

HeLa cancer cells in mice can be tagged in vivo with therapeutic moieties, thereby disrupting either tumor onset or growth.
Abstract
This study presents the early framework of selective cell tagging (SeCT) therapy, which is the concept of preferentially labeling specific cells in vivo with chemical moieties that can elicit a therapeutic response. Using glycosylated artificial metalloenzyme (GArM)–based protein labeling, this study reports two separate functional strategies. In one approach, early tumor onset can be suppressed by tagging cancer cells in living mice with an integrin-blocking cyclic–Arg-Gly-Asp (cRGD) moiety, thereby disrupting cell adhesion onto the extracellular matrix. In another approach, tumor growth in mice can be reduced by tagging with a cytotoxic doxorubicin moiety. Subsequent cell death occurs following internalization and drug release. Overall, experiments have shown that mouse populations receiving the mixture of SeCT labeling reagents exhibited a significant delay/reduction in tumor onset and growth compared with controls. Highlighting its adaptability, this work represents a foundational step for further development of SeCT therapy and its potential therapeutic applications.
INTRODUCTION
Chemical labeling methodologies for surface proteins of living cells in vivo have garnered much attention over the past decade (1). The upcoming challenge in this field, however, has slowly begun to shift to how these techniques can be adapted to manipulate biological processes for therapeutic benefit. For this to happen, in vivo labeling methodologies will likely need to be adaptable for whole cell–specific targeting using external reagents, which unfortunately precludes methods that require genetic modifications (2, 3), and protein-targeted approaches (4, 5). For methods such as metabolic glycoengineering (6–8) and glycan-targeted metal-catalyzed labeling (9), there is a clear opportunity to explore their therapeutic potential.
The concept of selective cell tagging (SeCT) therapy is based on the idea of tagging specific cells in vivo with molecules of biological interest. Some options include the use of nontoxic chemical moieties that can lead to impaired cellular function or the use of moieties that can locally release cytotoxic drugs without affecting surrounding tissue. This work aims to present a working methodology for SeCT therapy.
Two key concepts form the foundation of the design depicted in Fig. 1. The first aspect is based on the cancer-specific cell targeting observed with glycan-based targeting methods. Given that a well-known hallmark of cancer cells is their altered glycosylation pattern (10), researchers within the glycoscience field have long been interested in developing targeting methodologies capable of discriminating differences in cell surface glycosylation. Previous works done by our group have focused on constructing libraries of albumin glycoconjugates using complex N-glycans (glycoalbumins) to identify possible cancer cell–specific targeting interactions (11–15). As a result of these studies, glycoalbumins conjugated with a homogeneous composition of α(2,3)sialic acid–terminated complex N-glycans were identified for their ability to rapidly accumulate (<2 hours) in mice to implanted A431 epidermoid squamous carcinoma tumors (12) and HeLa229 cervical adenocarcinoma tumors (14).
Fig. 1. SeCT therapy is a concept based on tagging specific cells in vivo with chemical moieties of therapeutic benefit.
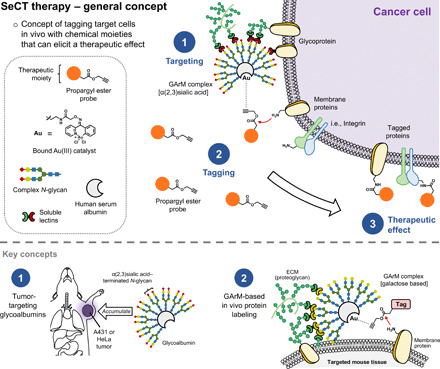
The approach presented in this study is based on two key areas of research. The first is the development of tumor-targeting glycoalbumins. Proteins decorated with certain assemblies of complex N-glycans (i.e., α(2,3)sialic acid–terminated glycans) have been shown to exhibit preferential accumulation to certain tumor types in mice. The second is the development of gold-mediated, PE-based protein labeling. This reaction has allowed GArM complexes to be used for in vivo protein labeling.
The second key aspect in the design approach relates to the in vivo labeling mechanism, which uses glycosylated artificial metalloenzyme (GArM)–based labeling (9, 16). Acting through an active intermediate formed via Au(III)–catalyzed aryl-alkynyl cross coupling between the 2-benzoylpyridine ligand complex and a propargyl ester (PE) probe (16), proteins can be labeled via lysine amidation. Targeted GArM-based labeling has previously been shown to be viable for the localized labeling of specific organs (e.g., liver and intestines) in living mice (9).
Combining these two principles, the focus of this study will principally investigate suitable chemical moieties for the in vivo labeling of cancer cells that can elicit a therapeutic effect. To do this, two separate strategies were explored that worked by either suppressing early tumor onset or reducing tumor growth.
In the first section, SeCT therapy will be used as a means to disrupt cancer cell adhesion at a cellular level. In general, a major component of cell adhesion is the interaction between integrins and extracellular matrix (ECM) proteins that contain the Arg-Gly-Asp (RGD) attachment site (e.g., fibronectin, vitronectin, fibrinogen, and van Willebrand factor) (17). Once bound, the cytoplasmic tail of integrins then initiates signaling cascades that regulate migration and adhesion via tyrosine phosphorylation of focal adhesion kinases (18). As a means to block these interactions, researchers have shown that cyclic–Arg-Gly-Asp (cRGD) pentapeptides are effective at inhibiting overexpressed αVβ3 and αVβ5 integrins on cancer cell surfaces, thereby preventing their attachment to the ECM (19–21). In addition, antagonizing integrins expressed on vascular endothelial cells have been shown to promote tumor regression through inhibition of angiogenesis (22, 23). To develop a means to suppress tumor onset via SeCT therapy in mouse models, an approach was devised so that cancer cells could be selectively tagged with a cRGD-linked PE (cRGD-PE). With surface integrins exposed to a higher localized concentration of cRGD, disruption of cell adhesion is expected to occur, which can lead to the suppression and delay of tumor onset.
In the second section of this study, another approach will focus on the use of SeCT therapy to reduce tumor growth and progression via labeling with a cytotoxic drug moiety. This strategy relies on selectively tagging cancer cell surface proteins with a doxorubicin-linked PE (doxo-PE). Widely known as an anticancer agent that blocks DNA synthesis via intercalation and inhibits topoisomerase II (24), it is expected that doxorubicin can cause cell death once internalized into tagged cancer cells. To facilitate release, the valine (Val)–citrulline (Cit) dipeptide was introduced into the linker moiety, which can undergo selective cleavage by overexpressed lysosomal proteases like cathepsin B (25). The active doxorubicin molecules are thus expected to elicit cell death, leading to the reduction in tumor growth and progression.
RESULTS
Flow cytometry studies to verify HeLa cell targeting
For these studies, HeLa cancer cells were chosen as the model cell line because of previous observations that glycoalbumins conjugated with α(2,3)sialic acid–terminated complex N-glycans could elicit preferential accumulation (14). As an extension, the HeLa targeting capacity of the Au-bound GArM complex was investigated with a series of flow cytometry studies. As an experimental note, the Au catalyst XV is anchored into the drug site I binding pocket of albumin via a high-affinity coumarin ligand, which itself undergoes increased fluorescence due to the hydrophobic environment (26). Under these conditions, fluorescence measured at λEx = 405 nm/λEm = 470 nm was deemed to be a good indicator of cells internalized with the Au-bound GArM complexes during analysis.
First, using cultured HeLa cells, flow cytometry experiments (Fig. 2A) showed a clear shift between HeLa cells treated with and without GArM. This distinction is further highlighted by analyzing a mixture of untreated and treated HeLa cells, which displayed two defined peaks. As a control, the ability of GArM to bind and internalize in macrophages extracted from mice abdominal cavities was next explored. By comparing histograms (Fig. 2B), an insubstantial change between macrophages treated with and without GArM was observed, which suggests little to no binding and internalization. In a final test to measure selectivity, a combination of HeLa cells and macrophages incubated together with and without GArM was monitored (Fig. 2C). For this experiment, the HeLa-V cell line, which expresses the Venus (V) yellow fluorescent protein (27), was used to allow observable differentiation between HeLa and macrophage cells. From the flow cytometry profile, a clear distinction can be made between Venus-expressing HeLa cells and macrophages. Once in the presence of GArM, the majority of HeLa-V cells were observed to move into the upper-right quadrant representative of GArM binding. Macrophages, on the other hand, do not show this similar shift. It should be noted that the small peaks observed around 105 in the upper-right quadrant are indicative of polymerized albumin bound to the Au catalyst XV. These impurities are typically the result of the commercial purification process (ethanol fractionation), which tends to promote trace amounts of albumin aggregation. From these data overall, it can be strongly suggested that GArM can bind and preferentially accumulate to HeLa cancer cells on a cellular level.
Fig. 2. Fluorescence-activated cell-sorting studies to validate the ability of the GArM complex to selectively accumulate toward HeLa cancer cells over peritoneal macrophages.
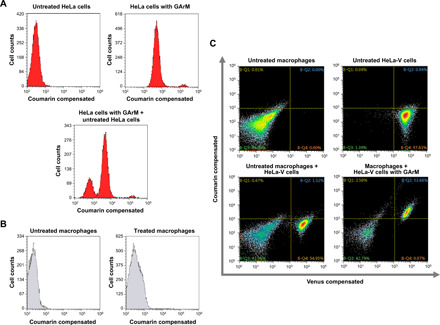
(A) Flow cytometry histograms of HeLa cells treated with and without GArM. (B) Flow cytometry histograms of macrophages (extracted from mice abdominal cavities) treated with and without GArM. (C) Flow cytometry histograms for the mixture of HeLa-V and macrophages cells incubated with and without GArM. For all flow cytometry studies, GArM is bound with a coumarin-linked Au catalyst, which forms a fluorescent protein complex that can be measured at λEx = 405 nm/λEm = 470 nm. Venus (V) protein can be measured at λEx = 515 nm/λEm = 528 nm.
Approach 1: Labeling of targeted cells with cRGD to prevent cell adhesion
In the first part of this study, the aim will be to develop the idea that selectively tagging cancer cells with cRGD can lead to their impaired capacity to seed onto the ECM, thereby suppressing tumor onset. Before carrying out the plan as depicted in (Fig. 3A), a series of control experiments were performed.
Fig. 3. Tumor onset suppression via cRGD-based SeCT therapy.
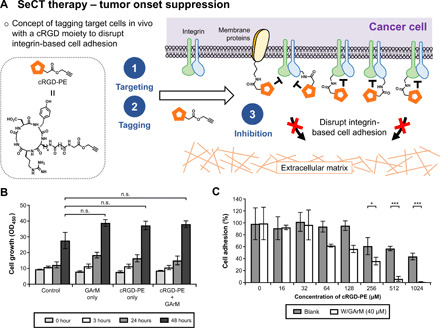
(A) Using the cRGD-PE reagent, this application is based on the indiscriminate tagging of cancer cell surface proteins with a cRGD moiety. Overexpressed integrins on cell surfaces will be exposed to a higher localized concentration of the inhibitor, leading to the disruption of integrin-based cell adhesion and the reduction in tumor onset and progression. (B) HeLa cell proliferation assay showing the nontoxic nature of the SeCT labeling reagents at various time points (0, 3, 24, and 48 hours) compared with a negative control. P values were determined by one-way analysis of variance (ANOVA). (C) Cell adhesion assay of HeLa cells to fibronectin-coated plates treated with SeCT labeling reagents. P values were determined by paired t test. All numerical data are presented as means ± SEM of three replicates. *P < 0.05, **P < 0.01, and ***P < 0.001; n.s., not significant.
One of the initial concerns was whether the cRGD labeling reagents could be intrinsically cytotoxic. To verify this, viability was confirmed under cell exposure to mixtures of either cRGD-PE, or GArM, or both (Fig. 3B). As expected, observations showed that throughout various time points, the labeling reagents did not cause any significant decrease in cell growth compared with the controls.
The next priority then shifted to obtaining in vitro confirmation that integrin-based cell adhesion could be disrupted with the cRGD-PE labeling reagents. To do this, a standard cell adhesion assay was performed using fibronectin-coated 96-well plates. HeLa cells, which are known to overexpress at least one to three RGD-specific integrins [α5β1 (28), αVβ3 (29, 30), and αVβ5 (28, 29)], were first incubated with and without GArM under varying concentrations of cRGD-PE. Following 1 hour, cells were spun down and washed twice to remove any unconjugated cRGD-PE. This was primarily done to ensure that disruption to integrin-based adhesion is due to surface-conjugated cRGD rather than the free reagent itself. The results of the assay are shown in Fig. 3C. As expected, decreases in cell adhesion were observed with HeLa cells exposed to both the labeling reagents (GArM/cRGD-PE). Because this reduced adhesion becomes more pronounced at higher concentrations of cRGD-PE, this is indicative of higher concentrations of cRGD being attached onto cell surfaces. As a further control, the effects to cell adhesion were also individually compared between cRGD and cRGD-PE alone (fig. S4), which confirms that the added PE moiety does not significantly interfere with RGD binding.
In the final control experiment, an investigation was done to verify whether cells tagged with integrin inhibitors would have a reduced ability to adhere onto the ECM in vivo. To do this, pretagged HeLa cells with cRGD were injected into mice to monitor their capacity to seed and advance into tumors (figs. S5 and S6). From these data, mouse populations exposed to condition 4 (HeLa cells preincubated with both GArM and cRGD-PE) were clearly shown to have a significant decrease in detected bioluminescence compared with control conditions (fig. S5B). This observation suggests that initial tumor seeding/onset had likely been impaired, leading to a slower rate of tumor development. As a result, this effect adequately translated into improved survival for mice exposed to these adhesion-impaired cancer cells (fig. S5C).
Tumor onset suppression via SeCT therapy
One of the main goals of this study was to determine whether in vivo labeling of cancer cells, otherwise known as SeCT therapy, can proceed with significant therapeutic effects. Because pretagged HeLa cells were found to have diminished tumorigenic ability in mice, the next step was to determine whether HeLa cells present inside mice could be tagged in vivo to elicit the same therapeutic response. The schedule of this experiment is depicted in Fig. 4A, where the experimental sequence begins with the intraperitoneal injection of HeLa-Luc cells into mice. After a 10-min wait, an injection of GArM was made, followed by another 15-min wait, and the final injection of cRGD-PE. In this manner, the effect of integrin-blocking SeCT therapy can be monitored well before cancer cells are given time to seed onto the ECM. Four different conditions were explored, where a saline solution was used to replace reagents in the general injection protocol as necessary. In terms of dosage, each mouse received approximately 3 nmol of GArM and 15 nmol of cRGD-PE, as indicated. Mice were monitored using an IVIS imaging system over the course of 4 weeks (once per week), as shown in Fig. 4B and fig. S7.
Fig. 4. Tumor onset suppression via cRGD-based SeCT therapy in mice.
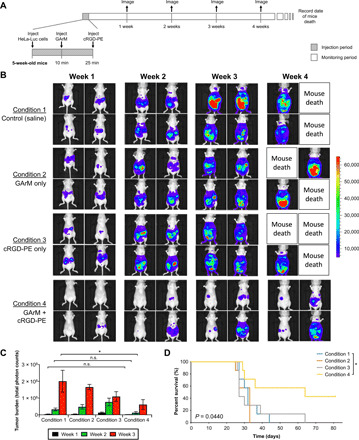
(A) Experimental timeline that details the injection schedule of HeLa-Luc cells and the labeling reagents. (B) IVIS imaging results showing a representative set of mice monitored for the onset and progression of HeLa tumors over a period of 4 weeks. (C) Comparison and quantification of bioluminescent signals emitted by HeLa-Luc cancer cells in IVIS imaging of mice (n = 7) over a period of 3 weeks. P values were determined by one-way ANOVA. (D) Comparison of mice survival for various conditions monitored over a period of 81 days. P values were determined using the log-rank test. All numerical data are presented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
For the control mouse populations (saline only), HeLa cells were shown to seed and develop into malignant tumors within 2 weeks, which was also matched by mice under conditions 2 and 3 (GArM only and cRGD-PE only, respectively). In the case of mouse populations exposed to condition 4 (injection with both GArM and cRGD-PE), a significant decrease in the detected bioluminescence was consistently observed (Fig. 4C). It should be noted that week 4 tumor burden data were excluded from analysis because of excessive death under conditions 1, 2, and 3. Overall, these data suggest that tumor onset and progression in mice receiving cRGD-based SeCT therapy had been significantly impaired. Following the initial 4-week period, mice were continually monitored for a total of 81 days to determine their rate of survival (Fig. 4D). For the mice exposed to condition 4, the survival rate is about 40% at the end of this period. On the other hand, mice exposed to conditions 1, 2, and 3 mostly dropped to a 0% survival well before the final time point.
Approach 2: Labeling of targeted cells with doxorubicin to prevent cell growth
In the second part of this study, an investigation was done to determine whether SeCT therapy could be used to reduce the growth of tumors already embedded onto the ECM. In a preliminary attempt, cRGD labeling was explored as a means to dislodge tumor cells in mice. However, because there was no observed growth rate reduction compared with controls (figs. S8 and S9), it was concluded that cRGD tagging via SeCT therapy is likely ineffective at disrupting preexisting fibronectin-integrin interactions, which has been reported in the literature to be stronger than the cRGD-integrin binding interaction (31).
Following this initial trial, a change in the approach was made to instead explore tagging tumor cells with a cytotoxic drug moiety. The hypothesis is that once surface-tagged proteins are internalized into cells, the active drug molecules can then be released to elicit a cytotoxic effect. The following experiments were conducted on the basis of the plan outlined in Fig. 5A.
Fig. 5. Tumor growth reduction via doxorubicin-based SeCT therapy.
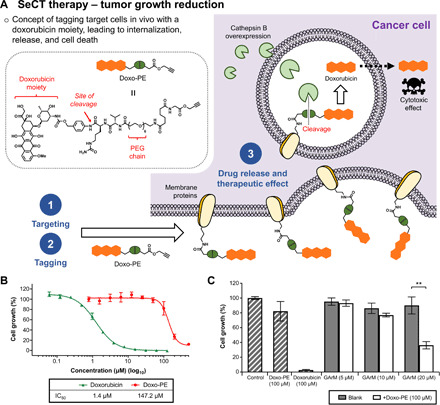
(A) Using the doxo-PE reagent, this application is based on the indiscriminate tagging of cancer cell surface proteins with a doxorubicin moiety. Following internalization, the Val-Cit-PAB linker is designed to be cleaved by overexpressed cathepsin B, leading to drug release and eventual cell death. (B) Cytotoxicity assay against HeLa cells showed that doxo-PE is roughly 100-fold less toxic than its active agent, doxorubicin. (C) Cytotoxicity assay that shows the combination of the SeCT labeling reagents (doxo-PE/GArM) causes significantly higher HeLa cell death compared with the addition of individual reagents (doxo-PE only or GArM only). P values were determined by paired t test. All numerical data are presented as means ± SEM of three replicates. *P < 0.05, **P < 0.01, and ***P < 0.001. IC50, median inhibitory concentration.
One key aspect to consider for any strategy is that the precursor labeling agent (i.e., doxo-PE) must be significantly less toxic compared with the active drug molecule. Considering a previous work that confirmed a hydrophilic polyethylene glycol (PEG) chain could be used to prevent doxorubicin-based cytotoxicity (32), doxo-PE was designed to include a short PEG chain to inhibit its passive uptake into cells. Another design choice was the inclusion of a Val-Cit-PAB linker, which can be used to facilitate drug release via lysosomal proteases. From subsequent cytotoxicity assays, doxo-PE was found to be roughly 100-fold less toxic than its active drug species (Fig. 5B).
To investigate the effects of doxorubicin labeling on cell surfaces, another in vitro assay was carried out using both the SeCT labeling reagents GArM and doxo-PE (Fig. 5C). At a concentration of 100 μM, observations showed doxo-PE to be relatively nontoxic compared with controls. However, with the supplementation of GArM (20 μM) to the mixtures, a reduction of >50% in HeLa cell growth was recorded. Given that GArM itself was previously tested to be noncytotoxic (at <40 μM), these results suggest that doxo-PE is likely labeled on cell surfaces, leading to the eventual release of doxorubicin and consequent tumor cell death.
Tumor growth reduction via SeCT therapy
Looking specifically at the capacity to reduce tumor growth in mice, SeCT therapy was next attempted for the in vivo labeling of cancer cells with doxorubicin. To conduct these experiments, the schedule depicted in Fig. 6A was followed. Beginning with the intraperitoneal injections of HeLa-Luc cells, tumors were allowed to develop in mice over the span of a week. Following this period, sequential injections of GArM and doxo-PE (done between a 15-min interval) were performed four times over 8 days. In terms of dosage, each treatment step saw mice receive approximately 5 nmol of GArM and 5 nmol of doxo-PE. Mice were then monitored using an IVIS imaging system for the next 3 weeks (once per week), as shown in Fig. 6B and fig. S10.
Fig. 6. Tumor growth reduction via doxorubicin-based SeCT therapy in mice.
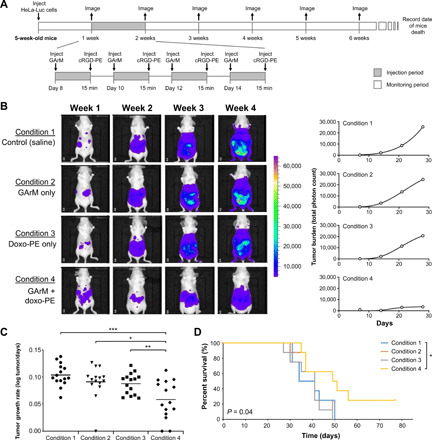
(A) Experimental timeline that details tumor development and the injection schedule of the labeling reagents. (B) IVIS imaging results to highlight tumor growth for a representative mouse from each group. Detected bioluminescent signals were then used to construct tumor growth curves over time, where the mean slope can be used to represent the rate of tumor growth. (C) Comparison of the calculated tumor growth rates for mice (n = 15) over a period of 3 weeks. P values were determined using a paired sample t test. (D) Comparison of mice survival for various conditions monitored over a period of 77 days. P values were determined using the log-rank test. *P < 0.05, **P < 0.01, and ***P < 0.001.
To investigate the efficacy of the various treatments on tumor growth over time, a rate-based treatment/control (T/C) test was applied (33). First, as expected, mice under conditions 1, 2, and 3 all developed tumors at similar rates with no statistical difference (Fig. 6C). However, when looking at mice that received both GArM and doxo-PE (condition 4), analysis shows that tumor growth rates are substantially reduced compared with the other groups. Between the vehicle group and mice treated under condition 4, calculation of the T/C ratio was found to be 0.11 (with a P value of 0.0003). As a note, T/C ratios below 0.4 are considered to be significant. Given these data, it can be suggested that the rate of tumor growth in mice receiving doxorubicin-based SeCT therapy had been significantly reduced. Mice were then continually monitored for a total of 77 days (Fig. 6D), which showed that the group receiving the SeCT labeling agents (condition 4) exhibited an overall improvement in survival (P value of 0.04 compared with the vehicle group).
DISCUSSION
In this study, we have demonstrated the concept of selectively tagging cells of interest in vivo to elicit a therapeutic response (SeCT therapy). This was principally done by introducing working examples based on two different approaches. The focus of the first strategy involved tagging cancer cells with a cRGD moiety to block integrin-dependent cell adhesion. The results of these experiments showed that tumor seeding and onset could be successfully suppressed with considerably long-lasting effects. For example, one dosage at the beginning of HeLa cell seeding in mice could delay tumor onset by up to 3 weeks. With the second strategy, the focus then shifted to tagging cancer cells with a doxorubicin moiety as a means to promote cell death. Data from these experiments showed that the observed reduction in tumor growth rates could eventually lead to an overall improvement in mouse survival rates.
Compared with traditional drug administration, the practice of SeCT therapy presents itself with several areas of potential benefit. First, given the physical linkage to the therapeutic moiety, tagged cells will become exposed to a higher localized concentration. This effect can be especially impactful for drug candidates that require high concentrations or suffer from issues of instability. For example, the cRGD peptide cilengitide was initially a promising candidate for anticancer therapy. Unfortunately, it was unable to move past phase 3 clinical trials because of insufficient evidence proving an improvement to overall patient survival (34, 35). One current opinion for the failure of cilengitide suggests that its short half-life and pharmacokinetic profile are the limiting factors (36). To compound this problem, studies have also shown that low dosages of cilengitide paradoxically stimulate angiogenesis and tumor growth (37), thereby necessitating the use of high concentrations to elicit a positive therapeutic effect. How SeCT therapy can overcome these limitations is best highlighted by experimental evidence shown in Fig. 4. Despite the expectation that the cRGD-PE reagent only (condition 3) could also exhibit integrin-inhibitory activity in vivo, only mice receiving the combination of GArM/cRGP-PE (condition 4) were found with suppressed tumor onset and progression.
Another potential benefit of SeCT therapy relates to the rising threat of drug resistance in cancer cells. Because GArM-based protein labeling is based on the indiscriminate labeling of surface proteins, the only perceived avenue for resistance would be to the glycan-based targeting mechanism, an effort that would require multiple concerted changes to signaling and glycosylation pathways. As such, GArM-based targeting and labeling could be theoretically more robust against resistance.
Not without its own set of limitations, the future development of SeCT therapy will require improving several aspects to make the leap as a viable therapeutic option. One challenging undertaking is the need to expand the library of targetable cancers. To do this, a larger library of targeting N-glycans should be identified and then verified in animal models. Also, given that this current study only deals with the HeLa cancer cell line, another goal will be to explore applicability for other cancer types. It is additionally recognized that issues may arise regarding administration, as two separate injections are currently used to introduce the labeling reagents. However, one can speculate that because the GArM complex is essentially an albumin-based protein therapeutic, subcutaneous injections could become a viable option, much like other albumin-based therapeutics (i.e., albiglutide).
Overall, this work represents an important step among the collected works focused on in vivo cell tagging, which remains a grossly underexplored concept considering its potential value to the pharmaceutical industry. By completing the principal aim of developing a working methodology that can validate SeCT therapy, the next step in this work will likely shift to exploring cell tagging with other chemical moieties of interest. Aside from cytotoxic drug molecules, one area of appeal is the ability to tag nontoxic chemical moieties, which can function by disrupting biological processes (ex., cell migration) or eliciting an immunological response (ex., antigens). As such, there remains much potential to develop other applications of SeCT-based therapeutics to elicit and address a wide array of biological responses and disorders.
MATERIALS AND METHODS
General chemicals
Reagents were purchased from Sigma-Aldrich (MO, USA), TCI Chemicals (Tokyo, Japan), or Wako Pure Chemicals (Osaka, Japan) without further purification. Human serum albumin and DBCO-NHS ester were purchased from Sigma-Aldrich (MO, USA). All complex N-glycan azides were supplied by GlyTech (Kyoto, Japan). Amicon Ultra Centrifugal Filters (30 kDa) and Durapore polyvinylidene difluoride (PVDF) 0.45-μm filters were purchased from MilliporeSigma (MA, USA). For peptide synthesis, Fmoc-Asp(OtBu)-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Gly-OH, HCTU, HOBt(6-Cl), and the H-Gly-Trt(2-Cl) resin were all purchased from Watanabe Chemical Industries (Hiroshima, Japan). Anhydrous dimethylformamide (DMF) was purchased from Wako Pure Chemicals (Osaka, Japan) and kept dried in the presence of molecular sieves. Dulbecco’s modified Eagle’s medium (DMEM), phosphate-buffered saline (PBS), and Hanks’ balanced salt solution (HBSS) were all purchased from Nacalai Tesque (Kyoto, Japan). Fetal bovine serum (FBS) was purchased from Equitech-Bio (TX, USA). Thin-layer chromatography (TLC) analyses (F-254) were performed with 60-Å silica gel from Merck Millipore (MA, USA).
General equipment
Nuclear magnetic resonance (NMR) analyses for 1H and 13C NMR spectra were measured on either a JEOL AL300 (300 MHz) or an AL400 (400 MHz) instrument. High-resolution mass spectroscopy (HRMS) was carried out on a Bruker MicrOTOF-QIII spectrometer by electrospray ionization time-of-flight (ESI-TOF). Matrix-assisted laser desorption ionization (MALDI-TOF) mass spectrometry analysis was obtained on a Bruker autoflex spectrometer using 2,5-dihydroxybenzoic acid as a matrix. Reversed-phase high-performance liquid chromatography (HPLC) was performed using a Shimadzu system (Kyoto, Japan), consisting of two LC-20AP pumps and a SPD-20AV photodiode array detector. The used column was a preparative 20 × 250–mm Cosmosil 5C18-AR-300 from Waters Corporation (MA, USA). Ultrapure water used for all experiments described in this paper was obtained from the Milli-Q Advantage A10 Water Purification System sold by Merck Millipore (Burlington, USA).
Preparation of cRGD-PE
The full synthetic scheme and structures are presented in fig. S1. To the stirred solution of IV (4.2 mg, 3.9 μmol) and compound V (1.1 mg, 7.8 μmol) in DMF (200 μl) was added N,N-Diisopropylethylamine (3.3 μl, 19.6 μmol). After the solution was stirred at room temperature for 24 hours, the resulting mixture was directly purified by reversed-phase HPLC using a linear gradient of 10 to 80% acetonitrile [0.1% trifluoroacetic acid (TFA)] in water (0.1% TFA) over a period of 40 min. Yield: 3.2 mg, 85%. 1H NMR (500 MHz, CD3OD, 25°C) δ 7.02 (d, J = 7.0 Hz, 2H), 6.71 (d, J = 7.0 Hz, 2H), 4.76 (t, J = 7.5 Hz, 1H), 4.75 (s, 2H), 4.44 (t, J = 7.5 Hz, 1H), 4.29 to 4.20 (m, 2H), 3.98 (s, 2H), 3.91 (dd, J = 9.5, 3.0 Hz, 1H), 3.25 to 3.20 (m, 1H), 3.18 to 3.11 (m, 1H), 3.11 (t, J = 7.0 Hz, 2H), 2.96 (s, 1H), 2.88 (d, J = 8.0 Hz, 2H), 2.85 (dd, J = 16.5, 8.0 Hz, 1H), 2.58 (dd, J = 16.5, 8.0 Hz, 1H), 2.29 (t, J = 7.0 Hz, 2H), 2.22 (t, J = 7.0 Hz, 2H), 1.92 to 1.84 (m, 1H), 1.74 to 1.60 (m, 6H), 1.59 to 1.42 (m, 3H), 1.41 (t, J = 8.0 Hz, 2H), and 1.09 to 0.80 (m, 2H). ESI-HRMS mass/charge ratio (m/z) calcd for C38H55N10O12 ([M + H]+) 843.3995, found 843.4006.
Preparation of doxo-PE
The full synthetic scheme and structures are presented in fig. S1. To the stirred solution of VIII (8.5 μmol) in DMF (500 μl) was added XIII (17 μmol) in CHCl3 (170 μl). After the solution was stirred at room temperature for 20 hours, the resulting mixture was diluted with CH3CN (2 ml) and purified by reversed-phase HPLC using a linear gradient of 10 to 100% acetonitrile (0.1% TFA) in water (0.1% TFA) over a period of 30 min. Yield: 6.7 mg, 53%. 1H NMR (400 MHz, CDCl3, 25°C) 9.93 (s, 1H), 8.26 (t, J = 7.2 Hz, 1H), 8.08 (d, J = 7.2 Hz, 1H), 7.90 (s, 1H), 7.89 (s, 1H), 7.77 to 7.88 (m, 2H), 7.60 to 7.67 (m, 1H), 7.42 to 7.58 (m, 2H), 7.15 to 7.28 (m, 2H), 7.23 (s, 1H), 7.10 (s, 1H), 6.96 (s, 1H), 6.82 (d, J = 9.0 Hz, 1H), 6.53 (br.s, 2H), 5.96 (br.s, 1H), 5.40 (br.s, 2H), 5.19 (s, 1H), 4.91 (s, 1H), 4.86 (s, 2H), 4.68 (d, J = 2.4 Hz 2H), 4.54 (s, 1H), 4.29 to 4.37 (m, 1H), 4.10 to 4.22 (m, 1H), 3.96 (s, 3H), 3.82 (d, J = 5.9 Hz, 2H), 3.53 to 3.59 (m, 3H), 3.44 to 3.48 (m, 20H), 3.32 to 3.38 (m, 2H, overlapped with water), 3.14 (dd, J = 11,4, 5.7 Hz, 2H) 2.81 to 3.02 (m, 4H), 2.39 to 2.41 (m, 1H), 2.28 to 2.38 (m, 2H), 2.14 to 2.21 (m, 1H), 2.09 (br.s, 2H), 2.03 (br.s, 2H), 1.92 (dd, J = 12.4, 5.8 Hz, 1H), 1.72 to 1.86 (m, 1H), 1.61 to 1.69 (m, 1H), 1.50 to 1.59 (m, 1H), 1.43 (br.s, 4H), 1.25 to 1.37 (m, 2H), 1.09 (d, J = 6.1 Hz, 3H), 0.81 (d, J = 6.3 Hz, 3H), 0.78 (d, J = 6.3 Hz, 3H); ESI-HRMS m/z calcd for C72H98N8O27 ([M + H]+) 1507.6614, found 1507.6596.
Preparation of GArM complex
The full preparatory steps and structures are presented in fig. S2. Previous reported procedures were used for the synthesis of the α(2,3)sialic acid–terminated glycan-aldehyde probe XIV (38), and for the coumarin-linked Au catalyst XV (9). To a solution of human serum albumin (5.3 ml of H2O solution, 66.7 nmol) was added XIV in dimethyl sulfoxide (1 μmol of a 3.8 mM stock solution, 15 eq, 260 μl) under air. The solution was mildly mixed and incubated overnight at 37°C. The solution was then concentrated and washed with H2O using Amicon Ultra Centrifugal Filters (30 kDa). The insoluble by-products were removed by filtering with Durapore PVDF 0.45-μm filter and diluted with water to give 1.0 mM stock solutions of glycoalbumin. Confirmation of glycan attachment was performed using MALDI-TOF-MS (positive mode), which detected the molecular weight of the glycoalbumin (92.3 kDa) indicating 8.6 molecules of complex N-glycans conjugated per albumin. In the next portion of the protocol, XV (66 nmol) in dioxane (6 μl) was added to the glycoalbumin solution (66 nmol) in PBS buffer (pH 7.4) (54 μl). The solution was mildly mixed and incubated at 37°C for 1 hour, which was then concentrated and washed with PBS buffer using Amicon Ultra Centrifugal Filters (30 kDa) to give the desired GArM complex.
Cell culture
Human cervix epithelial (HeLa) cells were obtained from the RIKEN Cell Bank, which were cultured in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin. The HeLa-Luc cell line was created by stable transfection of HeLa cells with firefly luciferase and puromycin acetyltransferase, which was cultured in DMEM supplemented with 10% FBS and 0.01% puromycin. The HeLa-V cell line was created by stable transfection of HeLa cells with firefly luciferase and Venus, which was cultured in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, and 0.8% Geneticin. All cell lines were grown in 37°C incubators with 5% CO2. Murine macrophages were harvested from the abdominal cavity of BALB/c-nu/nu mice with the injection of a 0.9% saline solution (6 ml), as described in the literature (39).
Transfection protocol
HeLa cells were stably transfected with expression vectors encoded with various genes expressing firefly luciferase reporter (Luc), yellow fluorescent protein variant Venus (V), and a puromycin acetyltransferase (Puro). Venus complementary DNA was gifted by A. Miyawaki (RIKEN BSI) (27). At 70 to 90% confluency, HeLa cells were transfected using Lipofectamine 3000 (Thermo Fisher Scientific, USA). For the HeLa-Luc stable cell line, a pCAG-Luciferase-IRES2-Puro plasmid vector was used, and puromycin selection (2 μg/ml) began 2 days after transfection. For the HeLa-V stable cell line, a pCAG-Luciferase-IRES2-Venus plasmid vector was used, and Geneticin selection (400 mg/ml) began 2 days after transfection.
Cell viability assay
Cell viability was determined using a WST-8 Cell Proliferation Assay, which is a colorimetric method to monitor the reduction in the water-soluble tetrazolium salt WST-8 to its formazan derivative via mitochondrial dehydrogenase of metabolically active cells. The commercial kit used in this study was the Cell Count Reagent SF Kit (Nacalai Tesque, Kyoto, Japan). For the cRGD-PE labeling study, approximately 10,000 cells per well of HeLa-Luc cells in 10 μl of HBSS media were first incubated with GArM (1 nmol in 1 μl of saline) for 15 min, followed by the addition of cRGD-PE (3 nmol of 1 μl saline), and then subsequently incubated for 30 min. Under control conditions, reagents were replaced with a 0.9% saline solution (Otsuka Pharmaceuticals, Japan) when necessary. Cells were then plated onto 96-well Falcon microplates along with the addition of HBSS media (90 μl) and grown for various time points (0, 3, 24, and 48 hours) at 37°C. For the doxo-PE labeling study, approximately 1000 cells per well of HeLa cells were plated and grown overnight on 96-well Falcon microplates. The media was then removed, followed by the incubation of various concentrations of labeling agents (GArM and/or doxo-PE). In general, 10 μl of compounds was added to 90 μl of media. Incubations were carried out for 3 hours to allow cell labeling to occur. The media was then replaced with HBSS media only (100 μl) and allowed to grow for a total of 4 days at 37°C. For all cases, quantification of cell viability was initialized by adding the Cell Count Reagent (10 μl). Following incubation at 37°C for 1 hour, end point absorbance was acquired at 450 nm using a Model 680 Microplate Reader (Bio-Rad Laboratories, USA).
Cell adhesion assay
The cell adhesion assay was performed using Human Fibronectin Coated 96-Well Microplates purchased from R&D Systems (Minneapolis, USA). Quantification of attached cells was measured via a commercial MTS assay, CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, USA). HeLa229 cells were serum starved 16 hours before the experiment by replacing its growth media to DMEM only (serum free). Cells were then subcultured to a stock concentration of 6 × 105 cells/ml, where an extra centrifugation step was applied to remove any remaining trypsin. Mixtures in Eppendorf tubes were then prepared using HeLa cells (360 μl from cell stock solution), cRGD-PE (45 μl from stock solutions of 0, 160, 320, 640, 1280, 2560, 5120, and 10240 μM in H2O), and GArM (45 μl of 400 μM in PBS buffer). In parallel, control mixtures were prepared where GArM was replaced with 45 μl of PBS buffer only. Incubations were carried out for 1 hour at room temperature on a rocking shaker. A washing step was then performed twice where cells were spun down (4 min at 0.8 rpm), supernatant removed, followed by resuspension in 450 μl of DMEM only (serum free). To the Fibronectin Coated 96-well Microplates, 130 μl of the labeled HeLa cell mixture was added into individual wells. Plates were then incubated at 37°C for 10 min. The media was removed, and attached cells were washed with PBS buffer three to four times. Intermittently, PBS buffer was pipetted up and down to dislodge any nonspecific binding cells. In the final step, 100 μl of DMEM (10% FBS + 1% penicillin-streptomycin) and 20 μl of MTS reagent were added. Following incubation at 37°C for 4 hours, end point absorbance was acquired at 490 nm using a SectraMax iD3 multimode microplate reader (Molecular Devices, USA).
Fluorescence-activated cell sorting assay
Flow cytometry and cell sorting were performed using standard procedures with a Sony SH800 Cell Sorter (Sony Corporation, Japan). The flow cytometer was equipped with 405-, 488-, 561-, and 638-nm lasers, where cells were gated at the excitation/emission wavelengths of λEx = 405 nm/λEm = 470 nm for GArM detection, and λEx = 515 nm/λEm = 528 nm for Venus (V) detection. Results were analyzed using the Sony SH800 software.
Mice
Five-week-old BALB/c-nu/nu mice were purchased from CLEA Japan (Tokyo, Japan). The mice were housed at the RIKEN Center for Life Science Technologies. All procedures involving mouse experiments were approved by the ethics committee of RIKEN (MAH21-19-17) and performed in accordance with the institutional and national guidelines.
Mouse model: Impaired tumor onset of cRGD prelabeled cancer cells
The injection schedule is depicted in fig. S5A. The treatment group (condition 4) was set up as follows: HeLa-Luc cells (100,000 cells suspended in 100 μl of HBSS buffer) were placed in a crytotube, followed by the addition of GArM (1 nmol in 100 μl of 10:90 H2O/saline). After an interval of 15 min, cRGD-PE (3 nmol in 100 μl of 5:95 H2O/saline) was added, and the solution was allowed to mix for an additional 30 min. This solution was then intraperitoneally injected into 5-week-old BALB/c-nu/nu mice (n = 8). The mice were then returned to their cages and monitored/imaged at varying time points (1, 2, 3, and 4 weeks). Various control groups were also used: condition 1 replaces both GArM and cRGD-PE with equivalent volumes of saline, condition 2 replaces cRGD-PE with an equivalent volume of saline, and condition 3 replaces GArM with an equivalent volume of saline.
Mouse model: Tumor onset suppression via cRGD-labeling SeCT therapy
The injection schedule is depicted in Fig. 4A. The treatment group (condition 4) was set up as follows: 5-week-old BALB/c-nu/nu mice (n = 7) were intraperitoneally injected first with HeLa-Luc cells (100,000 cells suspended in 100 μl of HBSS media), followed by an interval of 10 min, a second injection of GArM (3 nmol in 100 μl of 10:90 H2O/saline), another interval of 15 min, and a final injection of cRGD-PE (15 nmol in 100 μl of 5:95 H2O/saline). The mice were then returned to their cages and monitored/imaged at varying time points (1, 2, 3, and 4 weeks). Various control groups were also used: condition 1 replaces both GArM and cRGD-PE with equivalent volumes of saline, condition 2 replaces cRGD-PE with an equivalent volume of saline, and condition 3 replaces GArM with an equivalent volume of saline.
Mouse model: Tumor growth reduction via cRGD-labeling SeCT therapy
The injection schedule is depicted in fig. S8A. The treatment group (condition 4) was set up as follows: 5-week-old BALB/c-nu/nu mice (n = 8) were intraperitoneally injected first with HeLa-Luc cells (100,000 cells suspended in 100 μl of HBSS media). The mice were then returned to their cages and monitored for 7 days. Once tumor development was confirmed, an injection period was commenced where mice were injected daily for seven consecutive days first with GArM (3 nmol in 100 μl of 10:90 H2O/saline), followed by an interval of 15 min, and then with cRGD-PE (15 nmol in 100 μl of 5:95 H2O/saline). The mice were then returned to their cages and monitored/imaged at the 2- and 3-week time points. All mice were then euthanized to determine intestine tumor burden. Various control groups were also used: condition 1 replaces both GArM and cRGD-PE with equivalent volumes of saline, condition 2 replaces cRGD-PE with an equivalent volume of saline, and condition 3 replaces GArM with an equivalent volume of saline.
Mouse model: Tumor growth reduction via doxorubicin-labeling SeCT therapy
The injection schedule is depicted in Fig. 6A. The treatment group (condition 4) was set up as follows: 5-week-old BALB/c-nu/nu mice (n = 15) were intraperitoneally injected first with HeLa-Luc cells (100,000 cells suspended in 100 μl of HBSS media). The mice were then returned to their cages and monitored for 7 days. Once tumor development was confirmed, an injection period was commenced where mice were injected four times over the next 8 days first with GArM (5 nmol in 100 μl of 10:90 H2O/saline), followed by an interval of 15 min, and then with doxo-PE (5 nmol in 100 μl of 5:95 H2O/saline). The mice were then returned to their cages and monitored/imaged at varying time points (2, 3, or 4 weeks). Various control groups were also used: condition 1 replaces both GArM and doxo-PE with equivalent volumes of saline, condition 2 replaces doxo-PE with an equivalent volume of saline, and condition 3 replaces GArM with an equivalent volume of saline.
Bioluminescence imaging of mice and tumor burden analysis
As a method to monitor tumor formation and growth in living mice, HeLa-Luc cells were stably transfected with a firefly luciferase reporter gene. As such, tumor burden can be monitored via luciferase-catalyzed bioluminescence, which was confirmed to match well with visible tumor growth during dissection studies (fig. S3). To initiate in vivo detection, mice were injected with D-luciferin potassium salt (3 mg dissolved in 200 μl of saline). Following an interval of 10 min, mice were then anesthetized with pentobarbital or isoflurane and placed onto an IVIS kinetic fluorescence imager (Caliper Life Sciences, USA). Abdominal (anterior) images of the mouse specimens were then collected as described. To determine efficacy for tumor onset studies, this can be simply done by monitoring the total photon counts (photons per second), which correlates well with acquired tumor burden. To interpret efficacy in tumor reduction studies, changes to growth were analyzed using a rate-based T/C ratio (33). Tumor burden measured over time for each individual mouse was first transformed to logarithmic scale (to account for exponential growth). Rates of growth (slope) can then be calculated on the basis of the transformed tumor sizes and the corresponding time intervals. In this manner, the analysis can account for data taken from both early and late time points. The ratio of tumor growth rates between various conditions can be calculated using the following formula: rate-based T/C = 10(μT─μC) × #days, where μT is the mean growth rate of the treatment group, and μC is the mean growth rate of the control group.
Acknowledgments
We would like to thank Glytech Inc. for supplying various N-glycans. Funding: This work was supported by the JSPS KAKENHI grant numbers JP16H03287, JP18K19154, JP18K14347, and JP15H05843 in the Middle Molecular Strategy. Funding was also provided by the AMED grant JP15KM0908001, as well as with the support of the Russian Government Program for Competitive Growth (to Kazan Federal University) and JST PRESTO. Author contributions: T.T., K.V., and K.Ta. designed the experiments. K.Ts. and I.N. synthesized labeling reagents used in this study. T.T. and S.U. performed animal imaging experiments. T.T., S.U., and K.V. performed cell-based experiments. K.V. and K.Ta. wrote the manuscript. Y.N., A.K., H.O., and Y.W. provided guidance throughout the research. K.Ta. directed and supervised this research. All the authors commented on the manuscript and approved its content. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/17/eabg4038/DC1
REFERENCES AND NOTES
- 1.Yan Q., Bruchez M. P., Advances in chemical labeling of proteins in living cells. Cell Tissue Res. 360, 179–194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ernst R. J., Krogager T. P., Maywood E. S., Zanchi R., Beránek V., Elliott T. S., Barry N. P., Hastings M. H., Chin J. W., Genetic code expansion in the mouse brain. Nat. Chem. Biol. 12, 776–778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y., Ma J., Lu W., Tian M., Thauvin M., Yuan C., Volovitch M., Wang Q., Holst J., Liu M., Vriz S., Ye S., Wang L., Li D., Heritable expansion of the genetic code in mouse and zebrafish. Cell Res. 27, 294–297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsukiji S., Miyagawa M., Takaoka Y., Tamura T., Hamachi I., Ligand-directed tosyl chemistry for protein labeling in vivo. Nat. Chem. Biol. 5, 341–343 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Tamura T., Kioi Y., Miki T., Tsukiji S., Hamachi I., Fluorophore labeling of native FKBP12 by ligand-directed tosyl chemistry allows detection of its molecular interactions in vitro and in living cells. J. Am. Chem. Soc. 135, 6782–6785 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Xie R., Dong L., Du Y., Zhu Y., Hua R., Zhang C., Chen X., In vivo metabolic labeling of sialoglycans in the mouse brain by using a liposome-assisted bioorthogonal reporter strategy. Proc. Natl. Acad. Sci. U.S.A. 113, 5173–5178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H., Wang R., Cai K., He H., Liu Y., Yen J., Wang Z., Xu M., Sun Y., Zhou X., Yin Q., Tang L., Dobrucki I. T., Dobrucki L. W., Chaney E. J., Boppart S. A., Fan T. M., Lezmi S., Chen X., Yin L., Cheng J., Selective in vivo metabolic cell-labeling-mediated cancer targeting. Nat. Chem. Biol. 13, 415–424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prescher J. A., Dube D. H., Bertozzi C. R., Chemical remodelling of cell surfaces in living animals. Nature 430, 873–877 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Tsubokura K., Vong K. K. H., Pradipta A. R., Ogura A., Urano S., Tahara T., Nozaki S., Onoe H., Nakao Y., Sibgatullina R., Kurbangalieva A., Watanabe Y., Tanaka K., In vivo gold complex catalysis within live mice. Angew. Chem. Int. Ed. 56, 3579–3584 (2017). [DOI] [PubMed] [Google Scholar]
- 10.A. Varki, R. Kannagi, B. Toole, P. Stanley, Glycosylation changes in cancer, in Essentials of Glycobiology, A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart, M. E. Etzler, Eds. (Cold Spring Harbor Laboratory Press, 2009), pp. 617–632. [PubMed] [Google Scholar]
- 11.Ogura A., Tahara T., Nozaki S., Morimoto K., Kizuka Y., Kitazume S., Hara M., Kojima S., Onoe H., Kurbangalieva A., Taniguchi N., Watanabe Y., Tanaka K., Visualizing trimming dependence of biodistribution and kinetics with homo- and heterogeneous N-glycoclusters on fluorescent albumin. Sci. Rep. 6, 21797 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogura A., Tahara T., Nozaki S., Onoe H., Kurbangalieva A., Watanabe Y., Tanaka K., Glycan multivalency effects toward albumin enable N-glycan-dependent tumor targeting. Bioorganic Med. Chem. Lett. 26, 2251–2254 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Latypova L., Sibgatullina R., Ogura A., Fujiki K., Khabibrakhmanova A., Tahara T., Nozaki S., Urano S., Tsubokura K., Onoe H., Watanabe Y., Kurbangalieva A., Tanaka K., Sequential double “clicks” toward structurally well-defined heterogeneous N-glycoclusters: The importance of cluster heterogeneity on pattern recognition in vivo. Adv. Sci. 4, 1600394 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogura A., Urano S., Tahara T., Nozaki S., Sibgatullina R., Vong K., Suzuki T., Dohmae N., Kurbangalieva A., Watanabe Y., Tanaka K., A viable strategy for screening the effects of glycan heterogeneity on target organ adhesion and biodistribution in live mice. Chem. Commun. 54, 8693–8696 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Eda S., Nasibullin I., Vong K., Kudo N., Yoshida M., Kurbangalieva A., Tanaka K., Biocompatibility and therapeutic potential of glycosylated albumin artificial metalloenzymes. Nat. Catal. 2, 780–792 (2019). [Google Scholar]
- 16.Lin Y., Vong K., Matsuoka K., Tanaka K., 2-Benzoylpyridine ligand complexation with gold critical for propargyl ester-based protein labeling. Chem. Eur. J. 24, 10595–10600 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Ruoslahti E., RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 12, 697–715 (1996). [DOI] [PubMed] [Google Scholar]
- 18.Guan J.-L., Shalloway D., Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature 358, 690–692 (1992). [DOI] [PubMed] [Google Scholar]
- 19.Desgrosellier J. S., Cheresh D. A., Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 10, 9–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfaff M., Tangemann K., Müller B., Gurrath M., Müller G., Kessler H., Timpl R., Engel J., Selective recognition of cyclic RGD peptides of NMR defined conformation by αIIbβ3, αvβ3, and α5β integrins. J. Biol. Chem. 269, 20233–20238 (1994). [PubMed] [Google Scholar]
- 21.Aumailley M., Gurrath M., Muller G., Calvete J., Timpl R., Kessler H., Arg-Gly-Asp constrained within cyclic pentapeptides. Strong and selective inhibitors of cell adhesion to vitronectin and laminin fragment P1. FEBS Lett. 291, 50–54 (1991). [DOI] [PubMed] [Google Scholar]
- 22.Brooks P. C., Montgomery A. M. P., Rosenfeld M., Reisfeld R. A., Hu T., Klier G., Cheresh D. A., Integrin αvβ3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 79, 1157–1164 (1994). [DOI] [PubMed] [Google Scholar]
- 23.Stupack D. G., Cheresh D. A., Integrins and angiogenesis. Curr. Top. Dev. Biol. 64, 207–238 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Yang F., Teves S. S., Kemp C. J., Henikoff S., Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta Rev. Cancer 1845, 84–89 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubowchik G. M., Firestone R. A., Padilla L., Willner D., Hofstead S. J., Mosure K., Knipe J. O., Lasch S. J., Trail P. A., Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug. Chem. 13, 855–869 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Garg A., Mark Manidhar D., Gokara M., Malleda C., Suresh Reddy C., Subramanyam R., Elucidation of the binding mechanism of coumarin derivatives with human serum albumin. PLOS ONE 8, e63805 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagai T., Ibata K., Park E. S., Kubota M., Mikoshiba K., Miyawaki A., A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 20, 87–90 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Riikonen T., Vihinen P., Potila M., Rettig W., Heino J., Antibody against human α1β1 integrin inhibits HeLa cell adhesion to laminin and to type I, IV, and V collagens. Biochem. Biophys. Res. Commun. 209, 205–212 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Oba M., Fukushima S., Kanayama N., Aoyagi K., Nishiyama N., Koyama H., Kataoka K., Cyclic RGD peptide-conjugated polyplex micelles as a targetable gene delivery system directed to cells possessing αvβ3 and αvβ5 integrins. Bioconjug. Chem. 18, 1415–1423 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Xiong L., Yu M., Cheng M., Zhang M., Zhang X., Xu C., Li F., A photostable fluorescent probe for targeted imaging of tumour cells possessing integrin αvβ3. Mol. Biosyst. 5, 241–243 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Redick S. D., Settles D. L., Briscoe G., Erickson H. P., Defining fibronectin’s cell adhesion synergy site by site-directed mutagenesis. J. Cell Biol. 149, 521–527 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vong K., Yamamoto T., Chang T.-c., Tanaka K., Bioorthogonal release of anticancer drugs via gold-triggered 2-alkynylbenzamide cyclization. Chem. Sci. 11, 10928–10933 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hather G., Liu R., Bandi S., Mettetal J., Manfredi M., Shyu W. C., Donelan J., Chakravarty A., Growth rate analysis and efficient experimental design for tumor xenograft studies. Cancer Inform. 13, 65–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reardon D. A., Neyns B., Weller M., Tonn J. C., Nabors L. B., Stupp R., Cilengitide: An RGD pentapeptide ανβ3 and ανβ5 integrin inhibitor in development for glioblastoma and other malignancies. Future Oncol. 7, 339–354 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Stupp R., Hegi M. E., Gorlia T., Erridge S. C., Perry J., Hong Y.-K., Aldape K. D., Lhermitte B., Pietsch T., Grujicic D., Steinbach J. P., Wick W., Tarnawski R., Nam D.-H., Hau P., Weyerbrock A., Taphoorn M. J. B., Shen C.-C., Rao N., Thurzo L., Herrlinger U., Gupta T., Kortmann R.-D., Adamska K., Bain C. M., Brandes A. A., Tonn J. C., Schnell O., Wiegel T., Kim C.-Y., Nabors L. B., Reardon D. A., van den Bent M. J., Hicking C., Markivskyy A., Picard M., Weller M.; European Organisation for Research and Treatment of Cancer (EORTC); Canadian Brain Tumor Consortium; CENTRIC study team , Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 15, 1100–1108 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Tucci M., Stucci S., Silvestris F., Does cilengitide deserve another chance? Lancet Oncol. 15, e584–e585 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Reynolds A. R., Hart I. R., Watson A. R., Welti J. C., Silva R. G., Robinson S. D., Violante G. D., Gourlaouen M., Salih M., Jones M. C., Jones D. T., Saunders G., Kostourou V., Perron-Sierra F., Norman J. C., Tucker G. C., Hodivala-Dilke K. M., Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat. Med. 15, 392–400 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Nakamura K., Tsubokura K., Kurbangalieva A., Nakao Y., Murase T., Shimoda T., Tanaka K., Efficient route to RIKEN click probes for glycoconjugation. J. Carbohydr. Chem. 38, 127–138 (2019). [Google Scholar]
- 39.Ray A., Dittel B. N., Isolation of mouse peritoneal cavity cells. J. Vis. Exp., e1488 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/17/eabg4038/DC1