

Hydraulic resistance induces cell phenotype oscillations in confinement via the coupled dynamics of actin, myosin, and calcium.
Abstract
Cells penetrating into confinement undergo mesenchymal-to-amoeboid transition. The topographical features of the microenvironment expose cells to different hydraulic resistance levels. How cells respond to hydraulic resistance is unknown. We show that the cell phenotype shifts from amoeboid to mesenchymal upon increasing resistance. By combining automated morphological tracking and wavelet analysis along with fluorescence recovery after photobleaching (FRAP), we found an oscillatory phenotypic transition that cycles from blebbing to short, medium, and long actin network formation, and back to blebbing. Elevated hydraulic resistance promotes focal adhesion maturation and long actin filaments, thereby reducing the period required for amoeboid-to-mesenchymal transition. The period becomes independent of resistance upon blocking the mechanosensor TRPM7. Mathematical modeling links intracellular calcium oscillations with actomyosin turnover and force generation and recapitulates experimental data. We identify hydraulic resistance as a critical physical cue controlling cell phenotype and present an approach for connecting fluorescent signal fluctuations to morphological oscillations.
INTRODUCTION
Cells in vivo travel either by remodeling their surrounding three-dimensional (3D) extracellular matrix (ECM) to open up tunnel-like tracks or by migrating through 3D longitudinal tracks naturally occurring in various anatomical structures (1). During this journey, cells squeeze through confining pores ranging from 1 to 20 μm in diameter or channel-like tracks varying from 3 to 30 μm in width and hundreds of microns in length (2). Tumor cells move inside confining channels not only by uptaking and discharging water (3) but also by pushing the column of water ahead of them (4), which generates hydraulic resistance (5–7). Hydraulic resistance is a physiologically relevant cue, which regulates cell directional choices in confinement (4, 8, 9).
Plasticity enables tumor cells to adapt to diverse tissue microenvironments presenting different chemical and physical cues and to optimize their directed locomotion by tuning the underlying migration mechanisms. Cancer cells entering confining channels undergo a phenotypic mesenchymal-to-amoeboid transition (MAT) (10–14). Cells with a mesenchymal phenotype display an elongated spindle-like shape with actin-rich finger-like protrusions at the leading edge, which are stabilized by the interactions of focal adhesions (FAs) with ECM. On the other hand, amoeboid cells in confinement assume a pill-like morphology and exhibit membrane blebs promoted by elevated contractility, which are identified as discrete, sphere-like bulges localized primarily at the cell poles (14). It is unknown how confined cells respond to increasing hydraulic resistance.
Given the rapid interconversion between MAT and amoeboid-to-mesenchymal transition (AMT), we examined the migration mode of hundreds of LifeAct-GFP–tagged cells in confined channels of increasing length, and thus increasing hydraulic resistance, by quantifying the normalized cell protrusion length and the coefficient of variation of green fluorescent protein (GFP) signal, two parameters that define the cell morphological footprint. Mesenchymal cells relative to blebbing cells exhibit a larger protrusion length along with a uniform actin distribution (10) depicted by a low coefficient of variation, as opposed to blebbing cells that display actin polarization at the poles (10) and thus high coefficient of variation. Although confinement induces MAT, we found that the cell footprint shifts from amoeboid to mesenchymal upon increasing hydraulic resistance.
During motility, the intracellular fluorescence signal at the cell leading edge exhibits oscillatory patterns associated with changes in cell morphodynamics. These oscillations may encompass distinct intracellular processes, such as blebbing expansion/retraction, actin filament bundling, and cleavage. The objective is to decouple these oscillatory waves into individual components and determine their relative importance in frequency and time domains, thereby providing detailed, temporal information for the migration mode. Wavelet analysis provides a powerful platform for signal processing as it can detect, decompose, and classify various discrete oscillatory patterns. Wavelet analysis has been used in diverse fields of research such as radiometry, communication systems, cosmology, acoustics, fluid dynamics, etc. In biomedical research where the statistical characteristics are usually nonstationary, the wavelet transform algorithms are widely used for real-time detection of neural signals (15). We are herein using wavelet analysis to process fluorescence signals and correlate them to distinct morphological earmarks and ultimately to the mode of migration.
By applying fast Fourier transform (FFT), the oscillation frequency of distinct F-actin structures at the cell leading edge (blebbing and short, medium, and long actin filament) is identified on the basis of their characteristic turnover times, which are obtained from fluorescence recovery after photobleaching (FRAP). Using short-time Fourier transform (STFT), we obtained the spectrogram from the oscillation curves to identify the time dependence of frequency modulation and relative importance of distinct F-actin structures in the frequency domain. Our analysis reveals a periodic phenotypic change involving a cycle from blebbing to short, medium, and long actin filament formation that loops with bleb recurrence. The period of the unit cycle becomes shorter for blebbing cells at elevated resistances, which is consistent with their AMT. The period becomes independent of hydraulic resistance upon knocking out its key mechanosensor TRPM7 (transient receptor potential cation channel subfamily M member 7) or inhibiting its downstream targets Arp2/3 or formins. Hydraulic resistance increases actomyosin contractility through TRPM7 mechanosensing (9) that generates higher regional intracellular pressure to facilitate cell movement. The propagated pressure (16) then imposes compressive forces on the cell-lateral channel wall interfaces that foster FA maturation, actin-based filament formation, and bundling/extension, which favor the elongation of protrusion and AMT.Myosin motors are recruited to actin filaments from blebbing sites. The competition of myosin between actin-based filaments and blebs determines the cell phenotype. Examination of intracellular calcium signals reveals a similar periodicity pattern to that of actin, which is captured by a mathematical model of cytoplasmic Ca2+ dynamics and the cytoskeletal polymerization/depolymerization cycle.
RESULTS AND DISCUSSION
Hydraulic resistance regulates the phenotype of migrating cells
A microfluidic device, consisting of an array of confining channels of constant cross-sectional area (width, W = 4 μm and height, H = 10 μm) (3), was used to investigate the influence of hydraulic resistance on migration mode (Fig. 1A). Hydraulic resistance (5, 9) was tuned by varying the channel length from 150 to 1500 μm, thereby increasing the resistance by 10-fold (fig. S1A). Intriguingly, the migration speed of MDA-MB-231 cells increases with increasing hydraulic resistance (fig. S1B), which is counterintuitive, as resistance is expected to suppress motility. Moreover, hydraulic resistance induces a cell phenotypic switch from blebbing to mesenchymal (Fig. 1, B and C). Blebbing motile cells display a pill-like morphology and bear membrane blebs, which are identified as sphere-like bulges localized at the cell poles (Fig. 1C) (9, 13, 14), and represent 80% of confined MDA-MB-231 cells at 1× but only ~30% at 10× (Fig. 1B). On the other hand, mesenchymal cells exhibit finger-like protrusions (Fig. 1C). Using LifeAct-GFP–tagged MDA-MB-231 cells, we observed that blebbing cells display polarized actin distribution that is markedly enriched at the leading and trailing edges (10), whereas actin is more uniformly distributed in mesenchymal (protrusive) cells (fig. S1C). Because of their pill-like morphology, blebbing cells occupy the entire cross-sectional area of the channel along their longitudinal axis (fig. S1D). However, this is not the case for mesenchymal cells due to the presence of finger-like protrusion at the cell poles (Fig. 1C and fig. S1D).
Fig. 1. Hydraulic resistance regulates the phenotype of migrating cells.
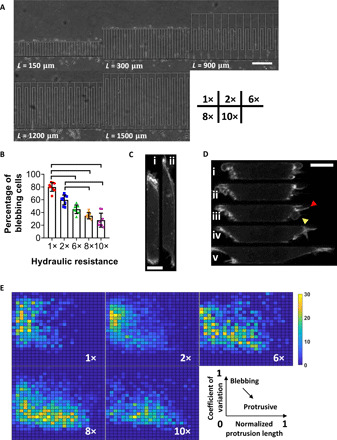
(A) Phase contrast image of the PDMS-based microfluidic device containing channels of prescribed dimensions (H = 10 μm, W = 4 μm, and L = 150 to 1500 μm). Their relative hydraulic resistance (1× to 10×) is listed at the bottom right. Scale bar, 50 μm. (B) Percentage of MDA-MB-231 cells exhibiting a blebbing phenotype inside channels of prescribed hydraulic resistances. Data represent the means ± SD for n > 40 cells per experiment from nine independent experiments. P < 0.05, assessed by Kruskal-Wallis test followed by Dunn’s post hoc. (C) Representative images of a LifeAct-GFP–tagged cell undergoing phenotypic transition from (i) blebbing to (ii) protrusive. Scale bar, 5 μm. (D) Representative image sequence depicting a LifeAct-GFP–tagged cell undergoing phenotypic transition. Arrowheads indicate the protrusion (red) and bleb (yellow) of a hybrid cell (iii). Scale bar, 5 μm. (E) 3D phase diagram showing normalized protrusion length over the cell length (x axis, from 0 to 1), coefficient of variation of LifeAct-GFP signal throughout the cell (y axis, from 0 to 1), and number of cell footprints (z axis, 10 frames or footprints collected per cell for n > 100 cells from three independent experiments) in each bin (24 × 24 bins, 0.05 step size) in channels of prescribed hydraulic resistances. Cells transitioning from blebbing to protrusive translocate from the top left (short protrusion, polarized actin at the poles) to the bottom right (long protrusion, uniform actin distribution) on the diagram.
To establish that hydraulic resistance regulates the cell phenotype, we analyzed the cell morphology footprint (Fig. 1D) inside channels of prescribed resistances and generated a phase diagram (Fig. 1E) with two parameters: the coefficient of variation of LifeAct-GFP signal versus the normalized protrusion length, which represents the cumulative length of cell parts whose widths were ≤70% of the channel’s width, divided by the overall cell length (fig. S1E). Blebbing cells, which exhibit a polarized actin distribution at the cell poles that corresponds to high values of coefficient of variation and a pill-like morphology that results in negligible protrusion lengths, are located at the top left of the phase diagram, whereas mesenchymal cells with relatively uniform actin signal and finger-like protrusions populate the bottom right (Fig. 1E). Cell treatment with the Rac inhibitor NSC23766, which promotes an amoeboid phenotype (12), shifted the cell population footprint inside channels of 10× hydraulic resistance from the bottom right to the top left corner (fig. S1F). In contrast, the ROCK inhibitor Y-27632, which supports a protrusive morphology (9, 14), concentrated cells migrating in channels of basal hydraulic resistance (1×) at the bottom right corner of the phase diagrams (fig. S1F).
Using the phase diagram, we examined the phenotypic transition of cells migrating in channels of different hydraulic resistances. A smooth transition of the cell population footprint from the top left toward the bottom right corner was observed upon gradually increasing the hydraulic resistance from 1× to 10× (Fig. 1E). This progressive transition is exemplified by the presence of a hybrid phenotype displaying both sphere-like blebs and finger-like protrusions (Fig. 1D, iii), which is located in the middle of the phase diagram. Thus, elevated hydraulic resistance promotes the growth of actin-based filaments, which mediate the phenotypic switch from blebbing to mesenchymal. We hypothesized that hydraulic resistance exacerbates branched actin formation at the cell leading edge consistent with the intrinsic load adaptation of actin network (17, 18). Consistent with this hypothesis, Arp2/3 inhibition caused a pronounced yet partial effect on suppressing phenotypic transition from blebbing to mesenchymal in channels of elevated (≥8×) resistance (fig. S1F). Similar effects were noted by knocking down the RhoA effector mDia1 (fig. S1F), which promotes the formation of bundled fibers. Both these interventions also reduce migration speeds at high hydraulic resistances (≥8×) (fig. S1, G and H). By introducing the migration speed as the z-axis variable in the phase diagram, we show that control cells with a protrusive phenotype display faster motility (fig. S1I).
TRPM7 was identified as the key mechanosensor of hydraulic resistance in confinement (9). Consistent with this finding, functional TRPM7 knockout (KO) (9) abolishes the regulation of cell phenotype by hydraulic resistance, resulting in a hybrid phenotype, as depicted on the phase diagram (fig. S1F), and fast migration irrespective of the hydraulic resistance (fig. S1J).
Hydraulic resistance promotes a thicker actomyosin cortex and larger FAs at the cell leading edge
Cells balance the external hydraulic pressure with cortical actomyosin at an intersection before a directional migration decision is made (9). We thus hypothesized that elevated hydraulic resistance promotes a thicker actomyosin cortex. To test this, we quantified the LifeAct-GFP and MIIA-GFP signals of cells displaying a blebbing or hybrid morphology in channels of different resistances (Fig. 2, A to C, and fig. S2, A to C). Both the actin and myosin signals at the cell leading edge increased with increasing hydraulic resistance (Fig. 2C and fig. S2C). TRPM7 KO abrogated the actin and myosin signal regulation by hydraulic resistance, consistent with its role as a mechanosensor of this cue (fig. S2, D and E). In addition, Arp2/3 inhibition or formin knockdown (KD) produced flat intensity values at all resistances (fig. S2, D and E) due to the suppression of branched and bundled actin network.
Fig. 2. Hydraulic resistance promotes a thicker actomyosin cortex and larger FAs at the cell anterior.
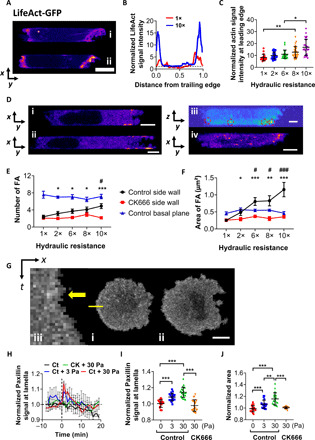
(A) LifeAct-GFP–tagged cells inside channels of (i) 1× and (ii) 10× hydraulic resistance. Scale bar, 5 μm. (B) Average line scanning profiles of representative cells in (A). All values were normalized by the corresponding cytosolic background values in the geometric cell center. (C) Normalized cortical LifeAct-GFP signal intensity at the cell anterior in channels of prescribed hydraulic resistances. Data represent the means ± SD (n > 20 cells from three experiments). *P < 0.05, **P < 0.01 assessed by Kruskal-Wallis test with Dunn’s post hoc. (D) Paxillin-GFP–tagged cells showing FA localization in a (i) blebbing and (ii) hybrid phenotype at middle focal planes, (iii) blebbing from side view (image reconstructed from z-stack scanning), or (iv) blebbing at basal plane. Scale bars, 5 μm. (E) The number and (F) area of FAs at basal or side walls in the presence or absence of CK666 at different hydraulic resistances. Data are means ± SEM, n > 10 cells from three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 between control and CK666 by two-tailed unpaired t test. #P < 0.05, and ###P < 0.001 relative to 1× using one-way ANOVA with Tukey’s post hoc. (G) Image sequences of paxillin-GFP–tagged cells on 2D (i) before and (ii) after the application of 3 Pa. Scale bar, 10 μm. (iii) Kymograph of line scan shown in (i). Yellow arrow indicates the first frame after hydrostatic pressure loading. (H) Normalized paxillin-GFP signal intensity of control and Arp2/3-inhibited cells on 2D following hydrostatic pressure loading (3 or 30 Pa) at t = 0 min. Signal intensities are normalized to the respective unstimulated controls. Data represent the means ± SEM. (I) Normalized fluorescence intensity and (J) spreading area of paxillin-GFP–expressing control or Arp2/3-inhibited cells immediately (I) and 10 min (J) after hydrostatic pressure exposure. Data represent means ± SD. **P < 0.01 and ***P < 0.001, assessed by two-tailed unpaired t test.
Hydraulic resistance is counteracted by actomyosin contractility at the cell leading edge, which generates high regional intracellular pressure that eventually dissipates throughout the cell body (9, 16). As the pressure wave propagates, the highest compressive forces are exerted at the cell-side channel wall interfaces at the leading edge (fig. S2F). Hence, during the phenotypic switch from blebbing to mesenchymal at elevated resistances, FAs and actin filaments are predicted to emerge and grow from these interfaces (fig. S2F). Live-cell imaging using paxillin-GFP reveals the presence of FAs primarily at the basal surface and at the lateral channel walls either intersecting or close to the basal plane at the cell front (Fig. 2D and fig. S2G). The FA number and area at the side channel walls increased with increasing hydraulic resistance, whereas the corresponding ones at the basal plane remained unchanged (Fig. 2, E and F).
The growth and dynamics of FAs and the recruitment of actin fibers at these sites are regulated by the Arp2/3 complex (19–21). In line with prior work, live-cell imaging using ARP3-mCherry shows that Arp2/3 displays a spatial distribution similar to that of FAs and localizes primarily at the side channel walls either intersecting or near the basal surface at the cell anterior (fig. S2, H and I). Moreover, Arp2/3 inhibition abolishes the hydraulic resistance–mediated increases in the number and area of FAs at the lateral (Fig. 2, E and F, and fig. S2I), but not basal, walls (fig. S2, J and K).
Arp2/3 is critical to the induction of larger FAs and lamella by hydrostatic pressure
The pressure load that cells experience at their leading edge inside confining channels is of the order of 1 Pa (4, 9). Hydrostatic pressure differentials were generated by pipetting prescribed amounts of media atop paxillin-GFP– or ARP3-mCherry–expressing cells seeded on collagen I–coated slides (9). The fluorescence signal intensities were quantified on the basis of the feature extraction from a thin layer of cell lamella at the basal 2D plane (Fig. 2G and fig. S2, L and M). Hydrostatic pressure induced a rapid albeit transient increase of both paxillin and ARP3 signals at the lamella (Fig. 2, H and I, and fig. S2, N and O), which faded away within 10 to 20 min. It also promoted the growth of lamella and cell spreading area in a magnitude-dependent manner (Fig. 2J and fig. S2P). Moreover, hydrostatic pressure loading fostered the colocalization of paxillin and Arp3, as evidenced by immunofluorescence staining (fig. S2, Q and R). Arp2/3 inhibition abolished the hydrostatic pressure–mediated increase of FA signal and cell spreading area (Fig. 2, I and J).
Using LifeAct-GFP–tagged cells, the lamella growth induced by hydrostatic pressure (30 Pa) was evident from the intense membrane extension and ruffling at the cell periphery (fig. S2S, ii and iii), which is indicative of strong actin polymerization and bundling (22), coupled with increased FA sites anchoring stress fibers at the periphery (fig. S2, iv). The hydrostatic pressure–induced elevation of LifeAct-GFP and MIIA-GFP signals was suppressed by Arp2/3 inhibition (fig. S2, T and U). Together, these data reveal the interplay of Arp2/3 and FAs in response to the elevated hydraulic/hydrostatic pressure to facilitate cell protrusions.
Actin filament dynamics is regulated by hydraulic resistance
During a typical phenotypic switch in confinement, the blebbing cell first experiences the shrinkage of blebs, then the polarization of actin at the lateral wall-cell interface, followed by the emergence of finger-like actin filaments, and last, the anchoring and elongation of these filaments along the side channel walls at the cell anterior (Fig. 3A). We split these distinct morphologies into four categories (Fig. 3A): (i) blebbing and (ii) short, (iii) medium, and (iv) long actin filaments. FRAP was performed at the leading edge of actin-GFP–tagged cells, and the recovery curve was fitted with an exponential function (fig. S3, A to C) to obtain the characteristic turnover time, t0 (23), which describes the period of molecular turnover of processes that occur within the affected region of interest (ROI). Using FRAP analysis, the characteristic turnover time of blebs in confinement (Fig. 3A, i) was determined to be ~60 s (Fig. 3B), which matches the life span of a bleb that was manually tracked in our experiments (fig. S3, D and E). This value obtained for confined cells is within the range reported from measurements on 2D, where expansion typically lasts 5 to 30 s and retraction 60 to 120 s (24).
Fig. 3. Actin filament dynamics is regulated by hydraulic resistance.
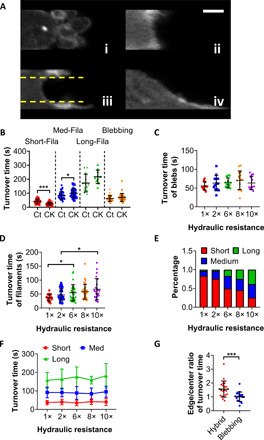
(A) Representative example of actin-GFP–tagged MDA-MB-231 cells in four distinct morphologies during phenotypic transition in confinement: (i) blebbing and (ii) short, (iii) medium, and (iv) long actin filaments. Yellow lines depict the separation of the cell edge near the side wall interface from the center area. Scale bar, 2 μm. (B) Characteristic turnover times obtained from the exponential fitting of FRAP recovery curves for the aforementioned four distinct morphologies of actin-GFP–tagged vehicle control and Arp2/3-inhibited (CK666; 100 μM) cells. Data represent means ± SD for n > 10 cells from three independent experiments. *P < 0.05 and ***P < 0.001, assessed by two-tailed unpaired t test. Characteristic turnover times of (C) blebs and (D) actin filaments, assessed by subjecting actin-GFP–tagged cells to FRAP, as a function of hydraulic resistance. Data represent means ± SD for n > 10 cells from three independent experiments. *P < 0.05 assessed by Kruskal-Wallis test with Dunn’s post hoc. (E) Percentage of cells displaying short, medium, or long actin filaments at different hydraulic resistances categorized during FRAP assays. Cells of >30 from three independent experiments were examined under each condition. (F) Characteristic turnover times of short, medium, and long actin filaments, determined by FRAP, at different hydraulic resistances. Data represent means ± SD for n > 10 cells from three independent experiments. (G) Edge-to-center ratio of characteristic turnover times for cells displaying blebbing versus filamentous/hybrid morphologies. Data represent means ± SD for n > 10 cells from three independent experiments. ***P < 0.001, assessed by two-tailed unpaired t test.
The turnover time of short actin filaments in confinement (Fig. 3A, ii) was calculated to be ~40 s (Fig. 3B), which is in line with published data (25, 26). Assembly and branching of short filaments are mediated by the Arp2/3 complex, and as such, inhibition of its activity via CK666 reduced their turnover time to 20 s (Fig. 3B), which is consistent with prior work showing that the turnover time of actin filaments scales with their length (23, 27). Along these lines, we determined the turnover time of the medium-sized filaments (Fig. 3A, iii) to be ~80 s (Fig. 3B). These filaments appear as wave-like extensions (Fig. 3A, iii) in close proximity but not anchored to the lateral channel walls because they lack FAs at their tip (fig. S3F). Their morphology resembles the peripheral ruffles found in membrane protrusions on 2D (22). The involvement of integrin-mediated FA complexes enables further elongation of actin fibers and the combination of multiple filaments into bundles (Fig. 3A, iv), thereby supporting an actin polymerization–based mode of migration (28) and cell transition to a mesenchymal phenotype. In line with published data (27), the characteristic turnover time of long filaments was on the order of hundreds of seconds. Arp2/3 inhibition moderately prolonged the turnover of medium-sized filaments, presumably due to the suppression of branching during filament elongation and/or bundling (Fig. 3B). It also had a modest, yet nonsignificant (P = 0.07), effect on the turnover of long filaments, and as expected, it did not alter blebbing turnover (Fig. 3B).
Hydraulic resistance did not regulate blebbing turnover as assessed by FRAP (Fig. 3C) or by manually tracking bleb lifetimes (fig. S3D). However, the turnover of actin filaments increased with hydraulic resistance (Fig. 3D), which is attributed to the higher frequency of longer actin filaments at elevated resistances (Fig. 3E) that exhibit larger turnover times (Fig. 3B). This is further substantiated by data showing that the turnover times of various actin filaments were not affected by hydraulic resistance (Fig. 3F). It is noteworthy that the turnover times of actin filaments near the lateral walls were substantially longer than those in the central protrusive region, whereas no spatial differences were detected for blebs (Fig. 3G), suggesting a polarized spatial distribution of actin-GFP at the cell leading edge during phenotypic transition.
Wavelet analysis of LifeAct-GFP signals reveals a periodic regulation during phenotypic transition
In light of the distinct characteristic turnover times of actin signals involved in different dominant processes, we monitored migrating LifeAct-GFP–expressing cells inside channels at a frame rate of 5 s, which is faster than the turnover time of any leading edge actin structure in confinement (>20 s). The average fluorescence intensity within the outlined cortical region (Fig. 4A) was normalized by the moving average to generate the oscillation curve of actin signal (fig. S4A). By performing FFT and plotting the amplitude of oscillations as a function of frequency, three distinct peaks were detected between 0 and 10, 10 and 20, and 20 and 40 mHz (fig. S4A), which correspond to the turnover times obtained by FRAP. A peak at 20 to 40 mHz represents an F-actin turnover period of 25 to 50 s, which correspond to the short actin filaments. A peak at 10 to 20 mHz is indicative of a medium-sized filament with an average turnover time of ~80 s. A further decrease in frequency (0 to 10 mHz) corresponds to a longer oscillation period (>100 s) found for long actin filaments. The representative peaks for blebbing fall in the frequency range of medium-sized filaments (10 to 20 mHz).
Fig. 4. Wavelet analysis of LifeAct-GFP signals reveals a periodic regulation during phenotypic transition.
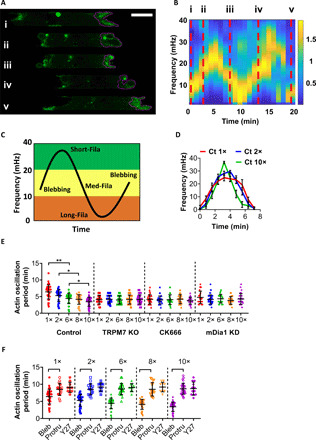
(A) Representative image sequences showing LifeAct-GFP–tagged MDA-MB-231 cells undergoing periodic phenotypic alterations: from (i) protrusive to (ii) blebbing, transition to (iii) hybrid phenotype with actin filaments growing along cell-side channel wall interface, and back to (iv) blebbing and (v) protrusive. The purple outlines were automatically generated from the algorithm detecting the cell cortex at the leading edge. Scale bar, 5 μm. (B) Modified spectrogram of average fluorescence LifeAct-GFP signal intensity of the cell cortex in (A). Red dashed lines pinpoint the time and frequency corresponding to the cell phenotypes in (A). (C) Typical pattern of oscillatory phenotypic transition revealed by the spectrogram of cortical LifeAct-GFP fluorescence intensity. The dominant morphological classification is shown in each of the three frequency domains. (D) Oscillatory pattern extracted from the spectrograms of wild-type LifeAct-GFP–tagged MDA-MB-231 cells migrating in channels of different hydraulic resistances. Data represent means ± SD for n > 10 cells from three independent experiments. (E) Oscillation period determined from the spectrograms of LifeAct-GFP–tagged wild-type, TRPM7 KO, CK666 (100 μM)–treated, and mDia1 KD cells. Data are means ± SD for n > 20 cells from three independent experiments. *P < 0.05 and **P < 0.01, assessed by Kruskal-Wallis test with Dunn’s post hoc. (F) Oscillation period, determined as in (E), at different hydraulic resistances for wild-type cells (distinguished in blebbing/hybrid versus protrusive phenotypes) and Y-27632–treated cells. Data represent means ± SD for n > 10 cells from three independent experiments. P < 0.001, assessed by two-tailed unpaired t test.
We next applied STFT on the oscillation curve to generate a spectrogram, which gives a visual representation of the power of the signal within a predefined range of frequency as a function of time (fig. S4B). A sinusoidal-like frequency modulation curve emerged from the optimized spectrogram (Fig. 4B and fig. S4B), where different frequency ranges were matched and verified with the distinct phenotypes observed in confocal images. An example cell initially displayed medium-sized filaments, as evidenced by frequency oscillations at ≥10 mHz (Fig. 4B, i) and verified by the presence of wave-like protrusions at the lateral wall-cell interfaces in confocal images (Fig. 4A, i). It then quickly assumed a blebbing phenotype, as suggested by the increased frequency at ~15 mHz (Fig. 4B, ii), which was visually verified by the presence of large spherical bulges at the leading edge (Fig. 4A, ii). Next, the cell switches to a protrusive phenotype exhibiting medium filaments at the side channel wall-cell interfaces coupled to a concave membrane curvature in the central protrusive region (Fig. 4A, iii, and fig. S4C). This phenotypic switch was captured in the spectrogram by the intense oscillations at ~15 mHz (Fig. 4B, iii), which was preceded by a peak at ~30 mHz, which corresponds to the assembly of small actin filaments during transition from a blebbing to protrusive phenotype with medium-sized filaments. In the next phenotype switch cycle, the cell assumed again a blebbing phenotype (Fig. 4A, iv) at 15 mHz (Fig. 4B, iv) before transitioning to protrusive (Fig. 4, A, v and B, v) with a convex membrane curvature at the central protrusive area (Fig. 4A, v, and fig. S4C). The overall cell length along the longitudinal axis varied with the cell phenotype and contracted during blebbing while it extended during transition to a protrusive phenotype (fig. S4D).
In a typical phenotype cycle (Fig. 4C), a blebbing cell at ~15 mHz will transition to a protrusive phenotype by retracting its blebs and initiating the assembly of small actin filaments (20 to 40 mHz), which will grow into medium (10 to 20 mHz) and then long (0 to 10 mHz) filaments. Representative spectrograms are shown for more cells before (fig. S4E) and after (fig. S4F) complete transition to a protrusive phenotype. Cumulatively, the spectrogram analysis reveals a periodic cycle of actin signals at the cell leading edge that controls migration phenotype (Fig. 4C).
We extracted the sinusoidal curves from the spectrogram of cells migrating in channels of different hydraulic resistances (Fig. 4D). The bell-shaped curve at 1× became narrower with increasing hydraulic resistance for control cells (Fig. 4D), reflecting a faster turnover (Fig. 4E). TRPM7 KO, which abrogates cell mechanosensitivity (9), abolishes the dependence of the phenotype cycle period on hydraulic resistance by promoting a faster turnover at lower (1× to 2×) resistances (Fig. 4E and fig. S4, G and H). Arp2/3 inhibition or mDia1 KD also results in actin cycles that are independent of hydraulic resistance (Fig. 4E and fig. S4I). The shorter period reflects a faster takeover of actin filaments by blebs at all resistances. In addition, posttransition cells exhibiting a protrusive phenotype and ROCK-inhibited cells display a robust long oscillation period at low frequencies (10 mHz) (Fig. 4F and fig. S4F).
Inside low-resistance channels, the distribution of actin oscillation period of blebbing cells overlapped quite extensively with those displaying a protrusive phenotype (Fig. 4F). However, these two distinct phenotypic subpopulations were split apart into discrete groups at elevated resistances (Fig. 4F). The disappearance of blebbing phenotypes with long oscillation period suggests that their successful transition to protrusive ones requires a shorter time window for sustained actin polymerization due to a reinforced basis for filamentous growth (i.e., elevated Arp2/3 activity and FA density) at higher resistances. The detected population of blebbing cells with short actin oscillation period at high resistances is indicative of survival bias.
The spatiotemporal distribution of myosin II at the cell anterior is regulated by hydraulic resistance and is a key determinant of cell phenotypic transition
The competition of the two migration modes, blebbing versus protrusive, is essentially a scramble for migration-related molecular resources, which include the cytoskeletal components actin and myosin. Cortical actomyosin is the key player in bleb-based migration (14, 24, 29, 30). The protrusive migration mode, which relies on actin bundles, requires myosin localization toward the bundled actin filaments to generate contractions and stabilize FAs (31, 32). We thus investigated the spatiotemporal localization of MIIA-GFP during actin oscillations by focusing on how blebbing cells lose the tug of war to a protrusive phenotype. When blebs were generated at the leading edge, the myosin signal was rather uniformly distributed at the cortex (Fig. 5A, i and iii). When the blebs collapsed, the growth of actin filaments at the cell-lateral channel wall interfaces was accompanied by MIIA translocation from the central protrusive region toward the actin filaments (Fig. 5A, ii and iv, and fig. S5, A and B). The presence of myosin at the center of the cell cortex facilitates the contraction of filaments at the cell-side channel wall interfaces, thereby promoting transition to a blebbing morphology (fig. S5, C to E).
Fig. 5. The spatiotemporal distribution of myosin II at the cell anterior regulated by hydraulic resistance is a key determinant of cell phenotypic transition.
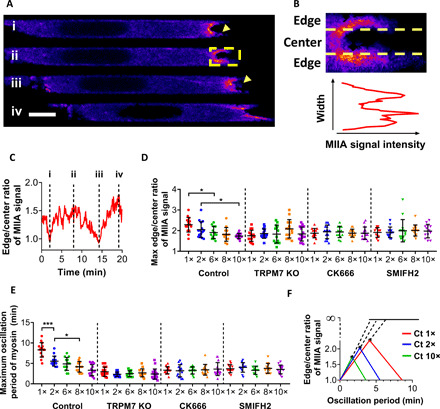
(A) Representative image sequence showing the spatiotemporal pattern of MIIA-GFP at the leading edge of MDA-MB-231 cells during phenotypic transition from blebbing (i and iii) to protrusion (ii and iv). The yellow box outlines the area analyzed in (B). Yellow arrowheads indicate retracting blebs. Scale bar, 5 μm. (B) Top: Yellow dashed lines depict the separation of the cell edge near the side channel wall interface from the center area. Bottom: Average fluorescence intensity profile of the cell cortex. (C) Dynamics of edge-to-center MIIA-GFP fluorescence intensity ratio during the phenotypic transition of the cell in (A). Labels (i) to (iv) correspond to those in (A). (D) Maximum edge-to-center ratio and (E) maximum period of cortical MIIA-GFP oscillation of MDA-MB-231 cells displaying a blebbing and/or hybrid phenotype during phenotypic transition at different hydraulic resistances. Data represent means ± SD for n ≥ 12 control, TRPM7 KO, CK666-inhibited, and formin-inhibited cells from three independent experiments. *P < 0.05 and ***P < 0.001, assessed by one-way ANOVA with Tukey’s post hoc. (F) Using the average values obtained for control cells in (D) and (E), the edge-to-center ratio versus oscillation period of MIIA-GFP was plotted. Asterisks indicate the maximum edge-to-center ratio necessary to retain a blebbing phenotype at different hydraulic resistances. Beyond that threshold, cells transition to a protrusive phenotype.
To quantify the MIIA-GFP signal, we split the cell cortex into three regions, edge-center-edge, and measured its intensity in each of them separately (Fig. 5B). The dynamics of edge/center MIIA intensity ratio exhibited a wave-like periodicity (Fig. 5C). It climbed up when filaments were growing at the cell-lateral channel wall interfaces, and at its apex (Fig. 5C, ii), the cell either started restoring the blebbing phenotype (Fig. 5C, iii) or transitioned to a protrusive phenotype (Fig. 5C, iv). The maximum edge/center ratio before phenotypic transition and the maximum time period of the oscillatory cycle decreased with increasing hydraulic resistance (Fig. 5, D and E). Similarly to actin, TRPM7 KO, Arp2/3, or formin inhibition abolished the dependence of edge/center ratio or MIIA period on hydraulic resistance (Fig. 5, D and E). Notably, the numerical values of the myosin II oscillation period match very well those obtained with actin.
The contractility-induced actin filament growth near the side channel walls facilitates myosin II localization at cell-channel interfaces, thereby increasing the MIIA edge/center ratio. Higher hydraulic resistances promote a faster molecular translocation, as depicted by the elevated slope (Fig. 5F). A stable blebbing phenotype would be maintained as long as the edge/center ratio remains below a threshold value, which is indicative of the cell’s ability to use the myosin II remaining at the central area to generate adequate contractility. This requirement is stricter at elevated hydraulic resistances because a higher local pressure from actomyosin is required for force balance. This implies that cells would tolerate a bigger loss of myosin at the central region in channels of lower hydraulic resistance to maintain the blebbing phenotype, whereas the same edge/center ratio would lead to a protrusive morphology in channels of higher resistance (Fig. 5F).
Calcium oscillations show reduced mechanosensitivity for blebbing cells at high hydraulic resistances
As downstream actuators, both actin and myosin fluorescence signals exhibit periodic oscillatory patterns in response to the hydraulic stimulus. We next focused on calcium as a potential upstream signaling molecule involved in phenotypic transition given its key role in cell motility (9) and cytoskeleton remodeling (33, 34). Using the fluorescent calcium indicator, Fluo-4 Direct, we monitored the intracellular Ca2+ distribution and the intensity of oscillations of nonprotrusive cells subjected to hydraulic resistance. As shown in representative examples, the intensity profile displayed smaller amplitude variations for cells inside channels of higher hydraulic resistance (Fig. 6, A and B), thereby suggesting a weaker regulation of intracellular Ca2+ and downstream signaling activities. By applying wavelet analysis, sinusoidal-like curves emerged from calcium spectrograms (fig. S6A). A decreasing pattern of calcium oscillation period with increasing resistance was observed (fig. S6B), which is consistent with data on actin and myosin signals. In addition, TRPM7 KO, Arp2/3, or formin inhibition resulted in oscillation periods that did not vary with hydraulic resistance (fig. S6B). The oscillatory power measured from the power spectral density (PSD) (fig. S6C), which represents a quantitative description of the signal variation amplitude and serves as a potential measurement of the intracellular activity, decreased with increasing hydraulic resistance in control cells (fig. S6D). TRPM7 KO, Arp2/3, or formin inhibition also lowered the power down to the levels noted at high (8× to 10×) hydraulic resistances (fig. S6D).
Fig. 6. Theory and experiments reveal the mutual regulation of calcium and actin dynamics governing phenotypic transition.
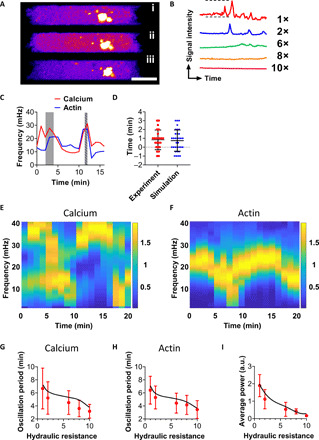
(A) Representative image sequence illustrating the fluctuation of calcium fluorescence intensity monitored by Fluo-4 Direct in a cell migrating inside a channel of 1× resistance. Calcium sparks appear in (ii). Scale bar, 5 μm. (B) Representative calcium signal intensity dynamics of Fluo-4 Direct–loaded MDA-MB-231 cells showing reduced variation of intracellular calcium activity at higher hydraulic resistances. The signal intensity between dashed lines is 100 a.u. (arbitrary units), whereas the time duration is 10 min. (C) Representative oscillation curves extracted from spectrograms generated by Fluo-4 Direct and LifeAct-Ruby2 colocalization confocal images. The phase lag between calcium and actin oscillation was quantified by comparing the locations of their respective peaks at high frequency (gray shadow shows that calcium oscillation precedes that of actin, whereas striped shadow shows the reverse). (D) Quantitation of the phase lag between calcium and actin oscillation over the entire range of hydraulic resistances from experimental and simulation data. Data represent means ± SD for n > 20 cells from three independent experiments/simulations. (E and F) Representative spectrograms generated from simulations of (E) cytosolic calcium concentration and (F) cortical actin concentration profiles of a cell inside a channel of 1× resistance. (G and H) Oscillation periods extracted from spectrograms of simulated (G) calcium and (H) actin (black lines). Red dots correspond to experimental data retrieved from fig. S6B for calcium and Fig. 5F for actin. (I) Average power from PSD of simulated calcium profiles (black line) superimposed by experimental data retrieved from fig. S6D.
The periods of cortical actin turnover, spatial translocation of MIIA, and Ca2+ oscillation collapsed numerically at each hydraulic resistance (fig. S6E). We thus tested the cross-talk of calcium and actin during phenotypic switch by generating spectrograms based on the wavelet signal obtained from cells tagged with LifeAct-Ruby2 and labeled with Fluo-4 Direct (fig. S6F). Upon overlaying the resulting curves, a synchronized oscillation behavior between calcium and actin became apparent (Fig. 6C) with a phase lag of less than 1 min on average (Fig. 6D), noted by comparing the location of oscillation peaks at ~30 mHz.
We developed a calcium-actin model (Materials and Methods), which quantitatively recapitulates the mutual regulation of actin and calcium dynamics observed experimentally (Fig. 6, E and F). The model computes the cytosolic contents for both Ca2+ and F-actin as a function of time. The model outputs are transformed using FFT to reveal how the frequencies of Ca2+ and F-actin components modulate over time and how Ca2+ and F-actin oscillations mutually influence each other (fig. S6, G and H). The frequency domain plots are quantitatively similar to those obtained from fluorescence tracking experiments. The computed spectrograms indicate that the model Ca2+ and actin oscillation periods matched with the experimental trends for all hydraulic conditions (Fig. 6, G and H). The observed power of intracellular Ca2+ in blebbing cells drops monotonically with increasing hydraulic resistance, which is captured quantitatively by the model (Fig. 6I). Moreover, a phase lag of <1 min emerged when comparing the synchronized behavior of simulated Ca2+ and actin (Fig. 6D).
The essential components of the model are described in fig. S6I, which shows the important Ca2+ channels and the mutual coupling of F-actin dynamics with Ca2+. The model contains two important variables, Jin and τ, that drive the changes in the Ca2+/F-actin content as well as the oscillatory patterns seen in the spectrogram. A fixed value of Ca2+ influx described by Jin (fig. S6J) would result in an oscillatory Ca2+ concentration curve (fig. S6K) and a constant frequency (flat line) on the spectrogram (fig. S6L). As Jin varies, the frequency of Ca2+ oscillation varies and a Hopf bifurcation transition is predicted, as observed in experiments. This suggests that Jin is a function of the hydraulic resistance and cortical forces that balance pressure differences across the cell surface. Moreover, Ca2+ influences F-actin dynamics and regulates actin polymerization/depolymerization through the time delay parameter τ (fig. S6, M and N). Therefore, the model describes the interdependence of τ and Jin: Ca2+ influx into the cell is influenced by the external hydraulic resistance and Jin that depends on cortical F-actin content; the F-actin dynamics, in turn, is influenced by Ca2+ concentration. This cross-talk between F-actin and Ca2+ determines the phenotypic structural assemblies at the cell leading edge. The model suggests that the cell uses an intricate mutual feedback between F-actin and Ca2+ that senses external hydraulic resistance and regulates cell phenotype and migration behavior.
Our model links intracellular Ca2+ dynamics with cytoskeletal remodeling and force generation and quantitatively explains experimental data. A key feature of the model is the Ca2+ influx, described by Jin, which depends on membrane tension and thus on contraction in the cortex and the pressure difference across the membrane (6, 34). The pressure difference, in turn, depends on the external hydraulic resistance (9). Increasing active contraction lowers membrane tension and decreases Ca2+ influx. Changes in the intracellular Ca2+, in turn, regulate actomyosin dynamics and cytoskeletal force generation by influencing actin polymerization and turnover through the parameter τ. This mutual coupling generates the observed coupled oscillations in Ca2+ and actin dynamics. The model correctly predicts that the intracellular Ca2+ concentration undergoes a Hopf bifurcation with increasing hydraulic resistance. The Ca2+ oscillation period as a function of resistance is consistent with experimental observations. Moreover, cortical actin content also oscillates, and the oscillation period is modulated by the hydraulic resistance and Ca2+. Therefore, the emerging physical picture is that Ca2+ influx is an important regulatory element in cell cytoskeletal organization, and under conditions of large hydraulic resistance, Ca2+ influx is a signal of local pressure change. The cell uses this signal to modulate cortical cytoskeleton organization and contractility to ensure proper force balance at the cell surface. The model currently accounts for a generic actomyosin network. However, distinct types of actin filaments (short, medium, and long) were observed experimentally, and thus, there is a rich behavior in their coupling to Ca2+. Ca2+ also directly influences cytoskeletal contraction through tropomyosin, and we have accounted for their involvement in a simple actomyosin model. Notably, the presence of FAs also influences the coupled oscillatory dynamics between actin and Ca2+ observed in our system.
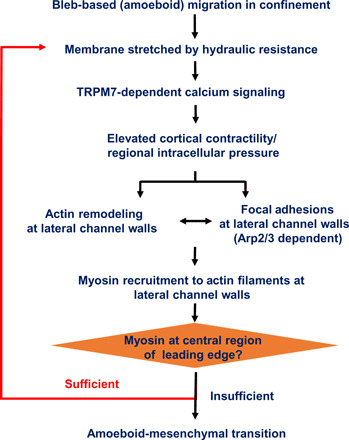
In summary, by combining automated morphological tracking and wavelet analysis, we found a periodic phenotypic switch in migrating MDA-MB-231 breast cancer cells in confinement involving a cycle from blebbing to short, medium, and long actin filament formation that loops back to blebbing and identified hydraulic resistance as a key parameter tuning migration phenotype (Fig. 7). Elevated hydraulic resistance promotes the growth of actin filaments at cell-side channel wall interfaces stabilized by FAs and recruits myosin motors from the cortical network, thereby lowering cortical contractility and driving AMT. Hydraulic resistance is sensed by TRPM7, which regulates intracellular Ca2+ and actomyosin turnover via a feedback mechanism that is quantitatively described by a novel mathematical model. A limitation of this study pertains to the use of a single, albeit widely used, human MDA-MB-231 breast cancer cell line.
Fig. 7. Signaling pathway regulating cell phenotype in response to hydraulic resistance.
MATERIALS AND METHODS
Cell culture
Human MDA-MB-231 breast cancer cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (Gibco) and 1% penicillin/streptomycin (10,000 U/ml; Gibco). Cells were maintained in an incubator at 37°C with 5% CO2 and passaged upon 60 to 80% confluency. Stable cell lines expressing LifeAct-GFP, LifeAct-Ruby2, Arp3-mCherry, Actin-GFP, or paxillin-GFP were generated via lentivirus transduction (9). MIIA-GFP and TRPM7 KO cell lines were generated as previously described (9).
Microfluidic device fabrication, cell seeding, cell treatment, and live-cell imaging
Polydimethylsiloxane (PDMS)–based microfluidic devices containing channels of prescribed height (10 μm), width (4 μm), and different lengths varying from 150 to 1500 μm, corresponding to distinct hydraulic resistances, were fabricated as described previously (9). The dimensions of all channels were verified by laser profilometer. In migration assays, all channels were incubated with collagen I (20 μg/ml; rat tail, Thermo Fisher Scientific) for at least 1 hour at 37°C in the presence of 5% CO2. Cells (105) suspended in serum-containing medium were added to the device inlet (9). No chemotactic gradient was applied in these experiments. In select experiments, cells were treated with the following pharmacological agents or corresponding vehicle controls: CK666 (100 μM; Sigma-Aldrich), SMIFH2 (25 μM; Sigma-Aldrich), NSC23766 (50 μM; Tocris Biosciences), and Y-27632 (30 μM; Sigma-Aldrich).
Cells were imaged every 10 min for ~20 hours in an inverted Nikon Eclipse Ti microscope (Nikon, Tokyo, Japan) equipped with a stage top incubator (Okolab, Pozzuoli, Italy or Tokai Hit, Shizuoka, Japan) at 37°C and 5% CO2, automated controls (NIS-Elements, Nikon), and a 10×/0.45 numerical aperture Ph1 objective. Cells were also imaged using a Nikon A1 confocal microscope (Nikon, Tokyo, Japan) every 1 to 5 s for 20 to 120 min with a 63× oil objective and fluorescein isothiocyanate and tetramethyl rhodamine isothiocyanate filters.
Hydraulic resistance measurements
The absolute value of hydraulic resistance for each channel was calculated using the formula (5)
where μ is the viscosity of the media; L, W, and H are the length, width, and height of the channel (with W/H << 1), respectively. α is a dimensionless parameter defined as (5)
Speed measurements and phenotypic characterization
Live-cell videos were analyzed using ImageJ (National Institutes of Health, Bethesda, MD). The tracks of cells with entire boundaries inside the channels were obtained manually via MTrackJ plugin. Only individual cells (lacking contacts with other cells) were analyzed. Cell speed was calculated via a custom MATLAB script (MathWorks, Natick, MA).
The percentage of cells displaying a blebbing phenotype was measured at a random time point 5 to 15 hours from the onset of migration experiments. Cells with a spherical bulge at their poles were considered as blebbing in the absence of any protrusions. Blebbing cells bearing actin-based protrusions are classified as hybrid. In Fig. 1B, the percentage of blebbing cells accounts for both purely blebbing and hybrid cells.
LifeAct-GFP–tagged cells captured by microscopy were detected and cropped by a custom-made MATLAB script. The overall length of the cell, which is defined as the distance between the leading and trailing edges, and the protrusion length, which represents the integrated length of cell parts that were ≤70% of the channel width, were recorded to generate a normalized protrusion length for x axis in the phase diagram. The fluorescence signals for each pixel inside the cell body were measured, and the SD was divided by the average value to obtain the coefficient of variation that represents the y axis. Consecutive frames (~10; corresponding to ~100 min) for each cell were selected. Cell footprints (>700) were used to generate the phase diagram for each channel of prescribed resistance with Parula colormap.
Cell exposure to hydrostatic pressure differentials
Cylindrical wells were generated by bonding PDMS-based cylinders (diameter, D = 6 mm; H = 1 cm) to a glass slide via oxygen plasma treatment (9) and then coated with collagen I. Prescribed hydraulic pressure differentials (3 or 30 Pa) were applied by adding 10 or 100 μl of medium atop the cells seeded at the bottom of the wells.
Line scan intensity profiling
For each cell, line scan LifeAct-GFP and MIIA-GFP intensity profiles were obtained and averaged from 20 lines across the cell with equal distance from the channel walls using a custom MATLAB script (29). The curve was smoothed by calculating the moving average (200 points of window size with ~2 million data points of sample size), and ~200 points were plotted for simplicity. All points were normalized by the base value in the cell cytosol between the nucleus and the leading edge. The peak value at the leading edge cortex was recorded.
Paxillin-GFP side view reconstruction and quantification of FAs
Z-stack images of >40 per paxillin-GFP–expressing cell were captured within ~1 min and imported into ImageJ. A 3- to 5-pixel thin layer near the side wall was cropped and averaged into a line profile. A custom-made MATLAB script was used to compile multiple line profiles into a 2D image to reconstruct the side view of the migrating cell (colormap: Parula). Standard image segmentation algorithms involving adaptive threshold binarization, Canny edge detection, dilation and thinning of closed contour, mask filtering, and image region property measurements were applied to characterize the number and the unit area of the FAs. The same algorithms were used for FA detection and quantification at the basal layer. In all images following adaptive segmentation, each pixel’s intensity value was assigned a color according to ImageJ’s fire heatmap or MATLAB Parula for visualization purposes.
Quantification of fluorescence intensity at cell periphery
Standard image segmentation algorithms discussed above were applied to analyze cell responses following the application of a hydrostatic pressure differential. All pixels from the outmost contour were recorded and then deleted from the original image. The processed image was further analyzed using the same procedure to record and remove the newly detected outline. After repeating this process for six times, the intensities of all recorded pixels were averaged to provide a representative value for data comparison. The spreading area is quantified using mask filtering and measuring image region properties.
Fluorescence recovery after photobleaching
FRAP experiments were conducted as reported in (35). A circular ROI with a diameter equal to the channel width was chosen at the leading edge of a β-actin–GFP–expressing cell. The cell was initially imaged for 2 min before laser ablation at the ROI. Next, the cell was monitored for another 15 min. The video was converted and analyzed by a customized MATLAB script, which quantifies the intensity profiles at three locations: leading edge, unaffected cytosolic region, and background signal inside the channel. The leading edge was further split into three regions—top 20% width (edge 1), middle 60% width (center), and bottom 20% width (edge 2)—and for each region, the intensity curve was calibrated as follows
The recovery part of the calibrated curve was fitted by the following exponential growth function
where I0, A, and t0 are the fitted parameters from input I, calibrated intensity, and t, time in seconds. The calculated t0 value represents the characteristic or the turnover time (seconds) of actin structures.
Wavelet analysis and spectrogram optimization
The cortical structure was extracted from LifeAct-GFP images by standard image segmentation algorithms. The average fluorescence signal intensity within the enclosed cortical area from each frame was quantified and normalized by the cytosolic value. The resulting curve was further normalized to the corresponding moving average, and then 1 was subtracted from the processed values to generate the oscillation curve, which was processed by discrete Fourier transform and STFT in customized MATLAB scripts with the following parameters (including pre- and post-analysis).
The spectrogram was optimized as detailed in fig. S4B. The sinusoidal wave-like curve was detected by tracking the maximum value per column. The oscillation period was obtained by averaging the time periods between local peaks and between local valleys. The average oscillation frequency was calculated by averaging the frequencies from the extracted curves.
Membrane curvature measurement
Image segmentation algorithms were applied to extract the cortical region of a moving LifeAct-GFP–tagged cell. The overall cortical curvature is measured by finding the skeleton line from the 2D geometry of the cortex. The membrane curvature at the cell leading edge is calculated by fitting the overall cortical curvature with a second-degree polynomial and obtaining the prefactor of the second order as the representative value.
Edge-to-center fluorescence ratio of myosin IIA
The leading edge of MIIA-GFP–tagged cells was extracted by standard image segmentation algorithms and split into three regions: top 20% width (edge 1), middle 60% width (center), and bottom 20% width (edge 2). The average intensity of the two edges combined versus the central region was calculated by integration of all pixel intensities and normalization over their respective areas. Local minima were automatically detected from the resulting edge-to-center ratio plot, and the oscillation period was obtained by finding the maximum distance between two adjacent points.
Calcium imaging and signal analysis
The calcium assay was performed using the Fluo-4 Direct Calcium Assay kit (9). A 0.4× reagent was added into the microfluidic device 2 hours after cell seeding to ensure complete cell entry into channels of prescribed resistance.
The overall calcium fluorescence intensity was measured using the aforementioned image segmentation algorithms and was used for wavelet analysis. The PSD was generated with the same parameter settings as those in Table 1 using MATLAB. From the PSD, the total power at each time domain was calculated by summing up the power of oscillations in the entire range of frequencies. The resulting curve was averaged over time to provide the average power.
Table 1. Parameter settings for spectrogram analysis in MATLAB.
| Variable | Value |
| Moving average neighbor data points | 10 |
| Time interval (s) | 5 |
| Window size (pixel) | 20 |
| Overlapping (%) | 50 |
| Frequency range (Hz) | [0.001, 0.04] |
| Number of frequency domains | 40 |
| Filter size for blurring (pixel) | 3 |
Statistical analysis
All data represent the means ± SD or means ± SEM from ≥3 independent experiments for each condition unless stated otherwise. Normality was determined using a D’Agostino and Pearson test. Two-tailed unpaired t test, Mann-Whitney U test, one-way analysis of variance (ANOVA) (with Tukey’s post hoc) test, and Kruskal-Wallis (with Dunn’s post hoc) test were used, wherever appropriate, to determine statistical significance. Statistical significance was identified as P < 0.05 in GraphPad Prism 8. Data analysis was also performed using MATLAB, GraphPad Prism 8, and OriginPro 2020 Software.
Theoretical model
Here, we present a phenomenological model to describe the oscillatory F-actin and calcium dynamics observed when cells migrate in different hydraulic resistance environments. We first describe the calcium model and the actin model separately and then combine them to model the observed mutual influence in experiments.
For the calcium model, we assume that during the time scale of the experiment, the concentration of relevant species is homogeneous throughout. We write c for the concentration of free Ca2+ ions in the cytoplasm, and denote ce as the homogeneous concentration of Ca2+ in the endoplasmic reticulum (ER). The rate of change c and ce is equal to the net flux of calcium in each compartment; therefore, for the cytoplasmic compartment
| (1) |
where we consider four major calcium fluxes: JIPR represents calcium released from the ER through the IP3 (inositol 1,4,5-trisphosphate) receptor (IPR), Jserca represents calcium pumped from the cytoplasm into the ER, Jin represents calcium entry from the extracellular environment through a variety of membrane channels, and Jout represents calcium pumped from the cytoplasm to the extracellular environment by active transport (Ca-ATPase).
For the ER Ca2+, we define the parameter , which is the ratio of the cytoplasmic volume to the ER volume. Then, we can write the ER Ca2+ as
| (2) |
For JIPR, we use Sneyd and Dufour’s saturating binding model of IPR (36); for Jserca, we use a four-state Markov model (36); for Jout, we use an early model of the Ca2+ adenosine triphosphatase (ATPase) pump, which uses a Hill equation for the flux with a Hill coefficient of about 2 (36). The detailed equations are
where
It is assumed that the IPR consists of four identical and independent subunits and that it allows calcium current when all four subunits are in either the O (open) or the A (activated) state. To derive the equation for all states, we use the sequential model of the IPR with saturating binding rates
where
The four-state Markov model for the SERCA calcium pump writes
Last, for the surface Ca-ATPase pump
The detailed parameters for these expressions have been established (36), and the key ones are listed in Table 2. These parameters follow previously published calcium models.
Table 2. List of parameters and their description.
| Parameters | Description | Values |
| r | The ratio of the cytoplasmic volume to the ER volume |
5.4 |
| a | Parameter that relates pressure to flux | 0.1 |
| K | Linear coefficient that relates F-actin and pressure inside of the cell |
0.1 |
| k+ | Constant for the polymerization rate | 0.2 s−1 |
| k− | Constant for the depolymerization rate | 0.2 |
| rh | Linear coefficient that relates calcium concentration and depolymerization rate of F-actin |
0.2 |
| α | Constant in the τ Hill equation | 15 s−n |
| β | Constant in the τ Hill equation | 0.9–1.32 μM |
| n | Exponent in the Hill equation | 2.5 |
| τ0 | Primary time delay parameter | 6 s |
For Ca2+ influx from the extracellular environment, Jin, we consider the possible action of surface mechanosensitive Ca2+ channels, which can open upon an increase in cell membrane tension. The cell membrane tension is approximately determined by the hydraulic pressure difference and active stress in the cell cortex generated by myosin contraction (34)
where T is the membrane tension, R is the cell surface mean curvature, and Pin and Pout are the hydraulic pressures inside and outside of the cell, respectively. σa is the active stress in the cytoskeletal cortex, and h is the cortical thickness. Note that Pout depends on the cell speed and the hydraulic resistance external to the cell. With increasing hydraulic resistance, Pout should increase (7, 9). Most of the time, the pressure difference is sustained by cortical stress, and T is small. When there is insufficient cortical actin and myosin to balance the pressure difference, the membrane tension can temporally increase, leading to Ca2+ influx through mechanosensitive ion channels such as TRPM7. Therefore, we model σa as being proportionally related to the actin content in the cortex, A(t). Therefore, Jin should be positively correlated with T and proportional to the difference of the internal and external hydraulic pressures. We phenomenologically write Jin as
| (3) |
where a and K are parameters. Note that, formally, Jin depends on other Ca2+ channels on the cell surface. However, those contributions are effectively included by parameters such as a. The physical meaning of Jin is simply that with increasing cortical actin and active contraction, membrane tension and Jin should decrease. This idea is roughly captured by Eq. 3.
Next, we consider F-actin cortical dynamics, which, in general, can be complex, involving the processes of actin polymerization/depolymerization, branching by Arp2/3, and myosin activity. For the sake of simplicity, to describe the oscillatory dynamics we observed in the experiment, we use a nonlinear time delay equation to describe actin oscillations. The change in F-actin concentration should be determined by two factors: polymerization and depolymerization. Hence, the magnitude of the depolymerization term should depend on the F-actin concentration in the past and the presence of F-actin at the current time. Since actin polymerization occurs on existing filaments, the polymerization term also should depend on the current F-actin content. Let k+ represent the effective polymerization rate and k− represent the effective depolymerization rate, we can write the F-actin equation as
Performing an expansion for the time-delayed term up to the second order, we rewrite the above equation as
| (4) |
In eukaryotic cells, Ca2+ pulses cause depolymerization of F-actin at the cortex (37), possibly through the action of actin disassembly factors such as gelsolin. Therefore, we can assume that the concentration of calcium inside the cell is linearly proportional to the actin depolymerization rate
| (5) |
where k and r are parameters. We also assume that k+ is a constant.
Equation 4 can generate oscillations in the F-actin oscillation as seen in experiments. It is easy to see that the time scale parameter τ influences the oscillation frequency, and calcium concentration will affect this frequency in multiple ways. We use a Hill function with exponent n to describe the influence of Ca2+ as
| (6) |
where τ0 is the baseline depolymerization time scale in the absence of calcium. In reality, Ca2+ also regulates myosin binding to F-actin through tropomyosin and affects active stress σa. Therefore, a detailed description of A(t) requires more complex models. For the sake of simplicity here, we introduce the simplest Ca2+ regulation of active stress in this model. All the relevant parameters for the model are listed in Table 2.
Equations 1 to 6 constitute our model of Ca2+ and F-actin dynamics, which can be solved using ODE solvers in MATLAB. First, our model shows that with increasing external hydraulic resistance, the cytoplasmic Ca2+ concentration increases until a bifurcation point at which oscillations in Ca2+ concentration appear. Next, we remove the DC component by subtracting the average of the normalized data to get the normalized concentration curves, which are similar to our experimental data. Last, we use the final normalized data to generate the FFT (fig. S6, G and H) and STFT (Fig. 6, E and F).
Acknowledgments
Funding: This work was supported, in part, by grants from the NIH R01CA183804 to K.K. and R01GM134542 and U54-CA210173 to S.X.S. and K.K. Author contributions: R.Z. designed the study, performed most experiments, analyzed data, and wrote the manuscript. S.C., Y.Z., K.B., and L.Z. performed select experiments and analyzed data. Z.G. and S.X.S. developed the theoretical model, extracted and analyzed the corresponding data, and edited the manuscript. K.K. designed and supervised the study and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/17/eabg4934/DC1
REFERENCES AND NOTES
- 1.Paul C. D., Mistriotis P., Konstantopoulos K., Cancer cell motility: Lessons from migration in confined spaces. Nat. Rev. Cancer 17, 131–140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weigelin B., Bakker G. J., Friedl P., Intravital third harmonic generation microscopy of collective melanoma cell invasion: Principles of interface guidance and microvesicle dynamics. IntraVital 1, 32–43 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stroka K. M., Jiang H., Chen S. H., Tong Z., Wirtz D., Sun S. X., Konstantopoulos K., Water permeation drives tumor cell migration in confined microenvironments. Cell 157, 611–623 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prentice-Mott H. V., Chang C. H., Mahadevan L., Mitchison T. J., Irimia D., Shah J. V., Biased migration of confined neutrophil-like cells in asymmetric hydraulic environments. Proc. Natl. Acad. Sci. U.S.A. 110, 21006–21011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuerstman M. J., Lai A., Thurlow M. E., Shevkoplyas S. S., Stone H. A., Whitesides G. M., The pressure drop along rectangular microchannels containing bubbles. Lab Chip 7, 1479–1489 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Li Y., Konstantopoulos K., Zhao R., Mori Y., Sun S. X., The importance of water and hydraulic pressure in cell dynamics. J. Cell Sci. 133, jcs240341 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y., Sun X., Transition from actin-driven to water-driven cell migration depends on external hydraulic resistance. Biophys. J. 114, 2965–2973 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moreau H. D., Blanch-Mercader C., Attia R., Maurin M., Alraies Z., Sanseau D., Malbec O., Delgado M. G., Bousso P., Joanny J. F., Voituriez R., Piel M., Lennon-Dumenil A. M., Macropinocytosis overcomes directional bias in dendritic cells due to hydraulic resistance and facilitates space exploration. Dev. Cell 49, 171–188.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Zhao R., Afthinos A., Zhu T., Mistriotis P., Li Y., Serra S. A., Zhang Y., Yankaskas C. L., He S., Valverde M. A., Sun S. X., Konstantopoulos K., Cell sensing and decision-making in confinement: The role of TRPM7 in a tug of war between hydraulic pressure and cross-sectional area. Sci. Adv. 5, eaaw7243 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balzer E. M., Tong Z. Q., Paul C. D., Hung W.-C., Stroka K. M., Boggs A. E., Martin S. S., Konstantopoulos K., Physical confinement alters tumor cell adhesion and migration phenotypes. FASEB J. 26, 4045–4056 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holle A. W., Devi N. G. K., Clar K., Fan A., Saif T., Kemkemer R., Spatz J. P., Cancer cells invade confined microchannels via a self-directed mesenchymal-to-amoeboid transition. Nano Lett. 19, 2280–2290 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hung W. C., Chen S. H., Paul C. D., Stroka K. M., Lo Y. C., Yang J. T., Konstantopoulos K., Distinct signaling mechanisms regulate migration in unconfined versus confined spaces. J. Cell Biol. 202, 807–824 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y. J., Le Berre M., Lautenschlaeger F., Maiuri P., Callan-Jones A., Heuze M., Takaki T., Voituriez R., Piel M., Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 160, 659–672 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Wisniewski E. O., Mistriotis P., Bera K., Law R. A., Zhang J., Nikolic M., Weiger M., Tuntithavornwat S., Afthinos A., Zhao R., Kalab P., Scarcelli G., Friedl P., Konstantopoulos K., Dorsoventral polarity directs cell responses to migration track geometries. Sci. Adv. 6, eaba6505 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim K. H., Kim S. J., A wavelet-based method for action potential detection from extracellular neural signal recording with low signal-to-noise ratio. IEEE Trans. Bio. Med. Eng. 50, 999–1011 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Charras G. T., Yarrow J. C., Horton M. A., Mahadevan L., Mitchison T. J., Non-equilibration of hydrostatic pressure in blebbing cells. Nature 435, 365–369 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bieling P., Li T. D., Weichsel J., McGorty R., Jreij P., Huang B., Fletcher D. A., Mullins R. D., Force feedback controls motor activity and mechanical properties of self-assembling branched actin networks. Cell 164, 115–127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mueller J., Szep G., Nemethova M., de Vries I., Lieber A. D., Winkler C., Kruse K., Small J. V., Schmeiser C., Keren K., Hauschild R., Sixt M., Load adaptation of lamellipodial actin networks. Cell 171, 188–200.e16 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Serrels B., Serrels A., Brunton V. G., Holt M., McLean G. W., Gray C. H., Jones G. E., Frame M. C., Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat. Cell Biol. 9, 1046–1056 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Chorev D. S., Moscovitz O., Geiger B., Sharon M., Regulation of focal adhesion formation by a vinculin-Arp2/3 hybrid complex. Nat. Commun. 5, 3758 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Swaminathan V., Fischer R. S., Waterman C. M., The FAK-Arp2/3 interaction promotes leading edge advance and haptosensing by coupling nascent adhesions to lamellipodia actin. Mol. Biol. Cell 27, 1085–1100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isogai T., van der Kammen R., Leyton-Puig D., Kedziora K. M., Jalink K., Innocenti M., Initiation of lamellipodia and ruffles involves cooperation between mDia1 and the Arp2/3 complex. J. Cell Sci. 128, 3796–3810 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Fritzsche M., Lewalle A., Duke T., Kruse K., Charras G., Analysis of turnover dynamics of the submembranous actin cortex. Mol. Biol. Cell 24, 757–767 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charras G., Paluch E., Blebs lead the way: How to migrate without lamellipodia. Nat. Rev. Mol. Cell Biol. 9, 730–736 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Miyoshi T., Tsuji T., Higashida C., Hertzog M., Fujita A., Narumiya S., Scita G., Watanabe N., Actin turnover-dependent fast dissociation of capping protein in the dendritic nucleation actin network: Evidence of frequent filament severing. J. Cell Biol. 175, 947–955 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai F. P., Szczodrak M., Block J., Faix J., Breitsprecher D., Mannherz H. G., Stradal T. E., Dunn G. A., Small J. V., Rottner K., Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J. 27, 982–992 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mogilner A., Rubinstein B., The physics of filopodial protrusion. Biophys. J. 89, 782–795 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pankova K., Rosel D., Novotny M., Brabek J., The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell. Mol. Life Sci. 67, 63–71 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mistriotis P., Wisniewski E. O., Bera K., Keys J., Li Y., Tuntithavornwat S., Law R. A., Perez N., Erdogmus E., Zhang Y., Zhao R., Sun S. X., Kalab P., Lammerding J., Konstantopoulos K., Confinement hinders motility by inducing RhoA-mediated nuclear influx, volume expansion, and blebbing. J. Cell Biol. 218, 4093–4111 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruprecht V., Wieser S., Callan-Jones A., Smutny M., Morita H., Sako K., Barone V., Ritsch-Marte M., Sixt M., Voituriez R., Heisenberg C. P., Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell 160, 673–685 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alieva N. O., Efremov A. K., Hu S., Oh D., Chen Z., Natarajan M., Ong H. T., Jegou A., Romet-Lemonne G., Groves J. T., Sheetz M. P., Yan J., Bershadsky A. D., Myosin IIA and formin dependent mechanosensitivity of filopodia adhesion. Nat. Commun. 10, 3593 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tojkander S., Gateva G., Schevzov G., Hotulainen P., Naumanen P., Martin C., Gunning P. W., Lappalainen P., A molecular pathway for myosin II recruitment to stress fibers. Curr. Biol. 21, 539–550 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Wollman R., Meyer T., Coordinated oscillations in cortical actin and Ca2+ correlate with cycles of vesicle secretion. Nat. Cell Biol. 14, 1261–1269 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tao J., Sun S. X., Active biochemical regulation of cell volume and a simple model of cell tension response. Biophys. J. 109, 1541–1550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sprague B. L., Pego R. L., Stavreva D. A., McNally J. G., Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys. J. 86, 3473–3495 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.J. P. Keener, J. Sneyd, Mathematical Physiology (Interdisciplinary applied mathematics, Springer, ed. 2, 2009). [Google Scholar]
- 37.Takeshita N., Evangelinos M., Zhou L., Serizawa T., Somera-Fajardo R. A., Lu L., Takaya N., Nienhaus G. U., Fischer R., Pulses of Ca2+ coordinate actin assembly and exocytosis for stepwise cell extension. Proc. Natl. Acad. Sci. U.S.A. 114, 5701–5706 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/17/eabg4934/DC1