

Abstract
The hepatitis B virus (HBV) ribonuclease H (RNaseH) is a promising but unexploited drug target. Inhibiting the RNaseH blocks viral reverse transcription by truncating the minus-polarity DNA strand, causing accumulation of RNA:DNA heteroduplexes, and abrogating plus-polarity DNA synthesis. Screening for RNaseH inhibitors is complicated by the presence of the minus-polarity DNA strand even when replication is fully inhibited because this residual DNA can be detected by standard screening assays that measure reduction in HBV DNA accumulation. We previously developed a strand-preferential qPCR assay that detects RNaseH replication inhibitors by measuring preferential suppression of the viral plus-polarity DNA strand. However, this assay employed cells grown in 6- or 12-well plates and hence was of very low throughput. Here, we adapted the assay to a 96-well format and conducted a proof-of-principle screen of 727 compounds. The newly developed assay is a valuable tool for anti-HBV drug discovery, particularly when screening for RNaseH inhibitors.
Keywords: Hepatitis B virus, replication inhibition assay, RIbonuclease H, assay optimization
1. Introduction
Hepatitis B virus (HBV) is a major global health problem. At least 250 million people are chronically infected worldwide, resulting in >870,000 deaths annually (Polaris Observatory Collaborators, 2018; Trepo, Chan, and Lok, 2014). Current anti-HBV drugs are limited to interferon-α (IFNα) or nucleoside and nucleotide analogs (NA), with the NAs tenofovir and entecavir dominating therapy (Trepo et al., 2014). These drugs have significantly improved patient outcomes, but they are not curative (Marcellin et al., 2008; van Bommel et al., 2010; Woo et al., 2010; Wursthorn et al., 2010). NA must be continued indefinitely, resulting in significant cost to patients, unknown side effects from prolonged use, and a risk of developing drug resistance (Levrero et al., 2018). Furthermore, the risk of developing hepatocellular carcinoma after 10 years of treatment with NAs is only reduced by 2- to 4-fold (Liaw, 2013).
Promising experimental therapies targeting many stages in the viral life cycle are under development. Examples include direct inhibitors of viral entry, reverse transcription, capsid assembly, release of viral particles, covalently closed circular DNA (cccDNA) formation, viral surface antigen production, viral mRNA accumulation, and immune modulators including TNF-α, TLR inhibitors, and PD-1 agonists (Fanning et al., 2019; Tang et al., 2017). We are developing replication inhibitors that target the viral ribonuclease H (RNaseH) because it is the only viral enzyme not currently targeted by approved drugs. Curing HBV is anticipated to require new drugs against new targets capable of further suppressing HBV. These drugs, including the RNaseH drugs we seek to develop, are expected to be used in combinations with drugs that target multiple host and viral functions simultaneously (Alter et al., 2017; Revill et al., 2019).
HBV replicates by reverse transcription within viral capsids catalyzed by the viral polymerase, yielding a partially double-stranded DNA genome (Summers and Mason, 1982). The polymerase has two enzymatically active domains, the reverse transcriptase (RT) and the RNaseH. The RT copies the pregenomic RNA (pgRNA) into the negative polarity (−) DNA strand and the RNaseH concomitantly degrades the pgRNA after it has been copied. The (−) DNA strand is then used as the template for positive polarity (+) DNA synthesis, which arrests early, resulting in the partially double-stranded DNA genome found in virions (Seeger, Zoulim, and Mason, 2013; Tavis and Badtke, 2009). Blocking either enzymatic activity prevents the synthesis of the viral genome, but the resulting phenotypes are different. When the RT is fully inhibited, synthesis of both DNA strands is blocked. In comparison, fully inhibiting the RNaseH causes early termination of most (−) polarity DNA strands and accumulation of RNA:DNA heteroduplexes inside viral capsids, preventing synthesis of the (+) polarity DNA strand. This blocks production of mature HBV DNA because the virus cannot remove the RNA:DNA heteroduplexes in the absence of RNaseH activity, preventing formation of the mature viral DNA. Failure to produce mature viral DNA blocks virion production and intracellular amplification of the intracellular cccDNA produced by reverse transcription that is the template for all HBV mRNAs. Blocking cccDNA formation will in turn prevent initiation of new rounds of reverse transcription and suppress production of all viral proteins.
Detecting RNaseH inhibitors is challenging because typical HBV replication inhibition assays detect total HBV DNA levels. These assays are poorly sensitive to RNaseH inhibitors due to the residual (−) polarity DNA present when the RNaseH is suppressed. Therefore, we developed assays capable of detecting RNaseH inhibitors. The oldest assay is the heteroduplex detection assay (HDA) (Gerelsaikhan, Tavis, and Bruss, 1996; Tavis et al., 2013). This assay is the gold standard for detecting RNaseH inhibition because it reveals the presence of RNA:DNA heteroduplexes from viral capsids that can only be made in the absence of RNaseH activity. However, the HDA assay is time consuming, very low throughput, and labor intensive. The second assay is a DNA strand-preferential quantitative polymerase chain reaction (qPCR) assay that is suitable for experiments done in 6- and 12-well plates (Lomonosova and Tavis, 2017). In this assay, RNaseH inhibitors are detected by their preferential suppression of the (+) polarity DNA strand. However, this assay is still too slow, laborious, and expensive for even mid-throughput screening. Here, we adapted the qPCR replication inhibition assay to a 96-well format to increase screening capacity and reduce costs.
2. Materials and Methods
2.1. Cells and cell culture
HepDES19 is a HepG2 human hepatoblastoma cell line derivative that is stably transfected with an HBV genotype D genome (GenBank accession number V01460) that cannot produce the viral surface proteins under the control of a tetracycline-repressible promoter (Guo et al., 2007). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F12 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) with 1 µg/mL tetracycline on 10 cm Biocoat collagen-coated dishes (Corning). Media was refreshed every 3 days. Expression of HBV pgRNA to launch HBV reverse transcription was induced by removing tetracycline from the culture medium.
2.2. Compound acquisition
The Tavis lab has an in-house library of >1200 compounds acquired from commercial vendors or synthesized by collaborators. All compounds are ≥95% pure. The NIH Clinical Collection Libraries 1 and 2 (NCC1 and NCC2) were obtained from the US NIH. All compounds in the Tavis library were dissolved at 10 mM in DMSO and stored in opaque tubes at −25°C, the NCC1 and NCC2 collections were stored at 10 mM in DMSO in 96-well plates protected from light at −80°C.
2.3. Plating HepDES19 cells into 96-well plates
Cell culture medium was removed from confluent 100 mm culture dishes. Cells were washed once with 10 mL phosphate buffered saline (PBS) prior to incubation with 1.5 mL 0.05% Trypsin-EDTA (Thermo Fisher Scientific) at 37°C for 5 min. Cells were diluted to 2 × 105 cells/ml and then the inner 60 wells of a 96-well plate were seeded with 200 µL of this suspension for a total of 4 × 104 cells per well. The outer wells were filled with 200 µL of PBS or medium as an evaporation buffer. Cells were incubated for 48 hours at 37°C in the presence of 5% CO2.
2.4. Screening assay setup
Qualitative screening assays were done at a final compound concentration of 20 µM with a final DMSO concentration of 1%. For quantitative assessment of efficacy, a series of nine threefold serial dilutions were performed. A 1% DMSO vehicle control was included in all experiments. The culture medium was removed from the 96-well plate by inverting the plate above a waste collection container, and then blotting the inverted plate on a clean tissue to remove residual medium. Cells were washed in 200 µL of PBS, which was then removed in the same manner. 200 µL of compound-containing medium was added to the appropriate wells. The outer wells were filled with 200 µL of PBS. Cells were incubated in the presence of compound-containing medium for 72 hours at 37°C in the presence of 5% CO2.
2.5. Quantitative-PCR
Four μL of crude lysate prepared using the procedure developed and described below was used as the template for strand-preferential quantitative polymerase chain reaction (qPCR) analysis. qPCR was performed in a final volume of 20 µL with an initial denaturation step of 98 °C for 3 minutes followed by 40 cycles of 95 °C for 15 seconds and 60 °C for 30 seconds employing the Kappa Probe Force universal PCR master mix (Roche KK4303). Both the annealing and elongation steps were done at 60 °C. The primers and probe (IDT Inc.) for the (+) DNA strand were 5’CATGAACAAGAGATGATTAGGCAGAG3’, 5’GGAGGCTGTAGGCATAAATTGG3’, and 5’/56-FAM/CTGCGCACC/ZEN/AGCACCATGCA/3IABkFQ. The primers and probe for the (−) DNA strand were 5’GCAGATGAGAAGGCACAGA3’, 5’CTTCTCCGTCTGCCGTT3’, and 5’/56-FAM/AGTCCGCGT/ZEN/AAAGAGAGGTGCG/3IABkFQ. The standard curve for the qPCR assays employed a PCR product amplified from the HBV isolate found in HepDES19 cells containing the primer binding sites for both the (+) and (–) strand primers. Fifty percent effective concentrations (EC50) were calculated from the (+) DNA data with GraphPad Prism using the three-parameter log(inhibitor)-versus-response algorithm with the bottom value set to zero.
2.6. Quality control assessment (Z’ factor)
Assay quality was assessed using the Z’ factor, which measures well-to-well variations in the assay relative to signals from positive and negative controls. In this assay, the negative control is the 1% DMSO vehicle control, and the positive control is treatment with the approved NA drug lamivudine at 20 µM, which suppresses both the (+) and (−) DNA strands to below the limit of detection. The Z’ factor was calculated using the formula:
where μ+ is the mean of the positive control wells, μ− is the mean of the negative control wells, σ+ is the standard deviation of the positive control wells, and σ− is the standard deviation of the negative control wells. The threshold for an acceptable assay was Z’ of ≥ 0.5.
2.7. Cytotoxicity assays
The effects of compounds on cell viability in HepDES19 cells were assessed with MTS assays using the CellTiter 96™ AQueous Non-Radioactive Cell Proliferation Assay (Promega 3580) as previously described (Edwards et al., 2017). Cells were seeded into a 96-well plate at 1 × 104 cells per well. The compounds were applied to cells 48 hours after removal of tetracycline from the cell culture media and the HBV-expressing cells were incubated for 72 hours in the presence of compound-containing media. Due to limited access to some compounds, cytotoxicity was assessed by doing four 3-fold dilutions from 50 µM. A compound was defined as cytotoxic if cell viability was reduced by 40% or more at 16.7 µM.
3. Results
The original qPCR HBV replication inhibition assay (Lomonosova and Tavis, 2017) involved collecting cells from 6- or 12-well plates, lysing the cells, removing non-encapsidated nucleic acids with micrococcal nuclease, digesting proteins in the sample to free the encapsidated HBV DNAs, purifying the DNAs, and then quantifying both DNA strands by qPCR. Modifying the assay to a 96-well plate format required optimizing the cell lysis procedure, confirming that cellular nucleic acids could be removed with micrococcal nuclease, identifying the best protease to digest the viral capsids, identifying contaminant-resistant qPCR conditions for use with crude lysates, and simplifying sample handling procedures.
3.1. Cell lysis and sample normalization
Well-to-well variation in cell plating and lysing efficiency were major sources of variability early during development of this assay, with initial assays having Z’ scores as low as −2.1. Therefore, we attempted to normalize the qPCR values to the levels of cellular proteins in each sample prior to protease digestion as reflected by absorbance at 280 nM. A high background from the 0.25% NP40 in the original lysis buffer masked differences in absorbance between samples. Changing the detergent to 0.25% Tween 20 led to equally effective lysis and permitted detection of absorbance differences between samples. Surprisingly, normalization in A280 levels among samples from multiple 96-well plates had a negligible effect on assay quality, with Z’ scores rising by <0.1. The high variability leading to low Z’ values was finally resolved by a combination of increasing lysis time from 30 to 40 min and using multi-channel pipets when plating cells and handling all reagents. This improved Z’ scores from <0.1 to routinely >0.5.
3.2. Removal of cellular nucleic acids
HepDES19 cells contain an integrated copy of the HBV genome under control of a tetracycline-repressible promoter (Guo et al., 2007) that would interfere with quantification of capsid-derived HBV DNAs. In the 12-well version of this assay, cellular nucleic acids, including the integrated HBV genome, are degraded by digestion with micrococcal nuclease. Encapsidated HBV DNAs are protected because the nuclease is too large to enter HBV capsids. Micrococcal nuclease (New England Biolabs, M0247S) can be inactivated by heat treatment at 70 °C for 10 minutes after digestion of the cellular nucleic acids. This step was tested for suitability in the 96- well assay by measuring HBV DNAs in the crude lysate and after treatment with micrococcal nuclease. Nuclease treatment eliminated the cellular DNA contamination without impacting the encapsidated DNA (Fig. 1), so this step was retained in the 96-well version of the assay.
Fig. 1. Removal of cellular DNAs with micrococcal nuclease.
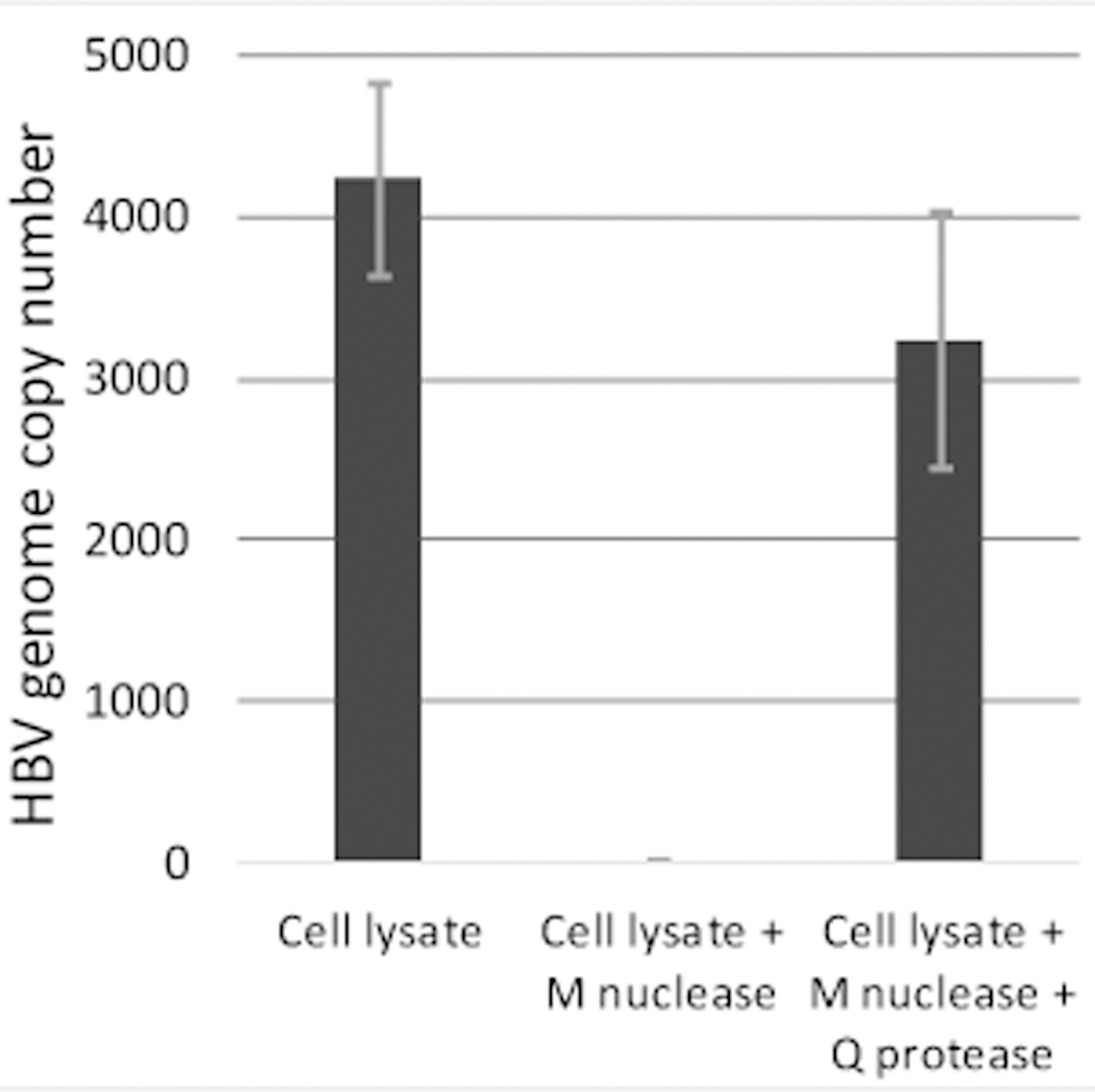
Cells expressing HBV were lysed, the lysates were treated with micrococcal nuclease (M nuclease), and capsids were then digested with Q protease. Equal fractions were removed at each step, DNAs were purified, and HBV genome copy number was determined by qPCR.
3.3. Capsid digestion
Prior to quantification of HBV DNA by qPCR, the viral capsids must be degraded to release the DNA, and the HBV polymerase that is covalently attached to the 5’ end of the HBV (−) DNA strand (Seeger et al., 2013; Tavis and Badtke, 2009) must be removed. The protease employed needs to be easily inactivated because the nucleic acids are not fully purified prior to qPCR, and retention of an active protease could degrade the DNA polymerase during qPCR. We choose heat treatment to inactivate the protease because it is cost effective and rapid. This necessitated shifting away from proteinase K (which was used in the 12-well version of the assay) because previous attempts in other contexts to completely heat inactivate the enzyme were unsuccessful. We evaluated Q protease (Qiagen 19157) based on product literature indicating it can readily be heat inactivated. Heat inactivation of Q-protease for 10 minutes at 70 °C failed to fully inactivate the enzyme, as revealed by failure of qPCR reactions using templates prepared in this manner. Raising the temperature to 90 °C for 10 minutes led to robust amplification (see Fig. 2 for example qPCR assays using these conditions), indicating that inactivation was sufficient for purposes of this assay. Therefore, Q-protease was used in all future experiments. Q-protease is stable at 4°C for only three weeks when dissolved in water at twice the working concentration. Therefore, careful attention must be paid to the age of aqueous Q-protease stocks.
Fig. 2. EC50 assays comparing the 12- and 96-well formats of the strand preferential HBV replication inhibition assay.
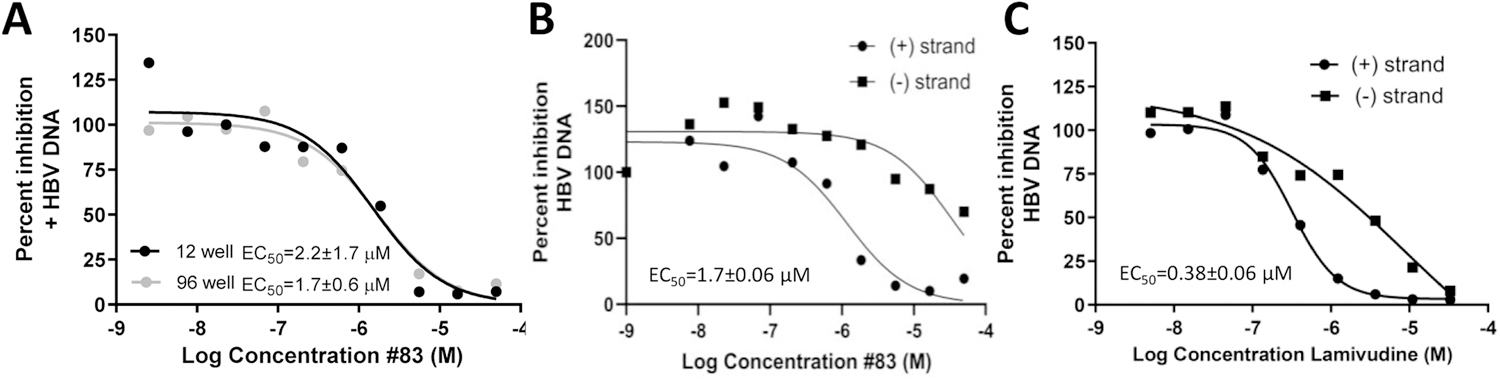
A. Comparison of EC50 curves for the (+) DNA strand using the 12- and 96-well versions of the assay. B. qPCR assay using the 96-well format showing the selective suppression of the (+) DNA strand from treatment with an RNaseH inhibitor. C. qPCR assay in 96-well format showing selective suppression of the (+) DNA for the chain terminating drug lamivudine. The curves show representative assays and the EC50 values are derived from three independent assays ± one standard deviation.
3.4. Contaminant-resistant qPCR conditions
Streamlining the strand-preferential RNaseH inhibitor assay requires using the crude mixture after protease digestion for the template during qPCR, and these samples still include the lysis buffer contents. Three qPCR enzyme mixes were assessed for activity in the buffer used to prepare these minimally processed samples. No amplification from a purified DNA template was observed when the GoTaq qPCR master mix (Promega A6101) used for the 12-well assay was tested. We next tested Kappa Probe Force master mix (Roche KK4303) and GoTaq qPCR master mix (Applied Biosystems 4444557). Both enzyme mixes were tolerant of buffer contaminants and yielded consistent amplification, but sensitivity with both enzyme mixes was suppressed relative to the HBV standard control in which the template was dissolved in water, and addition of BSA only modestly improved amplification (representative assay in Fig. 3). Kappa Probe Force master mix and was used for subsequent experiments because it yielded slightly higher signals and was less expensive.
Fig. 3. Evaluation of qPCR mixes for contamination resistance.
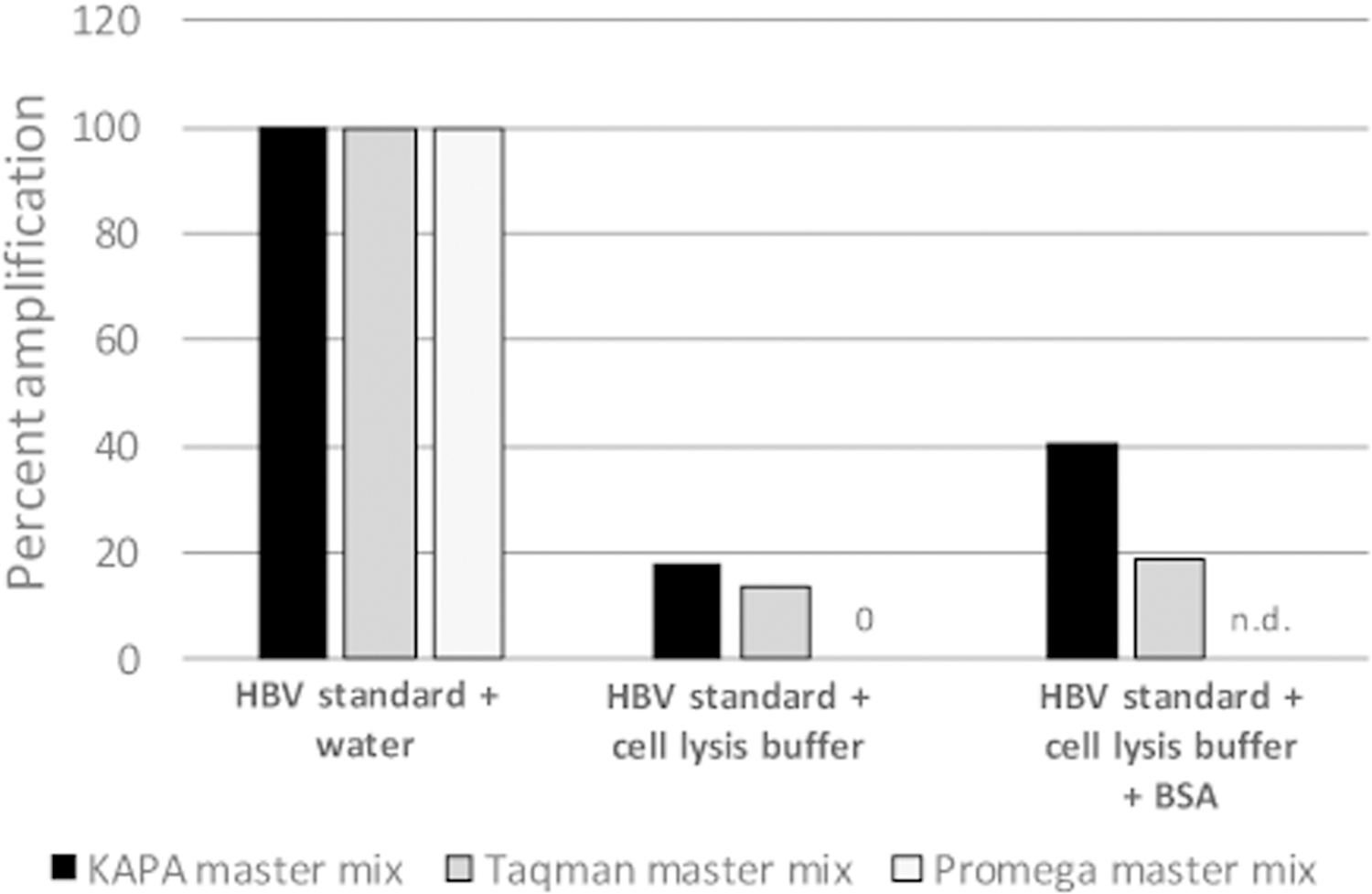
Three qPCR mixes were tested for amplification of a standard DNA (a HBV DNA PCR product) in the presence of added water, lysis buffer, and lysis buffer plus BSA. Data are normalized to amplification in the presence of added water. The figure shows a representative assay.
3.5. Final sample preparation protocol
Seed HepDES19 cells in 96-well plates at 4 × 104 cells per well and incubate in the absence of tetracycline for 48 hours to induce HBV replication.
Wash cells once with DMEM/F12 medium, add compounds in a final concentration of 1% DMSO, and incubate for 72 hours.
Wash cells two times with 200 µL PBS, incubate with 150 µL cell lysis buffer (10 mM Tris-HCl pH 7.4, 1% Tween and 150 mM NaCl) for 40 min at room temperature with shaking at 350 rpm on a microplate shaker.
Transfer the cell lysate to a clean 96-well qPCR plate and centrifuge at 4500 × g for 5 minutes at room temperature to sediment nuclei and other debris. Transfer 50 µL supernatant of each cell lysate into a non-skirted 96-well PCR plate, being very careful not to dislodge the pellet.
Digest non-encapsidated nucleic acids by adding 5 gel U/µL micrococcal nuclease (New England Biolabs) and incubating at 37 °C for 1 hour.
Inactivate the micrococcal nuclease by incubating sample at 70 °C in a thermocycler for 10 minutes.
Digest capsids and cellular proteins with 0.01AU Q protease per sample to overnight at 37°C.
Inactivate Q protease at 90 °C in a thermocycler for 10 minutes.
Conduct qPCR assays as described in the Methods section.
3.6. Performance of known HBV RNaseH inhibitors in the 96-well assay format
We next tested whether the newly designed 96-well strand-preferential qPCR assay could detect strand-preferential suppression of (+) polarity DNAs by RNaseH inhibitors that were previously identified using the 12-well assay. Ninety-three compounds were selected, 20 inhibitors and 73 inactive compounds. We defined inhibition as 50% inhibition of (+) DNA accumulation compared to the DMSO vehicle-treated controls. Each plate had a minimum of six positive control wells containing 20 µM lamivudine and 6 negative control wells containing 1% DMSO. Z’ factors for four replicate assays calculated from the lamivudine- and DMSO-treated wells were 0.61 – 0.74. There was a 97% (90/93) agreement with data from the 12-well assay (Table 1), indicating the two assays have similar sensitivity during compound screening. One discordant result was due to inhibition by compound 271 by 44%, just below the 50% threshold. To further validate the new assay format, we determined the EC50s for 8 compounds using both the 12- and 96-well replication inhibition assays (Table 2; example assay in Fig. 2).
Table 1.
Qualitative screen comparing the 12-well and 96-well formats for the strand-preferential HBV replication inhibition assay
| Assay format | |||
|---|---|---|---|
| Compound # | Formal Name | 12-well | 96-well |
| 2 | Sigma 74540 | − | − |
| 3 | Sigma n8164 | − | − |
| 4 | TimTec ST029023 | − | − |
| 5 | Enamine T0506–3483 | − | − |
| 6 | Chembridge 7929959 | − | − |
| 7 | Idofine 02030 | − | − |
| 9 | Sigma 70050 | − | − |
| 10 | Elvitegravir | − | − |
| 11 | Raltegravir | − | − |
| 19 | Sigma – 586862 | − | − |
| 23 | Sigma – 28605 | − | − |
| 30 | Chembridge – 7248520 | − | − |
| 31 | Chembridge – 5104346 | − | − |
| 34 | Indofine -D -009 | − | − |
| 35 | TCI America -D1118 | − | − |
| 39 | Asinex -BAS05223612 | − | − |
| 44 | Labotest 12243782 | − | − |
| 45 | TCI America H1040 | − | − |
| 46 | beta-thujaplicinol | + | + |
| 47 | beta-thujaplicin | − | − |
| 48 | gamma-thujaplicin | − | − |
| 50 | 5-nitrosotropolone | − | − |
| 51 | tropolone p-nitrobenzoate | − | − |
| 52 | NSC 79556 | − | − |
| 53 | Tropolone | − | − |
| 63 | Chembridge 5938894 | − | − |
| 67 | Sigma 17850 | − | − |
| 69 | Sigma R747092 | − | − |
| 72 | BMS-707035 | − | − |
| 106 | CM1012–6a | + | + |
| 107 | CM1012–6b | + | + |
| 108 | CM1012–6c | + | + |
| 109 | CM1012–6d | + | + |
| 110 | CM1012–6e | + | + |
| 112 | CM1012–6i | + | + |
| 113 | RM-YM-1–0613 | + | + |
| 121 | CWHM-000527 | − | − |
| 124 | CWHM-000554 | − | − |
| 128 | CNC_ID 100615760 | − | − |
| 131 | CNC_ID 408228831 | − | − |
| 132 | CNC_ID 389306767 | − | − |
| 138 | Sigma H53704 | − | − |
| 139 | Sigma 130672 | − | − |
| 141 | B2–10-1–8 | − | − |
| 142 | B2617 | − | − |
| 143 | MD-1–138 | + | + |
| 144 | DH-1–148 | − | − |
| 172 | 7-Hydroxytropolone | + | + |
| 191 | Piroctone olamine | − | − |
| 195 | Purpurogallin | − | − |
| 197 | Benzoyleneurea | − | − |
| 198 | 2,3-dihydroxyquinoxaline | − | − |
| 199 | TRC D416405 | − | − |
| 203 | TRC 700465 | − | − |
| 204 | AK-830/13217043 | − | − |
| 212 | Sigma 08445 | − | − |
| 214 | Sigma D5564 | − | − |
| 215 | Sigma 576441 | − | − |
| 218 | CNC_ID 110964057 | − | − |
| 232 | AB-1–45 | + | + |
| 233 | AB-1–46 | − | − |
| 234 | AB-1–51 | − | − |
| 236 | ZF4 | − | − |
| 238 | ZF18 | − | − |
| 240 | ZF24 | − | − |
| 241 | ZF29 | − | − |
| 243 | ZF47 | − | − |
| 244 | ZF65 | − | − |
| 245 | ZF66 | − | − |
| 246 | ZF73 | − | − |
| 247 | ZB5 | − | − |
| 248 | ZB10 | − | − |
| 249 | ZB15 | − | − |
| 253 | ZB54 | − | − |
| 260 | AG-I-84-P | − | − |
| 261 | AG40 | + | + |
| 262 | AG51 | + | + |
| 269 | DH-4–119 | + | + |
| 271 | DH-4–85 | + | − |
| 273 | DH-4–116 | − | − |
| 274 | DH-4–117 | + | + |
| 280 | AG77 | + | + |
| 287 | Sigma 343614692 | − | − |
| 310 | AG-II-21-P | + | + |
| 314 | AG-II-23-P | − | − |
| 316 | AG-1–187-P | + | + |
| 317 | AG-II-22-P | − | − |
| 320 | NBA-I-13 | + | + |
| 321 | AMS 149974043 | + | − |
| 322 | AMS 239435306 | − | − |
| 323 | AMS 388708402 | − | − |
| 324 | AMS 295104760 | − | − |
| 325 | AMS 295182442 | − | + |
+ = Inhibition >50% at 20 μM, - = Inhibition <50% at 20 μM.
Table 2.
EC50 values determined by the 12- and 96-well formats for the strand-preferential HBV replication inhibition assay
| EC50 Assay format |
|||
|---|---|---|---|
| Compound # | Compound Class | 12-well | 96-well |
| 83 | HID | 1.5 | 1.7 |
| 84 | HID | 8.1 | 8.0 |
| 86 | HID | 1.7 | 1.8 |
| 90 | HID | 19 | 27 |
| 208 | HPD | 0.6 | 0.7 |
| 266 | αHT | 1.4 | 0.7 |
| 271 | αHT | 50 | 44 |
Values in µM. HID = N-hydroxyisoquinolinedione, HPD = N-hydroxypyridinedione, αHT = α-hydroxytropolone.
3.7. Mid-throughput screening of the NIH clinical collection libraries 1 and 2
Finally, we conducted a proof-of-concept mid-throughput screen employing the 96-well assay with 727 compounds from the NCC1 and NCC2 libraries. A hit was defined as inhibiting the HBV (+) polarity DNA strand relative to the DMSO vehicle-treated control by 75%. We chose a more stringent hit definition for these assays to minimize false-positives. Fifteen plates were used. Each plate had five 1% DMSO vehicle controls, five 20 µM lamivudine controls, and 50 compounds diluted to 20 µM. Compounds were used at this relatively high concentration to maximize sensitivity for hits suitable for subsequent optimization, as is typical in drug screening. This assay identified 24 preliminary hits (Fig. 4a and 3b). Lamivudine was found twice because it is in both NCC libraries. Additionally, four hits are tetracycline analogues that were excluded from further analysis due to their potential to suppress transcription from the integrated HBV genome whose expression is under the control of a tetracycline repressible promoter (Guo et al., 2007). Therefore, the screen identified 18 preliminary hits for cytotoxicity analyses. These compounds were tested in duplicate for cytotoxicity using an MTS assay. Eight compounds were cytotoxic, reducing cell viability to ≤60% at 16.7 µM (Fig. 4c). Therefore, 10 hits were identified that suppressed HBV replication without excessive cytotoxicity.
Fig. 4. Proof-of-principle screen using the 96-well strand preferential HBV replication inhibition assay.
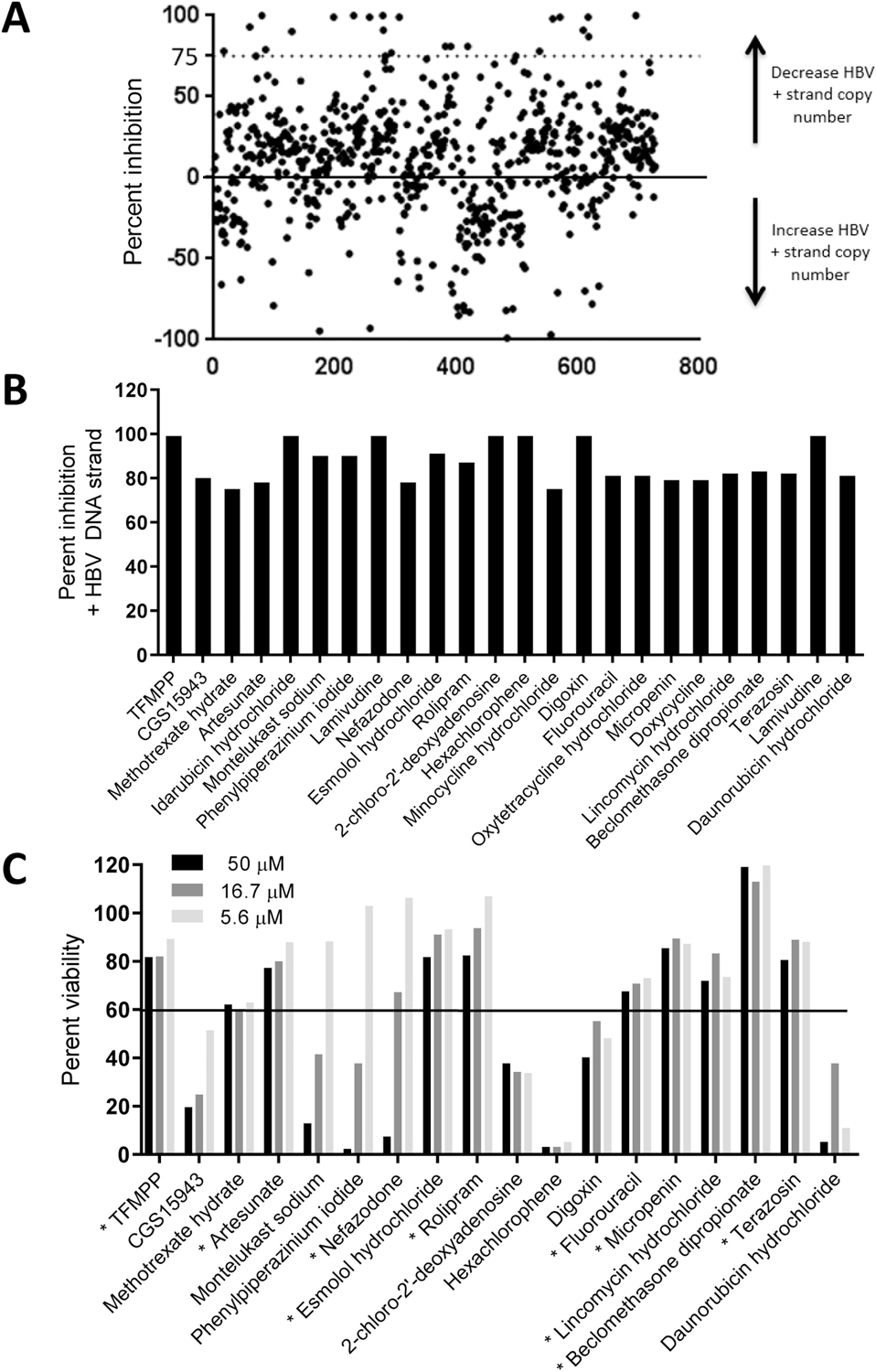
A. Results from the primary screen at 20 µM. Each compound is indicated by a dot and the cutoff for defining preliminary hits is indicated by a dotted line. B. Percent inhibition of HBV replication by the preliminary hits identified from the screen. C. Cytotoxicity of the preliminary hits measured by MTS assays at three compound concentrations. The cutoff for defining cytotoxicity is indicated by a solid line. Hits identified comparing data in panels B and C are indicated with a star next to the compound names in panel C.
4. Discussion
The 96-well plate strand-preferential qPCR assay has several advantages compared with the previous 12-well version. Most importantly, it increased screening capacity by >10-fold. Second, all pipetting is performed with multichannel pipettes, which both reduces hands-on time and errors from processing samples individually. Finally, this assay routinely gives Z’ factors of ≥0.6, whereas routinely calculating a Z’ score in 12-well assays is impractical due to throughput limitations.
Traditional HBV replication assays detect inhibitors by measuring suppression in total HBV DNA accumulation. These assays fail to efficiently detect RNaseH inhibitors because RNaseH inhibitors preferentially suppress (+) polarity HBV DNA strands but have only modest effects on total HBV DNAs due to the residual (−) polarity DNA (Gerelsaikhan et al., 1996; Pierra Rouviere, Dousson, and Tavis, 2020; Tavis et al., 2013). The assay described here quantifies both strands of the HBV DNAs separately and can therefore detect RNaseH inhibitors that would be missed in other assays. Note that this assay detects all compounds that inhibit HBV replication. Compounds inhibiting either the RNaseH or DNA elongation can be identified by their preferential inhibition of the (+) polarity strand, but the assay is not specific for RNaseH inhibitors because elongation inhibitors also preferentially inhibit (+) polarity DNA. Elongation inhibitors such as nucleos(t)ide analogs suppress the (−) polarity DNA by blocking chain elongation, whereas (+) DNA is blocked by both disrupting chain elongation and reduced accumulation of its (−) polarity template (an example assay with lamivudine is in Fig. 2c). Consequently, the testing funnel for hits identified by this assay must include studies to identify their mechanism of action, such as biochemical assays measuring inhibition of recombinant HBV RNaseH or that detect accumulation of RNA:DNA heteroduplexes in capsids. Another limitation is the assay employs a single cell line, which could affect the range of compounds it can detect. Such limitations are inherent in all primary screening assays and need to be addressed in follow-up studies. This assay is has been used in HepDE19 cells (Guo et al., 2007) without modification, and it is readily adaptable to other cell systems upon optimization of cell density, lysing conditions, and time of compound exposure.
To our knowledge there are only three other published assays capable of detecting RNaseH inhibitors. The limitations to the HDA and 12-well strand-preferential qPCR assays have already been presented. The remaining assay is a branch chain DNA quantification assay (bDNA assay) recently developed by Arbutus Biopharma. In this assay, HepDE19 cells containing a tetracycline-inducible HBV genomic cassette (analogous to the HepDES19 cells used in our assays) are incubated in tetracycline-free medium for 24 hours to initiate HBV replication, and are then treated with compound for seven days. HBV relaxed circular DNAs are purified and measured using a Quantigene 2.0 bDNA assay (Affymetrix) with HBV specific probes specific for each DNA strand. During a collaboration with Arbutus Biopharma, we tested seven compounds with both the strand-preferential qPCR assay and the bDNA assay. Both assays could detect RNaseH inhibitors, however the qPCR assay was more sensitive (Edwards et al., 2019). The strand-preferential qPCR assay appears to be superior in part because it requires incubation with compound for only three days, is more sensitive to inhibition of RNaseH activity, and can detect HBV DNAs using crude cellular lysates.
The 96-well strand-preferential qPCR assay was validated by screening 93 compounds from our in-house collection in both the 12- and 96-well assays. There was a 97% agreement between the two assays (Table 1), indicating that the 96-well plate assay is a suitable screening tool. The 96-well plate assay was used in combination with a cytotoxicity assay to screen the NCC1 and NCC2 libraries. This identified 10 compounds that inhibit HBV replication greater than 75% at 20 μM (Fig. 4). Therefore, the 96-well plate replication inhibition assay is suitable for both qualitative and quantitative assessment of HBV replication inhibitors, including RNaseH inhibitors.
There are several drawbacks to this new assay. First, we were unable to shorten its overall duration, primarily because HBV replicates slowly. Due to the five day incubation of the HepDES19 cells (two days for induction of HBV replication plus three days exposure to compounds), we are only able to use the inner 60 wells of the 96-well plate, with the outer wells being filled with medium as an evaporation barrier, reducing the assay capacity. Finally, the assay is suitable for mid-throughput applications, but it remains too expensive and labor intensive for high-throughput screening.
5. Conclusion
The HBV RNaseH is an attractive drug target as it is required for genome replication, but screening for inhibitors has been very slow due to the lack of higher throughput assays. Here, we adapted a 12-well replication inhibition assay to a 96-well format, increasing throughput ~10-fold. This new tool is a step forward in screening technologies to identify and develop inhibitors of HBV replication, particularly those that affect RNaseH activity.
Highlights.
The Hepatitis B Virus ribonuclease H is an attractive, unexploited drug target.
Identifying ribonuclease H inhibitors is hampered by challenges in developing screening assays.
We have optimized a 96-well screening assay that can identify ribonuclease H inhibitors.
A proof-of-concept compound screen validated utility of the 96-well screen.
Acknowledgments
This work was funded by NIH grants R01 AI122669, R01 AI104494, and R21 AI124672 to J.E.T.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
None of the authors have any competing interests.
References
- Alter H, Block T, Brown N, Brownstein A, Brosgart C, Chang KM, Chen PJ, Chisari FV, Cohen C, El-Serag H, Feld J, Gish R, Glenn J, Greten T, Guo H, Guo JT, Hoshida Y, Hu J, Kowdley KV, Li W, Liang J, Locarnini S, Lok AS, Mason W, McMahon B, Mehta A, Perrillo R, Revill P, Rice CM, Rinaudo J, Schinazi R, Seeger C, Shetty K, Tavis J and Zoulim F, 2017. A research agenda for curing chronic hepatitis B virus infection. Hepatology 67, 1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polaris Observatory Collaborators, 2018. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol. Hepatol 3, 383–403. [DOI] [PubMed] [Google Scholar]
- Edwards TC, Lomonosova E, Patel JA, Li Q, Villa JA, Gupta AK, Morrison LA, Bailly F, Cotelle P, Giannakopoulou E, Zoidis G and Tavis JE, 2017. Inhibition of hepatitis B virus replication by N-hydroxyisoquinolinediones and related polyoxygenated heterocycles. Antiviral Res 143, 205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards TC, Mani N, Dorsey B, Kakarla R, Rijnbrand R, Sofia MJ and Tavis JE, 2019. Inhibition of HBV replication by N-hydroxyisoquinolinedione and N-hydroxypyridinedione ribonuclease H inhibitors. Antiviral Res 164, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning GC, Zoulim F, Hou J and Bertoletti A, 2019. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nat Rev Drug Discov 18, 827–824. [DOI] [PubMed] [Google Scholar]
- Gerelsaikhan T, Tavis JE and Bruss V, 1996. Hepatitis B Virus Nucleocapsid Envelopment Does Not Occur without Genomic DNA Synthesis. J.Virol 70, 4269–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Jiang D, Zhou T, Cuconati A, Block TM and Guo JT, 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81, 12472–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levrero M, Subic M, Villeret F and Zoulim F, 2018. Perspectives and limitations for nucleo(t)side analogs in future HBV therapies. Curr Opin Virol 30, 80–89. [DOI] [PubMed] [Google Scholar]
- Liaw YF, 2013. Impact of therapy on the outcome of chronic hepatitis B. Liver international : official journal of the International Association for the Study of the Liver 33 Suppl 1, 111–5. [DOI] [PubMed] [Google Scholar]
- Lomonosova E and Tavis JE, 2017. In Vitro Enzymatic and Cell Culture-Based Assays for Measuring Activity of HBV RNaseH Inhibitors. Methods in molecular biology 1540, 179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcellin P, Heathcote EJ, Buti M, Gane E, De Man RA, Krastev Z, Germanidis G, Lee SS, Flisiak R, Kaita K, Manns M, Kotzev I, Tchernev K, Buggisch P, Weilert F, Kurdas OO, Shiffman ML, Trinh H, Washington MK, Sorbel J, Anderson J, Snow-Lampart A, Mondou E, Quinn J and Rousseau F, 2008. Tenofovir disoproxil fumarate versus adefovir dipivoxil for chronic hepatitis B. N.Engl.J.Med 359, 2442–2455. [DOI] [PubMed] [Google Scholar]
- Pierra Rouviere C, Dousson CB and Tavis JE, 2020. HBV replication inhibitors. Antiviral Res 179, 104815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revill PA, Chisari FV, Block JM, Dandri M, Gehring AJ, Guo H, Hu J, Kramvis A, Lampertico P, Janssen HLA, Levrero M, Li W, Liang TJ, Lim SG, Lu F, Penicaud MC, Tavis JE, Thimme R, Members of the ICEHBVWG, Chairs I-HSG, Advisors I-HS and Zoulim F, 2019. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol Hepatol 4, 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger C, Zoulim F and Mason WS 2013. Hepadnaviruses. In: Knipe DM and Howley PM (Eds), Fields Virology, Lippincott Williams & Wilkins, Philadelphia PA, pp. 2185–2221. [Google Scholar]
- Summers J and Mason WS, 1982. Replication of the Genome of a Hepatitis B-Like Virus by Reverse Transcription of an RNA Intermediate. Cell 29, 403–415. [DOI] [PubMed] [Google Scholar]
- Tang L, Zhao Q, Wu S, Cheng J, Chang J and Guo JT, 2017. The current status and future directions of hepatitis B antiviral drug discovery. Expert Opin Drug Discov 12, 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavis JE and Badtke MP 2009. Hepadnaviral Genomic Replication. In: Cameron CE, Goette M and Raney KD (Eds), Viral Genome Replication, Springer Science+Business Media, LLC, New York, pp. 129–143. [Google Scholar]
- Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R, Meyers MJ, Jacobsen EJ, Parniak MA and Sarafianos SG, 2013. The hepatitis B virus ribonuclease h is sensitive to inhibitors of the human immunodeficiency virus ribonuclease h and integrase enzymes. PLoS pathogens 9, e1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepo C, Chan HL and Lok A, 2014. Hepatitis B virus infection. Lancet 384, 2053–63. [DOI] [PubMed] [Google Scholar]
- van Bommel F, De Man RA, Wedemeyer H, Deterding K, Petersen J, Buggisch P, Erhardt A, Huppe D, Stein K, Trojan J, Sarrazin C, Bocher WO, Spengler U, Wasmuth HE, Reinders JG, Moller B, Rhode P, Feucht HH, Wiedenmann B and Berg T, 2010. Long-term efficacy of tenofovir monotherapy for hepatitis B virus-monoinfected patients after failure of nucleoside/nucleotide analogues. Hepatology 51, 73–80. [DOI] [PubMed] [Google Scholar]
- Woo G, Tomlinson G, Nishikawa Y, Kowgier M, Sherman M, Wong DK, Pham B, Ungar WJ, Einarson TR, Heathcote EJ and Krahn M, 2010. Tenofovir and entecavir are the most effective antiviral agents for chronic hepatitis B: a systematic review and Bayesian meta-analyses. Gastroenterology 139, 1218–1229. [DOI] [PubMed] [Google Scholar]
- Wursthorn K, Jung M, Riva A, Goodman ZD, Lopez P, Bao W, Manns MP, Wedemeyer H and Naoumov NV, 2010. Kinetics of hepatitis B surface antigen decline during 3 years of telbivudine treatment in hepatitis B e antigen-positive patients. Hepatology 52, 1611–1620. [DOI] [PubMed] [Google Scholar]