

Abstract
The systemic spread of tumor cells is the ultimate cause of the majority of deaths from cancer, yet few successful therapeutic strategies have emerged to specifically target metastasis. Here we discuss recent advances in our understanding of tumor-intrinsic pathways driving metastatic colonization and therapeutic resistance, as well as immune activating strategies to target metastatic disease. We focus on therapeutically exploitable mechanisms, promising strategies in preclinical and clinical development, and emerging areas with potential to become innovative treatments.
Introduction
Cancer metastasis, or the systemic spread and growth of tumor cells throughout the body, is the principal cause of cancer-related deaths1. Despite seventy years of drug development for cancer, survival rates for patients with metastatic disease remain abysmal, with five-year survival rates ranging from 5–30% across solid tumors2. This low survival rate has persisted because clinical results have consistently shown that preclinical therapeutic efficacy does not always translate to clinical benefit for patients suffering from metastatic disease3.
The majority of approved cancer drugs, including tyrosine kinase inhibitors (TKIs), systemic cytotoxic therapies (chemotherapy), and antibody-drug conjugates (ADCs), do not have a durable impact in the setting of established metastatic disease - often due to acquired mechanisms of therapeutic resistance3. On the other hand, the possibility of durable remissions after treatment with immunotherapies has begun a paradigm shift in Stage IV patients who previously received terminal diagnoses. These advances in immunotherapy have ushered in a new era where advanced cancer patients can now hope for long term remissions or chronic management of metastases4, 5. Despite this momentous advance, the majority of metastatic cancers have yet to see therapies with similar efficacy in inducing long-term, durable remissions.
Given this era of rapid progress in novel therapeutic modalities, multiple opportunities are under investigation that may have a transformative impact on cancer death rates. Rather than following the conventional development pipeline of prioritizing therapeutic leads by in vitro cytotoxicity, focusing on the mechanisms of cell plasticity and stress resistance have become key areas of promise for therapies tackling metastasis6. While targeting these interconnected pathways may not show strong efficacy in vitro¸ they are critical to therapeutic resistance as well as overcoming metastatic bottlenecks in vivo. In the tumor-extrinsic context, therapies to relieve immune suppression via reprogramming the local immune milieu may open a new avenues of metastasis immunotherapy.
In this Review we aim to i) Rationalize why cancer metastasis should be a principal consideration in future cancer drug development, ii) Appraise the therapies that have and have not worked in metastatic settings, and iii) Discuss emerging strategies of promise in treating metastatic disease.
Justifying a shift in drug development
The survival of patients diagnosed with localized or regional cancers has increased dramatically in the last decades, yet metrics for metastatic disease have remained constant for many solid tumors7–9. Although cancer death rates have declined by 29% since the mid-1990 peak2, most of this reduction is related to preventative lifestyle changes (smoking cessation10, HPV vaccination11, and Hepatitis C treatment), early screening12 and advances in adjuvant therapy for high-risk patients without clinically detectable metastatic disease13. Whereas these interventions have reduced the prevalence of macro-metastasis, few of them significantly affect survival in patients with established metastases.
The death toll caused by metastatic cancer is difficult to quantify as it manifests across multiple organs and mortality reporting is inconsistent across healthcare systems and cancer types14. For example, in a case study of breast cancer patients, 45% of deaths were attributed directly to metastatic disease that manifested as pulmonary insufficiency, central nervous system failures, hepatic failure and hypercalcemia. An additional 24% of deaths were caused by pneumonia or sepsis subsequent to extensive pulmonary metastases. Only a small minority of the cancer-related deaths could be traced to the primary tumor or drug treatment, and the remainder of deaths were attributed to unrelated causes15. Similar patterns were found in lung cancer, with metastasis as the direct cause of 44% of deaths, and an additional 32% of deaths attributed to pneumonia, sepsis and hemorrhaging subsequent to extensive tumor spread16. Comparable mortality burdens due to metastasis are observed across solid tumors, indicating that preventative and therapeutic approaches to lessen the impact of metastatic cancer must be undertaken to ensure a meaningful change in cancer death rates17.
Two generalized models of metastatic progression have emerged: 1) cancers that metastasize as a function of time and/or tumor size and 2) those that metastasize due to the specific cell of origin and/or mutational lineage18. Cancers with time-dependent metastasis include basal cell carcinoma, which is the most common cancer, yet fewer than 0.55% of cases develop metastatic disease. The low rate of metastasis in basal cell carcinoma reflects the ease of diagnosis and effective surgical interventions19. Pancreatic cancer is thought to be slow-growing and the formation of distant metastasis does not occur until advanced stages20, although cancer cell dissemination may also occur at earlier stages21. Unlike basal cell carcinoma, the difficulty of early diagnosis in pancreatic cancer means that the majority of clinical patients present with metastasis20. Population-wide measures for early detection of time-dependent cancers may present an ideal opportunity to reduce prevalence of metastasis. Common cancers such as breast, colorectal, renal, lung and prostate cancer belong to the early metastasizing group; while some cases might never spread, around 10–15% of breast cancers develop metastasis within three years, and genetic characterization reveals the existence of primary tumors that disseminate early22. This is supported by mouse studies showing that breast cancers can metastasize before the primary tumor is palpable18 or by prostate cancer patients showing molecular heterogeneity across bone metastases23. Mutational profiles of brain and liver metastases across cancer types also support a parallel progression model where metastasis can occur early and distinct metastatic clones convergently evolve24, 25. Organ transplants that later manifested donor-derived metastases in immune-suppressed recipients further suggest that these tumors metastasize early from undetectable primary cancers26. This is supported by the finding that 5–10% of tumor diagnoses are patients with unknown primary tumors presenting with systemic metastases27. For these parallel progression cancers, early detection may be less effective in preventing the development of metastasis or mortality, which has been witnessed by the controversies of implementing population-wide PSA and mammogram testing28, 29. Thus, while early screening and diagnosis may help to improve survival for some cancer types, effective therapies targeting metastatic disease will always be needed in the medical repertoire.
Limitations of targeted therapies
The 2020 NCI Surveillance, Epidemiology and End Results Program (SEER) report highlighted that some of the reduction in cancer mortality could be traced to key advances in targeted therapies and immunotherapies that are highly effective in treating metastatic melanoma and lung cancers (Figure 1)2. In contrast, the majority of the >200 drugs approved to treat cancer have done little to reduce mortality, revealing a glaring disconnect between approval and patient benefit. Improvements in cancer treatment, including surgery, radiation, chemotherapy and targeted therapy, contributed to only 4%−8% of the cancer mortality declines from 1991 to 201130, 31.
Figure 1. 5-Year survival rates of patients diagnosed with select cancer types over the time period from 2000–2017.
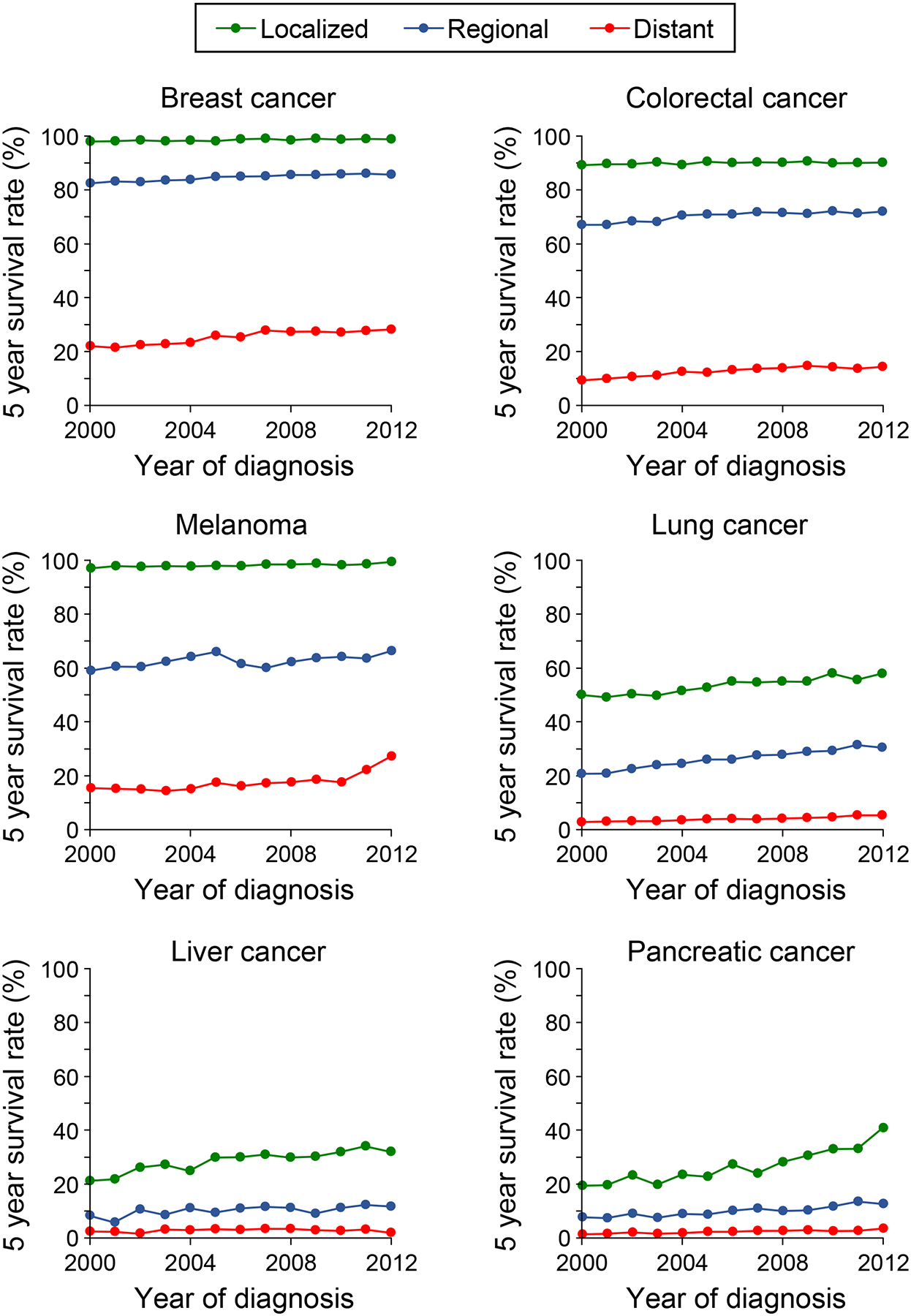
Patients were stratified into three groups according to the extent of invasion or metastasis upon the original diagnosis. The year in the x-axis indicates the year of diagnosis and the first year of the 5-year monitoring period with data last reported in 2017. Data adapted from the NIH-NCI SEER database2.
Much of the mortality reductions derived from therapeutics owe to advances in adjuvant treatment, which prevents metastatic relapse by eliminating disseminated tumor cells. Numerous conventional treatments have shown the ability to extend survival in the adjuvant setting, such as trastuzumab (anti-HER2) and chemotherapy in breast cancer32, apalutamide in prostate cancer33, chemotherapy in lung34 and colon cancers35, and imatinib in gastrointestinal stromal tumors. Recent trials demonstrate adjuvant efficacy for abemaciclib (CDK4/6) in HR+/Her2- breast cancers, osimertinib (EGFR) in early stage EGFR-mutant lung cancer, dabrafenib/trametinib (BRAF/MEK) in BRAF-mutant melanoma, and pembrolizumab (PD-1) in high-risk melanoma36.
While adjuvant therapy to prevent recurrence is currently the most effective strategy to reduce post-diagnosis cancer mortality, approval in this setting requires exceptionally high-powered clinical trials of long duration that are usually not feasible for studies attempting a first approval. A typical clinical development plan requires demonstration of efficacy according to RECIST.1 metrics in the advanced/metastatic tumor setting before justifying the expense of adjuvant trials, thus metastasis-preventing therapeutics are unlikely to ever reach the adjuvant testing space where they would be most effective. Moreover, identifying responsive sub-groups and predictive pharmacodynamics adds to these challenges. For example, adjuvant treatment with bone metastasis-specific resorption inhibitors is only effective in the post-menopausal subgroup of breast cancer patients37. Finally, long-term adjuvant treatment may be limited by chronic toxicity, such as cardiotoxicity from trastuzumab38. Thus, the current regulatory and financial framework for cancer drug development does not facilitate the development of metastasis-specific therapies.
Many treatment limitations in metastasis trace back to the shortcomings of the classical discovery of cancer therapeutics, which requires cytotoxicity in vitro, primary tumor shrinkage in preclinical models, and approval based on RECIST criteria39. This drug development strategy has worked for some exceptionally potent therapies showing dramatic responses in advanced or metastatic settings, such as cabozantinib (VEGFR, AXL, MET) in advanced/metastatic RCC40, endocrine therapies in HR+ breast cancers41 or vemurafenib (BRAF), inducing potent but short-lived responses in metastatic melanoma42. Newly emerging strategies targeting RET, NTRK and NRG1 have also recently demonstrated exceptional responses in NSCLC brain metastases harboring RET fusions (ORR >90%), leading to approval of selpercatinib43. Meanwhile, many other therapies targeting oncogenic drivers and dependencies, such as regorafenib (VEGFR)44 or cetuximab (anti-EGF)45 in metastatic CRC, show modest short-term responses but no impact on 5-year survival. Chemotherapy trials in metastatic breast cancer have similarly been futile; these cancers respond acutely to treatment, but >90% of metastatic cancers will develop resistance to cytotoxics, leading to death within 10 years46.
Antibody-drug conjugates (ADCs) have been enthusiastically pursued as potential one-two punch approaches. Examples of this class include ado-trastuzumab emtansine (anti-HER2), which extends 2-year survival in metastatic patients compared to lapatinib (HER2 inhibitor) and chemotherapy47. Alternately, ADCs targeting non-driver targets such as sacituzumab govitecan-hziy (anti-TROP2 linked SN-38) in metastatic TNBC yield only a modest response with short duration (5.5 months) and considerable toxicity48. Similarly, ADCs against non-driver surface proteins such as folate receptor in ovarian cancer (mirvetuximab soravtansine), DLL3 in lung cancer (rovalpituzumab tesirine) and EpCAM in bladder cancer (oportuzumab monatox) have met varying degrees of failure in phase II/III trials (Table 1)49.
Table 1.
Target, indication, and development stage of therapeutic agents against metastatic cancers
| Target | Cancer type | Latest stage | Survival benefit (i) or best efficacy (ii and iii) | Safety concerns | Citation or Trial | Status (Oncology) | |
|---|---|---|---|---|---|---|---|
| i. Select therapies approved in the metastatic setting | |||||||
| zoledronate | Osteoclasts | Breast | Phase III | Disease-free survival (HR: 0.66) | Osteonecrosis of the jaw | Brufsky et al. 2015 | NDA approval |
| denosumab | RANKL | Breast | Phase III | Disease-free survival (HR: 0.82) | Well-tolerated | NCT00556374 | BLA approval |
| tazemetostat | EZH2 | Epitheloid sarcoma (INI1-) | Phase II * | No OS endpoint | Asthenia/Anemia/Nausea | NCT02601950 | NDA approval |
| sacituzumab govitecan-hziy | TROP2 ADC | Breast (TNBC) | Phase II * | No OS endpoint | Myelotoxicity | NCT01631552 | BLA approval |
| ado-trastuzumab emtansine | HER2 ADC | Breast | Phase III | OS benefit vs Her2 mAb + Chemo (HR: 0.68) | Thrombocytopenia and Hepatotoxicity | NCT00829166 | BLA approval |
| olaparib | PARP | Ovarian | Phase III | OS benefit vs placebo (0.74) | Fatigue, Diarrhea and hypertension | NCT01874353 | NDA approval |
| vismodegib | SMO | Basal cell carcinoma | Phase II * | No OS endpoint | Pneumonia and syncope | NCT00833417 | NDA approval |
| pembrolizumab | PD-1 | Melanoma | Phase III | OS benefit vs Ipilimumab (HR:0.68) | Colitis, diarrhea, fatigue, hepatotox | NCT01866319 | BLA approval |
| ii. Investigational tumor-intrinsic targets | |||||||
| demcizumab | DLL4 (Notch) | Metastatic NSCLC | Phase II | 50% ORR | Severe cardiac toxicity | NCT01189968 | Terminated |
| tarextumab | Notch2/3 (Notch) | Metastatic PDAC | Phase II | Reduced OS | Severe GI toxicity | NCT01647828 | Terminated |
| rovalpituzumab tesirine | DLL3-ADC (Notch) | SCLC | Phase II | Terminated | Significant Grade 3–5 toxicity | NCT02674568 | Terminated |
| ipafricept | Wnt8 (Wnt) | Ovarian | Phase I | Terminated | Significant bone toxicity | NCT02092363 | Terminated |
| vantictumab | FZD1,2,5,7 (Wnt) | Metastatic PDAC | Phase I | Terminated | Significant bone toxicity | NCT02005315 | Terminated |
| fresolimumab | TGF-β | Melanoma and RCC | Phase I | <5% ORR | Reversible keratoacanthomas | NCT00356460 | Terminated |
| galunisertib | ALK5 (TGF-β) | metastatic PDAC | Phase II | DCR: 25% | Neutropenia and Hepatotoxicity | NCT02734160 | Combination trials |
| decitabine | DNMT1–4 | Metastatic prostate | Phase II | PR: 17% | Neutropenia | Thibault et al 1998 | Combination trials |
| tucidinostat | HDAC class 1 | Advanced HR+ breast | Phase III | PFS (HR: 0.75) | Neutropenia | NCT02482753 | Unknown |
| metarrestin | Perinucleolar envelope | Advanced solid tumors | Phase I | TBD | TBD | NCT04222413 | In development |
| nirogacestat | γ-secretase (Notch) | Desmoid | Phase III | TBD | TBD | NCT03785964 | In development |
| iii. Investigational immune activating strategies | |||||||
| NC318 | Siglec-15 | Advanced solid tumors | Phase II | TBD | TBD | NCT03665285 | In development |
| eftilagimod alpha | LAG3 | metastatic breast | Phase I | 50% ORR w/taxol | Asthenia | NCT00349934 | In development |
| tiragolumab | TIGIT | NSCLC | Phase II | TBD | TBD | NCT04294810 | In development |
| epacadostat | IDO1 | Melanoma | Phase III | None | None | NCT02752074 | Terminated |
| MBG453 | TIM3 | Advanced solid tumors | Phase II | TBD | Fatigue | NCT02608268 | In development |
| JNJ-61610588 | VISTA | Advanced solid tumors | Phase I | Terminated | Terminated | NCT02671955 | Terminated |
| Enoblituzumab | B7-H3 | Advanced solid tumors | Phase I | SD reported | Fatigue and infusion reactions | NCT01391143 | In development |
| Hu5F9-G4 | CD47/SIRP1 | Advanced solid tumors | Phase I | None | Myelotoxicity | NCT02216409 | In development |
| IPH5201 | CD39/73 | Advanced solid tumors | Phase I | TBD | TBD | NCT04261075 | In development |
single arm; TBD: to be determined
Perhaps the most notable shortfall for Stage IV patients are the numerous targeted therapies that have been approved despite showing only progression-free survival (PFS) benefits with no changes in overall survival50. Cancer patients receiving these therapies may have tumor shrinkage, but do not live longer or necessarily with a better quality of life. The discrepancies between PFS and OS are evident in two recent Phase III trials of nivolumab (anti PD-1) compared to everolimus (mTOR inhibitor) in advanced RCC or nivolumab compared to docetaxel in NSCLC: both trials showed compelling increases in overall survival for nivolumab with non-significant changes or even reverse trends in PFS51, 52.
Conversely, attempts to specifically target metastatic pathways have met with near-universal failure. At the forefront of this effort are the numerous trials targeting matrix metalloproteases (MMPs)53. Experimental models suggested MMPs were central to cancer metastasis by increasing motility and invasion. However, clinical trials of MMP inhibitors showed either no response or worse outcomes53. While failure has been chalked to an incomplete understanding of the specificity of the MMPs, inadequate clinical trial protocols and unintended effects on the immune system53, the fundamental reason is likely that therapeutics targeting invasion, migration and extravasation may not be effective in treating established metastatic disease. A myriad of evidence shows cancers non-specifically shed metastasis-competent cells54 and patients typically harbor metastasis-competent dormant cancer cells before diagnosis and treatment55, 56.
Targeting phenotypic plasticity and stress resistance
The concept of de-differentiation and phenotypic plasticity has been a central theme of metastasis research since pivotal early studies revealed that less differentiated cancers presented a higher risk of metastasis and therapeutic resistance57. This concept has fluidly evolved throughout time as tumor cell dedifferentiation, epithelial-mesenchymal transition (EMT)/mesenchymal-epithelial transition (MET), the cancer stem cell (CSC) hypothesis, and more recently, plasticity6, 58, 59. Controversies have surrounded these theories with studies claiming that EMT is either essential60 or dispensable for metastasis61, or that CSCs are only important to some cancer types62, are not present at all in others63, or are not yet defined well enough to be understood64.
Whereas plasticity has been recently linked to metastasis59, research has long established that cell state transitions toward stem-like states are central to therapeutic resistance65–70 and have been validated by discoveries of genes such as MTDH, which plays key roles in stemness, EMT-MET, metastasis, stress resistance and therapeutic resistance71, 72. It is therefore unsurprising that all these processes are intrinsically linked (Figure 2). These states describe the same underlying phenomenon of transcriptional and epigenetic plasticity, and while the phenotypic outputs are diverse, targeting the underlying regulators of plasticity has emerged as a critical therapeutic strategy.
Figure 2. Metastatic ability, therapeutic resistance and plasticity are intrinsically linked by genetic, epigenetic and metabolic pathways that offer promising therapeutic nodes for drug development.
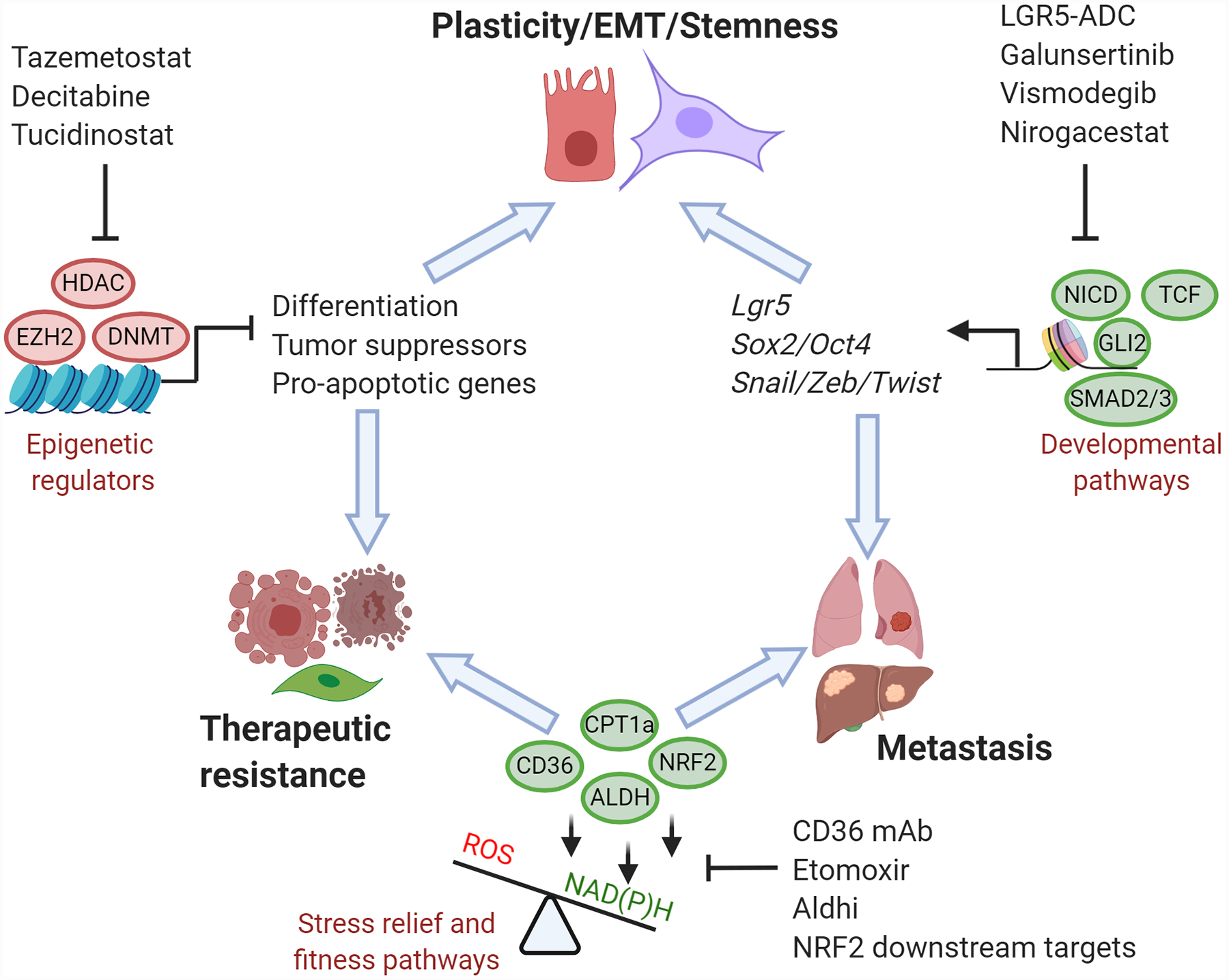
Epigenetic modifications created by DNA methyltransferases (DNMT1–4), histone methyltransferases (EZH2), and histone deacetylases (HDAC) repress tumor suppressors while promoting dedifferentiation to enhance both plasticity and therapeutic resistance. Metastatic ability and therapeutic resistance are linked by metabolic adaptations that enhance the reductive capacity of tumor cells via NAD(P)H generating pathways, such as lipid catabolism and antioxidant pathways. Cellular plasticity, epithelial-mesenchymal transition (EMT), stemness and metastasis are commonly driven by aberrant activities of key developmental pathways (TGF-β, Wnt, Notch and SHH). Therapeutic strategies to target shared mechanisms between these three hallmark characteristics of metastatic cancers may yield durable remissions either as monotherapies or when combined with classic cytotoxic therapies.
Developmental pathways in plasticity and therapy resistance
Several key developmental pathways driving metastasis, often through plasticity and stress resistance, are TGF-β, Wnt, Hedgehog and Notch signaling73. These pathways drive broad transcriptional changes and exhibit extensive cross-talk69, 73. Because of their central importance in human physiology, targeting these pathways has been limited by narrow therapeutic indices, but continued validation of pathway targets may yield success in treating metastatic disease.
Translational efforts have most extensively targeted the Notch signaling pathway due to its well characterized signaling cascade as well as its oncogenic role across tumor types. Notch signaling is initiated by Delta-like (DLL1–4) or Jagged ligands (JAG1–2) binding to the NOTCH1–4 receptors. This causes proteolytic cleavage and nuclear translocation of the Notch intracellular domain (NICD) where it can induce pleiotropic transcriptional activation73. Notch signaling plays a key role in development and its dysregulation is central to plasticity in cancer74. DLL1 is an important mammary stem cell marker75, and forced expression of NOTCH3 expands the CSC side population in pancreatic cancer76. Prostate cancer cells activate Notch to transdifferentiate into osteoblast-like cells that enhance bone metastasis77, and the interaction of Notch and Shh signaling drives docetaxel resistance in prostate cancer cells69. Notch is also critical for tumor-initiating properties downstream of p53- and RB1 deletion in HCC78. The JAG1-NOTCH interaction is a bona-fide driver in both bone and brain metastasis79, with both tumor and stromal cells expressing JAG1 and/or Notch receptors to sustain stemness and chemoresistance79, 80. Treatment of solid tumor PDXs with anti-NOTCH2/3 mAb plus chemotherapy depletes tumor-initiating cells and delays recurrence81. Gamma secretase inhibitors (GSI), which prevent Notch processing upon ligand binding, have similar effects in preventing HCC metastasis and MET82 or in sensitizing prostate CSCs to chemotherapy69. JAG1 is critical to the bone metastasis niche80 and whose therapeutic inhibition synergizes with chemotherapy to slow bone metastasis83.
Two therapeutic approaches targeting the Notch pathway have progressed into the clinic – GSI and mAbs against Notch membrane proteins73. GSI exhibited activity in early trials yet broad gastrointestinal toxicity prevented further development84. Thus other targeted approaches were attempted, including anti-DLL4 (demcizumab), which showed considerable toxicity and low efficacy in pancreatic and lung cancers85, or the NOTCH2/3 antibody tarextumab, which led to significantly worse survival outcomes compared to standard of care (Table 1)86. More recent development of an anti-DLL3 ADC, rovalpituzumab tesirine, was recently terminated in NSCLC due to a lack of survival benefit87. Interestingly, one oral GSI (nirogacestat) has progressed into Phase III trials for desmoid tumors after exhibiting moderate toxicity coupled with very promising results in multiple tumor types88 (Table 1).
The Wnt pathway has similarly been implicated in metastasis across tumor types89. APC mutations in colorectal cancers and amplification of multiple signaling proteins such as CTNNB1, FZD or LRP across solid tumors implicate Wnt signaling in tumor progression89. Wnt also drives the epithelial stem cell state; LGR5, a GPCR involved in Wnt signaling, has become a critical marker for colon crypt stem cells and has been similarly shown to enrich for and functionally induce EpCAM+ colorectal CSCs90. Wnt also maintains plasticity via expression of OCT4 to induce dedifferentiation91.
Wnt signaling has time-dependent and organ-tropic effects during metastasis92. Wnt signaling is induced in the vascular bone metastasis niche by E-selectin, leading to the acquisition of dual epithelial and stem cell properties93. Conversely, the Wnt inhibitor DKK1 has pro-metastatic roles by suppressing Wnt activity in dormant bone metastasis cells in order to evade NK cell-mediated clearance94 or by enhancing osteolysis in late-stage bone metastasis95.
Targeting the Wnt pathway is difficult due to the complexity of the signaling cascade as well as its central role in bone homeostasis. Initial safety data with a decoy receptor sponge for WNT8 (ipafricept)96 or anti-FZD1, 2, 5, 7 (vantictumab) in metastatic cancers identified severe bone toxicities97. Moreover, research has suggested that targeting the canonical Wnt pathway may induce the pro-metastatic non-canonical pathway98. Thus, continued exploration of the Wnt pathway or its downstream effectors is required. One potentially interesting route is via LGR5-ADCs that have shown compelling data across multiple models of tumorigenesis and metastasis. However, LGR5-ADCs have not progressed into the clinic due to a lack of enthusiasm for CSC-targeted treatments and potential toxicity to normal adult stem cell pools87.
The TGF-β pathway comprises multiple ligands, including the bone morphogenic proteins (BMPs), activins, and TGF-β1–3, that, when bound to TGF-β receptors (TGFβRI-II, ALK and BMPR), drives a SMAD-mediated transcriptional program that is highly context-dependent99. This ranges from acting as a tumor suppressor in early cancers100, a mediator of immune suppression across diseases, and a critical driver of mesenchymal traits, plasticity, and EMT in metastasis60, 99. TGF-β signaling is responsible for inducing and maintaining the mesenchymal de-differentiated state in metastatic cancers101, inducing pro-metastatic Jagged1 in bone metastasis83, IL11 in liver metastasis102, ANGPTL4 in lung metastasis103, and suppressing Wnt signaling via DKK1 induction94. TGF-β has been shown to be a critical pro-malignant pathway in late-stage cancers104 or for the supportive stroma for metastasis initiation102.
Preclinical testing of TGF-β inhibitors resulted in compelling evidence supporting the development of this class of therapies. For instance, inhibition of TGF-β in xenograft models prevents bone and lung metastasis while primary tumor growth remains unchanged105. Treatment can further deplete metastasis-initiating cells via stromal targeting102 or slow the vicious cycle of bone metastasis106. In contrast to toxicity associated with Wnt and Notch targeting, TGF-β pathway ablation is tolerated, with cardiac toxicity as the major on-target concern107. However, the multi-faceted role of TGF-β signaling in both tumor promotion and tumor suppression presents problems with clinical advancement, as one notable side effect of TGF-β inhibition is the transient and reversible onset of various neoplasms, mostly keratoacanthomas108.
Finally, Hedgehog signaling (SHH) is also associated with stemness and metastasis109, stromal activation during tumorigenesis110, and therapeutic resistance69. The central transcription factors of Shh signaling are GLI1/2, which show aberrantly high expression in bone metastatic tumor cells where it enhances PTHRP expression to induce osteolytic metastasis111. Multiple studies have explored the importance of SHH in metastasis. For instance, cyclopamine (SMO inhibitor) derivatives inhibited pancreatic cancer metastasis by depleting the ALDH-positive stem cell pool112. Importantly, long-term follow-up studies of vismodegib, an approved SMO inhibitor, yielded exceptional response rates in metastatic basal cell carcinoma, a cancer characterized by loss of function mutations in PTCH1. Vismodegib treatment results in durable responses exceeding one year and median survival nearing three years in metastatic patients113. However, SMO inhibitors resulted in worse patient outcomes across pancreatic cancer trials as SHH inhibition promoted progression via suppressing stromal populations114.
Epigenetic approaches to restrain tumor plasticity
Tumor cell plasticity often manifests as multiple hemi-states that do not co-exist in normal physiology. This is best illustrated by the gradient of epithelial-mesenchymal (E-M) states observed in circulating breast tumor cells that vary over time in response to PI3K targeting or systemic chemotherapy68. This gradient of E-M transitions has been observed across tumor types, and is ascribed to higher metastatic potential115 or increased therapeutic resistance116. Importantly, plasticity presents as phenotypic heterogeneity across the same tumor or metastases, and that this plasticity is controlled by the epigenome117. Large scale genomic studies further support the notion that metastasis is not driven by mutations distinct from the primary tumor118, while in vivo selection models suggest that metastasis may enrich for certain mutational combinations already present in the primary tumor without a requirement for de novo metastatic mutations119. Indeed, reversible switching between E-M states is dependent on concentrations of TGF-β ligand rather than genetic mutations101. This heterogeneity in cell transcription is functional to the metastatic process; epithelial to mesenchymal transition is observed from the core to the invasive edge of the primary tumor and then a reversion is observed in the metastatic site, with both phases deemed critical for metastatic progression93, 120–122.
Therapeutic targeting of the epigenome provides compelling evidence for the feasibility of altering the plastic, dedifferentiated state exhibited during the evolution of metastasis. Early efforts of drug development yielded the cytidine analogs (5-aza(deoxy)cytidine) initially developed as anti-neoplastic nucleoside analogs, however, studies revealed optimal biological activity at sub-cytotoxic doses through inhibition of DNA-methyltransferases123. Early trials in solid tumor metastasis revealed minor activity that was curtailed by myelosuppression. This limitation proved to be a clinical asset as these drugs were eventually approved to treat Myelodysplastic Syndromes123.
Owing to the toxicological limitations of hypomethylating therapies, yet stimulated by promising signs of efficacy, epigenetic therapies have slowly progressed through clinical trials. A key promising therapy is tazemetostat, an inhibitor of EZH2, the H3K27 methyltransferase that defines PRC2 complex activity. EZH2 activity is responsible for maintaining the dedifferentiated state in Ewing tumors124 and is an essential driver of melanoma metastasis through suppression of various tumor suppressors125. EZH2 activity leads to RB1 and p53 suppression in prostate cancers, this results in plasticity and metastasis while EZH2 inhibitors restore sensitivity to anti-androgens126. Tazemetostat was recently approved as the first targeted therapy for metastatic epitheloid sarcoma after demonstrating activity in INI1-negative solid tumors and follicular lymphoma127.
Another promising epigenetic strategy is targeting histone deacetylases (HDAC), as loss of H4K16 acetylation is a common hallmark of cancer128. Pan-HDAC inhibitors such as romidepsin, vorinostat and belinostat are approved for the treatment of cutaneous T-cell lymphomas128. Class-specific HDAC-inhibitors such as entinostat and tucidinostat have demonstrated modest efficacy in endocrine therapy-resistant breast cancer patients by increasing PFS129, 130, and class I HDAC inhibition suppresses cancer stem cell activity and metastasis initiating properties in TNBC131.
Fitness genes and stress resistance
At the simplest reduction, two types of pathways are responsible for cancer progression: 1) Driver genes that cause the cells to proliferate unchecked, and 2) Fitness genes that allow the cells to survive intrinsic and exogenous stressors. Alterations of driver genes are responsible for the competitive advantage of premalignant cells, including activating mutations in oncogenes such as BRAF, or loss-of-function mutations in tumor suppressors such as APC118. Stepwise accumulation of driver mutations leads to the linear progression of premalignant clones to clinically detectable tumors132. Only a small set of driver events are required for primary tumors to form, for example, APC deletion in colorectal cancer or BRAF and KRAS mutations in melanoma and pancreatic cancer, respectively118. TCGA analysis confirms that classic pathways such as PI3K signaling, MAPK signaling, cell cycle control and RTK signaling were some of the most highly mutated pathways133. Analyzing cancer genomes across patients suggests that most cancers are driven by only two to eight mutational events118 (Figure 3). Due to these oncogene-centric discoveries and the impact of these pathways in cancer initiation, most drug development efforts have focused on targeting driver genes despite limited their efficacy in the metastatic setting.
Figure 3. Development of metastasis competency requires additional malignant properties beyond primary tumorigenesis.

Distinct primary tumor types evolve in a stepwise fashion with each additional mutation in a tumor suppressor or oncogene leading to incremental increases in competitive advantage. Key oncogene and suppressor mutations driving common cancers are often distinct from other cancer types and relatively few mutations are required for tumor formation (estimated at 2–8 driver mutations). In comparison, mutations in common oncogenes are not sufficient for metastatic competence, which instead requires the acquisition of multiple cellular programs that must align in order for the cells to withstand the stresses imposed by the metastatic process. Distinct tumor types therefore convergently evolve to acquire these pro-metastatic programs, which in turn offer the opportunity for biomarker-driven tumor type-agnostic therapies in metastatic cancer patients. CIN: Chromosomal instability UPR: Unfolded protein response.
Conversely, metastasis is theorized as a discrete step in tumor evolution that may be independent of specific oncogene pathways or mutations, and instead co-opts cellular traits that mitigate immunologic, genotoxic and therapeutic stressors accreted during tumorigenesis. These adaptations enable a discontinuous jump to metastatic competence and survival in the early metastatic niche132 (Figure 3). The chromosomal instability response is a classic fitness program – metastatic cells exhibiting chromosomal instability activate a noncanonical NF-κB response rather than a lethal interferon response to circumvent cytotoxicity134. Supporting this discovery, screening for molecules that disassemble the tumor-specific perinucleolar compartment yielded Metarrestin, a molecule in early clinical testing to prevent or reverse metastasis135.
It has long been appreciated that oncogenes are not sufficient for metastasis, and furthermore, the genotoxic and metabolic stresses resulting from unchecked proliferation require specific compensatory pathways136. This idea parallels the synthetic lethality space; driver pathways such as ERK inhibition can be compensated via PI3K/mTOR signaling137, whereas BRCA-mutant cancers have few compensatory mechanisms to resist genotoxicity and PARP inhibition results in synthetic lethality. Unfortunately, PARP inhibitors only exhibited incremental improvements in PFS for advanced BRCA1/2-mutant ovarian cancers138 with resistance arising in most patients. Targeting survival adaptations in the metastatic niche could lead to effective treatments that are independent of specific genomic alterations in the cancer. For example, PD-L1 expression on tumor cells is not a driver mutation, but rather a stress resistance mechanism, and its therapeutic inhibition resulted in some of the most durable remissions seen to date (Table 1).
Whereas driver mutations offer an experimentally tractable approach for drug discovery, targeting cancer fitness genes requires the addition of an additional element – stress. By developing multiple stress resistance mechanisms, metastatic cells are able to overcome the oxidative and shear stresses of circulation, immune surveillance in the early metastatic niche, metabolic stresses of advanced metastatic lesions, and therapeutic challenge139 (Figure 3). Importantly, the metastatic process is incredibly inefficient132, suggesting that exploiting any survival liabilities utilized by these cells may have outsized effects on the early metastatic process.
Oxidative stress has emerged as a ubiquitous challenge to metastasizing cells. Whereas early studies assumed that oxidative stress led to cancer development, multiple clinical studies have shown that patients treated with antioxidants fare worse, particularly when treated concurrently with chemotherapy and/or radiotherapy140–142. Mouse studies have shown that antioxidant supplementation, often with N-acetylcysteine (NAC), is responsible for the initiation143, progression, and reduced survival from lung cancers by a mechanism that limits p53 induction144.
A landmark study demonstrated that patient-derived melanomas experience higher levels of oxidative stress during metastasis, that NAC supplementation was sufficient for inducing metastasis, and that NAPDH production via folate pathway enzymes ALDH1L2 or MTHFD2 was also necessary for metastasis145. Further studies have demonstrated that lactate utilization via the MCT1 transporter increases NADPH via the oxidative pentose phosphate pathway to enable melanoma metastasis146. Oxidative stress is also the main underlying cytotoxic mechanism of various chemo- and radiotherapies: NRF2-driven glutathione synthesis enables cisplatin resistance147 and antioxidant supplementation prevents the cytotoxic effects of paclitaxel148. Oxidative stress appears to be largely metastasis-specific; G6PD knockout to inhibit the oxidative pentose pathway does not affect primary tumor initiation or growth in breast, colorectal or lung cancers149. Cystine uptake via SLC7a11 provides cysteine for glutathione biosynthesis in an NADPH-dependent process, and some studies have identified this transporter as an essential component of redox homeostasis in tumors150.
This evidence suggests that oxidative stress is a critical node of synthetic lethality151 and that pro-oxidant therapy should be considered as a high priority drug development target141. Multiple enzymes have been validated as potential targets to induce oxidative stress such as superoxide dismutases, glutathione peroxidases, thioredoxins, catalases and others. These have shown some promise as cancer targets, yet are often redundant152. A key node in oxidative stress resistance is NRF2, the master redox transcriptional regulator whose induction by mutations or oxidative stress increases BACH1-mediated metastasis153 and cisplatin resistance147. NRF2 gain-of-function is observed across many cancers and pharmacologic targeting is currently in proof-of-concept154. Importantly, NRF2 expression is positively correlated to the stage of many cancers, showing the greatest levels in metastatic disease, and is further responsible for enhancing chemoresistance155.
An alternate pro-oxidant therapy is via targeting of metabolic processes that eliminate oxidative stressors and concomitantly generate reduced NAD+ or NADP+; one such strategy is via Aldehyde dehydrogenase (ALDH) activity. ALDH activity predicts tumorigenicity and worse clinical outcomes156 and is broadly used as a marker for stemness, metastatic behavior, and chemoresistance157. Among the 19 ALDH enzymes, ALDH1a3 has been shown as the main driver of ALDH activity158, and subsequent metastatic or chemoresistant traits159 via its role in lipid peroxidation or direct drug detoxification160. Serine biosynthesis via the folate pathway is a potential metastatic target as the ALDH1L1/1L2 enzymes have shown importance in metastasis via the generation of NADPH145, yet the redundancy of cytosolic and mitochondrial serine biosynthesis makes this metabolism difficult to target in tumors161. However, development of drug-like inhibitors of specific ALDH enzymes has not succeeded despite extensive efforts.157
Fatty acid oxidation (FAO) has received comparatively little interest in cancer drug development despite the numerous therapeutics available from cardiometabolic research. Increased FAO is a direct response to the Warburg effect to sustain ATP and NADH levels, particularly in the metastatic setting162. Ablation of FAO via targeting CPT1a with etomoxir prevents CRC metastasis in preclinical models163. Exogenous lipid sourcing is also critical to survival in hypoxic tumors where mTOR activity and TSC2 deficiency creates a synthetic dependence on desaturated lipids in hypoxic environments164. The lipid receptor CD36 was demonstrated as a critical component of metastasis-initiating cells through exogenous palmitate catabolism, and neutralizing antibodies could prevent metastasis165.
In the ever-evolving continuum of stress resistance pathways, ER stress and autophagy have similarly emerged as promising targets in the face of metabolic, genotoxic, therapeutic, and immune stresses. This intrinsic stress response is activated by nutrient limitation or the unfolded protein response, and is dependent on IRE1α, GCN2 and PERK166. A partial state of unfolded protein response was responsible for loss of immune surveillance in pancreatic cancer DTCs, allowing long-term persistence of pre-metastatic cells167. PERK signaling is pro-metastatic across solid tumors, yet clinical application of PERK inhibitors is limited by on-target toxicity. Recent studies have shed light on potential downstream targets of PERK such as CREB3L1168. Desaturated lipids are also critical to survival in hypoxic conditions where their absence causes UPR-mediated apoptosis164. ER stress and autophagy are linked, yet autophagy is not a selective cancer pathway and reports of its directionality of impact as well as therapeutic potential are conflicting169.
Immune checkpoint blockade
Immune checkpoint blocking therapies, chiefly PD-1/PD-L1 and CTLA-4, have become the central platform for treating a wide variety of metastatic cancers from which new therapies will be combined with or compared against. Despite extraordinary responses in a minority of patients, de novo or acquired resistance and non-responsiveness to the panoply of available PD-1 and CTLA4 therapies is the rule rather than exception170. In many carcinomas such as metastatic RCC and HCC, PD-1 inhibitors show only moderate activity, while immunologically suppressed cancers such as breast and pancreatic cancer have shown only marginal responses50. Where the combination of PD-1 inhibitors with various chemotherapies and TKIs has started to show compelling activity in various malignancies171, the discovery of next-generation immune activating therapies will be essential to expand the rate of durable remissions for metastatic patients (Figure 4).
Figure 4. Targeting the immune microenvironment of metastatic cancers.
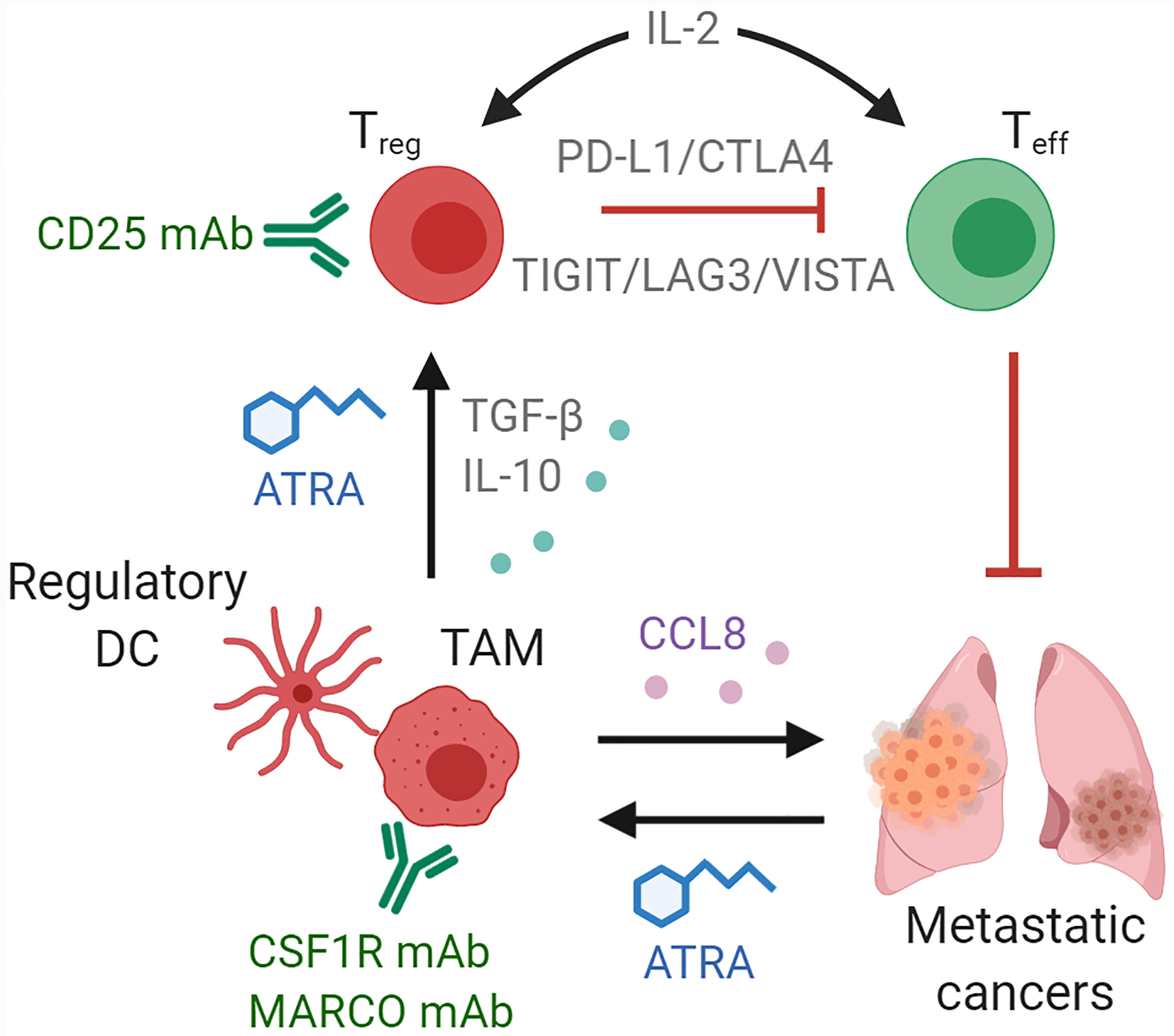
Metastatic lesions enforce immune suppression through the stimulation of key immunoregulatory cell types, including tumor-associated macrophages (TAM), regulatory dendritic cells (DC), regulatory T cells (Treg) and other myeloid-derived suppressive cell populations, that prevent effector T cells (Teff) from recognizing and destroying tumor cells. PD-1/PD-L1 inhibitors have already shown strong efficacy in alleviating Treg-imposed immune suppression at the metastatic site of immunologically-active tumors and additional immune checkpoint blocking therapies are in development (TIGIT, LAG3, VISTA). For immunologically-cold tumors, depletion of immunosuppressive cells through antibody treatment (CSF1R, MARCO, CD25) or inhibition of various differentiation/growth pathways (retinoid and cytokine) may enforce durable remissions of metastatic disease.
The most notable second generation of immune checkpoint receptors include TIM3, LAG3, TIGIT, B7-H3, Siglec-15, and VISTA, all of which have shown promise as anti-metastatic targets in various stages of clinical testing (Table 1)171. Early results with Siglec-15 therapies such as NC318 have shown little promise in expansion cohorts. On the other hand, therapeutics against LAG3, such as eftilagimod alpha, have shown promise in 1st-line NSCLC and metastatic HR+ breast cancer. Antibodies against TIGIT, whose blockade leads to dramatic anti-tumor responses and clearance of chronic viral infections172, have shown the most promise for clinical advancement. Recent results with anti-TIGIT tiragolumab demonstrate compelling efficacy in NSCLC patients, with Phase 3 trials currently underway. Therapies targeting TIM-3, VISTA and B7-H3 are also in early clinical testing (Table 1).
While immune checkpoint blocking therapies have demonstrated the most potent successes in metastatic tumors with high mutational burdens (melanoma, NSCLC) or harboring exogenous tumor viruses (Merkel cell carcinoma, EBV+ gastric cancer)173, less mutationally active tumors may develop alternate means to suppress the anti-tumor immune response. Chief targets under investigation have included immunometabolic targets such as indoleamine 2,3 dioxygenase (IDO1), glutaminase, arginase, and the ectonucleotidases CD39/73. IDO1, glutaminase, and arginase are each implicated in tumor immunosuppression via the depletion of their corresponding amino acid substrates (W, Q, R) on which cytotoxic T cells are thought to be dependent174. Generation of kynurenine via IDO1 or extracellular adenosine via the ectonucelotidases CD39/CD73 are further hypothesized as immune-suppressive factors174, 175. Despite considerable commercial activity in advancing these hypotheses, clinical data has not supported any of these immune-metabolic targets. In retrospect, preclinical data may have amplified the reported specificity of these pathways while clinical experience has highlighted the broad activity of each target across adult physiology176. Other immunometabolic pathways under investigation include the balance between glycolysis and FAO in T cell subsets: Lipid accumulation and resultant lipotoxicity is associated with reduced CD8+ T cell activity177 and increased Treg survival or differentiation via CD36-mediated lipid scavenging178.
Additional mechanisms, including therapies targeting tumor cell phagocytosis by macrophages, antibody-dependent B cell attack, and natural killer cell-mediated destruction, have shown little activity in the clinic despite preclinical support. For example, the CD47-SIRP1 complex is an important “don’t eat me signal” to prevent phagocytosis, yet early results in clinical trials do not support promising efficacy179. Chimeric antigen receptor T cell therapies (CAR-T) have similarly failed to translate from preclinical findings showing the elimination of metastases180 into a clinically viable solid tumor therapy, likely due to the same immunosuppressive features of solid tumors that tamp the natural anti-tumor immune response181.
Depletion of immune-regulatory cells (MDSCs, TAMs, Tregs)
Broad evidence supports the dominance of suppressor cell populations in the tumor microenvironment. Thus, depletion of regulatory T cells (Treg), myeloid-derived suppressor cells (MDSCs) or tumor-associated macrophages (TAMs) may be central to success in harnessing the immune system against metastasis. For example, 11% of patients with metastatic melanoma achieved a complete remission in response to autologous TIL infusion, this doubled to 22% when combined with a lymphodepletion regimen182. In mouse models, experimental depletion of Tregs is sufficient to eliminate both primary tumors and distant metastases183–185, and Treg depletion is further required to enhance anti-tumor response during anti-CTLA-4 treatment186.
Despite the well-established significance of Tregs in cancer immune tolerance, clinically viable strategies to deplete Tregs from tumors or metastatic lesions remain scant. Tregs differentiate from naïve CD4+ T cells via paracrine signaling of immunoregulatory factors from dendritic cells (DCs). Signaling via IL-2, TGF-β and all-trans retinoic acid (ATRA) secreted from these DCs is required for Treg maturation187–189. However, the anti-TGF-β antibody fresolimumab showed no objective response when combined with radiotherapy in advanced breast cancer, and dysfunctional CD8+ T cell signaling was not affected by TGF-β blockade190. Moreover, small molecule inhibitors of TGF-β have shown only minor effects in HCC191. Given the role of TGF-β in suppressing T helper 2 cell-mediated cancer immunity, bifunctional approaches depleting TGF-β signaling toward T cells provided promising preclinical and clinical activity: a TGF-β trap linked to a non-depleting anti-CD4 antibody has shown significant efficacy in mouse models192, and a ligand trap using the TGF-βII receptor domain fused to an anti-PD-1 antibody showed clinically meaningful responses in HPV+ cancers193.
Modulating IL-2 levels or the CD25 receptor on T cells has been broadly tested in both autoimmune disease and the metastatic setting. CD25-depleting antibodies show efficient depletion of Tregs194, yet also target CD4+ Th1 cells and NK cells, thus causing undesired immune suppression. This dual activity led to early abandonment of anti-CD25 approaches in metastatic cancer, however, an alternative approach maximizing the immune-stimulating effects of IL-2 while avoiding its immunosuppressive activity is via synthetic amino acid incorporation to tune receptor affinity. Promising therapies such as Synthorin IL-2 are under investigation in multiple solid tumors195.
Retinoid signaling is largely restricted to immune signaling in adults, as compared to TGF-β196. Retinoid signaling is dominant in inhibiting inflammatory Th17 maturation from naïve T cells while it is instructive in causing Tn maturation to FOXP3+, CTLA-4-expressing Treg cells197. In addition to its role in Treg differentiation, retinoic acid forces differentiation of monocyte populations into immune-suppressive TAMs in sarcoma198. Despite emerging support for inhibiting retinoid signaling to deplete immunosuppressive subsets, inhibitors of the RAR nuclear receptors exhibit broad toxicity and upstream targeting of the ALDH1 enzymes that catalyze the oxidation of retinal into bioactive retinoic acid has not progressed into viable therapies157. Given the ability of exogenous retinoids to suppress dysfunctions caused by T cell hyperactivation, such as atopic dermatitis199, therapeutic approaches to deplete this pathway may prove critical to future cancer immunotherapy.
TAM populations complement Tregs in establishing an immunosuppressive microenvironment that supports metastatic spread. Depletion of TAMs through anti-CSF1R in a p53−/− transgenic breast cancer model dramatically extends survival when combined with platinum therapies by restoring a type I interferon response200. Whereas numerous CSF1/CSF1R inhibitors and mAbs, such as pexidartinib, have failed as monotherapies, rational combinations based on new preclinical research may provide a powerful means to deplete regulatory immune populations and enhance responses to immune checkpoint blockade. Numerous studies have sought additional targetable molecules involved in TAM function, including those that deplete TAM populations or abrogate the downstream effects of TAMs. MARCO was identified as a pattern-recognition receptor whose targeting reprograms TAM populations to an inflammatory phenotype and could be combined with checkpoint inhibitors to block metastasis201. RON kinase was also recently described as a key immunomodulatory pathway via its role in TAM differentiation, and small molecule inhibitors of Ron prevented the outgrowth of micrometastatic colonies86. TAMs enhance regulatory T cell populations via TGF-β and IL-10 secretion, however recent studies show they can also promote the invasiveness of cancer cells via CCL8 stimulation of PITPNM3202. Altered lipid catabolism is furthermore important to TAM polarization via oleate-dependent respiration in myeloid cells203.
Future perspectives on treating metastatic disease
Cancer metastasis is a multifactorial process that relies both on intrinsic pathways that promote plasticity and stress responses as well as extrinsic pathways to establish the immunosuppressive stromal milieu. While this creates far more complex treatment demands compared to the driver-centric approaches to treating primary tumors, convergent evolution of distinct tumor types during the metastatic process offers compelling potential for tumor type-agnostic treatments. Despite the complexity and diversity in the pathways and mechanisms driving metastasis, some common themes emerge pointing to the importance of cellular plasticity and stress-relieving pathways for metastasis competency. Notably, a few candidate targets have been validated as key nodes of regulation and are shared across cancer types. Therefore, metastatic cancers should be approached through a biomarker-driven strategy that validates single agent or combinatorial strategies dependent upon the molecular make-up of individual tumors. As evidenced by recent studies, dual depletion of immune suppressive factors and tumor-intrinsic targeting will be a central paradigm to improve overall survival. Combinational therapies with distinct mechanisms of action will also be critical to preventing metastasis-associated resistance and promoting long term responses.
Relatively little emphasis has been placed on discovery of therapeutics enforcing metastatic regression in the preclinical setting. While this is the most difficult threshold to achieve in preclinical models, these proof-of-concept measures should become a central measure of efficacy to inform early drug development. Additionally, regulatory flexibility will be required to advance these novel classes of therapeutics in a resource-effective manner. For example, predictive pharmacodynamic markers may be used as key secondary outcome measures in addition to 1-year PFS/OS measures in high-need patients. This is particularly important as high-risk patients often cycle through multiple experimental regimens, making overall survival a difficult endpoint.
In the search for new therapeutics, the importance of lifestyle and diagnostic factors in reducing cancer mortality must also be prioritized. The greatest threat to our recent progress in cancer is the upswing in obesity rates becoming the greatest etiologic driver of cancer and metastasis initiation204. Other preventable risks meanwhile remain a major cause of cancer in much of the world. For those cancers with a slow onset of metastasis, improved diagnostic and early detection efforts will substantially improve survival. Ultimately, prevention, early diagnosis and treatment approaches must all be comprehensively optimized to further reduce cancer mortality.
Supplementary Material
Acknowledgments
We thank the members of our laboratory for helpful discussions. We also apologize to the many investigators whose important studies could not be cited directly here owing to space limitations. The work in the authors’ laboratories is supported by grants from the Brewster Foundation, American Cancer Society, Susan G. Komen Foundation, Breast Cancer Research Foundation, the NIH and the U. S. Department of Defense to YK. SG is supported by grants from the NCI, US Department of Defense, Breast Cancer Research Foundation, Hugs for Brady, AHEPA, Val Skinner Foundation and Gertrude Fogarty Trust. Illustrations created with Biorender.com.
Footnotes
Conflicts of interest
ME holds equity interest and a management position in KayoThera, a company developing cancer therapeutics. YK holds equity interest in KayoThera and Firebrand Therapeutics, and is a member of Scientific Advisory Boards of Kayothera, Firebrand Therapeutics, and Cytocares. YK has consulted for Merck, Amgen, Ono Pharma and has previously received funding support from Merck, Amgen, Johnson & Johnson, Janssen, Glycomimetics, and Ono Pharma. SG has consulted for Merck, Roche, Foundation Medicine, Foghorn Therapeutics, Novartis, Silagene, EQRX and Inspirata, has received research funding from M2GEN, and has equity interest in Inspirata and Silagene.
Data Availability
The human cancer mortality data in Figure 1 were derived from the SEER database with 2017 as the most recently annotated 5-year survival date: https://seer.cancer.gov/data/
The raw data are provided as a supplementary file.
References
- 1.Chaffer CL & Weinberg RA A perspective on cancer cell metastasis. Science 331, 1559–1564 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD & Jemal A Cancer statistics, 2020. CA Cancer J Clin 70, 7–30 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Ma B, Wells A & Clark AM The pan-therapeutic resistance of disseminated tumor cells: Role of phenotypic plasticity and the metastatic microenvironment. Semin Cancer Biol 60, 138–147 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei SC, Duffy CR & Allison JP Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 8, 1069–1086 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Litwin MS & Tan HJ The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 317, 2532–2542 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Kang Y & Pantel K Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell 23, 573–581 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun M et al. Age-adjusted incidence, mortality, and survival rates of stage-specific renal cell carcinoma in North America: a trend analysis. Eur Urol 59, 135–141 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Eloubeidi M Temporal trends (1973–1997) in survival of patients with esophageal adenocarcinoma in the United States: a glimmer of hope? The American Journal of Gastroenterology 98, 1627–1633 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Brenner H, Gondos A & Arndt V Recent major progress in long-term cancer patient survival disclosed by modeled period analysis. J Clin Oncol 25, 3274–3280 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Peto R et al. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ 321, 323–329 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lowy DR & Schiller JT Reducing HPV-associated cancer globally. Cancer Prev Res (Phila) 5, 18–23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clouston SAP et al. Fundamental causes of accelerated declines in colorectal cancer mortality: Modeling multiple ways that disadvantage influences mortality risk. Soc Sci Med 187, 1–10 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Davies C et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. The Lancet 381, 805–816 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riihimaki M, Hemminki A, Sundquist J & Hemminki K Patterns of metastasis in colon and rectal cancer. Sci Rep 6, 29765 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hagemeister FB Jr., Buzdar AU, Luna MA & Blumenschein GR Causes of death in breast cancer: a clinicopathologic study. Cancer 46, 162–167 (1980). [DOI] [PubMed] [Google Scholar]
- 16.Nichols L, Saunders R & Knollmann FD Causes of death of patients with lung cancer. Arch Pathol Lab Med 136, 1552–1557 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Talmadge JE & Fidler IJ AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res 70, 5649–5669 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hosseini H et al. Early dissemination seeds metastasis in breast cancer. Nature 540, 552–558 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walling HW, Fosko SW, Geraminejad PA, Whitaker DC & Arpey CJ Aggressive basal cell carcinoma: presentation, pathogenesis, and management. Cancer Metastasis Rev 23, 389–402 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Yachida S et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rhim AD et al. EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weigelt B, Peterse JL & van ‘t Veer LJ Breast cancer metastasis: markers and models. Nat Rev Cancer 5, 591–602 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Roudier MP et al. Phenotypic heterogeneity of end-stage prostate carcinoma metastatic to bone. Human Pathology 34, 646–653 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Brastianos PK et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov 5, 1164–1177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu Z et al. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat Genet 51, 1113–1122 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riethmuller G & Klein CA Early cancer cell dissemination and late metastatic relapse: clinical reflections and biological approaches to the dormancy problem in patients. Semin Cancer Biol 11, 307–311 (2001). [DOI] [PubMed] [Google Scholar]
- 27.van de Wouw AJ, Janssen-Heijnen ML, Coebergh JW & Hillen HF Epidemiology of unknown primary tumours; incidence and population-based survival of 1285 patients in Southeast Netherlands, 1984–1992. Eur J Cancer 38, 409–413 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Jorgensen KJ, Zahl PH & Gotzsche PC Breast cancer mortality in organised mammography screening in Denmark: comparative study. BMJ 340, c1241 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barry MJ Screening for prostate cancer--the controversy that refuses to die. N Engl J Med 360, 1351–1354 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Lichtenberg FR Has Medical Innovation Reduced Cancer Mortality? CESifo Economic Studies 60, 135–177 (2013). [Google Scholar]
- 31.Cutler DM Are we finally winning the war on cancer? J Econ Perspect 22, 3–26 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Piccart-Gebhart MJ et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med 353, 1659–1672 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Smith MR et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N Engl J Med 378, 1408–1418 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Arriagada R et al. Cisplatin-based adjuvant chemotherapy in patients with completely resected non-small-cell lung cancer. N Engl J Med 350, 351–360 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Group QC Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. The Lancet 370, 2020–2029 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Long GV et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N Engl J Med 377, 1813–1823 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Brufsky A & Mathew A Bisphosphonates, bone, and breast cancer recurrence. Lancet 386, 1319–1320 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Guarneri V, Barbieri E, Dieci MV, Piacentini F & Conte P Anti-HER2 neoadjuvant and adjuvant therapies in HER2 positive breast cancer. Cancer Treat Rev 36 Suppl 3, S62–66 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Anderson RL et al. A framework for the development of effective anti-metastatic agents. Nat Rev Clin Oncol 16, 185–204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choueiri TK et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. The Lancet Oncology 17, 917–927 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. The Lancet 365, 1687–1717 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Chapman PB et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364, 2507–2516 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan AC, Itchins M & Khasraw M Brain Metastases in Lung Cancers with Emerging Targetable Fusion Drivers. Int J Mol Sci 21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grothey A et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. The Lancet 381, 303–312 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Van Cutsem E et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360, 1408–1417 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Andreopoulou E & Sparano JA Chemotherapy in Patients with Anthracycline- and Taxane-Pretreated Metastatic Breast Cancer: An Overview. Curr Breast Cancer Rep 5, 42–50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lambert JM & Chari RV Ado-trastuzumab Emtansine (T-DM1): an antibody-drug conjugate (ADC) for HER2-positive breast cancer. J Med Chem 57, 6949–6964 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Bardia A et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N Engl J Med 380, 741–751 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Nagayama A, Ellisen LW, Chabner B & Bardia A Antibody-Drug Conjugates for the Treatment of Solid Tumors: Clinical Experience and Latest Developments. Target Oncol 12, 719–739 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Hess LM, Brnabic A, Mason O, Lee P & Barker S Relationship between Progression-free Survival and Overall Survival in Randomized Clinical Trials of Targeted and Biologic Agents in Oncology. J Cancer 10, 3717–3727 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Motzer RJ et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 373, 1803–1813 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Borghaei H et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med 373, 1627–1639 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vandenbroucke RE & Libert C Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov 13, 904–927 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Lindemann F, Schlimok G, Dirschedl P, Witte J & Riethmuller G Prognostic significance of micrometastatic tumour cells in bone marrow of colorectal cancer patients. Lancet 340, 685–689 (1992). [DOI] [PubMed] [Google Scholar]
- 55.Hüsemann Y et al. Systemic Spread Is an Early Step in Breast Cancer. Cancer Cell 13, 58–68 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Melchior SW et al. Early tumor cell dissemination in patients with clinically localized carcinoma of the prostate. Clin Cancer Res 3, 249–256 (1997). [PubMed] [Google Scholar]
- 57.Davis BW et al. Prognostic significance of tumor grade in clinical trials of adjuvant therapy for breast cancer with axillary lymph node metastasis. Cancer 58, 2662–2670 (1986). [DOI] [PubMed] [Google Scholar]
- 58.Scheel C & Weinberg RA Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol 22, 396–403 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang J et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 21, 341–352 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang J et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Fischer KR et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shackleton M, Quintana E, Fearon ER & Morrison SJ Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell 138, 822–829 (2009). [DOI] [PubMed] [Google Scholar]
- 63.Quintana E et al. Efficient tumour formation by single human melanoma cells. Nature 456, 593–598 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jaggupilli A & Elkord E Significance of CD44 and CD24 as cancer stem cell markers: an enduring ambiguity. Clin Dev Immunol 2012, 708036 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng X et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Auffinger B et al. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ 21, 1119–1131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Todaro M et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 1, 389–402 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Yu M et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Domingo-Domenech J et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell 22, 373–388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kerbel RS, Kobayashi H & Graham CH Intrinsic or acquired drug resistance and metastasis: are they linked phenotypes? J Cell Biochem 56, 37–47 (1994). [DOI] [PubMed] [Google Scholar]
- 71.Hu G et al. MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell 15, 9–20 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wan L et al. MTDH-SND1 interaction is crucial for expansion and activity of tumor-initiating cells in diverse oncogene- and carcinogen-induced mammary tumors. Cancer Cell 26, 92–105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takebe N, Harris PJ, Warren RQ & Ivy SP Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol 8, 97–106 (2011). [DOI] [PubMed] [Google Scholar]
- 74.Takebe N et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol 12, 445–464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chakrabarti R et al. Notch ligand Dll1 mediates cross-talk between mammary stem cells and the macrophageal niche. Science 360 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McAuliffe SM et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc Natl Acad Sci U S A 109, E2939–2948 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zayzafoon M, Abdulkadir SA & McDonald JM Notch signaling and ERK activation are important for the osteomimetic properties of prostate cancer bone metastatic cell lines. J Biol Chem 279, 3662–3670 (2004). [DOI] [PubMed] [Google Scholar]
- 78.Sun L et al. Modelling liver cancer initiation with organoids derived from directly reprogrammed human hepatocytes. Nat Cell Biol 21, 1015–1026 (2019). [DOI] [PubMed] [Google Scholar]
- 79.Xing F et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med 5, 384–396 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sethi N, Dai X, Winter CG & Kang Y Tumor-Derived Jagged1 Promotes Osteolytic Bone Metastasis of Breast Cancer by Engaging Notch Signaling in Bone Cells. Cancer Cell 19, 192–205 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yen WC et al. Targeting Notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clin Cancer Res 21, 2084–2095 (2015). [DOI] [PubMed] [Google Scholar]
- 82.Wu CX et al. Notch Inhibitor PF-03084014 Inhibits Hepatocellular Carcinoma Growth and Metastasis via Suppression of Cancer Stemness due to Reduced Activation of Notch1-Stat3. Mol Cancer Ther 16, 1531–1543 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Zheng H et al. Therapeutic Antibody Targeting Tumor- and Osteoblastic Niche-Derived Jagged1 Sensitizes Bone Metastasis to Chemotherapy. Cancer Cell 32, 731–747 e736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shih Ie M & Wang TL Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer Res 67, 1879–1882 (2007). [DOI] [PubMed] [Google Scholar]
- 85.McKeage MJ et al. Phase IB Trial of the Anti-Cancer Stem Cell DLL4-Binding Agent Demcizumab with Pemetrexed and Carboplatin as First-Line Treatment of Metastatic Non-Squamous NSCLC. Target Oncol 13, 89–98 (2018). [DOI] [PubMed] [Google Scholar]
- 86.Hu ZI et al. A randomized phase II trial of nab-paclitaxel and gemcitabine with tarextumab or placebo in patients with untreated metastatic pancreatic cancer. Cancer medicine 8, 5148–5157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marcucci F, Caserta CA, Romeo E & Rumio C Antibody-Drug Conjugates (ADC) Against Cancer Stem-Like Cells (CSC)-Is There Still Room for Optimism? Front Oncol 9, 167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kummar S et al. Clinical Activity of the gamma-Secretase Inhibitor PF-03084014 in Adults With Desmoid Tumors (Aggressive Fibromatosis). J Clin Oncol 35, 1561–1569 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Anastas JN & Moon RT WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 13, 11–26 (2013). [DOI] [PubMed] [Google Scholar]
- 90.Kemper K et al. Monoclonal antibodies against Lgr5 identify human colorectal cancer stem cells. Stem Cells 30, 2378–2386 (2012). [DOI] [PubMed] [Google Scholar]
- 91.Lee SH et al. Wnt/beta-catenin signalling maintains self-renewal and tumourigenicity of head and neck squamous cell carcinoma stem-like cells by activating Oct4. J Pathol 234, 99–107 (2014). [DOI] [PubMed] [Google Scholar]
- 92.DiMeo TA et al. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res 69, 5364–5373 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Esposito M et al. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat Cell Biol 21, 627–639 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Malladi S et al. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 165, 45–60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhuang X et al. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nat Cell Biol 19, 1274–1285 (2017). [DOI] [PubMed] [Google Scholar]
- 96.Moore KN et al. A phase 1b dose escalation study of ipafricept (OMP54F28) in combination with paclitaxel and carboplatin in patients with recurrent platinum-sensitive ovarian cancer. Gynecol Oncol 154, 294–301 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Davis SL et al. A phase 1b dose escalation study of Wnt pathway inhibitor vantictumab in combination with nab-paclitaxel and gemcitabine in patients with previously untreated metastatic pancreatic cancer. Invest New Drugs 38, 821–830 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ren DN et al. LRP5/6 directly bind to Frizzled and prevent Frizzled-regulated tumour metastasis. Nat Commun 6, 6906 (2015). [DOI] [PubMed] [Google Scholar]
- 99.Massague J TGFbeta signalling in context. Nat Rev Mol Cell Biol 13, 616–630 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Takaku K et al. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell 92, 645–656 (1998). [DOI] [PubMed] [Google Scholar]
- 101.Celia-Terrassa T et al. Hysteresis control of epithelial-mesenchymal transition dynamics conveys a distinct program with enhanced metastatic ability. Nat Commun 9, 5005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Calon A et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 22, 571–584 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Padua D et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 133, 66–77 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kang Y et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549 (2003). [DOI] [PubMed] [Google Scholar]
- 105.Bandyopadhyay A et al. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res 66, 6714–6721 (2006). [DOI] [PubMed] [Google Scholar]
- 106.Esposito M, Guise T & Kang Y The Biology of Bone Metastasis. Cold Spring Harb Perspect Med 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Colak S & Ten Dijke P Targeting TGF-beta Signaling in Cancer. Trends Cancer 3, 56–71 (2017). [DOI] [PubMed] [Google Scholar]
- 108.Morris JC et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One 9, e90353 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ruiz i Altaba A Hedgehog signaling and the Gli code in stem cells, cancer, and metastases. Sci Signal 4, pt9 (2011). [DOI] [PubMed] [Google Scholar]
- 110.Yauch RL et al. A paracrine requirement for hedgehog signalling in cancer. Nature 455, 406–410 (2008). [DOI] [PubMed] [Google Scholar]
- 111.Sterling JA et al. The hedgehog signaling molecule Gli2 induces parathyroid hormone-related peptide expression and osteolysis in metastatic human breast cancer cells. Cancer Res 66, 7548–7553 (2006). [DOI] [PubMed] [Google Scholar]
- 112.Feldmann G et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther 7, 2725–2735 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sekulic A et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: final update of the pivotal ERIVANCE BCC study. BMC Cancer 17, 332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee JJ et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U S A 111, E3091–3100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pastushenko I et al. Identification of the tumour transition states occurring during EMT. Nature 556, 463–468 (2018). [DOI] [PubMed] [Google Scholar]
- 116.Su Y et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc Natl Acad Sci U S A 114, 13679–13684 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yuan S et al. Global Regulation of the Histone Mark H3K36me2 Underlies Epithelial Plasticity and Metastatic Progression. Cancer Discov 10, 854–871 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vogelstein B et al. Cancer genome landscapes. Science 339, 1546–1558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jacob LS et al. Metastatic Competence Can Emerge with Selection of Preexisting Oncogenic Alleles without a Need of New Mutations. Cancer Res 75, 3713–3719 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brabletz T et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A 98, 10356–10361 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ocana OH et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 22, 709–724 (2012). [DOI] [PubMed] [Google Scholar]
- 122.Tsai JH, Donaher JL, Murphy DA, Chau S & Yang J Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22, 725–736 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jabbour E, Issa JP, Garcia-Manero G & Kantarjian H Evolution of decitabine development: accomplishments, ongoing investigations, and future strategies. Cancer 112, 2341–2351 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Richter GH et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci U S A 106, 5324–5329 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zingg D et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun 6, 6051 (2015). [DOI] [PubMed] [Google Scholar]
- 126.Ku SY et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Italiano A et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19, 649–659 (2018). [DOI] [PubMed] [Google Scholar]
- 128.Fraga MF et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37, 391–400 (2005). [DOI] [PubMed] [Google Scholar]
- 129.Yardley DA et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol 31, 2128–2135 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jiang Z et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Oncology 20, 806–815 (2019). [DOI] [PubMed] [Google Scholar]
- 131.Schech A, Kazi A, Yu S, Shah P & Sabnis G Histone Deacetylase Inhibitor Entinostat Inhibits Tumor-Initiating Cells in Triple-Negative Breast Cancer Cells. Mol Cancer Ther 14, 1848–1857 (2015). [DOI] [PubMed] [Google Scholar]
- 132.Vanharanta S & Massague J Origins of metastatic traits. Cancer Cell 24, 410–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bailey MH et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385 e318 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bakhoum SF et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Frankowski KJ et al. Metarrestin, a perinucleolar compartment inhibitor, effectively suppresses metastasis. Sci Transl Med 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gupta GP & Massague J Cancer metastasis: building a framework. Cell 127, 679–695 (2006). [DOI] [PubMed] [Google Scholar]
- 137.Shimizu T et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res 18, 2316–2325 (2012). [DOI] [PubMed] [Google Scholar]
- 138.Banerjee SN & Lord CJ First-line PARP inhibition in ovarian cancer - standard of care for all? Nat Rev Clin Oncol 17, 136–137 (2020). [DOI] [PubMed] [Google Scholar]
- 139.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 140.Jung AY et al. Antioxidant supplementation and breast cancer prognosis in postmenopausal women undergoing chemotherapy and radiation therapy. Am J Clin Nutr 109, 69–78 (2019). [DOI] [PubMed] [Google Scholar]
- 141.Gill JG, Piskounova E & Morrison SJ Cancer, Oxidative Stress, and Metastasis. Cold Spring Harb Symp Quant Biol 81, 163–175 (2016). [DOI] [PubMed] [Google Scholar]
- 142.Ambrosone CB et al. Dietary Supplement Use During Chemotherapy and Survival Outcomes of Patients With Breast Cancer Enrolled in a Cooperative Group Clinical Trial (SWOG S0221). J Clin Oncol 38, 804–814 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Breau M et al. The antioxidant N-acetylcysteine protects from lung emphysema but induces lung adenocarcinoma in mice. JCI Insight 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sayin VI et al. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med 6, 221ra215 (2014). [DOI] [PubMed] [Google Scholar]
- 145.Piskounova E et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tasdogan A et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577, 115–120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Silva MM, Rocha CRR, Kinker GS, Pelegrini AL & Menck CFM The balance between NRF2/GSH antioxidant mediated pathway and DNA repair modulates cisplatin resistance in lung cancer cells. Sci Rep 9, 17639 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ramanathan B et al. Resistance to paclitaxel is proportional to cellular total antioxidant capacity. Cancer Res 65, 8455–8460 (2005). [DOI] [PubMed] [Google Scholar]
- 149.Ghergurovich JM et al. Glucose-6-phosphate dehydrogenase is not essential for K-Ras-driven tumor growth or metastasis. Cancer Res (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhang Y et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol 20, 1181–1192 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pani G, Galeotti T & Chiarugi P Metastasis: cancer cell’s escape from oxidative stress. Cancer Metastasis Rev 29, 351–378 (2010). [DOI] [PubMed] [Google Scholar]
- 152.Harris IS et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222 (2015). [DOI] [PubMed] [Google Scholar]
- 153.Wiel C et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 178, 330–345 e322 (2019). [DOI] [PubMed] [Google Scholar]
- 154.Cuadrado A et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov 18, 295–317 (2019). [DOI] [PubMed] [Google Scholar]
- 155.Wang XJ et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29, 1235–1243 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ginestier C et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Koppaka V et al. Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol Rev 64, 520–539 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Marcato P et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells 29, 32–45 (2011). [DOI] [PubMed] [Google Scholar]
- 159.Luo Y et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells 30, 2100–2113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wu W et al. Lipid Peroxidation Plays an Important Role in Chemotherapeutic Effects of Temozolomide and the Development of Therapy Resistance in Human Glioblastoma. Transl Oncol 13, 100748 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ducker GS et al. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab 24, 640–641 (2016). [DOI] [PubMed] [Google Scholar]
- 162.van Weverwijk A et al. Metabolic adaptability in metastatic breast cancer by AKR1B10-dependent balancing of glycolysis and fatty acid oxidation. Nat Commun 10, 2698 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wang YN et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 37, 6025–6040 (2018). [DOI] [PubMed] [Google Scholar]
- 164.Young RM et al. Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev 27, 1115–1131 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pascual G et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45 (2017). [DOI] [PubMed] [Google Scholar]
- 166.Staschke KA & Wek RC Adapting to cell stress from inside and out. Nat Cell Biol 21, 799–800 (2019). [DOI] [PubMed] [Google Scholar]
- 167.Pommier A et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 360 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Feng YX et al. Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat Commun 8, 1079 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Thorburn A, Thamm DH & Gustafson DL Autophagy and cancer therapy. Mol Pharmacol 85, 830–838 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kalbasi A & Ribas A Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol 20, 25–39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Qin S et al. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer 18, 155 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Johnston RJ et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 26, 923–937 (2014). [DOI] [PubMed] [Google Scholar]
- 173.Ganesan S & Mehnert J Biomarkers for Response to Immune Checkpoint Blockade. Annual Review of Cancer Biology Vol. 4, 331–351 (2020). [Google Scholar]
- 174.Ramapriyan R et al. Altered cancer metabolism in mechanisms of immunotherapy resistance. Pharmacol Ther 195, 162–171 (2019). [DOI] [PubMed] [Google Scholar]
- 175.Allard D, Chrobak P, Allard B, Messaoudi N & Stagg J Targeting the CD73-adenosine axis in immuno-oncology. Immunol Lett 205, 31–39 (2019). [DOI] [PubMed] [Google Scholar]
- 176.Gunther J, Dabritz J & Wirthgen E Limitations and Off-Target Effects of Tryptophan-Related IDO Inhibitors in Cancer Treatment. Front Immunol 10, 1801 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Manzo T et al. Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J Exp Med 217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wang H et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol 21, 298–308 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sikic BI et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J Clin Oncol 37, 946–953 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Priceman SJ et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2(+) Breast Cancer Metastasis to the Brain. Clin Cancer Res 24, 95–105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Newick K, O’Brien S, Moon E & Albelda SM CAR T Cell Therapy for Solid Tumors. Annu Rev Med 68, 139–152 (2017). [DOI] [PubMed] [Google Scholar]
- 182.Rosenberg SA et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17, 4550–4557 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chen L et al. Rejection of metastatic 4T1 breast cancer by attenuation of Treg cells in combination with immune stimulation. Mol Ther 15, 2194–2202 (2007). [DOI] [PubMed] [Google Scholar]
- 184.Jang JE et al. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell reports 20, 558–571 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Marabelle A et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest 123, 2447–2463 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Peggs KS, Quezada SA, Chambers CA, Korman AJ & Allison JP Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med 206, 1717–1725 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Maldonado RA & von Andrian UH How tolerogenic dendritic cells induce regulatory T cells. Advances in immunology 108, 111–165 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mucida D, et al. Reciprocal TH17 ad regulatory T cell differnetiation mediated by retinoic acid. Science 317(5835): p. 1958–68 (2007). [DOI] [PubMed] [Google Scholar]
- 189.Galvin KC et al. Blocking retinoic acid receptor-alpha enhances the efficacy of a dendritic cell vaccine against tumours by suppressing the induction of regulatory T cells. Cancer immunology, immunotherapy : CII 62, 1273–1282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Formenti SC et al. Focal Irradiation and Systemic TGFbeta Blockade in Metastatic Breast Cancer. Clin Cancer Res 24, 2493–2504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kelley RK et al. A Phase 2 Study of Galunisertib (TGF-beta1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin Transl Gastroenterol 10, e00056 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Li S et al. Cancer immunotherapy via targeted TGF-beta signalling blockade in TH cells. Nature 587, 121–125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lan Y et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Sci Transl Med 10 (2018). [DOI] [PubMed] [Google Scholar]
- 194.Rech AJ & Vonderheide RH Clinical use of anti-CD25 antibody daclizumab to enhance immune responses to tumor antigen vaccination by targeting regulatory T cells. Ann N Y Acad Sci 1174, 99–106 (2009). [DOI] [PubMed] [Google Scholar]
- 195.Dien VT, Morris SE, Karadeema RJ & Romesberg FE Expansion of the genetic code via expansion of the genetic alphabet. Curr Opin Chem Biol 46, 196–202 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hall JA, Grainger JR, Spencer SP & Belkaid Y The role of retinoic acid in tolerance and immunity. Immunity 35, 13–22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Mucida D et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317, 256–260 (2007). [DOI] [PubMed] [Google Scholar]
- 198.Devalaraja S et al. Tumor-Derived Retinoic Acid Regulates Intratumoral Monocyte Differentiation to Promote Immune Suppression. Cell (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ruzicka T et al. Oral alitretinoin (9-cis-retinoic acid) therapy for chronic hand dermatitis in patients refractory to standard therapy: results of a randomized, double-blind, placebo-controlled, multicenter trial. Arch Dermatol 140, 1453–1459 (2004). [DOI] [PubMed] [Google Scholar]
- 200.Salvagno C et al. Therapeutic targeting of macrophages enhances chemotherapy efficacy by unleashing type I interferon response. Nat Cell Biol 21, 511–521 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Georgoudaki AM et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep 15, 2000–2011 (2016). [DOI] [PubMed] [Google Scholar]
- 202.Chen J et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 19, 541–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wu H et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol Med 11, e10698 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Quail DF et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat Cell Biol 19, 974–987 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.