

Abstract
The Biological Magnetic Resonance Data Bank (BioMagResBank or BMRB), founded in 1988, serves as the archive for data generated by nuclear magnetic resonance (NMR) spectroscopy of biological systems. NMR spectroscopy is unique among biophysical approaches in its ability to provide a broad range of atomic and higher-level information relevant to the structural, dynamic, and chemical properties of biological macromolecules, as well as report on metabolite and natural product concentrations in complex mixtures and their chemical structures. BMRB became a core member of the Worldwide Protein Data Bank (wwPDB) in 2007, and the BMRB archive is now a core archive of the wwPDB. Currently, about 10% of the structures deposited into the PDB archive are based on NMR spectroscopy. BMRB stores experimental and derived data from biomolecular NMR studies. Newer BMRB biopolymer depositions are divided about evenly between those associated with structure determinations (atomic coordinates and supporting information archived in the PDB) and those reporting experimental information on molecular dynamics, conformational transitions, ligand binding, assigned chemical shifts, or other results from NMR spectroscopy. BMRB also provides resources for NMR studies of metabolites and other small molecules that are often macromolecular ligands and/or nonstandard residues. This chapter is directed to the structural biology community rather than the metabolomics and natural products community. Our goal is to describe various BMRB services offered to structural biology researchers and how they can be accessed and utilized. These services can be classified into four main groups: (1) data deposition, (2) data retrieval, (3) data analysis, and (4) services for NMR spectroscopists and software developers. The chapter also describes the NMR-STAR data format used by BMRB and the tools provided to facilitate its use. For programmers, BMRB offers an application programming interface (API) and libraries in the Python and R languages that enable users to develop their own BMRB-based tools for data analysis, visualization, and manipulation of NMR-STAR formatted files. BMRB also provides users with direct access tools through the NMRbox platform.
Keywords: BioMagResBank, BMRB, NMR data, NMR-STAR, Visualization, Deposition, Retrieval, Search, Data formats, Analysis, Structures, PDB, NMR spectra, Time-domain data, Python, R
1. Introduction
The Biological Magnetic Resonance Data Bank (BioMagResBank or BMRB) [1] has served for the past 30 years as the primary archive for spectral and derived data generated by nuclear magnetic resonance (NMR) spectroscopy of biological systems. The BMRB archive is unique among biophysical data banks in that the archive contains primary time-domain data obtained by NMR spectrometers, processed spectra, spectral peak characteristics, assigned spectral peak chemical shifts, and derived data such as relaxation parameters, pKa values, and atomic coordinates for certain smaller molecules not covered by the Protein Data Bank Archive [2] (URL: http://wwpdb.org). BMRB has developed technology for annotating and processing the assigned chemical shift data archived at BMRB and the chemical shift and constraint data underlying NMR-based structures archived at the PDB. The field of biomolecular NMR is evolving continuously, and newly developed NMR techniques [3, 4] have the potential to enable NMR studies of various biological molecular systems, including larger proteins, nucleic acids, molecular machines, and membrane-bound biopolymers.
BMRB also offers support for studies of metabolomics and natural products through a library of a variety of 1D and two-dimensional (2D) NMR spectra of pure compounds (including metabolites, natural products, drugs, and compounds used for screening in drug discovery) and through its adoption of the ALATIS compound and atom identifiers [5], which are universal and based solely on the three-dimensional (3D) structure of the compound and the InChI convention. In addition, for a growing number of small molecules, BMRB is providing spin matrices in the GISSMO convention [6], which enables accurate simulation of spectra at any field strength. The combination of unique ALATIS naming and parameterized spectra offers the users of BMRB data a distinctive benefit in terms of robustness and reproducibility.
The importance of publicly accessible and persistent data archives such as BMRB for sustainable and reproducible research is embodied in the FAIR principles [7] espoused by the wwPDB [8], which are that data should be findable, accessible, interoperable, and reusable.
BMRB consists of the main archive at the University of Wisconsin-Madison and branches at UConn Health, Osaka, Japan (PDBj-BMRB), and Florence, Italy. BMRB is a core member of the Worldwide Protein Data Bank (wwPDB) along with the RCSB PDB, PDBe, and PDBj [2]. Currently, the core archives of the wwPDB consist of the PDB archive and the BMRB archive [8, 9]. In 2019, the Electron Microscopy Data Bank (EMDB) located at EMBL-EBI will become a core wwPDB member, and EMDB will become the third core archive. The core archives each share a common data format driven by a data dictionary, which enables coordinated searching of the combined resources.
BMRB mirror sites are supported at Osaka University, Japan, and at CERM in Florence, Italy, with the Osaka facility also being a data deposition and processing site. BMRB collaborates closely with the other groups in the wwPDB (RCSB PDB, PDBe, and PDBj). Over the years, BMRB has benefitted from interactions with many groups in the NMR community, including the CCPN group (now at the University of Leicester), the Northeast Structural Genomics (NESG) group, the Center for Eukaryotic Structural Genomics (CESG), the NMR metabolomic groups, and the National Magnetic Resonance Facility at Madison (NMRFAM). A wealth of experimental data from NMR studies linked to high-quality 3D structures of proteins was deposited to BMRB by centers funded by the NIH Protein Structure Initiative (PSI) [10]. Although the pace of structural NMR studies deposited declined after the end of the PSI in 2015, structural NMR data continue to comprise approximately half of BMRB depositions.
In addition, BMRB is a member of the Center for NMR Data Processing and Analysis, provider of the NMRbox [11] platform. NMRbox is a cloud-based computing platform providing the bio-NMR community access to existing NMR software tools and computational resources. Project goals include improving NMR data reproducibility, facilitating depositions to BMRB and other public databases, and developing new data analysis tools. As such, BMRB currently provides machine-to-machine (M2M) access to some services (e.g., CS-Rosetta structure determination) and is developing a computer-assisted deposition service that will gather data, metadata, and workflow information to facilitate and enrich depositions into BMRB for enhanced reproducibility. This chapter covers the usage of extant NMRbox-integrated BMRB tools where appropriate.
2. Resources
As an open access digital data resource that applies the FAIR data principles [7], BMRB houses and distributes the experimental and derived data from NMR experiments carried out on biologically relevant molecular systems. The archive consists of six main data depositories: (1) quantitative NMR spectral parameters for proteins, peptides, nucleic acids, carbohydrates, and ligands or cofactors (e.g., assigned chemical shifts, coupling constants, and peak lists) and derived data (e.g., relaxation parameters, residual dipolar couplings, hydrogen exchange rates, and pKa values); (2) time-domain spectral data from NMR experiments used to assign spectral resonances and determine the structures of biological macromolecules; (3) an archive of atomic coordinates for small molecules not accepted by the wwPDB; (4) a database for NMR constraints processed from original author depositions available from the PDB; (5) an archive of CS-Rosetta structures derived from BMRB chemical shift entries [12, 13]; and (6) a growing database of 1H and 13C 1D and 2D NMR spectra (including time-domain data) and assigned chemical shifts for over 1000 biological small molecules. Validation reports for BMRB chemical shift entries and MolProbity [14] validation reports for all PDB entries are available on the BMRB website. BMRB provides a variety of software services for querying the archive: for enabling interactive data visualizations of the archival data, user supplied data, and combinations of both; for carrying out file format conversions; for validating data; and for high-throughput calculation of structures using CS-Rosetta and HTCondor [15] in collaboration with the Center for High-Throughput Computing (CHTC) and the Open Science Grid (OSG). The data in the BMRB archives are linked to the literature citations related to the entries and to a number of public databases through BLAST sequence homology searches that are updated weekly. BMRB acquires data through depositor submissions by means of three deposition systems: (1) OneDep [16], through the wwPDB OneDep website https://deposit.wwpdb.org/; (2) ADIT-NMR, through either the Madison or Osaka BMRB branch (for other NMR data beyond those accepted by OneDep, as well as nonstructural NMR data); and (3) SMSDep, through the Osaka BMRB branch (for NMR-derived structures of molecules that do not fit the guidelines of the PDB archive). Additional data acquisition methods are (1) transfer of metabolite spectral data collected at the National Magnetic Resonance Facility at Madison (NMRFAM) to the Madison BMRB and (2) in-house generation of validation and structural data using third-party software like AVS, PANAV, and CS-Rosetta. A new version of ADIT-NMR, called BMRBdep, extends the capabilities of the original deposition system for biomacromolecules and supports the deposition of NMR data from small molecules of biological importance (e.g., metabolites, natural products, drugs). The Madison and Osaka branches of BMRB carry out processing and annotation of entries deposited at their sites. NMR data associated with structures deposited through the OneDep system are transferred to either of two BMRB annotation units, and BMRB annotators at these sites curate as the data as needed and convey any changes back to the OneDep team.
BMRB uses the NMR-STAR [17] data format to represent experiments, spectral and derived data, and supporting metadata. NMR-STAR was constructed along the object-relational data model using a subset of the Self-Defining Text Archival and Retrieval (STAR) specification [18]. The growth of the biological NMR field and the development of new experimental technologies have mandated the revision and enlargement of the NMR-STAR ontology [17]. BMRB provides users with tools to facilitate editing and handling of NMR-STAR files, whose use is explained below. The NMR-STAR ontology enhances the reusability (a FAIR goal) of NMR data by providing ample information on the experimental data being archived.
In terms of findability and accessibility, public uninterrupted access to BMRB services is provided through the BMRB website at http://www.bmrb.wisc.edu and its mirror websites in Japan (https://bmrb.pdbj.org) and Italy (http://bmrb.cerm.unifi.it). Due to ongoing development, the websites’ appearances may differ somewhat from the screenshots shown in this chapter. Nevertheless, the functionality of the tools and services described here will be maintained, and the website documentation will cover any future updates. The BMRB unit in Osaka (PDBj-BMRB) also maintains a website with access to other useful tools and extra documentation including a Japanese version at https://bmrbdep.pdbj.org (DOI:10.1002/pro.3273). PDBj-BMRB has been improving the inter-operability of BMRB archives with a particular focus on semantic web standards, represented by XML, RDF formats, of which data archives are accessible from each BMRB site (DOI: 10.1186/s13326-016-0057-1). For enhanced interoperability, further access to BMRB data is provided through BMRB’s application programming interface (API), described in the “Services for Programmers” section. Help is available through the “bmrbhelp” mailing list, by sending a message to bmrbhelp@bmrb.wisc.edu.
3. Methods
This section describes the methodology for accessing and using the tools and services available for each of four areas, data deposition, data retrieval, data analysis, and programmer services. Most of the included figures correspond to screenshots from the BMRB website at the time of writing this chapter. The website’s documentation pages will be kept up to date with any new developments, both in terms of new resources and updates to existing ones.
3.1. Data Deposition
BMRB accepts deposition containing many kinds of experimental and derived NMR data, including time-domain and processed data files. BMRB’s main deposition system (ADIT-NMR at the time of writing but soon to be replaced by BMRBdep which is described below) has been designed with the aim of providing researchers with a cloud-based data repository for an in-progress NMR project, allowing users to enter metadata and results as they are generated.
Since its inception, BMRB has worked closely with the PDB for the deposition and archiving of structural NMR data of biological macromolecules. Consequently, BMRB is considered a structural database, and many journals require depositions to both PDB and BMRB as a prerequisite for publication of an NMR-based structure. This close relationship has resulted in the integration of BMRB and PDB depositions for structural NMR data within the OneDep software system [16], which handles deposition of coordinates and associated data into the PDB archive, as well as chemical shifts, restraints, and associated data files into the BMRB archive.
3.1.1. PDB OneDep
The wwPDB partners, including BMRB, joined forces in creating OneDep, which replaced disparate depositions previously used by the wwPDB partners. The OneDep system unifies the deposition and annotation systems across all wwPDB deposition centers and focuses on improving data quality and completeness in the PDB archive while supporting growth in the number of depositions and increases in size and complexity of the structures deposited. The OneDep system at https://deposit.wwpdb.org serves as a single access point for biomolecular NMR data: if the user indicates that coordinates will be deposited, data will be collected through the OneDep site in the depositor’s geographical zone (RCSB PDB, PDBe, or PDBj); if no coordinates are associated, the user is transferred to the BMRB deposition system in the depositor’s geographical zone (BMRB or PDBj-BMRB). Alternatively, data not involving coordinates can be deposited by directly accessing either the BMRB or PDBj-BMRB site.
Depositions made through OneDep will generate both a PDB and a BMRB entry from the depositor’s data, but currently NMR data beyond those accepted by the PDB archive (chemical shifts and restraints are required; NOE peak lists are recommended) cannot be entered in this way and require a separate deposition at one of the BMRB sites. In response to feedback from the NMR community, the wwPDB has pledged in principle to integrate BMRB’s new deposition system (BMRBdep) with OneDep to allow NMR researchers to directly deposit more complete NMR data.
The wwPDB website provides ample documentation and tutorials on the use of OneDep. We provide here a summary of the instructions found at https://www.wwpdb.org/deposition/tutorial that are related to NMR depositions. Instructions for depositing coordinates are found at the same URL. As noted above, OneDep sends the depositor directly to the BMRB deposition system if no coordinates are being deposited. Even if the user is depositing coordinates, it is strongly recommended to access the BMRB deposition system after depositing the structure to PDB and to use the provided BMRB ID to continue the deposition of NMR data (e.g., time-domain data, spectra, relaxation parameters, pKa values, and other derived data). Once OneDep is fully integrated with BMRBdep, this step will not be necessary. As explained in Subheading 3.1.2, a practical and efficient way to deposit NMR structures would be to open a BMRB deposition early in the experimental work and gradually upload the data as the experiment proceeds, in effect using the BMRB depository as a lab notebook. This guarantees a complete BMRB deposition when depositing the final set of data.
At the start of deposition, OneDep will ask the depositor to provide information about the experimental methods employed to determine the structure. If “Solution NMR” or “Solid-state NMR” has been selected as the experimental method, you will be asked whether you are depositing coordinates. If “No” is selected, you will see that BMRB is the only requested accession code, and a “Deposit NMR data at BMRB” button will appear at the bottom of the screen. Once you click the button, you will then be redirected to the BMRB deposition start page. Please note that if you make a BMRB-only deposition, later deposition of associated coordinates will require that you complete a new PDB-only deposition. If “Yes” is selected in response to the coordinates question, then you will be asked to upload three mandatory files (coordinates in PDBx/mmCIF format [19], restraints in NEF [20] or NMR-STAR format, and chemical shifts in NMR-STAR format) and will be encouraged to upload another (peak lists in NMR-STAR format).
During file uploads, the following checks are performed: (1) the coordinates file is checked to ensure that each model has the same chemistry (identical atoms); (2) the chemical shift values are checked for outliers; and (3) the atom nomenclature in the chemical shift and coordinates files is compared for consistency.
If any warnings or errors are generated by these checks, they will be reported as follows:
Warning messages: Warnings encountered upon file upload will be presented in the “File format validation for model coordinates and data files” window. For NMR entries, warning messages provide information about chemical shift values outside of acceptable ranges. Warning messages are provided for depositors to review and either negate or correct as appropriate.
Error messages: Errors encountered upon file upload will be presented in two places: (1) on the diagnostic screen (headed by a graphic of red gears) that appears after the “Populate” or “Repopulate” button on the “File upload” page has been pressed and (2) on the “Upload Summary” page of the deposition interface. For NMR entries, error messages highlight atom nomenclature issues that must be corrected. If an error is present, new coordinates and/or chemical shift files must be uploaded before the deposition can be completed.
Entering NMR Data into the Deposition Interface
For NMR depositions, it is best to enter information starting from the top of the left-hand navigation panel and working downward sequentially, page by page, as some values on later pages (lower on the navigation panel) are dependent on information entered on earlier pages (higher on the navigation panel).
In particular, the chemical shift connection page cannot be completed before mandatory data items in the NMR experimental section and NMR software section have been completed. The chemical shift connection page links data used to assign the chemical shifts with information contained in other NMR sections, i.e., chemical shift filenames, chemical shift references, NMR samples, sample conditions, NMR experiments, and software. In addition, the spectral peak list section becomes mandatory when NMR peak list files are uploaded.
3.1.2. ADIT-NMR and BMRBdep
The ADIT-NMR deposition system was originally developed in collaboration with the RCSB-PDB, built upon PDB’s ADIT system for deposition of X-ray structures. The newly developed deposition system, BMRBdep, reproduces the ADIT-NMR functionality with a more responsive, easier-to-use interface, adding the capacity to handle deposition of small-molecule NMR data. Both deposition systems are driven by the NMR-STAR data dictionary, which automatically supports any changes in the NMR-STAR format.
BMRB encourages researchers to start a deposition early and to use ADIT-NMR/BMRBdep as a lab notebook that is filled in as the work proceeds. Incomplete sessions are not deleted, so a deposition session will remain accessible for a long time (up to 2 years since the last update). Please note that older deposition sessions may be required to be upgraded if the dictionary has changed significantly. The completed NMR-STAR files should then be ready for deposition.
Users from Asian countries (excluding Oceania) are encouraged to deposit to the regional BMRB mirror site PDBj-BMRB: http://deposit.bmrb.pdbj.org. Please note that depositions started at PDBj-BMRB cannot be continued on the UW-Madison server, and vice versa.
While ADIT-NMR is still the main deposition server at the time of writing, it will soon be replaced by the BMRBdep server. As a result, the procedures described below are for the new BMRBdep system, which was released recently. You can start a BMRBdep deposition at https://bmrbdep.bmrb.wisc.edu. PDBj-BMRB also provides the BMRBdep deposition service from the same URL, https://deposit.bmrb.pdbj.org, with appropriate changeover time.
The deposition procedure follows the following steps:
Step 1: Preparation for data deposition
Step 2: Creation of a BMRBdep session
Step 3: Upload of data files
Step 4: Entering relevant data
Step 5: Previewing and depositing the entry
Step 6: Receiving a report from BMRB/PDB
Step 7: Hold and release of the entry
Step 1: Preparation for Data Deposition
Before proceeding with your deposition, it will be useful to have on hand the following information:
Chemical description of the molecules in the system studied.
Residue sequences for polymers.
Sequence database reference for the biological molecule(s).
Atom and bond lists for ligands and nonstandard residues.
List of contents for at least a representative sample.
List of experimental conditions (temperature, pH, etc.).
A list of names to use for each sample and each set of experimental conditions.
ASCII file(s) containing chemical shift assignments or coupling constants, preferably in NMR-STAR format, but ASCII files containing tables with tab- or comma-delimited fields will be accepted.
Note that since a user can return to their deposition at any time, it is not necessary to have all or even any of the data mentioned above to begin a deposition, although they will be needed to complete it. The BMRB website provides tools for generating NMR-STAR files. To create NMR-STAR chemical shift assignment files, the user can access the template generators (available at http://www.bmrb.wisc.edu/software/tablegen/. See Fig. 1) or the STARch file converter, which takes different file formats and converts them into NMR-STAR (see Subheading 3.3.1).
Fig. 1.
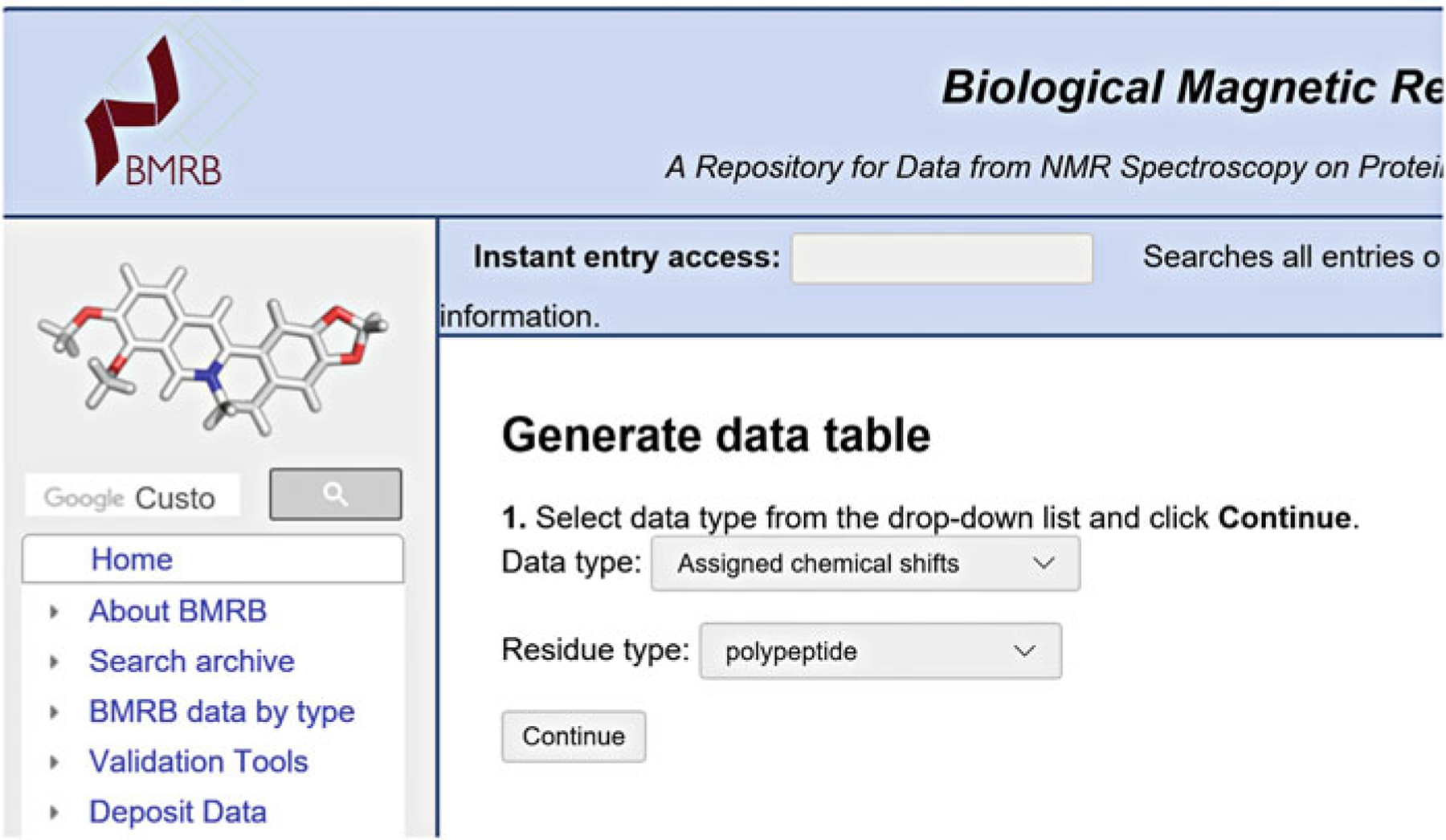
NMR-STAR template generator initial interface
The BMRB Template Generator
The template generator produces NMR-STAR data tables according to various selections from the user. The currently available data types are (1) assigned chemical shifts, (2) coupling constants, (3) H-exchange tables, (4) H-exchange protection factors, (5) heteronuclear NOE values, and (6) heteronuclear T1, T1rho, and T2 values. The type 1 (assigned chemical shifts) template generator is available for both proteins and polynucleotides, whereas template generators 2–6 are available only for proteins.
Each template generator has an input screen for entering a residue sequence string (in single letter format), and the user can make other selections, such as the atoms to include per residue. Figure 2 shows the screen template for chemical shift assignments.
Fig. 2.

NMR-STAR chemical shift assignment template generator interface
Step 2: Creation of an BMRBdep Session
To begin a BMRBdep session, a user must simply enter their e-mail address and a reference name for their deposition and specify the deposition type (new, from an existing entry, or from an uploaded file). By optionally filling out their ORCID ID, some of the fields in the deposition will be automatically populated using the data available in the ORCID database.
If the user wants to start the deposition from an existing NMR-STAR file or released BMRB entry, they can do so by selecting the appropriate “deposition type” and selecting the NMR-STAR file or BMRB ID to use to start the deposition. The NMR-STAR file can be edited before the upload. The best option is to use the JavaScript NMR-STAR viewer tool (see Subheading 3.3.4). Using this tool will ensure that your modifications do not violate the NMR-STAR format. Alternatively, it is possible to edit the file in a plain text editor like Notepad. In the future, an enhancement to the BMRBdep system will add a feature to existing depositions that will allow the user to start a new deposition pre-filled with the data contained within the existing deposition.
After clicking the new deposition button, the deposition session will be created and saved in the user’s browser, and an e-mail will be sent with a link used to verify the user’s e-mail. This verification link must be clicked prior to entering information about the deposition. To end a session, the user may click the “End session” button. A depositor can leave the site or close the browser at any time. As long as the original e-mail has been saved, the user can get back to the deposition session in the future by clicking the link in the original deposition e-mail. In the case of a lost e-mail, the user should contact BMRB help at bmrbhelp@bmrb.wisc.edu.
Step 3: Upload of Deposition Data Files
Once the session is initiated, the user is taken to the “data files” page (Fig. 3), where the user uploads one or more data files associated with the entry. After file upload, the user must select one or more data types contained within the file. The action of selecting the appropriate data types will enable additional data input fields in the deposition system related to the uploaded data types. The user can return to this page at any time to upload additional data files, and the interface will automatically and instantly update to reflect the changes.
Fig. 3.

BMRBdep interface for data files deposition
As explained above, BMRB uses the NMR-STAR format for all stored NMR data. File conversion instructions are described in Step 1. It is not necessary to have the data in NMR-STAR format before deposition, but it is very helpful.
Step 4: Entering Relevant Data
After uploading data files and proceeding, the layout of BMRBdep will be as shown in Fig. 4. To progress through the deposition, the user simply clicks the “Next section” or “Previous section” buttons above and below the data entry fields. Alternatively, they can navigate through the deposition and see which sections still need to be completed by opening the left panel menu. To do this, the user clicks the three-horizontal-line icon (called a hamburger menu) on the top left of the page. The navigation panel is illustrated in Fig. 4.
Fig. 4.
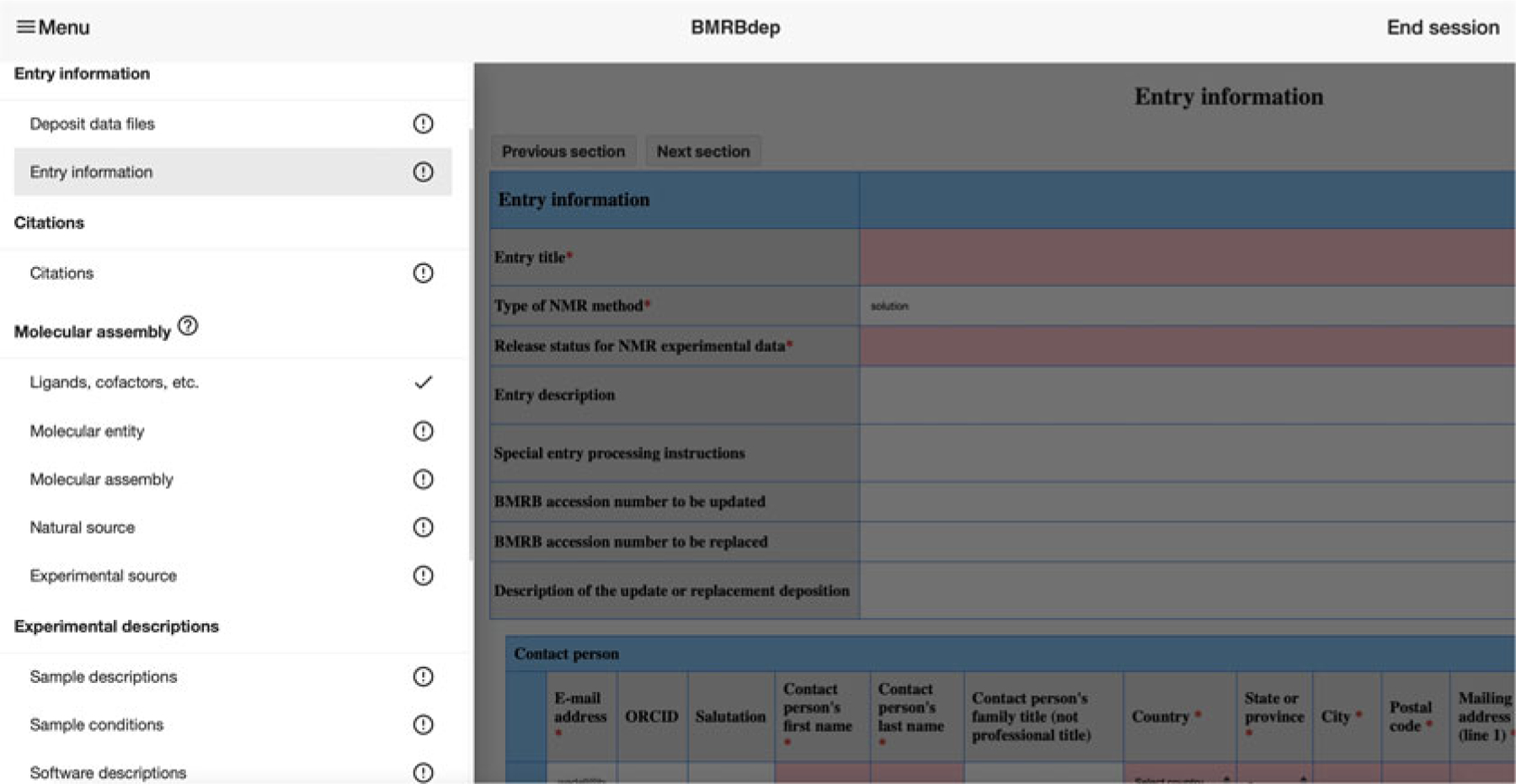
The BMRBdep navigation menu
The navigation panel indicates sections that still need to be completed with a circled exclamation mark symbol and indicates completed sessions with a check mark symbol.
To view help information for any data field, clicking on the data field name opens a help box. (This is indicated to the user by the mouse cursor turning into a question mark when hovering over this section of the interface.) Within any given category, a mandatory data field will have a red asterisk (*) next to it. In addition, the data input field will be highlighted with a light pink color if its value must be entered or if an invalid value has been entered.
Changes are automatically saved as they are made. The save operation has completed when the progress bar at the top of the screen disappears. If the user closes their browser before the data have uploaded to the server, unsaved data are preserved locally in the browser cache. Opening the page again (as long as the “End session” button has not been clicked) will allow the system to save all the changes to the server.
Step 5: Previewing and Depositing the Entry
After data entry is completed, the user can progress to the deposition step by clicking the “Deposit entry” button in the left navigation menu. After selecting that button, the user will be shown any mandatory fields that remain to be filled out and will be given an option to review the full deposition before submission. Once the deposition is complete and passes initial validation, the user will be given the option to submit the entry to the BMRB.
Step 6: Receiving a Report from BMRB/PDB
After a deposition has been submitted through BMRBdep, the authors will receive through e-mail a short notice of receipt with the BMRB accession number. BMRB’s annotators will check and validate the entry and then, usually within a few days, will send the depositor the full annotation reports from BMRB with comments for clarifications and/or updates, including the appropriate AVS and PANAV reports for the assignments of protein systems. A link to the processed entry in NMR-STAR format is included in the letter, and any corrections/updates can be sent to BMRB annotators.
Step 7: Hold and Release of the Entry
When the deposition is completed, the deposited data will be put on “Hold” status, until the publication of the associated paper or the date specified by the user (up to 1 year following deposition). The on-hold status of any entry can be confirmed by accessing “Entries on Hold” from the “Search Archive” section of the BMRB website’s navigation menu (http://www.bmrb.wisc.edu/data_library/held.shtml). BMRB and PDB accept revisions of entries at any time prior to release.
Upon publication of the associated paper or, in the absence of a publication, 1 year after deposition, the entry data will be released to the public on the BMRB and, if applicable, PDB archives. Users are encouraged to notify BMRB/PDB when the paper associated with a deposition is published so that its release is timely.
3.1.3. PDBj-BMRB SMSDep
In recognition of the fact that scientists have no place to archive information about NMR structures of biomolecules that fall outside the guidelines of the PDB (e.g., small cyclic peptides), the BMRB will consider accepting coordinate sets representing 3D structural models provided that the following criteria are met:
The molecule falls outside the guidelines of the PDB (i.e., the molecule is a peptide with 23 or fewer residues, a polynucleotide with three or fewer residues, a polysaccharide with three or fewer sugar residues, or a natural product).
The molecule is of biological interest.
The structural model(s) are based on experimental NMR data.
The coordinates are accompanied by a representation of the covalent structure of the molecule (atom connectivity), the assigned NMR chemical shifts for the molecule, and the structural restraints used in generating the structural model.
For depositions meeting these criteria, BMRB encourages authors to submit to PDBj-BMRB SMSDep (https://smsdep.pdbj.org), in addition to the primary (time-domain) data, peak lists, NOEs, and other relevant information.
3.2. Data Retrieval
Users have different options for accessing and downloading data from BMRB. Search options include a powerful “instant search” tool that searches the databases according to keywords entered by the user, an “advanced search” search page that lets the user control both the search criteria and the output requested (particularly useful for downloading subsets of the databases), and a “search grid” that provides single-click access to common searches by data types (chemical shifts, relaxation values, restraints, etc.). Data can also be downloaded from a web-accessible FTP server or kept up to date with the BMRB archive over time by connecting to the BMRB RSYNC server.
3.2.1. Instant Search
The BMRB “instant search” bar is present in the header of every BMRB web page. This search bar will search BMRB entries as you type and shows an automatically updating list of matching BMRB entries. The user can either press “enter” to go to a dedicated search results page or click on one of the suggested results to go directly to the entry summary page for the matched BMRB entry. Alternatively, entering a BMRB entry ID and pressing enter will take you directly to that entry.
The search is performed against multiple relevant fields in a BMRB entry: the citation authors, the entry title, the chemical formula and InChI key of any ligands, database IDs associated with the entry (PDB, PubChem, etc.), and a variety of other commonly searched fields. By hovering the mouse cursor over the suggested results, additional information about the entry will be displayed, including a description of exactly which field was matched.
3.2.2. Advanced Search
BMRB provides an “advanced search” interface that is well suited for generating tabular subsets of the databases, useful for data science projects. The interface provides access to most fields in either the BMRB macromolecule or small-molecule (metabolomics) database (represented by their corresponding NMR-STAR tag).
NMR-STAR 3.2 tags map to database tables and columns as _Table.Column, e.g., the value of _Entry.ID is stored in the “ID” column of the “Entry” table. In order to search for an _Entry. ID, select the top-level Entry information tab (section), then the middle-level Entry information tab (group), and then the Entry tab at the third level (Fig. 5).
Fig. 5.
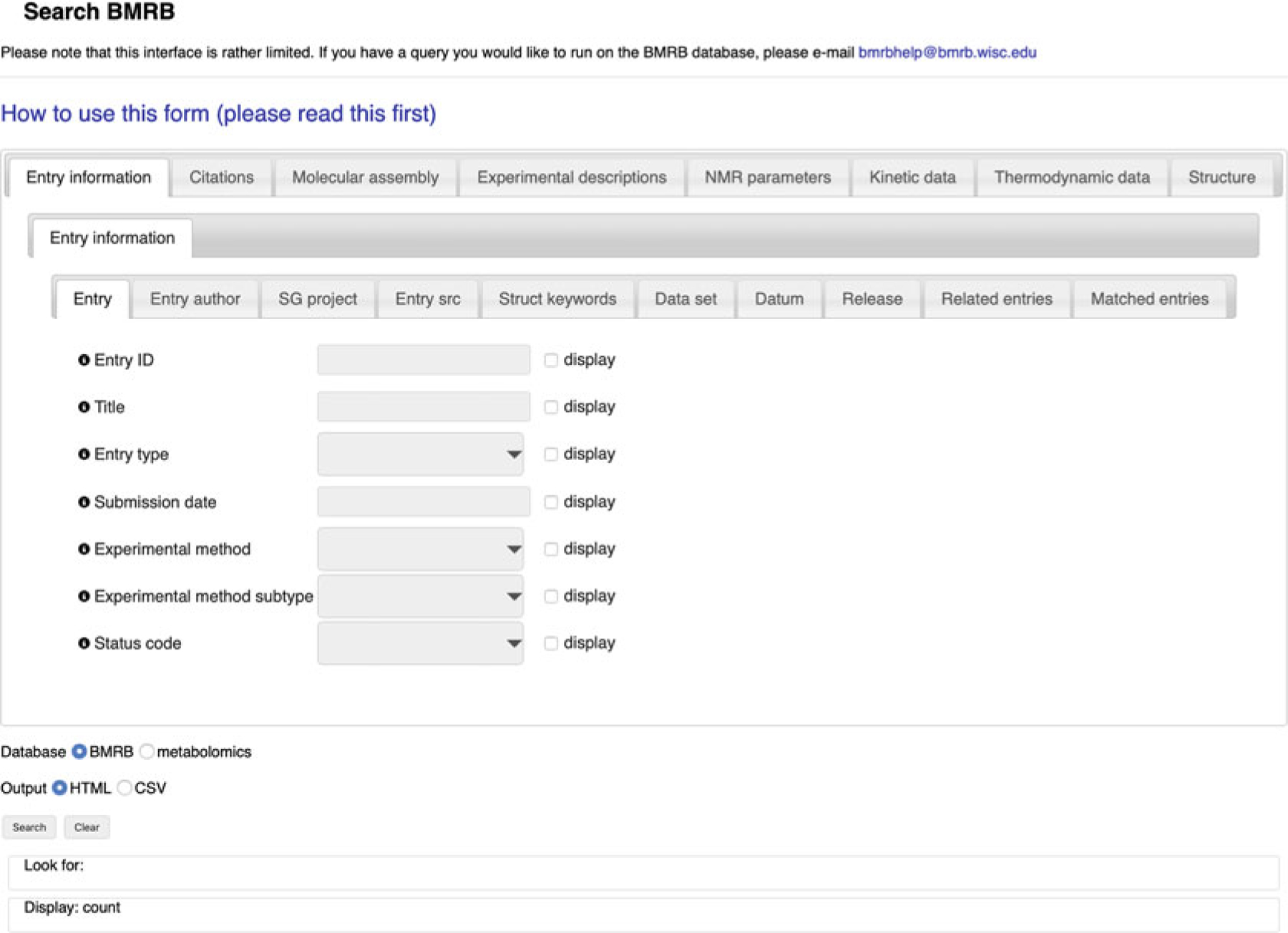
The advanced search interface
For each searchable tag, there is a “search term” text box and a “display” checkbox (Fig. 5). Select “display” to include the tag in the search result table. This arrangement provides the flexibility of using a tag as part of the search criteria or displaying the tag as a column in the resulting table or both: tags to display do not have to be the same ones searched on, and they need not be in the same tables either. If no tags are selected for display, the result will be the count of matching database rows.
Currently, all fields are treated as case-insensitive text and are searchable using POSIX regular expressions supported by PostgreSQL database engine, as explained in the PostgreSQL documentation: https://www.postgresql.org/docs/8.4/functions-matching.html#FUNCTIONS-POSIX-REGEXP
As an example, when looking for Entry.ID (and Entry.ID is selected for display):
| Search term | Result |
|---|---|
| .* | List of all BMRB IDs in the database (entries that have anything in ID tag) |
| 15.* | List of BMRB IDs with “15” anywhere in the ID |
| ^15.$ | List of BMRB IDs between 150 and 159 |
| ^15[0–3]$ | List of BMRB IDs between 150 and 153 |
All search terms are AND’ed. together.
Please note that the limitations of this interface include:
No support for numeric comparisons or range searches.
No OR searches.
An SQL JOIN clause is performed on the tables to search in and the tables to display. This may affect the results as JOIN, which may exclude some rows.
Join to bullet above some regular expressions supported by PostgreSQL do not work, for example, “advanced” REs starting with ***:(?options).
If you have a query you would like to run on the BMRB database that is not supported by this interface, contact bmrbhelp@bmrb.wisc.edu.
Search results are returned either as a webpage (in a separate window/tab) or a comma-delimited file. Note that BMRB’s metabolomics (i.e., small molecule) archive is maintained as a separate database; you can search in either macromolecule or metabolomics database (but not both at the same time).
3.2.3. BMRB Query Grid
BMRB’s query grid interface provides a mechanism for retrieving sets of entries that fit the criteria of predefined queries from the BMRB archive. It is accessible from the “Search Archive” sub-menu in the website navigation panel (Fig. 6). In the main query grid page, the query criteria are described by the headings for the rows and columns in each grid (see Fig. 7). For example, from the main grid, if one clicks on the link in the box in the grid that is at the intersection of the column labeled “DNA” and the row labeled “1H Chemical Shifts,” they will be taken to a page containing a listing of all the BMRB entries that have 1H chemical shifts for DNA polymers (Fig. 8). If one selects the grid box “Proteins/Peptides,” they will be taken to a page containing a link to an HTML page listing all the entries as well as a new query grid that can be used to further refine the query (Fig. 9). In many cases, the result of a query can be displayed with the entries sorted by the number of 1H, 13C, and 15N chemical shifts or BMRB accession number or PDB code. By selecting the “Compressed file” link, it is possible to download a single compressed file that contains all the entries returned by the current query.
Fig. 6.
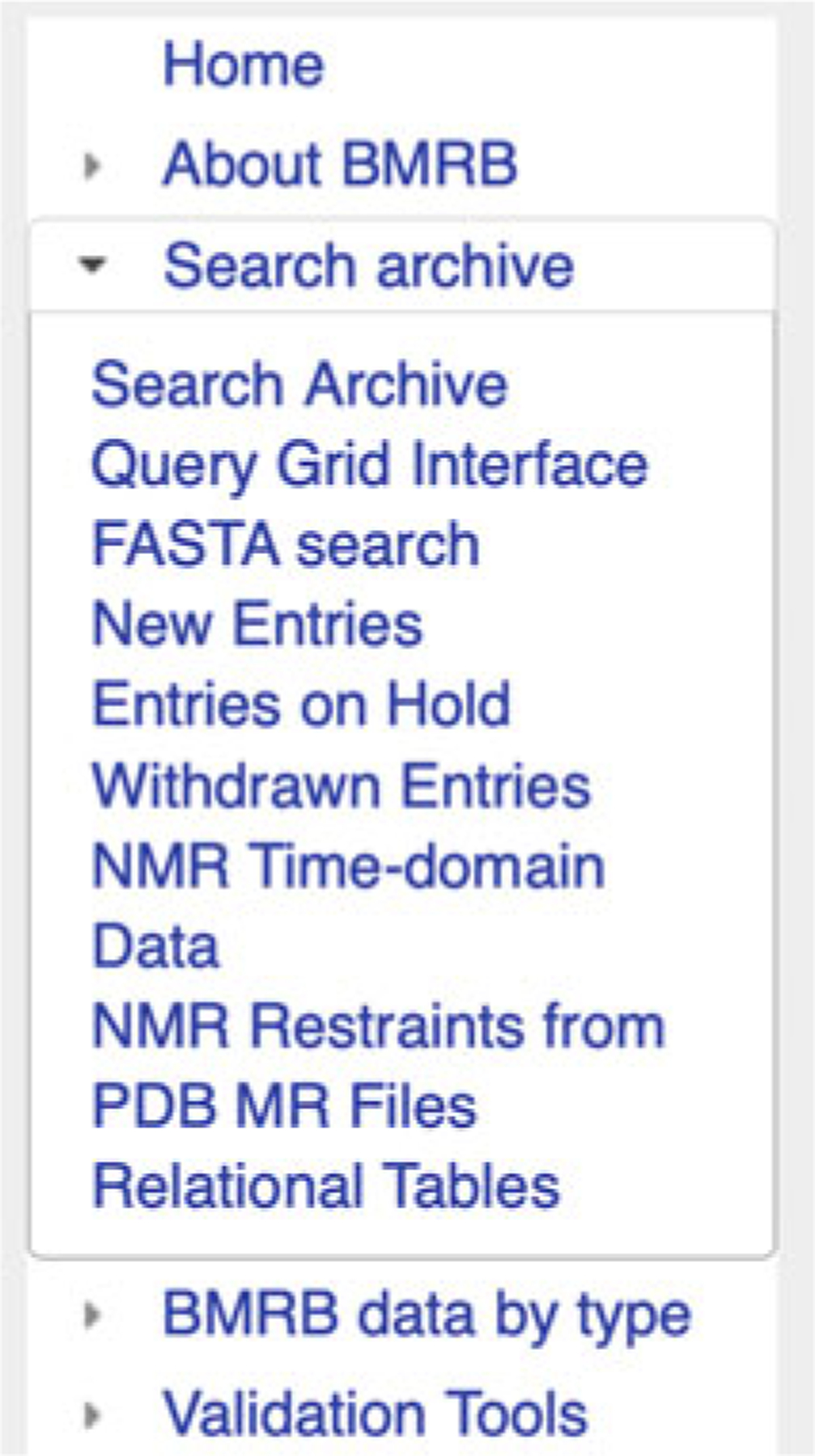
Search archive sub-menu in the navigation panel
Fig. 7.
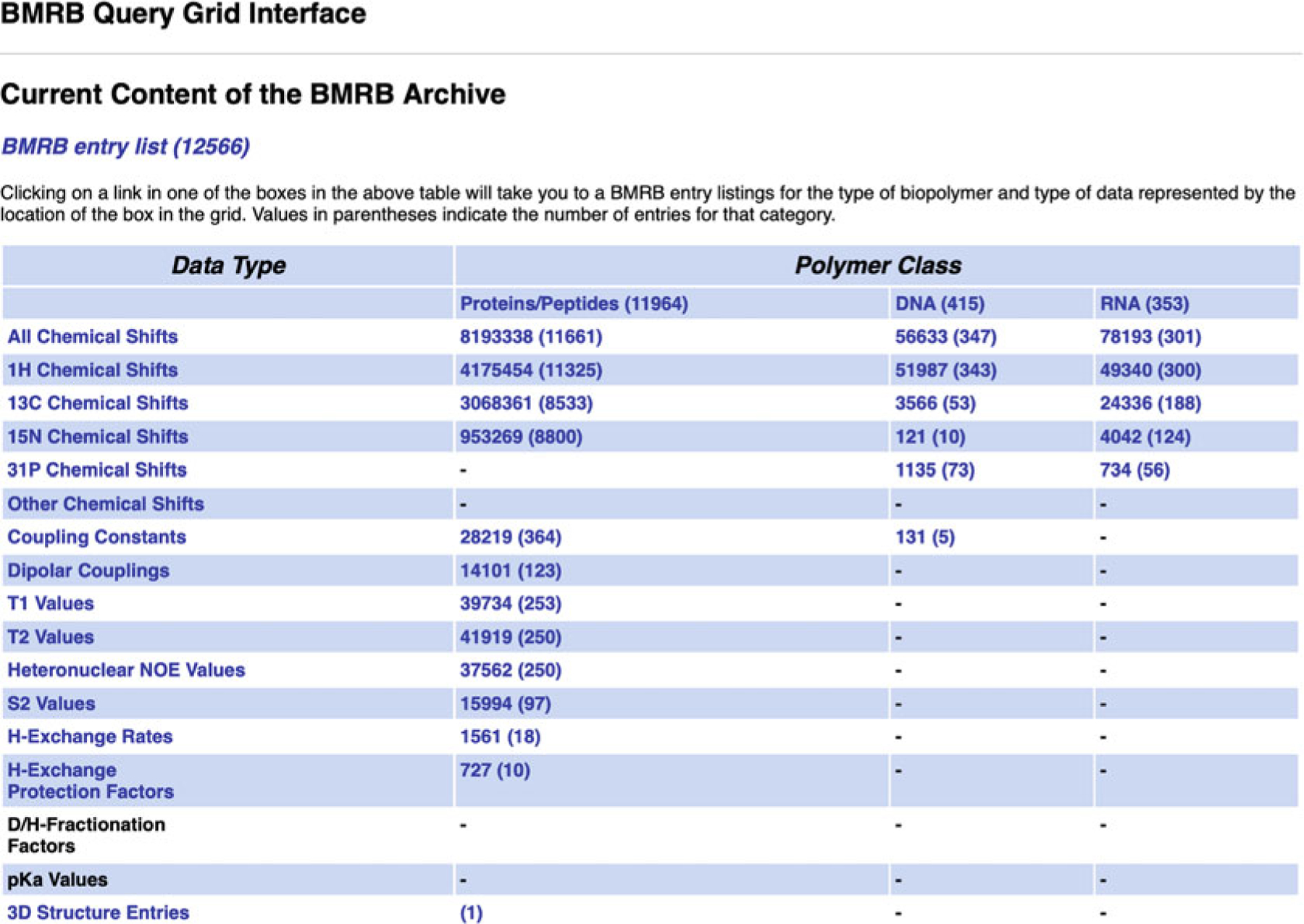
Main query grid page
Fig. 8.
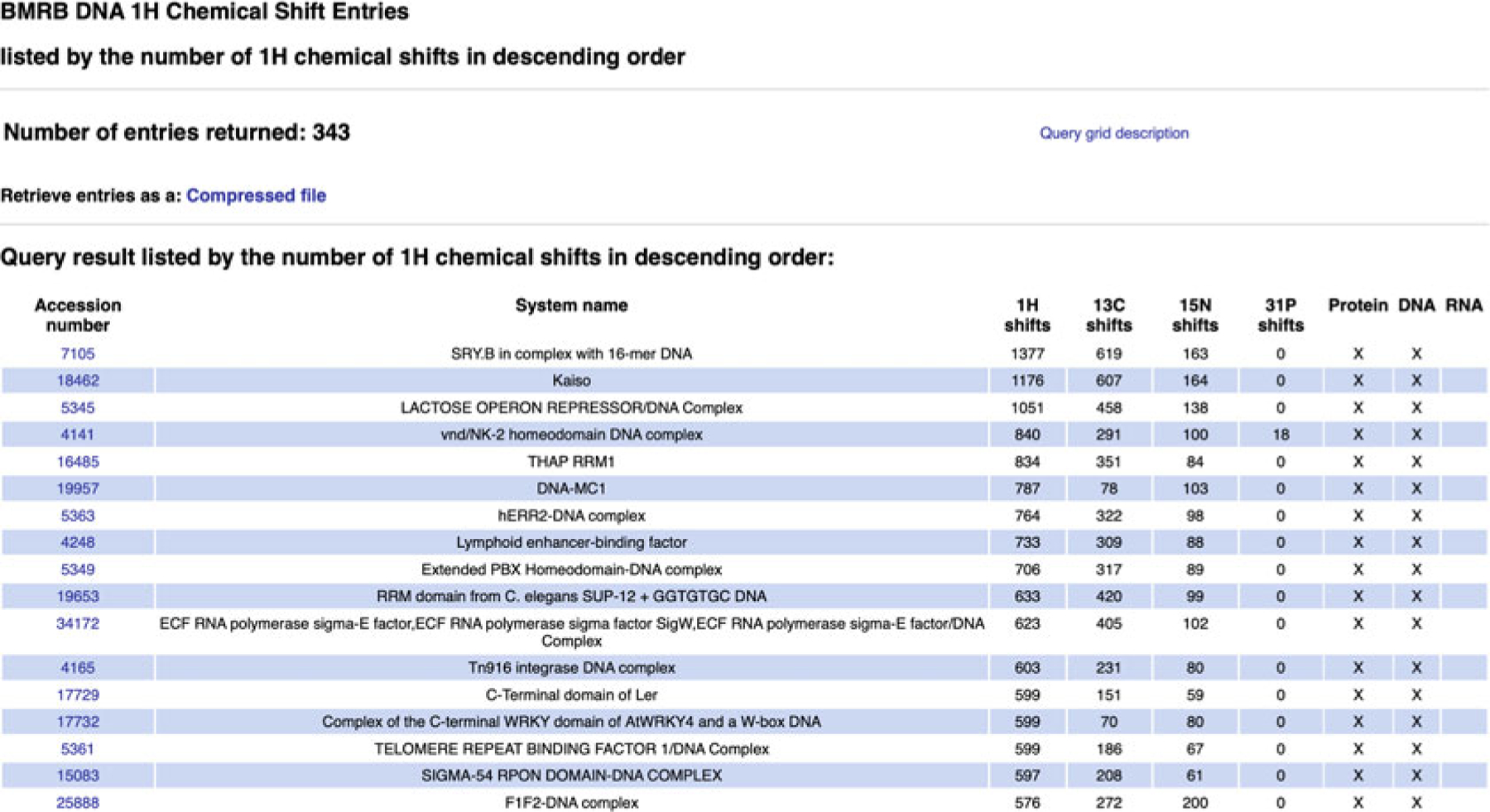
Listings obtain by selecting “DNA” and “1H Chemical Shifts” on the main query grid page
Fig. 9.
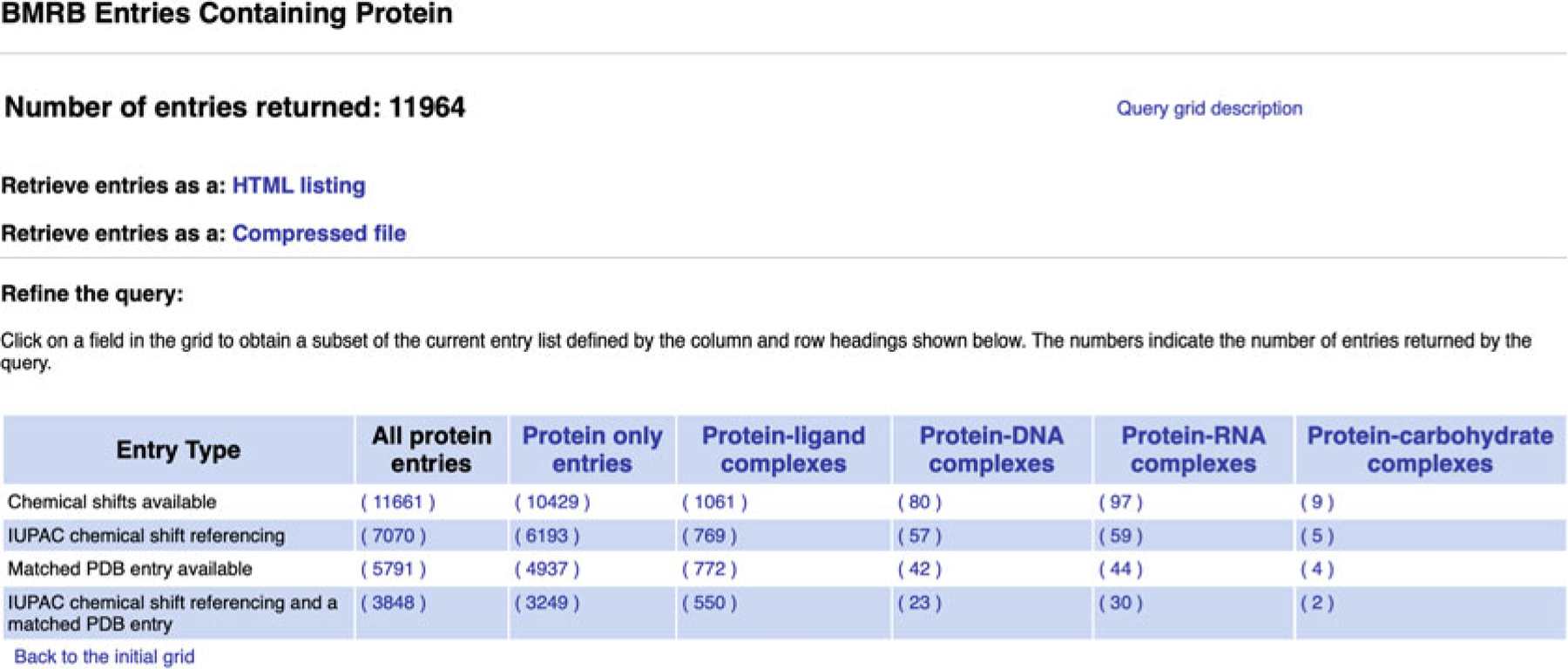
New query grid obtained by selecting “Proteins/Peptides” in the main query grid page
Other query grids by data type are available under the “BMRB Data by Type” item in the navigation panel. The current queryable data types are:
Macromolecular types.
NMR spectral parameters.
Restraints with atomic coordinates and chemical shifts.
Kinetics.
Thermodynamics.
Small-molecule structures.
Time-domain sets.
Solid-state NMR.
Unfolded proteins.
Binding data.
Entries relating to human diseases.
CS-Rosetta structures for BMRB entries.
Human genes.
By selecting an item in the sub-menu, the user gets directed to a query grid interface for the corresponding data type.
Cautionary Notes
The results obtained for any query will depend on the quality of the annotation for each entry. If ligands are not reported for a protein entry but do exist, the entry will still be included in the “Protein only” query result. In general, entries with accession numbers below 4000 may appear in inappropriate query results for these reasons.
In some cases, it is important to think about how a query may have been constructed. If one selects “DNA” from the main query grid, all of the entries where DNA was reported in the molecular system studied will be returned whether or not there is any quantitative data for the DNA. However, by selecting the link in the grid box corresponding to “DNA” and “All Chemical Shifts,” the entries containing DNA and where chemical shifts for the DNA were reported will be returned.
Currently a few BMRB entries (~25) in an older format are not included in the current queries.
The NMR Restraints Grid
A special query grid is the “NMR Restraints Grid” (Fig. 10), also accessible from the “Search Archive” sub-menu, under “NMR Restraints from PDB MR Files” (Fig. 6). The NMR Restraints Grid provides access to the restraints data from the PDB NMR structures contained in the BMRB archive.
Fig. 10.
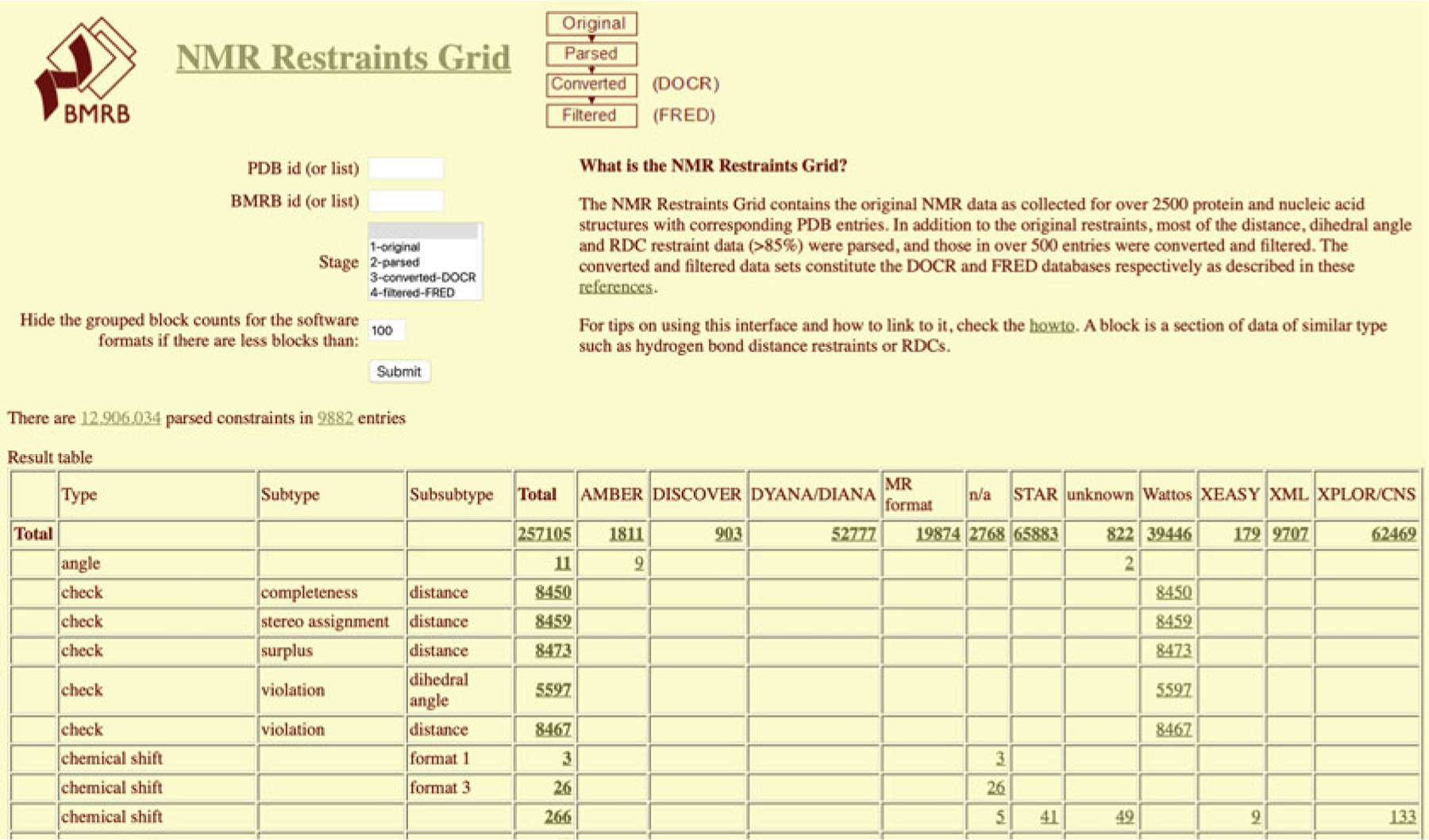
NMR Restraints Grid interface
In addition to the original restraints, most of the distance, dihedral angle, and RDC restraint data (>85%) were parsed, and those in over 500 entries were converted and filtered. The converted and filtered data sets constitute the DOCR and FRED databases, respectively [21].
To obtain the restraints for a specific PDB ID, follow these steps:
Start by selecting the NMR Restraints Grid so that the default options are used.
Specify a PDB entry to find data by entering its four-character code (e.g., 1A24).
Select 0 for “Hide the grouped block counts for the software formats if there are less blocks than.”
Submit by clicking on the Submit button.
A summary table with all the types of restraints archived for the entry will appear. The user can select different sets of restraints by clicking in the corresponding numbers or select all by clicking in the number under the row and column labeled “Total.”
In the resulting table, select one of the blocks by clicking on the number in the cell where the column is labeled mrblock_id. For example, select the block ID for where the stage column reads 3-converted-DOCR and the columns program, type, subtype, and format read XPLOR/CNS, distance, NOE, and ambi (Block ID 468407).
After selecting the mrblock_id, the NOE distance restraints of entry 1A24 from the DOCR database will be shown and can be saved in a ZIP file.
Other ways of downloading data are possible. Just check the “How to” page for the NMR Restraints Grid at http://restraintsgrid.bmrb.wisc.edu/NRG/wattos/MRGridServlet/html/howto.html.
3.2.4. Data Download
In general, data can be downloaded from BMRB through the website, either from the search tools described above or by clicking on appropriate download links from entry pages. BMRB users can also access data from our databases through our FTP server at http://www.bmrb.wisc.edu/ftp/pub/bmrb/.
The following directories are available at the FTP site:
Derived data.
Entry directories.
Entry lists.
Internal data.
Metabolomics.
NMR-STAR dictionary.
NMR PDB integrated data.
PDB MolProbity.
Relational tables.
Secondary metabolomics.
Sequence libraries.
Software.
Statistics
Time domain.
Validation reports.
Data in these directories comes in different formats, depending on the type of information accessed (e.g., entry data can be downloaded in NMR-STAR, RDF, and/or XML format). Entry pages in the website link directly to the FTP server for associated files, like time-domain data. The relational tables are used to recreate a local copy of the database using PostgreSQL and following the instructions at http://www.bmrb.wisc.edu/search/rdb31.shtml.
This can also be done with the small-molecule (metabolomics) database.
For users or labs that would like to keep a local updated copy of the BMRB databases, BMRB provides access to a public rsync server (for LINUX/UNIX-based systems). Contact BMRB through bmrbhelp@bmrb.wisc.edu for help on setting up a local database and keep it updated through rsync.
3.3. Data Analysis
3.3.1. Data Conversion
The STARch File Converter
The STARch file converter (http://www.bmrb.wisc.edu/software/starch) can take chemical shift tables in several formats (see Fig. 11), including ASCII tables in comma-separated or tab-delimited formats, and convert them into NMR-STAR version 3.2 format. For other data types (e.g., dipolar couplings, RDCs, relaxation values, etc.), STARch can convert NMR-STAR version 2.1 or ASCII tables (comma-separated or tab-delimited) into NMR-STAR 3.2.
Fig. 11.

The STARch file data converted interface
Information and recommendations for STARch users are as follows:
Many software packages can export NMR-STAR (e.g., NMRView, CCPN). For these packages, the export option should be used, rather than STARch, to avoid potential loss of information during conversion.
STARch only converts a given file format to NMR-STAR; it does not convert atom nomenclature. The user is advised to use IUPAC nomenclature in all tables, which will speed up the processing of the deposition.
For NMR structure depositions, residue and atom names must match those in the coordinates file.
STARch output is a “bare” data table (loop), not a complete STAR file.
When preparing a chemical shift table for NMR structure deposition, make sure residue and atom names match those in the coordinates. Otherwise, the table will be rejected by the deposition system.
3.3.2. Validation
During data curation, BMRB’s annotators validate the data files deposited. Under the “Validation Tools” item in the navigation panel, users will find links to many publicly available validation tools, as well as the possibility to download data validation software. Depositors of protein depositions are strongly encouraged to use validation software packages (in particular the wwPDB validation server and the PSVS (AVS) server) to check their NMR experimental data and structure files before uploading them.
3.3.3. CS-Rosetta High-Throughput Structure Estimation
CS-Rosetta is a software package that attempts to make de novo protein structure predictions from known chemical shifts and optionally RDC and NOE values [12, 13]. Using a library of protein fragments from proteins with experimentally determined structures, it uses Monte Carlo methods to make its structural predictions. Due to the computational effort involved in the Monte Carlo structure determination step, large computational resources can dramatically decrease the amount of time required to run CS-Rosetta. BMRB has utilized its computational resources, as well as those of the CHTC and OSG, to develop a web service that allows for the deposition of chemical shifts (and optionally RDC and NOE values) and returns the results of a CS-Rosetta run in an interactive fashion via a web page. Users who have submitted to the server are e-mailed status during the CS-Rosetta run. When it completes, they are e-mailed a link where they can inspect the generated structures.
When viewing the CS-Rosetta results, an interactive graph showing the RMS distance to the lowest energy structure and the relative energy of all structures is displayed (Fig. 12). Clicking on a point in the graph will pull up that specific structure in an interactive JSmol viewer [22].
Fig. 12.
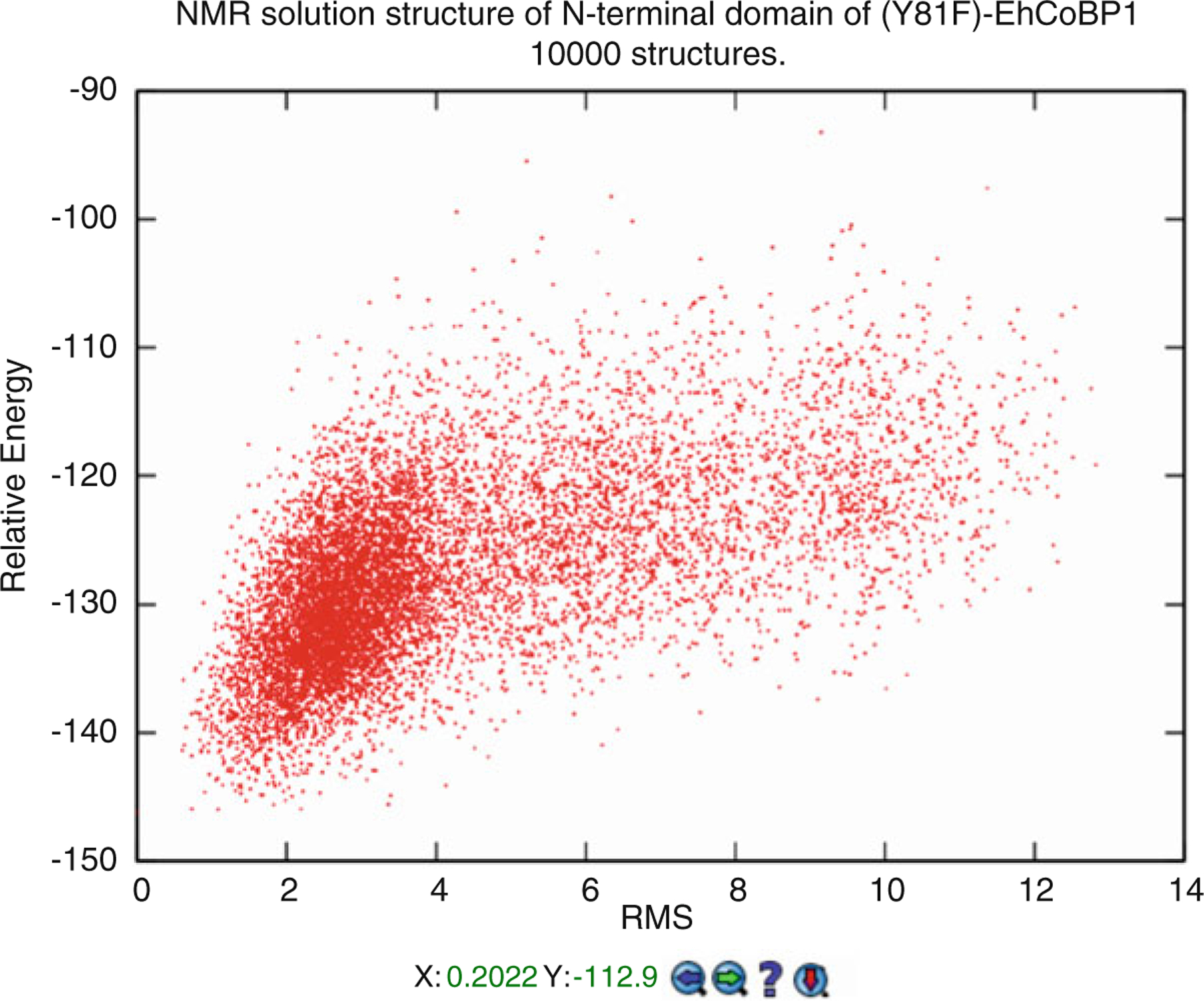
Calculated CS-Rosetta structures for BMRB entry 19193
In addition to a standard web interface, the CS-Rosetta server also provides an API that can be used to automatically make a submission to the server (http://www.bmrb.wisc.edu/tools/automated_csrosetta.shtml). BMRB has also developed a tool embedded in the NMRbox that allows a user to select input files via a GUI and then submits them to the CS-Rosetta server via this API. Moreover, the NMRFAM PINE [23] server has the option to automatically submit the chemical shifts to the CS-Rosetta server using the API.
The BMRB CS-Rosetta server is available at the following URL: https://csrosetta.bmrb.wisc.edu.
3.3.4. Working with NMR-STAR Files
As described above, NMR-STAR [17] is the archival and exchange format used by BMRB. NMR-STAR is available as input and/or output by several software packages that deal with the harvesting and processing of biomolecular data (CCPN [24], NMRView, TALOS [25], NMRFAM-SPARKY [26], PINE [23], ARECA [27], PONDEROSA [28], Integrative NMR [29], CSI [30], NMRFx [31], RCI [32], ABACUS [33], and PDBstat [34]) and with chemical shift prediction (SHIFTX2 [35] and SHIFTS [36]). NMR-STAR also is used as a data exchange format by the NMRbox project [11].
The NMR-STAR v3.2 ontology [17] provides an extensive controlled vocabulary for the description of NMR spectroscopic studies of biological systems. The ontology includes the description of experiments, the data generated, and the derived results such as molecular structures, dynamics, and functional properties. New NMR techniques and experiments are being developed continuously, and the NMR-STAR ontology grows consequently. BMRB has developed a JavaScript tool for interactively visualizing, validating, and editing NMR-STAR files, as well as a Python programming library for handling and generating NMR-STAR files programmatically (see Subheading 3.4.2). Here we describe how to access and use the NMR-STAR interactive viewer tool.
Using the NMR-STAR Interactive Viewer
The BMRB interactive NMR-STAR visualizer is located at the URL http://www.bmrb.wisc.edu/dictionary/starviewer. The page contains a form where you may select an NMR-STAR file from your computer to visualize and perform minor edits. (Alternatively, each entry in the BMRB archive has a link to load its contents in the interactive viewer.) When you select a file, it is processed in Java-Script inside your browser, and the contents of the NMR-STAR file you select are not uploaded to BMRB servers. When a file is selected, the page will automatically render the NMR-STAR file contents on the page and provide several features that will make it easier to inspect or edit the file (Fig. 13):
Any tags that do not contain values in the uploaded file are hidden by default in order to make the file more readable. You can toggle the display of empty tags by clicking the “Show tags without values” button at the top of the page.
All tags can be hovered over with a mouse in order to view a description of what data the tag references.
Data values are styled as plain text rather than an input field, but by clicking on any tag value, you can edit a value. Furthermore, if the tag is one that contains enumerations (suggested values) in the NMR-STAR dictionary, they will be auto-suggested as options when the first characters entered match an existing enumeration.
NMR-STAR data blocks (called saveframes and loops) can be collapsed to make it easier to view different portions of the entry. In addition, data in loops are lightly color coded in order to facilitate seeing which column a given data value belongs to.
Data type validation is performed instantaneously as changes are made. Tags with an invalid value (e.g., an invalid date) highlight in red to warn of the error.
In the NMR-STAR format, it is possible for a tag to reference data elsewhere in the file. Where these links are present, a hyperlink will display that can be clicked to jump directly to the referenced data.
Fig. 13.

The NMR-STAR viewer
When finished editing the file, click the “Download” button at the top of the page to save a copy with your changes.
Note that this tool is intended for minor edits and not major changes, as creating or deleting data rows is not supported. For more significant modification of NMR-STAR files, see the PyNMR-STAR tool (Subheading 3.4.2).
3.4. Programming Tools
The BMRB team has developed several tools for software developers accessing the BMRB databases or manipulating NMR-STAR files. Libraries and other tools described in this section are accessible through the BMRB GitHub page at https://github.com/uwbmrb.
3.4.1. BMRB API
BMRB has developed and deployed a RESTful API to enable rapid access to the most up-to-date version of the database for both metabolomics and macromolecule entries. This API enables programmatic access to the BMRB database from scripts and applications. Many internal BMRB tools use this API to provide access to the most up-to-date version of the BMRB database; PyNMR-STAR, PyBMRB, the Instant Search, the NMR-STAR interactive viewer, and other BMRB tools all use the API to retrieve data.
The API supports a variety of different query types to facilitate research, and new ones are being added in response to community feedback. Examples of supported queries are listed below:
Given a list of chemical shift values, return a list of BMRB entries that contain one or more of the queried shifts, ordered by number of shifts matched and the closeness of the match.
Return all chemical shifts in the BMRB, optionally filtered by residue, atom type, and chemical shift value. Optionally, also return the pH and temperature at which the chemical shift was observed.
Search a protein sequence in FASTA format against the BMRB archive and return a list of matching entries.
Return all entries that contain a given value for a given tag (e.g., entries that contain the “solid-state” tag).
Return information about the raw experimental data available for a given entry.
Return information about a given BMRB entry in either NMR-STAR or JSON format.
Many other query types are also available. For the most up-to-date list of query types, as well as instructions and extensive documentation, please view the GitHub page for the project: https://github.com/uwbmrb/BMRB-API.
3.4.2. PyNMR-STAR: NMR-STAR Handling Python Programming Library
BMBR entries are internally represented as NMR-STAR files, a custom file type for NMR study records that was modeled on the STAR format [18]. This format enables the capture of nearly all NMR-related data and is self-describing and text based, which means it is editable in any standard text editor. Despite that, editing the file while maintaining syntactical validity requires some degree of knowledge of the format. To ease the process of reading, creating, and modifying NMR-STAR files, BMRB has developed and released a Python module (at time of publishing, compatible with Python versions 2.6–3.7) called PyNMR-STAR. This software library significantly eases working with NMR-STAR files—it ensures that all files generated are syntactically valid; it provides tools to validate records against the BMRB NMR-STAR schema; and it provides a wide variety of additional features to ease working with NMR-STAR files.
There is extensive documentation and an introduction to working with the NMR-STAR format at the GitHub page for the project: https://github.com/uwbmrb/PyNMRSTAR.
3.4.3. PyBMRB and RBMRB: Data Retrieval and Visualization Libraries for Python and R
The information-rich data content in BMRB is a useful resource for understanding the properties of atoms in amino acids and nucleic acids in different sequences, conformational states, or sample conditions. Database-wide chemical shift statistics of a particular atom help us to understand the range of its properties. The numerous tags in the NMR-STAR dictionary precisely capture valuable metadata, including sample conditions, chemical shift referencing, and experiment type. The information-rich data content in NMR-STAR files is both human- and machine-readable format.
To help users visualize the data in different ways, BMRB has developed tools with similar functionalities in two languages popular among the bioinformatics community: Python and R. Under normal circumstances, one needs to download and parse the data before visualizing the data either as histogram or NMR spectrum. The BMRB-API supports direct access to the BMRB archive by tools in both Python (PyBMRB) and R (RBMRB) that enable database-level and entry-level data visualizations without the need for downloading and parsing steps. Both PyBMRB and RBMRB use the same visualization tool “plotly” in the backend, which generates interactive and portable visualizations. Interactive graphics visualizations open in any web browser and can be exported as a static image with a single click.
1. Database-Level Visualizations
Histograms are quite useful for understanding the effects of secondary structure on the chemical shifts of atoms in amino acids and nucleic acids. The chemical shift distributions of atoms in BMRB are sometimes bimodal or trimodal, which indicates that a particular atom is sensitive to secondary structural elements like helix, beta sheet, and coil. On the other hand, chemical shift outliers that are far away from expected values may indicate the presence of paramagnetic/metallic element or potential binding sites.
PyBMRB and RBMRB can generate chemical shift histogram of any amino acid or any atom or atom type using a single command. Figure 14 shows the chemical shift histogram of CB atoms generated by the command hist(atom = “CB”) using PyBMRB v1.2.6. Similar histogram can be generated using RBMRB v2.1.2 with the command chemical_shift_hist (atm = “CB”). Chemical shift correlations between any two atoms in an amino acid can be visualized by a 2D histogram. Figure 15 shows the chemical shift correlation between CB and N atoms in cysteine. Additional parameters are available to filter the data for chemical shift outliers using standard deviation-based statistical filtering and to convert the histograms into normalized density plots. PyBMRB v1.2.6 has an additional option that can be used to generate a conditional histogram for a particular atom in a residue filtered against a set of preassigned chemical shifts for other atoms in the same residue.
Fig. 14.
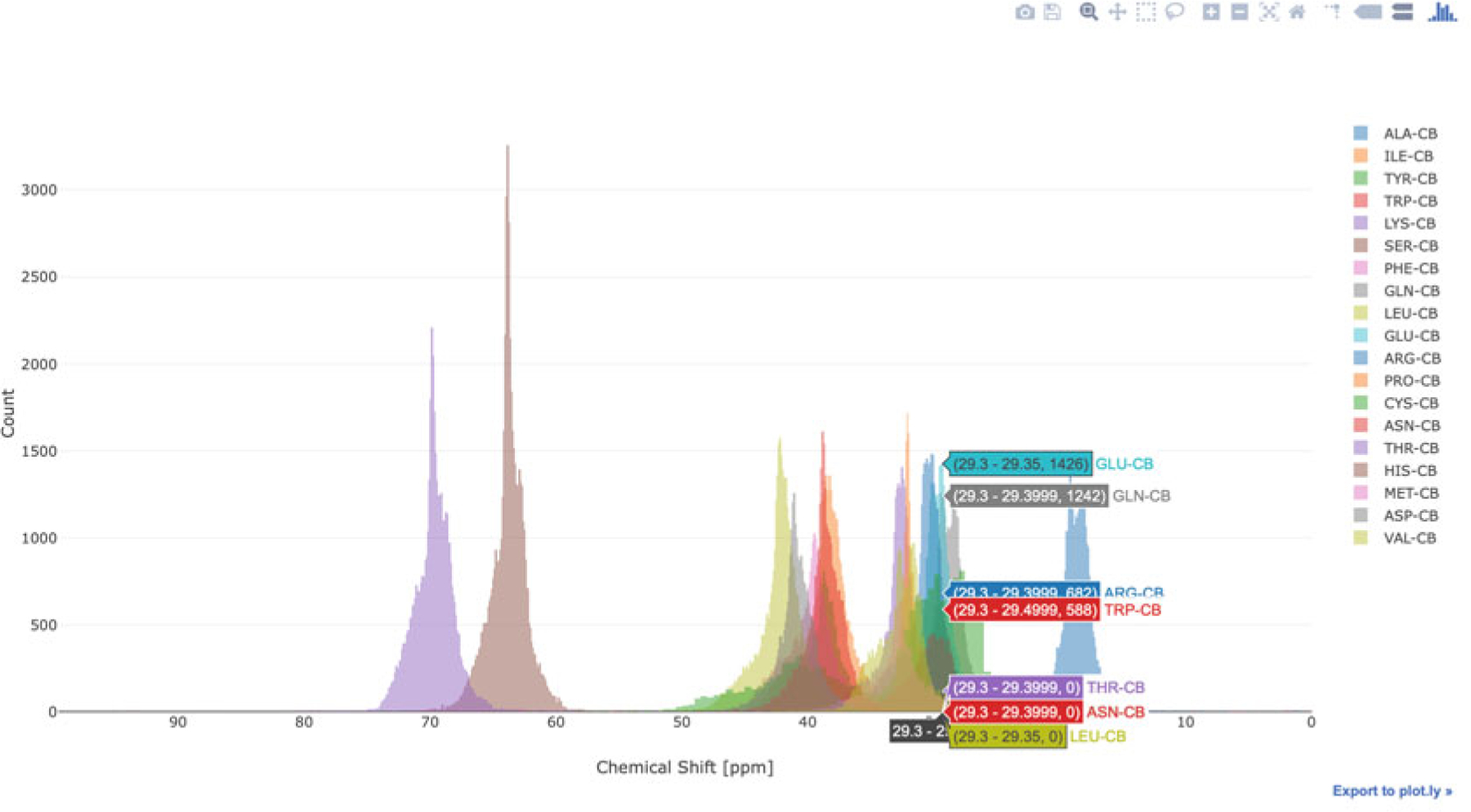
Chemical shift histogram for CB atoms in BMRB, generated using the PyBMRB library as explained in the text
Fig. 15.
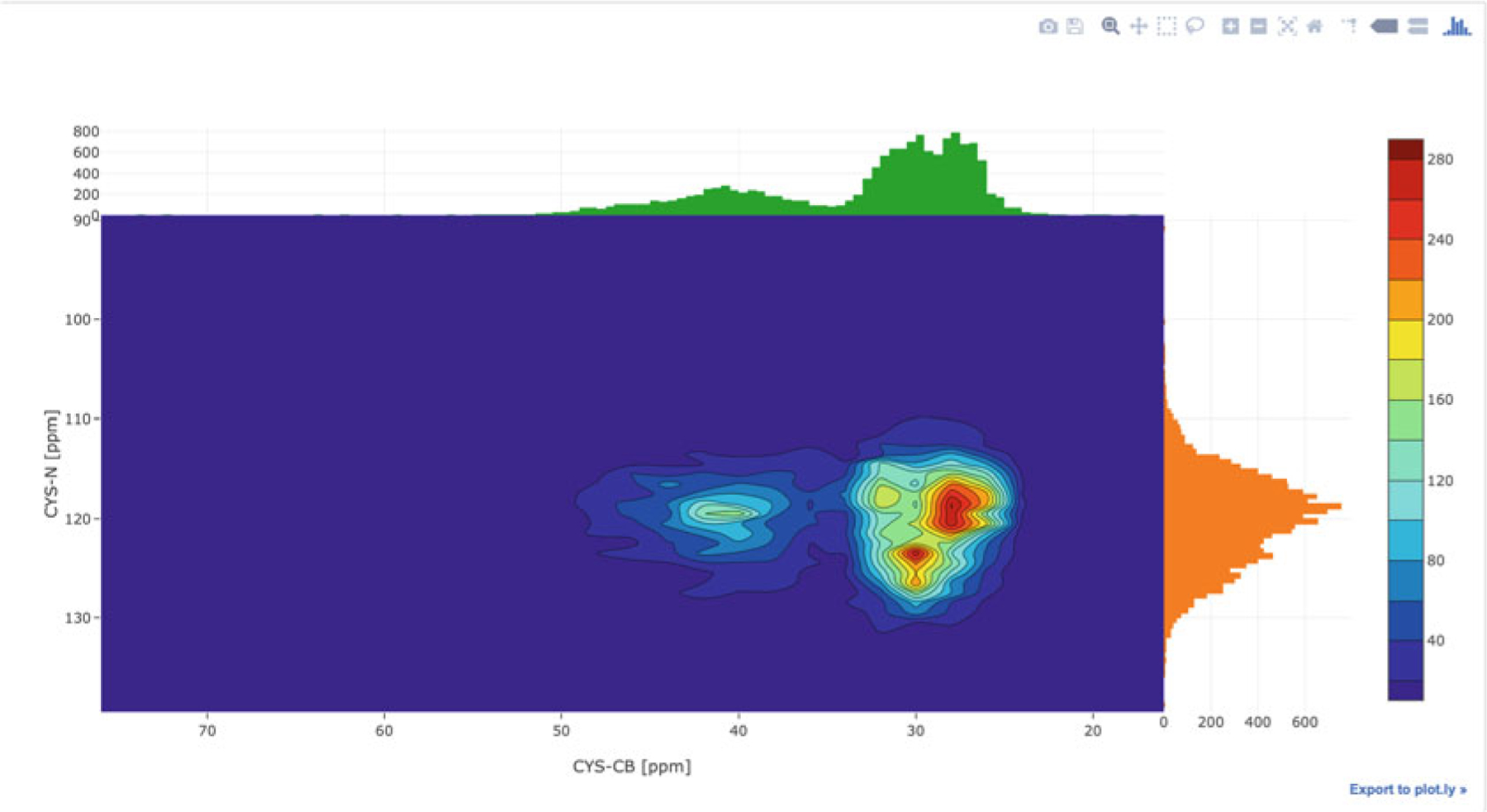
Interactive diagram showing the chemical shift correlation between CB and N atoms in cysteine across BMRB, generated using the PyBMRB library
2. Entry-Level Visualizations
1H–15N correlations from HSQC or other NMR experiments are commonly used in studies of folding, aggregation, ligand binding, or protein-protein interactions. PyBMRB and RBMRB have the functionality of simulating 1H–15N correlations from assigned chemical shifts in BMRB entries. The software can overlay 1H–15N correlations from multiple entries, and chemical shift changes can be easily tracked by connecting the peaks from resides in same position in the sequence. Figure 16 compares 1H–15N correlations for the enzyme arsenate reductase from three different BMRB entries; the visualization was generated by PyBMRBv1.2.6 with the command:
n15hsqc(bmrbid = [17074, 17076, 17077]
Fig. 16.
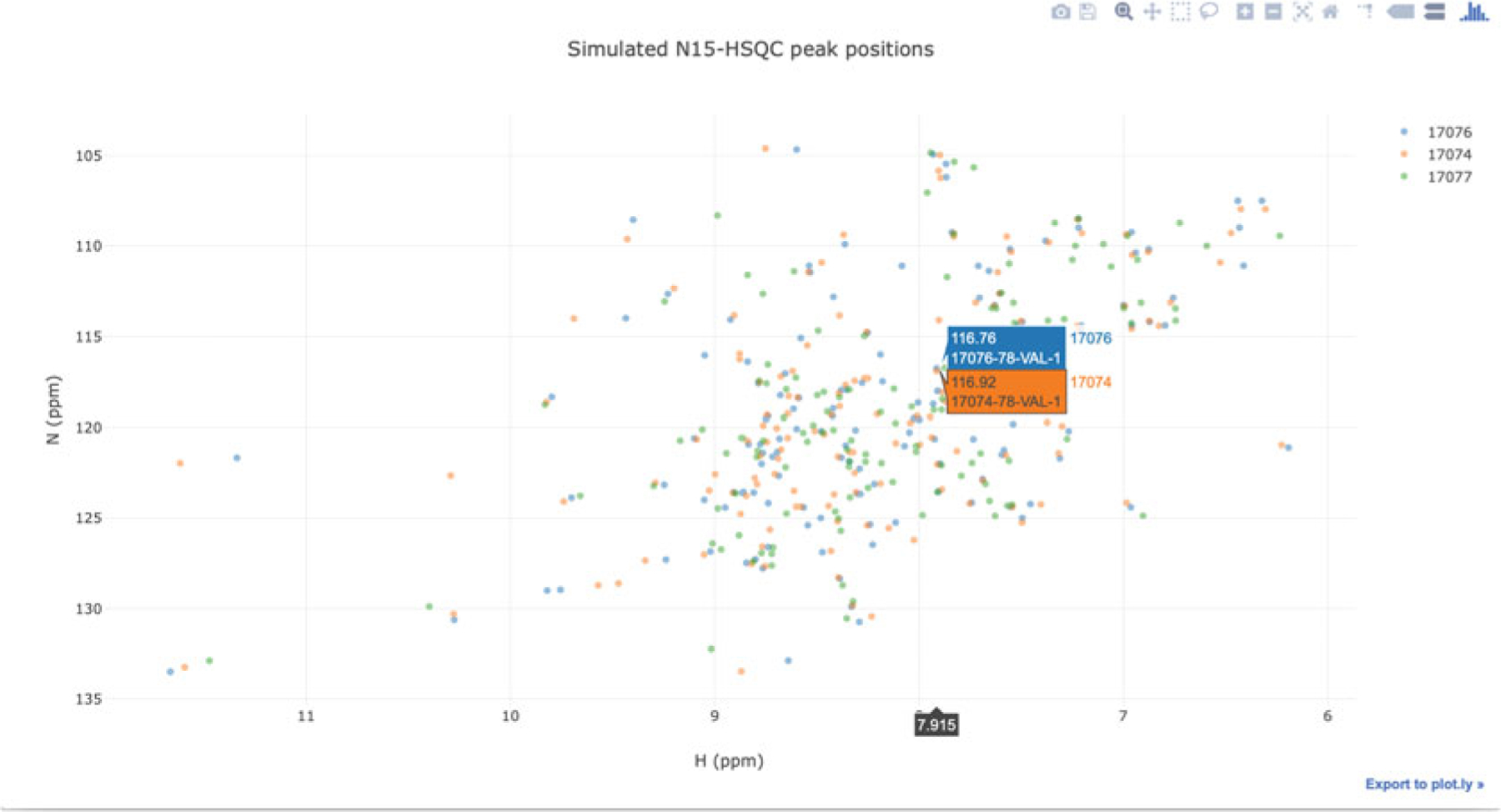
PyBMRB-generated simulated HSQC overlapping spectra comparing three BMRB entries
The software can be used in a local computer to compare data in an NMR-STAR file with data from a BMRB entry and to track the chemical shift changes using sequence numbers. Figure 17 shows such a comparison generated by PyBMRBv1.2.6 with the command:
‘n15hsqc(bmrbid = [17074, 17076, 17077], filename = ‘test.str’),
which compares local data (filename) with data from three BMRB entries (17074, 17076, and 17077).
Fig. 17.
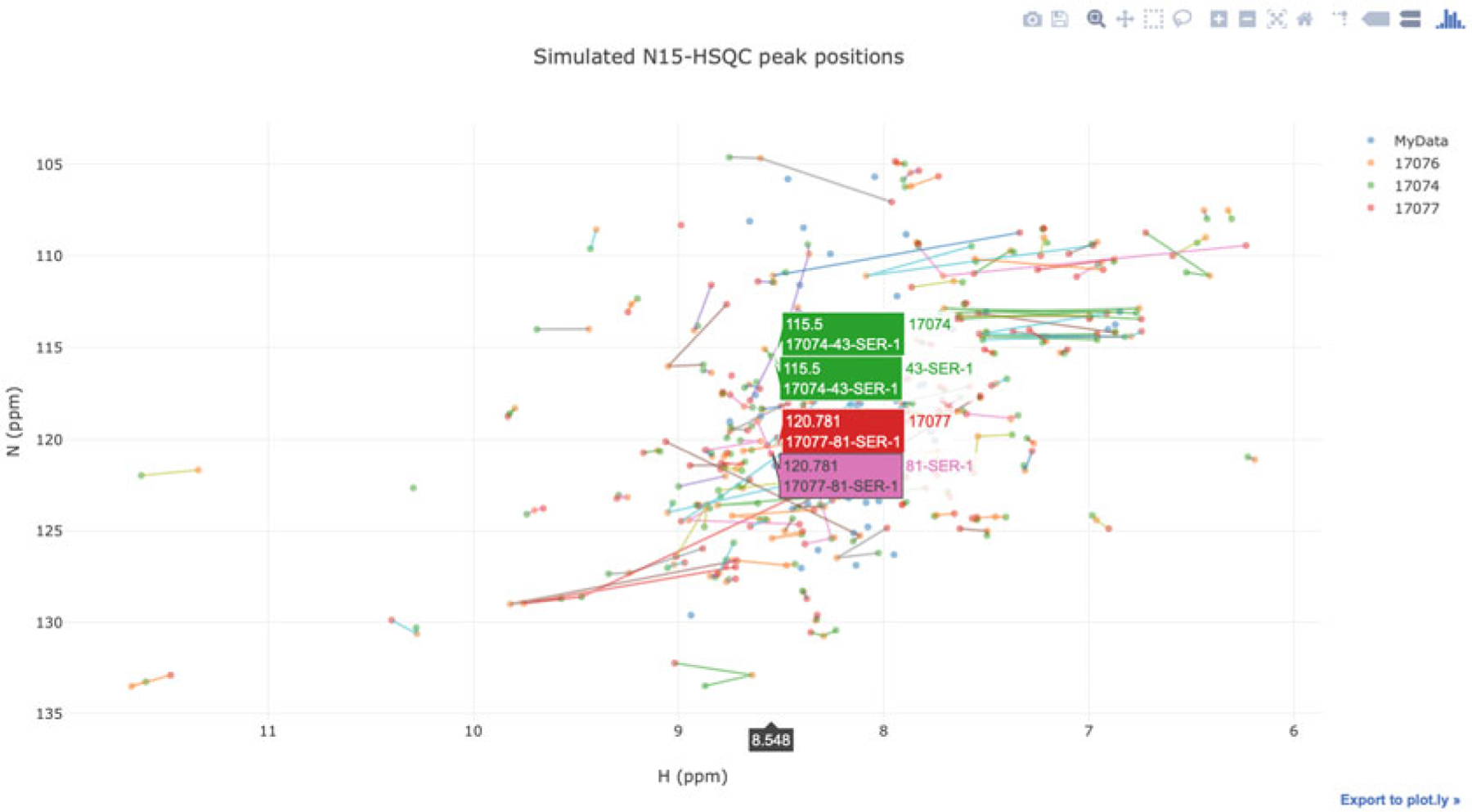
This simulated HSQC overlapping spectrum comparing a user-uploaded spectrum with three similar entries in BMRB was generated using PyBMRB
3. Installation and Availability
The source code and installation instructions for both packages are available from the BMRB GitHub repository (https://github.com/uwbmrb/RBMRB, https://github.com/uwbmrb/PyBMRB). These packages are also made available in their corresponding software package repositories, which enables users to easily install the packages using a single command. RBMRB v2.1.2 is available in CRAN repository (https://CRAN.R-project.org/package=RBMRB), and PyBMRB v1.2.6 is available in Python Package Index repository (https://pypi.org/project/pybmrb/). Documentation and example files are available in respective packages.
Jupyter Notebooks
Owing to their ease of use and reproducibility, Jupyter Notebooks (https://jupyter.org) are being widely adopted within the sciences; they provide a single environment that links computation and documentation in an interactive fashion. Both PyBMRB and RBMRB can be used in a notebook environment. The BMRB home page provides a link to a sample PyBMRB Jupyter Notebook, which was created by converting the PyBMRB GitHub repository into an interactive notebook by using a third-party server (https://mybinder.org), which does not require any installation and simply opens in a web browser. BMRB users can use this notebook to play with visualization tools without installing them.
References
- 1.Ulrich EL, Akutsu H, Doreleijers JF et al. (2008) BioMagResBank. Nucleic Acids Res 36:D402–D408. 10.1093/nar/gkm957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berman HM, Westbrook J, Feng Z et al. (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242. 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ladizhansky V (2017) Applications of solid-state NMR to membrane proteins. Biochim Biophys Acta Proteins Proteom 1865:1577–1586. 10.1016/j.bbapap.2017.07.004 [DOI] [PubMed] [Google Scholar]
- 4.Sugiki T, Kobayashi N, Fujiwara T (2017) Modern technologies of solution nuclear magnetic resonance spectroscopy for three-dimensional structure determination of proteins open avenues for life scientists. Comput Struct Biotechnol J 15:328–339. 10.1016/j.csbj.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dashti H, Westler WM, Markley JL et al. (2017) Unique identifiers for small molecules enable rigorous labeling of their atoms. Sci Data 4:170073. 10.1038/sdata.2017.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dashti H, Wedell JR, Westler WM et al. (2018) Applications of parametrized NMR spin systems of small molecules. Anal Chem 90:10646–10649. 10.1021/acs.analchem.8b02660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilkinson MD, Dumontier M, Aalbersberg IJ et al. (2016) The FAIR Guiding Principles for scientific data management and stewardship. Sci Data 3:160018. 10.1038/sdata.2016.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wwpdb (2019) Protein data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res 47:D520–D528. 10.1093/nar/gky949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berman H, Henrick K, Nakamura H (2003) Announcing the worldwide Protein Data Bank. Nat Struct Biol 10:980. 10.1038/nsb1203-980 [DOI] [PubMed] [Google Scholar]
- 10.Gifford LK, Carter LG, Gabanyi MJ et al. (2012) The protein structure initiative structural biology knowledgebase technology portal: a structural biology web resource. J Struct Funct Genom 13:57–62. 10.1007/s10969-012-9133-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maciejewski MW, Schuyler AD, Gryk MR et al. (2017) NMRbox: a resource for biomolecular NMR computation. Biophys J 112:1529–1534. 10.1016/j.bpj.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen Y, Lange O, Delaglio F et al. (2008) Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci U S A 105:4685–4690. 10.1073/pnas.0800256105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen Y, Vernon R, Baker D et al. (2009) De novo protein structure generation from incomplete chemical shift assignments. J Biomol NMR 43:63–78. 10.1007/s10858-008-9288-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen VB, Arendall WB, Headd JJ et al. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystal-logr D 66:12–21. 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thain D, Tannenbaum T, Livny M (2005) Distributed computing in practice: the Condor experience. Concurrency Comput Pract Ex 17:323–356. 10.1002/cpe.938 [DOI] [Google Scholar]
- 16.Young JY, Westbrook JD, Feng Z et al. (2017) OneDep: unified wwPDB system for deposition, biocuration, and validation of macromolecular structures in the PDB archive. Structure 25:536–545. 10.1016/j.str.2017.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ulrich EL, Baskaran K, Dashti H et al. (2018) NMR-STAR: comprehensive ontology for representing, archiving and exchanging data from nuclear magnetic resonance spectroscopic experiments. J Biomol NMR 73:5–9. 10.1007/s10858-018-0220-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall SR, Spadaccini N (1994) The Star File – detailed specifications. J Chem Inf Comput Sci 34:505–508. 10.1021/ci00019a005 [DOI] [Google Scholar]
- 19.Westbrook JD, Fitzgerald PM (2003) The PDB format, mmCIF, and other data formats. Methods Biochem Anal 44:161–179 [PubMed] [Google Scholar]
- 20.Gutmanas A, Adams PD, Bardiaux B et al. (2015) NMR Exchange Format: a unified and open standard for representation of NMR restraint data. Nat Struct Mol Biol 22:433–434. 10.1038/nsmb.3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doreleijers JF, Nederveen AJ, Vranken W et al. (2005) BioMagResBank databases DOCR and FRED containing converted and filtered sets of experimental NMR restraints and coordinates from over 500 protein PDB structures. J Biomol NMR 32:1–12. 10.1007/s10858-005-2195-0 [DOI] [PubMed] [Google Scholar]
- 22.Hanson RM, Prilusky J, Renjian Z et al. (2013) JSmol and the next-generation web-based representation of 3D molecular structure as applied to proteopedia. Isr J Chem 53:207–216. 10.1002/ijch.201300024 [DOI] [Google Scholar]
- 23.Bahrami A, Assadi AH, Markley JL et al. (2009) Probabilistic interaction network of evidence algorithm and its application to complete labeling of peak lists from protein NMR spectroscopy. PLoS Comput Biol 5:e1000307. 10.1371/journal.pcbi.1000307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vranken WF, Boucher W, Stevens TJ et al. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59:687–696. 10.1002/prot.20449 [DOI] [PubMed] [Google Scholar]
- 25.Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13:289–302 [DOI] [PubMed] [Google Scholar]
- 26.Lee W, Tonelli M, Markley JL (2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31:1325–1327. 10.1093/bioinformatics/btu830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dashti H, Tonelli M, Lee W et al. (2016) Probabilistic validation of protein NMR chemical shift assignments. J Biomol NMR 64:17–25. 10.1007/s10858-015-0007-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee W, Kim JH, Westler WM et al. (2011) PONDEROSA, an automated 3D-NOESY peak picking program, enables automated protein structure determination. Bioinformatics 27:1727–1728. 10.1093/bioinformatics/btr200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee W, Cornilescu G, Dashti H et al. (2016) Integrative NMR for biomolecular research. J Biomol NMR 64:307–332. 10.1007/s10858-016-0029-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hafsa NE, Arndt D, Wishart DS (2015) CSI 3.0: a web server for identifying secondary and super-secondary structure in proteins using NMR chemical shifts. Nucleic Acids Res 43: W370–W377. 10.1093/nar/gkv494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Norris M, Fetler B, Marchant J et al. (2016) NMRFx Processor: a cross-platform NMR data processing program. J Biomol NMR 65:205–216. 10.1007/s10858-016-0049-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berjanskii MV, Wishart DS (2007) The RCI server: rapid and accurate calculation of protein flexibility using chemical shifts. Nucleic Acids Res 35:W531–W537. 10.1093/nar/gkm328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grishaev A, Steren CA, Wu B et al. (2005) ABACUS, a direct method for protein NMR structure computation via assembly of fragments. Proteins 61:36–43. 10.1002/prot.20457 [DOI] [PubMed] [Google Scholar]
- 34.Tejero R, Snyder D, Mao B et al. (2013) PDBStat: a universal restraint converter and restraint analysis software package for protein NMR. J Biomol NMR 56:337–351. 10.1007/s10858-013-9753-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han B, Liu Y, Ginzinger SW et al. (2011) SHIFTX2: significantly improved protein chemical shift prediction. J Biomol NMR 50:43–57. 10.1007/s10858-011-9478-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu XP, Case DA (2002) Probing multiple effects on N-15, C-13 alpha, C-13 beta, and C-13′ chemical shifts in peptides using density functional theory. Biopolymers 65:408–423. 10.1002/bip.10276 [DOI] [PubMed] [Google Scholar]