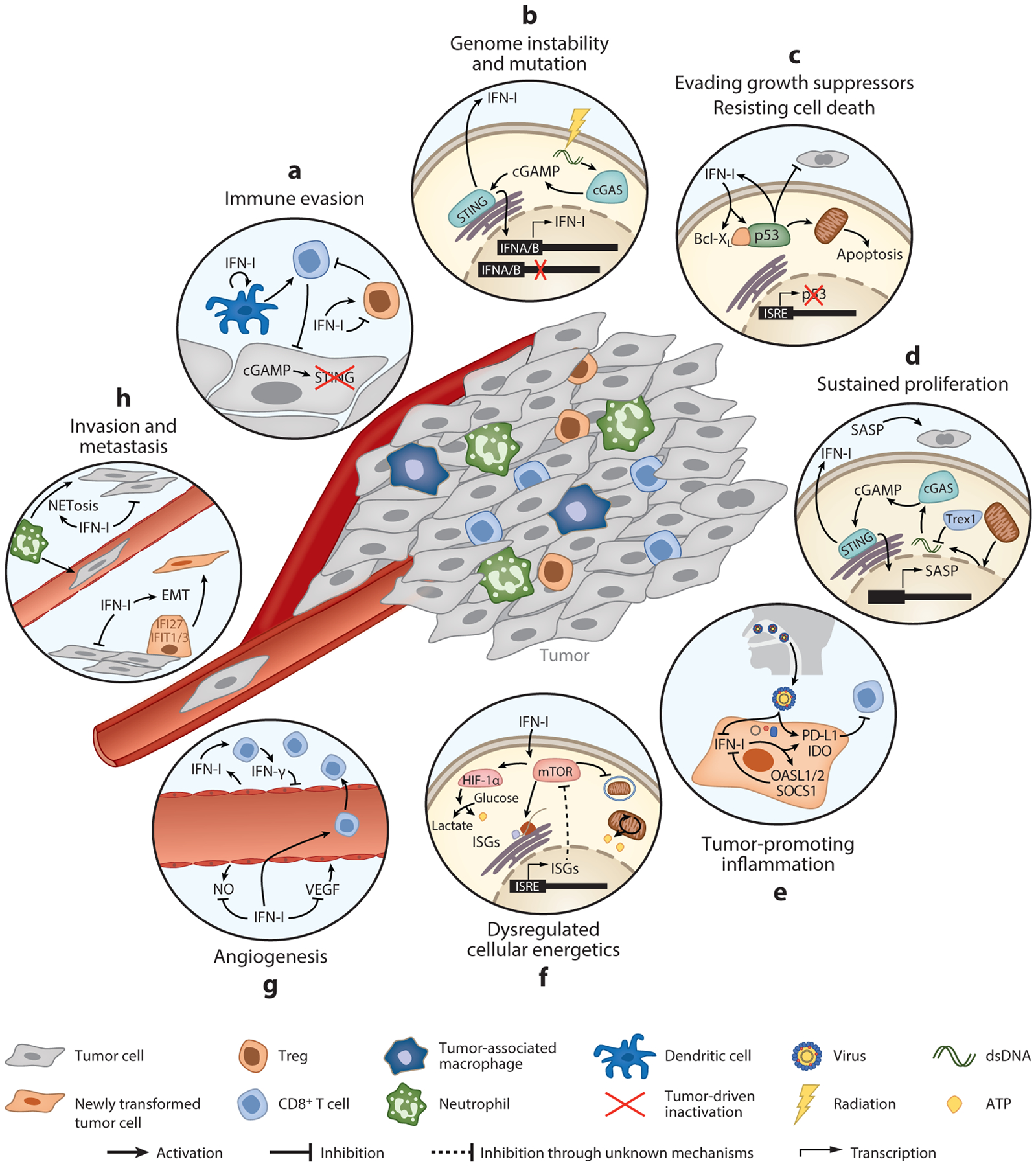
IFN-I-mediated regulation of the hallmarks of cancer. Key pathways highlighting examples of the opposing roles in IFN-Is in different pathological processes during cancer development. (a) Immune evasion. Inactivation of STING or IFNAR blunts IFN-I production, limiting the ability of dendritic cells to prime T cell responses and enhancing Treg infiltration. Cancer cells can also co-opt IFN-Is to promote Treg function in the tumor microenvironment. (b) Genome instability and mutation. DNA damage induced by accumulated mutations or by radiation therapy stimulates IFN-I responses through cGAS-STING-mediated expression of ISGs. IFN-Is are also commonly mutated in cancer. (c) Evading growth suppressors/resisting cell death. Mutual activation of IFN-Is and p53 inhibits tumor cell proliferation and promotes apoptosis. Tumors can overcome these effects by mutating p53. (d) Sustained proliferation. STING activated in senescent cells upregulates IFN-I and SASP. While senescent cells are replicatively paralyzed, SASP can promote tumor growth. (e) Tumor-promoting inflammation. Chronic inflammation caused, for example, by viral infection induces a skewed IFN-I response, favoring the expression of negative regulatory ISGs that facilitate continued tumor growth. (f) Dysregulated cellular energetics. Through regulation of mTORC1, IFN-Is activate glycolysis or autophagy, either of which promotes tumor cell growth and survival. (g) Angiogenesis. IFN-Is inhibit VEGF and promote vascular normalization, allowing T cell infiltration and antitumor immunity. (h) Invasion and metastasis. IFN-Is can facilitate EMT and also promote inflammation at distal sites that enhance metastasis. Abbreviations: EMT, epithelial-to-mesenchymal transition; IFNAR, IFN-α/β receptor; ISG, IFN-stimulated gene; mTORC1, mammalian target of rapamycin complex 1; SASP, senescence-associated secretory phenotype; VEGF, vascular endothelial growth factor.
An explanation for the pleiotropic effect of the IFN-I response is the ubiquitous expression of its receptor, allowing both immune and nonimmune cells to respond to IFN-Is. The effect of tumor-intrinsic IFN-I signaling is controversial, with some studies demonstrating that only IFN-Is acting on immune cells contribute to the initiation of antitumor immunity (10) and others demonstrating at least a partial requirement for tumor-intrinsic signaling (20). In the latter study, mutation of the cellular growth factor BRAF accompanied by loss of tumor-intrinsic IF-NAR expression in a melanoma model led to aggressive cancer cell growth, suggesting that the tumor-intrinsic role of IFN-Is depends at least in part on how driver mutations affect the ability of tumor cells to respond to IFN-Is (10, 20). Indeed, IFN-I signaling pathways converge on BRAF signaling, inhibiting proliferation by dephosphorylating key signaling molecules downstream of BRAF (21). Inactivation of IFNAR1 in the context of a gain-of-function mutation in BRAF therefore intensifies its proliferative effect (20), whereas this may not be the case in other oncogenic contexts. The interaction between IFN-I and BRAF pathways is further supported by the results of a clinical trial reported in 2019 wherein an intermediate dose of IFNα−2b was administered to patients with stage III melanoma, and both disease-free survival and overall survival were significantly higher in patients with BRAF-mutated tumors treated with IFNα−2b (22), highlighting that the tumor-intrinsic effect of IFN-I signaling and the outcome of IFN-I therapy are enhanced by the inflammatory by-products of tumor-driver mutations.
In addition to their tumor-intrinsic role, IFN-Is play a fundamental role in coordinating immune cell function to influence antitumor immunity. To fully understand the role IFN-Is play in counteracting cancer immune evasion, we must closely examine the numerous immune cells that protect against tumor cell invasion and how these cells sequentially become dysfunctional as tumors gain the ability to escape detection. In the following subsections, we explore the role of IFN-Is in dendritic cells, T cell subsets, and natural killer cells; immune cells such as macrophages, plasmacytoid dendritic cells (pDCs), neutrophils, and myeloid-derived suppressor cells are integrated into later sections of this review.
Dendritic Cells
Dendritic cells (DCs) are highly effective antigen-presenting cells (APCs). DCs efficiently process exogenously acquired antigen to activate naive CD4+ and CD8+ T cells. Further, DCs and particularly conventional type I DCs (cDC1s) have the unique ability to process exogenously acquired antigen for presentation on major histocompatibility complex class I (MHCI) (termed cross-presentation), making them potent stimulators of antitumor CD8+ T cell responses (18, 19). Not only are DCs among the most sensitive immune cells to IFN-Is but also pDCs are the most prolific producers of IFN-Is (reviewed in 23). As such, they are critical intermediates in IFN-I responses, bridging the innate and adaptive immune systems. DCs orchestrate both systemic immune responses necessary for initial and subsequent tumor recognition and, when possible, clearance (18, 19), as well as local inflammatory responses that contribute to T cell trafficking and the composition of the cytokine milieu (24). Importantly, both systemic and local DC responses depend on IFN-I signaling (18, 19, 24), which profoundly enhances their antigen-processing and antigen-presenting ability to activate CD8+ T cells (18, 19). In fact, in mouse models, IFNAR-deficient DCs are incapable of priming CD8+ T cells, leading to unrestricted tumor growth (18, 19). DCs directly perceive IFN-Is from tumor cells via cGAS-STING-mediated activation by tumor DNA (25). In addition, DCs can promote CD8+ T cell priming through cGAMP transferred from tumors that lack STING signaling (26, 27). Thus, tumors elicit IFN-I responses through both intrinsic and extrinsic mechanisms to stimulate DC-mediated antitumor immunity (Figure 2a).
Due to their powerful immune-activating capacity, DCs are highly sensitive to environmental stimuli. DC homeostasis is tightly regulated to restrain aberrant activation while maintaining the sensitivity to rapidly respond to emerging threats (28, 29). As such, DCs are sensitive to reprogramming by their specific environment and, in many cases, via temporal changes in IFN-I signaling. Consistent with the functional specialization of distinct DC subsets, not all DC subsets respond to IFN-Is in the same manner. Migratory cDC1s react locally to IFN-I in the TME to produce CXCL10, which recruits CD8+ T cells into the tumor (19, 24, 30), and carry antigen to lymph nodes, where IFN-I-activated tissue-resident cDC1s prime CD8+ T cell responses (30, 31). By contrast, IFN-Is can induce the suppressive activity of monocyte-derived DCs by promoting their expression of PD-L1, indoleamine 2,3-dioxygenase (IDO), interleukin-10 (IL-10) and other anti-inflammatory mediators during chronic virus infection and within the TME (13). Similarly, IFN-I-induced PD-L1 on migratory cDC1s tempers their antitumor efficacy (31), an adaptation that likely exists as a reciprocal mechanism to limit self-tissue damage and to maintain survival of immune cells in chronic inflammatory environments (32). This is perhaps an evolutionary feedback mechanism to limit progressive inflammation in autoimmune diseases; however, in the context of untreated anticancer immunity, this inherent protective strategy prevents elimination of tumor growth (33–36). It does present an opportunity for combination therapies to take advantage of synergy between IFN-I-inducing agents, DC function, and blockade of the PD-L1/PD-1 axis (31, 37, 38). Indeed, therapies that enhance the expansion or recruitment of migratory cDC1s to tumors and induce IFN-Is are more effective at promoting tumor clearance than expanding DCs alone, but only combinatorial anti-PD-L1/PD-1 provides complete control (31, 37, 38). These studies suggest that APCs are an important target of anti-PD-L1 therapy, with reports indicating that PD-L1 expression on APCs is needed for optimal PD-L1 blockade (39, 40). As discussed in the next section, the upregulation of PD-L1 (and potentially other negative regulatory factors) can be leveraged as an adjuvant to promote better responses to immunotherapy by maintaining IFN-I-induced activation while abrogating the reciprocal feedback inhibition.
CD8+ T Cells
T cells are central mediators of antitumor immunity and tumor elimination, and even in situations that augment other aspects of the immune response leading to cancer control, with rare exception, CD8+ T cells are the end point effectors of tumor control. Following activation by IFN-I-activated DCs (18, 19), cytolytic CD8+ T cells (CTLs), with the help of CD4+ T cells, acquire the potent killing capacity for tumor elimination. Many studies of viral infections and cancer have demonstrated the fundamental role of direct IFN-I signaling for the activation, proliferation, differentiation, and survival of antigen-activated CD8+ T cells (41). IFN-Is also directly enhance CD8+ T cell receptor sensitivity to cognate antigen (42–45) and, upon T cell activation, facilitate effector function by directly stimulating granzyme B (GzmB) and other effector molecules, leading to enhanced killing of tumor cells (43, 46, 47). Indeed, IFN-I promotes the formation of the GzmB+ effector CD8+ T cell subset in chronic viral infection (47, 48). Conversely, IFN-Is limit the development of the newly identified TCF1+ CXCR5+ memory-like CD8+ T cell subset that exhibits enhanced proliferative and renewal capacity to sustain long-term effector CD8+ T cell activity during chronic viral infections and potentially cancer, and that preferentially respond to anti-PD-L1 therapy (48–54). Yet the role of IFN-Is in driving TCF1+ memory-like CD8+ T cell differentiation into GzmB+ effector cells in cancer has not been well established. A study in 2020 indicated that in chronic viral infection, TCF1+ CD8+ T cells are maintained in lymph nodes by PD-1, which creates a protective niche for these cells to continually regenerate the ongoing CD8+ T cell response (55), suggesting that the lymph node may also be a protective site for these cells in patients with cancer. This could explain the differences in data identifying these cells in the TME, wherein they are retained in the lymph node (protected from IFN-I signaling) and may only exist in the TME in the presence of tertiary lymph node development. IFN-I signaling has been implicated in regulating stemness in other cell types (56, 57), and the maintenance of memory TCF1+ CD8+ T cells with functional potential in human lymph nodes was reliant on protection from IFN-I signaling (58). Thus, it will ultimately be interesting to determine whether IFN-Is are important in regulating differentiation of these TCF1+ cells in cancer and how these cells are protected from IFN-I signaling in environments rich with chronic IFN-I stimulation.
In addition to driving CD8+ T cell subset skewing and effector function, IFN-I signaling promotes the survival of antigen-specific CTLs. This is mediated primarily by increasing Bcl-XL expression and IL-2-mediated protection from cell death (41). Although IFN-Is have critical survival effects on antigen-specific T cells, they simultaneously lead to the depletion of bystander T cells (59). These differential survival effects occur in part because antigen-activated T cells also engage signaling pathways involving STAT4 and STAT3 downstream of IFN-Is, while naive T cells engage only STAT1 (46, 60). The depletion of bystander naive and memory T cell pools in response to IFN-Is allows for expansion of newly antigen-activated CD8+ T cells (59, 61) but may also affect responses to cancer therapy by reducing T cell clonal diversity. Indeed, in melanoma, sarcoma, and cervical cancers, T cell receptor diversity is decreased with increasing disease severity and is associated with resistance to anti-PD-1 immunotherapy (62, 63), potentially through IFN-I signaling.
Tumors evolve various mechanisms, including modulation of IFN-I signaling, to prevent CD8+ T cells from recognizing and eliminating tumor cells. In fact, spontaneous downregulation of IFNAR on CD8+ T cells infiltrating colon cancer and other tumor models contributes to aggressive cancer growth (41, 46). This early loss of IFN-I signaling on CTLs may push IFN-I signaling toward other immune cells such as regulatory T cells, where the effect of IFN-I signaling can be tumor protective (64, 65). In combination with T cell receptors (signal 1) and costimulation (e.g., CD28, signal 2), IFN-Is function as signal 3 in many situations to activate naive T cells to rapidly acquire effector function (44). Thus, the loss of IFNAR expression on these cells could subsequently affect the recognition of neoantigens that arise as cancer progresses. Further, whereas IFN-Is indirectly facilitate CD8+ T cell activation through enhanced DC antigen presentation (18), cancer cells and tumor-associated APCs respond to sustained IFN-I signaling by upregulating PD-L1 and other immunosuppressive molecules (13, 31, 33, 35, 66) to directly limit CTL functions. The mechanisms leading to loss of IFNAR expression on CD8+ T cells are not clear, but ultimately understanding how this loss occurs and overcoming it could lead to enhanced CD8+ T cell functions and tumor control.
Yet, sustained IFN-I signaling also leads to direct and, perhaps more important for the CTL response, indirect deleterious consequences (Figure 2a). The combination of (a) chronic antigen stimulation and immune activation, (b) increased inhibitory receptor signaling, and (c) interactions with other suppressive cells in the TME attenuates CD8+ T cell functions (termed T cell exhaustion), which is characterized by reduced proliferative potential, altered cytokine production and decreased cytotoxicity (67). Overcoming T cell exhaustion is one of the most important strategies for cancer immunotherapy. Paradoxically, although IFN-Is are largely indispensable for initial activation and potent effector function in response to virus or cancer, they also contribute to diminished CD8+ T cell function (11, 14). Therefore, both loss of IFN-I signaling and excessive IFN-I signaling on CD8+ T cells can lead to dysfunctional responses and promote immune evasion. This idea was first demonstrated in the lymphocytic choriomeningitis virus model of chronic infection, wherein IFN-I administration during the early phase of infection leads to improved viral control (68), whereas later in infection, IFN-I contributes to viral persistence by promoting chronic immune activation and T cell exhaustion (11, 14). Thus, in ways that have yet to be mechanistically understood, IFN-Is can positively and negatively regulate cancer pathogenesis and responsiveness to therapy by directly and indirectly modulating CD8+ T cell function and differentiation (13, 31, 33, 35, 59, 61, 66).
CD4+ T Cells
CD4+ T cells orchestrate overall immune direction at the onset of an immune response and license APCs to promote their ability to stimulate the immune response. CD4+ T cells then provide help in the form of cytokines and direct interactions that activate and sustain CD8+ T cells and B cells in infections and likely in cancer (although not as well studied) to control cancer progression. Following priming, CD4+ T cells can differentiate into multiple subsets depending on the signals they receive, and in viral infections they differentiate primarily into T helper 1 (Th1) cells to help CD8+ T cells or into T follicular helper (Tfh) cells to help B cell differentiation and class switching. IFN-Is shape CD4+ T cell differentiation, survival, and expansion, regulating the balance of helper subtypes and thereby fundamentally directing global immune responses (69). Inappropriate biasing of T cell helper subtypes, such as the Th2/Th1 imbalance in cancer or Tfh/Th1 in chronic infection, is altered by IFN-Is, contributing to chronic disease (70–73). The mechanisms of IFN-I-mediated influence on T cell help in infection have been well studied (6), whereas less is known about this regulation in cancer. In addition to influencing other cells, CD4+ T cells themselves can have cytolytic activity. In the past decade, growing evidence indicates these CD4+ CTLs control tumors in response to immunotherapy or Treg depletion (74–77) and, in the case of β2m-deficient Hodgkin lymphoma, are a primary cell type leading to control (78). The role of IFN-Is in the differentiation of these cells is less well known, but CD4+ CTLs are hypothesized to be a highly differentiated form of Th1 cells, and IFN-I signaling is critical for Th1 differentiation (7).
The timing of IFN-Is is critical for their effects on help from CD4+ T cells. During priming, IFN-Is can serve to program and reinforce Th1 lineage commitment (72) and can prevent subsequent conversion of Th1 cells to other subsets (e.g., Th17 cells) by inducing epigenetic modifications that prevent reprogramming when exposed to a changing cytokine milieu (79). Conversely, at later times, IFN-Is can prevent CD4+ Th1 differentiation (72), through the promotion of suppressive APCs expressing PD-L1 and IL-10 (13, 72, 73), and may elicit a similar outcome in priming of neoantigen-specific responses that arise with cancer progression.
Regulatory T Cells
A dominant mechanism of immunosuppression in the TME is the recruitment of regulatory T cells (Tregs), which dampen most if not all cells of the immune system. Because of their central role in driving immune evasion and their prognostic value in the clinic (80), Tregs are well studied in the context of cancer. The role of IFN-Is in directly modulating Treg function, homeostasis, and recruitment to tumors is also well established (81), but how IFN-I signaling by Tregs differentially affects subsequent tumor immunity is less well defined and more controversial.
The early events of tumorigenesis are unclear and difficult to study. However, it is possible that under normal physiological conditions the inflammation generated in the initial events of tumorigenesis and tumor growth is insufficient to mount a robust IFN-I response. Although model systems are critical to our understanding of the mechanisms of tumorigenesis and immunity, these earliest processes may not be mimicked completely accurately in adoptive transfer of tumor cells into mice (as the needle stick and injection likely induce inflammation themselves) or in spontaneous mouse models wherein large numbers of cells are simultaneously transformed by promoterdriven gene activation or deletion. IFN-Is typically dampen the expression of Treg chemoattractants like CCL17 and Treg activation, allowing induction of antitumor immunity (65, 82, 83). In the absence of IFN-I-driven regulation, Treg infiltration into tumors facilitates the generation of an immunosuppressive microenvironment (36). Indeed, reduced Treg frequency as a result of IFN-I treatment in the TME is accompanied by an increase in CD8+ T cell infiltration and enhanced tumor control (82). As tumors progress, the increased tumor burden and genomic instability would lead to the activation of IFN-Is through damage sensing such as the cGAS-STING pathway (84–86), but by the time this occurs, the TME is likely highly immunosuppressive, with IFN-Is likely contributing to the maintenance of this environment (35, 87). PD-L1 and IDO expressed by myeloid and tumor cells further enhance the ability of Tregs to restrain T cell function (36, 88), and IFN-Is drive IL-10 production by Tregs uniquely in the TME, potentiating their suppressive ability, highlighting that the TME can modulate how Tregs interpret local IFN-I signals (65).
Natural Killer Cells
Natural killer (NK) cells are cytotoxic innate lymphoid cells able to eliminate tumors that evade killing by CTLs by downregulating MHCI expression. IFN-Is maintain NK cell numbers and induce their cytotoxicity, contributing to their immune surveillance. Tumors that are normally rejected in an NK-dependent manner grow progressively in mice with specific deletion of IFNAR in NK cells (89), highlighting the important role of IFN-Is in promoting NK cell function. The initial rejection of tumors in this context seems to depend on IFN-I signaling to immature NK cells because specific deletion of IFNAR in mature cells expressing the activation receptor NKp46 still enables tumor rejection, despite reduced cytotoxicity (90). Therefore, a lack of IFN-Is prior to NK maturation would enable tumors to bypass immune surveillance. Indeed, IFN-Is produced by chronic lymphocytic choriomeningitis virus infection delayed tumor growth and prevented metastasis through enhanced NK cell cytotoxicity in infected mice compared with uninfected mice (91), indicating that systemic IFN-I-induced inflammation at the onset of tumorigenesis is beneficial for NK-mediated immune surveillance. Although mature NK cells did not require IFN-Is for immune surveillance (90), they did require IFN-Is to delay lung metastases in a model of spontaneous breast cancer (92). In addition to promoting NK cell cytotoxicity, IFN-Is simultaneously signal to T cells, which inhibits expression of the ligands for NKp46, protecting them from NK cell lysis (93); thus, IFN-Is work at multiple levels to promote tumor killing. In tumors such as colorectal cancer, where IFNAR is spontaneously silenced on CTLs in the TME (41), loss of IFN-I signaling would negatively affect T cell function as described in the previous sections on T cells and may concurrently lead to NK cells erroneously targeting those T cells that fail to downregulate NKp46 ligands.
Immune surveillance: the continuous elimination of neoplastic cells by the immune system
GENOME INSTABILITY, RESISTING CELL DEATH, AND REPLICATIVE IMMORTALITY
Cancer cells are highly adaptable and can survive through the successive accumulation of opportunistic genomic mutations, a process termed genomic instability. Many of these mutations are present in preneoplastic lesions (e.g., lung, breast, colon, and ovarian cancers) and are associated with increased DNA damage from inefficient DNA repair, owing in part to dysfunction or mutation of DNA damage repair components (e.g., BRCA1/BRCA2) or to regulators of cell cycle, growth, and differentiation (e.g., p53 and BRAF) (94, 95). In contrast to their tumor-driving effects, accumulated mutations can also yield neoantigens that can serve as CD8+ T cell targets and are associated with positive responses to immunotherapy (96). Thus, genomic instability is viewed as a characteristic that enables cancer development and growth but also provides new immune targets and immunotherapeutic opportunities (Figure 1).
DNA damage (leading to replication stress) associated with tumorigenesis or induced by genotoxic therapies can drive STAT1 activation and ISG expression (97). The earliest evidence for this emerged from experiments in which repeated treatment with ionizing radiation led to radioresistance, STAT1 overexpression, and a gene expression signature similar to that observed following IFN-I treatment (97). The molecular basis for this DNA damage-induced IFN-I response was gleaned both from ionizing radiation treatment and from cells with impaired ribonucleotide excision repair, wherein imperfect cell cycle checkpoint regulation allowed cells with DNA damage to progress into mitosis, resulting in DNA exposure to the cytoplasm (84–86). Cytoplasmic DNA is recognized by the viral pattern recognition receptor cGAS to initiate signaling through its cognate partner STING, resulting in STAT1 activation and ISG expression, which in theory should lead to enhanced immune activity and tumor killing (Figure 2b). Thus, a paradox emerges when exploring the contribution of cGAS-STING during early tumorigenesis. On the one hand, selective pressure must exist in a fraction of tumor cells to ablate cGAS-STING signaling and, consequently, its immune-promoting aspects. In some tumors, viral oncogenes (e.g., E7 from papillomavirus and E1A from adenovirus) can inactivate STING, and in a fraction of colon, melanoma, and ovarian cancers, cGAS or STING is epigenetically silenced, possibly through mitochondrial dysfunction (26, 98–101). This implies that cGAS-STING signaling, and likely the subsequent ISG induction, is a barrier to oncogenic transformation that must be overcome by the emerging tumor. On the other hand, cGAS-STING signaling is intact in various tumor types and can drive metastatic progression (102, 103), although exactly how is unclear. Ultimately, the role of cytoplasmic DNA sensing, and potentially IFN-I signaling, in tumorigenesis is likely dictated by the order in which the hallmarks of cancer arise and the tissues in which they do so (104).
Replication stress: activation of the DNA damage response by genomic stressors and oncogenes, which can exacerbate genomic instability
The above data suggest that selective pressure in tumorigenesis promotes loss of cGAS-STING signaling (and potentially other IFN-I-associated responses—see below). Whether this is directly attributable to pressure to ablate IFN-I signaling pathways driven by cGAS-STING is currently unknown. Alternative pattern recognition, for example, of double-stranded RNA via RIG-I/MDA5 that converge on similar ISG expression profiles, may remain intact in these cells, as was the case in ovarian cancer cell lines (26). Although the above discussion focuses on tumor-intrinsic signaling (or loss thereof) through cGAS-STING, normal tissues such as fibroblasts, endothelial cells, and immune cells within the TME may effectively maintain these signaling pathways. Indeed, the first evidence that cGAS-STING signaling (via IFN-I and IFNAR1) contributed to radiation-driven tumor regression emerged from the observation that STING-deficient mice did not exhibit tumor control in response to radiation therapy, and a similar observation was made for spontaneous tumor rejection (25, 105). Collectively, these studies traced radiation-induced tumor regression to cGAS-mediated sensing of double-stranded DNA and IFN-I production by DCs and the subsequent modulation of T cell activation. The connections between genomic instability, DNA damage-induced IFN-I signaling, and paracrine cytokine signaling in the TME pose a new opportunity for holistic understanding of IFN-I in early tumorigenesis. Disruption of the balance between protumorigenic and antitumorigenic influences in the TME is observed in many ways, including through inactivation of IFN-I-mediated antitumor effects (e.g., by epigenetic silencing of STING or STAT1) and through IFN-I-driven immunosurveillance and immunosuppression (10, 106). These IFN-I-driven outcomes are likely to be highly case and context specific, with links to the unique milieu of cytokines produced in nascent tumors, the mode of genomic stability dysregulation, and the order in which other tumor-suppressive mechanisms (such as senescence and apoptosis) are lost.
Perhaps the most characteristic features of cancer are resisting cell death and uncontrolled proliferation. Such unconstrained growth arises when a cell attains autonomy from typical growth signals, maintains intrinsic molecular profiles that stimulate proliferation, and evades death. Death evasion can result from mechanisms that downregulate death pathways (e.g., apoptosis) or by avoiding senescence, which would drive replicative death. As discussed above, these features ultimately arise through genomic instability. Induction of oncogenes, such as BrafV600E, can drive DNA damage in melanocytes via the replicative stress it puts on the cell, leading to cellular senescence and activation of a senescence-associated secretory phenotype (SASP), characterized by secretion of inflammatory and immune modulatory cytokines (20, 107). Loss of IFNAR1 in this BrafV600E model of SASP led to accelerated tumorigenesis, linking direct IFN-I signaling to tumor suppression (20). This also links the inherent driver mutations that enable tumor development and growth to direct alterations in the TME inflammatory state (Figures 1 and 2c,d). Another example of how unchecked proliferation can drive IFN-Is is provided by p53. Activation of p53 drives cell cycle checkpoints and cell death in response to ongoing genomic instability. TP53 transcription can be induced by and also subsequently indirectly induce IFN-I expression (108–110), intimately linking TP53 with IFN-I activation. However this mutual regulation is overcome in approximately 50% of tumors through TP53 mutation, loss, or inactivation by viral oncogenes (e.g., E6 from papillomavirus), which results in a significant impact on IFN-I responses and how they shape the TME (108, 110, 111). In addition to senescence and apoptosis, p53 contributes to cell cycle checkpoints after DNA damage (112); as a result, loss of p53-mediated checkpoints may increase the mitotic fraction and associated cGAS-STING signaling that further drives IFN (Figure 2c). Compounded with these driver mutations, loss-of-function mutations of IFN-Is and other IFN-I signaling components occur in multiple cancers (113, 114), potentially providing the combinatorial hit that enables unchecked tumor proliferation while eluding immune killing.
Accumulation of cytoplasmic DNA from excess damage activates cGAS-STING signaling. This leads to IFN-I production, which is required for senescence maintenance in fibroblasts and murine melanoma cells (115–117). In this context, mitochondria-to-nucleus retrograde signaling positively regulates the SASP, whereas DNases such as TREX1 degrade cytoplasmic DNA and antagonize cGAS-STING signaling during both senescence and radiation-induced DNA damage (118–120). SASP arises in cells induced to undergo senescence by a variety of stimuli (e.g., oncogenes, replicative senescence, and DNA damage) and is maintained long-term by IFN-Is (121). SASP involves the production of dozens of cytokines (e.g., IL-6) often considered to be protumorigenic (107). Thus, IFN-I-associated senescence acts as both a tumor-suppressive mechanism by limiting tumor growth (20) and a promoter of tumorigenesis by maintaining their senescent state, thereby enabling better immune escape, driving resistance to therapy (17), and promoting protumorigenic cytokines (107). These dichotomous implications of senescence probably reflect the precise milieu of the cytokines produced and the microenvironment of nascent tumors (Figure 2d). A unifying model for senescence in tumorigenesis has yet to emerge, but the connection between DNA damage and senescence and the associated SASP implicates IFN-Is in the initiation, progression, and treatment of cancer.
Cancer cell dormancy can be interpreted as a state of equilibrium in which host immune cells keep tumor cells in check (122). The balance can be tipped toward elimination or escape depending on the circumstances; for example, when T cells are depleted in MCA-induced sarcomas, tumors that had reached equilibrium begin to grow uncontrollably (122). The consequence of cancer cell dormancy is that it can go undetected for years, as evidenced by case studies in which secondary melanoma is diagnosed in kidney transplant recipients with no primary tumor (123). In hematopoietic stem cells (HSCs), acute exposure to IFN-Is can trigger escape from dormancy, inducing rapid proliferation and differentiation while rendering HSCs sensitive to chemotherapeutic agents targeting proliferating cells (56). However, repeated or chronic exposure to IFN-Is then leads to a significant decline in the ability of HSCs to self-renew, ultimately exhausting their ability to proliferate and give rise to new progenitors and potentially rendering them refractory to chemotherapy once again. There is evidence for such IFN-I-induced dormancy in cancer; for example in breast cancer, IRF7 expression is associated with delayed growth, metastasis, and resistance to chemotherapeutic drugs (17). Although this state of dormancy is antitumor, because tumor growth is delayed (17, 122), it could also provide a mechanism by which IFN-Is drive resistance to chemotherapies that target proliferating cells (17, 56).
TYPE I INTERFERON–DRIVEN TUMOR-PROMOTING INFLAMMATION
Tumor cells not only acquire the ability to evade immune destruction but also can harness cells of the immune system to actively support their growth and survival. Tumors take advantage of the feedback protective processes reciprocally induced by IFN-Is meant to limit host tissue damage in the face of chronic inflammation (35, 36, 87). As such, IFN-Is provide proinflammatory mediators needed for tumor progression while upregulating negative regulatory cells and factors that promote immune evasion (35, 124, 125), thus skewing immune responses at every level: danger sensing, transcription, costimulation, and cytokine production (13, 35, 124–127). Despite their antiproliferative function, IFN-Is can drive aggressive tumor growth in part through promoting survival and stemness of cancer stem cells (CSCs) (57, 128, 129). IFN-Is also promote expression of IL-6, a proinflammatory cytokine that contributes to the establishment of a tumor-permissive environment (130, 131). To secondarily promote tumor growth via immune obstruction, ISGs function via many mechanisms to allow cancer escape. For example, 2′−5′-oligoadenylate synthetase-like 1 and 2 (OASL1/2) and suppressor of cytokine signaling 1 (SOCS1) inhibit IFN-I signaling at the cellular level and prevent subsequent proinflammatory effects of IFN-I, and factors such as PD-L1, IDO, and IL-10 act more broadly and prevent immune killing of tumor cells (13, 35, 124–127). The details of how expression of these negative regulatory ISGs is induced during the early events of tumorigenesis are still not entirely clear. Certain cancers are strongly associated with prior viral/bacterial infections, which may not only directly promote mutagenesis and transformation but also drive the inflammation (including IFN-Is) (reviewed in 132). Thus, many tumorintrinsic and tumor-extrinsic mechanisms can induce IFN-I expression at the earliest stages of tumor development, setting the path for subsequent immune initiation and suppression.
Cancer stem cell (CSC): self-renewing cell that gives rise to tumor cells and seeds new tissues in metastasis; also called tumor-initiating cell
In the case of infection-driven cancers, the early events that modulate IFN-Is are better understood. In cervical cancer, human papilloma virus proteins E6 and E7 interact with IRFs and ISGs to attenuate IFN-I signaling and inhibit the constitutive IFN-I production that facilitates sensitivity to immune control (133, 134). Other tumor-promoting viral infections, such as Epstein-Barr virus, can skew IFN-I responses in tumors toward the production of PD-L1 and IDO (135), further supporting the above model whereby control of early IFN-I signaling differentially modulates tumorigenesis (Figure 2e).
Tumors themselves often adapt the ability to negatively regulate IFN-Is rather than support IFN-I production (20, 26, 41, 98–101), although temporally when these tumors develop resistance to IFN-Is and how this resistance affects different aspects of tumor and immune interactions are unclear. Despite this inactivation, other cells present in the TME (i.e., immune cells and fibroblasts) and adjacent tissue can still produce and respond to IFN-Is. That said, the role of IFN-Is in establishing the initial neoplasm is not well known and the early events in tumorigenesis remain elusive. Many studies attribute the initiation of negative regulatory ISGs largely through IFN-γ in response to infiltration of activated T cells (34, 36, 136). The induction of anti-inflammatory mediators by IFN-γ (and IFN-I) has a detrimental effect on effector T cell survival, proliferation, infiltration, and function in the TME, essentially depleting sources of IFN-γ, with perhaps less of an effect on IFN-I (13, 31, 33, 35, 66). Therefore, it is likely that IFN-Is help maintain inflammation and immunosuppressive factors once immune editing by tumors leads to widespread changes in the composition of the TME, potentially resembling the low but consistent IFN-I signaling that occurs in chronic infection and that simultaneously sustains immune cell survival and potentiates immune cell dysfunction (11, 14). Indeed, the expression of PD-L1 on myeloid-derived suppressive cells (MDSCs) is maintained by IFN-Is in the absence of IFN-γ and potentiates their suppressive function, leading to poor prognosis in cancer (137, 138), demonstrating along with findings from other studies that IFN-Is can compensate for decreased IFN-γ (36, 136). Concurrently, SOCS1 expressed by DCs and macrophages inhibits their ability to mount efficient antitumor immune responses, which is critical to promote inflammation-induced colon cancer progression (139, 140). By contrast, SOCS1 deficiency in mice leads to spontaneous colorectal carcinoma, driven by colitis and excessive inflammation from IFN-γ (141), suggesting that SOCS1 expression in normal tissue precedes tumorigenesis and may initially keep tumors at bay. In fact, SOCS1 prolongs the activity of the tumor suppressor p53 (125, 142), further supporting the notion that the effect of SOCS1 depends on the stage of tumor development. Taken together, these studies highlight an interesting temporal coordination between IFN-Is and IFN-γ, potentially suggesting that the outcome of IFN-I signaling depends on whether IFN-γ or IFN-Is signal first in the TME, with IFN-Is initially delaying tumor growth but later exacerbating immunosuppression driven by T cell–induced inflammation. Out-of-sequence signaling could trigger immunosuppression in order to protect cells from inappropriately activated T cells, a response that tumor cells could then take advantage of to prevent their elimination.
TYPE I INTERFERON–REGULATED ENERGETICS AND METABOLISM IN THE TUMOR MICROENVIRONMENT: PATHWAYS, TARGETS, AND OUTCOMES
Metabolic Reprogramming in Tumor Cells
To support the energetic demands of rapid and sustained proliferation, cancer cells undergo several metabolic adaptations. Perhaps the most well-known adaptation is a shift toward glycolytic metabolism, even in the presence of sufficient oxygen (Warburg metabolism) (143). Warburg metabolism occurs in part through hypoxia inducible factor-1α (HIF-1α), activated in oxygenlimited conditions to upregulate genes involved in glycolysis (143). HIF-1α is also activated by inflammatory signaling molecules such as IFN-Is to stimulate genes that help cells survive under pathogenic conditions in which nutrients may be limiting (144). IFN-I-activated HIF-1α in the TME triggers epithelial-to-mesenchymal transition (EMT), suggesting that IFN-I may mediate the tumor-intrinsic metabolic dysregulation that potentiates tumorigenesis (145). In the same study, IFN-I-driven HIF-1α occurred through activation of mammalian target of rapamycin complex 1 (mTORC1), a master inducer of anabolic cellular processes such as mRNA translation and induction of glycolysis (145, 146) (Figure 2f). The details of the widespread influence of mTORC1 on metabolism are beyond the scope of this review (see 147); however, the result of mTORC1 activation is the promotion of cell growth and survival and the inhibition of catabolic processes such as autophagy.
Glycolytic metabolism: anaerobic pathway that converts glucose to lactate, generating two ATP molecules
As tumors progress, selective pressure favors cells able to survive the harsh nutrient conditions in the TME, and tumors become increasingly dependent on mTORC1 activity to sustain glycolysis (148). IFN-Is can drive mTOR and potentially glycolysis; however, whether IFN-Is play a role in maintaining mTORC1 activity in this context is unclear, because IFN-Is also induce autophagy by inhibiting mTORC1 activity, allowing for the breakdown of cellular components that are used to feed various energetic pathways, including fueling protein synthesis, lipid biogenesis, and nucleotide biosynthesis (149–151). Autophagy can then help cancer cells survive by reciprocally counteracting IFN-I-stimulated proapoptotic functions (149–152), with inhibition of autophagy restoring IFN-I-induced toxicity and slowing tumor progression (149, 153). It is unclear exactly how IFN-Is inactivate mTORC1 (although it is clearly complex); however, this potentially occurs through initiation of ISGs that inhibit mTORC1 in a negative feedback loop, although specifically which ISGs are responsible is not yet clear. In support of this, exposure of cells to IFN-I transiently enhances mTORC1 and subsequently diminishes mTORC1 activity once these ISGs are expressed (149).
Autophagy: the catabolic process of recycling internal organelles to generate macromolecules for use in different energetic and metabolic pathways
Metabolic Reprogramming of Immune Cells in the Tumor Microenvironment
Several of the same metabolic adaptations first discovered in cancer cells, including Warburg metabolism, were also found to be adaptations that occur in T cells undergoing clonal expansion (147). Glycolysis induction in T cells is thought to be required to support the anabolic processes needed for adequate T cell activation and effector function. Indeed, glucose deprivation is detrimental to the capacity of CD8+ T cells to produce IFN-γ (154). Therefore, nutrient availability is likely an important way that cancer cells influence T cell activity. The role of IFN-Is in influencing T cell metabolism is understudied; however, one study suggests that IFN-I improves mitochondrial fitness in memory CD8+ T cells (155). Given that HIF-1α activation can drive function and migration of CTLs (156), and the importance of IFN-I in governing T cell activation and function particularly in viral infection and cancer, more work is needed to understand how IFN-Is directly contribute to T cell metabolic reprogramming. IFN-I-driven metabolic reprogramming is better studied in other immune cells such as APCs, indirectly impacting T cell priming and function.
Like many of their outcomes, IFN-Is differentially impact immune cell metabolism depending on cell type. A prime example of this is the simultaneous stimulation of fatty acid oxidation in pDCs and glycolysis in cDCs by IFN-Is, both programs proving necessary for the unique functions of each cell type (155, 157). In macrophages, IFN-Is inhibit cholesterol biosynthesis in a STING-dependent manner, promoting resistance to viral infection (158), in contrast to pDCs, which increase fatty acid synthesis downstream of IFN-I-inducing TLRs (155). That IFN-Is have such diverse effects on lipid metabolism suggests that it is not necessarily agnostic to these metabolic pathways but rather promotes the process most beneficial to the cell at the time. In support of this, stimulation of glycolysis in cDCs is needed to expand the cell membrane and intracellular compartments necessary for the synthesis of inflammatory mediators after TLR stimulation (159), and fatty acid oxidation in pDCs increases their mitochondrial fitness, making them resistant to IFN-I-induced apoptosis (155). By controlling IFN-I responses, cancer cells can create an unsuitable metabolic and nutrient environment that exacerbates disease. This is evidenced by tumor-intrinsic IDO overexpression discussed below (160) and lipid accumulation that occurs uniquely in DCs from patients with cancer, reducing their ability to prime T cell responses by interfering with antigen presentation (161). How a specific cell responds metabolically to IFN-I potentially occurs through collaboration with preexisting signaling/metabolic programs and distinct cytokine signals in a given cell. Owing to the importance of metabolic modulation to control cancer, further understanding of these metabolic fate decisions is critical to potentially design cell-directed metabolic reprogramming.
In early cancer research, altered tryptophan metabolism in cancer cells was attributed to IDO overexpression in tumors caused by IFN-Is and IFN-γ (162, 163). The increased IDO production by colorectal cancer cells correlated with reduced tumor infiltration (160). In addition to tumorintrinsic IDO production, IFN-Is stimulate production of IDO by APCs in chronic infections and cancer, amplifying their immunosuppressive function and further inhibiting T cell activity (13, 162, 164). IDO additionally potentiates the function of anti-inflammatory immune cells such as Tregs through the by-product of tryptophan breakdown, kynurenine (165). However, a 2019 phase III clinical trial did not reveal enhanced efficacy when an IDO1 inhibitor was combined with anti-PD-1 to treat metastatic melanoma, although there were concerns with appropriate dosing for complete inhibition (166). In combination with the immune restoration induced by PD-1 blockade, IDO inhibition may not be sufficiently expressed, may enhance immune functions similar to those of PD-1, or may not target the correct cells needed for immune restoration to overcome an advanced cancer. However, in combination with genotoxic agents that themselves kill cancer cells, such an approach may be effective. Radiation therapy can induce IFN-I production with the reciprocal upregulation of negative inhibitory factors (33, 34, 66). Consistent with this idea, preclinical studies this year suggest that IDO upregulation after radiation therapy is associated with poor outcome in patients with lung cancer (167), and IDO inhibition overcomes therapeutic resistance to radiation therapy in a mouse model of lung cancer (168). These studies suggest that secondarily targeting IDO (perhaps in conjunction with other reciprocal negative regulators induced by radiation therapy or IFN-I) could be an effective strategy to further enhance therapies that induce IFN-Is.
ANGIOGENESIS AND TYPE I INTERFERONS
Tumor cells stimulate de novo blood vessel formation, providing oxygen and nutrients critical for sustaining their rapid proliferation. IFN-Is interfere with angiogenesis in three ways: They act directly on vascular endothelial cells to inhibit their proliferation and function (169), they promote the infiltration and function of immune cells that remodel the tumor vasculature to benefit host immunity (170, 171), and they inhibit the function of immune and tumor cells that stimulate angiogenesis (172, 173).
Cancer cells and tumor-associated macrophages (TAMs) stimulate angiogenesis through production of vascular endothelial growth factor (VEGF) (172, 173), and IFN-Is downregulate VEGF, potently inhibiting endothelial cell proliferation needed to line the developing vessel walls (169, 174). Regression of tumor vessels essentially starves tumors, depriving them of nutrients crucial to their expansion. The role of IFN-I-mediated regulation of VEGF signaling is important, given that VEGF expressed by solid tumors has been associated with poor prognosis in melanoma, breast, colorectal, and other cancers (172). Additionally, negative feedback exists where VEGF signaling triggers downregulation of IFNAR, allowing for angiogenesis (175) and potentially representing a mechanism whereby tumor cells become resistant to IFN-Is, thus exacerbating disease. Whether the association of VEGF overexpression with negative outcome is due to resistance to IFN-I signaling is unknown; however, it would be interesting to understand whether VEGF could serve as an indirect indicator of impaired or dysfunctional responses to IFN-Is. Indeed, tumorbearing mice given anti-VEGF receptor in combination with anti-PD-1 showed delayed tumor growth and the upregulation of IFN-I signatures (176), suggesting that VEGF is a mechanism used by tumors to abrogate IFN-I-mediated tumor inhibition.
When tumor cells stimulate angiogenesis, the new tumor vasculature is often poorly developed. It lacks the structural integrity (such as pericytes) required to regulate permeability, making these vessels inefficient, particularly in allowing the infiltration of T cells (170). Therefore, induction of IFN-I responses to remodel or correct blood vessel architecture resulting from incomplete angiogenesis is a paradoxical strategy to facilitate better T cell infiltration and tumor regression (170). In this situation, STING expressed by endothelial cells can provide IFN-Is to promote this effect, culminating in increased CD8+ T cell infiltration (170, 171) and IFN-γ production to act on the endothelial cells and induce regression of the vasculature (177) (Figure 2g). Further, the recruitment of T cells likely occurs through enhanced production of CXCL10, an ISG that has been attributed to the antiangiogenic capability of IFN-Is in glioblastoma (174). Simultaneously, IFN-Is inhibit TAM-derived VEGF, which would otherwise contribute to angiogenesis by further facilitating the secretion of proangiogenic factors from tumor cells (178, 179). Thus, IFN-Is, through their inhibition of VEGF and their multispectral action on endothelial, tumor, and immune cells, are key mediators of a circuit by which angiogenesis is inhibited.
TISSUE INVASION AND METASTASIS
One of the most devastating outcomes of cancer is metastatic disease. As cancers progress, CSCs extravasate into the bloodstream and invade new tissues, generating secondary tumors and widespread organ dysfunction. Dynamic interactions between IFN-Is and host immune cells help maintain epithelial cell homeostasis, and dysregulation of this process can prompt EMT, a stepwise reprogramming of epithelial cells that enables tumor invasion and metastasis (180, 181). Overexpression of several ISGs in epithelial cells can spontaneously trigger EMT in several cancer types, implicating IFN-Is in this malignant process (128, 182, 183) (Figure 2h). Further, during chronic infection, epithelial cell turnover is accelerated by IFN-I-induced macrophage expression of apolipoprotein L9 (184), with a similar type of enhanced turnover contributing to the onset of malignancy (185). Importantly, under homeostatic conditions, transforming growth factor-β (TGF-β) slows epithelial turnover and inhibits epithelial cell transformation (186), consistent with studies showing that IFN-Is and TGF-β counteract each other (187). Thus, by regulating EMT at multiple levels, IFN-Is generate CSCs for subsequent tissue invasion and metastasis.
IFN-Is also regulate the functions of immune cells that help form a premetastatic niche for invading tumor cells (reviewed in 173). TAMs, neutrophils, and MDSCs can enable extravasation, implantation, and metastasis through the secretion of cytokines and growth factors that create a suitable environment for tumor growth (173) (Figure 2h). IFN-Is inhibit TAM differentiation and recruitment to tumors through interference with M-CSF signaling, thereby indirectly preventing metastasis (178, 179). Some tumor cells can counteract this effect by secreting granulocyte macrophage colony-stimulating factor (GM-CSF) or M-CSF, which then recruits TAMs, MDSCs, and other myeloid cell types into the tumors (173). TAMs then produce VEGF to stimulate angiogenesis needed for extravasation of tumor cells, EGF to condition tumor cells for continued growth and metastasis, and prostaglandin E2, a lipid mediator with anti-inflammatory functions, to limit inflammation in the TME (173). Therefore, by limiting recruitment of myeloid cells to primary tumors, IFN-Is prevent their ability to stimulate metastasis. Meanwhile, neutrophil infiltration into premetastatic lung tissue promotes metastatic lesions (188). IFN-I-driven neutrophil activities such as NETosis can then be detrimental to primary tumor growth but also beneficial to premetastatic niche formation in lung, esophageal, and gastric cancers (54, 173, 189–191). Thus, the IFN-I-driven effects of neutrophils can be protumor or antitumor depending on whether they occur systemically or in the TME. Surprisingly, depletion of pDCs in mouse models of breast cancer and head and neck squamous cell carcinoma also delayed tumor growth and prevented metastasis (192, 193). However, studies from the past two years have discovered that in the TME, pDCs polarized by TGF-β lose their ability to produce IFN-I, instead enhancing Treg activity and promoting tumor growth (194, 195), which is likely the mechanism by which they promote metastasis. This finding highlights how tumors employ multiple strategies to subvert IFN-I-driven antitumor immunity by either inactivating IFN-Is or enhancing recruitment of immune-suppressive cells, instead reinforcing their protumorigenic functions. Meanwhile, several studies discussed above show that IFN-Is themselves can directly drive CSC dormancy, which would restrain secondary tumor growth (56, 196), such that ablation of IFN-Is in the host could enable escape of metastatic cells. Therefore, it is reasonable to propose that rather than simply being protumor or antitumor, IFN-Is regulate a gradient of intermediate effects that depend on the balance of immune cells, tumor cells, and cytokines present both in the TME and systemically and on their stage of cellular differentiation at the time they encounter IFN-I signaling.
NETosis: the extrusion of DNA by neutrophils generating traps that can sequester pathogens or cancer cells
TYPE I INTERFERONS IN CANCER TREATMENT: MOVING FORWARD
The ultimate goal in understanding the mechanisms that drive tumor initiation, growth, metastasis, and immunity is to enhance early detection, better predict outcome, and develop more targeted therapies to eradicate cancers. Currently, many preclinical and clinical studies are aimed at harnessing the positive antitumor effects of IFN-Is and attenuating their negative effects to enhance tumor clearance and prevent metastasis. We have discussed mechanisms of IFN-I-driven resistance to therapy in other parts of this review. In brief, although IFN-Is are crucial for mounting a robust immune response, the immune-suppressive functions of IFN-I signaling, when prolonged, may outweigh their antitumor activity, termed adaptive resistance. Simultaneously, cancer cells can inactivate IFNAR and STING through several different mechanisms, rendering them insensitive to IFN-I and thus limiting IFN-I-driven toxicity and the efficacy of therapies designed to target IFN-Is to them (20, 41, 100). Cancer cells that can respond to IFN-I may enter a state of dormancy that allows them to resist antiproliferative chemotherapies (17, 111). Therefore, understanding when IFN-Is are beneficial and when they drive resistance is essential to better tailor therapies that directly rely on providing IFN-Is (or their induction) and therapies that induce IFN-Is as products of the therapy and may be reciprocally undermined by subsequent IFN-I induction (e.g., radiation therapy).
Adaptive resistance: an initial positive response to therapy followed by reciprocal negative feedback mechanisms that attenuate the therapeutic response
A growing body of literature is beginning to define IFN-related signatures associated with clinical response to and prognosis of multiple different types of cancer and therapies (17, 33, 66, 135, 197–199). Among the first signatures identified was an IFN-related DNA damage resistance pathway that conferred resistance to radiation therapy and chemotherapy (66). In line with the dual role of IFN-Is, in some cases an IFN-I signature, such as IFN-related DNA damage resistance pathway, is indicative of cancer progression and poor response to therapy (33, 34, 66, 200), whereas in other cases it is associated with positive outcome. Currently, there is a push toward reconciling these differences and recent advances in sequencing technology allow single-cell resolution of gene expression, providing exciting new opportunities to further improve our understanding of how IFN-I contributes to response and resistance to therapy. One study tracking peripheral CD8+ T cells in patients with metastatic melanoma discovered a prominent upregulation of IFN-I-related genes in response to anti-PD-1 and anti-cytotoxic T-lymphocyte antigen-4 (CTLA-4) combined compared with anti-PD-1 alone (201). Patients with an ongoing response to combined therapy had a decreased clonal diversity after therapy, with a high number of clones overrepresented in the repertoire (201). Whether the induction of IFN-Is was associated with and causative of the decreased diversity was not interrogated; however, it would be consistent with the selective expansion of antigen-specific CD8+ T cells and bystander cell death in response to IFN-Is (59, 61) discussed above. Another major ongoing effort is to define biomarkers to identify patients who will or will not respond to a given therapy. In the case of radiation therapy, Formenti and colleagues (5) observed that patients who developed systemic responses following radiation therapy and anti-CTLA-4 blockade correlated with induction of IFN-β, suggesting one potential indicator of response is induced IFN-β. How universal this biomarker will be remains to be seen and will rapidly evolve with changes in both immunotherapy regimen and complementary therapy.
In addition to the prognostic value of identifying IFN-I signatures, such analysis is also helping identify new potential therapeutic targets. One example from 2019 includes the identification of the IFN-I-induced RNA sensor ADAR-1 as a target for therapeutic intervention in patients with an existing high ISG signature (198, 199). Chronic ISG expression drives a unique dependence of cancer cells on ADAR-1 for their survival such that depleting ADAR-1 led to better tumor control (198, 199). This result highlights that targeting IFN-I-driven processes that promote cancer cell survival is potentially an effective therapeutic strategy. This type of synthetic lethality has been proposed to selectively target cancer cells without affecting normal, untransformed cells (202). Another emerging target for cancer therapy is the surface protein CD47, which delivers a don’t-eat-me signal to macrophages, protecting cancer cells from macrophage phagocytosis. A 2020 preclinical study showed that gut microbiota enhances responses to anti-CD47 through STING-mediated IFN-I production by DCs, improving CD8+ T cell priming (203). The enhanced IFN-I production may also allow macrophages to bypass CD47 through increased lipid biosynthesis, fueling more efficient phagocytosis (199). These studies highlight that stimulating IFN-I responses alongside therapeutic interventions is an important correlate of response.
Synthetic lethality: deficiency of one gene creates a unique dependency on another gene such that loss of both genes causes cell death
The potential for IFN-Is themselves as cancer-fighting agents was initially met with much excitement due to their crucial role in priming CTL responses. However, as researchers and clinicians began to understand the complicated biology, it became clear that their clinical application would be difficult. In clinical trials IFN-Is seemed to have consistent positive clinical benefit in patients with hematological malignancy such as chronic myeloid leukemia and myeloma (204–206), whereas in solid tumors the effect was mixed, with metastatic melanoma demonstrating reasonable clinical benefit but other cancers such as ovarian and breast showed minimal response and significant toxicity (207–210). Taken together, past studies suggest that IFN-Is are more effective in patients with early tumors, whereas patients with established tumors or metastatic lesions are less responsive and may experience more toxicity. Despite the mixed results of IFN-I therapy, the past few years have seen a revival in IFN-I trials, in part because of the growing body of research highlighting that many therapeutics induce IFN-Is as an important part of their antitumor activity (211, 212). Directly targeting IFN-Is or IFN-I-inducing agents such as poly(I:C) (polyinosinic:polycytidylic acid) to tumors holds clinical promise as a method to overcome the negative side effects of systemic delivery (83, 211, 213). By contrast, other studies propose using JAK inhibitors to attenuate IFN-I and IFN-γ signaling to prevent secondary therapeutic resistance driven by IFNs (33, 34). Many of these trials are still in the early phases; therefore, an assessment of their efficacy remains to be determined.
Incredible promise has also been put forth for new therapies based on immune checkpoint blockade (ICB), with hundreds of trials under way or planned. Importantly, combination therapy with ICB can synergize with IFN-Is by counteracting IFN-I-driven mechanisms of self-regulation (31, 213). Although enhanced posttherapy IFN-Is are generally thought to work by stimulating the ability of DCs to prime T cells (31, 105, 212), a DC vaccine trial in which autologous DCs were pulsed with tumor and adjuvanted with an IFN-I-inducing TLR ligand showed only limited evidence of CD8+ T cell priming in patients (214). One possible explanation could be that the TME is capable of desensitizing cells to IFN-I through downregulating IFNAR1 or signaling components (20, 41, 106). An approach that could overcome a TME where IFN-I responses are blunted through IFNAR1 downregulation is the induction of an infection-like state through oncolytic viruses. The use of a virus that is well controlled by IFN-Is could specifically target tumors where IFNAR1 is inactivated while leaving normal cells unaffected (215). Indeed, cells undergoing transformation progressively lose IFN-I responses, making them increasingly susceptible to oncolytic measles virus (216). In the same study, resistance to infection could be restored through expression of an ISG, IFITM1.
Collectively, these studies demonstrate how different therapeutic strategies can be used to selectively exploit the beneficial effects of IFN-Is or dampen their negative effects. The therapeutic benefit of both induction and inhibition of IFN-Is, depending on the context, highlights the need to identify personalized biomarkers to better predict the ideal therapy on a per-patient basis. It is also unclear whether established IFN-I-induced programs can be overcome or reversed by the addition of more IFN-Is to the system. Studies of chronic hepatitis C virus infection indicate that high preexisting IFN-I signatures are highly associated with treatment failure with IFN-α and ribavirin in the small percentage of patients who do not respond to the new direct-acting antivirals (7). Thus, in the presence of preexisting high IFN-I signaling, adding more IFN-Is to the system may effectively enhance negative ISG induction or selectively enhance the suppressive effects of IFN-Is by the target cells themselves. Ultimately, a deeper mechanistic understanding of IFN-I effects in both immune and nonimmune cells will help delineate the precise conditions under which IFN-Is are beneficial or detrimental to antitumor immunity.
CONCLUSION
IFN-Is are central players in cancer development and antitumor immunity. Because of their ability to signal on both immune and nonimmune cells and because they are stimulated in response to many environmental stressors, IFN-Is contribute to each hallmark of cancer. Early loss of IFN-I signaling in cancer is associated with tumor progression by enabling immune evasion and establishing a TME that is permissive to tumor growth. By contrast, certain ISGs are associated with EMT, tumor invasion, and metastasis. Furthermore, the most well-characterized function of IFN-Is is their regulation of immune responses. IFN-Is are potent stimulators of antitumor immunity but they also fuel aspects of immune cell dysfunction. The effects of IFN-Is are highly context dependent, and the outcome of IFN-Is depends on several factors including cell type, timing, dosage, and duration. The dual role of IFN-Is and the ability of essentially all cells to respond to IFN-I signaling make it difficult to predict when IFN-Is are beneficial to hosts. In cancer, this prediction is further complicated by mutation and epigenetic reprogramming that can modulate sensitivity to IFN-Is and the specific ISGs that are expressed. Treatment efforts centered around targeted delivery and combined with ICB and other therapeutic agents are an exciting new avenue that may lead to improved patient outcomes.
SUMMARY POINTS.
Type I interferons (IFN-Is) are central to mounting an efficient antitumor response. IFN-Is are needed by antigen-presenting cells to prime T cell responses, they directly promote CD8+ T cell function and cytotoxicity, they promote the differentiation of CD4+ Th1 cells, and they enhance natural killer cell cytotoxicity and restrict regulatory T cells. Simultaneously, IFN-Is stimulate the expression of negative regulatory molecules that can attenuate immune responses and promote exhaustion, enabling tumor growth.
Tumor mutational burden leads to the accumulation of cytosolic DNA, activating tumor-intrinsic STING and IFN-I responses. The downside of this signaling is that it drives senescence, slowing tumor growth but simultaneously prompting a senescence-associated secretory phenotype that enables the proliferation of other cancer cells in a paracrine fashion.
Cancers develop multiple ways to evade the antitumor effects of IFN-Is and as a result can use IFN-Is to benefit tumor growth and survival. Different strategies are used to control IFN-I responses; cancers can downregulate IFN-α/β receptor or mutate IFN-Is themselves, impairing signaling. They can alternatively skew IFN-I usage to benefit their own proliferation and survival, favoring genes that promote an immune-restricted tumor microenvironment (TME) and engaging distinct metabolic pathways to facilitate their survival.
IFN-Is affect nutrient balance, energetics, and stress responses in the TME. The cell-type-dependent and temporal changes in metabolism that are driven by IFN-Is suggest that they can serve to reinforce pathways that benefit the needs of the cells.
IFN-Is regulate the proliferation and stemness of cancer stem cells, in most cases inducing a state of dormancy. Although beneficial to the host in the short term, this dormancy along with the induction of catabolic pathways like autophagy helps cancer cells survive under harsh nutrient-limiting conditions and facilitates metastasis.
Metastasis is further regulated by immune cells that participate in premetastatic niche formation both in the TME and at distal metastatic sites. IFN-Is negatively regulate premetastatic niche formation by restricting the protumorigenic activity in the TME, but they promote premetastatic niche formation at secondary sites. Therefore, the effects of IFN-Is are highly dependent on cell type, timing, localization of responding cells, and other factors.
A preexisting high IFN-I signature is indicative of poor responses to radiation therapy and other therapies. By contrast, STING-mediated IFN-Is induced after many different therapies, including radiation therapy, are associated with clinical benefit and enhanced antitumor immunity, although these effects are not necessarily tumor intrinsic.
FUTURE ISSUES.
What is the precise role of tumor-intrinsic IFN-I signaling during the early events of transformation?
Which immune cells are responding to IFN-Is both in the TME and systemically? How does this evolve with cancer therapy? How does this evolve with cancer progression?
How can IFN-Is be effectively redirected toward antitumor immunity therapeutically, particularly without risking autoimmunity that could arise from dampening inherent host-protective feedback loops? Moreover, when designing therapeutic interventions, how can we consider differences in IFN-I function at primary and secondary sites in patients with metastatic disease?
There is a need to identify better ways to predict patient outcome and tailor immunotherapy on a per-patient basis. Although IFN-I signatures can be associated with response, we must better understand the key mechanisms and players that can reconcile the opposing roles of IFN-Is.
Little is understood about how tumor-specific responses are mounted and sustained in the draining lymph nodes and how stem-like T cells are potentially protected from IFN-I-induced differentiation in this niche. Because the lymph node represents a crucial site where immune responses are mounted and likely sustained, understanding how the immune response is regulated within the draining lymph nodes and how they are altered in response to therapies will be critical to understand correlating immunity within the TME.
Are memory-like CD8+ T cells preferentially maintained in the lymph node with tumor progression, and how are they specifically hidden from IFN-I signals in this environment?
ACKNOWLEDGMENTS
The authors would like to thank L. Snell, S. Lukhele, and L.K. Smith for thoughtful discussion of topics covered in this review. The authors are supported by the Canadian Institute of Health Research Foundation grant FDN148386 and the National Institutes of Health grant AI085043. Additional support for G.M.B. comes from the Hold’em for Life Cancer Research Fellowship from the Princess Margaret Cancer Centre. Work in the Harding lab is supported by the Canadian Institutes of Health Research (165926), the V Foundation, the Princess Margaret Cancer Centre, the National Sciences and Engineering Research Council of Canada (RGPIN-2019-04811), the Princess Margaret Cancer Foundation, and the Ontario Ministry of Health.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144(5):646–74 [DOI] [PubMed] [Google Scholar]
- 2.Pitroda SP, Chmura SJ, Weichselbaum RR. 2019. Integration of radiotherapy and immunotherapy for treatment of oligometastases. Lancet Oncol. 20:e434–42 [DOI] [PubMed] [Google Scholar]
- 3.Zappasodi R, Merghoub T, Wolchok JD. 2018. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell 33:581–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topalian SL, Taube JM, Anders RA, Pardoll DM. 2016. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 16:275–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, et al. 2018. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med 24:1845–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lukhele S, Boukhaled GM, Brooks DG. 2019. Type I interferon signaling, regulation and gene stimulation in chronic virus infection. Semin. Immunol 43:101277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snell LM, McGaha TL, Brooks DG. 2017. Type I interferon in chronic virus infection and cancer. Trends Immunol. 38:542–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaitin DA, Roisman LC, Jaks E, Gavutis M, Piehler J, et al. 2006. Inquiring into the differential action of interferons (IFNs): An IFN-α2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-β. Mol. Cell. Biol 26:1888–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Croze E, Russell-Harde D, Wagner TC, Pu H, Pfeffer LM, Perez HD. 1996. The human type I interferon receptor: identification of the interferon β-specific receptor-associated phosphoprotein. J. Biol. Chem 271:33165–68 [DOI] [PubMed] [Google Scholar]
- 10.Dunn GP, Bruce AT, Sheehan KCF, Shankaran V, Uppaluri R, et al. 2005. A critical function for type I interferons in cancer immunoediting. Nat. Immunol 6:722–29 [DOI] [PubMed] [Google Scholar]
- 11.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, et al. 2013. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340:202–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, et al. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–87 [DOI] [PubMed] [Google Scholar]
- 13.Cunningham CR, Champhekar A, Tullius MV, Dillon BJ, Zhen A, et al. 2016. Type I and type II interferon coordinately regulate suppressive dendritic cell fate and function during viral persistence. PLOS Pathog. 12:e1005356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, et al. 2013. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340:207–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhen A, Rezek V, Youn C, Lam B, Chang N, et al. 2017. Targeting type I interferon–mediated activation restores immune function in chronic HIV infection. J. Clin. Investig 127:260–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, et al. 2012. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med 18:1224–31 [DOI] [PubMed] [Google Scholar]
- 17.Lan Q, Peyvandi S, Duffey N, Huang Y-T, Barras D, et al. 2019. Type I interferon/IRF7 axis instigates chemotherapy-induced immunological dormancy in breast cancer. Oncogene 38:2814–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, et al. 2011. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med 208:1989–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, et al. 2011. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J. Exp. Med 208:2005–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katlinskaya YV, Katlinski KV, Yu Q, Ortiz A, Beiting DP, et al. 2016. Suppression of type I interferon signaling overcomes oncogene-induced senescence and mediates melanoma development and progression. Cell Rep. 15:171–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szabo A, Fekete T, Koncz G, Kumar BV, Pazmandi K, et al. 2016. RIG-I inhibits the MAPK-dependent proliferation of BRAF mutant melanoma cells via MKP-1. Cell. Signal 28:335–47 [DOI] [PubMed] [Google Scholar]
- 22.Tas F, Erturk K. 2019. BRAF mutation status might contribute an effect on both disease-free and overall survival in stage III cutaneous melanomas treated with intermediate dose interferon-alpha. Cancer Chemother. Pharmacol 84:521–26 [DOI] [PubMed] [Google Scholar]
- 23.Asselin-Paturel C, Trinchieri G. 2005. Production of type I interferons: plasmacytoid dendritic cells and beyond. J. Exp. Med 202:461–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spranger S, Dai D, Horton B, Gajewski TF. 2017. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31:711–23.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, et al. 2014. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41:830–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Queiroz N, Xia T, Konno H, Barber GN. 2019. Ovarian cancer cells commonly exhibit defective STING signaling which affects sensitivity to viral oncolysis. Mol. Cancer Res 17:974–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schadt L, Sparano C, Schweiger NA, Silina K, Cecconi V, et al. 2019. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep. 29:1236–48.e7 [DOI] [PubMed] [Google Scholar]
- 28.Boukhaled GM, Cordeiro B, Deblois G, Dimitrov V, Bailey SD, et al. 2016. The transcriptional repressor polycomb group factor 6, PCGF6, negatively regulates dendritic cell activation and promotes quiescence. Cell Rep. 16:1829–37 [DOI] [PubMed] [Google Scholar]
- 29.Dissanayake D, Hall H, Berg-Brown N, Elford AR, Hamilton SR, et al. 2011. Nuclear factor-κB1 controls the functional maturation of dendritic cells and prevents the activation of autoreactive T cells. Nat. Med 17:1663–67 [DOI] [PubMed] [Google Scholar]
- 30.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, et al. 2016. Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 30:324–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, et al. 2016. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 44:924–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Speiser DE, Utzschneider DT, Oberle SG, Munz C, Romero P, Zehn D. 2014. T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nat. Rev. Immunol 14:768–74 [DOI] [PubMed] [Google Scholar]
- 33.Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, et al. 2019. Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell 178:933–48.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, et al. 2016. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 167:1540–54.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gato-Cañas M, Zuazo M, Arasanz H, Ibañez-Vea M, Lorenzo L, et al. 2017. PDL1 signals through conserved sequence motifs to overcome interferon-mediated cytotoxicity. Cell Rep. 20:1818–29 [DOI] [PubMed] [Google Scholar]
- 36.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, et al. 2013. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci. Transl. Med 5:200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mistarz A, Komorowski MP, Graczyk MA, Gil M, Jiang A, et al. 2019. Recruitment of intratumoral CD103+ dendritic cells by a CXCR4 antagonist-armed virotherapy enhances antitumor immunity. Mol. Ther. Oncolytics 14:233–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williford J-M, Ishihara J, Ishihara A, Mansurov A, Hosseinchi P, et al. 2019. Recruitment of CD103+ dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Sci. Adv 5:eaay1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin H, Wei S, Hurt EM, Green MD, Zhao L, et al. 2018. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade–mediated tumor regression. J. Clin. Investig 128:805–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang H, Liang Y, Anders RA, Taube JM, Qiu X, et al. 2018. PD-L1 on host cells is essential for PD-L1 blockade–mediated tumor regression. J. Clin. Investig 128:580–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, et al. 2017. Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell 31:194–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, et al. 2006. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J. Immunol 176:4682–89 [DOI] [PubMed] [Google Scholar]
- 43.Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, et al. 2002. Critical role for STAT4 activation by type 1 interferons in the interferon-γ response to viral infection. Science 297:2063–66 [DOI] [PubMed] [Google Scholar]
- 44.Marshall HD, Prince AL, Berg LJ, Welsh RM. 2010. IFN-αβ and self-MHC divert CD8+ T cells into a distinct differentiation pathway characterized by rapid acquisition of effector functions. J. Immunol 185:1419–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richer MJ, Nolz JC, Harty JT. 2013. Pathogen-specific inflammatory milieux tune the antigen sensitivity of CD8+ T cells by enhancing T cell receptor signaling. Immunity 38:140–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu C, Klement JD, Ibrahim ML, Xiao W, Redd PS, et al. 2019. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J. Immunother. Cancer 7:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, et al. 2016. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol 1:eaai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Snell LM, MacLeod BL, Law JC, Osokine I, Elsaesser HJ, et al. 2018. CD8+ T cell priming in established chronic viral infection preferentially directs differentiation of memory-like cells for sustained immunity. Immunity 49:678–94.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, et al. 2016. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537:417–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, et al. 2019. Checkpoint blockade immunotherapy induces dynamic changes in PD-1−CD8+ tumor-infiltrating T cells. Immunity 50:181–94.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, et al. 2019. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20:326–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, et al. 2018. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175:998–1013.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, et al. 2019. Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50:195–211.e10 [DOI] [PubMed] [Google Scholar]
- 54.Wu C-F, Andzinski L, Kasnitz N, Kröger A, Klawonn F, et al. 2015. The lack of type I interferon induces neutrophil-mediated pre-metastatic niche formation in the mouse lung. Int. J. Cancer 137:837–47 [DOI] [PubMed] [Google Scholar]
- 55.Im SJ, Konieczny BT, Hudson WH, Masopust D, Ahmed R. 2020. PD-1+ stemlike CD8 T cells are resident in lymphoid tissues during persistent LCMV infection. PNAS 117:4292–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, et al. 2009. IFNα activates dormant haematopoietic stem cells in vivo. Nature 458:904–8 [DOI] [PubMed] [Google Scholar]
- 57.Qadir AS, Ceppi P, Brockway S, Law C, Mu L, et al. 2017. CD95/Fas increases stemness in cancer cells by inducing a STAT1-dependent type I interferon response. Cell Rep. 18:2373–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miron M, Kumar BV, Meng W, Granot T, Carpenter DJ, et al. 2018. Human lymph nodes maintain TCF-1hi memory T cells with high functional potential and clonal diversity throughout life. J. Immunol 201:2132–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bahl K, Hüebner A, Davis RJ, Welsh RM. 2010. Analysis of apoptosis of memory T cells and dendritic cells during the early stages of viral infection or exposure to Toll-like receptor agonists. J. Virol 84:4866–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gil MP, Ploquin MJ, Watford WT, Lee SH, Kim K, et al. 2012. Regulating type 1 IFN effects in CD8 T cells during viral infections: changing STAT4 and STAT1 expression for function. Blood 120:3718–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oh S-S, Moon C, Kim D-H, Song H, Park S, et al. 2012. Adenovirally delivered IFN-β exerts antitumor effects through transient T-lymphocyte depletion and Ag-specific T-cell proliferation. Int. J. Mol. Med 29:1153–57 [DOI] [PubMed] [Google Scholar]
- 62.Cui J-H, Lin K-R, Yuan S-H, Jin Y-B, Chen X-P, et al. 2018. TCR repertoire as a novel indicator for immune monitoring and prognosis assessment of patients with cervical cancer. Front. Immunol 9:2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, et al. 2014. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kawano Y, Zavidij O, Park J, Moschetta M, Kokubun K, et al. 2018. Blocking IFNAR1 inhibits multiple myeloma-driven Treg expansion and immunosuppression. J. Clin. Investig 128:2487–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stewart CA, Metheny H, Iida N, Smith L, Hanson M, et al. 2013. Interferon-dependent IL-10 production by Tregs limits tumor Th17 inflammation. J. Clin. Investig 123:4859–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DSA, Baker SW, et al. 2008. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. PNAS 105:18490–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wherry EJ, Kurachi M. 2015. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol 15:486–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y, Swiecki M, Cella M, Alber G, Schreiber RD, et al. 2012. Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe 11:631–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crouse J, Kalinke U, Oxenius A. 2015. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol 15:231–42 [DOI] [PubMed] [Google Scholar]
- 70.Huber JP, Ramos HJ, Gill MA, Farrar JD. 2010. Cutting edge: Type I IFN reverses human Th2 commitment and stability by suppressing GATA3. J. Immunol 185:813–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kusuda T, Shigemasa K, Arihiro K, Fujii T, Nagai N, Ohama K. 2005. Relative expression levels of Th1 and Th2 cytokine mRNA are independent prognostic factors in patients with ovarian cancer. Oncol. Rep 13:1153–58 [PubMed] [Google Scholar]
- 72.Osokine I, Snell LM, Cunningham CR, Yamada DH, Wilson EB, et al. 2014. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. PNAS 111:7409–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Snell LM, Osokine I, Yamada DH, De la Fuente JR, Elsaesser HJ, Brooks DG. 2016. Overcoming CD4 Th1 cell fate restrictions to sustain antiviral CD8 T cells and control persistent virus infection. Cell Rep. 16:3286–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kitano S, Tsuji T, Liu C, Hirschhorn-Cymerman D, Kyi C, et al. 2013. Enhancement of tumor-reactive cytotoxic CD4+ T cell responses after ipilimumab treatment in four advanced melanoma patients. Cancer Immunol. Res 1:235–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, et al. 2010. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med 207:637–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sledzinska A, Vila de Mucha M, Bergerhoff K, Hotblack A, Demane DF, et al. 2020. Regulatory T cells restrain interleukin-2- and Blimp-1-dependent acquisition of cytotoxic function by CD4+ T cells. Immunity 52:151–66.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yan H, Hou X, Li T, Zhao L, Yuan X, et al. 2016. CD4+ T cell-mediated cytotoxicity eliminates primary tumor cells in metastatic melanoma through high MHC class II expression and can be enhanced by inhibitory receptor blockade. Tumor Biol. 37:15949–58 [DOI] [PubMed] [Google Scholar]
- 78.Roemer MGM, Redd RA, Cader FZ, Pak CJ, Abdelrahman S, et al. 2018. Major histocompatibility complex class II and programmed death ligand 1 expression predict outcome after programmed death 1 blockade in classic Hodgkin lymphoma. J. Clin. Oncol 36:942–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Curtis MM, Rowell E, Shafiani S, Negash A, Urdahl KB, et al. 2010. Fidelity of pathogen-specific CD4+ T cells to the Th1 lineage is controlled by exogenous cytokines, interferon-γ expression, and pathogen lifestyle. Cell Host Microbe 8:163–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, et al. 2005. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. PNAS 102:18538–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Metidji A, Rieder SA, Glass DD, Cremer I, Punkosdy GA, Shevach EM. 2015. IFN-α/β receptor signaling promotes regulatory T cell development and function under stress conditions. J. Immunol 194:4265–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hashimoto H, Ueda R, Narumi K, Heike Y, Yoshida T, Aoki K. 2014. Type I IFN gene delivery suppresses regulatory T cells within tumors. Cancer Gene Ther. 21:532–41 [DOI] [PubMed] [Google Scholar]
- 83.Hirata A, Hashimoto H, Shibasaki C, Narumi K, Aoki K. 2019. Intratumoral IFN-α gene delivery reduces tumor-infiltrating regulatory T cells through the downregulation of tumor CCL17 expression. Cancer Gene Ther. 26:334–43 [DOI] [PubMed] [Google Scholar]
- 84.Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, et al. 2017. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548:461–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. 2017. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548:466–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bartsch K, Knittler K, Borowski C, Rudnik S, Damme M, et al. 2017. Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum. Mol. Genet 26:3960–72 [DOI] [PubMed] [Google Scholar]
- 87.Chen J, Cao Y, Markelc B, Kaeppler J, Vermeer JAF, Muschel RJ. 2019. Type I IFN protects cancer cells from CD8+ T cell-mediated cytotoxicity after radiation. J. Clin. Investig 129:4224–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sharma MD, Hou DY, Baban B, Koni PA, He Y, et al. 2010. Reprogrammed Foxp3+ regulatory T cells provide essential help to support cross-presentation and CD8+ T cell priming in naive mice. Immunity 33:942–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Swann JB, Hayakawa Y, Zerafa N, Sheehan KC, Scott B, et al. 2007. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J. Immunol 178:7540–49 [DOI] [PubMed] [Google Scholar]
- 90.Mizutani T, Neugebauer N, Putz EM, Moritz N, Simma O, et al. 2012. Conditional IFNAR1 ablation reveals distinct requirements of type I IFN signaling for NK cell maturation and tumor surveillance. Oncoimmunology 1:1027–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oh JH, Kim MJ, Choi SJ, Ban YH, Lee HK, et al. 2019. Sustained type I interferon reinforces NK cell–mediated cancer immunosurveillance during chronic virus infection. Cancer Immunol. Res 7:584–99 [DOI] [PubMed] [Google Scholar]
- 92.Rautela J, Baschuk N, Slaney CY, Jayatilleke KM, Xiao K, et al. 2015. Loss of host type-I IFN signaling accelerates metastasis and impairs NK-cell antitumor function in multiple models of breast cancer. Cancer Immunol. Res 3:1207–17 [DOI] [PubMed] [Google Scholar]
- 93.Crouse J, Bedenikovic G, Wiesel M, Ibberson M, Xenarios I, et al. 2014. Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity 40:961–73 [DOI] [PubMed] [Google Scholar]
- 94.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, et al. 2005. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434:907–13 [DOI] [PubMed] [Google Scholar]
- 95.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, et al. 2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434:864–70 [DOI] [PubMed] [Google Scholar]
- 96.Castle JC, Uduman M, Pabla S, Stein RB, Buell JS. 2019. Mutation-derived neoantigens for cancer immunotherapy. Front. Immunol 10:1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. 2004. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. PNAS 101:1714–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lau L, Gray EE, Brunette RL, Stetson DB. 2015. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350:568–71 [DOI] [PubMed] [Google Scholar]
- 99.Xia T, Konno H, Barber GN. 2016. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 76:6747–59 [DOI] [PubMed] [Google Scholar]
- 100.Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, et al. 2019. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 9:34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xia T, Konno H, Ahn J, Barber GN. 2016. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 14:282–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.An X, Zhu Y, Zheng T, Wang G, Zhang M, et al. 2019. An analysis of the expression and association with immune cell infiltration of the cGAS/STING pathway in pan-cancer. Mol. Ther. Nucleic Acids 14:80–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, et al. 2018. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553:467–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li J, Bakhoum SF. 2019. Expanding the role of STING in cellular homeostasis and transformation. Trends Cancer 5:195–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Deng L, Liang H, Xu M, Yang X, Burnette B, et al. 2014. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41:843–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhan X, Guo S, Li Y, Ran H, Huang H, et al. 2020. Glioma stem-like cells evade interferon suppression through MBD3/NuRD complex–mediated STAT1 downregulation. J. Exp. Med 217:e201913140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis 5:99–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, et al. 2003. Integration of interferon-α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature 424:516–23 [DOI] [PubMed] [Google Scholar]
- 109.Muñoz-Fontela C, Macip S, Martínez-Sobrido L, Brown L, Ashour J, et al. 2008. Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med 205:1929–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Miciak J, Bunz F. 2016. Long story short: p53 mediates innate immunity. Biochim. Biophys. Acta Rev. Cancer 1865:220–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu Y, Lv J, Liu J, Liang X, Jin X, et al. 2018. STAT3/p53 pathway activation disrupts IFN-β–induced dormancy in tumor-repopulating cells. J. Clin. Investig 128:1057–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Halazonetis TD, Gorgoulis VG, Bartek J. 2008. An oncogene-induced DNA damage model for cancer development. Science 319:1352–55 [DOI] [PubMed] [Google Scholar]
- 113.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, et al. 2017. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 7:188–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ye Z, Dong H, Li Y, Ma T, Huang H, et al. 2018. Prevalent homozygous deletions of type I interferon and defensin genes in human cancers associate with immunotherapy resistance. Clin. Cancer Res 24:3299–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, et al. 2017. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550:402–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, et al. 2017. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol 19:1061–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yang H, Wang H, Ren J, Chen Q, Chen ZJ. 2017. cGAS is essential for cellular senescence. PNAS 114:E4612–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, et al. 2018. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat. Commun 9:1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, et al. 2017. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun 8:15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vizioli MG, Liu T, Miller KN, Robertson NA, Gilroy K, et al. 2020. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 34:428–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, et al. 2019. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566:73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, et al. 2007. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450:903–7 [DOI] [PubMed] [Google Scholar]
- 123.Suranyi MG, Hogan PG, Falk MC, Axelsen RA, Rigby R, et al. 1998. Advanced donor-origin melanoma in a renal transplant recipient: immunotherapy, cure, and retransplantation. Transplantation 66:655–61 [DOI] [PubMed] [Google Scholar]
- 124.Lee MS, Kim B, Oh GT, Kim Y-J. 2013. OASL1 inhibits translation of the type I interferon-regulating transcription factor IRF7. Nat. Immunol 14:346–55 [DOI] [PubMed] [Google Scholar]
- 125.Tobelaim WS, Beaurivage C, Champagne A, Pomerleau V, Simoneau A, et al. 2015. Tumour-promoting role of SOCS1 in colorectal cancer cells. Sci. Rep 5:14301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zitzmann K, Brand S, De Toni EN, Baehs S, Göke B, et al. 2007. SOCS1 silencing enhances antitumor activity of type I IFNs by regulating apoptosis in neuroendocrine tumor cells. Cancer Res. 67:5025–32 [DOI] [PubMed] [Google Scholar]
- 127.Sim CK, Cho YS, Kim BS, Baek I-J, Kim Y-J, Lee MS. 2016. 2′−5′ Oligoadenylate synthetase-like 1 (OASL1) deficiency in mice promotes an effective anti-tumor immune response by enhancing the production of type I interferons. Cancer Immunol. Immunother 65:663–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li S, Xie Y, Zhang W, Gao J, Wang M, et al. 2015. Interferon alpha-inducible protein 27 promotes epithelial-mesenchymal transition and induces ovarian tumorigenicity and stemness. J. Surg. Res 193:255–64 [DOI] [PubMed] [Google Scholar]
- 129.Tjandra SS, Hsu C, Goh I, Gurung A, Poon R, et al. 2007. IFN-β signaling positively regulates tumorigenesis in aggressive fibromatosis, potentially by modulating mesenchymal progenitors. Cancer Res. 67:7124–31 [DOI] [PubMed] [Google Scholar]
- 130.Nan J, Wang Y, Yang J, Stark GR. 2018. IRF9 and unphosphorylated STAT2 cooperate with NF-κB to drive IL6 expression. PNAS 115:3906–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Grivennikov S, Karin M. 2008. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell 13:7–9 [DOI] [PubMed] [Google Scholar]
- 132.Goldszmid RS, Dzutsev A, Trinchieri G. 2014. Host immune response to infection and cancer: unexpected commonalities. Cell Host Microbe 15:295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Beglin M, Melar-New M, Laimins L. 2009. Human papillomaviruses and the interferon response. J. Interferon Cytokine Res 29:629–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gough DJ, Messina NL, Clarke CJP, Johnstone RW, Levy DE. 2012. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity 36:166–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chakravorty S, Yan B, Wang C, Wang L, Quaid JT, et al. 2019. Integrated pan-cancer map of EBV-associated neoplasms reveals functional host-virus interactions. Cancer Res. 79:6010–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, et al. 2017. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 19:1189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ostrand-Rosenberg S, Sinha P. 2009. Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol 182:4499–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xiao W, Klement JD, Lu C, Ibrahim ML, Liu K. 2018. IFNAR1 controls autocrine type I IFN regulation of PD-L1 expression in myeloid-derived suppressor cells. J. Immunol 201:264–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hashimoto M, Ayada T, Kinjyo I, Hiwatashi K, Yoshida H, et al. 2009. Silencing of SOCS1 in macrophages suppresses tumor development by enhancing antitumor inflammation. Cancer Sci. 100:730–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shen L, Evel-Kabler K, Strube R, Chen S-Y. 2004. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat. Biotechnol 22:1546–53 [DOI] [PubMed] [Google Scholar]
- 141.Hanada T, Kobayashi T, Chinen T, Saeki K, Takaki H, et al. 2006. IFNγ-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J. Exp. Med 203:1391–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Calabrese V, Mallette FA, Deschenes-Simard X, Ramanathan S, Gagnon J, et al. 2009. SOCS1 links cytokine signaling to p53 and senescence. Mol. Cell 36:754–67 [DOI] [PubMed] [Google Scholar]
- 143.Hsu PP, Sabatini DM. 2008. Cancer cell metabolism: Warburg and beyond. Cell 134:703–7 [DOI] [PubMed] [Google Scholar]
- 144.Fritsch SD, Weichhart T. 2016. Effects of interferons and viruses on metabolism. Front. Immunol 7:630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yeh Y-H, Hsiao H-F, Yeh Y-C, Chen T-W, Li T-K. 2018. Inflammatory interferon activates HIF-1α-mediated epithelial-to-mesenchymal transition via PI3K/AKT/mTOR pathway. J. Exp. Clin. Cancer Res 37:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sharma A, Janocha AJ, Hill BT, Smith MR, Erzurum SC, Almasan A. 2014. Targeting mTORC1–mediated metabolic addiction overcomes fludarabine resistance in malignant B cells. Mol. Cancer Res 12:1205–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Guertin DA, Sabatini DM. 2007. Defining the role of mTOR in cancer. Cancer Cell 12:9–22 [DOI] [PubMed] [Google Scholar]
- 149.Ambjørn M, Ejlerskov P, Liu Y, Lees M, Jäättelä M, Issazadeh-Navikas S. 2013. IFNB1/interferon-β-induced autophagy in MCF-7 breast cancer cells counteracts its proapoptotic function. Autophagy 9:287–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schmeisser H, Fey SB, Horowitz J, Fischer ER, Balinsky CA, et al. 2013. Type I interferons induce autophagy in certain human cancer cell lines. Autophagy 9:683–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhu S, Cao L, Yu Y, Yang L, Yang M, et al. 2013. Inhibiting autophagy potentiates the anticancer activity of IFN1@/IFNα in chronic myeloid leukemia cells. Autophagy 9:317–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Du Y, Duan T, Feng Y, Liu Q, Lin M, et al. 2018. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J. 37:351–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhan Z, Xie X, Cao H, Zhou X, Zhang XD, et al. 2014. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy 10:257–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, et al. 2015. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42:41–54 [DOI] [PubMed] [Google Scholar]
- 155.Wu D, Sanin DE, Everts B, Chen Q, Qiu J, et al. 2016. Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity 44:1325–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Palazon A, Tyrakis PA, Macias D, Veliça P, Rundqvist H, et al. 2017. An HIF-1α/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell 32:669–83.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP. 2014. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLOS Biol. 12:e1001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.York AG, Williams KJ, Argus JP, Zhou QD, Brar G, et al. 2015. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell 163:1716–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Everts B, Amiel E, Huang SC-C, Smith AM, Chang CH, et al. 2014. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat. Immunol 15:323–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Brandacher G, Perathoner A, Ladurner R, Schneeberger S, Obrist P, et al. 2006. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin. Cancer Res 12:1144–51 [DOI] [PubMed] [Google Scholar]
- 161.Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, et al. 2010. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med 16:880–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, et al. 2003. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med 9:1269–74 [DOI] [PubMed] [Google Scholar]
- 163.Yasui H, Takai K, Yoshida R, Hayaishi O. 1986. Interferon enhances tryptophan metabolism by inducing pulmonary indoleamine 2,3-dioxygenase: its possible occurrence in cancer patients. PNAS 83:6622–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. 1999. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med 189:1363–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, et al. 2006. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor ζ -chain and induce a regulatory phenotype in naive T cells. J. Immunol 176:6752–61 [DOI] [PubMed] [Google Scholar]
- 166.Muller AJ, Manfredi MG, Zakharia Y, Prendergast GC. 2019. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin. Immunopathol 41:41–48 [DOI] [PubMed] [Google Scholar]
- 167.Wang W, Huang L, Jin J-Y, Pi W, Ellsworth SG, et al. 2020. A validation study on IDO immune biomarkers for survival prediction in non–small cell lung cancer: radiation dose fractionation effect in early-stage disease. Clin. Cancer Res 26:282–89 [DOI] [PubMed] [Google Scholar]
- 168.Liu M, Li Z, Yao W, Zeng X, Wang L, et al. 2020. IDO inhibitor synergized with radiotherapy to delay tumor growth by reversing T cell exhaustion. Mol. Med. Rep 21:445–53 [DOI] [PubMed] [Google Scholar]
- 169.Jia H, Thelwell C, Dilger P, Bird C, Daniels S, Wadhwa M. 2018. Endothelial cell functions impaired by interferon in vitro: insights into the molecular mechanism of thrombotic microangiopathy associated with interferon therapy. Thromb. Res 163:105–16 [DOI] [PubMed] [Google Scholar]
- 170.Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, et al. 2019. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Investig 129:4350–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, et al. 2015. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. PNAS 112:15408–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Berger DP, Herbstritt L, Dengler WA, Marmé D, Mertelsmann R, Fiebig HH. 1995. Vascular endothelial growth factor (VEGF) mRNA expression in human tumor models of different histologies. Ann. Oncol 6:817–25 [DOI] [PubMed] [Google Scholar]
- 173.Kitamura T, Qian BZ, Pollard JW. 2015. Immune cell promotion of metastasis. Nat. Rev. Immunol 15:73–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Takano S, Ishikawa E, Matsuda M, Yamamoto T, Matsumura A. 2014. Interferon-β inhibits glioma angiogenesis through downregulation of vascular endothelial growth factor and upregulation of interferon inducible protein 10. Int. J. Oncol 45:1837–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zheng H, Qian J, Carbone CJ, Leu NA, Baker DP, Fuchs SY. 2011. Vascular endothelial growth factor–induced elimination of the type 1 interferon receptor is required for efficient angiogenesis. Blood 118:4003–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kato Y, Tabata K, Kimura T, Yachie-Kinoshita A, Ozawa Y, et al. 2019. Lenvatinib plus anti-PD-1 anti-body combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLOS ONE 14:e0212513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tian L, Goldstein A, Wang H, Lo HC, Kim IS, et al. 2017. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 544:250–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tong Y, Zhou L, Yang L, Guo P, Cao Y, et al. 2019. Concomitant type I IFN and M-CSF signaling reprograms monocyte differentiation and drives pro-tumoral arginase production. EBioMedicine 39:132–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.U’Ren L, Guth A, Kamstock D, Dow S. 2010. Type I interferons inhibit the generation of tumor-associated macrophages. Cancer Immunol. Immunother 59:587–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, et al. 2008. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133:704–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tian L, Li L, Xing W, Li R, Pei C, et al. 2015. IRGM1 enhances B16 melanoma cell metastasis through PI3K-Rac1 mediated epithelial mesenchymal transition. Sci. Rep 5:12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wenzel J, Tomiuk S, Zahn S, Küsters D, Vahsen A, et al. 2008. Transcriptional profiling identifies an interferon-associated host immune response in invasive squamous cell carcinoma of the skin. Int. J. Cancer 123:2605–15 [DOI] [PubMed] [Google Scholar]
- 183.Pidugu VK, Wu M-M, Yen A-H, Pidugu HB, Chang K-W, et al. 2019. IFIT1 and IFIT3 promote oral squamous cell carcinoma metastasis and contribute to the anti-tumor effect of gefitinib via enhancing p-EGFR recycling. Oncogene 38:3232–47 [DOI] [PubMed] [Google Scholar]
- 184.Sun L, Miyoshi H, Origanti S, Nice TJ, Barger AC, et al. 2015. Type I interferons link viral infection to enhanced epithelial turnover and repair. Cell Host Microbe 17:85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ahn WS, Bae SM, Lee JM, Namkoong SE, Han SJ, et al. 2004. Searching for pathogenic gene functions to cervical cancer. Gynecol. Oncol 93:41–48 [DOI] [PubMed] [Google Scholar]
- 186.Guasch G, Schober M, Pasolli HA, Conn EB, Polak L, Fuchs E. 2007. Loss of TGFβ signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell 12:313–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Grunwell JR, Yeligar SM, Stephenson S, Ping XD, Gauthier TW, et al. 2018. TGF-β1 suppresses the type I IFN response and induces mitochondrial dysfunction in alveolar macrophages. J. Immunol 200:2115–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wculek SK, Malanchi I. 2015. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528:413–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, et al. 2016. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med 8:361ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Andzinski L, Kasnitz N, Stahnke S, Wu C-F, Gereke M, et al. 2016. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int. J. Cancer 138:1982–93 [DOI] [PubMed] [Google Scholar]
- 191.Rayes RF, Mouhanna JG, Nicolau I, Bourdeau F, Giannias B, et al. 2019. Primary tumors induce neutrophil extracellular traps with targetable metastasis-promoting effects. JCI Insight 5:e128008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sawant A, Hensel JA, Chanda D, Harris BA, Siegal GP, et al. 2012. Depletion of plasmacytoid dendritic cells inhibits tumor growth and prevents bone metastasis of breast cancer cells. J. Immunol 189:4258–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yang L-L, Mao L, Wu H, Chen L, Deng W-W, et al. 2019. pDC depletion induced by CD317 blockade drives the antitumor immune response in head and neck squamous cell carcinoma. Oral Oncol. 96:131–39 [DOI] [PubMed] [Google Scholar]
- 194.Raychaudhuri D, Bhattacharya R, Sinha BP, Liu CSC, Ghosh AR, et al. 2019. Lactate induces pro-tumor reprogramming in intratumoral plasmacytoid dendritic cells. Front. Immunol 10:1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Terra M, Oberkampf M, Fayolle C, Rosenbaum P, Guillerey C, et al. 2018. Tumor-derived TGFβ alters the ability of plasmacytoid dendritic cells to respond to innate immune signaling. Cancer Res. 78:3014–26 [DOI] [PubMed] [Google Scholar]
- 196.Lee J, Sayed N, Hunter A, Au KF, Wong WH, et al. 2012. Activation of innate immunity is required for efficient nuclear reprogramming. Cell 151:547–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yang CA, Huang HY, Chang YS, Lin CL, Lai IL, Chang JG. 2017. DNA-sensing and nuclease gene expressions as markers for colorectal cancer progression. Oncology 92:115–24 [DOI] [PubMed] [Google Scholar]
- 198.Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, et al. 2019. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565:43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Liu H, Golji J, Brodeur LK, Chung FS, Chen JT, et al. 2019. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat. Med 25:95–102 [DOI] [PubMed] [Google Scholar]
- 200.Jacquelot N, Yamazaki T, Roberti MP, Duong CPM, Andrews MC, et al. 2019. Sustained type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 29:846–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Fairfax BP, Taylor CA, Watson RA, Nassiri I, Danielli S, et al. 2020. Peripheral CD8+ T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat. Med 26:193–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zecchini V, Frezza C. 2017. Metabolic synthetic lethality in cancer therapy. Biochim. Biophys. Acta Bioenerg 1858:723–31 [DOI] [PubMed] [Google Scholar]
- 203.Shi Y, Zheng W, Yang K, Harris KG, Ni K, et al. 2020. Intratumoral accumulation of gut microbiota facilitates CD47-based immunotherapy via STING signaling. J. Exp. Med 217:e20192282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Guilhot F, Chastang C, Michallet M, Guerci A, Harousseau JL, et al. 1997. Interferon alfa-2b combined with cytarabine versus interferon alone in chronic myelogenous leukemia. French Chronic Myeloid Leukemia Study Group. N. Engl. J. Med 337:223–29 [DOI] [PubMed] [Google Scholar]
- 205.Mandelli F, Avvisati G, Amadori S, Boccadoro M, Gernone A, et al. 1990. Maintenance treatment with recombinant interferon alfa-2b in patients with multiple myeloma responding to conventional induction chemotherapy. New Engl. J. Med 322:1430–34 [DOI] [PubMed] [Google Scholar]
- 206.Osterborg A, Björkholm M, Björeman M, Brenning G, Carlson K, et al. 1993. Natural interferon-alpha in combination with melphalan/prednisone versus melphalan/prednisone in the treatment of multiple myeloma stages II and III: a randomized study from the Myeloma Group of Central Sweden. Blood 81:1428–34 [PubMed] [Google Scholar]
- 207.Gogas H, Abali H, Ascierto PA, Demidov L, Pehamberger H, et al. 2015. Who benefits most from adjuvant interferon treatment for melanoma? Am. J. Ther 22:54–60 [DOI] [PubMed] [Google Scholar]
- 208.Alberts DS, Hannigan EV, Liu P-Y, Jiang C, Wilczynski S, et al. 2006. Randomized trial of adjuvant intraperitoneal alpha-interferon in stage III ovarian cancer patients who have no evidence of disease after primary surgery and chemotherapy: an intergroup study. Gynecol. Oncol 100:133–38 [DOI] [PubMed] [Google Scholar]
- 209.Hall GD, Brown JM, Coleman RE, Stead M, Metcalf KS, et al. 2004. Maintenance treatment with interferon for advanced ovarian cancer: results of the Northern and Yorkshire gynaecology group randomised phase III study. Br. J. Cancer 91:621–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Muss HB, Kempf RA, Martino S, Rudnick SA, Greiner J, et al. 1984. A phase II study of recombinant alpha interferon in patients with recurrent or metastatic breast cancer. J. Clin. Oncol 2:1012–16 [DOI] [PubMed] [Google Scholar]
- 211.Bald T, Landsberg J, Lopez-Ramos D, Renn M, Glodde N, et al. 2014. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 4:674–87 [DOI] [PubMed] [Google Scholar]
- 212.Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, et al. 2011. The efficacy of radiotherapy relies upon induction of type I interferon–dependent innate and adaptive immunity. Cancer Res. 71:2488–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Liang Y, Tang H, Guo J, Qiu X, Yang Z, et al. 2018. Targeting IFNα to tumor by anti-PD-L1 creates feedforward antitumor responses to overcome checkpoint blockade resistance. Nat. Commun 9:4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Mehrotra S, Britten CD, Chin S, Garrett-Mayer E, Cloud CA, et al. 2017. Vaccination with poly(IC:LC) and peptide-pulsed autologous dendritic cells in patients with pancreatic cancer. J. Hematol. Oncol 10:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Bell J, McFadden G. 2014. Viruses for tumor therapy. Cell Host Microbe 15:260–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Aref S, Castleton AZ, Bailey K, Burt R, Dey A, et al. 2020. Type 1 interferon responses underlie tumor-selective replication of oncolytic measles virus. Mol. Ther 28(4):1043–55 [DOI] [PMC free article] [PubMed] [Google Scholar]