

Tiny silicon microchip–based high-throughput hanging drop arrays allow drug screening and geometric control.
Abstract
Existing three-dimensional (3D) culture techniques are limited by trade-offs between throughput, capacity for high-resolution imaging in living state, and geometric control. Here, we introduce a modular microscale hanging drop culture where simple design elements allow high replicates for drug screening, direct on-chip real-time or high-resolution confocal microscopy, and geometric control in 3D. Thousands of spheroids can be formed on our microchip in a single step and without any selective pressure from specific matrices. Microchip cultures from human LN229 glioblastoma and patient-derived mouse xenograft cells retained genomic alterations of originating tumors based on mate pair sequencing. We measured response to drugs over time with real-time microscopy on-chip. Last, by engineering droplets to form predetermined geometric shapes, we were able to manipulate the geometry of cultured cell masses. These outcomes can enable broad applications in advancing personalized medicine for cancer and drug discovery, tissue engineering, and stem cell research.
INTRODUCTION
Cell culture has been extensively applied in drug discovery, tissue engineering, and stem cell research (1–6). However, conventional two-dimensional (2D) monolayer cultures do not replicate the important features of 3D conditions, and much work has been done to elucidate the differences in cellular morphology, behavior, and molecular signaling between the classic cell monolayer approaches and analogous 3D cultures (1, 2). 3D culture systems offer the unique opportunity to grow cells such that the resulting tissues acquire morphological and cellular characteristics relevant to in vivo conditions (7–15). Although several 3D culture techniques exist that deploy protein-based or synthetic polymer–based gel environments (7), rotation-based bioreactors (16), magnetic levitation (17), and hanging drop techniques (18, 19), their broad practical application has been limited because of several reasons (1). Matrix and hydrogel-based techniques are often used to generate organotypic cultures from isolated stem cells or tumor-initiating cells. These organotypic cultures have increasingly become an invaluable tool in cancer biology as these matrices allow more control over the cell environment and signaling during culture. However, it should be noted that they also require cell proliferation and matrix engineering, as the scaffolds can introduce unpredictable cell-polymer interactions that influence and bias downstream applications of the organoids (1). Aggregation techniques such as magnetic levitation using nanoparticles to cause spheroid formation have demonstrated a limited number of spheroids and require cells to be pretreated with magnetic beads at high concentrations adding complexity and potential toxicity (7).
A special consideration is that all proliferative models of drug testing, including 2D, 3D organoids, and patient-derived xenograft (PDX) models, are subject to selective pressure and tumor evolution (20, 21). Because the tumor microenvironment has been increasingly recognized as a key contributor to cancer progression and resistance to therapy, removing selective pressure by optimizing culture conditions to maintain the tumor microenvironment are essential for accurate prediction of response to treatment. Because of its rapid reaggregation of dispersed tumor cells, ideally suited for this is the hanging drop technique where inverted 20- to 40-μl droplets containing cells held by capillary forces form tumor spheroids, and commercial hanging drop plates in 96- and 384-well formats are now available (18, 22, 23). However, the current technology for performing hanging drop cultures is very laborious and low throughput and requires the disruption of culture conditions and transfer of formed spheroids into a secondary plate for end point analysis. The current techniques are not compatible with direct live microscopy-based characterizations and also suffer from elevated osmolarity caused by evaporation of media from the droplets (7), which also limits the time for growth and analysis.
To address these challenges, we introduce a modular microchip hanging drop platform that can be used for drug screening, live imaging, and geometric control applications. In our platform, the individual droplets with cells are held by capillary forces in nanoliter-sized wells. For drug screening applications, the miniaturized and optimized culture conditions allow one cell seeding step to produce an array of hundreds to thousands of uniform tumor spheroids with diameter in the hundreds of micrometers (and, hence, we use the term “microcancers” or “microcancer spheroids”).We have shown that microcancer size and volume can be controlled and easily varied through both chip design and cell seeding densities. Formed spheroids were characterized in real time through upright fluorescence microscopy for tumor spheroid formation, cell death, and other processes. We validate this technique with LN229 human glioblastoma (GBM) cells and PDX glioblastoma cells, also demonstrating compatibility with automated live cell imaging showing formation of the microcancers. We also show capability to perform high-resolution confocal analysis to study the expression of protein markers directly on-chip without the need for extraction of the tumor spheroids. To demonstrate that our platform does not bias tumor evolution by putting selective pressure, we show, through the use of mate pair sequencing, a complete overlap in chromosomal rearrangement (CR) events between the PDX and the corresponding microcancers grown in our platform. As one of the main applications of the technology, we show direct drug testing on LN229 and PDX GBM tumor spheroids formed in our platform as characterized by real-time optical microscopy. Our nanodroplet culture model allows simultaneous testing of hundreds to thousands of 3D microcancer cultures in real time where each microcancer captures the salient characteristics of the tumor. Because of the small size of the platform, smaller biopsies can be tested with many drugs. Drug testing can be done in days, and the system also allows for sequential (staggered time point) drug testing. Therefore, this system makes it possible to rapidly test many drugs in real time even from clinical scenarios where only small biopsies are available.
Furthermore, geometric control in a 3D culture platform can be advantageous because tissue structures including size and shape facilitate the physiological function of the tissue by allowing cells and their microenvironment to exchange chemical, electrical, and mechanical cues (3, 4, 24–33). Engineering of tissues with tuneable morphologies has been studied in 3D culture techniques such as bioprinting and micropatterning, but these techniques are known to cause high shear stress on cells during seeding (3, 34). Guided assembly using magnetic levitation and nanoparticles has also been used to demonstrate the formation of annular ring structures using human GBM cells (17). However, the formed structures in this platform are unable to retain shape during cellular compaction, and pretreatment with magnetic beads at high concentrations adds complexity and potential toxicity (7). In this paper using human GBM cells, we demonstrate the use of hanging drop culture for geometric control of 3D cultured structures. Because our platform uses a microchip with etched channels, we fabricated a chip with circular, square, and triangular annular wells, which allowed the media droplet shape to conform to the shape of channel cavity because of capillary forces. Using this approach, we were able to generate dense and continuous annular circle, square, and triangle 3D shapes ranging in sizes from a few hundred micrometers to several millimeters. We believe that our platform will open up applications of hanging drop platform where different cell types can be cocultured in 3D on-chip to generate self-assembled and self-organized tissues with the salient in vivo–like features naturally formed via cell-cell interactions.
RESULTS
Experimental design
We fabricated several microchip designs containing an array of silicon microwells of varying dimensions etched to a depth of 120 μm. The silicon surface was oxidized after the formation of the wells. Figure 1A shows the platform design and schematic of on-chip cell seeding protocol. The entire chip is less than the size of a quarter, and Fig. 1B shows two chip designs, one with an array of 900 microwells with a constant well size of 300 μm (side length of well) and the other with a well size gradient in which the well side lengths were varied from 100 to 500 μm. This design can easily be scaled to a larger size and a higher number of individual wells. The spacing between each well was kept at 20 μm. Each well in this array will downstream act as an isolated incubation chamber for 3D hanging drop cell culture. A detachable polydimethylsiloxane (PDMS) well (or well array) was assembled on top of the silicon chip to select a subset of the silicon wells to customize the number of microcancers required. The temporary adhesion between the PDMS well and the chip surface forms a removable reservoir over the chip where the cell solution can be loaded and centrifuged. The cell seeding protocol began by first making the surface of the chips hydrophilic by exposing to oxygen plasma for 3 min at 300 W (Fig. 1A, 1). This step serves two goals. First, the hydrophilicity helps in homogeneous cell seeding by allowing easy access of media and cell solution inside the wells, and, second, it makes the chip surface and environment sterile for downstream culture. After making the chip hydrophilic, the cell solution is loaded in the PDMS reservoir, and the cells centrifuged at 200g for 2 min. The centrifugation step brings down the cells into the underlying wells and loads the entire array in a single step (Fig. 1A, 2 and 3). Once the cell loading is complete, the PDMS reservoir is removed and the individual microwells are partitioned using mineral oil and shear force from air or oil, as described in our previous work (35) (Fig. 1A, 4). This step removes well-well connections and forms individual droplets in wells. Last, the chip was inverted in mineral oil in the cell culture chamber to create inverted hanging drops, thus increasing cell-cell interactions and resulting in the tumor spheroid formation (Fig. 1A, 5 and 6). The volume of each well is kept constant by the mineral oil layer that serves as an evaporation barrier allowing culture for an extended period. No additional surface coatings or gel formulations are needed, which might put selective pressure on tumor evolution.
Fig. 1. On-chip cell culture schematic and spheroid characterization in a well size gradient chip.
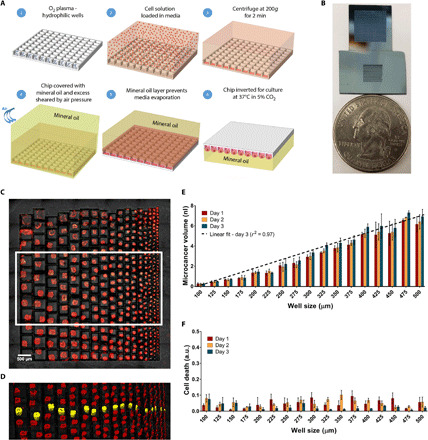
(A) Overall process flow schematic. (B) Optical image of two microarray chips beside a quarter. The first chip (top) is an array of 900 microwells of 300-μm well size. The second chip (bottom) includes a gradient of well sizes for spheroid formation. (C) Fluorescence image of microcancer spheroid formation of gradient sizes seen 1 day after cell seeding. Easy optical characterization with an upright microscope is shown. Scale bar, 500 μm. (D) Volumetric mapping of spheroids in the white box seen in (C). One of each microcancer size is highlighted in yellow. Scale bar, 500 μm. (E) Bar graph of microcancer volumes for spheroids in each well size variation for days 1 to 3 after cell seeding. The data encompasses volumes of three microcancers (n = 3) for each condition. Linear fit of day 3 mean volumes has an r-squared value of 0.97, indicating a linear relationship and control between well and spheroid sizes. (F) Bar graph of cell death (green volume normalized to red volume) for spheroids in each well size variation for days 1 to 3 after cell seeding. Bar graphs and error bars indicate mean and SD, respectively. a.u., arbitrary units.
Cell culture on chip and microcancer size control
To evaluate our platform’s ability to form and culture microcancer spheroids on chip of different sizes, we first seeded LN229 cells on the well size gradient chip and examined the survival profiles of the formed microcancers over time. A real-time imaging compatible cell death indicator green dye (CellTox, Promega) and a cell membrane staining red dye (PKH, Sigma-Aldrich) were added to the media to quantify cell death in the microcancer tumor volume with time. Tile and z-stack imaging was performed for each day of culture to volumetrically map the green and red volumes within a microwell. Volumetric mapping of the green and red dyes was performed on Imaris software, and final individual well tracking was performed using MATLAB. The details of analyses are in Materials and Methods and fig. S1. The cell death in each well was normalized to the microcancer volume in that same well to account for well-to-well variations in cell numbers. Figure 1C shows a fluorescence microscope image of a high-throughput culture of LN229 human glioblastoma microcancers after 1 day of culture after cell seeding on the well size gradient chip. Loading a solution of cell density at 1.87 million cells/ml resulted in a range of microcancer sizes from 7.6 nl for 500-μm wells to 0.36 nl for 100-μm wells. Details on cell seeding can be found in Materials and Methods. Figure 1D shows the volumetric mapping of spheroids seen in the white box in Fig. 1C. One of each microcancer size has been highlighted in yellow. Three microcancers of each size were analyzed for microcancer volume and cell death. In Fig. 1E, we see a linear relationship of well size to microcancer volume with an r2 value of 0.97. In addition, in current 3D culture approaches, variations in spheroid sizes and morphology leads to challenges in creating standard culture and assay protocols, as well as in analyzing data (36). Our data show that the size of spheroids can be well controlled. Over the 3 days of culture, the 100-μm spheroids (smallest size) remain relatively constant in size with an average volume increase of about 1.5%. In contrast, the 500-μm spheroids show an average volume increase of 12.5% over 3 days of culture. As seen in Fig. 1F, cell death remains below 13% over the 3 days of culture independent of the spheroid size. Being able to control and test a range of spheroid sizes in a high-throughput manner will be useful in cancer drug screening and other biological applications (36–38). Microcancer size can also be controlled in our platform for a constant well size by varying the seeding cell density. The box-and-whiskers plot (n = 12) of the microcancer volume and cell death for varying cell numbers per well over 3 days of culture for LN229 human glioblastoma cells cultured in 300-μm well array is shown in fig. S2. We found that 300 cells per well did not seem to aggregate as well as the other higher cell numbers per well, evidenced by a higher range of volumes on day 1. Note that for 1000 cells per well, the microcancer spheroid volume is ~4 nl on day 1, which is still less than 50% of the microwell volume (10.8 nl), leaving additional volume for further cell growth.
Next, to demonstrate the ability to culture primary tumor cells on our platform, we loaded 300-μm well array with cells harvested from GBM8 PDXs implanted in mouse brain (Fig. 2, A to D). The harvest cells from the xenograft represented the cellular diversity of the PDX brain tumor (39). Figure 2 (E and F) shows the box-and-whiskers plot (n = 12) of GBM8 microcancer volume and cell death for varying PDX cell numbers cultured on chip for 3 days. For PDX cells, 300 cells per well formed dense spheroids as opposed to their LN229 cell line counterpart. Also, the PDX spheroids had lower day 1 volumes for 700 (~1.5 nl) and 1000 cells (~2 nl) per well compared to their LN229 cell line counterparts (3.5 and 3.75 nl, respectively). These differences likely reflect differential expression of adhesion receptors and increased compaction of cells in the PDX tumor. It is important to note that our technique can optically track individual wells for changes in cell death or microcancer volume over time and can automatically normalize for any variations in cell loading process. Figures S3 and S4 show individual traces of volume and cell death versus time for LN229 and PDX microcancer spheroids, respectively.
Fig. 2. On-chip PDX culture.
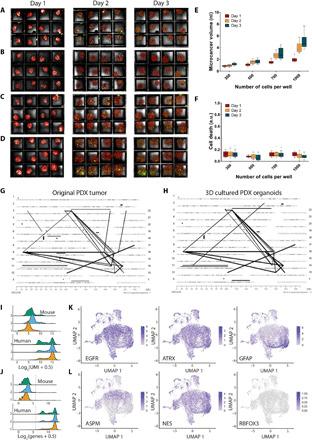
(A to D) Maximum projections of formed microcancers in 12-well culture for days 1 to 3 after cell seeding of 300 (A), 500 (B), 700 (C), and 1000 cells per well (D). Each well has a side length of 300 μm (for scale). Box-and-whiskers plot of microcancer volumes (E) and cell death (green volume normalized to red volume) (F) for each cell variation type for days 1 to 3 after cell seeding (n = 12). Mate pair sequencing demonstrates high degree of relatedness and similar genomic abnormalities in original PDX (G) and 3D cultures (H) grown in our array platform. Assessment of cellular identity and diversity of microcancers (n = 7653) was evaluated through single-cell RNA sequencing. (I) Distributions of UMI counts in GEM cells. Summed UMI counts from mouse genes (top) and human genes (bottom). (J) Distributions of numbers of genes with >1 UMI in each GEM cell. Total number of mouse genes with >1 UMI (top) and human genes with >1 UMI. Line shows threshold of 64 genes detected (6 on log2 scale). Each dot represents the transcriptome of a single cell from dissociated microcancers. (K) and (L) show the single-cell gene expression of key markers for gliomas and glioma stem cells. Scale bars represent z-test–normalized gene counts.
Harvesting 3D cultures from chip for genomic analysis
Because our system is in an open format, we can also readily harvest the formed 3D cultures from the chip. To do this, we first submerged the chip in 1 ml of media to remove the residual oil layer from the top of the array in a 12-well culture. Then, the 3D cultures are removed directly by pipetting and aspirating the volume from the chip.
Mate pair sequencing demonstrates high degree of relatedness between the PDX and 3D cultures
To further explore the biological attributes afforded by our platform and to show that our culture platform does not bias tumor evolution by putting selective pressure, we harvested PDX microcancers after 3 days of culture on chip and performed genomic analysis on microcancers and the PDX source for comparison. Figure 2 (G and H) shows the genome plots of data merged from three independent 3D experiments from our platform. Each line on the plot represents a CR event supported by at least six sequencing fragments chosen as threshold. These plots show high degree of similarity. Significantly, over 60% (38 of 63) of all CRs were shared between the two samples. However, it should be noted that for the CR events that appeared to be unique to one sample, we could find sequencing fragments corresponding to the same event in the 3D cultures but at lower than threshold levels. Hence, we can conclude that there was a complete overlap in CR events between the two samples, suggesting high degree of relatedness between the PDX and the 3D cultures.
Single-cell RNA sequencing shows cell diversity in PDX microcancers cultured on our platform
To thoroughly analyze the phenotypical composition of the microcancers grown on chip, we conducted single-cell RNA sequencing on microcancers extracted from three separate chips after 3 days of culture. Details on library preparation and sequencing can be found in Materials and Methods. To determine the ratio of mouse to human cells within our cell clusters, we looked at both total UMI counts and total numbers of genes detected. Across our three libraries, only 86 of 10,300 gel bead emulsion (GEM) cells had mouse UMI counts > human UMI (Unique Molecular Identifier) counts, suggesting a very low percentage of mouse cells in our samples. In addition, the distributions of mouse UMIs in all GEM cells were much lower than most human UMIs (median UMIs: mouse = 62 and human = 28,250; Fig. 2I). We also looked at the number of mouse and human genes with at least 1 UMI per GEM cell (Fig. 2J). There is a much bigger separation between mouse and human, with only 1.1% of GEM cells having >64 mouse genes detected (log2 scale = 6) but 99.9% of GEM cells having >64 human genes detected. Therefore, the true number of mouse cells in our samples is on the order of 1 to 2%. We then discarded the mouse genes and used R v4.0.3 package Seurat v3.2.1 to do further quality-based cell filtering, normalization, principal components analysis, and, last, a uniform manifold approximation and projection (UMAP) plot of the three combined chips (final cell n = 7653; details in Materials and Methods). Projections of select genes confirmed high expression of critical markers related to glioblastoma (Fig. 2K). GBM8 cells exhibit epidermal growth factor receptor (EGFR) amplification and express ATRX. Consistent with this, we confirmed abundant expression of both EGFR and ATRX. Furthermore, localization in the expression profile of a known neurogenesis and glioma stem cell regulator, ASPM; a marker of astrocytic lineage, glial fibrillary acidic protein (GFAP); a neural progenitor marker often associated with glioma stem cells, NES (nerolidol synthase); along with the absence of expression of the mature neuron marker RBFOX3 (RNA Binding Fox-1 Homolog), indicates the presence of heterogeneous cell populations within the GBM8 microcancers, including both progenitor and more differentiated cell populations. (Fig. 2L).
On-chip live cell imaging of microcancer formation
To observe the microcancer formation in real time, we performed automated live-cell fluorescence microscopy on a Zeiss Axio Observer Z1 inverted microscope. Figure 3A and movie S1 show the time-lapsed images over 72 hours from a live cell imaging setup shown in fig. S5A. We tracked four cell clusters initially located at the well periphery for three wells and plotted the change in the 2D projected distance between these regions over time (Fig. 3, B to D, and fig. S5, B and C). The rate of aggregation of cells was slower for the first 9 hours after seeding as seen in Fig. 3D possibly because of the time it takes to reexpress adhesion molecules following trypsinization or for the secretion of soluble factors that enhance cell-cell adhesion. Separate small local cell aggregates form initially at different locations within a well, followed by joining of these aggregates over time, and, last, movement of these aggregates toward the center of the well to form the final microcancer spheroid. Once the formation was complete in 24 hours, the microcancers maintained their shape and integrity for the next 48 hours of the experiment. This imaging capability can extend the applications of our platform to a range of studies of spheroid, organoid, and acini formation in microenvironments and various tissue-on-a-chip applications.
Fig. 3. Capturing real-time microcancer spheroid formation with live cell imaging on an inverted microscope and molecular characterizations of spheroids in microwell array.
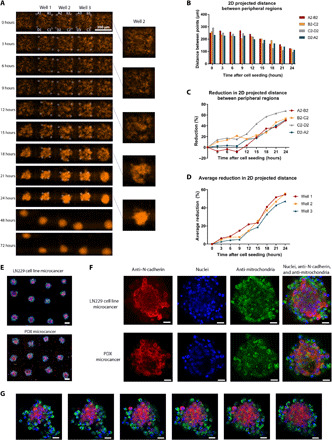
(A) Time progression of formation of five microcancers (LN229 cells) on our platform, imaged on an automated inverted microscope culture setup every 3 hours. Scale bar, 300 μm. (B) 2D projected distance between peripheral cell clusters in well 2 over 24 hours. These regions are shown in white circles and labeled in (A). (C) Reduction (%) from time “0” in 2D projected distance between peripheral cell clusters in well 2 over 24 hours. (D) Average reduction (%) from time “0” in 2D projected distance between peripheral cell clusters for all three wells over 24 hours. (E to G) Immunofluorescence detection of N-cadherin (red), 4′,6-diamidino-2-phenylindole nuclear staining (blue), and human mitochondria staining (green) in microcancers formed with LN229 cells and PDX. Tile of 12-well culture of LN229 microcancers (top) and PDX microcancers (bottom) imaged directly on chip (scale bar, 100 μm) (E). High-resolution images of individual microcancers for LN229 and PDX (scale bar, 20 μm) (F). Confocal z-stack images of PDX microcancer 3 days after cell seeding. Distance between each z-stack slice is 10 μm (scale bar, 20 μm) (G).
Morphological and molecular characterization
We also performed morphological analysis of these 3D spheroids using scanning electron microscopy to reveal the architectural integrity of these structures (fig. S6). It is important to note that the samples could be directly fixed and imaged on chip without the need for any extraction. We characterized the spheroids formed in our platform, both structurally and molecularly, to show their similarity with in vivo tumor characteristics and hence propose their utility as in vitro cancer models for drug screening in glioblastoma. We cultured human glioblastoma cell (LN229) and PDX microcancers on chip for 3 days and then imaged them using a high-resolution confocal microscope directly on chip after fixing and staining the microcancers. We evaluated the expression of N-cadherin, a transmembrane protein that mediates cell-cell contact through homotypic cell adhesion. Consistent with reported cellular localization in GBM, other 3D cultures, and xenograft models (17, 40), we observed N-cadherin expression both in the membrane at areas of cell-cell contact and in the cytoplasm (Fig. 3, E to G). This is in contrast with 2D cultures in which N-cadherin expression was found in the cytoplasm and the nucleus but absent from the membrane (17, 40). Figure S7 (A and B) shows confocal images of LN229 tumor spheroids with high N-cadherin expression at the cell junctions and cytoplasm within 1 day of culture on-chip. Figure S7 (C and G) shows a large confocal tile of an array of tumor spheroids on chip and high-resolution confocal analysis of individual tumor spheroids, and fig. S8 shows a split view of confocal z-stack of a PDX microcancer. These results demonstrate the versatility of our platform to perform high-throughput and high-resolution protein expression analysis to elucidate and study heterogeneity in the 3D cultures directly on chip.
The high-resolution confocal analysis presented above was performed directly on chip without the need to extract the 3D cultures. This feature allows users to select and analyze specific microcancers on the basis of specific criteria and to track individual microcancers over the course of analysis. This is important when heterogeneity within the microcancers that originated from the same patient-derived tumor cell suspension could be expected because of tumor heterogeneity or because of subsampling of different cell populations. The tracking is not possible in prior techniques, which require users to extract the 3D cultures in bulk and analyze them downstream. This direct on-chip characterization can be even more important for primary cell cultures, studies on acini, lumen formation, etc., where 3D structures can be very fragile and break upon shear stress from pipetting and handling. In summary, we show direct compatibility of our platform with several forms of high-resolution optical characterization techniques.
Application: Drug testing directly on chip
We next evaluated the possibility of drug testing directly on our platform by taking real-time measurements of response to drugs, as opposed to the current hanging drop methods used to extract the formed tumor spheroids and then performing an end point–only analysis in a separate well plate (41). We chose dasatinib for drug testing and introduced it after 3 days of culture in the chip. Dasatinib is a tyrosine kinase inhibitor that has previously been shown to reduce cell viability in several conventional and PDX GBM cell lines, including LN229 (42, 43). The process for introducing the drugs on chip is as follows. The chip was kept upright inside mineral oil and centrifuged briefly to bring down the spheroids. Next, an appropriate volume of media with drugs is loaded on chip through oil and incubated for 30 min to reach a homogeneous concentration. It is important to note that in the entire process of media exchange or drug loading, the microcancers are always immersed in culture media held inside the wells with capillary forces and never physically come in contact with oil. The volume of drugs loaded depends on the number of spheroids being cultured or wells filled with media on chip, which, in turn, are defined by the size and numbers of PDMS reservoirs initially used for cell seeding. The volume of media with drugs added is usually five times the volume of wells containing spheroids. The concentration of the drug is increased by an appropriate factor to account for the media without drugs already present in wells, resulting in the desired final drug concentration in the wells after diffusion. The chip was partitioned as before by using oil, inverted, and daily measurements of green and red fluorescence were taken. It is of note that this process can be used to periodically change media on chip for long-term 3D culture. Figure S9A shows a timeline of drug testing experiments on chip.
Figure 4A and figure S9 (B to E) show the results of drug testing for LN229 microcancer spheroids. Figure 4A shows alternate day maximum projection images for concentrations of 10 μM (top), 100 nM (middle), and no drug control (bottom). Figure S9 (B to E) shows the same for concentrations of 1 μM, 10, 1 nM, and 100pM of dasatinib drug, respectively. Figure 4 (B and C) show the box-and-whiskers plots (n = 12) of tumor spheroid volume and normalized cell death. It can be observed that for higher concentrations of drugs (10, 1 μM, and 100 nM), the cell death starts to increase as soon as 4 days after culture with drugs (day 7 overall) compared to lower drug concentrations and the no drug control. The median inhibitory concentration (IC50) value for dasatinib on LN229 microcancers was found to be 95.9 nM as measured on day 9, which is within the reported range found by others (Fig. 4D) (44, 45). Because of excessive cell death and destruction of spheroids for these high drug concentrations, the entire well starts to appear green in days 7 to 9, and hence, the overall green volume captured exceeds the spheroid volume (red), and the normalization results in values greater than 1. We also performed drug testing on PDX microcancers. Figure 4E shows alternate day maximum projection images of drug administered PDX spheroids for concentrations 10 μM (top), 100 nM (middle), and no drug control (bottom), and fig. S10 (A to D) shows the same for concentrations of 1 μM, 10, 1 nM, and 100pM of dasatinib drug, respectively. Figure 4 (F and G) show the box-and-whiskers plot (n = 12) of PDX microcancer volume and normalized cell death. The IC50 value for dasatinib on PDX microcancers was found to be 13.8 nM as measured on day 9, which is very similar to what has been previously observed (43) (Fig. 4H). However, the overall response to dasatinib in terms of normalized cell death was significantly lower for PDX, as the PDX microcancers overall remained intact and viable even for higher drug concentrations. This result is in agreement with the inability of dasatinib to affect tumor growth in GBM8 PDX in vivo, although it is highly effective in 2D culture (43). Figures S11 and S12 show individual traces of volume and cell death as response to drug over time for 12 LN229 and PDX microcancer spheroids, respectively.
Fig. 4. On-chip LN229 and PDX drug testing.
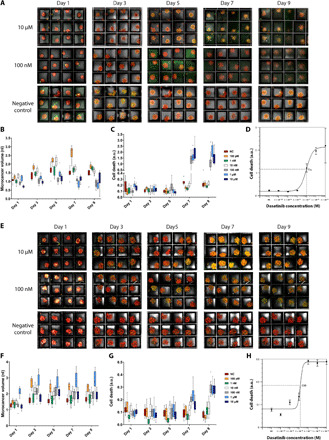
(A and E) Maximum projections of 12-well cultures of LN229 spheroids (A) and PDX spheroids (E) for days 1 to 9 after cell seeding (initial cell concentration: 500 cells per well). On day 3 after imaging, dasatinib was loaded onto the chips. Final concentrations of drug added were 10 μM (top row), 100 nM (middle row), and negative control (bottom row). For negative control, only media and CellTox green dye were added. Each well has a side length of 300 μm (for scale). (B, C, F, and G) Box-and-whiskers plot of volumes and cell death (green volume normalized to red volume per well) of LN229 microcancer spheroids (B and C) and PDX spheroids (F and G) from each drug concentration type for days 1 to 9 after cell seeding. The data encompass volumes and cell death information of microcancers for the 12 wells (n = 12) seen above for each condition. NC, negative control. (D and H) The IC50 value for LN229 spheroids (D) and PDX spheroids (H) was calculated to be 95.9 and 13.8 nM, respectively, as measured on day 9. The IC50 curve shown is based on 12-well data for each concentration of drug (10 μM to 100 pM and negative control), where the error bars represent the SEM.
Application: Geometric control in 3D culture
To evaluate the shape control capabilities of our platform, we fabricated a silicon microchip with etched annular circle, square, and triangle channels. Figure 5A shows the etched chip and describes the process flow for geometric shape formation in an annular circular channel. Briefly, the inside surface of the etched channel was made hydrophilic before cell seeding using oxygen plasma, following our previous protocol. For more details on geometric control experimental protocol, please see Materials and Methods. Seeding of cells in a single step was performed similar to the previous experiments by assembling a detachable PDMS well. This was followed by droplet formation using oil shear as previously discussed. Droplet shapes conformed to the shape of the channel cavity due to capillary forces, allowing us to generate dense and continuous annular circular, square, and triangular 3D shapes. These shapes were cultured and imaged for 3 days. Media exchange for 3D shapes was performed every day in the same manner as the drug loading procedure described in previous sections, and more details can be found in Materials and Methods. On the microchip, the diameter of circular shaped channels and side length of square-shaped channels ranged from 450 μm to 1.4 mm, whereas the side length of triangular shaped channels ranged from 780 μm to 2.4 mm. In our experiments, the channel width (between the internal post and outer boundary) was kept constant at 200 μm. Figure 5B shows the fluorescent microscopic images of these 3D shapes formed. We found that 1 day after cell seeding, continuous shapes were formed with widths of 70 to 100 μm for annular circles and squares and 120 to 160 μm for the annular triangles, which were the largest of the shapes. Fluorescence images taken on days 2 and 3 after cell seeding can be seen in fig. S13. It can be noted that by day 3, minor fragmentation in the 900-μm and 1.4-mm circular shapes begin to occur possibly because of continued cellular compaction and the absence of any external matrices in the system. Furthermore, volumetric mapping and analysis of these shapes were conducted to determine the shape volume (PKH dye mapping) and cell death (CellTox dye mapping) (Fig. 5C). The bar graphs for the volume and cell death of different annular shapes for 3 days of culture on chip are shown in Fig. 5 (D and E). Because of the large range of sizes that can be simultaneously tested on our platform, the volumes on chip spanned from 0.6 to 10 nl on day 1 to 0.86 to 11.8 nl on day 3. Cell death of these shapes, as can be seen in Fig. 5E, remained less than 8% for all 3 days of culture. Our platform allows geometric control in hanging drop format with sizes ranging from a few hundred micrometers to several millimeters while allowing characterization of multiple shapes on a single chip. The geometry control is a technology feature and enhancement that could be useful when different stress and gradients of stresses are to be examined or in applications where precise shape control of tissue structures are needed such as for regenerative medicine.
Fig. 5. Geometric control in microchip hanging drop 3D culture.
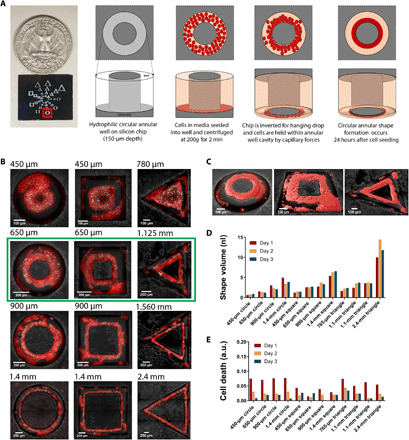
(A) Process flow 3D shape formation. Chip with annular circle, square, and triangle channels is fabricated such that the depth of the channel is ~150 μm. Internal well surfaces are made hydrophilic through oxygen plasma, after which cells with media are loaded in a single step and centrifuged. Oil shear is performed for droplet formation, as the cavity between outer border of the channel and inner post causes the droplet to conform to the channel shape. The chip is inverted for hanging drop, and after 24-hour incubation of cells, dense 3D shapes are generated. (B) Fluorescence image of shape formation of different sizes seen 1 day after cell seeding. Outer diameter (circle) or side lengths (square and triangle) of shape channels are mentioned on top left corner of each image. (C) Volumetric mapping of the shapes in the green box seen in (B). (D) Bar graph of volumes of annular circle, square, and triangle shapes for each channel size for days 1 to 3 after cell seeding. (E) Bar graph of cell death (green volume normalized to red volume) in annular circle, square, and triangle shapes for each channel size for days 1 to 3 after cell seeding.
DISCUSSION
We have described a modular and highly versatile microchip 3D hanging drop culture platform that allows high replicates and high content for drug screening compatibility with direct on-chip real-time or high-resolution confocal microscopy and geometric control of formed cell mass in 3D. Our high-throughput culture platform can be combined with commercially available automated micro-/nanoinjection systems to allow for testing hundreds of drug combinations in a single chip or deploying additive manufacturing to build microfluidic concentration gradients integrated with our chip and testing a reasonable and appropriate number of drugs on the platform (46, 47). We show that microcancers of similar size and density can be formed in a single step, without the need for individual well pipetting, thus reducing error and increasing reproducibility. We demonstrate quantitative analysis of 3D culture based on initial cell density both for a conventional glioblastoma cell line and for PDX-derived microcancers. We show the ability to perform real-time fluorescence observations while tracking individual cultures directly on chip and thus eliminating the need for any extractions of the microcancers. The capability of high-resolution scanning electron microscopy and confocal microscopy directly on chip using our demonstrated platform can allow for simplicity and ease of use for downstream analysis. The miniaturized hanging drop platform described here does not require specific matrices, specialized gels or nanoparticles, engineered scaffolds, or any form of synthetic surface coatings to form the tumor spheroids. This is especially useful when any of the above agents can be suspected of creating selective pressure and biasing tumor evolution during 3D culture. Growth media can be adapted for a variety of potential applications. In comparison to the classical hanging drop culture (41), cell aggregation into multicellular spheroids is significantly faster in the miniaturized platform (1 day versus 4 in the classical assay), likely an effect of the smaller well size that affects the diffusion of soluble factors and enhanced cell-cell adhesion. Moreover, our technique can also be adapted to replenish the role of various macromolecules, including extracellular matrix components and polysaccharides, as reported by others (48).
Our nanohanging drop culture model allows simultaneous testing of hundreds to thousands of 3D microcancer spheroids in real time where each microcancer appears to capture the salient characteristics of the tumor as evidenced by mate pair sequencing and confocal analysis. Differential responses of different cell subpopulations in a PDX spheroid can also potentially be measured using our platform and will be pursued in our future studies. The small size of the platform is also critical when available tumor tissue is limited, as is often the case in biopsies. In addition, the platform also allows for sequential (staggered time point) drug testing. Therefore, this system makes it possible to rapidly (within days) examine the effect of multiple drugs in real time even in cases where small biopsies might be available.
Last, we also demonstrate shape control of the cell mass using our microchip hanging drop 3D culture platform. Because our platform uses a microchip with etched channels, we fabricated a chip with circular, square, and triangular annular wells, which allowed the media droplet shape to conform to the shape of channel cavity because of capillary forces. We show that 3D shape formation can occur using our single-step loading and droplet formation technique by forming dense and continuous annular shapes ranging in sizes from a few hundred micrometers to several millimeters. In future studies, our microchip hanging drop platform could be used to coculture different cell types, allowing for self-organized tissue formation with salient in vivo–like features. This simple, scalable, and customizable platform may be suitable for a broad range of applications in drug discovery, regenerative medicine, stem cell research, and biotechnology.
MATERIALS AND METHODS
Off chip culture
The LN229 cells were cultured in the Dulbecco modified Eagle medium without sodium pyruvate (Gibco) including 10% (v/v) fetal bovine serum (FBS) (Thermo Fisher Scientific), 1% (v/v) nonessential amino acids (Thermo Fisher Scientific), and 2 mM l-glutamine (Thermo Fisher Scientific). After getting 70 to 80% of cell confluence, they were trypsinized with 0.25% (w/v) trypsin and 0.53% (w/v) EDTA solution (Gibco) and loaded to chips. For the PDX cells, 1% of penicillin-streptomycin (Lonza) was added to the media for LN229 cell culture. They were cultured for 2 days after thawing the PDX cells from liquid nitrogen tank to get the estimated counts and then loaded to chips. For the automated live cell imaging on the inverted microscope, the culture media also included 10 mM Hepes buffer.
Chip fabrication
Three types of chips were created. The first silicon chip has a microarray of wells of gradient sizes between 100 to 500 μm (each side of the well). The second chip (10 mm by 10 mm) has microarray of wells (each 300 μm by 300 μm) with a depth of 120 μm. Last, the third chip that was fabricated was for 3D shape formation (for geometric control experiments). This chip included annular circle, square, and triangle shapes of different sizes. To further explain, each shape was fabricated with an outer and inner boundary. For the circle and square shapes, the diameter/side lengths of the outer boundary were 450, 650, 900, and 1400 μm. The inner boundary diameter/side lengths (inner posts) of these shapes were 50, 250, 500, and 1000 μm, respectively. The triangle shapes were of the size such that the circle shape could be circumscribed within its boundaries; so the side dimensions of the outer boundaries were 780, 1125, 1560, and 2450 μm, and the inner boundaries (inner posts) were 318, 663, 1098, and 1988 μm.
Chip fabrication of all oxidized silicon chips were done in the same method. A 100mm diameter <100> silicon wafer (UniversityWafer, South Boston, MA) with one side polished was thoroughly cleaned and used as the substrate in the photolithography process. The wafer was dehydrated on a heating plate at 140°C for 2 min and cooled for 30 s before loading into a Molecular Vapor Deposition (Applied MicroStructures, San Jose, CA). A single layer of hexamethyldisilazane was deposited under low pressure to the polished side of the wafer, increasing the hydrophobicity of the wafer surface. After the deposition, the wafer was unloaded and dehydrated on an aluminum hot plate at 110°C for 2 min and cooled for 30 s. Thereafter, positive photoresist SPR 220 (MicroChem, Newton,MA) was spin-coated on the polished side of the wafer to form a 4.5-μm covering layer, followed by a soft bake at 60°C for 2 min and 110°C for 1 min. The photoresist was then exposed with an i-line (365 nm) mask aligner (EVG 620) in a hard contact with an expose dose of 210 J/cm2. The exposed regions with the outline of the microarray pattern were subsequently removed by immersing the wafer in an AZ 400K developer diluted 1:5 with deionized water for 45 s. For the anisotropic etching of the silicon substrate, a Bosch process reactive-ion etching with alternating steps of SF6/O2 etching and C4F8 passivation was used to create a 120-μm deep trench. After the Bosch process, the remaining photoresist was cleaned with acetone and isopropanol rinses. The photoresist was stripped with heated (100°C) PR Stripper 1165 for 10 min and sonicated for another 10 min, leaving the bare silicon exposed. Last, the wafer was thermally oxidized in a furnace (1150°C) for 60 min to grow 137-nm SiO2 and, subsequently, scribed into individual chips.
Cell seeding on chip
To prepare for cell seeding, the chips were cleaned using a piranha etch to rid of any organic residues on the surface of the wells. About 45 min before use, the chips were rinsed with acetone and isopropanol and dipped in ethanol (200 proof) for 2 min. Thereafter, the chips were blow-dried with nitrogen gas and kept in a covered petri dish. Before cell seeding, a cured PDMS polymer with a reservoir of a size according to the required number of exposed wells was attached to each chip, and the chips were made hydrophilic through O2 plasma treatment at 300 W for 3 min.
The total number of cells to be loaded on a single chip was equal to the product of the number of wells to be filled with cells (in the PDMS reservoir) and the number of cells needed per well (organoid size control). The cell density was calculated by dividing the total number of cells by the area of the exposed wells. The cell density was kept constant for the well gradient and 300-μm array chips and was found to be 4883 cells/mm2.
The total number of cells were extracted from the off-chip culture into a 1.7-ml tube and then centrifuged at 200g for 5 min so that all cells settle to the bottom of the tube. After aspirating the supernatant, the cells were washed with serum free media and centrifuged at 400g for 5 min. After, a PKH26 Red Fluorescent Cell Linker Kit for general cell membrane labeling (Sigma-Aldrich) was prepared according to product instructions and mixed with the cells. To stop the PKH26 Red Fluorescence staining, media with FBS was added to the cell and dye solution. The cells were again centrifuged at 400g for 5 min to bring all cells to the bottom of the tube, and subsequently, the supernatant was discarded. CellTox Green Cytotoxicity Assay (Promega), a green dye required to determine cytotoxicity and compatible with real-time imaging, was prepared according to product instructions (1:500 ratio of dye to media) in media with serum. This media and green dye solution was then added to the cell suspension in the total volume required to seed on to the chips. For PDX cells, the cell seeding process was the same, except all media used included 1% penicillin-streptomycin.
The cells were then seeded on chip and centrifuged in a petri dish at 200g for 2 min. Next, the PDMS reservoir was removed and the chip was dipped in mineral oil, after which an air pressure was applied to the chip to shear off the excess media from the top of the wells. This process is done to digitize or partition the chip and create individual disconnected wells with cells inside them. The shearing process can also be done with a mineral oil flow from top to bottom of the chip. The chip was then inverted in mineral oil to form inverted hanging nanodroplets. These inverted chips were then incubated in 37°C, RH (Relative Humidity) 95%, and 5% CO2. The cells were incubated for 9 days in this condition and taken out for 10 min daily for imaging and drug loading on day 3 after cell seeding.
Spheroid imaging
Real-time imaging was done daily after seeding with an upright Olympus BX63 fluorescence microscope with a 10× objective [0.3 numerical aperture (NA)]. For imaging, the chips were kept in oil upright for the duration of imaging and then inverted back for culture once the imaging was complete. Tiles and z-stacks of 10-μm slice thickness of the area with spheroids or shapes were taken off chips in bright-field, green fluorescent protein (GFP), and red fluorescent protein (RFP) fluorescence wavelengths. The CellTox green fluorescence dye was captured using the GFP fluorescence, and the PKH Red dye was captured using the RFP. The spatial pixel size was 1 μm by 1 μm for a depth of 10 μm.
Automated live-cell microscopy with inverted microscope
Live-cell fluorescence microscopy was performed using wide-field illumination on a Zeiss Axio Observer Z1 inverted microscope with a 20× 0.50-NA Plan-Neofluar air objective and 10-W halogen lamp illumination. The microscope was equipped with a temperature and 5% CO2 environmental chamber. The Zeiss ZEN software was used for data acquisition via a Photometrics eXcelon Evolve 512 EMCCD camera. Excitation and emission light was filtered using Zeiss filters (BP 550/25 and BP 605/70 nm respectively). Images were acquired every 3 hours after cell seeding for 72 hours.
Mate pair library preparation and the bioinformatics pipeline
To generate mate pair libraries from the 3D cultures, we used an in situ amplification protocol that was developed previously in our laboratory (49–51). In this protocol, cultured cells were directly applied to a modified Repli-g whole-genome amplification (WGA) protocol (50). WGA DNA (1 μg) from 3D cultures and tumor DNA from the original PDX tumor were fragmented to 3 to 5 kb using the Covaris E210 and used in the standard Illumina mate pair protocol (version 2) followed by sequencing on the Illumina HiSeq 4000 platform. Sequencing data were processed through our bioinformatics algorithms including the SVAtools utility to detect CRs and summarize the results in “Genome Plots.” In these plots, all chromosomes are displayed and arranged in a U shape. Each line represents a junction identified by the SVAtools. The thickness of the line is relative to the number of fragments supporting the junction. The end points of the line indicate the position of the junction’s two breakpoints. Diamonds identify a junction where one breakpoint maps to the position shown on the genome plot and the other breakpoint maps to sequences included in reference genome GRCh38 (Genome Reference Consortium Human genome build 38) but not within chromosomes 1 to 22, X, or Y, such as unplaced or unlocalized contigs, alternate sequences, or mitochondrial DNA. The final plot of 3D cultured cells merged the results of three individually cultured chips with arrays of 3D spheroids.
Drug loading and media exchange protocol
On day 3 after cell seeding, the chips were loaded with drugs diluted to the desired concentration in media (with serum and dye), except for the negative control chip in which only media was loaded. Media with serum was prepared with CellTox Green Cytotoxicity Assay as explained above. Media with serum for PDX cells included 1% penicillin-streptomycin. The drug stock of 10 mM dasatinib in 100% dimethyl sulfoxide was diluted in the media and dye solution to achieve the desired concentration of drug (10 μM to 100 pM). Compensating for the volume of media already in the wells, the concentration of the drug was increased. For example, if the cell culture had 100 wells loaded with spheroids, then the volume of media already present in the wells equaled volume of 1 well × 100 = 10.8 nl × 100 = 1080 nl. During drug loading, if 50 μl of media with drugs was added on this chip, then the final total volume = 50 + 1.08 μl = 51.08 μl. So the concentration of the drugs in the 50 μl of media added is equal to (51.08/50) × X; where X is the desired final concentration.
Before drug loading, the chips were first kept upright and centrifuged at 200g for 5 min. Then, 10 to 50 μl of the drugs/media solution were added in the cell area through the mineral oil layer on the chips. Density of the drugs/media is greater than density of mineral oil. Thus, the increased density allows the drugs/media to settle through the mineral oil and make contact with the wells. Hydrophilic media already inside the wells allows the drug to be spread throughout the area. The chips were then left upright for incubation at room temperature for 30 min. Thereafter, the chips were centrifuged again at 400g for 2 min. The chips were then dipped in mineral oil, and the excess media and drugs were sheared off with a mineral oil flow. The chips were then inverted in mineral oil and incubated in 37°C, RH 95%, and 5% CO2. The chips were imaged for six more days as explained above. Note that, in this entire process, the spheroids are always immersed in culture media held inside the wells with capillary forces and never physically come in contact with oil.
Cell seeding and media exchange protocol in geometric control experiments
For geometric control experiments, the cell seeding protocol for annular circle, square, and triangle shapes were performed in a single step in the same method as described previously. Briefly, a cured PDMS polymer was attached to the chips, after which the annular channels were made hydrophilic through O2 plasma treatment at 300 W for 3 min. Thereafter, off chip–cultured cells were stained with PKH dye, and the final suspension was added to media prepared with CellTox dye, ready to be added to the chip. A cell density of 14,648 cells/mm2 was seeded on chip and centrifuged in a petri dish at 200g for 2 min. After centrifugation, the PDMS reservoir was removed, and oil shear was performed to create droplet shapes that conform to the shape of the channel cavity because of capillary forces. Last, the chips were then inverted in mineral oil to form inverted hanging droplet shapes. These inverted chips were then incubated in 37°C, RH 95%, and 5% CO2. The cells were incubated for 3 days in this condition, taken out for 20 + 35 min daily for imaging.
Media exchange for 3D shapes was performed every day in the same manner as the drug loading procedure described above. First, the chips were kept upright for 15 min. Thereafter, fresh media was loaded on chip through mineral oil, incubating it for 20 min. Excess media was removed by oil shear after incubation, and the chip was inverted back for further culture.
Image analysis
Each of the z-stack images were converted to Tiff Series, in which a single tiff image of the series was of a single z-stack slice. Autoquant X3 software was then used for 3D deconvolution of the bright-field, green, and red channels of these tiff series images. The 3D deconvolution algorithm uses multiple iterations to develop a theoretical point spread function for the fluorescent points for each image. The optical parameters for the deconvolution included pixel spacing of 1 μm by 1 μm by 10 μm. The objective lens had an NA of 0.3. The emission wavelengths were 508 and 565 nm for the green channel (CellTox dye) and the red channel (PKH Red dye), respectively. The resulting deconvolved images were then analyzed using Imaris (Bitplane) software. In this software, the green and red dye were used to track and calculate the dead cells and analyze the spheroid volumes, respectively. This software uses algorithms to identify the local contrast in intensity to determine a threshold for color capture. The cell function was used to determine the green volume of the dead cells, and the surface function was used to calculate the volume of the spheroids. Cell and surface files were exported as Excel files, which were used to map the cell death and spheroid volumes in each well. A MATLAB script was created to extract the positional data from each of the cell information and volume information, and last, the total volume of the spheroid and the total cell death (green volume normalized with red volume) in each well were calculated. The results of each well were plotted against time in line graphs in Excel, and box-and-whisker plots of the cell death were generated according to drug concentration in Excel as well. Maximum projections were also obtained from Imaris software.
Confocal imaging
Samples were fixed in 4% (v/v) of paraformaldehyde overnight in 4°C, then washed with phosphate-buffered saline (PBS) three times, and used 0.25% (v/v) diluted Triton-X to permeabilize the cell membrane for 15 min. After washing with PBS, samples were blocked and stored in 1% (w/v) bovine serum albumin (Sigma-Aldrich) at 4°C overnight. The primary antibodies, rabbit N-cadherin monoclonal antibody (Thermo Fisher Scientific) and mouse mitochondria monoclonal antibody (Thermo Fisher Scientific), were used to stain for cadherin and mitochondria, respectively, at a 1:50 dilution and then incubated overnight at 4°C. Both primary antibodies are human specific. Samples were then washed three times before staining with secondary antibodies. Alexa Fluor 568 anti-rabbit (Thermo Fisher Scientific) and Alexa Fluor 488 anti-mouse (Thermo Fisher Scientific) antibodies were used to stain N-cadherin and mitochondria primary antibodies, respectively, and incubated overnight at 4°C. 4′,6-Diamidino-2-phenylindole (Thermo Fisher Scientific) was used for staining of nucleus and incubated for 1 hour at room temperature. After washing with PBS three times, samples were mounted on the cover glasses by using ProLong Antifade (Thermo Fisher Scientific). The LSM 710 microscope was used for the confocal fluorescent imaging.
Construction and sequencing of 10× V3.1 single-cell libraries
Single-cell 3′ complementary DNA (cDNA) libraries were prepared at the DNA Services laboratory of the Roy J. Carver Biotechnology Center at the University of Illinois at Urbana-Champaign. Three single-cell suspensions with an average viability of 70 to 75% by acridine orange and propidium iodide staining on the Nexcelom K2 (Nexcelom Bioscience, Lawrence, MA) were converted into individually barcoded cDNA libraries with the Chromium Next GEM Single-Cell 3′ dual-index Kit version 3.1 from 10X Genomics (Pleasanton, CA) following the manufacturer’s protocols. The target number of cells per library was 6000. The 10X Chromium instrument separates thousands of single cells into GEMs that add a barcode to the mRNA from each cell. Following double-stranded cDNA synthesis, individually barcoded libraries compatible with the Illumina chemistry were constructed. The final libraries were quantitated on Qubit, and the average size was determined on the AATI Fragment Analyzer (Advanced Analytics, Ames, IA). Libraries were pooled evenly, and the final pool was diluted to 5 nM concentration and further quantitated by quantitative polymerase chain reaction on the Bio-Rad CFX Connect Real-Time System (Bio-Rad Laboratories Inc., CA). The final library pool was sequenced on two lanes of an Illumina NovaSeq 6000 SP flowcell as paired reads with 28 cycles for read 1, 10 cycles for each index read, and 90 cycles for read 2. Basecalling and demultiplexing of raw data were done with the mkfastq command of the software Cell Ranger 4.1 (10X Genomics).
Quantification and analysis of single-cell data
The three demultiplexed fastq files were aligned to 10X Genomics’ prebuilt reference for human (GRCh38) and mouse (mm10) combined (version 2020-A) using the count command of Cell Ranger 4.0.0 with –expect-cell = 6000. When using a mixed-species reference, Cell Ranger runs a multiplet detection algorithm on all GEM barcodes associated with ≥1 cell (GEM cells). The algorithm starts by separating GEM cells into those with total mouse UMIs > total human UMIs (“mouse” cells) and those with mouse < human (“human” cells). It then uses the 10th percentile of mouse UMI counts in mouse cells and human UMI counts in human cells to define thresholds to say whether each GEM cell contains both a human and mouse cell (multiplet). Next, the algorithm runs a maximum likelihood estimator for the number of additional GEM cells expected to contain two human cells or two mouse cells based on the observed number of mixed species multiplets and the inferred ratio of cells from each species. Last, it estimates the total number of individual human and mouse cells, which, combined, is greater than the number of GEM cells due to multiplets. This algorithm works best when the species ratio is close to 1:1 and there is little ambient RNA from either species in the background. However, our samples greatly skewed toward human cells, and there was a small amount of ambient mouse RNA in all called cells (~8 to 256 UMIs), leading to the algorithm estimating an improbable >90% multiplet rate overall and overestimating the number of mouse cells at 14.8% of mouse + human cells. Instead of using 10X’s algorithm, we input the UMI counts for all GEM cells into R (v4.0.3) using the Seurat package (v3.2.2) and started the same way by summing the total UMI from mouse versus human in each GEM. In addition, we calculated the total number of mouse versus human genes detected (≥ 1 UMI) in each GEM. For both total UMI counts and detected genes, we calculated the number of GEMs with human > mouse, and we also compared the distributions of values for mouse versus human to estimate the number of GEMs containing a mouse cell versus a human cell.
Further analyses on human cells were performed. First, we excluded all mouse genes and any human genes that were not detected in at least 10 GEMs. Next, we filtered out GEMs with less than 300 human genes detected and where the percentage of UMIs from mitochondrial genes was greater than 3 median absolute deviations (threshold = 9.208%). The remaining 7653 GEMs were then normalized with Seurat’s SCTransform method. The scaled, normalized values of the top 3000 most variable genes were run through principal components analysis, and then the top 40 PC (Principal Components) scores were input to UMAP to represent expression variation among the cells in 2D space. Expression values of marker genes of interest were visualized on the UMAP plots.
Acknowledgments
We thank the staff at the Micro and Nanotechnology Laboratory at UIUC for facilitating the chip fabrication. We also thank the Mayo-Illinois Alliance for Technology-Based Healthcare. Funding: R.B. acknowledges the funding from University of Illinois, NIGMS grant number 1R01GM129709 to support A.G. P.Z.A. is supported by R01 NS101721-01A1. P.L. was supported by NIBIB T32EB019944 and the NSF grant 0965918 IGERT. K.P.R.-C. and R.B. also acknowledge NRT-UtB: Training the Next Generation of Researchers in Engineering and Deciphering of Miniature Brain Machinery (grant number 1735252). Research reported in this publication was also supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under award number T32EB019944. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Author contributions: A.G., P.Z.A., G.V., and R.B. conceived the idea and designed the study. A.G., A.M., C.S., J.B. performed the on-chip experiments. Y.K., K.P.R.-C., and O.A. performed off-chip culture. Y.K. and G.J.P.D. performed on-chip confocal imaging. P.L. performed the live cell imaging with the inverted microscope. A.M.S. and F.K. provided intellectual input with experimental design. A.G., A.M., F.K., P.Z.A., G.V., and R.B. wrote the manuscript. All authors edited the manuscript. Competing interests: A.G., R.B., P.Z.A., and G.V. are inventors on a provisional patent application related to this work filed by University of Illinois and Mayo Clinic (number 63/019,823, filed 4 May 2020]. G.V. is the owner of WholeGenome LLC. The authors declare that they have no other competing interests. Data and materials availability: All data are available in the main text or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/17/eabc1323/DC1
REFERENCES AND NOTES
- 1.Astashkina A., Grainger D. W., Critical analysis of 3-D organoid in vitro cell culture models for high-throughput drug candidate toxicity assessments. Adv. Drug Deliv. Rev. 69–70, 1–18 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Pickl M., Ries C. H., Comparison of 3D and 2D tumor models reveals enhanced HER2 activation in 3D associated with an increased response to trastuzumab. Oncogene 28, 461–468 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Todhunter M. E., Jee N. Y., Hughes A. J., Coyle M. C., Cerchiari A., Farlow J., Garbe J. C., LaBarge M. A., Desai T. A., Gartner Z. J., Programmed synthesis of three-dimensional tissues. Nat. Methods 12, 975–981 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moroni L., Burdick J. A., Highley C., Lee S. J., Morimoto Y., Takeuchi S., Yoo J. J., Biofabrication strategies for 3D in vitro models and regenerative medicine. Nat. Rev. Mater. 3, 21–37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryu N.-E., Lee S.-H., Park H., Spheroid culture system methods and applications for mesenchymal stem cells. Cell 8, 1620 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yahya W. N. W., Kadri N. A., Ibrahim F., Cell patterning for liver tissue engineering via dielectrophoretic mechanisms. Sensors 14, 11714–11734 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nath S., Devi G. R., Three-dimensional culture systems in cancer research: Focus on tumor spheroid model. Pharmacol. Ther. 163, 94–108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma H., Jiang Q., Han S., Wu Y., Tomshine J. C., Wang D., Gan Y., Zou G., Liang X.-J., Multicellular tumor spheroids as an in vivo–like tumor model for three-dimensional imaging of chemotherapeutic and nano material cellular penetration. Mol. Imaging 11, 10.2310/7290.2012.00012 (2012). [PubMed] [Google Scholar]
- 9.Baker L. A., Tiriac H., Clevers H., Tuveson D. A., Modeling pancreatic cancer with organoids. Trends Cancer 2, 176–190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boj S. F., Hwang C.-I., Baker L. A., Chio I. I. C., Engle D. D., Corbo V., Jager M., Ponz-Sarvise M., Tiriac H., Spector M. S., Gracanin A., Oni T., Yu K. H., van Boxtel R., Huch M., Rivera K. D., Wilson J. P., Feigin M. E., Öhlund D., Handly-Santana A., Ardito-Abraham C. M., Ludwig M., Elyada E., Alagesan B., Biffi G., Yordanov G. N., Delcuze B., Creighton B., Wright K., Park Y., Morsink F. H. M., Molenaar I. Q., Rinkes I. H. B., Cuppen E., Hao Y., Jin Y., Nijman I. J., Iacobuzio-Donahue C., Leach S. D., Pappin D. J., Hammell M., Klimstra D. S., Basturk O., Hruban R. H., Offerhaus G. J., Vries R. G. J., Clevers H., Tuveson D. A., Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clevers H., Modeling development and disease with organoids. Cell 165, 1586–1597 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Gao D., Vela I., Sboner A., Iaquinta P. J., Karthaus W. R., Gopalan A., Dowling C., Wanjala J. N., Undvall E. A., Arora V. K., Wongvipat J., Kossai M., Ramazanoglu S., Barboza L. P., Di W., Cao Z., Zhang Q. F., Sirota I., Ran L., MacDonald T. Y., Beltran H., Mosquera J.-M., Touijer K. A., Scardino P. T., Laudone V. P., Curtis K. R., Rathkopf D. E., Morris M. J., Danila D. C., Slovin S. F., Solomon S. B., Eastham J. A., Chi P., Carver B., Rubin M. A., Scher H. I., Clevers H., Sawyers C. L., Chen Y., Organoid cultures derived from patients with advanced prostate cancer. Cell 159, 176–187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baker B. M., Chen C. S., Deconstructing the third dimension – How 3D culture microenvironments alter cellular cues. J. Cell Sci. 125, 3015–3024 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pampaloni F., Reynaud E. G., Stelzer E. H. K., The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 8, 839–845 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Mueller-Klieser W., Three-dimensional cell cultures: From molecular mechanisms to clinical applications. Am. J. Physiol. 273, C1109–C1123 (1997). [DOI] [PubMed] [Google Scholar]
- 16.Kunz-schugharT L. A., Kreutz M., Knuechel R., Multicellular spheroids: A three-dimensional in vitro culture system to study tumour biology. Int. J. Exp. Pathol. 79, 1–23 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Souza G. R., Molina J. R., Raphael R. M., Ozawa M. G., Stark D. J., Levin C. S., Bronk L. F., Ananta J. S., Mandelin J., Georgescu M.-M., Bankson J. A., Gelovani J. G., Killian T. C., Arap W., Pasqualini R., Three-dimensional tissue culture based on magnetic cell levitation. Nat. Nanotechnol. 5, 291–296 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tung Y.-C., Hsiao A. Y., Allen S. G., Torisawa Y., Ho M., Takayama S., High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array. Analyst 136, 473–478 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frey O., Misun P. M., Fluri D. A., Hengstler J. G., Hierlemann A., Reconfigurable microfluidic hanging drop network for multi-tissue interaction and analysis. Nat. Commun. 5, 4250 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Anderson A. R. A., Weaver A. M., Cummings P. T., Quaranta V., Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell 127, 905–915 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Smalley K. S. M., Brafford P. A., Herlyn M., Selective evolutionary pressure from the tissue microenvironment drives tumor progression. Semin. Cancer Biol. 15, 451–459 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Jørgensen A., Young J., Nielsen J. E., Joensen U. N., Toft B. G., Rajpert-De Meyts E., Loveland K. L., Hanging drop cultures of human testis and testis cancer samples: A model used to investigate activin treatment effects in a preserved niche. Br. J. Cancer 110, 2604–2614 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.GravityPLUS(TM) 3D Culture and Assay Platform (2015); https://www.selectscience.net/products/gravityplus-hanging-drop-system/?prodID=116710.
- 24.Ingber D. E., Mechanical control of tissue growth: Function follows form. Proc. Natl. Acad. Sci. U.S.A. 102, 11571–11572 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson C. M., Jean R. P., Tan J. L., Liu W. F., Sniadecki N. J., Spector A. A., Chen C. S., Emergent patterns of growth controlled by multicellular form and mechanics. Proc. Natl. Acad. Sci. U.S.A. 102, 11594–11599 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedl P., Gilmour D., Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 10, 445–457 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Murphy W. L., McDevitt T. C., Engler A. J., Materials as stem cell regulators. Nat. Mater. 13, 547–557 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gomez E. W., Chen Q. K., Gjorevski N., Nelson C. M., Tissue geometry patterns epithelial-mesenchymal transition via intercellular mechanotransduction. J. Cell. Biochem. 110, 44–51 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boghaert E., Gleghorn J. P., Lee K., Gjorevski N., Radisky D. C., Nelson C. M., Host epithelial geometry regulates breast cancer cell invasiveness. Proc. Natl. Acad. Sci. U.S.A. 109, 19632–19637 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee J., Abdeen A. A., Wycislo K. L., Fan T. M., Kilian K. A., Interfacial geometry dictates cancer cell tumorigenicity. Nat. Mater. 15, 856–862 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Sil H., Sen T., Chatterjee A., Fibronectin-integrin (α5β1) modulates migration and invasion of murine melanoma cell line B16F10 by involving MMP-9. Oncol. Res. 19, 335–348 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Long K. R., Huttner W. B., How the extracellular matrix shapes neural development. Open Biol. 9, 180216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aizawa Y., Owen S. C., Shoichet M. S., Polymers used to influence cell fate in 3D geometry: New trends. Prog. Polym. Sci. 37, 645–658 (2012). [Google Scholar]
- 34.Yang W., Yu H., Li G., Wang Y., Liu L., High-throughput fabrication and modular assembly of 3D heterogeneous microscale tissues. Small 13, 1602769 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Ganguli A., Ornob A., Spegazzini N., Liu Y., Damhorst G., Ghonge T., Thornton B., Konopka C. J., Dobrucki W., Clare S. E., Bhargava R., Smith A. M., Kosari F., Bashir R., Pixelated spatial gene expression analysis from tissue. Nat. Commun. 9, 202 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fang Y., Eglen R. M., Three-dimensional cell cultures in drug discovery and development. SLAS Discov. 22, 456–472 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwak B., Lee Y., Lee J., Lee S., Lim J., Mass fabrication of uniform sized 3D tumor spheroid using high-throughput microfluidic system. J. Control. Release 275, 201–207 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Glicklis R., Merchuk J. C., Cohen S., Modeling mass transfer in hepatocyte spheroids via cell viability, spheroid size, and hepatocellular functions. Biotechnol. Bioeng. 86, 672–680 (2004). [DOI] [PubMed] [Google Scholar]
- 39.B. L. Carlson, J. L. Pokorny, M. A. Schroeder, J. N. Sarkaria, in Current Protocols in Pharmacology (John Wiley & Sons Inc., 2011), vol. 52, pp. 14.16.1–14.16.23. [Google Scholar]
- 40.Ofek G., Revell C. M., Hu J. C., Allison D. D., Grande-Allen K. J., Athanasiou K. A., Matrix development in self-assembly of articular cartilage. PLOS ONE 3, e2795 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vasmatzis G., Liu M. C., Reganti S., Feathers R. W., Smadbeck J., Johnson S. H., Klein J. L. S., Harris F. R., Yang L., Kosari F., Murphy S. J., Borad M. J., Thompson E. A., Cheville J. C., Anastasiadis P. Z., Integration of comprehensive genomic analysis and functional screening of affected molecular pathways to inform cancer therapy. Mayo Clin. Proc. 95, 306–318 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nehoff H., Parayath N. N., McConnell M. J., Taurin S., Greish K., A combination of tyrosine kinase inhibitors, crizotinib and dasatinib for the treatment of glioblastoma multiforme. Oncotarget 6, 37948–37964 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis-Tuffin L. J., Feathers R., Hari P., Durand N., Li Z., Rodriguez F. J., Bakken K., Carlson B., Schroeder M., Sarkaria J. N., Anastasiadis P. Z., Src family kinases differentially influence glioma growth and motility. Mol. Oncol. 9, 1783–1798 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Avril T., Etcheverry A., Pineau R., Obacz J., Jegou G., Jouan F., Le Reste P. J., Hatami M., Colen R. R., Carlson B. L., Decker P. A., Sarkaria J. N., Vauléon E., Chiforeanu D. C., Clavreul A., Mosser J., Chevet E., Quillien V., CD90 expression controls migration and predicts dasatinib response in glioblastoma. Clin. Cancer Res. 23, 7360–7374 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Benezra M., Hambardzumyan D., Penate-Medina O., Veach D. R., Pillarsetty N., Smith-Jones P., Phillips E., Ozawa T., Zanzonico P. B., Longo V., Holland E. C., Larson S. M., Bradbury M. S., Fluorine-labeled dasatinib nanoformulations as targeted molecular imaging probes in a PDGFB-driven murine glioblastoma model. Neoplasia 14, 1132–1143 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duarte C., Salm E., Dorvel B., Reddy B. Jr., Bashir R., On-chip parallel detection of foodborne pathogens using loop-mediated isothermal amplification. Biomed. Microdevices 15, 821–830 (2013). [DOI] [PubMed] [Google Scholar]
- 47.NANOJECT III W/UNIVERSAL ADAPTER (Item no. 3-000-207) - Drummond Scientific Company; https://shop.drummondsci.com/nanoject-iii-w-universal-adapter-item-3-000-207/?gclid=CjwKCAjwlID8BRAFEiwAnUoK1eG6XCgzn7VX8rujpUU3Ex0qqz5J3fLq2OgYdro-hFdRKhb_W9Pk0BoCwBEQAvD_BwE.
- 48.Tao F., Sayo K., Sugimoto K., Aoki S., Kojima N., Development of a tunable method to generate various three-dimensional microstructures by replenishing macromolecules such as extracellular matrix components and polysaccharides. Sci. Rep. 10, 6567 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kovtun I. V., Murphy S. J., Johnson S. H., Cheville J. C., Vasmatzis G., Chromosomal catastrophe is a frequent event in clinically insignificant prostate cancer. Oncotarget 6, 29087–29096 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murphy S. J., Cheville J. C., Zarei S., Johnson S. H., Sikkink R. A., Kosari F., Feldman A. L., Eckloff B. W., Karnes R. J., Vasmatzis G., Mate pair sequencing of whole-genome-amplified DNA following laser capture microdissection of prostate cancer. DNA Res. 19, 395–406 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murphy S. J., Karnes R. J., Kosari F., Castellar B. E. R. P., Kipp B. R., Johnson S. H., Terra S., Harris F. R., Halling G. C., Klein J. L. S., Nasir A., Bergstrahl E., Rangel L. J., Sukov W. R., Vasmatzis G., Cheville J. C., Integrated analysis of the genomic instability of PTEN in clinically insignificant and significant prostate cancer. Mod. Pathol. 29, 143–156 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/17/eabc1323/DC1