

Abstract
Objective
High-grade glioma (HGG) rarely spreads outside the CNS. To test the hypothesis that the lesions were metastases originating from an HGG, we sequenced the relapsing HGG and distant extraneural lesions.
Methods
We performed whole-exome sequencing of an HGG lesion, its local relapse, and distant lesions in bone and lymph nodes.
Results
Phylogenetic reconstruction and histopathologic analysis confirmed the common glioma origin of the secondary lesions. The mutational profile revealed an IDH1/2 wild-type HGG with an activating mutation in EGFR and biallelic focal loss of CDKN2A (9p21). In the metastatic samples and the local relapse, we found an activating PIK3CA mutation, further copy number gains in chromosome 7 (EGFR), and a putative pathogenic driver mutation in a canonical splice site of FLNA.
Conclusions
Our findings demonstrate tumor spread outside the CNS and identify potential genetic drivers of metastatic dissemination outside the CNS, which could be leveraged as therapeutic targets or potential biomarkers.
Patients with high-grade glioma (HGG) (WHO grade III and IV) have a dismal prognosis with a median survival of 16.0 months for grade IV tumors.1 Distant metastasis of glioma outside the CNS remains a rare event, occurring in less than 2% of gliomas,2 and the molecular features of metastatic dissemination in these tumors are not characterized. In this report, we performed whole-exome sequencing on the primary tumor of a patient with HGG and on multiple secondary lesions to study potential metastatic drivers outside the CNS.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
The family of the patient signed the informed consent form and approved the disclosure of this publication. The collection of biological samples and health-related data was approved by the Ethics Committee of the state of Geneva. The study was conducted in accordance with the 1975 Declaration of Helsinki.
Data Analysis
A small part of the tissue from all 4 lesions was frozen and stored at −80°C, whereas the largest part was fixed in 4% buffered formalin and paraffin embedded (FFPE). The bone biopsy was decalcified with ethylenediaminetetraacetic acid before embedding. From the FFPE material, 2–3-µm-thick slides were stained with H&E. Immunohistochemistry according to standard protocols was then performed using the following antibodies: GFAP, IDH1 (R132H), Ki67, MAP2, NOGO-A, p53, and synaptophysin in the primary and secondary brain lesions and synaptophysin, Ki67, MAP2, NOGO-A, chromogranin A, protein S100, Melan-A, HMB45, epithelial membrane antigen, O13, NB84, INI1, MAP2, EGR, granzyme B, myogenin, keratin, CD3, CD5, CD20, CD79a, CD30, CD34, CD56, and CD117 in the lymph node metastasis and the bone biopsy.
For the genomic analysis, DNA was extracted from blood and FFPE tumor samples with the Qiagen DNeasy Blood and Tissue Kit and Qiagen QIAmp DNA FFPE Tissue Kit, respectively. DNA quality was evaluated with Agilent TapeStation genomic DNA ScreenTape. Exome capture and sequencing libraries were prepared with the Agilent SureSelect Human All Exon V5 with 1 μg DNA. Libraries were sequenced in an Illumina HiSeq2000 to obtain 100-bp paired-end read at an average coverage of 110 (SD 26). Sequencing reads were mapped to the reference genome GRCh38 with the Burrows-Wheeler Aligner and then processed with the bcbio cancer variant calling pipeline. Sequencing quality and target enrichment were verified with Picard tools metrics (broadinstitute.github.io/picard/). MuTect2, VarScan, VarDict, and Freebayes somatic (arxiv.org/abs/1207.3907) with a majority voting approach were used to identify somatic standard normal variables and indels.3 Stop-gain, splice site, or probably damaging nonsynonymous mutations and variants reported in the COSMIC v71 database were considered putative drivers. Somatic copy number aberrations were estimated from exome sequencing data with CNVkit.
Data Availability
Deidentified patient data are available on request.
Results
A 16-year-old patient presented in July 2013 with a right fronto-insular lesion (figure 1, A and B), which was subtotally resected, and a second diffuse lesion infiltrating the pons (not shown). Histopathologic analysis revealed an IDH wild-type HGG without 1p/19q codeletion, compatible with a rare anaplastic pediatric-type oligodendroglioma.4 Chemoradiation with temozolomide, followed by 6 cycles of temozolomide, was administered from August 2013 to March 2014. The lesion in the pons responded favorably; no relapse was observed around the initial resection cavity.
Figure 1. Radiologic Features of the Brain Tumors.
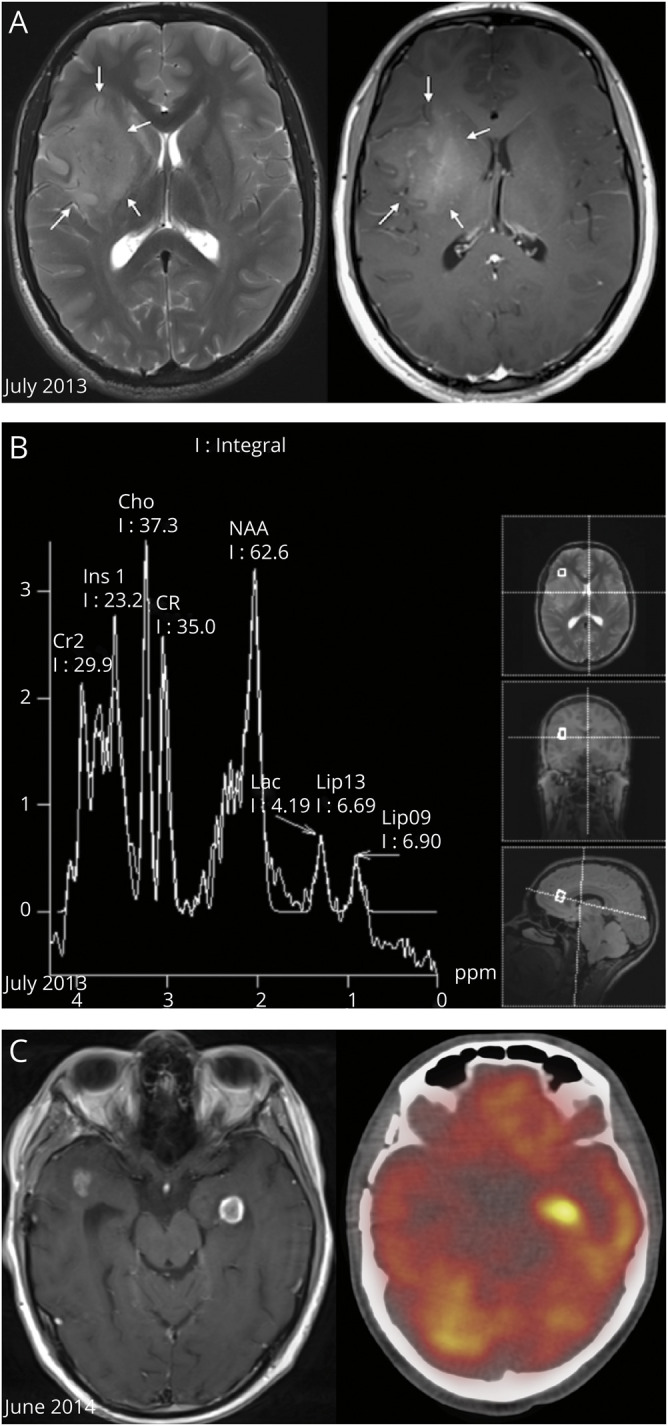
(A) Axial planes of T2 turbo spin echo-weighted brain MRI (left image) and T1 gadolinium sequence (right image) showing a right fronto-insular lesion with high signal on T2 (arrows) and with inhomogeneous enhancement on T1, corresponding to a high-grade lesion. (B) Magnetic resonance spectroscopy (MRS) shows a raised choline peak and N-acetyl aspartate peak (resonating at 3.7 and 6.2 ppm, respectively), suggestive of anaplastic glioma. (C) Gadolinium T1-weighted brain MRI (left image) showing tumor relapse with 2 new enhancing left temporal lesions (the left one was biopsied stereotactically) and the corresponding hypermetabolic image on PET CT (right).
In April 2014, the patient presented with rapidly worsening inflammation of the right knee, fever, and tachycardia. A total-body FDG PET CT scan revealed generalized lymphadenopathy involving multiple mediastinal and retroperitoneal lymph nodes, a distal femoral heterogeneous lesion (figure 2A.b), and a pattern of carcinomatous lymphangitis in the lungs (figure 2A.a, figure e-1, links.lww.com/NXG/A377). We obtained tumor biopsies from the popliteal lymph node and the right femur. Histopathology assessment showed that the 3 lesions (brain, lymph node, and bone) appeared similar (figure 2, B and C) with intense MAP2 and CD56 staining, confirming their glial origin, and weak or focal staining for Olig2, GFAP (figure 2D), synaptophysin, and TP53.
Figure 2. Radiologic and Pathologic Features of the Serially Collected Tumors.

(A, A.a) Whole-body fluorodeoxyglucose PET (FDG PET) imaging with strong FDG uptake in various nodal localizations, notably in the mediastinum. (A.b) Axial plane, FDG PET-CT imaging of both knees; note the hypermetabolic lymphadenopathies behind the right knee. (B) Histology shows a dense, poorly differentiated primary neuroepithelial tumor (B.a, HE 400×), with numerous secondary structures of Scherer (B.b, satellitosis of tumor cells around neurons, HE 400×). (C) Histology of the popliteal fossa lymph node (C.a) and bone (C.b) (HE 400×). (D) Both biopsies show a poorly differentiated neuroepithelial lesion with variable GFAP positivity (400×).
Unfortunately, we observed rapid multifocal disease progression in the brain (figure 1C). A biopsy of a new left temporal lobe lesion was performed in June 2014, showing the same histology as the initial brain lesion, but with additional aggressiveness features compatible with a glioblastoma multiforme (GBM) (pseudopalisading necrosis and high proliferation index). Further treatment lines, including ifosfamide and doxorubicin, were attempted without eliciting a response, and the patient died in August 2014.
Exome sequencing of the primary lesion, the brain relapse, and the distant bone and lymph node metastasis showed biallelic focal loss of CDKN2A (9p21) and in-frame insertion in the EGFR locus (p.Asn771_His773dup) (figure e-2, links.lww.com/NXG/A378). Phylogenetic reconstructions revealed a common clonal origin (figure 3, A and B). Eleven high-confidence variants were shared between all tumors. Seventeen additional mutations were shared by the bone and lymph node metastases and the brain relapse but not by the initial HGG (Additional files 1 and 2, links.lww.com/NXG/A386 and links.lww.com/NXG/A387). Deletions in chr.9, chr.6, and chr.1q gain (3 copies) were present in the initial tumor and subsequent lesions. A gain in Chr.7 was shared between the subsequent lesions (figure 3C). The brain relapse had many more copy number alterations than the primary lesion (164 copy-altered segments vs 27, respectively, figure 3C), possibly a consequence of double-strand breaks from radiochemotherapy. Despite the high secondary mutation rate in the recurrent tumor, the patient did not present any family history of malignancies, and tumor mutation burden (TMB) was less than 6/Mb, suggesting an absence of MSH2 and MSH6 germline alterations, which are associated with a TMB of 40/Mb.5
Figure 3. Whole-Exome Sequencing and Phylogenetic Tree Reconstruction, Compatible With the Glial Origin of the Distant Lesions.

(A) Phylogenetic tree with parallel evolution events harboring different mutations occurring between the primary and secondary tumors. (B) Map summarizing all standard normal variable (SNV), insertions and deletions per lesion, and their pathogenic role in known tumor driver genes. (C) A heatmap of copy number variations (CNVs) showing losses (blue) and gains (red).
The results suggest that the secondary lesions derive from the same subclone of the original HGG. Among the shared mutations, we observed 3 putative driver events (figure 3A): an activating mutation in PIK3CA (p.His1047Arg) known for its oncogenicity,6 gain of 2 copies of the EGFR mutation (p.Asn771_His773dup, chr.7, 4n), confirmed by variant allele frequency analysis in each specimen, and a putative pathogenic driver mutation in a canonical splice site of FLNA. In addition, a frameshift variant in the FHOD1 gene was common in all secondary lesions.
Discussion
Of interest, mutations in EGFR and PIK3CA are usually mutually exclusive and are found together in less than 2% of HGG. Both are part of the RTK/Ras/phosphatidylinositide 3-kinase (PI3K) signaling cascade, which is altered in 90% of adult HGG.7 The EGFR exon 20 insertion is typically associated with resistance to tyrosine kinase inhibitors,8,9 and it is rare in GBM,10 where the most common variant is EGFRvIII.11 The additional copy gain of mutated EGFR could be another spreading trigger. The PIK3CA mutation (p.His1047Arg) is a well-described activating mutation in cancer and is present in 10% of GBM.7 Moreover, the association of CDKN2A deletion and EGFR amplification was found in all 4 lesions and is known to be a marker of tumor aggressiveness.12 A third putative driver is the variant in the FLNA gene. Mutations causing a noncanonical splicing event can lead to aberrant transcripts that cause many diseases, including cancer.13 FLNA is an actin-binding protein involved in cell motility and invasion of glioblastoma cells,14 and its overexpression is associated with progression to metastasis and/or poor prognosis in several cancers.15 Its effect could be synergistic with the mutation of the formin FHOD1, also involved in migration of glioma cells in vitro.16,17
Conclusion
Our findings confirm the metastatic hematogenous dissemination of a primary CNS tumor,2 as previously described by Hamilton et al.,18 suggesting that the main vascular route of GBM metastasis is reflux into the systemic venous system. Our results indicate that both the brain relapse and the distant metastatic lesions derive from the same subclone of the original HGG. Besides molecular features known to be associated with GBM aggressiveness, other genes (FLNA and FHOD1) could be novel drivers of metastatic spread. A previous study on medulloblastoma reported molecular evidence of hematogenous dissemination, showing that specific drivers contribute to cell invasion outside the CNS.19 Functional validation of these candidates using gene editing may lead to the discovery of therapeutic targets.
Acknowledgment
The authors acknowledge the continuous support and trust of the patient and his family. They also thank the hospital personnel who took care of the patient with professionalism and humanity and Prof. David N. Louis for helpful comments and discussions.
Glossary
- FFPE
formalin and paraffin embedded
- GBM
glioblastoma multiforme
- HGG
high-grade glioma
- TMB
tumor mutation burden
Appendix. Authors
Contributor Information
Eliana Marinari, Email: eliana.marinari@unige.ch.
Valerie Dutoit, Email: valerie.dutoit@unige.ch.
Sergey Nikolaev, Email: serg.nikolaev@gmail.com.
Maria-Isabel Vargas, Email: maria.i.vargas@hcuge.ch.
Karl Schaller, Email: karl.schaller@hcuge.ch.
Johannes Alexander Lobrinus, Email: johannes.a.lobrinus@hcuge.ch.
Pierre-Yves Dietrich, Email: pierre-yves.dietrich@hcuge.ch.
Petros Tsantoulis, Email: petros.tsantoulis@hcuge.ch.
Study Funding
No targeted funding reported.
Disclosure
D. Migliorini is supported by the ISREC Foundation, the Swiss Bridge Foundation, Innosuisse, Ligue Genevoise contre le Cancer, and Ernst and Lucie Schmidheiny Foundation. P. Tsantoulis is supported by the Ligue Genevoise contre le Cancer. P.-Y. Dietrich is supported by Fondation Privée des Hôpitaux Universitaires de Genève, Fondation Lionel Perrier, Association Marietta, Association Frédéric Fellay, and Fondaction. Go to Neurology.org/NG for full disclosure.
References
- 1.Stupp R, Taillibert S, Kanner A, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 2017;318:2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muller C, Holtschmidt J, Auer M, et al. Hematogenous dissemination of glioblastoma multiforme. Sci Transl Med 2014;6:247ra101. [DOI] [PubMed] [Google Scholar]
- 3.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mackay A, Burford A, Carvalho D, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 2017;32:520–537 e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salem ME, Grothey A, Kim ES, et al. Impact of MLH1, PMS2, MSH2, and MSH6 alterations on tumor mutation burden (TMB) and PD-L1 expression in 1,057 microsatellite instability-high (MSI-H) tumors. J Clin Oncol 2018;36:abstr 3572. [Google Scholar]
- 6.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A 2005;102:802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell 2013;155:462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qin YR, Jian H, Tong XL, et al. Variability of EGFR exon 20 insertions in 24 468 Chinese lung cancer patients and their divergent responses to EGFR inhibitors. Mol Oncol 2020;14:1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Remon J, Hendriks LEL, Cardona AF, Besse B. EGFR exon 20 insertions in advanced non-small cell lung cancer: a new history begins. Cancer Treat Rev 2020;90:102105. [DOI] [PubMed] [Google Scholar]
- 10.Gao Y, Vallentgoed WR, French PJ. Finding the right way to target EGFR in glioblastomas; lessons from lung adenocarcinomas. Cancers (Basel) 2018;10:489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Binder ZA, Thorne AH, Bakas S, et al. Epidermal growth factor receptor extracellular domain mutations in glioblastoma present opportunities for clinical imaging and therapeutic development. Cancer Cell 2018;34:163–177.e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayashi Y, Ueki K, Waha A, Wiestler OD, Louis DN, von Deimling A. Association of EGFR gene amplification and CDKN2 (p16/MTS1) gene deletion in glioblastoma multiforme. Brain Pathol 1997;7:871–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sibley CR, Blazquez L, Ule J. Lessons from non-canonical splicing. Nat Rev Genet 2016;17:407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chantaravisoot N, Wongkongkathep P, Loo JA, Mischel PS, Tamanoi F. Significance of filamin A in mTORC2 function in glioblastoma. Mol Cancer 2015;14:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamil M, Shinsato Y, Higa N, et al. High filamin-C expression predicts enhanced invasiveness and poor outcome in glioblastoma multiforme. Br J Cancer 2019;120:819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chesarone MA, DuPage AG, Goode BL. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol 2010;11:62–74. [DOI] [PubMed] [Google Scholar]
- 17.Heuser VD, Kiviniemi A, Lehtinen L, et al. Multiple formin proteins participate in glioblastoma migration. Bmc Cancer 2020;20:710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamilton JD, Rapp M, Schneiderham TM. Glioblastoma multiforme metastasis outside the CNS: three case reports and possible mechanisms of escape (vol 32, pg e80, 2014). J Clin Oncol 2014;32:abstr 3462. [DOI] [PubMed] [Google Scholar]
- 19.Garzia L, Kijima N, Morrissy AS, et al. A hematogenous route for medulloblastoma leptomeningeal metastases (vol 172, pg 1050, 2018). Cell 2018;173:1549–1550. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deidentified patient data are available on request.