

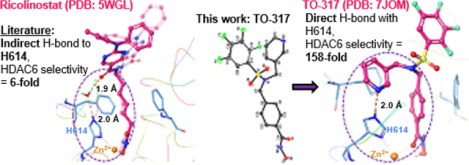
Abstract
HDAC6 is involved in multiple regulatory processes, ranging from cellular stress to intracellular transport. Inhibition of aberrant HDAC6 activity in several cancers and neurological diseases has been shown to be efficacious in both preclinical and clinical studies. While selective HDAC6-targeting has been pursued as an alternative to pan-HDAC drugs, identifying truly selective molecular templates has not been trivial. Herein, we report an SAR study yielding TO-317, which potently binds the HDAC6 catalytic domain 2 (Ki = 0.7 nM) and inhibits enzyme function (IC50 = 2 nM). TO-317 exhibits 158-fold selectivity for HDAC6 over other HDAC isozymes by binding the catalytic Zn2+ and, uniquely, making a never seen before, direct hydrogen bond with the Zn2+ coordinating residue, His614. This novel structural motif targeting the second sphere His614 interaction, observed in a 1.84 Å-resolution crystal structure with drHDAC6 from zebrafish, can provide new pharmacophores for identifying enthalpically driven, high affinity, HDAC6-selective inhibitors
Graphical Abstract
Introduction
Histone Deacetylases (HDACs) are widely investigated as clinical targets due to their role as epigenetic modulators.1–6 There are 18 known HDACs, with 11 having Zn2+-dependent catalytic deacetylation activity, and the remaining 7, referred to as sirtuins (Class III HDACs), exhibiting NAD+-dependent deacetylation. The 11 Zn2+-dependent HDACs are further differentiated into four distinct classes, based on phylogenetic analysis.7,8 HDACs 1, 2, 3, and 8 are grouped into class I. HDACs 4, 5, 7, and 9 are grouped as class IIA. HDACs 6 and 10 belong to class IIB, while HDAC 11 individually occupies group IV.
HDAC6 is unique among other Zn2+-dependent HDACs. While some HDACs reside in the nucleus and regulate gene transcription, HDAC6 is predominantly found in the cytoplasm and directly engages with a host of cytosolic proteins and substrates, including α-tubulin and β-tubulin, assembled microtubules, cortactin, and heat shock proteins.9–11 In addition, while histones are the primary substrates for most Zn2+-dependent HDACs, this is not the case for HDAC6 as α-tubulin is proposed to be the major substrate for HDAC6.12–14 Notably, HDAC6 is structurally distinct from other HDACs, with two unique and independent catalytic domains (CDs) which have diverse roles, and different substrate preference and activity.15,16 Catalytic domain 2 (CD2) is established as the tubulin deacetylase17 and a critical target for drug design. HDAC6 also contains a Zn2+-finger ubiquitin binding domain through which it binds to polyubiquitinated proteins and shuttles them to dynein motors associated with microtubule cargo transport.16 From a therapeutic perspective, several knockout studies in mice lacking the HDAC6 gene have shown no survival dependency and that no lethality is associated with HDAC6 deletion.18,19
Currently, the US FDA has approved several HDAC inhibitors for clinical treatment of different hematological cancers including cutaneous T-cell lymphoma (CTCL), peripheral T-cell lymphoma (PTCL), and multiple myeloma (MM). Vorinostat (SAHA), was the first FDA-approved HDAC inhibitor in 2006 for CTCL patients whose disease had persisted or worsened after two systemic therapies. As a result of severe side effects, all clinical HDAC inhibitors are approved only for cases where the patient does not respond to first option treatment, or at least two prior standard therapies (Romidepsin, approved in 2009 for CTCL patients and in 2011 for PTCL patients, both only after at least one prior systemic therapy; Panobinostat, approved in 2016 for multiple myeloma patients after two treatment regimens), or in cases of relapse or recurring disease (Belinostat, approved in 2014 for relapsed PTCL). These inhibitors all exhibit pan-HDAC inhibition which may contribute to their side effects.
Since no lethal effects have been observed in HDAC6 knock-out mice, it was hypothesized that selective inhibition may attenuate the toxicity of current clinical HDAC inhibitors while retaining therapeutic efficacy. HDAC6 has been validated as an attractive therapeutic target with modestly selective inhibitors such as Citarinostat and Ricolinostat currently at different phases of clinical trials, as single agents or combination therapies for several conditions, including refractory multiple myeloma, malignant melanoma, non-small cell lung cancer, lymphoid malignancies, metastatic breast cancer, and diabetic neuropathic pain.20 These inhibitors achieve selective HDAC6 inhibition by incorporating large bulky cap groups to a lysine hydroxamate mimic, similar to the first non-selective/pan HDAC inhibitor, SAHA (Fig. 1) This scaffold is well tolerated at the outer surface of the HDAC6 binding pocket, unlike other HDAC homologs which are unable to favorably accommodate the cap groups due to steric clashes with the outer surfaces of the binding pockets.21 The selectivity afforded by these designs does not always translate to a safe therapeutic index and some exhibit moderate to advanced toxicities at clinically efficacious doses.22–24
Figure 1:
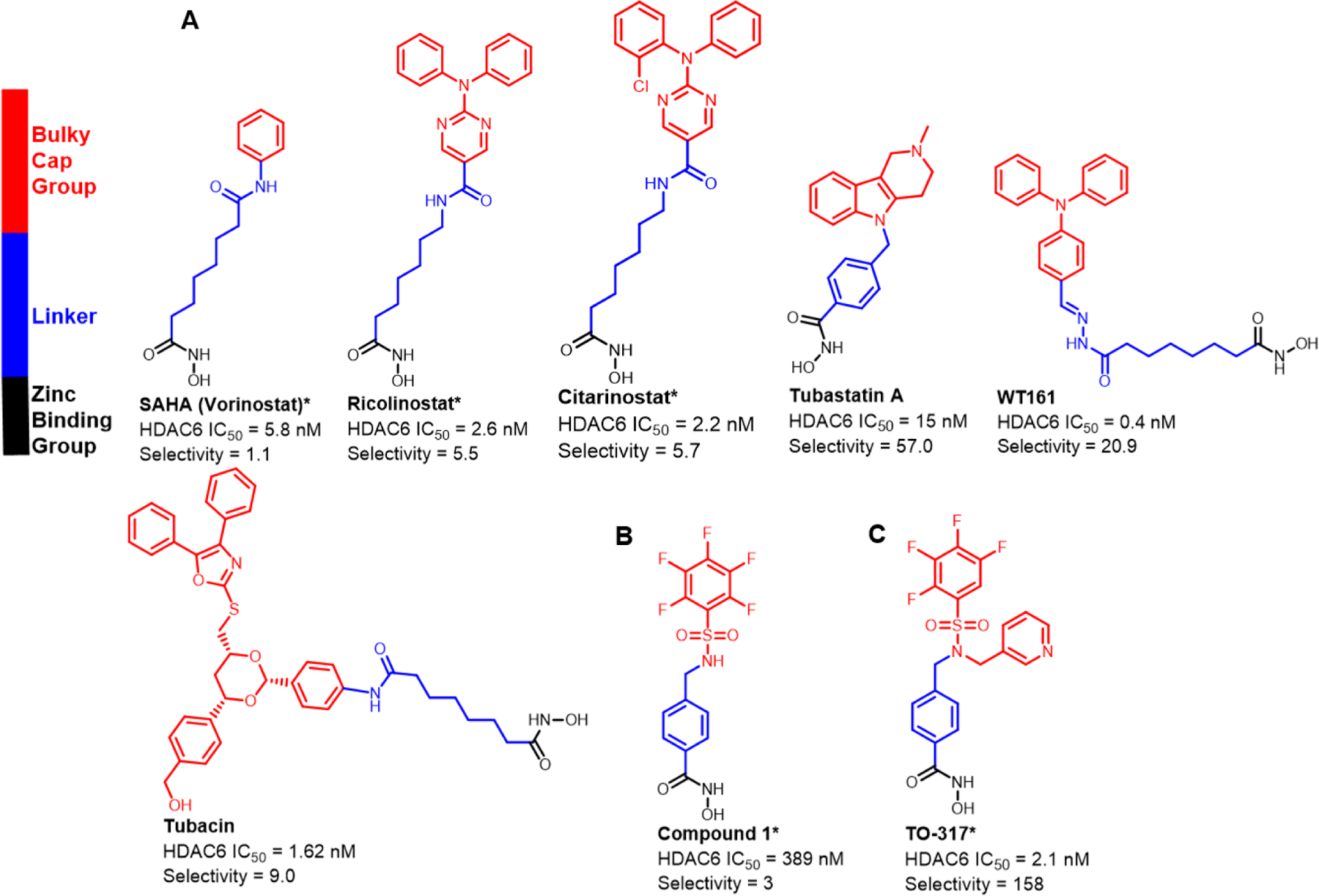
Currently known HDAC6 inhibitors. The structural features involve a hydroxamic acid which is required for Zn2+ binding, and a linker that connects the hydroxamic acid to a cap group. a) Currently known inhibitors achieved HDAC6 selectivity by replacing the benzene substituent on SAHA with a bulkier, usually hydrophobic cap group. b) Previous studies showed compound 1 is a nM inhibitor against HDAC6 exhibiting limited selectivity c) Current studies show TO-317 adopts a rotatable cap group with two aromatic substituents that occupy the HDAC6 surface facilitating specific residue interactions.
*IC50 values were determined using an activity-based electrophoretic mobility shift assay (EMSA) by Nanosyn Inc., USA.
The lead compound described in this study, TO-317, exhibits low nanomolar affinity for HDAC6 (Ki = 0.7 nM), low nanomolar inhibition of HDAC6 function (IC50 = 2 nM), and over 150-fold selectivity for HDAC6 across a panel of 11 Zn2+-dependent HDACs. The in vitro selectivity of TO-317 for HDAC6 is translated in cellulo by Western blot analyses of both pan-HDAC and HDAC6-selective pharmacodynamic (PD) targets. Most interestingly, the X-ray crystal structure determination of TO-317 bound to HDAC6 CD2 from Danio rerio (zebrafish, a well-studied surrogate of human HDAC6 CD2, and henceforth designated drHDAC6) reveals a unique binding mode in which the inhibitor simultaneously coordinates to the catalytic Zn2+ ion and participates in a “second shell” interaction with Zn2+ by forming a direct hydrogen bond with metal-coordinating residue, H614. A direct enzyme-inhibitor hydrogen bond with this residue has not been previously observed. Exploitation of this structural finding may lead to a new generation of HDAC6-targeting ligands.
Results
Discovery of TO-317, A Potent and Highly Selective HDAC6 Inhibitor:
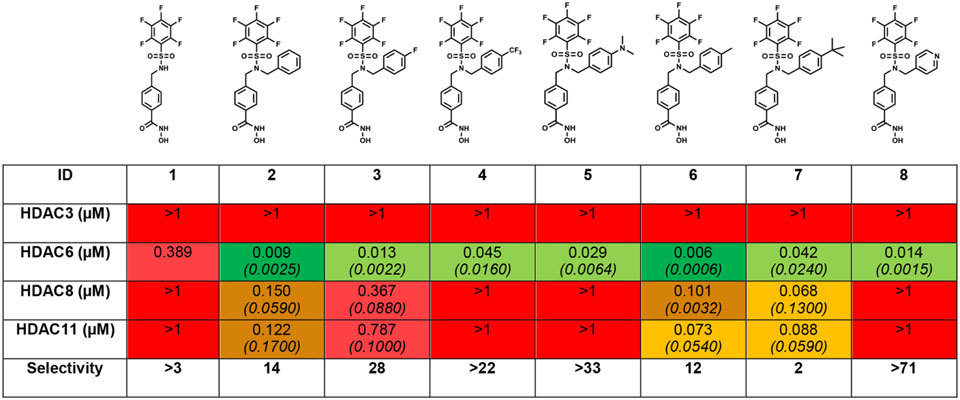
Herein, we describe a rational, iterative approach to identifying potent and selective HDAC6 inhibitors with drug-like properties and acceptable PK profiles. Identification of novel inhibitors lacking the high molecular weight cap groups, which contribute to reduced ligand efficiency (LE) and increased molecular weight while not affording selectivity, was considered critical.25 Examining the binding pocket of HDAC6 revealed a hydrophobic tunnel which opens to an outer cleft region flanked by two outer regions (L1 and L2, Fig. S1B) and an uneven surface topology consisting of polar residues.26,27 This outer surface region is more expansive in HDAC6 (the rough dimensions of the tunnel entrance are 5.6 Å × 10.8 Å; PDB 5WGI, Fig. S1C) compared to other homologs: the rough dimensions of the tunnel entrance in HDAC3 are 5.6 Å × 5.5 Å (PDB 4A69) and in HDAC8 are 4.9 Å × 6.4 Å (PDB: 1T64) (Fig. S1C). As a result, HDAC6 appears to tolerate inhibitors bearing sterically bulky cap groups, and affords selectivity, as was found for Ricolinostat and Citarinostat.28 However, bulky cap groups are predominantly solvent exposed with minimal residue interactions, thereby reducing their contributions to HDAC6 binding. From internal hit-to-lead HDACi development and lead optimization,29,30 compound 1 (Fig 1B, Table 1) was identified, which exhibited low micromolar activity against HDAC6 in vitro. Compound 1, while limited by poor HDAC6 selectivity, poor cellular potency, and an undesirable pharmacokinetic profile, afforded a suitable starting point for optimization due to the presence of a free sulfonamide -NH. Virtual docking of N-alkylated derivatives was hypothesized to afford improved binding compounds with better HDAC6-selectivity. Initial docking studies with 1 suggested that F583 and F643 in drHDAC6 CD2 (PDB 5WGI, corresponding to residues F620 and F680 in Homo sapiens (human) HDAC6, henceforth designated hHDAC6, PDB 5EDU), which line the hydrophobic HDAC6 binding tunnel, form an offset aromatic π-π stacking interaction with the benzyl linker of compound 1(Fig. S1A). Maintaining this hydrophobic interaction required retention of the benzyl linker group. Linking groups in HDAC6 inhibitors usually direct the cap groups toward the L1 crevice, found in the exposed surface of the enzyme, to facilitate interactions with nearby residues.26,28,31 With the exception of a small subset of compounds, all reported HDAC6 inhibitors engage only at the L1 region, and in some rare cases, can alternate the positioning of the cap groups between the L1 and L2 crevices.26,31 (Fig. S1B) The pentafluorobenzene (PFB) ring of 1 was shown to be directed into the L2 region of HDAC6 and is nested on the outer surface with the ring partially exposed to water (Fig. S1A). The PFB ring is predicted to make a π–π aromatic interaction with F643. The sulfonamide -NH does not participate in binding to the protein, despite the predicted interaction with S531, a residue which has previously been proposed to afford selectivity.32 N-Alkylation was therefore not expected to have any disruptive consequences for HDAC6 binding. Water-mediated hydrogen bonding of the sulfonyl oxygen with L712 and the Zn2+-binding ligand, H614, completed the interactions observed with 1 and drHDAC. Structural insights into the binding pockets and the outer surface crevices of HDAC6 (drHDAC6 CD2, PDB: 5WGI) identified several aromatic residues lining the L1 outer surfaces (F642, F643, F583, H463, H573, H614, and Y745) and HDAC6 binding pocket. N-alkylation of 1 with a benzyl substituent was proposed to improve binding by these hydrophobic residues via a windmill-type conformation (Fig. S2). Compound 2 was synthesized (Table 1, Fig. S2) and assessed for its inhibitory activity against 4 representative HDAC homologs. HDACi 2 exhibited an improved selectivity window as compared to 1 and both Citarinostat and Ricolinostat. Next, scaffold optimization was attempted by differential substitution of the benzene ring of 2. Compounds 4 (-CF3) and 5 (-N(CH3)2) provided insights into the electronic effects on activity and selectivity, while 6 (-CH3) and 7 (-tBu) elucidated the role of incorporating bulk at the para-position. To investigate electron deficient rings, compound 8, containing a pyridine ring, was synthesized, and was suggested to be the most promising HDAC6 selective inhibitor (>71 fold).
Table 1:
Activity of 1 and derivatives against 4 HDAC homologs. IC50 values were determined by EMSA using full-length hHDAC6 and a 6-carboxy fluorescein-aminohexyl amidite (FAM) labeled acetylated peptide substrate of HDAC6 catalytic domain 2. IC50 values are an average of duplicate experiments and are reported in μM, and the 95% confidence interval is italicized and reported in brackets close to the IC50 values. Sigmoidal curves of inhibitor concentration (12 concentrations, 5.6 × 10−6 μM – 1 μM) vs % inhibition was fitted using XLDB software (a four-parameter sigmoidal dose–response model). Full protocol is described in detail in the supplementary procedure. Full IC50 curves are shown in supplementary data.
|
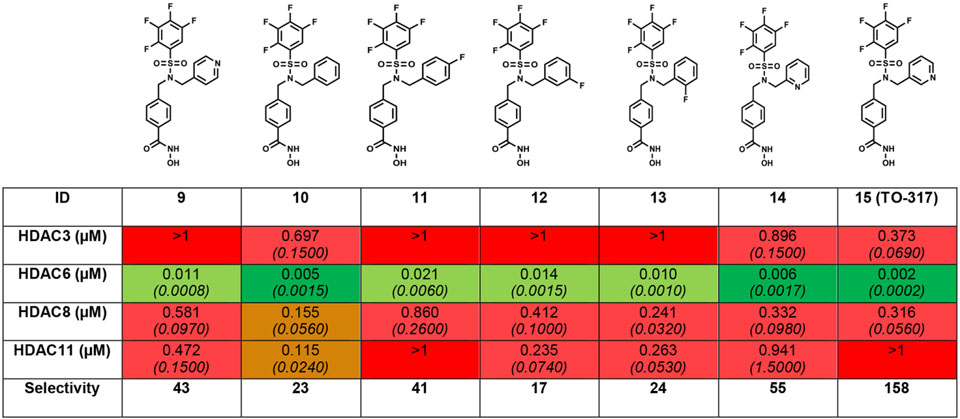
Tetrafluorobenzene (TFB) was previously identified as a bio-isostere of PFB, improving phase II metabolic stability, and contributing to improved selectivity for HDAC6.25,33 PFB was replaced on 8, 2, and 3 with a TFB to yield 9, 10, and 11, respectively (Table 2). The location of the fluorine substituent was varied to both the ortho- (13) and meta- (12) positions. Potency was marginally improved (2-fold) in both cases, although selectivity was lost. Subsequently, modifications to the pyridine analog were explored. The 2-picolyl substituted inhibitor (14) is 2-fold more potent against HDAC6, with only minimal improvement in selectivity when compared to 9. The 3-picolyl analog, TO-317 (15), showed significant improvements in both potency and selectivity.
Table 2:
Focused SAR highlights the substitution of the PFB ring with TFB analogs showing improvements in selectivity for HDAC6. We have also shown that TFB analogs have superior metabolic profiles compared to their PFB isotypes. Substituent location on the cap group aromatic ring plays a significant role in targeted HDAC6 binding. The pyridine motifs displayed the most potent and discriminative activity against HDAC6. IC50 values were determined as described in Table 1 caption and supplementary procedures. IC50 values are an average of duplicate experiments and are reported in μM, and their 95% confidence interval is italicized and reported in brackets under each IC50 value. Full IC50 curves are shown in supplementary data.
|
In addition to the inhibitory activity in an EMSA assay, binding was measured by an orthogonal fluorescence polarization (FP) assay (Fig. S3), which confirmed an interaction with 4 nM potency between TO-317 and drHDAC6 CD2. This interaction was substantially tighter when compared to other compounds in the SAR (>20-fold stronger compared to the nearest analog, 11). Based on the promising activity and binding data, the broader HDAC selectivity of TO-317 against a panel of all 11 Zn2+-based HDACs was investigated (Fig. 2). TO-317 was determined to be selective for HDAC6 across all 11 HDAC homologs, showing relatively modest affinity for HDACs 3, 8, and 10 as compared to HDAC6. To further characterize HDAC6 selectivity, TO-317 was evaluated against the other HDAC homologs at a top treatment concentration of 10 μM. The results, shown in Table 3, support the hypothesis that TO-317 is a highly selective HDAC6 inhibitor. Further investigation of the selectivity of TO-317 and its utility as a lead candidate was pursued in a series of structural and cellular biology experiments outlined below.
Figure 2:
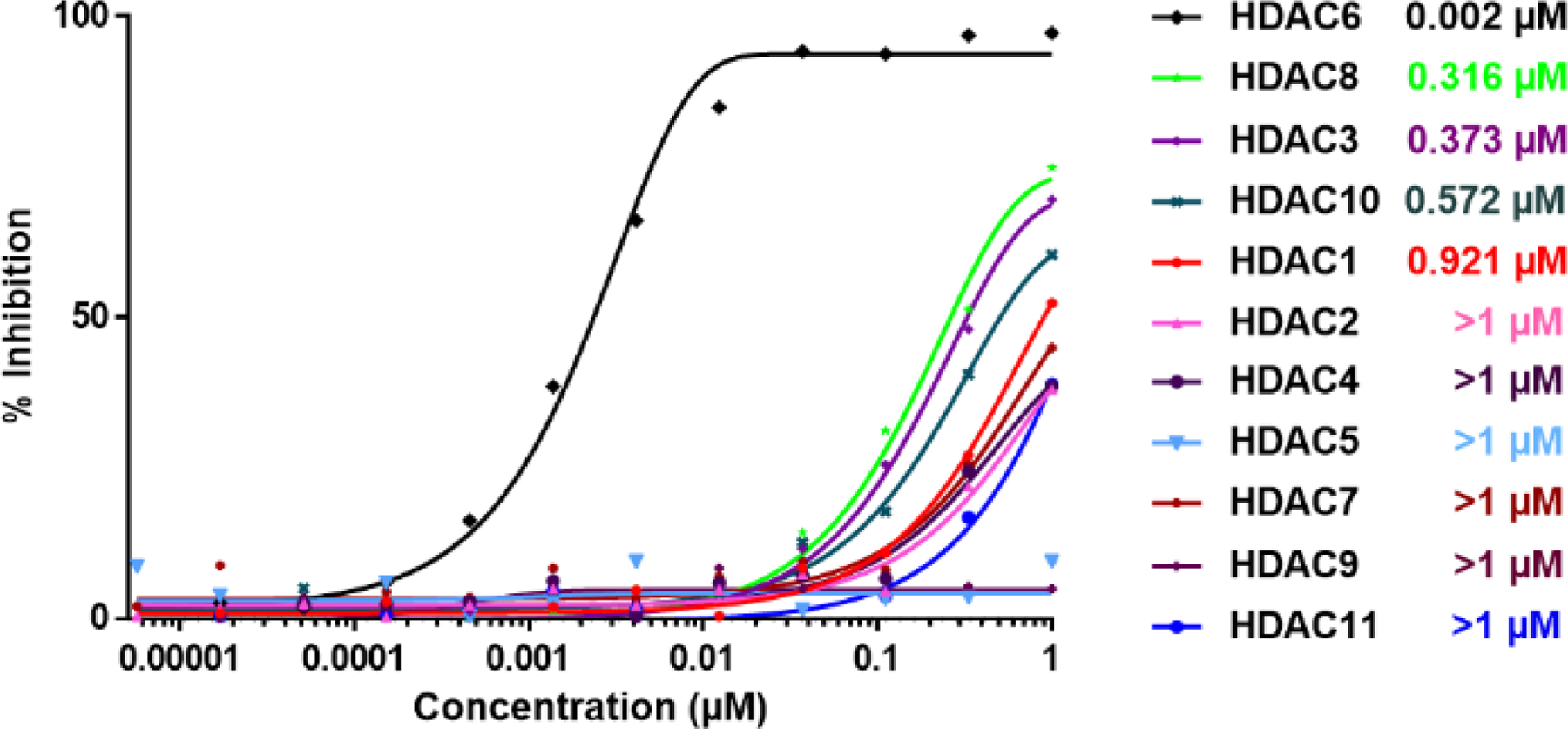
Overlaid Inhibition curves for TO-317 against a panel of 11 Zn2+-based HDAC homologs. TO-317 shows in vitro HDAC6 selectivity across all Zn2+-dependent HDAC homologs and becomes a viable candidate for further biological investigation of the HDAC6 enzyme.
Table 3:
IC50 values for TO-317 against all HDAC isoforms determined with a top concentration of 10 μM. TO-317 maintains strong in vitro inhibitory preference for HDAC6 indicating a promising selectivity profile that is worthy of further biological characterization. IC50 values are an average of duplicate experiments, and their 95% confidence interval is italicized and in brackets under each IC50 value.
| ID | HDAC1 | HDAC2 | HDAC3 | HDAC4 | HDAC5 | HDAC6 | HDAC7 | HDAC8 | HDAC9 | HDAC10 | HDAC11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TO-317 IC50 | 0.921 (0.1400) | 4.970 (0.8000) | 0.373 (0.0690) | >10 | 8.650 (1.5200) | 0.002 (0.0002) | 7.290 (1.4100) | 0.316 (0.0560) | 2.750 (0.6000) | 0.572 (0.1400) | 4.870 (2.2200) |
HDAC6 selectivity mediated by unique catalytic domain interactions:
Computational modelling was employed to investigate the conformation of TO-317 that afforded HDAC6 selectivity. For comparison, Citarinostat and TO-317 were docked with drHDAC6 (PDB 5WGI, Fig. 3 and Fig. S4-A, respectively). Docking Citarinostat showed the cap group to be largely solvent-exposed and directed away from the L1 and L2 outer surface. The crystal structure of Ricolinostat with the CD2 of drHDAC6 (PDB 5WGL) was obtained and an overlay of our docking image of Citarinostat was performed (Fig. S4-A). Both inhibitors adopt identical conformations with the tricyclic pyrimidine cap groups extending away from the L1 crevice without making any apparent interaction with nearby residues, validating the docking results. The authors indicated that water-mediated hydrogen bonding is possible with S531 and D460 residues of the L1 crevice.28 In contrast, TO-317 was found to adopt a ligand-enzyme fit that showed scaffold engagement with both L1 & L2 outer crevices (PDB 5WGI, Fig. 3). Furthermore, TO-317 was predicted to adopt a conformation that engaged several residues through aromatic π-π interactions and hydrogen bonding. The benzyl linker retained the π-π interaction with F643 and F583 observed with compound 1. The pyridyl ring was predicted to occupy the L2 crevice and engage in a π-π stacking interaction with F642 and H-bonding with the protonated Zn2+-bound, H614. These in silico HDAC6 enzyme engagements are absent in comparative HDAC8 studies (PDB 1T64, Fig. S4-B, Table S1).
Figure 3:
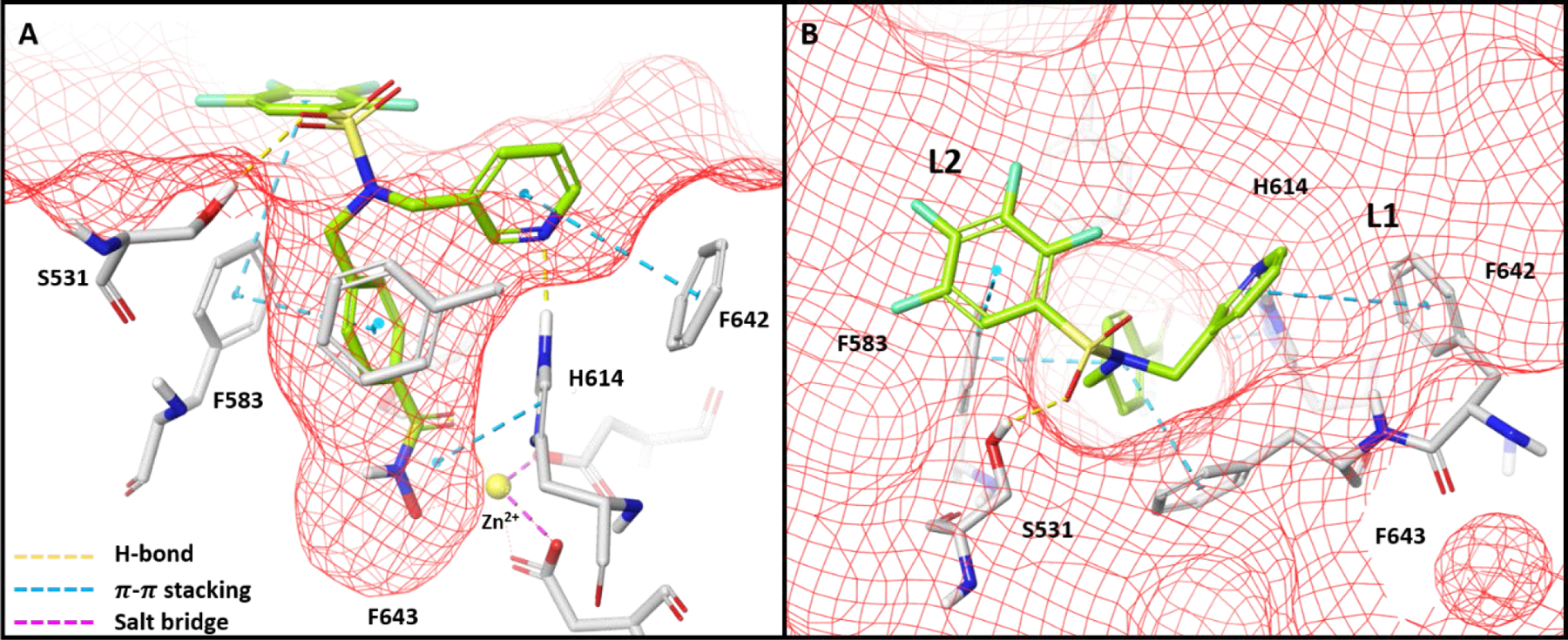
Docking conformation of TO-317 with drHDAC6 CD2 (PDB 5WGI) reveals occupancy of both L1 and L2 crevices which facilitates a plethora of interactions. Deck A shows the pocket view of these interactions, while deck B shows how TO-317 adopts a windmill conformation that allows both aromatic ring cap groups to engage with the L1 and L2 crevices of the HDAC6 outer surface.
To validate these docking predictions, the X-ray crystal structure of the HDAC6–TO-317 complex was solved at 1.84 Å resolution (Fig. 4). There are 2 monomers in the asymmetric unit in this orthorhombic crystal form, and the structures of each are essentially identical (root-mean-square deviation = 0.07 Å for 296 Cα atoms). Similarly, the inhibitor binding mode is essentially identical in both monomers, with the hydroxamate moiety coordinating to the catalytic Zn2+ ion in a monodentate fashion as first observed for the binding of N-hydroxy-4-(2-[(2-hydroxyethyl)(phenyl)amino]-2-oxoethyl)benzamide (HPOB).15,34 The hydroxamate N-O– group coordinates to Zn2+ with an average separation of 2.4 Å and is hydrogen bonded with Y745. The hydroxamate C=O hydrogen bonds with the Zn2+-bound water molecule, which further engages H573 and H574 in hydrogen bonding.
Figure 4.
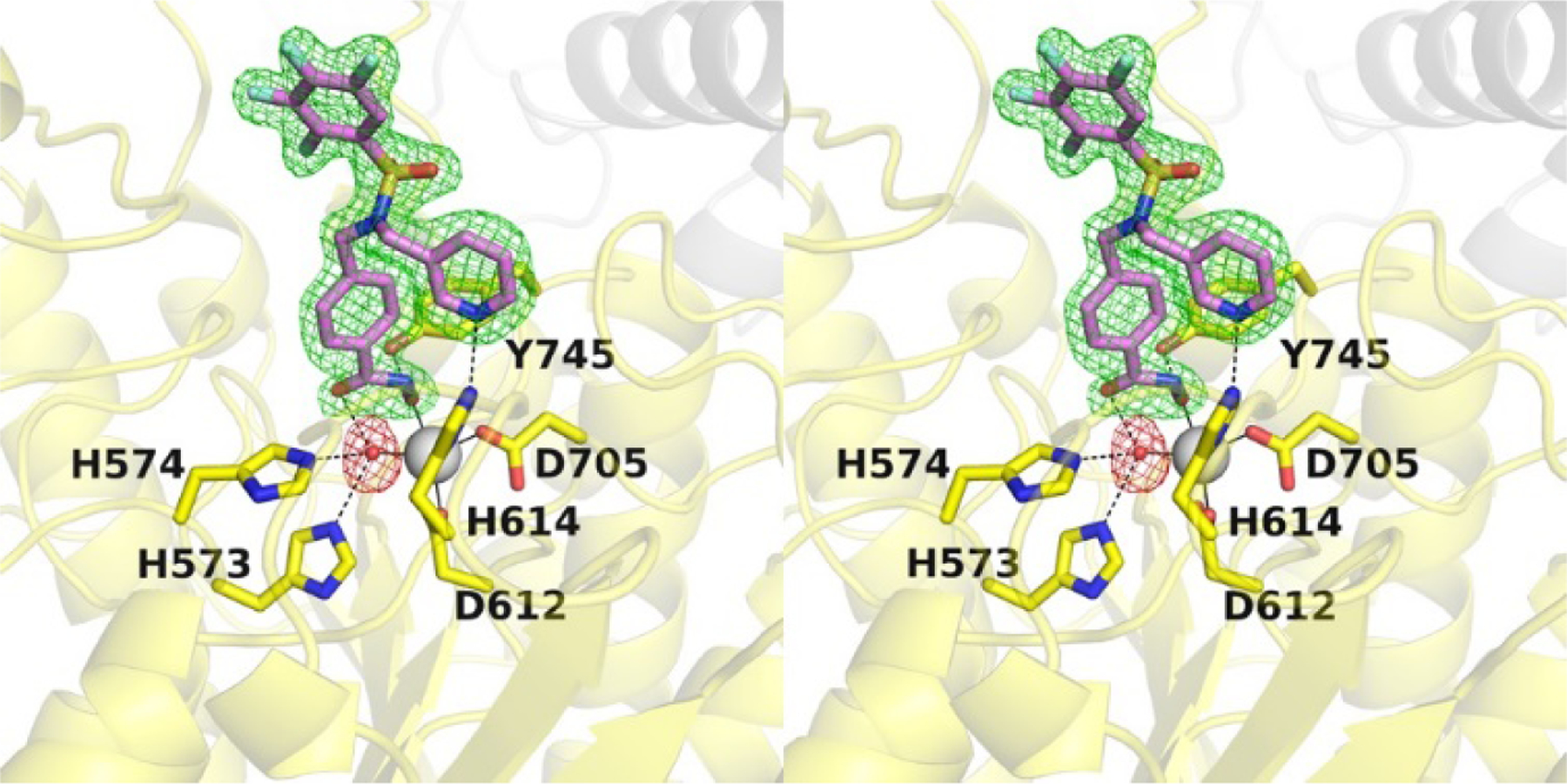
Stereo-view of a Polder omit map (contoured at 4.0σ) showing the binding of TO-317 in the active site of HDAC6 (PDB code: 7JOM). The catalytic Zn2+ ion is shown as a grey sphere, the Zn2+ bound water molecule is shown as a red sphere; metal coordination and hydrogen bond interactions are shown as solid and dashed black lines, respectively.
Outside the active site cleft, the benzyl group of the phenylhydroxamate moiety packs between F583 and F643 and engages in favorable offset π-π interactions. Desolvation of this region may contribute an entropic advantage to the binding of inhibitors with aromatic linker groups.35 The capping group is bifurcated via the dialkyl sulfonamide nitrogen, with each substituent directed to different pockets in the enzymes binding pocket outer surface. Bifurcated capping groups enable the capture of additional affinity and selectivity interactions in the HDAC6 active site.32 Notably, the pyridine ring binds in the L1 pocket and accepts a hydrogen bond from Zn2+ ligand H614. While water-mediated enzyme-inhibitor hydrogen bonds with H614 have been observed,28 a direct enzyme-inhibitor hydrogen bond with this residue has not been previously observed. As such, the inhibitor pyridine group serves as a “second shell” or indirect ligand to the catalytic Zn2+ ion,36,37 and chelation of the Zn2+ ion by direct and indirect interactions through the hydroxamate group and the pyridine ring, respectively, presumably makes a substantial contribution to .affinity. The sulfonamide group does not participate in any hydrogen bond interactions and is oriented such that the TFB ring is directed towards solution. The closest residue to the TFB ring is S531, which at its closest point is 3.4 Å away.
The molecular residency of TO-317 at the HDAC6 active site was investigated by kinetic experiments and mass spectrometry analysis. In a jump dilution experiment, TO-317 exhibited a Ki of 0.7 nM and a residence time of 142 min in a two-step reversible inhibition model (Fig. S5A). This occupancy at the catalytic pocket could rationalize the IC50 values exhibited by TO-317 in enzyme pre-incubation studies (No pre-incubation IC50 = 4 nM, 3-hr pre-incubation IC50 = 2 nM; IC50’s an average of duplicate studies, Fig. S5B) as intact mass spectrometry analysis confirmed that no covalent modification of the enzyme was observed on treatment with TO-317 (Fig. S5C). These experiments suggest a reversible, yet thermodynamically driven interaction between TO-317 and HDAC6 whose stability is presumably driven by the observed dual Zn2+-coordination at the active tunnel.
TO-317 exhibits in cellulo HDAC6 target engagement and mediates mechanistic cell proliferation in leukemic cancers:
The cellular potency of TO-317 was evaluated in a selection of indications where HDAC6 activity has been implicated and treated with HDAC6 inhibitors as single agents. Although HDAC6 has been validated as a clinical target in hematological cancers such as MM, HDAC6 knockout/deletion studies with siRNA or CRISPR/CAS9 show discrepancies in dependency. TO-317 was assessed across five disease indications (12 cancer cell lines) previously validated by several research groups for their dependency on HDAC6 in growth and survival.1,38–44 Assessment in acute myeloid leukemia (AML) cell lines was prioritized owing to growing evidence of HDAC6 playing a critical role in survival and resistance to various treatment regimens.1,42 As can be seen in Fig. 5., TO-317 was active in several cancer cell lines but showed minimal activity in ‘normal’ MRC9 lung fibroblasts. TO-317 was shown to have anti-proliferative activity in all six leukemic cancer cells tested, having an average IC50 of <2.0 μM. Sub-μM anti-proliferative activity was observed in both MM cell lines where HDAC6 inhibitors have been deployed clinically.2,38,40,45
Figure 5:
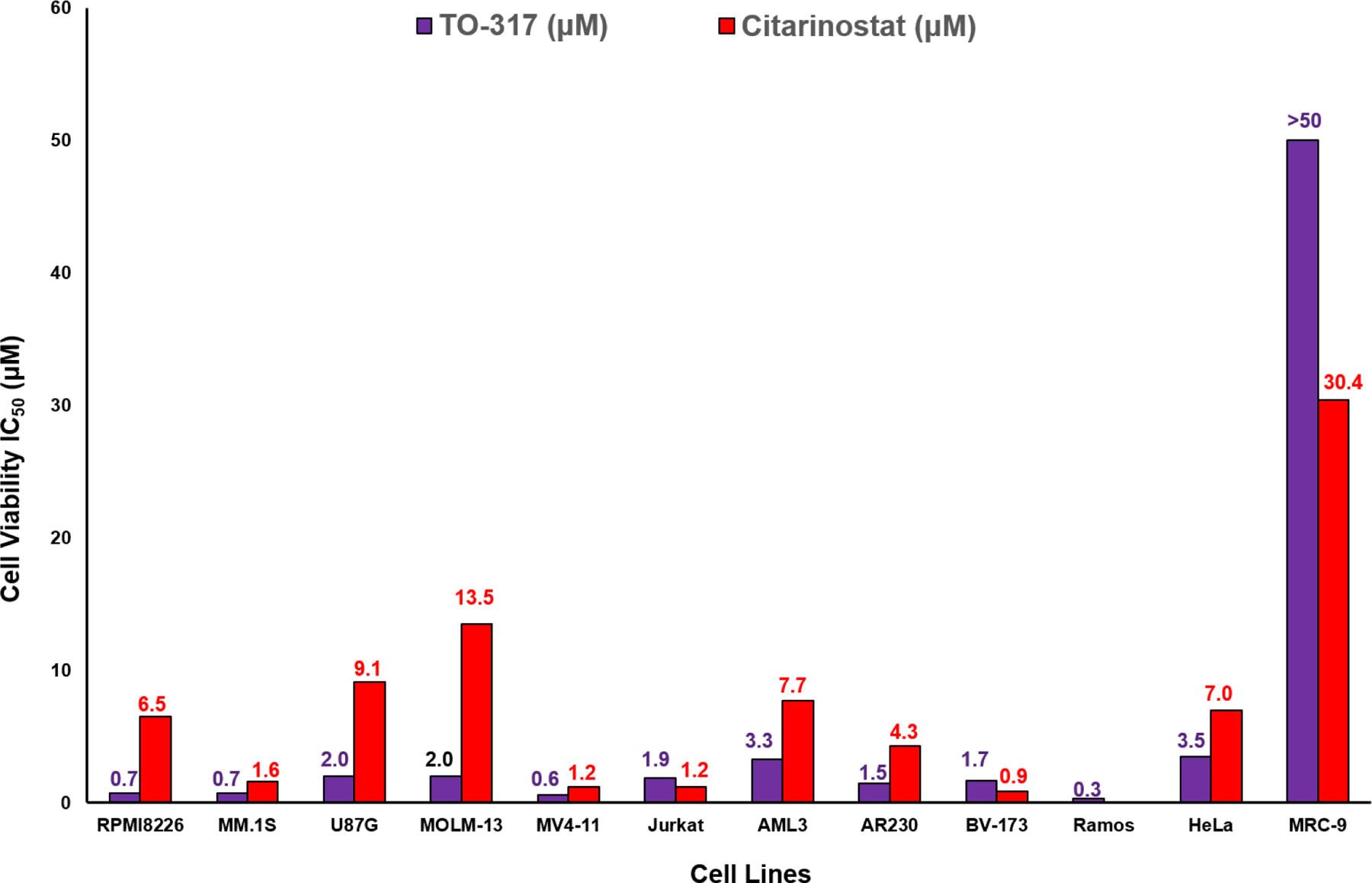
Cellular potency of TO-317 and Citarinostat (positive control) in 12 cell lines (1 heathy cell, MRC-9 included). TO-317 shows anti-proliferative potency across different cancer cells with minimal activity in healthy cells. IC50 values are indicated above each graph and reported in μM. IC50 values with their 95% confidence intervals are reported in Table S3.
Acetylation levels of α-tubulin are used as a clinical biomarker for HDAC6 inhibition.46,47 Inhibition of other HDACs leads to an increase in histone H3K9 and H3K27 acetylation.48,49 Comparative acetylation of both substrates provides a robust biomarker of HDAC6 inhibition. Treatment of MV4–11 cells with TO-317 induced a significant increase in Ac-α-tubulin levels, even at 0.25 μM, with only minimal accumulation of Ac-histones (Fig. 6A). From comparative Western blots in TO-317 and Citarinostat treated MV4–11 cells (0.25 −1.0 μM), it was determined that TO-317 is marginally more potent for HDAC6 (Fig. 6A) with strong increases in Ac-α-tubulin at concentrations of 0.25 μM. Citarinostat appears more selective than TO-317 in a cellular environment, with accumulation of Ac-Histone H3K27 between 0.75 −1.00 μM in both AML and MM cells. Similar results were observed in multiple myeloma cell lines (MM.1S cell lines, Fig. 6B) where TO-317 was more potent for HDAC6 over the range of concentrations assayed. These results recapitulate the selectivity and potency of TO-317 for HDAC6 in cellulo. However, at higher concentrations the effects and cytotoxicity observed may be partially due to nuclear HDAC inhibition.
Figure 6:
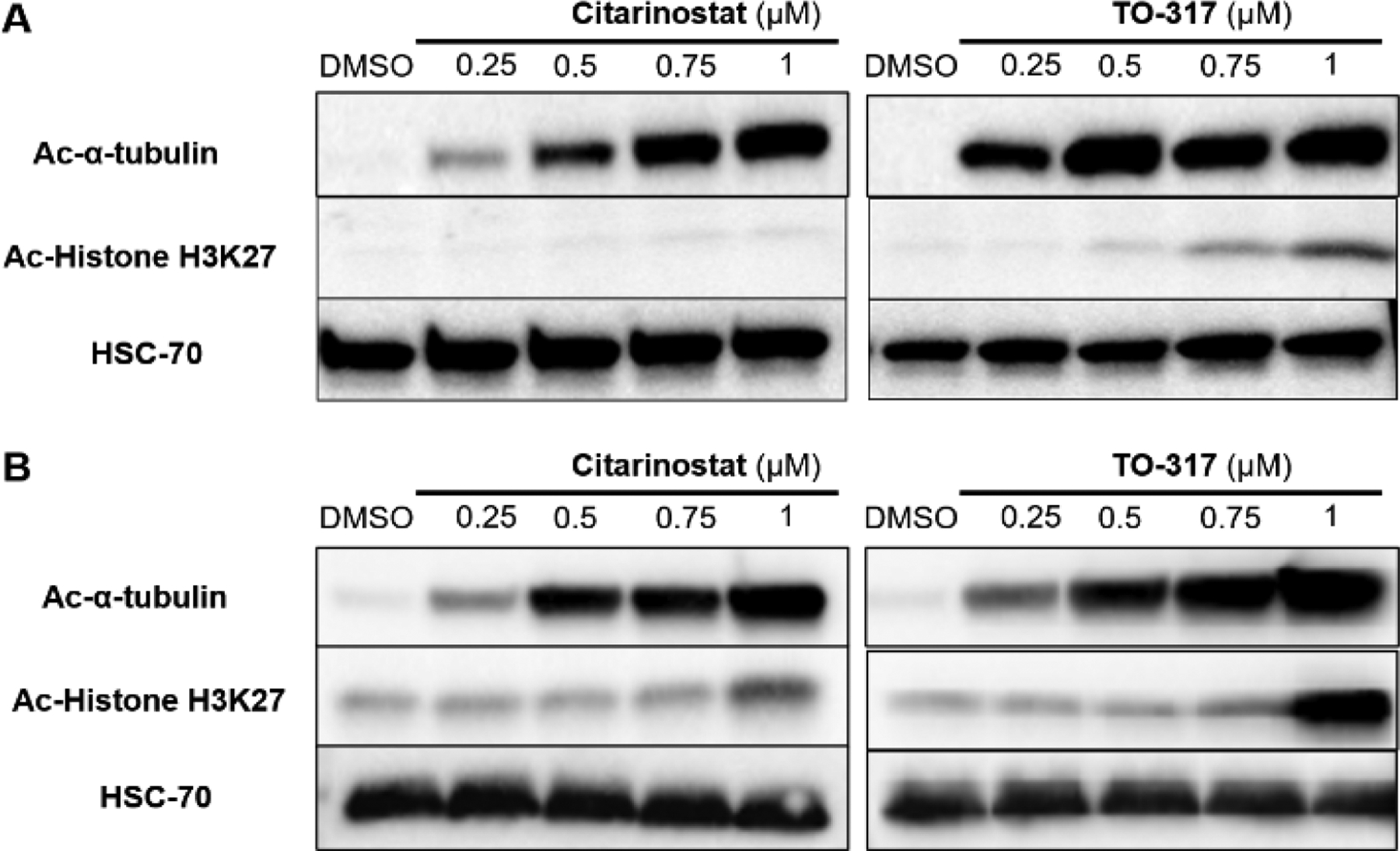
TO-317 shows superior HDAC6 selectivity and potency in MV4–11 and MM.1S cells. (A) MV4–11 cells treated with increasing doses (0.25 μM - 1 μM) of Citarinostat and TO-317 show accumulation of Ac-α-tubulin for TO-317 at 0.25 μM but not for Citarinostat. (B) Similar dose-dependent responses were observed for both inhibitors in MM.1S cells.
Immunofluorescence was employed as a secondary biorthogonal assay to validate the in cellulo HDAC6 selectivity of TO-317. HeLa cells were incubated with two different concentrations of TO-317 or Citarinostat at 0.1 μM and 2 μM. Treatment with TO-317 led to significant accumulation of Ac-α-tubulin (red stain) at 2 μM. These effects were slightly less prominent with Citarinostat at the same concentration (Fig. 7 & Fig. S7). The levels of Ac-histone (green stain) do not change significantly upon treatment with either 0.1 μM or 2 μM concentration of TO-317 (Fig. 7). A DAPI (4′,6-diamidino-2-phenylindole) nuclear stain (blue, Fig. S6) confirmed cell viability during the 6 h incubation period assessed. An overlay of both acetylated tubulin and histone stains also showed preferential Ac-α-tubulin accumulation over Ac-H3K27 histones at all concentrations of TO-317 (Fig. S6). In conjunction with initial Western blots, immunofluorescence further recapitulated the in cellulo HDAC6 target engagement exhibited by TO-317.
Figure 7:
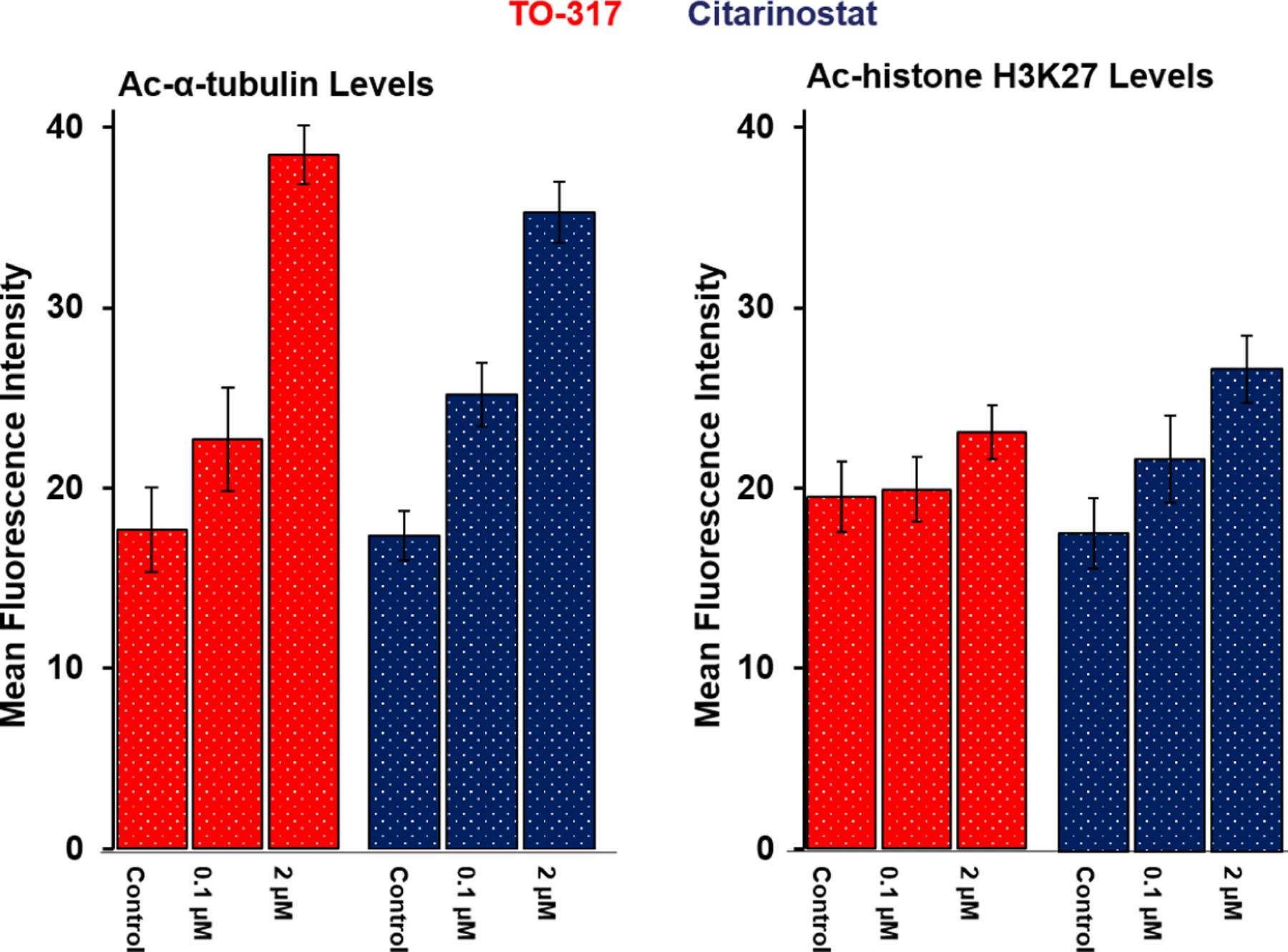
Quantification of Ac-α-tubulin and Ac-histone levels in immunofluorescence assay. DMSO was used as a negative control in each cohort, and Citarinostat was the positive control. Result indicates that TO-317 induces a clear dose-dependent increase in acetylated α-tubulin with minimal cellular accumulation of acetylated histones under the same dosing concentrations. Data reported above are an average of duplicate experiments.
To characterize the mechanism of inhibition and cell death by TO-317, fluorescence activated cell sorting (FACS) flow cytometry was employed to sort the populations of treated leukemic cells at different stages of the cell cycle via their differential staining with annexin and PI fluorophore. Cells in the sub-population UL (upper level, red stain) are necrotic cells and an increase in cell population in this quadrant is indicative of a toxic inhibitor lacking a defined mechanism. TO-317 does not show statistically different changes to necrosis as this sub-population is consistent across the range of inhibitor concentration tested and comparable to the DMSO control (Fig. 8). The LL sub-population (lower level, green stain) shows viable cells where the fluorophore cannot access the cytosol (green stain) and is representative of intact and healthy cancer cells. The healthy cell count dramatically declines from 88% with no inhibitor present to 35% at 2 μM inhibitor concentration. The loss in healthy cells is compensated in the LR and UR quadrants (blue stains) showing cell populations undergoing early and late apoptosis, respectively. These quadrants show an increase in respective populations as the concentration of TO-317 is increased. Early and late apoptotic cell populations increased from a combined 11% at 0.25 μM to 65% at 2 μM, and to 73% at 4 μM (Fig. S8-A). A similar trend was observed for Citarinostat (Fig 8, Panel B; Fig S8-B), albeit with reduced potency as was seen with TO-317. This systematic disruption and arrest of cell cycle is the signature of a mechanistic-based inhibitors, and strongly supports the hypothesis that TO-317 achieves cell death in leukemia cells through programmed cell death.
Figure 8:
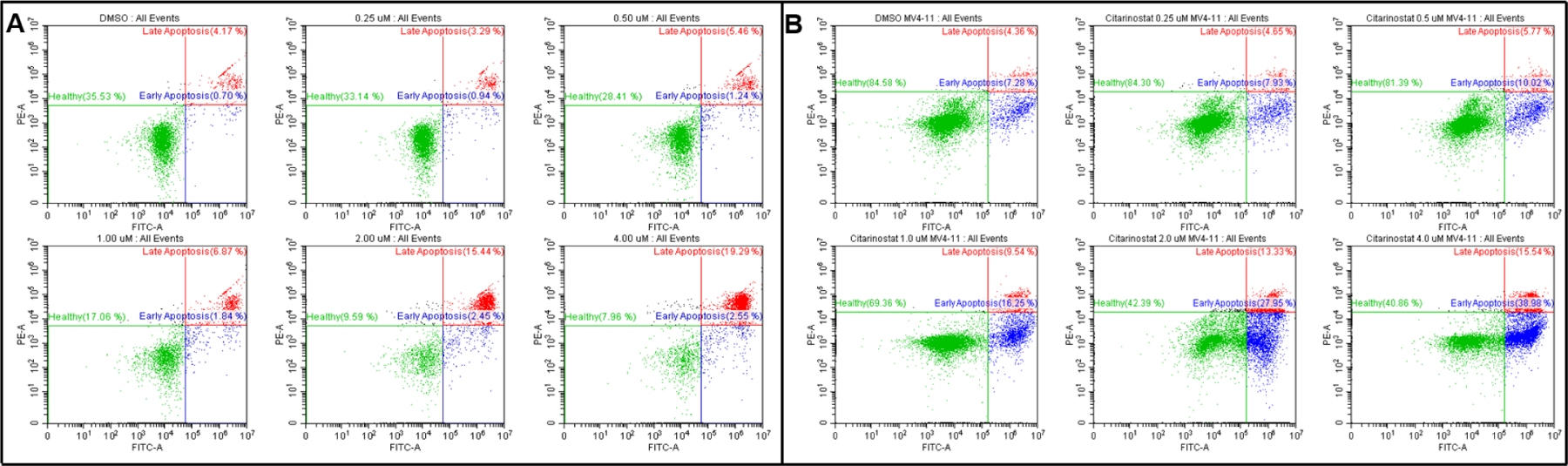
MV4–11 treated with TO-317 (Panel A) and Citarinostat (Panel B) leads to dose-dependent programmed cell death. Death by necrosis (Q1) is minimal and consistent at all doses. Dose-dependent cell death by TO-317 is consistent with a mechanistic approach of cell cycle arrest leading to apoptosis and corroborates the previously observed superior anti-proliferative activity of TO-317 over Citarinostat, the positive control. Data reported above are an average of duplicate experiments, and quantification of each cell population with the associated standard deviation is reported graphically in Fig. S8-A & Fig. S8-B.
The role of reactive oxygen species (ROS) in the progression and survival of leukemic cells has been very contentious.50 NADPH oxidases (NOX) are elevated in several AML mutations including FLT3-ITD AML and Ras.51 These mutations are known to be associated with poor prognosis and a high resistance and relapse rates whose mechanism depends on over-secretion of ROS. Several models of AML also show increased ROS generation-induced redox dysregulation and oxidative stress that is strongly linked to promotion of cellular proliferation, survival, and immune evasion.52 However, several studies also indicate that the persistence of elevated ROS levels may significantly strengthen oxidative DNA damage leading to single and double strand breaks and lipid peroxidation, which further leads to cellular damage. Since HDAC inhibitors are known to generate ROS and induce cell cycle arrest in cancer cells,53–55 TO-317 treated MV4–11 cells were investigated. As expected, TO-317-treated cells showed increased ROS generation when compared to the DMSO control over a 4 h period (Fig. 9). This is consistent with previously reported HDAC inhibitors and may implicate ROS accumulation as a possible mechanism for the induction of apoptosis observed with TO-317.
Figure 9:
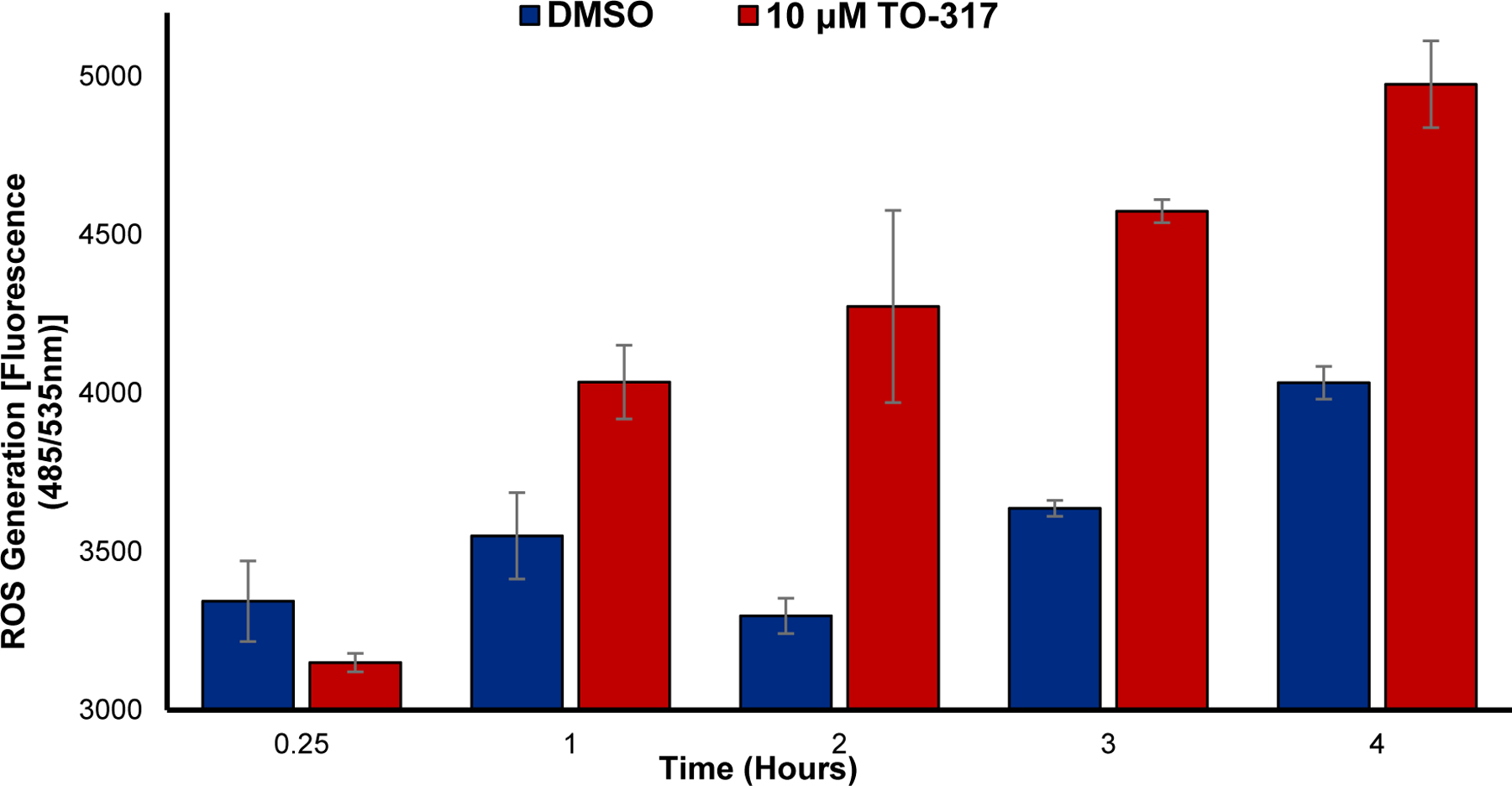
ROS generation in MV4–11 following incubation with 10 μM TO-317 for up to 4 h. TO-317 shows markedly increased levels of ROS after 2 h incubation which continues to increase up to the 4 h incubation time tested. Data reported is an average of duplicate studies.
TO-317 in vitro & in vivo stability profiles:
Pharmacokinetic profiles of TO-317 were assessed in both in vitro and in vivo experiments. Plasma and whole blood stability assays were used to determine the stability of TO-317 in biological fluids. Several studies have shown reduction of hydroxamic acids in whole blood, but not in plasma.56,57 After 1 h incubation in mouse plasma, 68% of TO-317 remained with a calculated t1/2 of 110 min (Table. 4). In pooled human whole blood samples, ~69% of TO-317 was present after 1 h with a calculated t1/2 of 108 min, indicating collectively, that TO-317 does not suffer from the previously reported high clearance rates and metabolic degradation of N-hydroxamic acids in biological fluids.58,59 TO-317 was advanced for preclinical pharmacokinetic study employing a dose of 50 mg/kg via intraperitoneal (i.p.) injection in male MALB/c mouse. Consistent with previous plasma and whole blood studies, TO-317 displayed modest stability in vivo (t1/2 = 1.7 h) as compared to known HDAC inhibitors, reaching a Cmax of 95.1 ng/mL in 1 h (Fig. 10). However, given the dose, 50 mg/kg, significantly higher plasma concentrations were anticipated, indicating either a metabolic instability in vivo, or poor solubility / permeability, which limits systemic bioavailability and further progression as a therapeutic agent.
Table 4:
TO-317 showed extended lifetimes in both mouse plasma and pooled human blood stability assays. Assay values reported was an average of triplicate studies.
| Remaining Percentage TO-317 Concentration | |||||
|---|---|---|---|---|---|
| Assay | 0 min | 15 min | 30 min | 60 min | T1/2 (min) |
| Mouse Plasma* | 100% | 88.1% | 79.8.0% | 68.0% | 110 |
| Human Whole Blood* | 100% | 88.0% | 80.8% | 68.9% | 108 |
Figure 10:
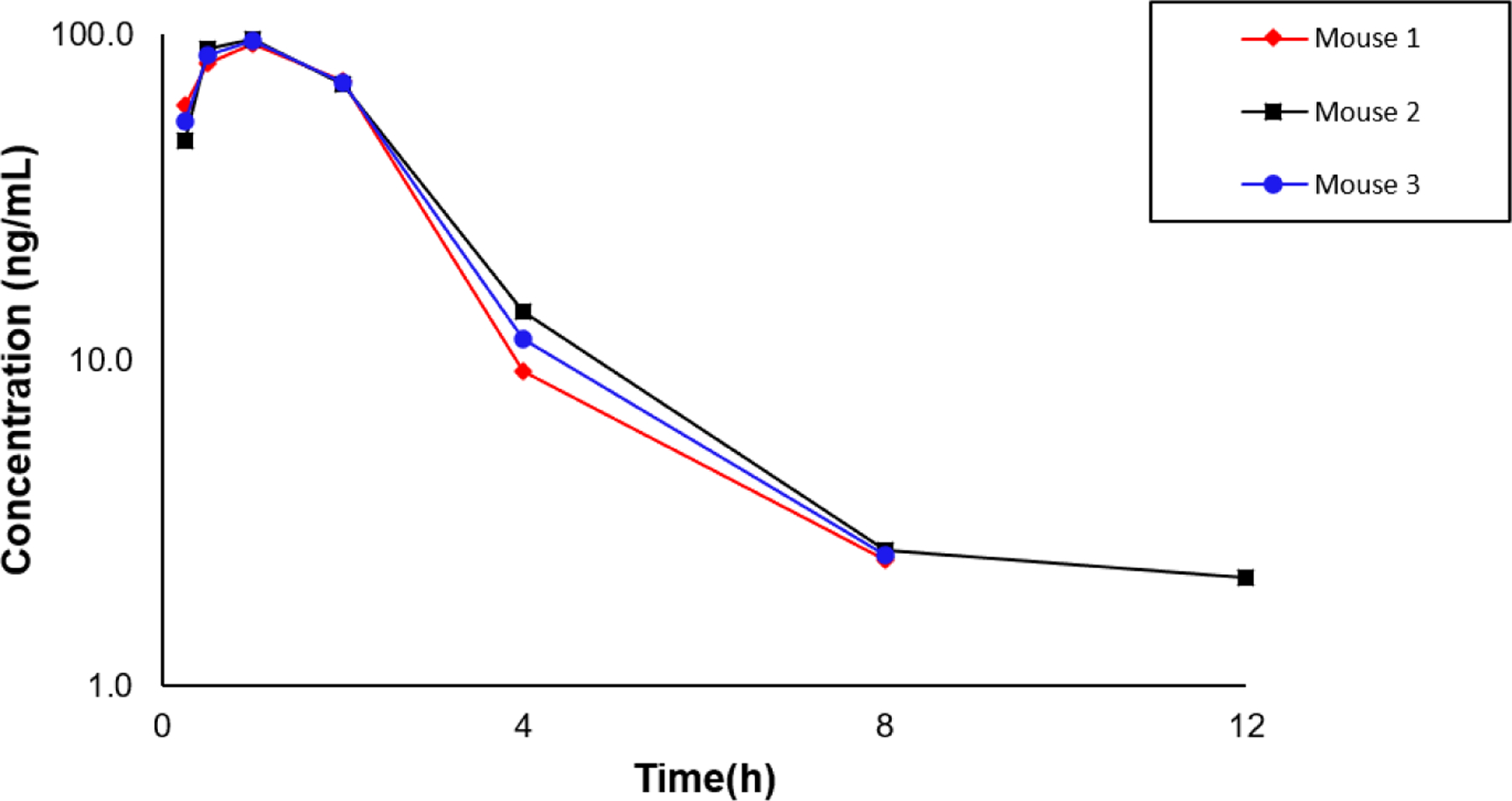
TO-317 is stable in vivo with a t1/2 (avg) = 1.7 h. Male BALB/c mice were dosed TO-317 intraperitoneally (50 mg/mL solution 5% DMSO, 30% PEG400, 1% Tween80 & 64% saline) and peaks were detected and quantified from collected blood samples at different time concentrations using a triple-quad LC-MS/MS.
Kinetic solubility experiments (Fig. S9) showed TO-317 to exhibit solubility in PBS, pH 7.4 with a saturation concentration of 161.50 ± 33.20 μM (n = 3), ruling out solubility as a likely cause for its poor in vivo exposure. PAMPA studies (Table S2) revealed poor permeability profiles which may be responsible for the limited availability observed in vivo with TO-317, with an average permeability score of -logPe = 6.71. Membrane permeable molecules generally exhibit permeability scores with -logPe ≤ 6.0. It is interesting that TO-317 can elicit PD in proliferating cells such as AML and MM but is unable to passively diffuse in the i.p. dosed preclinical experiments.
Future studies will therefore be directed towards improving the permeability properties of TO-317 in vivo, while retaining HDAC6 potency, selectivity, and biological activity.
Discussion
The push for selective HDAC6 inhibitors has been on the rise due to the promising clinical efficacy and lower toxicity profile exhibited in trial. For example, Citarinostat (ACY-241, in combination with lenadomide) is currently in clinical trials for smoldering multiple myeloma; Ricolinostat (ACY-1215) is enrolled in multiple clinical trials for a variety of conditions such as breast cancer, multiple myeloma, diabetic neuropathic pain amongst others.20,39,43,60 Recent clinical candidates have selectivity windows that are 5–6-fold for HDAC6 as compared to off-target HDACs. While it has been previously reported that HDAC6 has conformational predilections that discriminate between different structural motifs and imposes requirements for inhibitors to achieve maximum ligand-enzyme fit and interactions,26,31 the clinical HDAC6 inhibitors have instead opted for bulky cap groups to confer selectivity. In this study, by X-ray crystallography, we have shown that TO-317, employing a bifurcated approach, using a pyridyl cap group in concert with the traditional N-hydroxamic acid occupies the L1 outer crevice and makes a strong direct H-bonding with the Zn2+-ligand, H614. To the best of our knowledge, there has been no previous report of a direct H-bonding interaction with a Zn2+-ligand at the HDAC6 active site. TO-317 is shown to adopt an induced fit model which may facilitate this direct residue interaction and thereby stabilize the enzyme-inhibitor complex. This unique structural motif, which engages the second shell interactions with the catalytic Zn2+ metal can provide novel pharmacophores for identifying tighter binding HDAC6-selective inhibitors.
The selectivity of TO-317 was demonstrated in vitro through several orthogonal cell biology experiments. We showed in a dose-dependent manner the preferential increase in acetylated α-tubulin on incubation with concentrations as low as 50 nM of TO-317. Furthermore, ~0.75 μM was required to observe distinct acetylation of H3 histone, validating the in vitro preferential HDAC6 selectivity of TO-317. TO-317 was found to be biologically active in several cancer cell lines, while not eliciting any meaningful cell-killing activity in normal cells, indicating an appreciable therapeutic window, which was attributed to the observed HDAC6 selectivity. TO-317 mediates programmed cancer cell apoptosis, which may be associated with ROS generation along the mechanistic pathway. Preliminary stability assays in various biological fluids highlighted potential bioavailability issues in vivo with this inhibitor. Future studies will attempt to improve the poor bioavailability exhibited by TO-317.
Collectively, we have demonstrated a novel approach to selective HDAC6 inhibition, achieving low nM potency against HDAC6. Structural studies have revealed a unique binding mode in which TO-317 simultaneously coordinates to Zn2+ and the H614 residue in the catalytic tunnel, affording >150-fold selectivity for HDAC6. Optimized PK properties might yield a preclinical candidate HDAC6 inhibitor from this compound. Finally, the TO-317 model presents a structural model that can be incorporated for the selective targeting of HDAC6.
Experimental Section
Chemistry:
All chemicals and reagents were purchased from any of the following suppliers – Combi-Blocks, Sigma-Aldrich, Enamine, Alfa-Aesar, and Oakwood Chemicals. Solvents such as DCM were obtained in anhydrous forms from our in-house Mbraun solvent dispensary system. Other solvents such as acetonitrile and DMF were used in their dry forms from 100 mL Sigma-Aldrich bottles. Reactions were carried out in oven-dried glassware, and reaction progress monitored using a TLC silica gel 60 F254 and a 254 nM UV lamp. Purification of intermediates were carried out using a Biotage automated flash chromatography instrument, in pre-packed SNAP KP-SIL cartridges supplied by Biotage in sizes of 12G, 25G, 50G, and 100G packings. Characterization for intermediates include 1H, 19F, and 13C NMR obtained using a 400 MHz Bruker NMR spectrometer, low-res mass spectrometry obtained using a Waters LC-MS Micromass ZQ using ESI positive and negative ionization modes. Purifications of final compounds were carried out using a Waters prep-HPLC system equipped with Waters 2545 quaternary gradient pump system, XSelect 300G CSH Phenyl-Hexyl C18 column, 2489 dual absorbance UV detector, and ran using mobile phases acetonitrile and water spiked with 0.1% TFA. Characterization for final compounds include 1H, 19F, and 13C NMR and are obtained like the intermediates, high-res mass spectrometry collected using a quadrupole time of flight LC/MS system at the Advanced Instrumentation for Molecular Structure (AIMS) facility at the University of Toronto, purity check for all final compounds satisfy a ≥95% purity requirement and traces were collected using a HP 1100 series analytical HPLC using gradient 5% - 100% Acetonitrile in water for 40 mins / 60 mins. The retention times (TR) and purity as calculated from peak area of compound / total area of all peaks in spectra X 100% were reported as automatically calculated by the analytical HPLC instrument.
Details for protein crystallographic methods are provided in detail in the online supplementary information.
All intermediates and final product characterizations and their yields are reported in the online supplementary information.
Synthesis of Product A’ (Tetrafluorobenzenesulfonyl Chloride):
Tetrafluorobenzene (1.0 g, 6.6 mmol) was carefully added into chlorosulfonic acid (3.88 g, 33.3 mmol) and the mixture was refluxed for 3 hrs. Reaction vessel was cooled to 0 °C, and the solution was slowly pipetted into ice-cold water. This aqueous solution was extracted with 20 mL EtOAc twice. Combined EtOAc fractions were washed with brine and dried over MgSO4. Clear solution was concentrated under vacuum and purified using flash chromatography, isocratic condition 10% EtOAc in Hexane. Pure product was collected as a clear oil, 95% yield. 1H NMR (400 MHz, Acetonitrile-d3) δ 7.92 – 7.66 (m, 1H).,19F NMR (376 MHz, Acetonitrile-d3) δ −133.82 (dtd, J = 20.7, 12.0, 5.9 Hz), −135.65 – −136.10 (m), −142.87 (tdd, J = 19.2, 12.0, 7.8 Hz), −151.06 – −151.57 (m). 13C NMR (101 MHz, Acetonitrile-d3) δ 147.8 (ddd, J = 10.7, 3.8, 1.8 Hz), 147.3 (ddd, J = 15.8, 12.3, 3.1 Hz), 146.7 (dt, J = 12.7, 3.3 Hz), 145.3 (ddd, J = 10.8, 3.9, 1.8 Hz), 144.6 (ddd, J = 15.9, 12.3, 3.1 Hz), 144.1 (dt, J = 12.7, 3.3 Hz), 143.1 (ddd, J = 16.0, 13.0, 3.2 Hz), 140.5 (ddd, J = 16.1, 12.9, 3.2 Hz), 127.4 (ddd, J = 11.6, 6.8, 4.4 Hz), 116.6, 111.8 (dd, J = 23.4, 3.5 Hz). MS ESI- Found 229.04 corresponding to M-1 for the acid showing rapid compound hydrolysis under MS conditions. Also found 459.0 (M+M+1 peak)
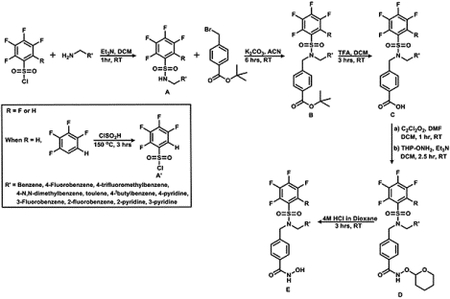
Synthesis of Product A (Secondary Sulfonamides):
In an oved-dried round bottom flask charged with a magnetic stirring bar was added 1 molar equivalence of the corresponding sulfonyl chloride and 1.1 molar equivalent of the corresponding amine in 0.1M DCM solution and the mixture is stirred at RT under inert conditions for 1 hr or until TLC confirms complete consumption of starting materials. An insoluble precipitate is usually formed corresponding to the protonated quaternary sulfonamide product which is neutralized on addition of 2 molar equivalence of the triethylamine base usually leading to dissolution of the precipitate and a clear solution. This clear solution is further stirred for an additional 10 minutes. Reaction solvents are removed under vacuum and residue obtained is dissolved in 2:1 mixture of EtOAc : H2O. The organic layer is washed with brine and dried over MgSO4. The organic solution is evaporated and purified by flash chromatography using conditions as determined by TLC.
Synthesis of Product B (Tertiary Sulfonamide, Ester Derivatives):
To the corresponding secondary sulfonamides (1 eq.) was added under inert conditions 1.1 molar equivalent of tert-butyl 4-(bromomethyl) benzoate and K2CO3 (2 eq.) in 0.25M acetonitrile solution. Mixture was stirred for 6 hours at room temperature, then the acetonitrile solvent is removed, and the solid residue obtained is dissolved in EtOAc and washed with H2O. Organic layer is further washed with brine and dried over MgSO4. The clear solution obtained is concentrated and purified using flash chromatography at the appropriate conditions as determined by TLC.
Synthesis of Product C: (Tertiary Sulfonamides, Acid Derivatives):
The corresponding ester is dissolved in 1:4 TFA : DCM (0.1M solution) and stirred for 3 hrs or until TLC indicates complete deprotection of the ester. Acidic solution is then neutralized with sodium bicarbonate to a pH of ~8 before extracting 3x using appropriate amounts of DCM. The combined organic phase is washed once with brine and dried over MgSO4. Clear organic solution is evaporated under vacuum to give the required acid which is carried forward to the next step without any further purification.
Synthesis of Compound D: O-(Tetrahydro-2H-pyran-2-yl) Protected Hyrodoxamate Esters:
In an oven-dried round bottom flask was added the corresponding acid dissolved in 0.2M DCM containing few drops of DMF. This solution is cooled to 0 °C, and 5 molar equivalent oxalic chloride is added resulting in wild evolution of CO2 gas. On complete effervescence, the solution is warmed to room temperature and then allowed to stir for 1 hr. After stirring for 1 hr, the solvents areremoved by evaporation and the residue usually obtained is dried under vacuum for 1 hr. The dried chloride residue is further taken up in 0.5M DCM and 1.5 molar equivalent of O-(Tetrahydro-2H-pyran-2-yl)hydroxylamine resulting in a cloudy mixture which is stirred for 15 minutes before the addition of 2 molar equivalent triethylamine which results in a clear solution. This mixture is further stirred for 1 hr or as judged to be completion of reaction by TLC. The DCM solvent is then removed under vacuum, dissolved in H2O and extracted with EtOAc. The organic layer is washed with brine then dried over MgSO4 before being purified using flash chromatography based on conditions as determined by TLC.
Synthesis of Final Compounds: Corresponding Hydroxamic Acids:
The precursor hydroxamate ester is dissolved in 0.1M solution of 4M HCl in Dioxane and the acidic solution was stirred for 3 hrs. or until TLC indicates complete consumption of the starting material. The acid solution is removed under vacuum and the crude hydroxamic acid is taken up in 2 – 5 mL mixture of H2O: Acetonitrile in proportions that is suitable for dissolution. A 200 mL aliquot is subjected to an analytical HPLC trial run (5% - 100% acetonitrile in H2O) to determine appropriate conditions for preparatory HPLC purification runs. About 2 – 5 mL of reaction mixture is then subjected to a preparatory HPLC run, 5% - 95% acetonitrile or gradient conditions as determined from analytical HPLC trial run. Fractions from prep-HPLC are further checked by analytical HPLC for purity check, and then analyzed by LCMS for identity check before pure fractions are pooled together and lyophilized overnight to give the pure dry hydroxamic acids.
Cytotoxicity assays:
HeLa cells were grown in Dulbecco’s Modified Eagles Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) (Sigma-Aldrich). MV4–11 and AML3 were maintained in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% FBS. MOLM-13, MRC-9, MM.1S, MM.1R, Jukat, BV-173 and RPMI 8226 cells were maintained in RPMI-1640 and supplemented with 10% FBS. U87G cells were maintained in Eagle’s Minimum Essential Medium (EMEM) and supplemented with 10% FBS.AR230 cells were maintained in RPMI-1640 and supplemented with 10% FBS, 2 mM L-Glutamine and 1 μM Imatinib. Normal Human Fibroblasts (NHF) were purchased from Cell System and grown in Cell System growth medium, supplemented with Culture Boost. HUVEC cells were purchased from ATCC and cultured in Vascular Cell Basal medium supplemented with Endothelial Cell Grow Kit-VEGF. Appropriate number of cells were plated per well in 96-well flat-bottom sterile culture plates with low-evaporation lids (Costar #3997) for these cell lines. After 24 h, inhibitors and a vehicle control (0.5% DMSO) were added. Wells were treated with Cell Titer-Blue® (Promega #G808A) (20 μL/well) after 72 h and fluorescence was recorded at 560/590 nm using a Cytation S63 spectrophotometer. IC50 values were determined using non-linear regression analysis with GraphPad Prism 6.0 (GraphPad Software Inc.).
FACs Apoptosis Detection Assay:
MV4–11 cells were cultured, dosed, and washed twice with cold 1X PBS. Using FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen), cell pellets were resuspended in 1X binding buffer at a density of 1 × 106 cells/ml. 5 μL FITC Annexin V and 5 μL Propidium Iodide (PI) were added to 250 μL of solution (2.5 × 105 cells). Cells were vortexed and incubated in the dark for 15 min, followed by addition of 250 μL of 1X binding buffer. Cells were analyzed by flow cytometry within 1 hour using Cytoflex S (Beckman Coulter).
Western blotting:
MV4–11 and MM.1S cells were incubated with compounds for 6 hours, before lysing with radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris pH 7.4, 150 mM NaCl, 0.5% deoxycholate, 1% Triton X-100, and 0.1% sodium dodecyl sulfate (SDS)). Total protein was measured using a BCA assay (ThermoFisher), in which clarified protein was resolved on a 4 – 20% polyacrylamide SDS gel and transferred to a nitrocellulose membrane (Bio-Rad). The membranes were blocked with a 5% solution of skimmed milk powder in PBST, followed by an overnight incubation at 4 °C in primary antibody (1:1000 dilution). Blots were probed with antibodies against acetylated alpha-tubulin mouse monoclonal (EMD Millipore), acetylated histone H3 (Ac-Lys18, Sigma) and HSC70 (Santa Cruz). Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG secondary antibody (Cell Signaling) or HRP-linked anti-rabbit IgG secondary antibody (Cell Signaling) were applied to the membrane (1:5000 dilution) and bands were visualized using clarity western ECL substrate luminal/enhancer solution and peroxide solution 1:1 ratio for HRP-conjugated secondary antibody (Bio-Rad) and analyzed using Image lab software (Bio-Rad).
Detection of ROS Generation:
MV4–11 cells were treated with the relevant compounds and analyzed for production of ROS using DCFDA (Abcam 113851 DCFDA Cellular ROS detection Assay Kit). Briefly, cells were collected and washed with 1× PBS once and incubated with DCFDA (33 μM) at 37 °C for 30 min in the dark. The cells were then washed and resuspended in 1X supplemental buffer, seeded in a clear bottom black 96-well plate (Fisher Scientific), dosed with compound and fluorescence at Ex/Em=485/535 was measured using a Cytation S63 spectrophotometer.
Immunofluorescence Assay:
HeLa cells were plated to sub-confluency on a clear bottom black 96-well plate (Fisher Scientific) and treated with compound after 24 h. Samples were washed with 1× PBS, fixed with 4% formaldehyde (Millipore Sigma), permeabilized with 1% Triton X-100 (Millipore Sigma), and blocked in 5% bovine serum albumin (BSA) (BioShop) for 1 hour at room temperature. The cells were incubated in an antibody cocktail made up of acetylated alpha-tubulin mouse monoclonal (1:100 dilution, MD Millipore) and acetylated histone H3 (1:50 dilution, Ac-Lys18, Sigma). Cells were counterstained for nucleic acids using 4′,6-diamidino-2-phenylindole (DAPI) (ThermoFisher Scientific). Images were acquired using a Cytation S63 spectrophotometer.
Further protocols have been provided in supplementary methods
Supplementary Material
Funding Sources
P.T.G is supported by research grants from NSERC (RGPIN-2014-05767), CIHR (MOP-130424, MOP-137036), Canada Research Chair (950-232042), Canadian Cancer Society (703963), Canadian Breast Cancer Foundation (705456), Leukaemia and Lymphoma Society of Canada and infrastructure grants from CFI (33536) and the Ontario Research Fund (34876). D.W.C. is supported by grant GM49758 from the US National Institutes of Health. O.O.O. is supported by an OGS Fellowship. Y.S.R and N.N are supported by grants from Jesse’s Journey and A.D.C is supported by NSERC CGS. This work utilized beamline 17-ID-1 (AMX) of the National Synchroton Light Source II, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE office of Science by Brookhaven National Laboratory under contract DE-SC0012704. The Center for BioMolecular Structure (CBMS) is primarily supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health through a Center Core P30 Grant (P30GM133893) and by the DOE Office of Biological and Environmental Research (KP1605010). The authors would also like to thank Andrew Sedmihradsky and family, who have raised funds, awareness for DMD and generated support for Max’s Big Fellowship.
Abbreviations:
- CAS9
CRISPR associated protein 9
- CD
catalytic domain
- CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats
- CTCL
Cutaneous T-Cell Lymphoma
- DAPI
4′,6-diamidino-2-phenylindole
- drHDAC6
danio rerio Histone Deacetylase 6
- EMSA
Electrophoretic Mobility Shift Assay
- FP
Fluorescence Polarization
- HDAC
Histone Deactylase
- HDAC6
Histone Deacetylase 6
- LE
Ligand Efficiency
- LL
Lower Left
- LR
Lower Right
- MM
Multiple Myeloma
- PFB
Pentafluoro Benzene
- PI
Propidium Iodide
- PTCL
Peripheral T-Cell Lymphoma
- TFB
Tetrafluoro Benzene
- UL
Upper Left
- UR
Upper Right
Footnotes
Supporting Information
The supporting information is available free of charge at:
HDAC binding and inhibition profiles using fluorescence polarization and enzymatic activity assays; PAMPA, kinetic solubility, and pharmacokinetic data; cell population sorting; cell viability screens; mass spectrometry; immunofluorescence; synthetic schemes, experimental procedures and compound characterization (PDF)
Molecular formula strings (CSV)
Docking of TO-317 with drHDAC6 (PDB)
Docking of 1 with drHDAC6 (PDB)
Docking of 2 with drHDAC6 (PDB)
Docking of Citarinostat with drHDAC6 (PDB)
Docking of TO-317 with HDAC8 (PDB)
Data Availability
Atomic coordinates and structure factor amplitudes for the drHDAC6-TO-317 complex are deposited in the Protein Data Bank (www.rcsb.org) with accession code 7JOM.
Authors will release the atomic coordinates and experimental data upon article publication.
Competing Interests
The authors declare no competing interests.
References
- (1).Hackanson B; Rimmele L; Benkißer M; Abdelkarim M; Fliegauf M; Jung M; Lübbert M HDAC6 as a Target for Antileukemic Drugs in Acute Myeloid Leukemia. Leukemia Research 2012, 36 (8), 1055–1062. 10.1016/j.leukres.2012.02.026. [DOI] [PubMed] [Google Scholar]
- (2).Santo L; Hideshima T; Kung AL; Tseng JC; Tamang D; Yang M; Jarpe M; Van Duzer JH; Mazitschek R; Ogier WC; et al. Preclinical Activity, Pharmacodynamic, and Pharmacokinetic Properties of a Selective HDAC6 Inhibitor, ACY-1215, in Combination with Bortezomib in Multiple Myeloma. Blood 2012, 119 (11), 2579–2589. 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wang Z; Tang F; Hu P; Wang Y; Gong J; Sun S; Xie C HDAC6 Promotes Cell Proliferation and Confers Resistance to Gefitinib in Lung Adenocarcinoma. Oncology Reports 2016, 36 (1), 589–597. 10.3892/or.2016.4811. [DOI] [PubMed] [Google Scholar]
- (4).Ho TCS; Chan AHY; Ganesan A Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. Journal of Medicinal Chemistry 2020, 63 (21), 12460–12484. 10.1021/acs.jmedchem.0c00830. [DOI] [PubMed] [Google Scholar]
- (5).Wang P; Wang Z; Liu J Role of HDACs in Normal and Malignant Hematopoiesis. Molecular Cancer. 2020. 10.1186/s12943-019-1127-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhao C; Dong H; Xu Q; Zhang Y Histone Deacetylase (HDAC) Inhibitors in Cancer: A Patent Review (2017-Present). Expert Opinion on Therapeutic Patents 2020, 30 (4), 263–274. 10.1080/13543776.2020.1725470. [DOI] [PubMed] [Google Scholar]
- (7).Leipe DD; Landsman D Histone Deacetylases, Acetoin Utilization Proteins and Acetylpolyamine Amidohydrolases Are Members of an Ancient Protein Superfamily. Nucleic Acids Research 1997, 25 (18), 3693–3697. 10.1093/nar/25.18.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gregoretti IV; Lee YM; Goodson HV Molecular Evolution of the Histone Deacetylase Family: Functional Implications of Phylogenetic Analysis. Journal of Molecular Biology 2004, 338 (1), 17–31. 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- (9).Rossaert E; Van Den Bosch L HDAC6 Inhibitors: Translating Genetic and Molecular Insights into a Therapy for Axonal CMT. Brain Research. 2020. 10.1016/j.brainres.2020.146692. [DOI] [PubMed] [Google Scholar]
- (10).Auzmendi-Iriarte J; Saenz-Antoñanzas A; Mikelez-Alonso I; Carrasco-Garcia E; Tellaetxe-Abete M; Lawrie CH; Sampron N; Cortajarena AL; Matheu A Characterization of a New Small-Molecule Inhibitor of HDAC6 in Glioblastoma. Cell Death and Disease 2020, 11 (6). 10.1038/s41419-020-2586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Park JK; Jang YJ; Oh BR; Shin J; Bae D; Ha N; Choi Y Il; Youn GS; Park J; Lee EY; et al. Therapeutic Potential of CKD-506, a Novel Selective Histone Deacetylase 6 Inhibitor, in a Murine Model of Rheumatoid Arthritis. Arthritis Research and Therapy 2020, 22 (1). 10.1186/s13075-020-02258-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Boyault C; Sadoul K; Pabion M; Khochbin S HDAC6, at the Crossroads between Cytoskeleton and Cell Signaling by Acetylation and Ubiquitination. Oncogene. 2007, pp 5468–5476. 10.1038/sj.onc.1210614. [DOI] [PubMed] [Google Scholar]
- (13).Moreno-Gonzalo O; Mayor F; Sánchez-Madrid F HDAC6 at Crossroads of Infection and Innate Immunity. Trends in Immunology. 2018, pp 591–595. 10.1016/j.it.2018.05.004. [DOI] [PubMed] [Google Scholar]
- (14).Depetter Y; Geurs S; De Vreese R; Goethals S; Vandoorn E; Laevens A; Steenbrugge J; Meyer E; de Tullio P; Bracke M; et al. Selective Pharmacological Inhibitors of HDAC6 Reveal Biochemical Activity but Functional Tolerance in Cancer Models. International Journal of Cancer 2019, 145 (3), 735–747. 10.1002/ijc.32169. [DOI] [PubMed] [Google Scholar]
- (15).Hai Y; Christianson DW Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nature Chemical Biology 2016, 12 (9), 741–747. 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Miyake Y; Keusch JJ; Wang L; Saito M; Hess D; Wang X; Melancon BJ; Helquist P; Gut H; Matthias P Structural Insights into HDAC6 Tubulin Deacetylation and Its Selective Inhibition. Nature Chemical Biology 2016, 12 (9), 748–754. 10.1038/nchembio.2140. [DOI] [PubMed] [Google Scholar]
- (17).Haggarty SJ; Koeller KM; Wong JC; Grozinger CM; Schreiber SL Domain-Selective Small-Molecule Inhibitor of Histone Deacetylase 6 (HDAC6)-Mediated Tubulin Deacetylation. Proceedings of the National Academy of Sciences of the United States of America 2003, 100 (8), 4389–4394. 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang Y; Kwon S; Yamaguchi T; Cubizolles F; Rousseaux S; Kneissel M; Cao C; Li N; Cheng H-L; Chua K; et al. Mice Lacking Histone Deacetylase 6 Have Hyperacetylated Tubulin but Are Viable and Develop Normally. Molecular and Cellular Biology 2008, 28 (5), 1688–1701. 10.1128/mcb.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Messaoudi K; Ali A; Ishaq R; Palazzo A; Sliwa D; Bluteau O; Souquère S; Muller D; Diop KM; Rameau P; et al. Critical Role of the HDAC6-Cortactin Axis in Human Megakaryocyte Maturation Leading to a Proplatelet-Formation Defect. Nature Communications 2017, 8 (1). 10.1038/s41467-017-01690-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Shen S; Kozikowski AP A Patent Review of Histone Deacetylase 6 Inhibitors in Neurodegenerative Diseases (2014–2019). Expert Opinion on Therapeutic Patents. 2020, pp 121–136. 10.1080/13543776.2019.1708901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Mackwitz MKW; Hamacher A; Osko JD; Held J; Schöler A; Christianson DW; Kassack MU; Hansen FK Multicomponent Synthesis and Binding Mode of Imidazo[1,2- a]Pyridine-Capped Selective HDAC6 Inhibitors. Organic Letters 2018, 20 (11), 3255–3258. 10.1021/acs.orglett.8b01118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chia PL; Mukhopadhyay P; Kelly S; Wu R; Fenn K; Trivedi MS Network-Based Assessment of HDAC6 Activity Is Highly Predictive of Pre-Clinical and Clinical Responses to the HDAC Inhibitor Ricolinostat. medRxiv 2020. [Google Scholar]
- (23).Jochems J; Boulden J; Lee BG; Blendy JA; Jarpe M; Mazitschek R; Van Duzer JH; Jones S; Berton O Antidepressant-like Properties of Novel HDAC6-Selective Inhibitors with Improved Brain Bioavailability. Neuropsychopharmacology 2014, 39 (2), 389–400. 10.1038/npp.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Vergani B; Sandrone G; Marchini M; Ripamonti C; Cellupica E; Galbiati E; Caprini G; Pavich G; Porro G; Rocchio I; et al. Novel Benzohydroxamate-Based Potent and Selective Histone Deacetylase 6 (HDAC6) Inhibitors Bearing a Pentaheterocyclic Scaffold: Design, Synthesis, and Biological Evaluation. Journal of Medicinal Chemistry 2019, 62 (23), 10711–10739. 10.1021/acs.jmedchem.9b01194. [DOI] [PubMed] [Google Scholar]
- (25).Shouksmith AE; Shah F; Grimard ML; Gawel JM; Raouf YS; Geletu M; Berger-Becvar A; De Araujo ED; Luchman HA; Heaton WL; et al. Identification and Characterization of AES-135, a Hydroxamic Acid-Based HDAC Inhibitor That Prolongs Survival in an Orthotopic Mouse Model of Pancreatic Cancer. Journal of Medicinal Chemistry 2019, 62 (5), 2651–2665. 10.1021/acs.jmedchem.8b01957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Porter NJ; Osko JD; Diedrich D; Kurz T; Hooker JM; Hansen FK; Christianson DW Histone Deacetylase 6-Selective Inhibitors and the Influence of Capping Groups on Hydroxamate-Zinc Denticity. Journal of Medicinal Chemistry 2018, 61 (17), 8054–8060. 10.1021/acs.jmedchem.8b01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shen S; Svoboda M; Zhang G; Cavasin MA; Motlova L; McKinsey TA; Eubanks JH; Bařinka C; Kozikowski AP Structural and in Vivo Characterization of Tubastatin A, a Widely Used Histone Deacetylase 6 Inhibitor. ACS Medicinal Chemistry Letters 2020, 11 (5), 706–712. 10.1021/acsmedchemlett.9b00560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Porter NJ; Mahendran A; Breslow R; Christianson DW Unusual Zinc-Binding Mode of HDAC6-Selective Hydroxamate Inhibitors. Proceedings of the National Academy of Sciences of the United States of America 2017, 114 (51), 13459–13464. 10.1073/pnas.1718823114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gawel JM; Shouksmith AE; Raouf YS; Nawar N; Toutah K; Bukhari S; Manaswiyoungkul P; Olaoye OO; Israelian J; Radu TB; et al. PTG-0861: A Novel HDAC6-Selective Inhibitor as a Therapeutic Strategy in Acute Myeloid Leukaemia. European Journal of Medicinal Chemistry 2020, 201. 10.1016/j.ejmech.2020.112411. [DOI] [PubMed] [Google Scholar]
- (30).Shouksmith AE; Gawel JM; Nawar N; Sina D; Raouf YS; Bukhari S; He L; Johns AE; Manaswiyoungkul P; Olaoye OO; et al. Class I/IIb-Selective HDAC Inhibitor Exhibits Oral Bioavailability and Therapeutic Efficacy in Acute Myeloid Leukemia. ACS Medicinal Chemistry Letters 2020, 11 (1), 56–64. 10.1021/acsmedchemlett.9b00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Osko JD; Christianson DW Structural Basis of Catalysis and Inhibition of HDAC6 CD1, the Enigmatic Catalytic Domain of Histone Deacetylase 6. Biochemistry 2019, 58 (49), 4912–4924. 10.1021/acs.biochem.9b00934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Osko JD; Christianson DW Structural Determinants of Affinity and Selectivity in the Binding of Inhibitors to Histone Deacetylase 6. Bioorganic and Medicinal Chemistry Letters. 2020. 10.1016/j.bmcl.2020.127023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ali AM; Gómez-Biagi RF; Rosa DA; Lai PS; Heaton WL; Park JS; Eiring AM; Vellore NA; De Araujo ED; Ball DP; et al. Disarming an Electrophilic Warhead: Retaining Potency in Tyrosine Kinase Inhibitor (TKI)-Resistant CML Lines While Circumventing Pharmacokinetic Liabilities. ChemMedChem 2016, 11 (8), 850–861. 10.1002/cmdc.201600021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Porter NJ; Christianson DW Structure, Mechanism, and Inhibition of the Zinc-Dependent Histone Deacetylases. Current Opinion in Structural Biology. 2019, pp 9–18. 10.1016/j.sbi.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Porter NJ; Wagner FF; Christianson DW Entropy as a Driver of Selectivity for Inhibitor Binding to Histone Deacetylase 6. Biochemistry 2018, 57 (26), 3916–3924. 10.1021/acs.biochem.8b00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Christianson DW; Fierke CA Carbonic Anhydrase: Evolution of the Zinc Binding Site by Nature and by Design. Accounts of Chemical Research 1996, 29 (7), 331–339. 10.1021/ar9501232. [DOI] [Google Scholar]
- (37).Dudev T; Lin Y. lin; Dudev M; Lim C First-Second Shell Interactions in Metal Binding Sites in Proteins: A PDB Survey and DFT/CDM Calculations. Journal of the American Chemical Society 2003, 125 (10), 3168–3180. 10.1021/ja0209722. [DOI] [PubMed] [Google Scholar]
- (38).Ray A; Das DS; Song Y; Hideshima T; Tai YT; Chauhan D; Anderson KC Combination of a Novel HDAC6 Inhibitor ACY-241 and Anti-PD-L1 Antibody Enhances Anti-Tumor Immunity and Cytotoxicity in Multiple Myeloma. Leukemia. 2018, pp 843–846. 10.1038/leu.2017.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Yee AJ; Bensinger W; Voorhees PM; Berdeja JG; Richardson PG; Supko J; Tamang D; Jones SS; Wheeler C; Markelewicz RJ; et al. Ricolinostat (ACY-1215), the First Selective HDAC6 Inhibitor, in Combonation with Lenalidomide and Dexamethasone in Patients with Relapsed and Relapsed-and-Refractory Multiple Myeloma: Phase 1b Results (ACE-MM-101 Study). Blood 2015, 126 (23), 3055–3055. 10.1182/blood.v126.23.3055.3055. [DOI] [Google Scholar]
- (40).Hideshima T; Qi J; Paranal RM; Tang W; Greenberg E; West N; Colling ME; Estiu G; Mazitschek R; Perry JA; et al. Discovery of Selective Small-Molecule HDAC6 Inhibitor for Overcoming Proteasome Inhibitor Resistance in Multiple Myeloma. Proceedings of the National Academy of Sciences of the United States of America 2016, 113 (46), 13162–13167. 10.1073/pnas.1608067113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wang Z; Hu P; Tang F; Lian H; Chen X; Zhang Y; He X; Liu W; Xie C HDAC6 Promotes Cell Proliferation and Confers Resistance to Temozolomide in Glioblastoma. Cancer Letters 2016, 379 (1), 134–142. 10.1016/j.canlet.2016.06.001. [DOI] [PubMed] [Google Scholar]
- (42).Maharaj K; Powers JJ; Achille A; Deng S; Fonseca R; Pabon-Saldana M; Quayle SN; Jones SS; Villagra A; Sotomayor EM; et al. Silencing of HDAC6 as a Therapeutic Target in Chronic Lymphocytic Leukemia. Blood Advances 2018, 2 (21), 3012–3024. 10.1182/bloodadvances.2018020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Cosenza M; Civallero M; Quayle SN; Sacchi S; Pozzi S Ricolinostat (ACY-1215), a Selective HDAC6 Inhibitor, Alone and in Combination with Bendamustine Is Effective in Preclinical Studies in Lymphoma Cell Lines. Blood 2016, 128 (22), 2772–2772. 10.1182/blood.v128.22.2772.2772. [DOI] [Google Scholar]
- (44).Cosenza M; Civallero M; Marcheselli L; Sacchi S; Pozzi S Ricolinostat, a Selective HDAC6 Inhibitor, Shows Anti-Lymphoma Cell Activity Alone and in Combination with Bendamustine. Apoptosis 2017, 22 (6), 827–840. 10.1007/s10495-017-1364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Guha M HDAC Inhibitors Still Need a Home Run, despite Recent Approval. Nature Reviews Drug Discovery. 2015, pp 225–226. 10.1038/nrd4583. [DOI] [PubMed] [Google Scholar]
- (46).Hubbert C; Guardiola A; Shao R; Kawaguchi Y; Ito A; Nixon A; Yoshida M; Wang XF; Yao TP HDAC6 Is a Microtubule-Associated Deacetylase. Nature 2002, 417 (6887), 455–458. 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- (47).Catley L; Weisberg E; Kiziltepe T; Tai YT; Hideshima T; Neri P; Tassone P; Atadja P; Chauhan D; Munshi NC; et al. Aggresome Induction by Proteasome Inhibitor Bortezomib and α-Tubulin Hyperacetylation by Tubulin Deacetylase (TDAC) Inhibitor LBH589 Are Synergistic in Myeloma Cells. Blood 2006, 108 (10), 3441–3449. 10.1182/blood-2006-04-016055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sanchez GJ; Richmond PA; Bunker EN; Karman SS; Azofeifa J; Garnett AT; Xu Q; Wheeler GE; Toomey CM; Zhang Q; et al. Genome-Wide Dose-Dependent Inhibition of Histone Deacetylases Studies Reveal Their Roles in Enhancer Remodeling and Suppression of Oncogenic Super-Enhancers. Nucleic Acids Research 2018, 46 (4), 1756–1776. 10.1093/nar/gkx1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Lue JK; Prabhu SA; Liu Y; Gonzalez Y; Verma A; Mundi PS; Abshiru N; Camarillo JM; Mehta S; Chen EI; et al. Precision Targeting with EZH2 and HDAC Inhibitors in Epigenetically Dysregulated Lymphomas. Clinical Cancer Research 2019, 25 (17), 5271–5283. 10.1158/1078-0432.CCR-18-3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Yang H; Villani RM; Wang H; Simpson MJ; Roberts MS; Tang M; Liang X The Role of Cellular Reactive Oxygen Species in Cancer Chemotherapy. Journal of Experimental and Clinical Cancer Research. 2018. 10.1186/s13046-018-0909-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Jayavelu AK; Müller JP; Bauer R; Böhmer SA; Lässig J; Cerny-Reiterer S; Sperr WR; Valent P; Maurer B; Moriggl R; et al. NOX4-Driven ROS Formation Mediates PTP Inactivation and Cell Transformation in FLT3ITD-Positive AML Cells. Leukemia 2016, 30 (2), 473–483. 10.1038/leu.2015.234. [DOI] [PubMed] [Google Scholar]
- (52).Zhou F; Shen Q; Claret FX Novel Roles of Reactive Oxygen Species in the Pathogenesis of Acute Myeloid Leukemia. Journal of Leukocyte Biology 2013, 94 (3), 423–429. 10.1189/jlb.0113006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Eckschlager T; Plch J; Stiborova M; Hrabeta J Histone Deacetylase Inhibitors as Anticancer Drugs. International Journal of Molecular Sciences. 2017. 10.3390/ijms18071414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Debeb BG; Lacerda L; Larson R; Wolfe AR; Krishnamurthy S; Reuben JM; Ueno NT; Gilcrease M; Woodward WA Histone Deacetylase Inhibitor-Induced Cancer Stem Cells Exhibit High Pentose Phosphate Pathway Metabolism. Oncotarget 2016, 7 (19), 28329–28339. 10.18632/oncotarget.8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Valdez BC; Brammer JE; Li Y; Murray D; Liu Y; Hosing C; Nieto Y; Champlin RE; Andersson BS Romidepsin Targets Multiple Survival Signaling Pathways in Malignant T Cells. Blood Cancer Journal 2015, 5 (10). 10.1038/bcj.2015.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Freisleben A; Brudny-Klöppel M; Mulder H; De Vries R; De Zwart M; Timmerman P Blood Stability Testing: European Bioanalysis Forum View on Current Challenges for Regulated Bioanalysis. Bioanalysis 2011, 3 (12), 1333–1336. 10.4155/bio.11.121. [DOI] [PubMed] [Google Scholar]
- (57).Sugihara K; Kitamura S; Ohta S; Tatsumi K Reduction of Hydroxamic Acids to the Corresponding Amides Catalyzed by Rabbit Blood. Xenobiotica 2000, 30 (5), 457–467. 10.1080/004982500237479. [DOI] [PubMed] [Google Scholar]
- (58).Hermant P; Bosc D; Piveteau C; Gealageas R; Lam B; Ronco C; Roignant M; Tolojanahary H; Jean L; Renard PY; et al. Controlling Plasma Stability of Hydroxamic Acids: A MedChem Toolbox. Journal of Medicinal Chemistry 2017, 60 (21), 9067–9089. 10.1021/acs.jmedchem.7b01444. [DOI] [PubMed] [Google Scholar]
- (59).Flipo M; Charton J; Hocine A; Dassonneville S; Deprez B; Deprez-Poulain R Hydroxamates: Relationships between Structure and Plasma Stability. Journal of Medicinal Chemistry 2009, 52 (21), 6790–6802. 10.1021/jm900648x. [DOI] [PubMed] [Google Scholar]
- (60).M.S. G; G. S; J. S; D. J; B. L; P. C; J. C; Y. LB; R.D. H A Phase 1b Study of the Safety, Pharmacokinetics, and Preliminary Antitumor Activity of Citarinostat (ACY-241) in Combination with Paclitaxel (Pac) in Patients (Pts) with Advanced Solid Tumors (AST). Journal of Clinical Oncology 2018. https://doi.org/10.1200/JCO.2018.36.15_suppl.2547 LK - https://doi.org/10.1200/JCO.2018.36.15_suppl.2547http://sfx.library.uu.nl/utrecht?sid=EMBASE&issn=15277755&id=doi:10.1200%2FJCO.2018.36.15_suppl.2547&atitle=A+phase+1b+study+of+the+safety%2C+pharmacokinetics%2C+and+preliminary+antitumor+activity+of+citarinostat+%28ACY-241%29+in+combination+with+paclitaxel+%28Pac%29+in+patients+%28pts%29+with+advanced+solid+tumors+%28AST%29&stitle=J.+Clin.+Oncol.&title=Journal+of+Clinical+Oncology&volume=36&issue=15&spage=&epage=&aulast=Gordon&aufirst=Michael+S.&auinit=M LK - http://sfx.library.uu.nl/utrecht?sid=EMBASE&issn=15277755&id=doi:10.1200%2FJCO.2018.36.15_suppl.2547&atitle=A+phase+1b+study+of+the+safety%2C+pharmacokinetics%2C+and+preliminary+antitumor+activity+of+citarinostat+%28ACY-241%29+in+combination+with+paclitaxel+%28Pac%29+in+patients+%28pts%29+with+advanced+solid+tumors+%28AST%29&stitle=J.+Clin.+Oncol.&title=Journal+of+Clinical+Oncology&volume=36&issue=15&spage=&epage=&aulast=Gordon&aufirst=Michael+S.&auinit=M . [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.