

Abstract
Polycystic Kidney Disease (PKD) triggers a robust immune system response including changes in both innate and adaptive immunity. These changes involve immune cells (e.g., macrophages and T cells) as well as cytokines and chemokines (e.g., MCP-1) that regulate the production, differentiation, homing, and various functions of these cells. This review is focused on the role of the immune system and its associated factors in the pathogenesis of PKDs as evidenced by data from cell-based systems, animal models, and PKD patients. It also highlights relevant pre-clinical and clinical studies that point to specific immune system components as promising candidates for the development of prognostic biomarkers and therapeutic strategies to improve PKD outcomes.
Keywords: autosomal dominant polycystic kidney disease, ADPKD, autosomal recessive polycystic kidney disease, ARPKD, immune response, chemokines, macrophages, T cells, kidney function
An abnormal immune response is among the most notable hallmarks of polycystic kidney diseases (PKDs)[1]. Since these changes were initially attributed to renal cystic burden, their impact, if any, was thought to be limited to advanced stages of disease progression. This view was changed by Cowley and Grantham who pointed to the pro-inflammatory chemokine monocyte chemoattractant protein 1 (MCP-1) as a regulator of PKD-associated interstitial inflammation and renal function loss[2, 3]. Multiple follow-up experiments in lines, animal models, and PKD patients revealed complex PKD-associated changes in innate and adaptive immunity, and in some cases, immune factors were defined as regulators of PKD progression (Figure 1). Some of these immune factors even emerged as promising candidates for the development of prognostic markers as well as future PKD therapeutics. In this review, we address three key aspects of PKD-associated immune response abnormalities and their relevance to the disease pathogenesis: chemokines and their regulators, innate immune cells, and adaptive immune cells.
Figure 1. Immune responses in PKD.

This model is based on data from PKD patients and animal models showing abnormal production of cytokines, chemokines and complement factors by epithelial cells carrying the cystogenesis-inducing defect, the ensuing changes in recruitment and differentiation of effector cells of innate and adaptive immunity, and the cystogenesis-promoting effects these immune cells exert by further altering the intrarenal cytokine milieu. Such positive feedback loops are expected to exacerbate the impact of immune responses on cystic disease progression over time.
CHEMOKINES AND THEIR REGULATORS IN PKD
Chemokines, a family of chemotactic cytokines, regulate migration, as well as cell proliferation and differentiation of diverse immune cell types including monocytes and macrophages, neutrophils, dendritic cells, and T cells. Therefore, they are critical for the function of both innate and adaptive immune systems and act as disease pathogenesis modulators in a wide range of renal conditions (reviewed, e.g., in[4–6]).
The abnormal expression of chemokines is one of the early phenotypes induced by PKD gene defects. For example, data from isolated cells of normal human kidney, human autosomal dominant PKD (ADPKD) non-cystic nephrons, and ADPKD cystic nephrons showed that cystic nephrons express abnormal levels of genes encoding a diverse range of immune factors including cytokines/chemokines, the complement system, and genes associated with lymphocyte activation[7]. These changes include over-expression of genes encoding Interleukin 6 (IL6), Interleukin 1β (IL1β), MCP-1, Interleukin 8 (IL8), Interleukin 17D (IL17D), Colony-stimulating factor 1 (CSF1), (C-X-C motif) ligand 1 (CXCL1), and Chemokine ligand 28 (CCL28). Similarly, overexpression of cytokines/chemokines and many components of the complement system was described in renal cysts from ADPKD kidneys[8]. Also, once renal cysts or PKD-induced vascular abnormalities trigger focal renal ischemic injury, a more prominent focal cytokine/chemokine and complement system response begins to resemble that observed in renal injury (reviewed in [5, 9]). Such changes and their impact on the severity of renal PKD outcomes are expected to increase as cystic renal disease progresses over time.
The remaining parts of this section are focused on MCP-1, the most extensively studied chemokine in PKD. Also discussed are TNFα and the complement system that regulate both chemokine expression, as well as renal cystic disease progression in animal models. The involvement of other chemokines/cytokines associated with PKD is discussed throughout the innate and adaptive immune sections
Monocyte Chemoattractant Protein-1 (MCP-1)
MCP-1, a chemokine encoded by the gene Ccl2, is a critical regulator of monocyte and macrophage migration[10, 11]. It is secreted by multiple cell types, including tubular epithelial cells and interstitial cells, as well as inflammatory cells such as macrophages[12]. MCP-1 was one of the first immune factors associated with PKD[2, 3] and it has become one of the most extensively studied immune factors in animal models and patients with PKD.
MCP-1 participates in the pathogenesis of multiple renal disorders (reviewed in[13, 14]). It was also identified as a potential biomarker of acute kidney injury (AKI;[15]). AKI is a broadly accepted trigger of renal cystogenesis (the `third hit`;[16–18]) that induces PKD phenotypes even in Pkd1 haploinsufficient[19] and wild-type mice[20, 21]. It is conceivable that in addition to the AKI-induced cystogenic effects, MCP-1 also modulates injury effects caused by chronic, focal, renal parenchyma damage in response to increasing renal cystic burden. This effect is suggested by the exponential increase in MCP-1 expression in animal models with advanced cystic disease, which occurs despite the progressive loss of the functional renal parenchyma that is responsible for expression of these chemoattractant factors[22]. However, the increase in MCP-1 expression might also be injury-independent, triggered directly by a PKD gene defect. Such dysregulated MCP-1 expression was observed in cell lines deficient in polycystin-1 (PC-1)[23], polycystin-2 (PC-2), and kinesin 3a[24, 25]. Also, the expression of the MCP-1 encoding gene Ccl2 in cystic kidneys precedes the expression of renal injury markers such as Lcn2 (a neutrophil gelatinase-associated lipocalin; NGAL) [26].
The central role of MCP-1 in the pathogenesis of PKD was demonstrated in a mouse model with a knockout of both Pkd1 and Ccl2 (MCP-1). In this double knockout mouse, MCP-1 deficiency led to a reduction in macrophage numbers and initial tubular cell injury, slower cyst growth, and improved renal function[27]. The MCP-1 effects on renal cyst growth in this model were modulated by both proliferation-independent and proliferation-dependent mechanisms. Similar improvement of renal function and morphology was achieved by treatment of Pkd1 knockout mice with an inhibitor of the MCP-1 receptor (C-C Motif Chemokine Receptor 2; CCR2)[27].
In patients with ADPKD, the levels of MCP-1 were increased in renal cyst fluid (vs. urine)[3]. Urinary MCP-1 excretion levels were also associated with PKD1 (vs. PKD2) genotype, higher height-adjusted total kidney volume (TKV), and worse renal function outcomes[3, 28]. Further, the urinary MCP-1 levels were strongly associated with more rapidly declining renal function (glomerular filtration rate; GFR), and it’s levels served as a predictor of chronic kidney disease stage 3 (GFR<45 ml/min/1.73m2) in multivariable models[28–30]. These findings corroborate observations from animal models and point to urinary MCP-1 as a predictive biomarker in patients with ADPKD.
Interestingly, in contradiction to the Pkd1;MCP-1 double knockout animals, genetic deletion of MCP-1 in a mouse model that phenocopies Autosomal Recessive PKD (ARPKD) due to a defect in the Cys1 gene (Cys1cpk or cpk mouse;[31]) had no effect on renal macrophage concentration, cystic disease, or kidney function[4]. However, MCP-1 deficiency substantially increased the survival of cpk mice by preventing the development of pulmonary edema and decreasing resting heart rate [4]. Similarly, treatment with bindarit, an MCP-1 inhibitor, did not alter renal and liver cystic severity and renal function[32] in a rat model with a mutation in Pkhd1 (Pkhd1pck or pck rat; [33]), the principal gene affected in patients with ARPKD. However, this treatment did reduce renal macrophage numbers as well as proteinuria by limiting podocyte foot process effacement and by ameliorating slit diaphragm frequency in this model[32]. It remains unknown why bindarit did not inhibit cystogenesis in the Pkhd1pck rat. A possible explanation for the lack of cystogenesis-inhibiting effects include insufficient efficacy when compared to an MCP-1-encoding gene knockout in mice (this question might be resolved once a rat knockout for this gene becomes available). Alternatively, MCP-1 effects might differ in animal models of ARPKD/ciliopathies (vs. ADPKD), a hypothesis that would be supported by the data of genetic Ccl2 loss in the cpk mouse[4]. A lack of adequate clinical samples due to lower ARPKD/Ciliopathy prevalence has complicated systematic assessment of the relevance of these observations in patients with recessive PKD.
Together, the studies in animal models and patients point to MCP-1 as a critical component of ADPKD pathogenesis that might be valuable in predicting ADPKD outcomes and in developing ADPKD therapeutics. Animal studies of MCP-1 effects on the renal cystic disease in ARPKD/ciliopathy models appear less consistent; however, they point to the unexpected MCP-1-dependent effects in cardiopulmonary manifestations.
Tumor Necrosis Factor - alpha (TNFα)
TNFα, a canonical immune cytokine, is a potent inducer of chemokine expression, including MCP-1[34]. In addition, it is involved in the activation of multiple signal transduction pathways involved in inflammatory signaling such as FAS-associated signals via death domain (FADD)/caspase 8/caspase 3, mitogen-activated kinase (MAPK), Jun kinase (JNK)/activation protein (AP-1), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways (reviewed in [35]). TNFα is also candidate inducer of MCP-1 expression in PKD and the cyst fluid levels of TNFα correlate with renal cyst size[36–38]. In addition, TNFα regulates cystic epithelial proliferation through Akt/mTOR and ERK/MAPK/Cdk2-mediated Id2 signaling[39, 40]; it also interferes with processing and membrane presentation of polycystin-2[36]. In mouse embryonic kidney organ cultures, treatment with TNFα triggered cyst formation, and this effect was exacerbated in the Pkd2+/− cultures. TNFα also induced cystogenesis in vivo in Pkd2+/− mice and TNFα inhibition with etanercept attenuated cytogenesis in the same model. However, these results should be evaluated with caution due to the high phenotypic variability of the Pkd2+/− model[36]. Further, this treatment was ineffective in animal models with established cystic kidney disease (Pkhd1PCK rat and Pkd2(w s25/w183) mouse)[41].
Complement system
Complement system activation induces expression and release of multiple chemokines, including MCP-1[42, 43]. Also, many complement components are split into smaller fragments that directly regulate a wide range of PKD-relevant innate and adaptive immune responses[44, 45]. The activation of the complement system occurs mainly through three independent activation pathways (classical, alternative, and lectin) that converge on complement component 3 (C3)[46]. After its activation, the C3 convertase cleaves C5, which in turn activates the common complement pathway (C6-C9).
PKD1 mutations result in over-expression of multiple complement components in epithelial cells [7, 8] as well as in cystic kidneys from animal models[26]. As noted above, eleven complement system components were also over-expressed in mildly affected ADPKD kidneys, including all three components of C1 complex (C1q, C1r, and C1s), complement factor D, and C3. The C1 complex is the first component of the classical complement activation pathway, complement factor D catalyzes factor B cleavage (the rate-limiting step of the alternative pathway of complement activation), and C3 is the central component of the complement cascade that is activated by all complement activation pathways to form the C3 convertase (C3b). Notably, this protein remains biologically active even after its inactivation by C3b cleavage to a smaller fragment iC3b. iC3b is a major ligand that binds complement receptor 3 [CR3, also known as CD11b/CD18 or Macrophage Receptor 1; Mac-1[47]]. Mac-1 is an essential regulator of monocyte and macrophage survival and alternative (M2; repair and fibrosis-promoting) activation[48]. Such activation was associated with cystic disease progression in PKD models[23]. Also, mass spectrometry identified C3 as the most abundant protein in the cyst fluid from Pkd1 deficient mice and ADPKD patients [49, 50]. While C3 is expressed in an inactive form, cyst fluid and urine from ADPKD patients contain C3 fragments that are activated[51]. So far, the value of urinary excretion of C3 or other activated complement fragments as markers of the disease activity was not systematically evaluated. An association between a hypoactive C3 variant genetic polymorphism and slower progression to ESRD in a limited set of ADPKD patients [52] points to the potential utility of C3 genotyping in early prediction of ADPKD outcomes; however, this study will require validation in a larger cohort of ADPKD patients. The functional relevance of C3 in PKD pathogenesis was demonstrated in crosses of murine PKD models with C3-deficient (C3−/−) mice[25] and by the administration of non-selective C3 inhibitors in several PKD models[53, 54].
INNATE IMMUNE CELLS AND CYSTIC KIDNEY DISEASES
Introduction to renal innate immune system cells
The renal innate immune system is comprised of multiple cell types that are responsible for maintaining tissue homeostasis, fighting infection, and repairing damaged tissue. Innate cells are believed to be distinct from their adaptive counterparts due to their non-specificity and lack of long-term memory responses, although this paradigm has recently been challenged[55–57]. During the initial stages of disease, innate immune cells become activated, phagocytose foreign debris, and produce pro-inflammatory cytokines that may lead to tissue damage[58, 59]. In contrast, during the resolution phase, innate immune cells produce reparative cytokines that drive epithelial proliferation and tissue repair[60]. Collectively, the cells of the innate immune system are responsible for maintaining homeostasis and promoting the functional recovery of the kidney following injury.
Evolutionary conservation of the innate immune system
The innate immune system in mammals is thought to be comprised of neutrophils, natural killer cells (NK cells), dendritic cells (DCs), innate lymphoid cells, macrophages, basophils, eosinophils, and mast cells. It is commonly accepted that these cells are present in all mammalian species. However, the lack of evolutionarily conserved cell surface markers that can be used to identify a similar population of cells across species has prevented the validation of this hypothesis. With the advent of single-cell RNA sequencing, it is now possible to provide a more granular assessment of cells of the renal innate immune system in mice, humans, and other species. Further, the ability to sequence a large number of single cells has negated the need for antibody-dependent enrichment of the population of interest. Using single cell RNA sequencing, Zimmerman et al. recently showed that the cells of the mouse renal innate immune system were indeed comprised of macrophages, neutrophils, natural killer (NK) cells, innate lymphoid cells, dendritic cells, and a small population of other cells possibly consisting of eosinophils, basophils, and mast cells[61]. Cell surface protein markers commonly used to identify these cells are summarized in Figure 2. The authors also showed that a majority of renal innate immune cells are evolutionarily conserved across mammalian species, including humans, although the proportion of each cell as a percentage of CD45+ cells was different between species (Figure 3)[62]. Despite the broad evolutionary conservation, there were a few striking differences in the composition of the renal innate immune system from each species. For example, Ly6chi macrophages identified in the mouse using Ly6c2 were believed to be similar to CD14+ macrophages in the human[63, 64]; however, the data indicate that a majority of genes enriched in mouse Ly6chi infiltrating macrophages were not expressed by CD14+ monocytes in normal human kidney tissue[61]. The data also showed that the mouse kidney contained two distinct clusters of dendritic cells, while only one cluster was present in human kidney tissue. These data suggest that while cells of the innate immune system are broadly conserved across species, there are apparent discrepancies that must be recognized when trying to extrapolate data from mouse studies to human patients.
Figure 2. Murine cell surface markers and transcription factors commonly used to identify major immune cell types in the kidney.
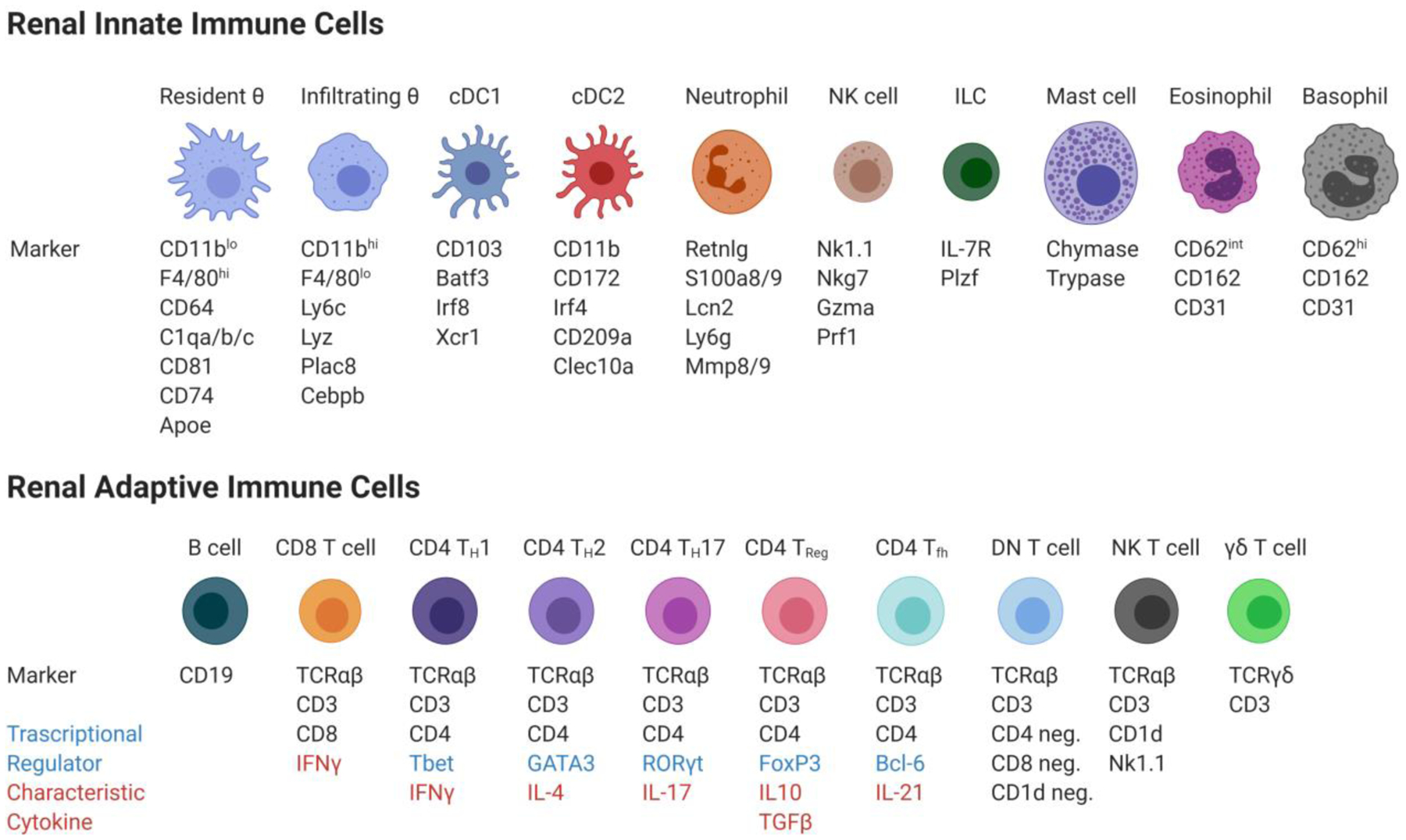
The renal innate immune cells include resident and infiltrating macrophages (Resident θ and Infiltrating θ), two dendritic cell populations (cDC1 and cDC2), neutrophils, natural killer cells (NK cells), innate lymphoid cells (ILC), mast cells, eosinophils, and basophils. The renal adaptive immune cells include B cells, CD8+ T cells, CD4+T helper type I cells (CD4 TH1), type 2 cells (CD4 TH1), type 17 cells (CD4 TH17), regulatory T cells (CD4 TReg), follicular helper cells (CD4 Tfh), double negative T cells (DN T cell), natural killer T cells (NK T cell) and Gamma delta T cells (γδ T cells).
Figure 3. Most renal innate immune cell types are evolutionarily conserved across mammalian species.
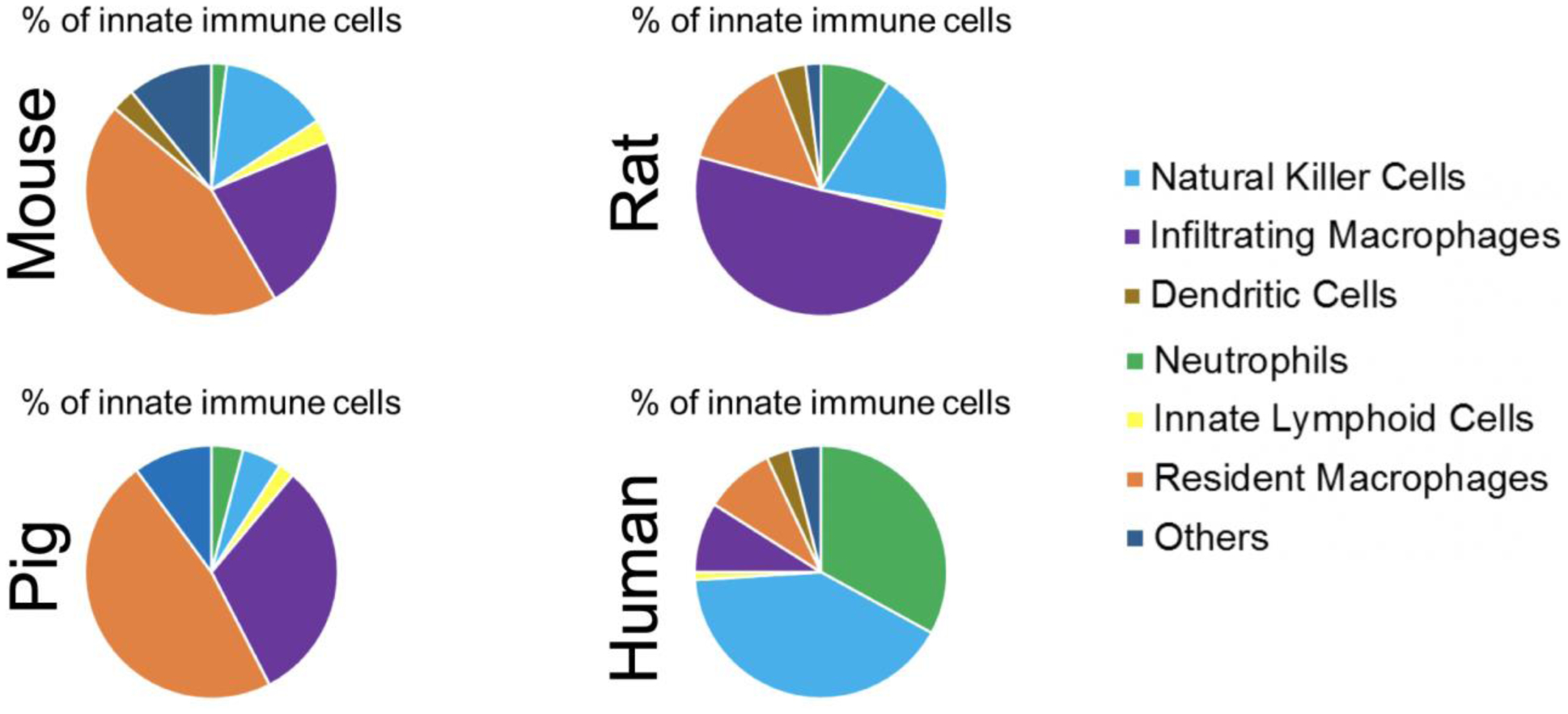
The above proportions were obtained from unaffected (wild type) kidneys. Renal immune cells are conserved among species although their proportion is different, even when comparing rat versus mouse. It is possible that these differences in immune cell proportions partially explain why altering immune cell function via genetic loss or pharmacologic intervention has a differing effect on PKD pathogenesis in mouse versus rat models.
The mononuclear phagocyte system
The mononuclear phagocyte system in the kidney is comprised of macrophages and dendritic cells, although the number, function, and location of each population differ depending on the marker used and the reporting investigator. For example, CD11c (encoded by the Itgax gene) was originally believed to be a lineage-defining cell surface marker of dendritic cells[65]. However, recent data indicate that CD11c is expressed by both dendritic cells and macrophages in the kidney, clouding the interpretation of previous studies that relied on this marker[65, 66]. Using the single-cell RNA sequencing approach, Zimmerman et al. provided substantial evidence, at the transcriptional level, that there are separate and distinct populations of macrophages and dendritic cells in the mouse kidney[61]. They also identified distinct transcriptional markers that could separate these cells into each compartment. This data is supported by a recent study indicating that renal macrophages and dendritic cells have distinct morphologies and functions in the kidney[67]. In this study, dendritic cells could be identified using Snx22 and Zbtb46 and these markers were not present in renal macrophages. Further, the deletion of cDC1 or cDC2 dendritic cells using Batf3-deficient and Zbtb46-DTR mice, respectively, resulted in opposite outcomes during antibody-induced kidney injury[67]. Together, these data indicate that macrophages and dendritic cells are functionally discrete populations in the kidney.
Origin of renal macrophages
For several decades, it was hypothesized that all macrophages in the kidney were derived from the bone marrow. However, seminal studies in the macrophage field challenged this paradigm and showed that renal macrophages can be separated based on their ontological origin into bone-marrow-derived infiltrating and embryonic derived tissue-resident macrophages[68, 69]. Resident macrophages were initially identified by Schulz and colleagues who showed that mice deficient in myb, a transcription factor that is required for hematopoiesis, did not have reduced numbers of CD11blo F4/80hi resident macrophages in the kidney[68]. In contrast, the number of CD11bhi, F4/80lo infiltrating macrophages was substantially reduced in these animals. Follow up studies showed that CD11blo, F4/80hi resident macrophages have minimal replacement from hematopoietic bone marrow precursors in adult mice, resulting in the term resident macrophages[70, 71]. It was also shown that in adult mice, resident macrophages maintained their population through self-proliferation. However, this paradigm has recently been challenged by Salei and colleagues who demonstrated using a Clec9acre fate mapper mouse that a significant proportion of resident macrophages in adult mice had Clec9a expression history, indicating that these cells came from a common dendritic precursor (CDP)[72]. This was in contrast to F4/80hi macrophages that were isolated from postnatal day 2 and 14 mice that lacked Clec9a expression history, suggesting that these cells are embryonically derived. These data suggest that not all resident macrophages in adult mice have an embryonic origin and that a proportion of these cells are derived from bone marrow precursors. Despite the controversy in the origin and turnover of kidney resident macrophages in adult mice, it is accepted that CD11bhi, F4/80lo infiltrating macrophages are continually turned over in an adult kidney due to replacement from bone marrow progenitor cells. Infiltrating and resident macrophages work together to maintain homeostasis and to properly respond following an infection or injury.
Function of infiltrating and resident macrophage in the kidney
CD11bhi F4/80lo infiltrating macrophages in the kidney can be further subdivided based on cell surface expression of Ly6c. Ly6chi macrophages infiltrate into the kidney within 24–28 hours following injury, produce pro-inflammatory cytokines, and initiate clearance and removal of foreign debris[73]. These cells are derived from common monocytic precursors (CMP) in the bone marrow, which are distinct from the CDP lineage[74]. Upon reaching the kidney, inflammatory Ly6chi infiltrating macrophages downregulate Ly6c expression and transition to Ly6clo cells[75]. Ly6clo infiltrating macrophages produce wound healing and reparative cytokines that promote tissue repair and resolution of injury. A third predominant population of infiltrating macrophages expressing intermediate levels of Ly6c (Ly6cint) has also been reported[76]. In these studies, a distinct cluster of CD11bhi Ly6cint cells peaked 1 day after ischemia-reperfusion injury (IRI) followed by a gradual reduction over the 35 day time course[76].
It has been proposed that macrophages in the kidney are involved in developmental processes, including phagocytosis and clearance of apoptotic debris, ureteric bud branching morphogenesis, nephron formation, and blood and lymphatic vessel development[77]. As embryonic-derived resident macrophages are the major macrophage population present in the kidney during developmental periods, it is believed that resident macrophages have a critical role in regulating renal development. In support of a trophic role for resident macrophages during kidney development, mice lacking the CSF1 receptor that is expressed by resident macrophages have reduced kidney size compared to littermate controls[78]. In contrast, mice treated with CSF1 have an increased kidney weight to body weight ratio[78]. Since the CSF1 receptor is also expressed by the epithelium[79], it is possible that the putative role of CSF1/CSF1R signaling in kidney growth and development is not resident macrophage-specific. Studies using approaches that more specifically target kidney resident macrophages are needed to determine the importance of these cells in kidney development.
In addition to the proposed functions of resident macrophages during kidney development, it is thought that these cells are important in regulating the injury and repair process in the kidney. Data from Lever et al showed that the number of kidney resident macrophages is increased following IRI and that the transcriptional profile of these cells is similar to developing kidney resident macrophages [80]. As developing kidney resident macrophages produce factors that drive epithelial growth, it is likely that the reprogramed resident macrophages promote tubular proliferation, growth, and repair following IRI. Further, it would suggest that resident macrophages maintain a level of developmental plasticity that allows them to alter their function during disease settings. Kidney resident macrophages have also been implicated in Ift88, cpk, and Pkd1 models of renal cystic disease[81, 82].
Macrophage polarization in the kidney
Macrophages are involved in tissue development, repair, and homeostasis[83, 84]. They express surface markers, including F4/80, CD68, and CD11b in mice and CD14 and CD16 in humans, which are used to identify different macrophage populations. An interesting feature of macrophages is their ability to become polarized towards a pro-inflammatory or anti-inflammatory phenotype in response to signals found in the tissue microenvironment. This paradigm is supported by data showing two distinct responses to treatment with cytokines in vitro[85, 86]. Macrophages stimulated with LPS, or interferon-gamma demonstrate a pro-inflammatory Th1-like phenotype with potent antimicrobial and antitumor activity and are referred to as M1-like macrophages. In the kidney, M1 macrophages express the high levels of the cell surface marker Ly6c (Ly6chi), which is associated with the production of pro-inflammatory cytokines[87]. On the other hand, naïve macrophages treated with interleukin-4 or 13 produce an anti-inflammatory response and are referred to as alternatively activated or M2-like macrophages[88]. These macrophages are Ly6clo and produce anti-inflammatory cytokines. However, several studies demonstrated that macrophages cannot be clearly separated into M1 or M2 like states in vivo, but have a wide range of plasticity depending on their environment[8, 89–94]. It is now believed that macrophages that exist in vivo display a range of phenotypes that fall somewhere in between the M1 and M2 spectrum and that they can switch these phenotypes based on external cues. Intriguingly, evidence suggests that M1 macrophages can transition to an M2-like polarization following phagocytosis of necrotic or apoptotic cells[95, 96], demonstrating the importance of the tissue microenvironment on macrophage polarization.
Relevance of individual renal innate immune cell types in PKD
The presence of innate immune cells in cystic kidney disease was first discovered several decades ago. Since this initial observation, the majority of studies have focused on the involvement of macrophages in cystic disease. However, until recently, it was not known whether macrophages were causative in cystic kidney disease or accumulated in response to the expanding cyst. Despite the growing body of literature suggesting that macrophages directly cause cyst formation and progression, the involvement of other innate and adaptive immune cells remains relatively unexplored (Table 1). In this review, we will describe the relevant data connecting each innate and adaptive immune cell with cystic kidney disease in both mouse and human and discuss the relevant data supporting a causative or correlative role for each cell.
Table 1.
Involvement of immune cell types in PKD
| Cell type | Reported in mouse models of cystic disease | Reported in human PKD patients | Functional role in cystic kidney disease pathogenesis |
|---|---|---|---|
| Innate lymphoid cells (ILCs) | No | No | Unknown |
| Natural killer cells | No | No | Unknown |
| Eosinophils | No | No | Unknown |
| Basophils | No | No | Unknown |
| Dendritic cells (cDC1 and cDC2) | No | No | Unknown |
| Mast cells | No | Yes | Unknown |
| Neutrophils | Yes | Yes | Unknown |
| Non-specified macrophages | Yes | Yes | Yes |
| Infiltrating macrophages | Yes | Yes | Yes |
| Resident macrophages | Yes | No | Yes |
| B cells | Yes | Yes | Unknown |
| CD8 T cells | Yes | Yes | Yes |
| CD4 T cells | Yes | Yes | Unknown |
| DN T cells | Yes | Yes | Unknown |
| NK T cells | No | No | Unknown |
| ɣδ T cells | No | No | Unknown |
Mast cells
Mast cells are derived from hematopoietic bone marrow precursors and seed the kidney in regions adjacent to blood vessels and epithelial cells[97]. Although the number of mast cells is sparse in a normal healthy kidney, their numbers rapidly increase in diseases, including glomerulonephritis, diabetic nephropathy, and allograft rejection. In the kidney, mast cells produce tryptase and chymase, two proteases that promote inflammation and fibrosis[98]. In addition, mast cells are capable of producing cytokines, chemokines, and leukotrienes that drive recruitment and activation of inflammatory leukocytes[99]. The presence of mast cells in interstitial regions of end-stage kidney tissue from ADPKD patients has been reported[100]. In these studies, chymase protein was found in regions adjacent to interstitially localized mast cells. Further, 13 out of 14 kidney extracts from ADPKD tissues had chymase-like angiotensin II (AngII) generating capacity. Interestingly, AngII is implicated in the generation of inflammatory and chemoattractant cytokines[101]. These data suggest that mast cells may promote cyst formation in an AngII-dependent manner. However, mouse models deficient in mast cells, such as the C57BL/6-KitW−sh/W−sh mouse[102], are necessary to determine the involvement of these cells in cystic disease.
Neutrophils
Neutrophils are also derived from hematopoietic stem cell progenitors in the bone marrow and comprise the main immune cell type in human blood. These cells are the first responders following kidney injury, accumulating in the kidney within hours following ischemic injury[103, 104]. Upon entering the kidney, neutrophils participate in the phagocytosis of bacteria, the release of serine proteases and matrix metalloproteases, the production of reactive oxygen species, and the release of neutrophil extracellular traps (NETs). The presence of excess neutrophils in human patients[105] and mouse models [106] of cystic disease has been reported. A potential limitation of these studies is the use of CD68 to show that neutrophils were located in the interstitial region of human patients with ADPKD. While this protein is likely to be expressed by neutrophils based on single-cell RNAseq data[107], it is also expressed by multiple other myeloid populations. Thus, further studies using more specific markers of human neutrophils in patients with cystic kidney disease are warranted. It has also been reported that blood neutrophil counts are significantly elevated in ADPKD patients compared to healthy controls[108] and that kidney neutrophil numbers are increased in a canine model of cystic kidney disease models[109]. Future studies using neutrophil depleting antibodies in mouse models of cystic disease are needed to delineate the functional role of these cells in PKDs.
Macrophages
The presence of macrophages in cystic diseases has been recognized for almost three decades. Until recently, these cells were considered to be a homogenous population that could be characterized as M1 or M2 based on their in vitro polarization status. However, recent data indicate that macrophages are not M1 or M2 in vivo, but rather, exist in a spectrum between these two distinct states. The recent re-characterization of macrophages based on ontological origin (infiltrating and resident) allows researchers to better identify, functionally characterize, and pharmacologically target these cells during disease progression. Therefore, we will separate the macrophage section of this review into studies that identify cells as general macrophages, infiltrating macrophages, or resident macrophages.
Studies of general macrophages in cystic disease
The first report claiming an association between macrophages and cystic kidney disease in patients came in 1992 when Zeier and colleagues identified CD68+ macrophages in interstitial regions of human ADPKD kidneys[110]. The authors speculated that these cells may be involved in the interstitial fibrosis that was observed in these patients. Although this was the first study describing the involvement of macrophages in cystic disease, it should be noted that the macrophage-derived cytokines IL-1β and TNFα were detected in cyst fluid from ADPKD patients the previous year[37].
The first report of macrophages in an animal model of cystic disease came from Takahashi et al. who showed enhanced interstitial infiltrates, including macrophages and lymphocytes in the cortico-medullary region of DBA/2-pcy mice[111], which grossly and histologically resemble patients with ADPKD[112]. Follow up studies demonstrated the DBA/2-pcy mice also had increased expression of monocyte/macrophage chemoattractant cytokines leading to the hypothesis that these cells may be both producing and responding to these cytokines in cystic disease[113]. Although these were the first reports to study macrophages in genetic mouse models of cystic disease, previous data identified an association between interstitial inflammation and cystic disease in non-genetic models of renal cyst formation[114].
The first data suggesting that macrophages were more than simple bystanders in cystic disease came from Cowley and colleagues who showed that the level of interstitial inflammation correlated with the severity of fibrocystic disease in Han:SPRD rats[115]. In these studies, it was shown that the level of interstitial inflammation was increased in male Han:SPRD rats compared to female Han:SPRD rats and that male rats had worsened fibrocystic disease. These data led the researchers to hypothesize that inflammatory cells, including macrophages, may produce factors that influence the outcome of fibrocystic disease. Additional reports of macrophages in cystic regions of kat2J/kat2J mice were also reported[116]. The first conclusive immunohistochemical staining for macrophages in an animal model of cystic disease came in 2001 when Cowley and colleagues showed increased ED-1+ macrophages in interstitial regions of Han:SPRD rats[2].
The number of ED-1+ interstitial macrophages was also increased in the autosomal recessive Lewis polycystic kidney rat model of disease[117]. Although these data indicated that the number of interstitial macrophages were not significantly increased until large cysts were present, it must be noted that the number of ED-1+ cells was nearly doubled at the early stage of cyst development. More recently, Mrug and colleagues showed that genes involved in the innate immune response were significantly upregulated in severely cystic cpk mice, a nonorthologous model of ARPKD[26]. Of interest, several genes that were recently identified as being specific for resident macrophages by single-cell RNA sequencing (C1q, Cxcl16)[107] were among the most significantly upregulated in cpk mice[61]. The authors went on to show that Cd14 expression correlated better with total kidney volume than did MCP-1 expression in cpk mice[22]. Interestingly, although CD14 is typically associated with monocytes/macrophages, the increased expression of Cd14 in cpk mice was driven mainly by the epithelium and not interstitial inflammatory cells. Interstitial CD14+ mononuclear cells were also observed in the renal parenchyma of end-stage PKD patients [22]. Similar to the gene expression studies [26], Song and colleagues used an Affymetrix array to show that ADPKD kidneys had an enrichment of genes associated with immune/inflammatory pathways[8].
The first data showing that macrophages could promote cyst progression came from Karihaloo and colleagues[23]. In these seminal studies, the authors showed that F4/80+ macrophages accumulated in regions adjacent to cysts in both the C57Bl6 Pkd1fl/fl;Pkhd1-Cre and Pkd2WS25/− mouse models of ADPKD[23]. Based on flow cytometry data, the authors showed that the number of CD45+ F4/80+ CD11c- Ly6clo macrophages in Pkd1fl/fl;Pkhd1-Cre+ mice was increased compared to controls and that expression of both MCP-1 and Cxcl16 was significantly increased in Pkd1−/− tubular epithelial cells compared to controls. Notably, treatment of Pkd1fl/fl;Pkhd1-Cre+ mice with the phagocytic poison, liposomal clodronate, significantly reduced macrophage number (95% reduction), cystic index, %KW/BW, and BUN compared to liposomal vehicle-treated mice[23]. Treatment with liposomal clodronate also reduced cystic epithelial proliferation compared to liposomal vehicle treated mice suggesting that kidney macrophages promoted cystic epithelial proliferation, as originally hypothesized by Cowley.
These data were supported by follow-up studies from Swenson-Fields et al. who looked at the involvement of macrophages in human patients and mouse models of ARPKD[118]. Their data showed that CD163+ macrophages were scattered throughout the interstitium and in cystic regions of ADPKD and ARPKD human kidneys. The ability of macrophages to differentiate into M2-like cells was enhanced by co-culture of bone-marrow-derived macrophages with the cystic epithelium. Further, the M2-like macrophages produced soluble factors that promoted enhanced proliferation in the ADPKD cystic epithelium. Similar to the previous study in an ADPKD mouse model, the number of F4/80+ CD11c- Ly6clo macrophages were significantly increased in cpk mouse kidneys compared to controls[118]. Treatment of cpk mice with liposomal clodronate once again reduced KW/BW ratio, F4/80+ macrophage number, cystic index, and BUN.
Additional data indicate that the cyst lining epithelium produces macrophage migratory inhibitory factor (MIF) and that this protein was required for the migration and retention of macrophages in Pkd1 deficient mice[119]. Genetic deletion or pharmacological inhibition of MIF in Pkhd1cre or Kspcre Pkd1f/f mice delayed cyst growth. Finally, recent data indicate that Arginase 1 expressing macrophages are involved in direct cell-cell communication with the cilia-dysfunctional epithelium[120] in conditional Pkd1 mice induced at postnatal day 10 (P10). Inhibition of Arg1 activity significantly reduced epithelial proliferation and cyst growth in this model. Finally, using in vitro approaches, the authors showed that cyst lining epithelial cells produced L-lactic acid that promoted the formation of Arg1+ macrophages and that Arg1+ macrophages could directly stimulate cyst lining epithelial proliferation[120].
A recent study looked at the involvement of micro RNAs in modulating inflammatory signaling, macrophage accumulation, and cyst growth in Pkd1 and Pkd2 mutant mice and patients. In these studies, the authors showed that micro RNA 214 (miR-214) and its host lncRNA DNM3OS were upregulated in mouse models and patients with ADPKD and localized to interstitial cells[121]. Genetic deletion of miR-214 enhanced cyst-associated inflammation, Mrc1+ macrophage accumulation in cystic regions, and worsened cystic outcome. In these studies, it was proposed that the miR-214 host gene was activated in a TLR4/INF-γ/Stat4 dependent manner[121]. This study brings about the exciting possibility that macrophages produce or respond to miRNAs to modulate inflammation and cystic kidney disease.
Studies on infiltrating macrophages in PKD
As noted above, infiltrating macrophages are bone-marrow-derived cells expressing high levels of CD11b and low levels of F4/80 (CD11bhi, F4/80lo). These cells use chemoattractant gradients to migrate to the site of tissue injury or infection. The most common chemoattractant that promotes infiltrating macrophage recruitment is MCP-1 (encoded by Ccl2 gene). This chemoattractant binds to its cognate receptor (CCR2; Ccr2) expressed on monocytes to mediate trafficking to the site of injury. Previous literature indicates that genetic deletion of MCP-1 or Ccr2 results in a significant reduction in monocyte recruitment[122]. Importantly, the mechanism through which each genetic knockout inhibits infiltrating macrophage accumulation is different. Ccr2 deficient mice have reduced infiltrating macrophage accumulation in the kidney due to the inability of Ccr2 deficient monocytes to exit the bone marrow[123]. In contrast, the inability of MCP-1 deficient mice to recruit infiltrating macrophages to the site of kidney injury is due to the lack of the chemoattractant gradient[124]. Since the MCP-1/Ccr2 interaction regulates the recruitment and bone-marrow emigration of Ly6chi infiltrating macrophages, this is the major population that would be predicted to be affected in both models. Although significant data supports the idea that Ly6chi infiltrating macrophages transition to Ly6clo infiltrating macrophages, loss of MCP-1 or Ccr2 has a minimal effect on this population. This suggest that some Ly6clo infiltrating macrophages can accumulate in the kidney independent of Ly6chi cell recruitment.
The first study that directly evaluated infiltrating macrophages in cystic kidney disease was done in the pck rat (Pkhd1pck)[32]. In this study, bindarit, a compound that inhibits MCP-1 synthesis, was used to test the effect of blocking infiltrating macrophage recruitment on cystic kidney disease. The studied pck rats were treated with bindarit from 5 to 15 weeks of age. This treatment resulted in a ~50% reduction in ED-1+ interstitial macrophages, reduced proteinuria, and fewer tubular casts[32]. Surprisingly, bindarit did not improve renal or hepatic cystic indices. Based on these data, the authors concluded that the improved renal function observed in bindarit treated rats was due to reduced podocyte damage. These initial studies dampened the enthusiasm for studying infiltrating macrophages in cystic disease and brought back doubts about whether macrophages were causative or correlative in cystic kidney disease.
To better study infiltrating macrophages in the context of cystic kidney disease, Viau and colleagues used inducible Lkb1 and Pkd1 mouse models of renal cystogenesis crossed to the MCP-1 deficient mouse[125]. Lkb1 is a cilia-localized protein that serves as a metabolic sensor, specifically in the cilium. Crossing the Lkb1f/f mouse to the Kspcre line results in cortico-medullary renal cysts and interstitial fibrosis approximately 14 weeks after birth. Tubular deletion of Lkb1 resulted in increased MCP-1 expression and increased numbers of both CD11bhi F4/80lo infiltrating macrophages and CCR2+ CD11blo F4/80hi resident macrophages at 10 weeks of age[125]. Whether the latter are infiltrating macrophages masquerading as resident macrophages or simply resident macrophages that have upregulated CCR2 expression independent of bone marrow recruitment is unknown. The authors also showed that an intact primary cilium was required for induction of MCP-1 expression in the context of cilia dysfunction. Tubular deletion of Pkd1 using Pax8 rtTA tetocre resulted in increased MCP-1 expression which was significantly reduced when primary cilia were subsequently deleted[125]. In parallel, the conditional deletion of Pkd1 resulted in a significant increase in both CCR2+ infiltrating and resident macrophages, and the deletion of cilia in this model substantially reduced the number of both macrophage groups and the severity of renal cysts. Finally, inhibition of monocyte recruitment using an MCP-1 knockout mouse on the conditional Pkd1 background significantly reduced cystic index, F4/80+ macrophages, and CD11b mRNA expression. It remains unknown if the deletion of MCP-1 reduced cystic severity by inhibiting the recruitment of CCR2+ infiltrating macrophages, decreasing CCR2+ resident macrophage function or survival, some additional mechanism, or a combination of all possibilities.
The extensive mechanistic data presented by Viau and colleagues was supported by an additional publication from Cassini et al.[27]. This study showed that tubular deletion of both MCP-1 and Pkd1 significantly reduced cystic index compared to tubular deletion of just Pkd1. Of interest, the authors showed that the macrophages found in regions adjacent to early developing cysts promoted tubular injury, oxidative damage, and proliferation, independent of cyst formation[27]. In contrast, during the later stages of cyst progression, the macrophages became alternatively activated and promoted cystogenesis in a proliferation dependent manner.
Together, these data suggest that the recruitment of inflammatory cells is required for cyst progression. However, a recent manuscript questions this conclusion. In these studies, genetic disruption of CCR2, the receptor for MCP-1, did not reduce macrophage recruitment or impact cystic disease in a cpk mouse model[126]. However, CCR2 deficient cpk mice did survive longer compared to controls[126]. Likewise, genetic deletion of CCR2 did not impact liver cysts in an Ift88Orpk mouse but did reduce the severity of fibrosis[127].
Resident macrophages in cystic disease
Zimmerman et al provided the first evidence for the involvement of resident macrophages in cystic disease using conditional Ift88 mice and renal IRI to model accelerated cyst formation. They showed that a specific subpopulation of CD11blo, F4/80hi resident macrophages expressing low levels of CD11c were increased in conditional Ift88 mice compared to controls prior to and during cyst formation[82]. These macrophages were located directly adjacent to renal cysts and expressed Ki67 indicating that they were undergoing rapid proliferation. Further, the conditional Ift88 epithelium had increased expression of cell surface CSF1, a protein that promotes the proliferation of kidney resident macrophages in a CSF1R dependent manner. Inhibition of CSF1/CSF1R dependent signaling using GW2580 significantly reduced CD11clo resident macrophage accumulation without impacting the number or proliferation of infiltrating macrophages[82]. The data also showed that GW2580 treatment could reduce cyst progression in the injured conditional Ift88 model and in the rapidly progressive cpk mouse model.
These data were followed up by studies using a unilateral nephrectomy model of accelerated cystogenesis in conditional Pkd1 mice[81]. Using these mice, Zimmerman and colleagues showed that the number of infiltrating and resident macrophages are increased in conditional Pkd1 mice receiving unilateral nephrectomy compared to sham operated mice and that the increase occurs prior to the onset of severe cystogenesis. In addition, the authors found that a transcription factor that is known to induce cytokine production in macrophages, IRF5, is increased in infiltrating and resident macrophages from conditional Pkd1 mice undergoing unilateral nephrectomy[81]. IRF5 antisense oligo treatment of conditional Pkd1 mice receiving unilateral nephrectomy reduced expression of Irf5 and Il6 specifically in resident macrophages, which was associated with reduced Stat3 phosphorylation in the cystic epithelium and reduced expression of p-Stat3 target genes in whole kidney tissue[81]. These data suggest that IRF5 is a master regulator of IL-6 production in resident macrophages and that resident macrophage localized IL-6 promotes epithelial cell activation in an IL-6, Stat3 dependent manner.
THE IMPACT OF ADAPTIVE IMMUNE CELLS ON RENAL PATHOLOGIES INCLUDING POLYCYSTIC KIDNEY DISEASE
Fundamental Features of the Adaptive Immune Response
Increasing amounts of evidence point to the adaptive immune system as an important modulator of renal pathobiology. In addition, awareness of the interplay between immune cells and renal epithelial cells has opened up new avenues for renal disease treatment[128–130]. Adaptive immunity is characterized by antigen specificity/diversity and immunological memory. It is historically subdivided into humoral and cell-mediated responses induced by B and T lymphocytes, respectively; each type having specific subtypes and marker (Figure 2). However, B cells have also been shown to play a critical role in cell-mediated immunity.
Humoral immunity is initiated by B cell recognition of antigen through the B cell receptor (BCR) and can occur dependent or independent of T cell involvement[131, 132]. A T cell-independent antibody response is predominantly elicited to multivalent (e.g., polysaccharide) antigens, produces low-affinity, short-lasting IgM antibodies, and is preferentially executed by marginal zone (MZ) B cells in the spleen/secondary lymphoid organs or B1 B cells in mucosal tissue and the peritoneum. In contrast, follicular (FO) B cells in secondary lymphoid organs are considered the conventional B cell and elicit a T cell-dependent antibody response by presenting its activating protein antigen on MHCII to CD4+ follicular helper T cells (TfH). This developing interaction produces high-affinity, long-lasting IgG, IgA, and IgE antibodies through isotype switching[131, 132]. Beyond antibody production leading to extracellular microbe neutralization, phagocytosis, and complement activation, B cells are also known enhancers of cell-mediated immunity. Due to their endocytic/antigen processing capacity, B cells are able to act as antigen-presenting cells (APCs) to both CD4+ and CD8+ T cells, a function that has been shown to ensure optimal T cell activation, cytokine production, and memory T cell generation[133, 134]. Further, B cells can release a variety of pro- and anti-inflammatory cytokines that have been associated with immune-mediated renal pathologies as well as T cell polarization/activation[131, 135, 136]. Lastly, all B cell subpopulations have been shown to differentiate into regulatory B cells (Bregs), predominantly characterized by the production of IL-10[137]. Bregs are able to modulate innate cells, suppress TH1 and TH17 cell responses, and promote TReg proliferation, all hampering an ongoing immune response[131, 138, 139].
Cell-mediated immunity is initiated by naïve T lymphocytes recognizing through their T cell receptor (TCR) an antigen presented by MHC I or II. This encounter triggers an activation signaling cascade in the T cell, which generally occurs in secondary lymphoid organs, and promotes proliferation to produce effector T cells. The family of effector T cells is large, containing multiple subtypes (CD4+, CD8+, γδ, Natural Killer-like [NK] T cells), and subtypes of subtypes (e.g., CD4+ TH1, TH2, TH17; Figure 2); all with vastly different characteristics and functions depending on the costimulatory and cytokine milieu[132, 140]. Generally, CD4+ helper T cells, TH, recognize antigen presented by MHC II on activated APCs and aid in coordinating and promoting polymorphonuclear and innate immune cell function[132, 141]. CD4+ TH1 cells produce the pro-inflammatory cytokine INFγ, which acts on macrophages promoting their differentiation into M1-like macrophages that ingest and destroy microbes. They also secrete IL-2, which helps effector T cells to become fully activated after antigen recognition. CD4+ TH2 cells secrete IL-4 and IL-13, stimulating M2-like macrophage activation, which are anti-inflammatory and involved in tissue repair. IL-4, together with IL-5, another TH2 produced cytokine, are further responsible for eosinophil recruitment and activation, which specialize in the killing of parasites. CD4+ TH17 cells primarily secrete IL-17, which induces neutrophil-rich inflammation responsible for destroying extracellular bacteria and fungi. However, IL-17 also promotes M1-like macrophage activation. Beyond these subtypes, other TH subtypes have been described, and their above described simplified function and sub-classification is ever-evolving. Another small but potent subpopulation of CD4+ T cells are regulatory T cells (TRegs), marked by the expression of the transcription factor Foxp3. Through the production of IL-10 and TGFβ their primary function is to suppress immune responses and maintain self-tolerance. Although TRegs respond to a specific antigen, their suppression is not antigen-specific[132, 142]. In contrast to CD4+ T cells, CD8 expressing T cells directly act on MHC I cells expressing their recognized cognate antigen. Upon engagement, CD8+ T cells induce apoptosis of the infected cell, hence their name cytotoxic T lymphocyte (CTL), via two pathways; release of cytotoxic granules containing granzyme B and perforin, or engagement of the FasL/Fas ligand/receptor pair on CTLs/target cell, respectively. CTLs also secrete INFγ and, like TH1, contribute to phagocytic clearance of ingested microbes by M1-like macrophages[132].
Beyond the classical CD4+ or CD8+ T cells expressing CD3 and TCRαβ, other T cell subtypes have been shown to be important in kidney disease. These include γδ T cells expressing CD3 and TCRγδ, which recognize antigen in an MHC independent way by responding to a non-processed antigen such as lipids and heat chock proteins and produce INFγ and IL-4 in response to TH1 or TH2 stimulating pathogens, respectively[143, 144]. Further, natural killer T (NKT) cells, another renal T cell subset, express TCRαβ, and recognize lipids bound to CD1, a class I MHC-like molecule. NKT cells help marginal zone B cells to produce antigens against lipids and have, like γδ T cells, been shown to secrete INFγ and IL-4[145]. Double negative (DN) T cells, expressing TCRαβ but lacking CD4 and CD8, have also been found in the kidney and shown to produce the anti-inflammatory cytokine IL-10[146]. Lastly, it is important to note that the kidney is somewhat unique in that many different T cell subtypes establish local residency, even though, typically, lymphocytes are known to be highly mobile cells[147]. In fact, in the kidney, it has been suggested that a substantial proportion of adaptive lymphocytes display surface markers of tissue-residency (i.e. CD69+)[147]; however, the reason for and impact on renal disease of this local lymphocyte hub needs further investigation.
Adaptive Immunity-Kidney Crosstalk in Acute Kidney Injury
Subclinical AKI or ischemic stress has been proposed as an important factor contributing to ADPKD progression and heterogeneity. The crucial link between the two was demonstrated by multiple murine studies highlighting that tubular injury accelerates renal cystogenesis[16–18, 20] and that heterozygous ADPKD gene loss sensitizes the kidney to interstitial inflammation and fibrosis[106]. Along those lines, ADPKD kidneys and kidneys responding to ischemic damage share multiple pathophysiological characteristics, including renal epithelial cell dedifferentiation, tubular basement membrane remodeling, and immune cell infiltration[148]. Further, signaling cascades such as Hif1α induction[20, 149], mTOR[150, 151] or STAT6[148] activation, and Wnt/β-catenin deregulation[152, 153] are common between the two pathologies. Hence, it is not surprising that injury repair processes are a hallmark gene signature of ADPKD[154] and that AKI is seen as an epistatic modulator of the disease.
Adaptive immunity has been shown to be a key regulator of AKI response, although the current literature of whether B and T cells are protective or pathogenic is conflicting. In respect to B cells, it has been reported that, in mice, renal B cell numbers significantly increase during the AKI repair phase (~3–10 days post IRI) but not the acute phase (~3–72h post-IRI)[155, 156]. However, their functional role is controversial. Two studies suggested that B cells impair renal repair, as B cell deficiency either in μMT mice or via anti-CD126 treatment increased epithelial cell proliferation and reduced tubular atrophy[155, 157]. Here, the suggested pathogenic mechanism of B cells was via secretion of IgM and its deposition[157, 158]. However, another study showed that only depletion of peritoneal B cells prevents the deposition of IgM and attenuates renal injury post-IRI while μMT mice, lacking all B cells, sustain worse renal injury post-IRI, suggestive of a protective role[159]. It is important to note that the time point when kidney injury was assessed, acute vs. repair phase, and the mouse strain varied in these studies, but the findings advocate that different B cell subsets function distinctly during AKI response.
Like for B cells, delineating distinct T cell functions in AKI is complicated. The literature agrees that T cells accumulate in the kidney within a few hours to days after AKI[160], but contradictory observations have been made regarding their functional role. For CD4+ T cells, multiple studies using genetic Cd4 knock-out animals or adaptive transfer of CD4+ T cells in T cell-deficient mice suggested that CD4+ T cells are a major pathogenic factor in ischemia or cisplatin-induced AKI[161–163]. However, other studies found that Cd4 knock-out mice are not protected against cisplatin-induced renal dysfunction[164] or that anti-CD4 treatment worsens aristolochic acid-induced acute tubular necrosis[165]. Hence, whether CD4+ T cells are pathogenic or protective remains unclear, and it is likely that different CD4+ T cell subtypes have distinct functions during AKI response. This has been proven using Stat4 and Stat6 knock-out mice that lack proteins regulating CD4+ TH1 or TH2 effector subtype differentiation, respectively. Stat6−/− mice presented with worse renal function and marked increase in tubular injury post-IRI compared to wildtype mice, while Stat4−/− mice were modestly protected from renal IRI compared to wildtype, highlighting that CD4+ T cells of the TH1 phenotype are potentially pathogenic and those of the TH2 phenotype are likely protective[166]. Another CD4+ T cell subtype that has been found to be protective are CD4+ TRegs, which are known for their anti-inflammatory action. Using genetic models or antibody-based depletion strategies pre- or post-renal IRI or cisplatin injury, TReg deficiency most often resulted in enhanced renal inflammation, tubular necrosis, and increased loss of renal function[167–170]. One exception is a study of septic AKI where TReg depletion protected mice from renal dysfunction and improved survival[169]. The critical role of TRegs has also been proven in ischemic preconditioning, a powerful intervention to protect kidneys from reperfusion injury[171, 172]. Importantly, TRegs are a powerful therapeutic target as TReg adoptive transfer has been performed in humans[173], and because agents promoting TReg recruitment have been demonstrated to protect against IRI[174, 175].
The functional role of CD8+ T cells in ischemic or cisplatin-induced renal injury is less well known. This is primarily due to initial studies suggesting that they are dispensable during AKI response[161, 163, 176]. However, CD8+ T cells have also been found to be either detrimental or protective in AKI depending on the model used[162, 165]. Hence, further studies are required to elucidate more precisely their roles in AKI. Similarly, future studies are needed to understand the functional importance of γδ, NK, and DN T cells in AKI. Current literature suggests that NKT cells are renoprotective[177, 178] while γδ T cells are pathogenic[179] in renal IRI. Renal DN T cells have been shown to expand significantly post IRI and the adoptive transfer of these cells has been shown to reduce ischemic AKI in mice[146, 176, 180].
Adaptive Immunity in PKD
To date, there is limited literature on the role of the adaptive immune system in PKD. Historic histopathology studies of non-orthologous ADPKD models[111, 115, 116, 181] as well as end-stage renal disease (ESRD) ADPKD patients[110] report that lymphocytes together with monocytes infiltrate the cystic renal cortex either as individual cells or in small aggregates. Further, cyst fluid of ADPKD patients contains lymphokines such as IL-2[37], suggesting that adaptive immune cells may play a role in disease pathology. More recently, Kleczko et al. studied the role of T cells in the Pkd1 p.R3277C (Pkd1RC/RC) mouse model in more detail and found that both renal CD4+ and CD8+ T cell numbers increase correlative with disease severity[182]. Interestingly, even in kidneys with only a few cysts, renal T cell infiltrate was two-fold higher compared to wildtype and T cells specifically localized to cystic lesions. Further, T cell recruiting chemokines (Cxcl9/Cxcl10) were significantly increased in PKD compared to wildtype murine kidneys and found to be secreted by cystic epithelial cells as well as cells within the renal interstitium. This observation was confirmed in ADPKD and ARPKD patients. Importantly, immunodepletion of CD8+ T cells resulted in the worsening of ADPKD, coinciding with decreased epithelial apoptosis and increased epithelial proliferation. Hence, this study was the first to suggest a functional renoprotective role of CD8+ T cells in ADPKD pathology. Concurrent with the prior studies, Zimmerman and colleagues recently reported peri-cystic localization of T cells as well as an increase of CD4+ and CD8+ T cells in ADPKD patient kidneys compared to normal controls [183]. Interestingly, T cells were also identified in the urine of the majority of tested ADPKD patients (n=30). Urinary CD4+ T cell count correlated moderately with eGFR decline slope, similar to kidney length. Next to providing additional support for the potential role of T cells in ADPKD pathology, this study suggests that urinary T cells may be a potential biomarker of disease activity[183], as has been reported for proliferative lupus nephritis[184, 185].
Utilizing branches of adaptive immunity as an ADPKD biomarker has also been proposed earlier. For example, Soleimani et al. showed that in a population of 54 ADPKD patients and 54 healthy controls, serum or urine levels of the lymphokine IL-17 is highly accurate in predicting ADPKD[186]. Similarly, Lee et al. showed that in their population of 90 ADPKD patients and 21 healthy controls, SEMA7A expression on immune cells might be a potential disease biomarker[187]. SEMA7A is a membrane-bound glycosylphosphatidylinositol-linked protein expressed on various leukocytes, including activated T cells, and has been shown by others and Lee et al. to play a critical role in lung and renal fibrosis[187–189]. Interestingly, in this study, Lee at al found increased SEMA7A levels on circulating monocytes as well as CD4+ T cells, including TRegs of ADPKD patients compared to controls. Further, SEMA7A levels decreased in tolvaptan treated versus tolvaptan-naïve ADPKD patients. This suggests, that in ADPKD, the SEMA7A pathway may play a role in renal fibrosis and that SEMA7A level on innate or adaptive immune cells may indicate disease severity or therapy response. However, more research is needed to substantiate these findings.
Beyond these studies, nothing is known about the functional role of B cells, CD4+ T cell subpopulations, or other T cell types (NKT, γδ T cells, DN T cells) in ADPKD. Zimmerman et al highlights that renal DN T cell numbers increase in ADPKD patients, but that increase is correlative with an overall increase in infiltrating immune cells and T cells[183]. The same was found to be true in the Pkd1 p.R3277C model, with the additional observation that renal DN T cell numbers decrease as percent of total renal T cells in the C57Bl/6 strain, which presents with milder disease compared to Balb/C Pkd1RC/RC mice in which DN T cell numbers as percent of total renal T cells remained constant when compared to wildtype[190].
PC-1 and PC-2 have also been shown to regulate immune response and proliferation of B cells and T cells; although, the relevance of these changes to PKD pathogenesis remains unknown. For example, B-lymphoblastoid cells (LCLs) derived from ADPKD patients express both PC-1 and PC-2-encoding genes and induce Ca2+ release upon platelet-activating factor (PAF) stimulation. LCLs from ADPKD patients also have reduced proliferation when compared to LCLs derived from healthy controls[191]. Similarly, circulating B cell counts are lower but still within the normal range in ADPKD transplant patients (n=126) vs. non-PKD transplant recipients (n=574)[192]. T cells also express PC-1 and PC-2 but at a lower level than LCLs [193]; however, the Ca2+ release was only compromised in circulating T cells from patients carrying PC-2, but not PC-1 mutations. Interestingly, T cells derived from both patients with PC-1 and PC-2 mutations showed increased proliferation and increased homotypic T cell aggregation, a mechanism involved in T cell activation[193]. Further, expression of NFκB and macrophage migration inhibitory factor (MIF) was increased in patient-derived T cells[193]. The effects of fibrocystin in adaptive immune cells remain unknown; although, a fibrocystin-like protein, Pkhdl1, is predominantly expressed in T lymphocytes[194].
CONCLUSIONS AND PERSPECTIVES
Based on the data presented in this review, there are several unanswered questions regarding the involvement of immune cells in cystic disease. Perhaps most obvious is the glaring lack of data investigating the importance of non-macrophage immune cells in cystic disease. Although data from multiple labs suggest that macrophages are critical in the process of cyst initiation and progression, this does not mean that other immune cells are not important in promoting or inhibiting cystic disease.
One interesting area for debate is the importance of different macrophage populations in cystic disease. Based on seminal studies, it is presumed that M2-like macrophages are important promoters of cyst formation or progression. The question is, how do these cells stratify into the new macrophage classification system? Are all M1 macrophages derived from the bone marrow while all M2-like macrophages are resident macrophages? If this is the case, it may be possible to harness the novel genetic and pharmacological approaches that have been developed to inhibit the accumulation of these cells for the treatment of cystic disease. For example, available data support the use of CCR2 antagonists to reduce the recruitment of bone marrow monocytes to the site of injury (cysts). Further, data indicate that it is possible to use the CSF1R kinase inhibitor to prevent the accumulation of kidney resident macrophages[82]. Since robust pharmacological approaches exist to target each population, it will be important to rigorously characterize the involvement of each population in cystic disease using both ADPKD and ARPKD mouse models.
It is also interesting to debate the importance of infiltrating monocyte-derived macrophages in cystic kidney disease. Pioneering studies in the field of macrophages and cystic disease form Karihaloo and Swenson-Fields showed that global deletion of all macrophages significantly attenuated cyst formation[23, 118]. After seminal studies from Zoja et al. showed that inhibition of infiltrating macrophage accumulation using Bindarit had no impact on cyst formation in the pck rat, it was presumed that infiltrating macrophages were dispensable for cyst formation[32]. However, two recent papers question this result as genetic deletion of MCP-1 significantly attenuated cystic disease in conditional Pkd1 mice[27, 125]. Interestingly, mechanistic studies from Viau showed that mice lacking primary cilia (Pax8 rtTA Kif3af/f mice) and Pkd1 had minimal increases in MCP-1 mRNA compared to mice lacking just Pkd1[125]. These data suggest that a dysfunctional primary cilium (i.e. one lacking PKD1/PKD2) is required for MCP-1 induction and that the mechanism of cyst formation may be different in mice lacking primary cilia compared to mice with primary cilia dysfunction. However, studies in the pck rat (Pkhd1 mutation)[32] and recent work in the cpk mouse (Cys1 mutation)[4] show that inhibition of infiltrating macrophage accumulation does not prevent cyst formation even though these animals contain dysfunctional primary cilia. This would suggest an additional layer of complexity with each type of cilia mutation resulting in a different mechanism of cystogenesis. For example, it is conceivable that conditional Pkd mice, which model ADPKD, have increased expression of MCP-1, resulting in increased infiltrating macrophage-dependent cyst progression. In contrast, mutation of Cys1 or Pkhd1, which result in an ARPKD-like phenotype, may induce cyst formation by recruiting and activating a different subtype of macrophage (such as resident macrophages) or cyst formation in these models may be macrophage independent. Additionally, the differential effects of MCP-1 inhibition/knockout on cyst growth in mouse and rat models of cystogenesis may be due to the level of macrophage inhibition achieved by either approach (i.e. inhibitors are less effective at inhibiting macrophages than genetic knockout animals) or the composition of immune populations across species (Figure 3). However, before any conclusion can be drawn, a careful examination of infiltrating and resident macrophage involvement in dominant and recessive models of cystogenesis using flow cytometry and immunofluorescence microscopy approaches must be performed.
The involvement of adaptive immunity in cystic kidney disease is starting to be uncovered. Much like the macrophage field, a majority of studies at the early stage of this emerging field are descriptive and report correlations between T cell numbers and disease severity[183]. However, a recent study by Kleczko et al. provided the first mechanistic data and suggested that CD8 T cells could have a direct impact on cystic outcome[195]. In these studies, it was hypothesized that CD8 T cells were recruited to cystic regions in response to an epithelia-produced chemoattractant gradient, which included Cxcl9 and Cxcl10. Once in the cystic region, the authors proposed that activated CD8 T cells could restrict cyst growth, possibly through production of IFNγ. These groundbreaking studies lead to a plethora of additional questions: What are the chemoattractants that drive T cell recruitment? Are other T cell populations including NK T cells, CD4 T cells, and DN T cells involved in cystic disease? Do T cells in a cystic kidney recognize a cognate antigen? Do T cells from cystic kidneys become exhausted? What is the mechanism by which T cells modulate cyst growth? While several open questions remain, the recent discovery of drugs that induce activation of exhausted T cells, termed checkpoint inhibitors, brings about the exciting possibility that these drugs may be useful in PKD patients.
Overall, this review highlights the abnormal immune responses that are observed in cystic kidney disease including abnormal production of inflammatory mediators (e.g., cytokines, chemokines, and complement factors) that participate in the recruitment, proliferation, and differentiation of inflammatory cells (e.g., macrophages and T-cells). In turn, innate and adaptive immune cells cross-talk with renal tubular cells and can alter the severity of the renal cystic phenotype. Since PKD-associated immune factors also play a central role in the pathogenesis of renal tissue injury and repair, a factor that induces and accelerates cystogenesis, it is conceivable that PKD-induced immune responses modulate renal cystogenesis through participation in a PKD-dependent cycle of failed injury repair. As postulated by Thomas Weimbs, such a futile repair may be central to the pathogenesis of renal cystic phenotypes induced by different genetic defects[196]. Chronic activation of PKD-induced immune responses may also contribute to the loss of functional renal parenchyma due to alterations in vascular function and acceleration of interstitial fibrosis. While multiple cystogenesis-modulating components of the immune response have been identified, these findings point to a highly entangled network of biological processes and cellular interactions that alter renal cystic phenotypes. A better understanding of these complex interactions may lead to the discovery of more reliable markers of cystic kidney disease activity, more accurate predictors of the disease course in PKD patients, and identification of druggable targets for PKD.
Highlights.
Chemokines and their regulators that modulate progression of renal cystic diseases
The role of innate immune cells in cystic kidney diseases
The impact of adaptive immune cells on renal pathologies including polycystic kidney disease
Acknowledgements
Support was provided in part by the National Institutes of Health (NIH)-funded University of Alabama at Birmingham (UAB) Hepato/Renal Fibrocystic Disease Core Center P30DK074038 (to M.M) and K01DK114164 (to K.H.), 1I01BX004232-01A2 from the Office of Research and Development, Medical Research Service, Department of Veterans Affairs (to M.M.), a PKD Foundation Research Award (to K.H.), by the Zell Family Foundation (to K.H.), by the Detraz Endowed Research Fund in Polycystic Kidney Disease (to M. M.), by the National Institutes of Health T32 training grant in Basic Immunology and Immunologic disease 2T32AI007051-38 (to K.A.Z) and two Pilot and Feasibility Grants from the Baltimore PKD Center 2P30DK090868 (to K.H and K.A.Z) and the UAB Hepato/Renal Fibrocystic Disease Core Center 5P30DK074038 (to K.A.Z).
Disclosures
M. M. reports grants and consulting fees outside the submitted work from Otsuka Pharmaceuticals, Sanofi and Chinook.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Song CJ, Zimmerman KA, Henke SJ, Yoder BK, Inflammation and Fibrosis in Polycystic Kidney Disease, Results Probl Cell Differ 60 (2017) 323–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cowley BD Jr., Ricardo SD, Nagao S, Diamond JR, Increased renal expression of monocyte chemoattractant protein-1 and osteopontin in ADPKD in rats, Kidney Int 60(6) (2001) 2087–96. [DOI] [PubMed] [Google Scholar]
- [3].Zheng D, Wolfe M, Cowley BD Jr., Wallace DP, Yamaguchi T, Grantham JJ, Urinary excretion of monocyte chemoattractant protein-1 in autosomal dominant polycystic kidney disease, J Am Soc Nephrol 14(10) (2003) 2588–95. [DOI] [PubMed] [Google Scholar]
- [4].Salah SM, Meisenheimer JD, Rao R, Peda JD, Wallace DP, Foster D, Li X, Li X, Zhou X, Vallejo JA, Wacker MJ, Fields TA, Swenson-Fields KI, MCP-1 promotes detrimental cardiac physiology, pulmonary edema, and death in the cpk model of polycystic kidney disease, Am J Physiol Renal Physiol 317(2) (2019) F343–F360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chung AC, Lan HY, Chemokines in renal injury, J Am Soc Nephrol 22(5) (2011) 802–9. [DOI] [PubMed] [Google Scholar]
- [6].Fischereder M, Schroppel B, The role of chemokines in acute renal allograft rejection and chronic allograft injury, Front Biosci (Landmark Ed) 14 (2009) 1807–14. [DOI] [PubMed] [Google Scholar]
- [7].de Almeida RM, Clendenon SG, Richards WG, Boedigheimer M, Damore M, Rossetti S, Harris PC, Herbert BS, Xu WM, Wandinger-Ness A, Ward HH, Glazier JA, Bacallao RL, Transcriptome analysis reveals manifold mechanisms of cyst development in ADPKD, Hum Genomics 10(1) (2016) 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Song X, Di Giovanni V, He N, Wang K, Ingram A, Rosenblum ND, Pei Y, Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks, Hum Mol Genet 18(13) (2009) 2328–43. [DOI] [PubMed] [Google Scholar]
- [9].Zheng L, Gao W, Hu C, Yang C, Rong R, Immune Cells in Ischemic Acute Kidney Injury, Curr Protein Pept Sci 20(8) (2019) 770–776. [DOI] [PubMed] [Google Scholar]
- [10].Deshmane SL, Kremlev S, Amini S, Sawaya BE, Monocyte chemoattractant protein-1 (MCP-1): an overview, Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research 29(6) (2009) 313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yadav A, Saini V, Arora S, MCP-1: chemoattractant with a role beyond immunity: a review, Clinica chimica acta; international journal of clinical chemistry 411(21–22) (2010) 1570–9. [DOI] [PubMed] [Google Scholar]
- [12].Cao Q, Wang Y, Wang XM, Lu J, Lee VW, Ye Q, Nguyen H, Zheng G, Zhao Y, Alexander SI, Harris DC, Renal F4/80+ CD11c+ mononuclear phagocytes display phenotypic and functional characteristics of macrophages in health and in adriamycin nephropathy, J Am Soc Nephrol 26(2) (2015) 349–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Segerer S, Nelson PJ, Schlondorff D, Chemokines, chemokine receptors, and renal disease: from basic science to pathophysiologic and therapeutic studies, J Am Soc Nephrol 11(1) (2000) 152–76. [DOI] [PubMed] [Google Scholar]
- [14].Viedt C, Orth SR, Monocyte chemoattractant protein-1 (MCP-1) in the kidney: does it more than simply attract monocytes?, Nephrol Dial Transplant 17(12) (2002) 2043–7. [DOI] [PubMed] [Google Scholar]
- [15].Munshi R, Johnson A, Siew ED, Ikizler TA, Ware LB, Wurfel MM, Himmelfarb J, Zager RA, MCP-1 gene activation marks acute kidney injury, J Am Soc Nephrol 22(1) (2011) 165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Happe H, Leonhard WN, van der Wal A, van de Water B, Lantinga-van Leeuwen IS, Breuning MH, de Heer E, Peters DJ, Toxic tubular injury in kidneys from Pkd1-deletion mice accelerates cystogenesis accompanied by dysregulated planar cell polarity and canonical Wnt signaling pathways, Hum Mol Genet 18(14) (2009) 2532–42. [DOI] [PubMed] [Google Scholar]
- [17].Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, Igarashi P, Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia, Hum Mol Genet 17(11) (2008) 1578–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Takakura A, Contrino L, Zhou X, Bonventre JV, Sun Y, Humphreys BD, Zhou J, Renal Injury is a Third Hit Promoting Rapid Development of Adult Polycystic Kidney Disease, Hum Mol Genet (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bastos AP, Piontek K, Silva AM, Martini D, Menezes LF, Fonseca JM, Fonseca II, Germino GG, Onuchic LF, Pkd1 haploinsufficiency increases renal damage and induces microcyst formation following ischemia/reperfusion, J Am Soc Nephrol 20(11) (2009) 2389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kurbegovic A, Trudel M, Acute kidney injury induces hallmarks of polycystic kidney disease, Am J Physiol Renal Physiol 311(4) (2016) F740–F751. [DOI] [PubMed] [Google Scholar]
- [21].Liu J, Kumar S, Dolzhenko E, Alvarado GF, Guo J, Lu C, Chen Y, Li M, Dessing MC, Parvez RK, Cippa PE, Krautzberger AM, Saribekyan G, Smith AD, McMahon AP, Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion, JCI Insight 2(18) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhou J, Ouyang X, Cui X, Schoeb TR, Smythies LE, Johnson MR, Guay-Woodford LM, Chapman AB, Mrug M, Renal CD14 expression correlates with the progression of cystic kidney disease, Kidney Int 78(6) (2010) 550–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Karihaloo A, Koraishy F, Huen SC, Lee Y, Merrick D, Caplan MJ, Somlo S, Cantley LG, Macrophages Promote Cyst Growth in Polycystic Kidney Disease, J Am Soc Nephrol (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Flores D, Battini L, Gusella GL, Rohatgi R, Fluid shear stress induces renal epithelial gene expression through polycystin-2-dependent trafficking of extracellular regulated kinase, Nephron Physiol 117(4) (2011) p27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhou J, Ouyang X, Schoeb TR, Bolisetty S, Cui X, Mrug S, Yoder BK, Johnson MR, Szalai AJ, Mrug M, Kidney injury accelerates cystogenesis via pathways modulated by heme oxygenase and complement, J Am Soc Nephrol 23(7) (2012) 1161–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mrug M, Zhou J, Woo Y, Cui X, Szalai AJ, Novak J, Churchill GA, Guay-Woodford LM, Overexpression of innate immune response genes in a model of recessive polycystic kidney disease, Kidney Int 73(1) (2008) 63–76. [DOI] [PubMed] [Google Scholar]
- [27].Cassini MF, Kakade VR, Kurtz E, Sulkowski P, Glazer P, Torres R, Somlo S, Cantley LG, Mcp1 Promotes Macrophage-Dependent Cyst Expansion in Autosomal Dominant Polycystic Kidney Disease, J Am Soc Nephrol 29(10) (2018) 2471–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Grantham J, Torres V, Chapman A, Bae K, Tao C, Guay-Woodford L, Harris P, Mrug M, Bennett W, Moxey-Mims M, Bost J, Urinary Monocyte Chemotactic Protein-1 (MCP1) Predicts Progression in Autosomal Dominant Polycystic Kidney Disease (ADPKD) J Am Soc Nehrol 21(Suppl) (2010) 526A. [Google Scholar]
- [29].Chapman AB, Bost JE, Torres VE, Guay-Woodford L, Bae KT, Landsittel D, Li J, King BF, Martin D, Wetzel LH, Lockhart ME, Harris PC, Moxey-Mims M, Flessner M, Bennett WM, Grantham JJ, Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease, Clin J Am Soc Nephrol 7(3) (2012) 479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mrug M, Mrug S, Landsittel D, Torres V, Bae K, Harris P, Guay-Woodford L, Flessner M, Bennett W, Grantham J, Chapman A, Prediction of GFR Endpoints in Early Autosomal Dominant Polycystic Kidney Disease, Am J Nephrol 24 (2013) 59A. [Google Scholar]
- [31].Hou X, Mrug M, Yoder BK, Lefkowitz EJ, Kremmidiotis G, D’Eustachio P, Beier DR, Guay-Woodford LM, Cystin, a novel cilia-associated protein, is disrupted in the cpk mouse model of polycystic kidney disease, J Clin Invest 109(4) (2002) 533–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zoja C, Corna D, Locatelli M, Rottoli D, Pezzotta A, Morigi M, Zanchi C, Buelli S, Guglielmotti A, Perico N, Remuzzi A, Remuzzi G, Effects of MCP-1 inhibition by bindarit therapy in a rat model of polycystic kidney disease, Nephron 129(1) (2015) 52–61. [DOI] [PubMed] [Google Scholar]
- [33].Ward C, Hogan M, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham J, Bacallao R, Ishibashi M, Milliner D, Torres V, Harris P, The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein, Nat Genet 30 (2002) 259–269. [DOI] [PubMed] [Google Scholar]
- [34].Park SK, Yang WS, Han NJ, Lee SK, Ahn H, Lee IK, Park JY, Lee KU, Lee JD, Dexamethasone regulates AP-1 to repress TNF-alpha induced MCP-1 production in human glomerular endothelial cells, Nephrol Dial Transplant 19(2) (2004) 312–9. [DOI] [PubMed] [Google Scholar]
- [35].Aggarwal BB, Gupta SC, Kim JH, Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey, Blood 119(3) (2012) 651–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Li X, Magenheimer BS, Xia S, Johnson T, Wallace DP, Calvet JP, Li R, A tumor necrosis factor-alpha-mediated pathway promoting autosomal dominant polycystic kidney disease, Nat Med 14(8) (2008) 863–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gardner KD Jr., Burnside JS, Elzinga LW, Locksley RM, Cytokines in fluids from polycystic kidneys, Kidney Int 39(4) (1991) 718–24. [DOI] [PubMed] [Google Scholar]
- [38].Partsch G, Steiner G, Leeb BF, Dunky A, Broll H, Smolen JS, Highly increased levels of tumor necrosis factor-alpha and other proinflammatory cytokines in psoriatic arthritis synovial fluid, The Journal of rheumatology 24(3) (1997) 518–23. [PubMed] [Google Scholar]
- [39].Zhou JX, Fan LX, Li X, Calvet JP, Li X, TNFalpha signaling regulates cystic epithelial cell proliferation through Akt/mTOR and ERK/MAPK/Cdk2 mediated Id2 signaling, Plos One 10(6) (2015) e0131043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Alcalay NI, Sharma M, Vassmer D, Chapman B, Paul B, Zhou J, Brantley JG, Wallace DP, Maser RL, Vanden Heuvel GB, Acceleration of polycystic kidney disease progression in cpk mice carrying a deletion in the homeodomain protein Cux1, Am J Physiol Renal Physiol 295(6) (2008) F1725–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Roix J, Saha S, TNF-alpha blockade is ineffective in animal models of established polycystic kidney disease, BMC Nephrol 14 (2013) 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang C, Wang C, Li Y, Miwa T, Liu C, Cui W, Song WC, Du J, Complement C3a signaling facilitates skeletal muscle regeneration by regulating monocyte function and trafficking, Nat Commun 8(1) (2017) 2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Torzewski J, Oldroyd R, Lachmann P, Fitzsimmons C, Proudfoot D, Bowyer D, Complement-induced release of monocyte chemotactic protein-1 from human smooth muscle cells. A possible initiating event in atherosclerotic lesion formation, Arterioscler Thromb Vasc Biol 16(5) (1996) 673–7. [DOI] [PubMed] [Google Scholar]
- [44].Bohana-Kashtan O, Ziporen L, Donin N, Kraus S, Fishelson Z, Cell signals transduced by complement, Mol Immunol 41(6–7) (2004) 583–97. [DOI] [PubMed] [Google Scholar]
- [45].Fischer MB, Ma M, Goerg S, Zhou X, Xia J, Finco O, Han S, Kelsoe G, Howard RG, Rothstein TL, Kremmer E, Rosen FS, Carroll MC, Regulation of the B cell response to T-dependent antigens by classical pathway complement, J Immunol 157(2) (1996) 549–56. [PubMed] [Google Scholar]
- [46].Zhou W, Farrar CA, Abe K, Pratt JR, Marsh JE, Wang Y, Stahl GL, Sacks SH, Predominant role for C5b-9 in renal ischemia/reperfusion injury, J Clin Invest 105(10) (2000) 1363–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ueda T, Rieu P, Brayer J, Arnaout MA, Identification of the complement iC3b binding site in the beta 2 integrin CR3 (CD11b/CD18), Proc Natl Acad Sci U S A 91(22) (1994) 10680–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gordon S, Alternative activation of macrophages, Nat Rev Immunol 3(1) (2003) 23–35. [DOI] [PubMed] [Google Scholar]
- [49].Bakun M, Niemczyk M, Domanski D, Jazwiec R, Perzanowska A, Niemczyk S, Kistowski M, Fabijanska A, Borowiec A, Paczek L, Dadlez M, Urine proteome of autosomal dominant polycystic kidney disease patients, Clinical proteomics 9(1) (2012) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wu Y, Xu J, Li S, Hsieh T, Lu T, Kong T, The role of complement C3 in focal inflammation and development of kidney cysts induced by Pkd1 inactivation FASEB J 28(1 Suppl) (2014) 690.3. [Google Scholar]
- [51].Mrug M, Zhou J, Mrug S, Guay-Woodford LM, Yoder BK, Szalai AJ, Complement C3 activation in cyst fluid and urine from autosomal dominant polycystic kidney disease patients, J Intern Med 276(5) (2014) 539–40. [DOI] [PubMed] [Google Scholar]
- [52].Mrug M, Zhou J, Mannon RB, C3 polymorphisms and outcomes of renal allografts, N Engl J Med 360(23) (2009) 2477–8. [DOI] [PubMed] [Google Scholar]
- [53].Hong Y, Zhou W, Li K, Sacks SH, Triptolide is a potent suppressant of C3, CD40 and B7h expression in activated human proximal tubular epithelial cells, Kidney Int 62(4) (2002) 1291–300. [DOI] [PubMed] [Google Scholar]
- [54].Su Z, Wang X, Gao X, Liu Y, Pan C, Hu H, Beyer RP, Shi M, Zhou J, Zhang J, Serra AL, Wuthrich RP, Mei C, Excessive activation of the alternative complement pathway in autosomal dominant polycystic kidney disease, J Intern Med (2014). [DOI] [PubMed] [Google Scholar]
- [55].Yao Y, Jeyanathan M, Haddadi S, Barra NG, Vaseghi-Shanjani M, Damjanovic D, Lai R, Afkhami S, Chen Y, Dvorkin-Gheva A, Robbins CS, Schertzer JD, Xing Z, Induction of Autonomous Memory Alveolar Macrophages Requires T Cell Help and Is Critical to Trained Immunity, Cell 175(6) (2018) 1634–1650 e17. [DOI] [PubMed] [Google Scholar]
- [56].Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, Jacobs L, Jansen T, Kullberg BJ, Wijmenga C, Joosten LAB, Xavier RJ, van der Meer JWM, Stunnenberg HG, Netea MG, Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes, Cell Host Microbe 12(2) (2012) 223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Leopold Wager CM, Hole CR, Campuzano A, Castro-Lopez N, Cai H, Caballero Van Dyke MC, Wozniak KL, Wang Y, Wormley FL Jr., IFN-gamma immune priming of macrophages in vivo induces prolonged STAT1 binding and protection against Cryptococcus neoformans, PLoS Pathog 14(10) (2018) e1007358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Cao Q, Harris DC, Wang Y, Macrophages in kidney injury, inflammation, and fibrosis, Physiology (Bethesda) 30(3) (2015) 183–94. [DOI] [PubMed] [Google Scholar]
- [59].Huen SC, Cantley LG, Macrophages in Renal Injury and Repair, Annu Rev Physiol 79 (2017) 449–469. [DOI] [PubMed] [Google Scholar]
- [60].Chen T, Cao Q, Wang Y, Harris DCH, M2 macrophages in kidney disease: biology, therapies, and perspectives, Kidney Int 95(4) (2019) 760–773. [DOI] [PubMed] [Google Scholar]
- [61].Zimmerman KA, Bentley MR, Lever JM, Li Z, Crossman DK, Song CJ, Liu S, Crowley MR, George JF, Mrug M, Yoder BK, Single-Cell RNA Sequencing Identifies Candidate Renal Resident Macrophage Gene Expression Signatures across Species, J Am Soc Nephrol 30(5) (2019) 767–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Stewart BJ, Ferdinand JR, Young MD, Mitchell TJ, Loudon KW, Riding AM, Richoz N, Frazer GL, Staniforth JUL, Vieira Braga FA, Botting RA, Popescu DM, Vento-Tormo R, Stephenson E, Cagan A, Farndon SJ, Polanski K, Efremova M, Green K, Del Castillo Velasco-Herrera M, Guzzo C, Collord G, Mamanova L, Aho T, Armitage JN, Riddick ACP, Mushtaq I, Farrell S, Rampling D, Nicholson J, Filby A, Burge J, Lisgo S, Lindsay S, Bajenoff M, Warren AY, Stewart GD, Sebire N, Coleman N, Haniffa M, Teichmann SA, Behjati S, Clatworthy MR, Spatiotemporal immune zonation of the human kidney, Science 365(6460) (2019) 1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhu YP, Thomas GD, Hedrick CC, 2014 Jeffrey M. Hoeg Award Lecture: Transcriptional Control of Monocyte Development, Arterioscler Thromb Vasc Biol 36(9) (2016) 1722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, Lang R, Haniffa M, Collin M, Tacke F, Habenicht AJ, Ziegler-Heitbrock L, Randolph GJ, Comparison of gene expression profiles between human and mouse monocyte subsets, Blood 115(3) (2010) e10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Weisheit CK, Engel DR, Kurts C, Dendritic Cells and Macrophages: Sentinels in the Kidney, Clin J Am Soc Nephrol 10(10) (2015) 1841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Viehmann SF, Bohner AMC, Kurts C, Brahler S, The multifaceted role of the renal mononuclear phagocyte system, Cell Immunol 330 (2018) 97–104. [DOI] [PubMed] [Google Scholar]
- [67].Brahler S, Zinselmeyer BH, Raju S, Nitschke M, Suleiman H, Saunders BT, Johnson MW, Bohner AMC, Viehmann SF, Theisen DJ, Kretzer NM, Briseno CG, Zaitsev K, Ornatsky O, Chang Q, Carrero JA, Kopp JB, Artyomov MN, Kurts C, Murphy KM, Miner JH, Shaw AS, Opposing Roles of Dendritic Cell Subsets in Experimental GN, J Am Soc Nephrol 29(1) (2018) 138–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F, A lineage of myeloid cells independent of Myb and hematopoietic stem cells, Science 336(6077) (2012) 86–90. [DOI] [PubMed] [Google Scholar]
- [69].Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR, Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors, Nature 518(7540) (2015) 547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, Garcia-Sastre A, Stanley ER, Ginhoux F, Frenette PS, Merad M, Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes, Immunity 38(4) (2013) 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lever JM, Yang Z, Boddu R, Adedoyin OO, Guo L, Joseph R, Traylor AM, Agarwal A, George JF, Parabiosis reveals leukocyte dynamics in the kidney, Lab Invest 98(3) (2018) 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Salei N, Rambichler S, Salvermoser J, Papaioannou NE, Schuchert R, Pakalniskyte D, Li N, Marschner JA, Lichtnekert J, Stremmel C, Cernilogar FM, Salvermoser M, Walzog B, Straub T, Schotta G, Anders HJ, Schulz C, Schraml BU, The Kidney Contains Ontogenetically Distinct Dendritic Cell and Macrophage Subtypes throughout Development That Differ in Their Inflammatory Properties, J Am Soc Nephrol 31(2) (2020) 257–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Bonventre JV, Yang L, Cellular pathophysiology of ischemic acute kidney injury, J Clin Invest 121(11) (2011) 4210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, Jardine L, Dixon D, Stephenson E, Nilsson E, Grundberg I, McDonald D, Filby A, Li W, De Jager PL, Rozenblatt-Rosen O, Lane AA, Haniffa M, Regev A, Hacohen N, Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors, Science 356(6335) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S, Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis, Immunity 38(1) (2013) 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Clements M, Gershenovich M, Chaber C, Campos-Rivera J, Du P, Zhang M, Ledbetter S, Zuk A, Differential Ly6C Expression after Renal Ischemia-Reperfusion Identifies Unique Macrophage Populations, J Am Soc Nephrol 27(1) (2016) 159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Munro DAD, Hughes J, The Origins and Functions of Tissue-Resident Macrophages in Kidney Development, Front Physiol 8 (2017) 837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Alikhan MA, Jones CV, Williams TM, Beckhouse AG, Fletcher AL, Kett MM, Sakkal S, Samuel CS, Ramsay RG, Deane JA, Wells CA, Little MH, Hume DA, Ricardo SD, Colony-stimulating factor-1 promotes kidney growth and repair via alteration of macrophage responses, Am J Pathol 179(3) (2011) 1243–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Menke J, Iwata Y, Rabacal WA, Basu R, Yeung YG, Humphreys BD, Wada T, Schwarting A, Stanley ER, Kelley VR, CSF-1 signals directly to renal tubular epithelial cells to mediate repair in mice, J Clin Invest 119(8) (2009) 2330–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lever JM, Hull TD, Boddu R, Pepin ME, Black LM, Adedoyin OO, Yang Z, Traylor AM, Jiang Y, Li Z, Peabody JE, Eckenrode HE, Crossman DK, Crowley MR, Bolisetty S, Zimmerman KA, Wende AR, Mrug M, Yoder BK, Agarwal A, George JF, Resident macrophages reprogram toward a developmental state after acute kidney injury, JCI Insight 4(2) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zimmerman KA, Huang J, He L, Revell DZ, Li Z, Hsu J-S, Fitzgibbon WR, Hazard ES, Hardiman G, Mrug M, Bell PD, Yoder BK, Saigusa T, Interferon Regulatory Factor 5 in Resident Macrophage Promotes Polycystic Kidney Disease, Kidney 360 (2020) 10.34067/KID.0001052019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Zimmerman KA, Song CJ, Li Z, Lever JM, Crossman DK, Rains A, Aloria EJ, Gonzalez NM, Bassler JR, Zhou J, Crowley MR, Revell DZ, Yan Z, Shan D, Benveniste EN, George JF, Mrug M, Yoder BK, Tissue-Resident Macrophages Promote Renal Cystic Disease, J Am Soc Nephrol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK, New insights into the multidimensional concept of macrophage ontogeny, activation and function, Nat Immunol 17(1) (2015) 34–40. [DOI] [PubMed] [Google Scholar]
- [84].Wynn TA, Chawla A, Pollard JW, Macrophage biology in development, homeostasis and disease, Nature 496(7446) (2013) 445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Gordon S, Taylor PR, Monocyte and macrophage heterogeneity, Nat Rev Immunol 5(12) (2005) 953–64. [DOI] [PubMed] [Google Scholar]
- [86].Mantovani A, Sozzani S, Locati M, Allavena P, Sica A, Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes, Trends Immunol 23(11) (2002) 549–55. [DOI] [PubMed] [Google Scholar]
- [87].Lin SL, Castano AP, Nowlin BT, Lupher ML Jr., Duffield JS, Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations, J Immunol 183(10) (2009) 6733–43. [DOI] [PubMed] [Google Scholar]
- [88].Adams DO, Hamilton TA, The cell biology of macrophage activation, Annual review of immunology 2 (1984) 283–318. [DOI] [PubMed] [Google Scholar]
- [89].Helm O, Held-Feindt J, Grage-Griebenow E, Reiling N, Ungefroren H, Vogel I, Kruger U, Becker T, Ebsen M, Rocken C, Kabelitz D, Schafer H, Sebens S, Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis, Int J Cancer 135(4) (2014) 843–61. [DOI] [PubMed] [Google Scholar]
- [90].Kratochvill F, Neale G, Haverkamp JM, Van de Velde LA, Smith AM, Kawauchi D, McEvoy J, Roussel MF, Dyer MA, Qualls JE, Murray PJ, TNF Counterbalances the Emergence of M2 Tumor Macrophages, Cell Rep 12(11) (2015) 1902–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Stables MJ, Shah S, Camon EB, Lovering RC, Newson J, Bystrom J, Farrow S, Gilroy DW, Transcriptomic analyses of murine resolution-phase macrophages, Blood 118(26) (2011) e192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino-Castro A, Mallmann MR, Labzin L, Theis H, Kraut M, Beyer M, Latz E, Freeman TC, Ulas T, Schultze JL, Transcriptome-based network analysis reveals a spectrum model of human macrophage activation, Immunity 40(2) (2014) 274–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ginhoux F, Jung S, Monocytes and macrophages: developmental pathways and tissue homeostasis, Nat Rev Immunol 14(6) (2014) 392–404. [DOI] [PubMed] [Google Scholar]
- [94].Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I, Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment, Cell 159(6) (2014) 1312–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Huen SC, Cantley LG, Macrophage-mediated injury and repair after ischemic kidney injury, Pediatr Nephrol 30(2) (2015) 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM, Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF, J Clin Invest 101(4) (1998) 890–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Holdsworth SR, Summers SA, Role of mast cells in progressive renal diseases, J Am Soc Nephrol 19(12) (2008) 2254–61. [DOI] [PubMed] [Google Scholar]
- [98].Owens EP, Vesey DA, Kassianos AJ, Healy H, Hoy WE, Gobe GC, Biomarkers and the role of mast cells as facilitators of inflammation and fibrosis in chronic kidney disease, Transl Androl Urol 8(Suppl 2) (2019) S175–S183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Tani K, Ogushi F, Kido H, Kawano T, Kunori Y, Kamimura T, Cui P, Sone S, Chymase is a potent chemoattractant for human monocytes and neutrophils, J Leukoc Biol 67(4) (2000) 585–9. [DOI] [PubMed] [Google Scholar]
- [100].McPherson EA, Luo Z, Brown RA, LeBard LS, Corless CC, Speth RC, Bagby SP, Chymase-like angiotensin II-generating activity in end-stage human autosomal dominant polycystic kidney disease, J Am Soc Nephrol 15(2) (2004) 493–500. [DOI] [PubMed] [Google Scholar]
- [101].Ruiz-Ortega M, Ruperez M, Esteban V, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J, Angiotensin II: a key factor in the inflammatory and fibrotic response in kidney diseases, Nephrol Dial Transplant 21(1) (2006) 16–20. [DOI] [PubMed] [Google Scholar]
- [102].Reber LL, Marichal T, Galli SJ, New models for analyzing mast cell functions in vivo, Trends Immunol 33(12) (2012) 613–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Bolisetty S, Agarwal A, Neutrophils in acute kidney injury: not neutral any more, Kidney Int 75(7) (2009) 674–6. [DOI] [PubMed] [Google Scholar]
- [104].Awad AS, Rouse M, Huang L, Vergis AL, Reutershan J, Cathro HP, Linden J, Okusa MD, Compartmentalization of neutrophils in the kidney and lung following acute ischemic kidney injury, Kidney Int 75(7) (2009) 689–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Bernhardt WM, Wiesener MS, Weidemann A, Schmitt R, Weichert W, Lechler P, Campean V, Ong AC, Willam C, Gretz N, Eckardt KU, Involvement of hypoxia-inducible transcription factors in polycystic kidney disease, Am J Pathol 170(3) (2007) 830–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Prasad S, McDaid JP, Tam FW, Haylor JL, Ong AC, Pkd2 dosage influences cellular repair responses following ischemia-reperfusion injury, Am J Pathol 175(4) (2009) 1493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Zimmerman KA, Bentley MR, Lever JM, Li Z, Crossman DK, Song CJ, Liu S, Crowley MR, George JF, Mrug M, Yoder BK, Single-Cell RNA Sequencing Identifies Candidate Renal Resident Macrophage Gene Expression Signatures across Species, J Am Soc Nephrol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Turkmen K, Tufan F, Selcuk E, Akpinar T, Oflaz H, Ecder T, Neutrophil-to lymphocyte ratio, insulin resistance, and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease, Indian J Nephrol 23(1) (2013) 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].O’Leary CA, Mackay BM, Malik R, Edmondston JE, Robinson WF, Huxtable CR, Polycystic kidney disease in bull terriers: an autosomal dominant inherited disorder, Aust Vet J 77(6) (1999) 361–6. [DOI] [PubMed] [Google Scholar]
- [110].Zeier M, Fehrenbach P, Geberth S, Mohring K, Waldherr R, Ritz E, Renal histology in polycystic kidney disease with incipient and advanced renal failure, Kidney Int 42(5) (1992) 1259–65. [DOI] [PubMed] [Google Scholar]
- [111].Takahashi H, Calvet J, Dittemore-Hoover D, Yoshida K, Grantham J, Gattone V, A hereditary model of slowly progressive polycystic kidney disease in the mouse, J Am Soc Nephrol 1 (1991) 980–989. [DOI] [PubMed] [Google Scholar]
- [112].Takahashi H, Ueyama Y, Hibino T, Kuwahara Y, Suzuki S, Hioki K, Tamaoki N, A new mouse model of genetically transmitted polycystic kidney disease, J Urol 135(6) (1986) 1280–3. [DOI] [PubMed] [Google Scholar]
- [113].Nakamura T, Ebihara I, Nagaoka I, Tomino Y, Nagao S, Takahashi H, Koide H, Growth factor gene expression in kidney of murine polycystic kidney disease, J Am Soc Nephrol 3(7) (1993) 1378–86. [DOI] [PubMed] [Google Scholar]
- [114].Gardner KD Jr., Reed WP, Evan AP, Zedalis J, Hylarides MD, Leon AA, Endotoxin provocation of experimental renal cystic disease, Kidney Int 32(3) (1987) 329–34. [DOI] [PubMed] [Google Scholar]
- [115].Cowley B, Gudapaty S, Kraybill A, barash B, Harding M, Calvet J, Gattone V, Autosomal-dominant polycystic kidney disease in the rat, Kidney Int 49 (1993) 522–534. [DOI] [PubMed] [Google Scholar]
- [116].Vogler C, Homan S, Pung A, Thorpe C, Barker J, Birkenmeier EH, Upadhya P, Clinical and pathologic findings in two new allelic murine models of polycystic kidney disease, J Am Soc Nephrol 10(12) (1999) 2534–9. [DOI] [PubMed] [Google Scholar]
- [117].Phillips JK, Hopwood D, Loxley RA, Ghatora K, Coombes JD, Tan YS, Harrison JL, McKitrick DJ, Holobotvskyy V, Arnolda LF, Rangan GK, Temporal relationship between renal cyst development, hypertension and cardiac hypertrophy in a new rat model of autosomal recessive polycystic kidney disease, Kidney Blood Press Res 30(3) (2007) 129–44. [DOI] [PubMed] [Google Scholar]
- [118].Swenson-Fields KI, Vivian CJ, Salah SM, Peda JD, Davis BM, van Rooijen N, Wallace DP, Fields TA, Macrophages promote polycystic kidney disease progression, Kidney Int 83(5) (2013) 855–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Chen L, Zhou X, Fan LX, Yao Y, Swenson-Fields KI, Gadjeva M, Wallace DP, Peters DJ, Yu A, Grantham JJ, Li X, Macrophage migration inhibitory factor promotes cyst growth in polycystic kidney disease, J Clin Invest 125(6) (2015) 2399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Yang Y, Chen M, Zhou J, Lv J, Song S, Fu L, Chen J, Yang M, Mei C, Interactions between Macrophages and Cyst-Lining Epithelial Cells Promote Kidney Cyst Growth in Pkd1-Deficient Mice, J Am Soc Nephrol 29(9) (2018) 2310–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Lakhia R, Yheskel M, Flaten A, Ramalingam H, Aboudehen K, Ferre S, Biggers LM, Mishra A, Chaney CP, Wallace DP, Carroll T, Igarashi P, Patel V, Interstitial microRNA miR-214 attenuates inflammation and polycystic kidney disease progression, JCI Insight (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Braga TT, Correa-Costa M, Silva RC, Cruz MC, Hiyane MI, da Silva JS, Perez KR, Cuccovia IM, Camara NOS, CCR2 contributes to the recruitment of monocytes and leads to kidney inflammation and fibrosis development, Inflammopharmacology 26(2) (2018) 403–411. [DOI] [PubMed] [Google Scholar]
- [123].Serbina NV, Pamer EG, Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2, Nat Immunol 7(3) (2006) 311–7. [DOI] [PubMed] [Google Scholar]
- [124].Fuentes ME, Durham SK, Swerdel MR, Lewin AC, Barton DS, Megill JR, Bravo R, Lira SA, Controlled recruitment of monocytes and macrophages to specific organs through transgenic expression of monocyte chemoattractant protein-1, J Immunol 155(12) (1995) 5769–76. [PubMed] [Google Scholar]
- [125].Viau A, Bienaime F, Lukas K, Todkar AP, Knoll M, Yakulov TA, Hofherr A, Kretz O, Helmstadter M, Reichardt W, Braeg S, Aschman T, Merkle A, Pfeifer D, Dumit VI, Gubler MC, Nitschke R, Huber TB, Terzi F, Dengjel J, Grahammer F, Kottgen M, Busch H, Boerries M, Walz G, Triantafyllopoulou A, Kuehn EW, Cilia-localized LKB1 regulates chemokine signaling, macrophage recruitment, and tissue homeostasis in the kidney, EMBO J (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Salah SM, Meisenheimer JD, Rao R, Peda JD, Wallace DP, Foster D, Li X, Li X, Zhou X, Vallejo JA, Wacker MJ, Fields TA, Swenson Fields KI, MCP-1 Promotes Detrimental Cardiac Physiology, Pulmonary Edema and Death in the cpk model of Polycystic Kidney Disease, Am J Physiol Renal Physiol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Zimmerman KA, Song CJ, Gonzalez-Mize N, Li Z, Yoder BK, Primary cilia disruption differentially affects the infiltrating and resident macrophage compartment in the liver, Am J Physiol Gastrointest Liver Physiol 314(6) (2018) G677–G689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Rodriguez-Iturbe B, Pons H, Herrera-Acosta J, Johnson RJ, Role of immunocompetent cells in nonimmune renal diseases, Kidney Int 59(5) (2001) 1626–40. [DOI] [PubMed] [Google Scholar]
- [129].Oleinika K, Mauri C, Salama AD, Effector and regulatory B cells in immune-mediated kidney disease, Nature reviews. Nephrology 15(1) (2019) 11–26. [DOI] [PubMed] [Google Scholar]
- [130].Rabb H, The T cell as a bridge between innate and adaptive immune systems: implications for the kidney, Kidney Int 61(6) (2002) 1935–46. [DOI] [PubMed] [Google Scholar]
- [131].Hoffman W, Lakkis FG, Chalasani G, B Cells, Antibodies, and More, Clin J Am Soc Nephrol 11(1) (2016) 137–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Abbas AK, Lichtman AH, Pillai S, Baker DL, Baker A, Cellular and molecular immunology, Ninth edition. ed., Elsevier, Philadelphia, PA, 2018. [Google Scholar]
- [133].Crawford A, Macleod M, Schumacher T, Corlett L, Gray D, Primary T cell expansion and differentiation in vivo requires antigen presentation by B cells, J Immunol 176(6) (2006) 3498–506. [DOI] [PubMed] [Google Scholar]
- [134].Giles JR, Kashgarian M, Koni PA, Shlomchik MJ, B Cell-Specific MHC Class II Deletion Reveals Multiple Nonredundant Roles for B Cell Antigen Presentation in Murine Lupus, J Immunol 195(6) (2015) 2571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Harris DP, Haynes L, Sayles PC, Duso DK, Eaton SM, Lepak NM, Johnson LL, Swain SL, Lund FE, Reciprocal regulation of polarized cytokine production by effector B and T cells, Nat Immunol 1(6) (2000) 475–82. [DOI] [PubMed] [Google Scholar]
- [136].Haas C, Ryffel B, Le Hir M, IFN-gamma is essential for the development of autoimmune glomerulonephritis in MRL/Ipr mice, J Immunol 158(11) (1997) 5484–91. [PubMed] [Google Scholar]
- [137].Ding Q, Yeung M, Camirand G, Zeng Q, Akiba H, Yagita H, Chalasani G, Sayegh MH, Najafian N, Rothstein DM, Regulatory B cells are identified by expression of TIM-1 and can be induced through TIM-1 ligation to promote tolerance in mice, J Clin Invest 121(9) (2011) 3645–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Carter NA, Vasconcellos R, Rosser EC, Tulone C, Munoz-Suano A, Kamanaka M, Ehrenstein MR, Flavell RA, Mauri C, Mice lacking endogenous IL-10-producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells, J Immunol 186(10) (2011) 5569–79. [DOI] [PubMed] [Google Scholar]
- [139].Moulin V, Andris F, Thielemans K, Maliszewski C, Urbain J, Moser M, B lymphocytes regulate dendritic cell (DC) function in vivo: increased interleukin 12 production by DCs from B cell-deficient mice results in T helper cell type 1 deviation, J Exp Med 192(4) (2000) 475–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Hall BM, T Cells: Soldiers and Spies--The Surveillance and Control of Effector T Cells by Regulatory T Cells, Clin J Am Soc Nephrol 10(11) (2015) 2050–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Zhu J, Paul WE, Heterogeneity and plasticity of T helper cells, Cell Res 20(1) (2010) 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Vignali DA, Collison LW, Workman CJ, How regulatory T cells work, Nat Rev Immunol 8(7) (2008) 523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Hayday AC, [gamma][delta] cells: a right time and a right place for a conserved third way of protection, Annual review of immunology 18 (2000) 975–1026. [DOI] [PubMed] [Google Scholar]
- [144].Ferrick DA, Schrenzel MD, Mulvania T, Hsieh B, Ferlin WG, Lepper H, Differential production of interferon-gamma and interleukin-4 in response to Th1- and Th2-stimulating pathogens by gamma delta T cells in vivo, Nature 373(6511) (1995) 255–7. [DOI] [PubMed] [Google Scholar]
- [145].Godfrey DI, Kronenberg M, Going both ways: immune regulation via CD1d-dependent NKT cells, J Clin Invest 114(10) (2004) 1379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Martina MN, Noel S, Saxena A, Bandapalle S, Majithia R, Jie C, Arend LJ, Allaf ME, Rabb H, Hamad AR, Double-Negative alphabeta T Cells Are Early Responders to AKI and Are Found in Human Kidney, J Am Soc Nephrol 27(4) (2016) 1113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Turner JE, Becker M, Mittrucker HW, Panzer U, Tissue-Resident Lymphocytes in the Kidney, J Am Soc Nephrol 29(2) (2018) 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Weimbs T, Polycystic kidney disease and renal injury repair: common pathways, fluid flow, and the function of polycystin-1, Am J Physiol Renal Physiol 293(5) (2007) F1423–32. [DOI] [PubMed] [Google Scholar]
- [149].Tanaka S, Tanaka T, Nangaku M, Hypoxia as a key player in the AKI-to-CKD transition, Am J Physiol Renal Physiol 307(11) (2014) F1187–95. [DOI] [PubMed] [Google Scholar]
- [150].Wu MJ, Wen MC, Chiu YT, Chiou YY, Shu KH, Tang MJ, Rapamycin attenuates unilateral ureteral obstruction-induced renal fibrosis, Kidney Int 69(11) (2006) 2029–36. [DOI] [PubMed] [Google Scholar]
- [151].Ibraghimov-Beskrovnaya O, Natoli TA, mTOR signaling in polycystic kidney disease, Trends Mol Med 17(11) (2011) 625–33. [DOI] [PubMed] [Google Scholar]
- [152].Zhou D, Tan RJ, Fu H, Liu Y, Wnt/beta-catenin signaling in kidney injury and repair: a double-edged sword, Lab Invest 96(2) (2016) 156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Wuebken A, Schmidt-Ott KM, WNT/beta-catenin signaling in polycystic kidney disease, Kidney Int 80(2) (2011) 135–8. [DOI] [PubMed] [Google Scholar]
- [154].Malas TB, Formica C, Leonhard WN, Rao P, Granchi Z, Roos M, Peters DJ, t Hoen PA, Meta-analysis of polycystic kidney disease expression profiles defines strong involvement of injury repair processes, Am J Physiol Renal Physiol 312(4) (2017) F806–F817. [DOI] [PubMed] [Google Scholar]
- [155].Jang HR, Gandolfo MT, Ko GJ, Satpute SR, Racusen L, Rabb H, B cells limit repair after ischemic acute kidney injury, J Am Soc Nephrol 21(4) (2010) 654–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Ascon DB, Lopez-Briones S, Liu M, Ascon M, Savransky V, Colvin RB, Soloski MJ, Rabb H, Phenotypic and functional characterization of kidney-infiltrating lymphocytes in renal ischemia reperfusion injury, J Immunol 177(5) (2006) 3380–7. [DOI] [PubMed] [Google Scholar]
- [157].Burne-Taney MJ, Ascon DB, Daniels F, Racusen L, Baldwin W, Rabb H, B cell deficiency confers protection from renal ischemia reperfusion injury, J Immunol 171(6) (2003) 3210–5. [DOI] [PubMed] [Google Scholar]
- [158].Linfert D, Chowdhry T, Rabb H, Lymphocytes and ischemia-reperfusion injury, Transplant Rev (Orlando) 23(1) (2009) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Renner B, Strassheim D, Amura CR, Kulik L, Ljubanovic D, Glogowska MJ, Takahashi K, Carroll MC, Holers VM, Thurman JM, B cell subsets contribute to renal injury and renal protection after ischemia/reperfusion, J Immunol 185(7) (2010) 4393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Friedewald JJ, Rabb H, Inflammatory cells in ischemic acute renal failure, Kidney Int 66(2) (2004) 486–91. [DOI] [PubMed] [Google Scholar]
- [161].Burne MJ, Daniels F, El Ghandour A, Mauiyyedi S, Colvin RB, O’Donnell MP, Rabb H, Identification of the CD4(+) T cell as a major pathogenic factor in ischemic acute renal failure, J Clin Invest 108(9) (2001) 1283–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Liu M, Chien CC, Burne-Taney M, Molls RR, Racusen LC, Colvin RB, Rabb H, A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity, J Am Soc Nephrol 17(3) (2006) 765–74. [DOI] [PubMed] [Google Scholar]
- [163].Tapmeier TT, Fearn A, Brown K, Chowdhury P, Sacks SH, Sheerin NS, Wong W, Pivotal role of CD4+ T cells in renal fibrosis following ureteric obstruction, Kidney Int 78(4) (2010) 351–62. [DOI] [PubMed] [Google Scholar]
- [164].Ravichandran K, Wang Q, Ozkok A, Jani A, Li H, He Z, Ljubanovic D, Weiser-Evans MC, Nemenoff RA, Edelstein CL, CD4 T cell knockout does not protect against kidney injury and worsens cancer, J Mol Med (Berl) 94(4) (2016) 443–55. [DOI] [PubMed] [Google Scholar]
- [165].Baudoux T, Husson C, De Prez E, Jadot I, Antoine MH, Nortier JL, Hougardy JM, CD4(+) and CD8(+) T Cells Exert Regulatory Properties During Experimental Acute Aristolochic Acid Nephropathy, Sci Rep 8(1) (2018) 5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Yokota N, Burne-Taney M, Racusen L, Rabb H, Contrasting roles for STAT4 and STAT6 signal transduction pathways in murine renal ischemia-reperfusion injury, Am J Physiol Renal Physiol 285(2) (2003) F319–25. [DOI] [PubMed] [Google Scholar]
- [167].Kinsey GR, Sharma R, Huang L, Li L, Vergis AL, Ye H, Ju ST, Okusa MD, Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury, J Am Soc Nephrol 20(8) (2009) 1744–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Lee H, Nho D, Chung HS, Lee H, Shin MK, Kim SH, Bae H, CD4+CD25+ regulatory T cells attenuate cisplatin-induced nephrotoxicity in mice, Kidney Int 78(11) (2010) 1100–9. [DOI] [PubMed] [Google Scholar]
- [169].Lee SY, Lee YS, Choi HM, Ko YS, Lee HY, Jo SK, Cho WY, Kim HK, Distinct pathophysiologic mechanisms of septic acute kidney injury: role of immune suppression and renal tubular cell apoptosis in murine model of septic acute kidney injury, Crit Care Med 40(11) (2012) 2997–3006. [DOI] [PubMed] [Google Scholar]
- [170].Gandolfo MT, Jang HR, Bagnasco SM, Ko GJ, Agreda P, Satpute SR, Crow MT, King LS, Rabb H, Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury, Kidney Int 76(7) (2009) 717–29. [DOI] [PubMed] [Google Scholar]
- [171].Kinsey GR, Huang L, Vergis AL, Li L, Okusa MD, Regulatory T cells contribute to the protective effect of ischemic preconditioning in the kidney, Kidney Int 77(9) (2010) 771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Cho WY, Choi HM, Lee SY, Kim MG, Kim HK, Jo SK, The role of Tregs and CD11c(+) macrophages/dendritic cells in ischemic preconditioning of the kidney, Kidney Int 78(10) (2010) 981–92. [DOI] [PubMed] [Google Scholar]
- [173].Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, Defor T, Levine BL, June CH, Rubinstein P, McGlave PB, Blazar BR, Wagner JE, Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics, Blood 117(3) (2011) 1061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Lai LW, Yong KC, Lien YH, Pharmacologic recruitment of regulatory T cells as a therapy for ischemic acute kidney injury, Kidney Int 81(10) (2012) 983–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Kim MG, Lee SY, Ko YS, Lee HY, Jo SK, Cho WY, Kim HK, CD4+ CD25+ regulatory T cells partially mediate the beneficial effects of FTY720, a sphingosine-1-phosphate analogue, during ischaemia/reperfusion-induced acute kidney injury, Nephrol Dial Transplant 26(1) (2011) 111–24. [DOI] [PubMed] [Google Scholar]
- [176].Sadasivam M, Noel S, Lee SA, Gong J, Allaf ME, Pierorazio P, Rabb H, Hamad ARA, Activation and Proliferation of PD-1(+) Kidney Double-Negative T Cells Is Dependent on Nonclassical MHC Proteins and IL-2, J Am Soc Nephrol 30(2) (2019) 277–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Yang SH, Lee JP, Jang HR, Cha RH, Han SS, Jeon US, Kim DK, Song J, Lee DS, Kim YS, Sulfatide-reactive natural killer T cells abrogate ischemia-reperfusion injury, J Am Soc Nephrol 22(7) (2011) 1305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Bajwa A, Huang L, Ye H, Dondeti K, Song S, Rosin DL, Lynch KR, Lobo PI, Li L, Okusa MD, Dendritic cell sphingosine 1-phosphate receptor-3 regulates Th1-Th2 polarity in kidney ischemia-reperfusion injury, J Immunol 189(5) (2012) 2584–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Hochegger K, Schatz T, Eller P, Tagwerker A, Heininger D, Mayer G, Rosenkranz AR, Role of alpha/beta and gamma/delta T cells in renal ischemia-reperfusion injury, Am J Physiol Renal Physiol 293(3) (2007) F741–7. [DOI] [PubMed] [Google Scholar]
- [180].Ascon DB, Ascon M, Satpute S, Lopez-Briones S, Racusen L, Colvin RB, Soloski MJ, Rabb H, Normal mouse kidneys contain activated and CD3+CD4- CD8- double-negative T lymphocytes with a distinct TCR repertoire, J Leukoc Biol 84(6) (2008) 1400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Nakamura T, Ebihara I, Nagaoka I, Tomino Y, Nagao S, Takahashi H, Koide H, Growth factor expression in the kidney of murine polycystic kidney disease, J Am Soc Nephrol 3 (1993) 1378–1386. [DOI] [PubMed] [Google Scholar]
- [182].Kleczko EK, Marsh KH, Tyler LC, Furgeson SB, Bullock BL, Altmann CJ, Miyazaki M, Gitomer BY, Harris PC, Weiser-Evans MCM, Chonchol MB, Clambey ET, Nemenoff RA, Hopp K, CD8(+) T cells modulate autosomal dominant polycystic kidney disease progression, Kidney Int (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Zimmerman KA, Gonzalez NM, Chumley P, Chacana T, Harrington LE, Yoder BK, Mrug M, Urinary T cells correlate with rate of renal function loss in autosomal dominant polycystic kidney disease, Physiol Rep 7(1) (2019) e13951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Enghard P, Humrich JY, Rudolph B, Rosenberger S, Biesen R, Kuhn A, Manz R, Hiepe F, Radbruch A, Burmester GR, Riemekasten G, CXCR3+CD4+ T cells are enriched in inflamed kidneys and urine and provide a new biomarker for acute nephritis flares in systemic lupus erythematosus patients, Arthritis Rheum 60(1) (2009) 199–206. [DOI] [PubMed] [Google Scholar]
- [185].Kopetschke K, Klocke J, Griessbach AS, Humrich JY, Biesen R, Dragun D, Burmester GR, Enghard P, Riemekasten G, The cellular signature of urinary immune cells in Lupus nephritis: new insights into potential biomarkers, Arthritis Res Ther 17 (2015) 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Soleimani A, Adabavazeh R, Nikoueinejad H, Sharif MR, Faraji S, Otroshi Shahreza B, Akbari H, Einollahi B, Evaluation of Th17 pathway in the diagnosis of autosomal dominant polycystic kidney disease, Iran J Kidney Dis 9(2) (2015) 105–12. [PubMed] [Google Scholar]
- [187].Lee Y, Blount KL, Dai F, Thompson S, Scher JK, Bitterman S, Droher M, Herzog EL, Moeckel G, Karihaloo A, Dahl NK, Semaphorin 7A in circulating regulatory T cells is increased in autosomal-dominant polycystic kidney disease and decreases with tolvaptan treatment, Clin Exp Nephrol 22(4) (2018) 906–916. [DOI] [PubMed] [Google Scholar]
- [188].Suzuki K, Okuno T, Yamamoto M, Pasterkamp RJ, Takegahara N, Takamatsu H, Kitao T, Takagi J, Rennert PD, Kolodkin AL, Kumanogoh A, Kikutani H, Semaphorin 7A initiates T-cell-mediated inflammatory responses through alpha1beta1 integrin, Nature 446(7136) (2007) 680–4. [DOI] [PubMed] [Google Scholar]
- [189].Reilkoff RA, Peng H, Murray LA, Peng X, Russell T, Montgomery R, Feghali-Bostwick C, Shaw A, Homer RJ, Gulati M, Mathur A, Elias JA, Herzog EL, Semaphorin 7a+ regulatory T cells are associated with progressive idiopathic pulmonary fibrosis and are implicated in transforming growth factor-beta1-induced pulmonary fibrosis, Am J Respir Crit Care Med 187(2) (2013) 180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Nemenoff RA, Kleczko EK, Hopp K, Renal double negative T cells: unconventional cells in search of a function, Ann Transl Med 7(Suppl 8) (2019) S342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Aguiari G, Banzi M, Gessi S, Cai Y, Zeggio E, Manzati E, Piva R, Lambertini E, Ferrari L, Peters DJ, Lanza F, Harris PC, Borea PA, Somlo S, Del Senno L, Deficiency of polycystin-2 reduces Ca2+ channel activity and cell proliferation in ADPKD lymphoblastoid cells, FASEB J 18(7) (2004) 884–6. [DOI] [PubMed] [Google Scholar]
- [192].Van Laecke S, Kerre T, Nagler EV, Maes B, Caluwe R, Schepers E, Glorieux G, Van Biesen W, Verbeke F, Hereditary polycystic kidney disease is characterized by lymphopenia across all stages of kidney dysfunction: an observational study, Nephrol Dial Transplant 33(3) (2018) 489–496. [DOI] [PubMed] [Google Scholar]
- [193].Magistroni R, Mangolini A, Guzzo S, Testa F, Rapana MR, Mignani R, Russo G, di Virgilio F, Aguiari G, TRPP2 dysfunction decreases ATP-evoked calcium, induces cell aggregation and stimulates proliferation in T lymphocytes, BMC Nephrol 20(1) (2019) 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Hogan MC, Griffin MD, Rossetti S, Torres VE, Ward CJ, Harris PC, PKHDL1, a homolog of the autosomal recessive polycystic kidney disease gene, encodes a receptor with inducible T lymphocyte expression, Hum Mol Genet 12(6) (2003) 685–98. [PubMed] [Google Scholar]
- [195].Kleczko EK, Marsh KH, Tyler LC, Furgeson SB, Bullock BL, Altmann CJ, Miyazaki M, Gitomer BY, Harris PC, Weiser-Evans MCM, Chonchol MB, Clambey ET, Nemenoff RA, Hopp K, CD8(+) T cells modulate autosomal dominant polycystic kidney disease progression, Kidney Int 94(6) (2018) 1127–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Weimbs T, Regulation of mTOR by polycystin-1: is polycystic kidney disease a case of futile repair?, Cell Cycle 5(21) (2006) 2425–9. [DOI] [PubMed] [Google Scholar]