

Abstract
The exact mechanism of retinal ganglion cell loss in the pathogenesis of glaucoma is yet to be understood. Mitochondrial damage-associated molecular patterns (DAMPs) resulting from mitochondrial dysfunction have been linked to Leber's hereditary optic neuropathy and autosomal dominant optic atrophy, as well as to brain neurodegenerative diseases. Recent evidence shows that, in conditions where mitochondria are damaged, a sustained inflammatory response and downstream pathological inflammation may ensue. Mitochondrial damage has been linked to the accumulation of age-related mitochondrial DNA mutations and mitochondrial dysfunction, possibly through aberrant reactive oxygen species production and defective mitophagy. The present review focuses on how mitochondrial dysfunction may overwhelm the ability of neurons and glial cells to adequately maintain homeostasis and how mitochondria-derived DAMPs trigger the immune system and induce neurodegeneration.
1. Introduction
Glaucoma is a complex and multifactorial neurodegenerative disease characterized by the irreversible loss of retinal ganglion cells' (RGCs) soma and degeneration of the optic nerve axons [1]. Glaucoma is the most common optic neuropathy and the leading cause of irreversible blindness worldwide [2, 3]. It is generally accepted that elevated intraocular pressure (IOP) is a major risk factor [2].
The exact mechanisms by which elevated IOP triggers axonal degeneration and RGC death are yet to be known. IOP-induced mechanical compression to the optic nerve head (ONH), at the level of the lamina cribrosa, might directly lead to ischemia-hypoxia damage, blockage of axonal transport, and deprivation of growth factors [4–7]. However, indirectly, damage to RGCs may also result from the action of factors released by activated glial cells located in the lamina cribrosa [8–11]. Regardless of the mechanisms that initiate damage, there is evidence that, in glaucoma, these events converge into axonal degeneration, RGC death, and clinical overlap between the different glaucoma subphenotypes.
Notwithstanding the major role of IOP in the progression of glaucoma, in patients with normal-tension glaucoma (NTG), the elevation of IOP is not necessary for the development of glaucomatous damage [12]. Moreover, the reduction of IOP does not always prevent neurodegeneration, and many patients progress with the disease despite having IOP within normal range [13, 14]. This suggests that mechanisms other than IOP biomechanical and/or ischemic injury may be responsible for the neurodegenerative process. Although the precise mechanisms that lead to RGC insult and loss have not been identified, accumulating evidence supports a primary role of inflammation and the immune system [15–17]. Despite these indications, ocular hypertension remains the only target for current glaucoma therapies.
The central nervous system (CNS), which also includes the retina and optic nerve, is an immune-privileged site where immune functions are tightly regulated and are mediated by a limited number of cell types [18]. The onset of inflammation in glaucoma is hypothesized to be triggered by an altered crosstalk between RGCs and glial cells that involves the release of proinflammatory mediators, such as reactive oxygen species (ROS), nitric oxide (NO), tumor necrosis factor-alpha (TNF-α), and interleukin-1β (IL-1β) [9, 19–23]. Despite the recognized role of inflammation in glaucoma, to date, the key inflammatory signals and events that lead to polarization of microglia and astrocytes in the disease progression are not known. Transcriptomic studies showed that many inflammatory genes are upregulated in the retina and ONH in the early stage of the disease [17, 24, 25]. Some of the pathogenic events in glaucoma have been attributed to modifications of neurotrophin and glutamate signaling, excitotoxicity, oxidative stress, mitochondrial dysfunction, protein misfolding, hypoxia, ischemia, autoimmunity, and autophagy dysfunction [26–28].
The present review focuses on the role of the immune system in glaucomatous neurodegeneration and its contribution to disease progression. In particular, it discusses how mitochondrial dysfunction and concomitant neuronal and glial pathological processes can induce and perpetuate sterile inflammation in glaucoma, and which crossroads may be implicated in that. In the first part of the review, potential causes of neuronal mitochondrial dysfunction are addressed. In the second and third parts, the discussion will focus on currently known mechanisms of neuroglial dysfunction and the immune response secondary to mitochondrial damage.
To obtain a comprehensive collection of publications dealing with the neurodegenerative inflammatory response in glaucoma, multiple unrestricted PubMed searches specifying the occurrence of the term “glaucoma” in combination with “inflammation,” “neurodegeneration,” “immune response,” “mitochondrial dysfunction,” “oxidative stress,” “mitophagy,” and “mutation” were performed. The search was limited to primary open-angle glaucoma (POAG). Human studies that dealt with primary closed-angle glaucoma or secondary glaucoma were not considered for this review.
2. Mitochondrial Dysfunction in Retinal Ganglion Cells
The retina is one of the most metabolically active tissues in the body and requires a precise regulation of energy production to meet its consumption needs [29, 30]. Energy in the form of adenosine triphosphate (ATP) is required to synthesize neurotransmitters, organize synaptic vesicles, restore ion gradients, buffer calcium, and transport cargo bidirectionally along axons [30, 31]. Due to the absence of saltatory conduction, the unmyelinated portion of RGC axons within the retina requires more energy for the generation of action potentials [32]. In response to this high metabolic demand, a large proportion of mitochondria populate the unmyelinated portion of RGC axons [32–35].
Strict coordination between mitochondrial biogenesis, dynamics, transport, and degradation is essential to preserve the integrity of mitochondria within RGCs [30, 35]. These events are tightly regulated to ensure that mitochondria can adapt to fluctuations in energy requirements [36, 37]. Even though these metabolic processes are not unique to RGCs, these cells have lower tolerance for mitochondrial damage, and an inadequate supply of healthy mitochondria or the accumulation of defective mitochondria may be the origin of an energy crisis in RGCs [28, 30]. Among other potential causes, mitochondrial dysfunction has been linked to oxidative stress, mutations in mitochondrial DNA (mtDNA), and deficient mitophagy [38, 39].
2.1. Oxidative Stress
Oxidative stress is broadly defined as an imbalance that favors the production of ROS over antioxidant defenses. A consequence of electron transport through mitochondrial oxidative phosphorylation (OXPHOS) complexes is the generation of ROS, such as superoxide anions (O2−) and hydrogen peroxide (H2O2). Although ROS are key second messengers in various redox-sensitive signaling pathways, they can damage cellular proteins, lipids, and nucleic acids via oxidation [40]. As mitochondria are a significant source of ROS in many eukaryotic cells, especially in the context of age-related deterioration of mitochondrial electron chain transfer, mitochondrial ROS have been suggested to be an important immunostimulatory stimulus in glaucoma [40, 41].
Oxidative stress occurs as a result of malfunctions in one or more of the mitochondria's four main functions: generation of energy in the form of ATP; regulation of ROS production; regulation of cytosolic calcium levels; and modulation of apoptosis via mitochondrial permeability [42, 43]. In normal conditions, the generation of ROS in low levels is blocked by antioxidants, such as glutathione peroxidase, superoxide dismutase, and catalase. The most active sites of mitochondrial ROS production (i.e., sites of electron leakage) are complex I and complex III of the OXPHOS chain [44]. Although these sites do not constitute a major source of ROS within the cell, the damage inflicted by mitochondrial ROS can be very detrimental due to a particular vulnerability of the mitochondria to oxidative stress, notably to mtDNA. ROS, when in excess, can also induce lipid peroxidation and apoptosis by increasing mitochondrial membrane permeability and by inhibiting the mitochondrial respiratory chain [45–48].
The accumulation of neurotoxic levels of glutamate is another effect of oxidative stress. Glutamine synthase, which converts retinal glutamate into a nontoxic form, and glutamate transporter proteins were modified by ROS in experimental models of ocular hypertension [49–51]. Many of these cellular pathways and components that are damaged by oxidative stress become themselves the cause of further ROS production, perpetuating a cycle of cellular injury.
The cellular accumulation of ROS, regardless of the triggering stress condition, can mediate the induction of autophagy [52]. Autophagy is a nonselective degradation pathway that primarily promotes cellular protection through clearance of damaged organelles and protein aggregates. The autophagic process is fundamental to neuronal homeostasis and can be distinguished into more organelle-specific pathways, such as mitophagy for selective removal of mitochondria (see Section 2.3). In RGCs, ROS serve as signaling molecules in the induction of autophagosomes by regulating the activity of the cysteine protease ATG4, which seems to be a key element in the activation of autophagy [53]. Yoshimura et al. found that mRNA levels of ATG4B are high in brain tissues and retina, among other sites [54]. Dysfunction of autophagy has also been linked to brain chronic neurodegenerative diseases, suggesting common mechanisms of autophagy-related neuronal loss [52].
A long list of factors has been linked to ROS formation in glaucoma. To some extent, these factors seem to be related to intrinsic abnormal function of mitochondria (primary) or to external events that expose the RGCs and neuroglial mitochondria to stress (secondary), such as compromised blood flow, hypoxia, nutrient deficiency, calcium dysregulation, or mechanical injury (Figure 1) [49, 55–59].
Figure 1.
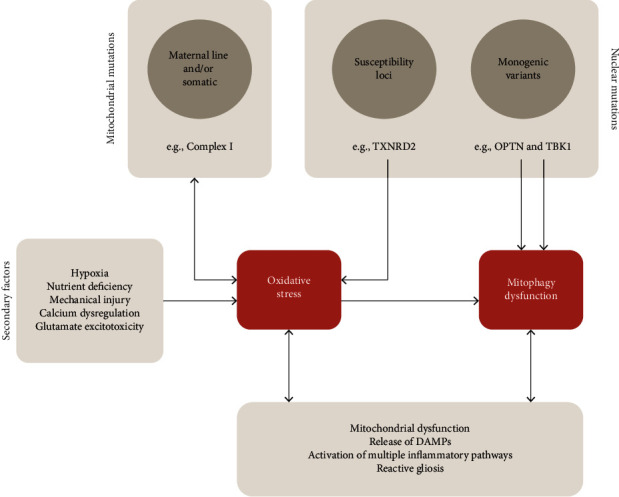
Mechanisms of mitochondrial dysfunction in retinal ganglion cells (RGCs). Mutations in mitochondrial and nuclear genomes can cause mitochondrial dysfunction via oxidative stress or defective mitophagy. Oxidative stress can also be generated through secondary causes external to the mitochondria, such as hypoxia. Mitochondrial-derived components can act as damage-associated molecular patterns (DAMPs) and trigger multiple inflammatory pathways that cause reactive gliosis and RGC loss.
Oxidative stress in POAG has been linked to specific OXPHOS defects, in particular to defects in complex I. Using a lymphoblast-based model to measure systemic mitochondrial function, Lee et al. and Van Bergen et al. showed, in two different glaucoma patient cohorts, decrease rates of ATP synthesis related to complex I defects [60, 61]. Although those defects had a low impact on the in vitro growth of lymphoblasts, they possibly rendered RGCs more susceptible to oxidative stress and metabolic crisis due to the high energetic requirements of RGCs and the presence of multiple cellular stressors in glaucoma. These results are in line with similar findings for complex I defects in hereditary optic neuropathies and brain neurodegenerative diseases [62, 63].
Oxygen and nutrient deficits not only cause RGCs to become bioenergetically compromised but also dramatically increase ROS production. This increase is due to slower electron transportation in the OXPHOS, which augments the reduction state of electron carriers and favors superoxide production at low oxygen concentrations [64]. Studies have also shown the upregulation of hypoxia-inducible factor 1 (HIF1) in glaucoma as a cellular response to reduced oxygen levels in the retina and ONH [65]. HIF1-alpha is a transcriptional activator that induces the expression of several proteins whose main function is to increase oxygen availability in hypoxic tissues [43]. These findings suggest the importance of hypoxia signaling mechanisms in the pathogenesis of glaucoma.
Oxidative stress has also been linked to abnormal cellular calcium influx [66]. Even though calcium serves a major role in normal intracellular metabolism and signaling (including the generation of ROS), an uncontrolled influx may occur through a variety of insults, such as loss of cell membrane integrity and inhibition of calcium ATPase activity [67]. Once the cell has been exposed to an excess of calcium, a synergic effect between calcium and ROS may occur, as ROS can also inhibit calcium ATPase activity in neurons [68]. An excessive influx of calcium and neuronal damage has also been linked to homocysteine accumulation. The mechanism of this homocysteine-induced RGC toxicity seems to take place through direct overstimulation of N-methyl-D-aspartate (NMDA) receptors and a subsequent increase in intracellular calcium and formation of ROS [69].
In addition to retinal and optic nerve damage caused by oxidative stress, the trabecular meshwork in the anterior segment of the eye is also vulnerable to ROS [70, 71]. This event, which has been connected in some cases to genetic mutations in myocilin, may lead to increased resistance of the aqueous humor outflow and cause further retinal exposure to high IOP [71–73]. Mechanical injury to the ONH caused by increased IOP induces neuronal oxidative stress and lipid peroxidation, which increments the risk of neurodegeneration [49, 74, 75]. In a study conducted in aged mice by Kong et al., even short-term elevations of IOP were shown to significantly increase oxidative stress [76].
Increasing evidence also points out the impact of oxidative stress and mitochondrial dysfunction in Müller cells, which may contribute to the pathogenesis neurodegenerative conditions in the retina. As Müller cells have numerous supporting functions to maintain RGC homeostasis, with many of them being metabolic-related, the impact of restricted energy availability in Müller cells has been implicated in glaucomatous RGC loss [77, 78]. A published review by Toft-Kehler et al. on mitochondrial function in Müller cells can provide an in-depth discussion on the topic [28].
As oxidative stress also represents a failure of endogenous antioxidant defenses to meet an increase of ROS, many studies have been demonstrating an accumulation oxidative stress markers in the serum, aqueous humor, and retina of glaucoma patients, as well as suggesting a general compromise of antioxidant defense in these patients [41, 48, 49, 75, 79, 80]. In a case-controlled study conducted by Yuki et al., the incidence of NTG was significantly correlated with high serum total antioxidant levels and low urinary 8-hydroxy-2′-deoxyguanosine/creatinine (a marker of DNA damage from oxidative stress), suggesting systemic oxidative stress [81]. Similarly, a long prospective study in a cohort of 3500 patients with glaucoma showed an association between low intake of antioxidant nutrients and a higher risk of POAG [82]. Identical results have been also documented in experimental models of glaucoma [83].
Several works have assessed the impact of antioxidant treatment in the reduction of oxidative stress in RGCs. Brimonidine, an alpha-adrenergic receptor agonist currently used to lower IOP, has been found to have a neuroprotective effect beyond IOP lowering [84–86]. Brimonidine seems to prevent abnormal elevations of cytosolic calcium and RGC apoptosis through mechanisms not yet fully elucidated [85]. Tempol, a nitroxyl antioxidant, resulted in increased survival of RGCs exposed to TNF-α and hypoxia in the presence of a caspase inhibitor. Crocin, a carotenoid, has antioxidant properties that are capable of suppressing ROS production, increasing mitochondrial membrane potential, and enhancing RGC viability upon H2O2 treatment [45, 87]. Coenzyme Q10 and N-acetyl cysteine are antioxidant compounds that directly target mitochondria and have shown beneficial effects in experimental models of glaucoma. Coenzyme Q10, as an essential cofactor in mitochondrial OXPHOS, afforded retinal protection in an ischemia-reperfusion rat model [88]. Similarly, the administration of N-acetyl cysteine, which enhances mitochondrial OXPHOS, decreased oxidative stress in a rat model of ocular hypertension [84].
The incorporation of antioxidants into mitochondrial membranes has also been shown to prevent lipid peroxidation, a process known to be initiated by ROS that can lead to cell membrane damage and RGC loss. In a rabbit model of glaucoma, 10-(6′-plastoquinonyl) decyltriphenylphosphonium (SkQ1), a mitochondrial-targeted plastoquinone-containing antioxidant, was able to reverse signs of glaucomatous injury [47]. Likewise, in a mouse model of glaucoma, choric acid and 3,5-dicaffeoylquinic acid of ethanol extract of Crepidiastrum denticulatum seemed to reduce lipid peroxidation and protect against RGC death [89]. Yokota et al. also showed that molecular hydrogen, through peroxynitrite scavenging capacity, protected lipid peroxidation and prevented retinal cell apoptosis [69].
2.2. Mitochondrial DNA Mutations
Glaucoma has not been clearly linked to nuclear genome mutations that result in mitochondrial dysfunction [90]. Large genome-wide association studies (GWAS) identified, with limited success, susceptibility loci associated with mitochondrial dysfunction [60, 61, 91]. A large meta-analyzed GWAS conducted by Bailey et al. identified, for example, a single nucleotide polymorphism in thioredoxin reductase 2 (TXNRD2), a mitochondrial protein required for redox homeostasis [92]. The effects of TXNRD2 in vivo were reported by Caprioli et al. who observed reduced RGC death after optic nerve axotomy in experimental models of glaucoma overexpressing TXNRD2 when treated with pharmacologically induced oxidative stress [93].
Mutations with Mendelian inheritance have only been associated with rare cases of POAG with early disease onset. These monogenic variants have been identified in myocilin (MYOC), optineurin (OPTN), and TANK binding kinase 1 (TBK1) [94–99]. Mutations in MYOC, as mentioned above, primarily cause oxidative stress abnormalities in the trabecular meshwork leading to increased IOP. Mutations in OPTN and TBK1 have been linked to dysregulation of mitophagy and apoptosis, and activation of inflammation with RGC loss (Figure 1). A more detailed discussion about the role of OPTN and TBK1 mutations in POAG is provided in the next section.
Several attempts to sequence and identify mutations in mitochondrial DNA have been hindered by mtDNA heteroplasmy, i.e., the multiple variants of mtDNA that can be present within a cell or a tissue. However, massive parallel sequencing tools have allowed better recognition of large deletions in the mitochondrial genome in a few studies using small cohorts of patients with glaucoma [100–102]. Even though these studies extrapolated mitochondrial variants from peripheral blood leukocytes, they provide evidence of an association between systemic mtDNA defects and glaucoma. Moreover, from peripheral blood leukocytes, Sundaresan et al. identified mtDNA mutations in complex I in approximately one-third of POAG patients [102]. These results are in line with previous studies that addressed the possibility of defects in complex I resulting in impaired respiration rates and ATP production in lymphocytes of patients with POAG [60, 61]. To what proportion mtDNA variants are inherited through maternal line or acquired throughout life as somatic mutations is currently not known.
Somatic mutations are prone to accumulate more frequently in the mitochondrial genome due to the lack of protective histones and an efficient DNA repair system associated with the nuclear genome [103]. Mitochondrial abnormalities are associated with a number of optic neuropathies, and accumulating evidence indicates that age-related mitochondrial defects play a central role in the pathogenesis of glaucoma [44, 104, 105]. Mitochondrial genetic variants have also been linked to other neurodegenerative diseases, including Parkinson's disease and Alzheimer's disease, which are thought to have some overlapping pathologic features with glaucomatous neurodegeneration [106–109].
Exposure of mtDNA to ROS can induce its degradation, and mtDNA degradation products are found in human cerebrospinal fluid and plasma [110]. Transfection of mouse primary astrocytes with degraded mitochondrial polynucleotides was shown to cause a proinflammatory response, which was characterized by the upregulation of TNF-α, IL-1β, IL-6, and monocyte chemotactic protein 1 [110]. This observation suggests a mechanism for mtDNA degradation and downstream activation of proinflammatory phenotypes in glial cells (Figure 1). It was also previously reported that mtDNA deteriorates in response to hydrogen peroxide in HA-1 hamster ovarian cells, an effect that was not observed with nuclear DNA or cytoplasmic RNA [111]. Degradation of the mitochondrial genome was apparent in both mtDNA and mitochondrial RNA species.
Even though an immunostimulatory release of mtDNA may occur through the formation of mitochondrial-derived vesicles or necrotic cell death, a more controlled and less immunogenic mechanism of mtDNA processing has also been proposed [40, 112]. PTEN-induced kinase 1 (PINK1) and Parkin, two mitochondrial proteins linked to mitophagy and Parkinson's disease, have been shown to actively inhibit mitochondrial-derived vesicles and mitochondrial antigen presentation in favor of mitophagy [112]. This suggests that impairments in PINK1-Parkin signaling may contribute to inflammation in neurodegenerative conditions.
2.3. Deficient Mitophagy
Mitophagy is the process in which damaged mitochondria are degraded by the autophagy system. This process is vital for removing debris and aggregating proteins from the cells, thereby protecting cells of potential cell-damaging proteins. For postmitotic cells like neurons, mitophagy is an essential survival mechanism for neuroprotection and elimination of toxic compounds. Damaged mitochondria are singled out and degraded, through the activities of PINK1 and Parkin [113].
In physiological conditions, PINK1, imported from the cytosol via mitochondrial translocases, is usually found within the inner mitochondrial membrane, where it is exported, proteolytically cleaved, and degraded. PINK1 has a mitochondrial targeting sequence that directs the protein into the correct mitochondrial subcompartment [114]. During mitochondrial damage, the mitochondrial membrane potential is altered and PINK1 is prevented from entering through the outer mitochondrial membrane (OMM) [114]. As PINK1 molecules accumulate on the OMM, they start a cascade of events to recruit and activate cytosolic Parkin. Parkin initiates the process of degradation of the mitochondrion, which involves ubiquitination of the mitochondria, autophagosome engulfment, and lysosomal fusion. Both Parkin and PINK1, therefore, contribute to the build-up of phosphorylated ubiquitin and abundant accumulation of PINK1 on the OMM, which acts as an indicator to the cell where the mitochondrion is damaged and will need to be removed [115].
Mitophagy can be activated by ROS in response to various conditions, such as oxidative stress, starvation, and mechanical injury [52, 116]. Wang et al. showed that ROS have a direct effect in PINK1-Parkin signaling and these effects can be reversed with antioxidant treatment using superoxide dismutase-2 [116]. Independently from the direct effects of ROS in mitophagy, autosomal recessive forms of glaucoma, familial Parkinson's disease, and amyotrophic lateral sclerosis (ALS) have been associated with dysfunctions of PINK1-Parkin pathway.
Specific mutations in OPTN and TBK1 have been implicated in the pathogenesis of a subgroup of NTG with early onset (Figure 1) [99]. OPTN is a ubiquitously expressed protein involved in neuroinflammation, Golgi maintenance, vesicular trafficking, and autophagy. In mitophagy, OPTN acts as a receptor protein, downstream of PINK1-Parkin, that is translocated to damaged mitochondria via binding to ubiquitinated mitochondrial proteins and through interaction with microtubule-associated protein 1 light chain 3 (LC3) to couple the mitochondria with an autophagosome for degradation [99, 117]. Several hypotheses about the role of OPTN in glaucoma have indicated a neuroprotective function and shown that mutations may lead to RGC loss through downregulation or dysfunction of this protein. Due to a relatively high expression of OPTN in the retina, mutations in this protein may increase RGC vulnerability. Chernyshova et al. showed that several glaucoma OPTN-mutant proteins (E50K, A377T, H486R, H26D, E103D, T202R, and A336G) seemed to restore mitophagy in HeLa cells where Parkin-dependent mitophagy was previously inhibited [117]. Such results were not seen in OPTN-mutant proteins associated with ALS, which suggests that glaucoma associated with OPTN mutations may occur through a mechanism independent from mitophagy dysfunction. A different study, conducted by Shim et al., found that acute overexpression of OPTN E50K mutant in rat RGCs caused increased mitophagy, ROS production, and activation of apoptosis via Bax pathway [118]. A possible explanation for an increased mitophagy was elucidated by reports that observed that the OPTN E50K and M98K mutants displayed striking affinity to TBK1 [119–121]. As TBK1 stimulates autophagy by phosphorylating and activating OPTN, abnormal TBK1 activation of OPTN may explain the increase in mitophagy [98]. Minegishi et al. also reported that the OPTN E50K mutant forms aggregates of insoluble protein in neuronal cells derived from induced pluripotent stem cells, which seem to lead to cell death [121].
Copy number variation mutations of the TKB1 gene have also been found in a subgroup of NTG [98]. A duplication of TBK1 has been found to increment TBK1 transcription, which is linked to a gain of function role of TBK1. Transgenic mice that had the TBK1 gene duplicated or triplicated showed progressive loss of RGCs, proportional to the number of gene copies, with no increase in IOP [122]. Apart from OPTN phosphorylation, TBK-1 is also able to activate mitophagy in an OPTN-independent manner through p62 phosphorylation, suggesting that OPTN and p62 regulate mitophagy by different mechanisms [123].
Apart from a direct role of TBK1 and OPTN in mitophagy, both proteins are also involved in innate immune inflammatory signaling, such as through the activation of nuclear factor-kappa B (NF-κB) [124]. NF-κB is a family of transcription factors that are key regulators of cytokine production and respond to a wide variety of inflammatory stimuli.
The role of OPTN in NF-κB regulation has been somewhat controversial due to divergent results that came from different cell types and stimuli [125, 126]. OPTN was shown to interact with cylindromatosis (CYLD), an enzyme that deubiquitinates receptor-interacting protein (RIP) to inhibit TNF-α-induced NF-κB activation [127]. Studies with the glaucoma-associated OPTN H486R mutant demonstrated loss of interaction of OPTN and CYLD, which resulted in increased NF-κB signaling induced by inflammatory cytokines [128, 129]. Tanishima et al. also found another binding partner of OPTN, which is involved in NF-κB signaling [128]. The interaction of OPTN with IL-1β receptor-associated kinase 1 (IRAK1) also prevents NF-κB activation, induced not by TNF-α but also by IL-1β and TLR signaling. Taken together, these studies suggest multiple links between OPTN and NF-κB pathway, although not yet clearly characterized for glaucoma. It is important also to point out that OPTN expression is regulated by NF-κB itself by binding to an OPTN promoter and making a negative feedback loop to NF-κB activation [130]. The impact of glaucoma-associated OPTN mutants in this NF-κB negative feedback loop is currently not known.
TBK1 plays a key role in innate immunity by regulating the expression of inflammatory factors, such as NF-κB, IRF3, and IRF7, which has been linked to type-I interferon production [131, 132]. Despite TBK1 immune regulatory capacity, direct inflammatory effects derived from the glaucoma-associated TBK1 duplication have not been documented to date.
3. Dysfunction of Neuroglial Cells
Similar to many other neurodegenerative diseases, glial activation is recognized as a hallmark of neuroinflammation in glaucoma. When proinflammatory stimuli arise during injury, astrocytes, Müller cells, and microglia become activated to produce cytokines and chemokines. Although Müller cells, astrocytes, and microglia each have a different developmental origin, they share many functions within the retina, and there is an intricate interrelationship among these cells in the induction of an inflammatory phenotype [133, 134]. However, along with prolonged inflammatory activation of glial cells, there is also a failure in the regulation of immunity, which may ultimately tilt an initial beneficial inflammatory response towards a dysfunctional immune response and neuronal injury.
3.1. Astroglia
Astrocytes and Müller cells facilitate the interface between neurons, endothelia, and other glial cells to mediate and modulate metabolic functions, synaptic activity, and homeostasis of the blood-retina barrier [135–138]. These neurosupportive cells also contribute to the defense and homeostasis of neurons by recognizing and responding to local insults. However, under pathological conditions, astrocytes and Müller cells can undergo a pronounced transformation termed gliosis [19, 139–141].
Previous studies revealed that reactive astroglial cells can have both protective and detrimental influences on neuronal survival in glaucoma and other neurodegenerative conditions [19]. Astroglia was shown to become highly reactive in the retina and ONH in glaucomatous human donor eyes, and experimental models of glaucoma [141–143]. Sun et al. demonstrated that a brief period of ocular hypertension may be sufficient to initiate astroglial reactivity in experimental glaucoma [144]. This activation, seen in early stages of the disease, is characterized by morphological alterations and molecular responses that may be detectable even before the damage of RGCs and axons [145]. Although gliosis has the primary aim of circumscribing injured tissues to protect uninjured neurons, astroglial activation may lead to tissue remodeling that result in further biomechanical stress on optic nerve axons and in inadequate metabolic support to RGCs [143–145]. A proinflammatory signature acquired by astrocytes upon stress signals may, therefore, lead to tissue destruction and RGC death [146, 147]. Along with the capacity to trigger innate immunity signaling pathways, reactive astroglia may also compromise the blood-retina barrier, which further increases the access of systemic immune cells into the retina and the optic nerve [137, 145].
3.2. Microglia
Microglial cells are myeloid-derived cells that reside in the CNS, providing neurotrophic support and promoting tissue renewal through their phagocytic functions [148]. In inflammatory conditions, microglial cells may also contribute to neuron injury [27, 149, 150]. Despite having a more limited antigen-presentation capacity than that of peripheric professional antigen-presenting cells, microglial cells also act as the first and main form of active immune response in the CNS [138, 148].
Similar to other glial cells, microglia show increased reactivity in the retina and ONH in experimental models and human donor eyes with glaucoma [133, 149, 150]. Microglia react to neural injury with morphological changes, proliferation, migration, and production of inflammatory cytokines that further propagate neuroinflammation. This reaction also includes the release of ROS, NO, and TNF-α, leading to neurotoxic effects and aggravated neuronal loss [22, 23, 151]. It has been reported that microglial activation is one of the first events in glaucomatous neural damage occurring prior to RGC loss [152]. In a mouse model of inherited glaucoma (DBA/2J mice), the extent of neurodegeneration correlated with early microglial alterations in vivo [152]. Treatments with minocycline, which inhibit microglial activation, reduced RGC death in the same mouse model [153]. In addition to resident microglia, data from DBA/2J glaucoma model also indicate that glaucomatous proinflammatory state may be amplified by monocytes and other circulating immune cells that invade the ONH and contribute to neurodegeneration [17].
Nitric oxide (NO) is also known to be secreted by microglia in inflammatory conditions [154]. Upregulation of inducible nitric oxide synthase (iNOS) and increased NO levels were found in the ONH of glaucomatous patients and in the retina and ONH of experimental models of glaucoma [155–158]. Inhibition of iNOS by aminoguanidine provided protection to RGCs in a glaucoma model, supporting the possibility of a role of NO in the pathophysiology of glaucoma [159].
4. Activation of the Immune System
Neuronal metabolic dysfunction and reactive gliosis are known to activate glial Toll-like receptor (TLR) signaling and induce complement activation (Figure 2) [20, 160]. In addition to the costimulatory role of ROS in antigen presentation of glial cells, oxidative stress may modify the antigenic features of retina and optic nerve proteins with accumulation of advanced glycation end-products, activation of NF-κB transcriptional program, and stimulation of glial cytokine production in glaucomatous tissues [50, 161, 162].
Figure 2.
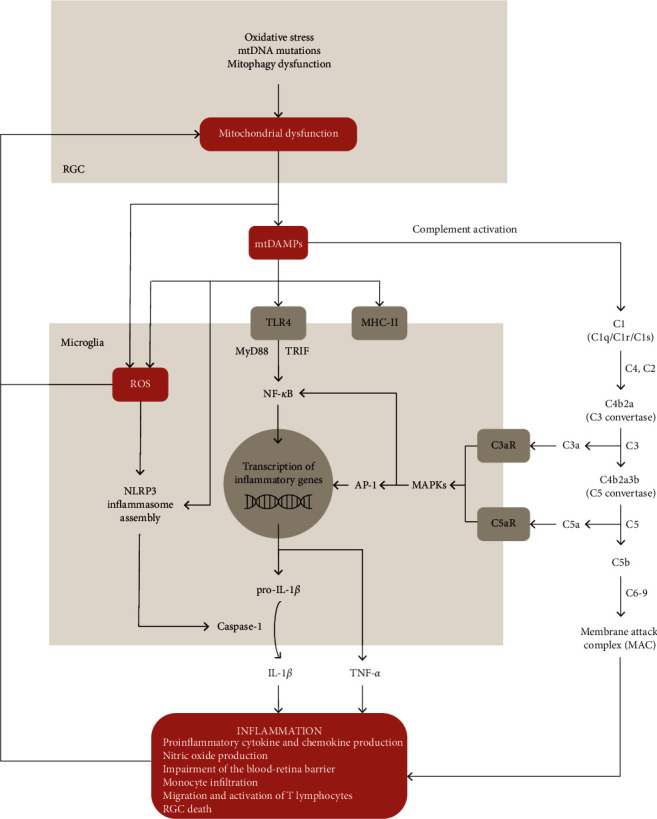
Neuronal mitochondrial damage-associated molecular patterns (mtDAMPs) can act as inducers of chronic inflammation in glaucoma. Mitochondrial-derived components from retinal ganglion cells (RGCs) can trigger inflammatory responses when recognized by complement molecules (classical pathway) and microglial pattern-recognition receptors, such as toll-like receptors (TLRs). TLR signaling induces the transcription of proinflammatory cytokines and chemokines (e.g., pro-IL-1β and TNF-α) through NF-κB. Inflammation triggered by mtDAMPs can further induce mitochondrial dysfunction, thereby amplifying a vicious cycle of inflammation. In addition to TLR signaling, the presentation of DAMPs through Class II MHC (MHC-II) molecules can further promote the induction of inflammation through T-cell activation. Mitochondrial products can also function directly as NLRP3 activators, which allows caspase-1-dependent release of IL-1β. AP-1: activator protein-1; MAPKs: mitogen-activated protein kinases; C3aR: C3a receptor; C5aR: C5a receptor.
In addition to the activation of innate immunity in the retina and ONH, glial cells are capable of stimulating systemic immune responses by displaying and releasing signals that favor the recruitment of circulating T lymphocytes. Several factors seem to contribute to the increment of immunogenicity and the activation of inflammatory cues for autoimmunity, namely, an increased exposure of antigens due to neuronal injury, increased expression of immunostimulatory stress proteins, and increased antigenicity due to protein modifications [25, 50, 162–166]. In a similar fashion to peripheral antigen-presenting cells, reactive glia in human glaucoma and animal models display high surface levels of major histocompatibility complex II molecules and stress-associated costimulatory molecules that may enhance antigen presentation to T lymphocytes [161, 167–169].
4.1. Release of Damage-Associated Molecular Patterns (DAMPs)
The sterile inflammatory response following glaucomatous damage activates innate immunity mechanisms that are also triggered in infection-induced inflammation. Following infection, microorganisms are initially sensed by pattern-recognition receptors (PRRs) of the innate immune system, which bind conserved molecular patterns that are shared by different classes of microorganisms. These pathogen-associated molecular patterns (PAMPs) include microbial structural components, nucleic acids, and proteins [170]. The list of PRRs that are known to be able to sense PAMPs is extensive and is comprised most notably of four families: TLRs, nucleotide oligomerization domain (NOD)-like receptors (NLRs), C-type lectin receptors (CLRs), and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) [170, 171]. PRR ligation triggers multiple signaling pathways that may culminate in the activation of NF-κB, mitogen-activated protein kinases (MAPKs), and interferon regulatory factors (IRFs), which control the expression of proinflammatory cytokines, chemokines, and costimulatory molecules [170–172]. The resulting proinflammatory state is necessary for the generation of a robust antimicrobial response and for the activation of the adaptive immune system.
In addition to recognizing PAMPs, PRRs can be triggered by cellular damage and stress in the absence of microbial infection. Sterile tissue injury and cellular necrosis elicit robust responses characterized by proinflammatory cytokine production and leukocyte recruitment, which are triggered by TLR-, NLR-, and RLR-dependent sensing of “alarmins” or damage-associated molecular patterns (DAMPs) (Figure 2) [170, 173]. DAMPs are endogenous molecules that are isolated within intracellular compartments (e.g., DNA and N-formylated peptides) or are subject to robust metabolism and/or editing in healthy cells (e.g., DNA and double-stranded RNA) [170]. These molecules often exhibit similarities with PAMPs and can be recognized by PRRs during pathological injury [174]. Therefore, this “hidden-self” recognition serves to alert the immune system of cellular or tissue dysfunction.
Due to ancestral bacterial origin, eukaryotic mitochondria maintain prokaryotic features, including a double-membrane structure, the unique cell membrane lipids (e.g., cardiolipin), a circular genome containing CpG DNA, the absence of histones, the ability to replicate independently of the nucleus, the ability to form N-formyl peptides, which are distinct byproducts of mitochondrial translation that reflect their prokaryotic origin, and the use of separate sets of rRNAs and tRNAs encoded by the mitochondrial genome [40, 175]. Thus, cellular damage leading to the release of prokaryotic-like mitochondrial constituents through different modes of cell death can engage PRRs and act as a potent trigger of innate immune responses during stress and injury [40, 175].
Astroglia and microglia are known to participate in early stages of glaucoma but, to date, it is not known which deleterious events contribute the most to the pathogenesis of the disease. A possibility is that stressed or damaged RGCs in the ONH are the seminal source of DAMPs. Heat shock proteins (HSPs), a type of DAMPs, were shown to be upregulated in response to an elevation in IOP and increased in human glaucomatous retinas [20, 138, 176]. More recently, Chen et al. identified both bacterial and host HSPs as possible key natural antigens and demonstrated that commensal microflora induces HSP-specific memory T cells, which are then activated by host HSPs released in the retina after IOP elevation [177]. A second and still valid possibility is that astrocytes and/or microglia initiate gliosis and produce DAMPs independently of RGCs or preceding neuronal dysfunction [178]. Experimental models of glaucoma have identified reactive morphologic changes that precede axonopathy [179–181]. Along with morphologic changes derived from biomechanic causes, neuronal insult in glaucoma may be initiated or aggravated by the absence of the critical glial support, such as in astroglial metabolic substrate transfers (e.g., lactate), neurotrophin secretion, and transforming growth factor-beta (TGF-β) production, combined with a detrimental production of TNF-α [178, 182]. Speculatively, early glial responses may also constitute a glial effort to guarantee their own survival, with consequential detrimental effects on neuronal support. The identification of initiators of inflammatory events is a relevant area of study in glaucoma.
4.2. Activation of Pattern-Recognition Receptors and Inflammatory Signaling
Once stress products are recognized by PRRs such as TLRs, TNF receptor, and inflammasome, specific pathways initiate cascades of events that involve NF-κB (Figure 2) [151]. NF-κB, one of the key regulators of inflammatory immune responses, has been shown to be activated in human glaucoma and in animal models of glaucoma [20, 160].
TLR signaling pathways are among the first to be upregulated in the retinas of patients with glaucoma [19, 20, 183]. Different TLRs display increased expression on astroglia and microglia in human glaucoma, experimental ocular hypertension, and DBA/2J mice with hereditary glaucoma [19, 20]. TLR2, TLR3, and TLR4 were shown to be expressed in microglia and astrocytes of DBA/2J mice retinas, and 11 of 13 TLRs were upregulated in the ONH in early stages of the disease [183]. From these, TLR4 is the most studied Toll-like receptor and is widely expressed in the CNS. Upon DAMPs recognition, TLRs recruit adaptor proteins such as myeloid differentiation primary-response protein 88 (MyD88) and/or Toll/interleukin-1 receptor domain-containing adaptor including interferon-β (TRIF) to activate downstream transcription factors. These transcription factors include members of the NF-κB, AP-1, and interferon regulator factor families, which allow the initiation of the transcription of amplifiers and effectors such as TNF-α, IL-1β, IL-6, and an array of chemokines (e.g., CCL2, CXCL1, and CXCL10) [138, 171, 184].
Another group of PRRs that can modulate microglial inflammatory response is the NOD-like receptors, which form protein complexes commonly known as inflammasomes. Cooperative downstream cross-talks between TLRs and NOD-like receptors can lead to the maturation and release of cytokines like IL-1β and IL-18 [185–187]. Mitochondria can directly activate inflammasome signaling. Mitochondria-derived or DAMPs-induced ROS activate the NLR family pyrin domain-containing 3 (NLRP3) inflammasome pathway [188]. NLRP3 is normally associated with the endoplasmic reticulum membrane but, upon activation, is redistributed to nuclear and mitochondrial membranes, where it oligomerizes with apoptosis-associated speck-like protein containing a CARD (ASC) and procaspase-1 to form the NLRP3 inflammasome (Figure 2) [185–187]. NLRP3 inflammasome activation leads to caspase-1-dependent secretion of IL-1β and IL-18 and an inflammatory form of cell death termed as pyroptosis [188]. Studies in neurodegeneration in the CNS revealed that mitochondrial dysfunction, including the blockade of mitophagy, is sensed by the NLRP3 inflammasome [188, 189]. Several molecules were specifically identified as DAMPs by these complexes, namely, mtDNA, ATP, and cytochrome C [114, 188]. Despite this evidence, it has not been identified yet how the various aspects of mitochondrial dysfunction converge to a common pathway to activate NLRP3 inflammasome [188, 189]. On the other hand, in bone marrow-derived macrophages, caspase-1 was shown to increase mitochondrial disassembly through the activation of multiple deleterious pathways that amplify ROS production, as well as through the inhibition mitophagy mediated by cleavage of Parkin [190]. NLRP1 and NLRP3 inflammasomes seem to be involved in the pathogenesis of glaucoma in models of acute glaucoma, and, to date, only one study has demonstrated the presence of NLRP3 inflammasome in human glaucomatous eyes [160, 191, 192]. More studies are required to better understand the role of inflammasomes in the progression of glaucoma.
In addition to stress/damage sensors (e.g., TLRs and NLRP3) and inflammatory transducers (e.g., MyD88 and NF-κB), effectors and amplifiers of inflammation are also upregulated in glaucoma. Proteomic analysis of human and experimental glaucomatous retinas revealed an upregulation of kinases that are involved in the activation of the NF-κB pathway, such as RIPK, NIK, and IκK [19, 160]. Activation of NF-κB results in the transcription of IL-1 cytokine family, which can further amplify inflammation by inducing a secondary release of cytokines in microglia and astrocytes. IL-1 cytokines were shown to be upregulated in the ONH at early stages of DBA/2J glaucoma in mice, and antioxidant treatment could afford the downregulation of proinflammatory cytokines and NF-κB [183, 193].
The proinflammatory imbalance seen in glaucomatous tissues is characterized by a marked increase of TNF-α production, which is also linked to RGC death [23, 151, 161, 194, 195]. TNF-α release can itself affect mitochondrial function by impairing the function of mitochondrial components, reducing ATP production, increasing ROS, and depolarizing the mitochondrial membrane potential [39]. Increased ROS can then have further detrimental effects by maintaining the activation of NF-κB and the production of proinflammatory signaling. The stimulation of neuroinflammation can therefore also have a damaging effect on RGC mitochondrial function itself, thus creating a vicious cycle.
4.3. Activation of the Complement System
The complement system is part of the innate immune defense, and it consists of a number of small proteins that provide immune surveillance both peripherally and in the CNS. Complement proteins are expressed in normal physiological processes of the retina and become increased in pathological conditions like inflammation, aging, and trauma and in neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and glaucoma [138, 196]. In addition to its role in pathogen recognition and removal, the complement system is also involved in homeostasis by synapse elimination and clearance of potential mediators of damage or injury [197, 198]. Thus, the decisive element that tilts the balance between a homeostatic or a proinflammatory complement-mediated response appears to be the presence of specific danger signals associated with chronic inflammation.
The complement system comprises three distinct and tightly regulated activatory cascades known as the classical, alternative, and lectin pathways [197]. All pathways, which converge in the activation of C3 convertase, contribute to the opsonization of foreign pathogens or apoptotic cell/cellular compartments, the release of inflammatory signaling through anaphylatoxins, and the formation of the membrane attack complex (MAC) [199, 200]. The classical pathway (Figure 2) is initiated when the C1 complex recognizes, among others, antigen-antibody complexes, mitochondrial membrane components, apoptotic cells, or amyloid fibrils [201]. This recognition leads to the formation of C3 convertase and cleavage of C3 protein into C3a and C3b. C3a is an anaphylatoxin that attracts phagocytes to sites of inflammation, and C3b acts as opsonin, which further amplifies complement activation by inducing the cleavage of C5 into C5a and C5b by C5 convertase. C5a is also an anaphylatoxin, and C5b leads to the formation of a lysis-inducing channel in the targeted cell together with C6, C7, C8, and C9 [138, 200].
Increased complement activation has been linked to decreased RGC survival in humans and in animal models of glaucoma, suggesting an involvement of the complement system in the progression of the disease [24, 183, 202–204]. Studies in rodents and nonhuman primates indicated an increased expression of multiple components of the classical complement cascade, which seem to constitute one of the earliest signaling responses to high IOP in the retina and ONH [183, 205, 206]. In DBA/2J glaucomatous mice, an increment of the C1 complex protein C1QA was observed in the ONH before the detection of axonal damage, indicating a potential causal contribution [183]. Deposition of C1QA was also observed in RGC dendrites in glaucomatous retinas of DBA/2J mice, nonhuman primates, and humans, suggesting an involvement of the complement cascade in pathological synapse elimination and/or dendrite remodeling in glaucoma [198, 202–204]. Ablation of C1QA in DBA/2J mice or the viral overexpression of Crry, a C3 inhibitor, were both effective in decreasing RGC loss [183, 207, 208].
Downstream components of the complement cascade that are necessary for the formation of MAC seem to also contribute to glaucoma [197]. In human glaucomatous retinas and in experimental models of glaucoma, RGCs were shown to have a marked deposition of MAC [202, 208, 209]. Drug inhibition of complement activation or knockout of the C5 gene reduced MAC deposition as well as RGC loss in rodent models of ocular hypertension and glaucoma [208, 209]. More recently, the intravitreal administration of a monoclonal antibody against the C5 could also afford the preservation of RGCs in an experimental autoimmune model of glaucoma [210]. Together these findings indicate that the complement system can be activated in early stages of RGC injury, which can result in a net increase of retinal damage. Although poorly defined, astrocytes and Müller cells are also able to produce complement proteins and therefore may be conducive to degeneration in a similar fashion [203, 211].
4.4. Leukocyte Infiltration
Leukocyte infiltration is a common event in CNS that occurs after injury, disease, and chronic stress [18, 138]. Although early immune responses are likely to be natural attempts to minimize damage after an injury, later immune responses are prone to evolve into chronicity and become more detrimental. In some cases, these beneficial and detrimental events involve molecules in the same pathways [138].
Leukocyte infiltration comprises a cascade of sequential steps that are initiated by the production of cytokines and chemokines in glial cells [23, 138, 212]. The molecular markers involved in leukocyte infiltration include various classes of adhesion/activation/migration proteins expressed on the blood-retina barrier and migrating leukocytes, such as selectins (e.g., P-selectin) and integrin ligands (e.g., VCAM-1 and ICAM-1) on endothelial cells, and selectin ligands (e.g., PSGL1) and integrins (e.g., LFA1) on the surface of leukocytes [18, 138]. These interactions between endothelial cells and leukocytes result in the loosening of endothelial tight junctions to grant access of leukocytes to the CNS [18, 213]. The mechanisms of transendothelial migration in the CNS during injury and disease are very complex, and recent work has shown that the molecules involved in this process are more abundant than previously thought [213].
In the DBA/2J glaucoma model, aberrant upregulation of P-selectin and VCAM-1 was observed in early stages of the disease, suggesting that transendothelial migration of leukocytes takes part in the initiation of glaucomatous damage [17]. Reactive gliosis seems to be able to weaken the perivascular barriers and facilitate access of circulating immune cells and other components such as autoantibodies into the retina and ONH [214]. Breaches in the blood-retina barrier may become visible and result in small optic disc hemorrhages or parapapillary chorioretinal atrophy areas, both of which are commonly detectable in glaucomatous eyes [215, 216]. In line with these results, a longitudinal study revealed a gradual increment of serum autoantibodies in glaucoma patients with optic disc hemorrhages and no detectable change in patients with no optic disc hemorrhages [217]. In addition, the transfer of mononuclear cells from patients with optic disc hemorrhages into immune-deficient mice resulted in increased RGC loss [145, 217]. The tracing of circulating monocytes with an inflammatory profile in the DBA/2J model confirmed the entry of these cells into the ONH and its contribution to glaucomatous damage [17]. These observations are consistent with the higher titers of proinflammatory cytokines found in the blood of patients with glaucoma, as well as the reduction of monocyte infiltration when DBA/2J mice were treated with radiation [17, 218, 219].
Recent findings shed light on the involvement of T-cell-mediated mechanisms in the pathogenesis of glaucoma. This notion is supported by the observation that local inflammation in the retina and ONH is required for T cell crossing of the blood-retina barrier and T cell accumulation is more prominent in inflammatory areas that are more susceptible to high IOP [177, 220–222]. Mice deficient in T cells, but not B cells, displayed a dramatically attenuated RGC and axon damage [177]. One report, however, pointed out that neurodegeneration could be induced in mice, with IOP being within normal or elevated range, after the adoptive transfer of T cells isolated from genetic mouse models of glaucoma [177, 223]. These results suggest that activated T cells of glaucomatous mice are also capable of entering the retina with an intact blood-retina barrier, although probably at a much slower rate or under certain conditions [177, 223]. Studies of blood samples from patients with glaucoma have also detected a shift in the T lymphocyte signature towards a proinflammatory Th1 phenotype and an imbalance of regulatory T cells, indicating a possible lack of efficient T-cell suppression and a minor role of B cells in the disease process [218, 219]. Despite these observations, it remains unclear which subsets of T cells predominate as effector cells or act as initiators of glaucomatous neurodegeneration.
5. Conclusion
Numerous forms of endogenous and environmental stressors may disrupt mitochondrial function by impacting mitochondrial homeostasis. Mitochondrial function in RGCs may decline progressively in association with physiologic aging, leading to the release of multiple mitochondrial DAMPs. These misplaced or altered mitochondria-derived molecules may subsequently trigger innate immune responses and result in the onset or progression of inflammatory neurodegenerative diseases, such as in glaucoma. However, the precise mechanism of how mitochondrial DAMPs lead to glaucomatous neurodegeneration is yet to be fully dissected.
There are also mechanisms of glial dysfunction that contribute to neurodegeneration in the early stages of glaucoma. Despite the evident role of inflammation in the disease, to date, it is still not known which exact inflammatory signals lead to activation of glia during the disease progression and how intricate these processes are to neuronal dysfunction, retina-blood barrier impairment, or systemic inflammation. Several experimental challenges hinder a comprehensive understanding of glaucoma. Unlike neurons, microglia and astrocytes are challenging to study in vitro as these cells acquire different phenotypes that hardly resemble in vivo conditions. Several important questions, therefore, remain open, such as, which and how mitochondria-derived molecules contribute to neuroinflammation, and how upstream or downstream this process is in the progression of the disease.
An answer to these questions will facilitate the design of better therapeutic options that are not merely supportive. As glaucoma has many subphenotypes, finding a common therapeutic solution for all poses many challenges. Nonetheless, considering that RGC death is the final common pathway for a very complex pathology, identifying and targeting the key events of neuronal, glial, and immune dysfunction may help halt the neurodegenerative process in the early stages of the disease.
Acknowledgments
The author apologizes for any studies that may have been omitted in this review. This work was supported by the Lundbeck Foundation (grant R266-2017-3984) and Novo Nordisk Foundation (grant NNF18SA0034956).
Conflicts of Interest
The author declares no conflicts of interest.
References
- 1.Quigley H. A., Dunkelberger G. R., Green W. R. Chronic human glaucoma causing selectively greater loss of large optic nerve fibers. Ophthalmology. 1988;95(3):357–363. doi: 10.1016/s0161-6420(88)33176-3. [DOI] [PubMed] [Google Scholar]
- 2.Quigley H. A. The number of people with glaucoma worldwide in 2010 and 2020. British Journal of Ophthalmology. 2006;90(3):262–267. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kingman S. Glaucoma is second leading cause of blindness globally. Bulletin of the World Health Organization. 2004;82(11):887–888. [PMC free article] [PubMed] [Google Scholar]
- 4.Nickells R. W. From ocular hypertension to ganglion cell death: a theoretical sequence of events leading to glaucoma. Canadian Journal of Ophthalmology. 2007;42(2):278–287. doi: 10.3129/can.j.ophthalmol.i07-036. [DOI] [PubMed] [Google Scholar]
- 5.Hirt J., Porter K., Dixon A., McKinnon S., Liton P. B. Contribution of autophagy to ocular hypertension and neurodegeneration in the DBA/2J spontaneous glaucoma mouse model. Cell Death Discovery. 2018;4:p. 14. doi: 10.1038/s41420-018-0077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crish S. D., Sappington R. M., Inman D. M., Horner P. J., Calkins D. J. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proceedings of the National Academy of Sciences. 2010;107(11):5196–5201. doi: 10.1073/pnas.0913141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rakic P., Riley K. Overproduction and elimination of retinal axons in the fetal rhesus monkey. Science. 1983;219(4591):1441–1444. doi: 10.1126/science.6828871. [DOI] [PubMed] [Google Scholar]
- 8.Quigley H. A., McKinnon S. J., Zack D. J., et al. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Investigative Ophthalmology & Visual Science. 2000;41(11):3460–3466. [PubMed] [Google Scholar]
- 9.Almasieh M., Wilson A. M., Morquette B., Cueva Vargas J. L., Di Polo A. The molecular basis of retinal ganglion cell death in glaucoma. Progress in Retinal and Eye Research. 2012;31(2):152–181. doi: 10.1016/j.preteyeres.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen J. V., Soto I., Kim K.-Y., et al. Myelination transition zone astrocytes are constitutively phagocytic and have synuclein dependent reactivity in glaucoma. Proceedings of the National Academy of Sciences. 2011;108(3):1176–1181. doi: 10.1073/pnas.1013965108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chidlow G., Ebneter A., Wood J. P. M., Casson R. J. The optic nerve head is the site of axonal transport disruption, axonal cytoskeleton damage and putative axonal regeneration failure in a rat model of glaucoma. Acta Neuropathologica. 2011;121(6):737–751. doi: 10.1007/s00401-011-0807-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iester M., De Feo F., Douglas G. R. Visual field loss morphology in high- and normal-tension glaucoma. Journal of Ophthalmology. 2012;2012:8. doi: 10.1155/2012/327326.327326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shields M. B. Normal-tension glaucoma: is it different from primary open-angle glaucoma? Current Opinion in Ophthalmology. 2008;19(2):85–88. doi: 10.1097/icu.0b013e3282f3919b. [DOI] [PubMed] [Google Scholar]
- 14.Leske M. C., Heijl A., Hyman L., Bengtsson B., Dong L., Yang Z. Predictors of long-term progression in the early manifest glaucoma trial. Ophthalmology. 2007;114(11):1965–1972. doi: 10.1016/j.ophtha.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 15.Tezel G. Immune regulation toward immunomodulation for neuroprotection in glaucoma. Current Opinion in Pharmacology. 2013;13(1):23–31. doi: 10.1016/j.coph.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rizzo M. I., Greco A., De Virgilio A., et al. Glaucoma: recent advances in the involvement of autoimmunity. Immunologic Research. 2017;65(1):207–217. doi: 10.1007/s12026-016-8837-3. [DOI] [PubMed] [Google Scholar]
- 17.Howell G. R., Soto I., Zhu X., et al. Radiation treatment inhibits monocyte entry into the optic nerve head and prevents neuronal damage in a mouse model of glaucoma. Journal of Clinical Investigation. 2012;122(4):1246–1261. doi: 10.1172/jci61135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ransohoff R. M., Brown M. A. Innate immunity in the central nervous system. Journal of Clinical Investigation. 2012;122(4):1164–1171. doi: 10.1172/jci58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tezel G., Yang X., Luo C., Cai J., Powell D. W. An astrocyte-specific proteomic approach to inflammatory responses in experimental rat glaucoma. Investigative Opthalmology & Visual Science. 2012;53(7):4220–4233. doi: 10.1167/iovs.11-9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo C., Yang X., Kain A. D., Powell D. W., Kuehn M. H., Tezel G. Glaucomatous tissue stress and the regulation of immune response through glial Toll-like receptor signaling. Investigative Opthalmology & Visual Science. 2010;51(11):5697–5707. doi: 10.1167/iovs.10-5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chua J., Vania M., Cheung C. M., et al. Expression profile of inflammatory cytokines in aqueous from glaucomatous eyes. Molecular Vision. 2012;18:431–438. [PMC free article] [PubMed] [Google Scholar]
- 22.Madeira M. H., Boia R., Santos P. F., Ambrósio A. F., Santiago A. R. Contribution of microglia-mediated neuroinflammation to retinal degenerative diseases. Mediators of Inflammation. 2015;2015:15. doi: 10.1155/2015/673090.673090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakazawa T., Nakazawa C., Matsubara A., et al. Tumor necrosis factor- mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. Journal of Neuroscience. 2006;26(49):12633–12641. doi: 10.1523/jneurosci.2801-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmed F., Brown K. M., Stephan D. A., Morrison J. C., Johnson E. C., Tomarev S. I. Microarray analysis of changes in mRNA levels in the rat retina after experimental elevation of intraocular pressure. Investigative Opthalmology & Visual Science. 2004;45(4):1247–1258. doi: 10.1167/iovs.03-1123. [DOI] [PubMed] [Google Scholar]
- 25.Yang Z., Quigley H. A., Pease M. E., et al. Changes in gene expression in experimental glaucoma and optic nerve transection: the equilibrium between protective and detrimental mechanisms. Investigative Opthalmology & Visual Science. 2007;48(12):5539–5548. doi: 10.1167/iovs.07-0542. [DOI] [PubMed] [Google Scholar]
- 26.Tombran-Tink J., Barnstable C. J., Bruce Shields M. Mechanisms of the Glaucomas: Disease Processes and Therapeutic Modalities. Berlin, Germany: Springer Science & Business Media; 2008. [Google Scholar]
- 27.Ramirez A. I., de Hoz R., Salobrar-Garcia E., Salazar J. J., Rojas B., Ajoy D. The role of microglia in retinal neurodegeneration: Alzheimer’s disease, Parkinson, and glaucoma. Frontiers in Aging Neuroscience. 2017;9:p. 214. doi: 10.3389/fnagi.2017.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toft-Kehler A. K., Skytt D. M., Svare A., et al. Mitochondrial function in Müller cells-does it matter? Mitochondrion. 2017;36:43–51. doi: 10.1016/j.mito.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Yu D.-Y., Cringle S. J. Retinal degeneration and local oxygen metabolism. Experimental Eye Research. 2005;80(6):745–751. doi: 10.1016/j.exer.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 30.Ito Y. A., Di Polo A. Mitochondrial dynamics, transport, and quality control: a bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion. 2017;36:186–192. doi: 10.1016/j.mito.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 31.Morgan J. E. Circulation and axonal transport in the optic nerve. Eye. 2004;18(11):1089–1095. doi: 10.1038/sj.eye.6701574. [DOI] [PubMed] [Google Scholar]
- 32.Barron M. J., Griffiths P., Turnbull D. M., Bates D., Nichols P. The distributions of mitochondria and sodium channels reflect the specific energy requirements and conduction properties of the human optic nerve head. British Journal of Ophthalmology. 2004;88(2):286–290. doi: 10.1136/bjo.2003.027664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andrews R. M., Griffiths P. G., Johnson M. A., Turnbull D. M. Histochemical localisation of mitochondrial enzyme activity in human optic nerve and retina. British Journal of Ophthalmology. 1999;83(2):231–235. doi: 10.1136/bjo.83.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bristow E. A., Griffiths P. G., Andrews R. M., Johnson M. A., Turnbull D. M. The distribution of mitochondrial activity in relation to optic nerve structure. Archives of Ophthalmology. 2002;120(6):791–796. doi: 10.1001/archopht.120.6.791. [DOI] [PubMed] [Google Scholar]
- 35.Wang L., Dong J., Cull G., Fortune B., Cioffi G. A. Varicosities of intraretinal ganglion cell axons in human and nonhuman primates. Investigative Opthalmology & Visual Science. 2003;44(1):2–9. doi: 10.1167/iovs.02-0333. [DOI] [PubMed] [Google Scholar]
- 36.Rambold A. S., Kostelecky B., Elia N., Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proceedings of the National Academy of Sciences. 2011;108(25):10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gomes L. C., Benedetto G. D., Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nature Cell Biology. 2011;13(5):589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilkins H. M., Carl S. M., Greenlief A. C. S., Festoff B. W., Swerdlow R. H. Bioenergetic dysfunction and inflammation in Alzheimer’s disease: a possible connection. Frontiers in Aging Neuroscience. 2014;6 doi: 10.3389/fnagi.2014.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilkins H. M., Weidling I. W., Ji Y., Swerdlow R. H. Mitochondria-derived damage-associated molecular patterns in neurodegeneration. Frontiers in Immunology. 2017;8:p. 508. doi: 10.3389/fimmu.2017.00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.West A. P., Shadel G. S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nature Reviews Immunology. 2017;17(6):363–375. doi: 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang B., Li S., Cao W., Sun X. The association of oxidative stress status with open-angle glaucoma and exfoliation glaucoma: a systematic review and meta-analysis. Journal of Ophthalmology. 2019;2019:14. doi: 10.1155/2019/1803619.1803619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanamori A., Catrinescu M.-M., Kanamori N., Mears K. A., Beaubien R., Levin L. A. Superoxide is an associated signal for apoptosis in axonal injury. Brain. 2010;133(9):2612–2625. doi: 10.1093/brain/awq105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alqawlaq S., Flanagan J. G., Sivak J. M. All roads lead to glaucoma: induced retinal injury cascades contribute to a common neurodegenerative outcome. Experimental Eye Research. 2019;183:88–97. doi: 10.1016/j.exer.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 44.Chrysostomou V., Rezania F., Trounce I. A., Crowston J. G. Oxidative stress and mitochondrial dysfunction in glaucoma. Current Opinion in Pharmacology. 2013;13(1):12–15. doi: 10.1016/j.coph.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 45.Tezel G. L N., Yang X. Caspase-independent component of retinal ganglion cell death, in vitro. Investigative Opthalmology & Visual Science. 2004;45(11):4049–4059. doi: 10.1167/iovs.04-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li G.-Y., Osborne N. N. Oxidative-induced apoptosis to an immortalized ganglion cell line is caspase independent but involves the activation of poly(ADP-ribose)polymerase and apoptosis-inducing factor. Brain Research. 2008;1188:35–43. doi: 10.1016/j.brainres.2007.10.073. [DOI] [PubMed] [Google Scholar]
- 47.Iomdina E. N., Khoroshilova-Maslova I. P., Robustova O. V., et al. Mitochondria-targeted antioxidant SkQ1 reverses glaucomatous lesions in rabbits. Frontiers in Bioscience (Landmark Edition) 2015;20:892–901. doi: 10.2741/4343. [DOI] [PubMed] [Google Scholar]
- 48.Inman D. M., Lambert W. S., Calkins D. J., Horner P. J. α-Lipoic acid antioxidant treatment limits glaucoma-related retinal ganglion cell death and dysfunction. PLoS One. 2013;8 doi: 10.1371/journal.pone.0065389.e65389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moreno M. C., Campanelli J., Sande P., Sáenz D. A., Keller Sarmiento M. I., Rosenstein R. E. Retinal oxidative stress induced by high intraocular pressure. Free Radical Biology and Medicine. 2004;37(6):803–812. doi: 10.1016/j.freeradbiomed.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 50.Tezel G. L. N., Yang X., Cai J. Proteomic identification of oxidatively modified retinal proteins in a chronic pressure-induced rat model of glaucoma. Investigative Opthalmology & Visual Science. 2005;46(9):p. 3177. doi: 10.1167/iovs.05-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin K. R., Levkovitch-Verbin H., Valenta D., Baumrind L., Pease M. E., Quigley H. A. Retinal glutamate transporter changes in experimental glaucoma and after optic nerve transection in the rat. Investigative Ophthalmology & Visual Science. 2002;43(7):2236–2243. [PubMed] [Google Scholar]
- 52.Lin W.-J., Kuang H.-Y. Oxidative stress induces autophagy in response to multiple noxious stimuli in retinal ganglion cells. Autophagy. 2014;10(10):1692–1701. doi: 10.4161/auto.36076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scherz-Shouval R., Shvets E., Fass E., Shorer H., Gil L., Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. The EMBO Journal. 2007;26(7):1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshimura K., Shibata M., Koike M., et al. Effects of RNA interference of Atg4B on the limited proteolysis of LC3 in PC12 cells and expression of Atg4B in various rat tissues. Autophagy. 2006;2(3):200–208. doi: 10.4161/auto.2744. [DOI] [PubMed] [Google Scholar]
- 55.Satilmis M. Ü. C., OrgÜl S., Doubler B., Flammer J. Rate of progression of glaucoma correlates with retrobulbar circulation and intraocular pressure. American Journal of Ophthalmology. 2003;135(5):664–669. doi: 10.1016/s0002-9394(02)02156-6. [DOI] [PubMed] [Google Scholar]
- 56.Yanagi M., Kawasaki R., Wang J. J., Wong T. Y., Crowston J., Kiuchi Y. Vascular risk factors in glaucoma: a review. Clinical & Experimental Ophthalmology. 2011;39(3):252–258. doi: 10.1111/j.1442-9071.2010.02455.x. [DOI] [PubMed] [Google Scholar]
- 57.Malone P. E., Hernandez M. R. 4-Hydroxynonenal, a product of oxidative stress, leads to an antioxidant response in optic nerve head astrocytes. Experimental Eye Research. 2007;84(3):444–454. doi: 10.1016/j.exer.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McElnea E. M., Quill B., Docherty N. G., et al. Oxidative stress, mitochondrial dysfunction and calcium overload in human lamina cribrosa cells from glaucoma donors. Molecular Vision. 2011;17:1182–1191. [PMC free article] [PubMed] [Google Scholar]
- 59.Vohra R., Dalgaard L. M., Vibæk J., et al. Potential metabolic markers in glaucoma and their regulation in response to hypoxia. Acta Ophthalmologica. 2019;97(6):567–576. doi: 10.1111/aos.14021. [DOI] [PubMed] [Google Scholar]
- 60.Lee S., Sheck L., Crowston J. G., et al. Impaired complex-I-linked respiration and ATP synthesis in primary open-angle glaucoma patient lymphoblasts. Investigative Opthalmology & Visual Science. 2012;53(4):2431–2437. doi: 10.1167/iovs.12-9596. [DOI] [PubMed] [Google Scholar]
- 61.Van Bergen N. J., Crowston J. G., Craig J. E., Burdon K. P., Kearns L. S., Sharma S. Measurement of systemic mitochondrial function in advanced primary open-angle glaucoma and leber hereditary optic neuropathy. PLoS One. 2015;10 doi: 10.1371/journal.pone.0140919.e0140919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chevrollier A., Guillet V., Loiseau D., et al. Hereditary optic neuropathies share a common mitochondrial coupling defect. Annals of Neurology. 2008;63(6):794–798. doi: 10.1002/ana.21385. [DOI] [PubMed] [Google Scholar]
- 63.Schapira A. H. V. Complex I: inhibitors, inhibition and neurodegeneration. Experimental Neurology. 2010;224(2):331–335. doi: 10.1016/j.expneurol.2010.03.028. [DOI] [PubMed] [Google Scholar]
- 64.Chidlow G., Wood J. P. M., Casson R. J. Investigations into hypoxia and oxidative stress at the optic nerve head in a rat model of glaucoma. Frontiers in Neuroscience. 2017;11:p. 478. doi: 10.3389/fnins.2017.00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tezel G., Wax M. B. Hypoxia-inducible factor 1α in the glaucomatous retina and OpticNerve head. Archives of Ophthalmology. 2004;122(9):1348–1356. doi: 10.1001/archopht.122.9.1348. [DOI] [PubMed] [Google Scholar]
- 66.Guo D., Bi H., Wang D., Wu Q. Zinc oxide nanoparticles decrease the expression and activity of plasma membrane calcium ATPase, disrupt the intracellular calcium homeostasis in rat retinal ganglion cells. The International Journal of Biochemistry & Cell Biology. 2013;45(8):1849–1859. doi: 10.1016/j.biocel.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 67.Rohowetz L., Kraus J., Koulen P. Reactive oxygen species-mediated damage of retinal neurons: Drug development targets for therapies of chronic neurodegeneration of the retina. International Journal of Molecular Sciences. 2018;19(11):p. 3362. doi: 10.3390/ijms19113362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zaidi A., Michaelis M. L. Effects of reactive oxygen species on brain synaptic plasma membrane Ca2+-ATPase. Free Radical Biology and Medicine. 1999;27(7-8):810–821. doi: 10.1016/s0891-5849(99)00128-8. [DOI] [PubMed] [Google Scholar]
- 69.Yokota T., Kamimura N., Igarashi T., Takahashi H., Ohta S., Oharazawa H. Protective effect of molecular hydrogen against oxidative stress caused by peroxynitrite derived from nitric oxide in rat retina. Clinical & Experimental Ophthalmology. 2015;43(6):568–577. doi: 10.1111/ceo.12525. [DOI] [PubMed] [Google Scholar]
- 70.Alvarado J., Murphy C., Polansky J., Juster R. Age-related changes in trabecular meshwork cellularity. Investigative Ophthalmology & Visual Science. 1981;21(5):714–727. [PubMed] [Google Scholar]
- 71.Saccà S. C. Oxidative DNA damage in the human trabecular meshwork. Archives of Ophthalmology. 2005;123(4):p. 458. doi: 10.1001/archopht.123.4.458. [DOI] [PubMed] [Google Scholar]
- 72.Joe M. K., Tomarev S. I. Expression of myocilin mutants sensitizes cells to oxidative stress-induced apoptosis. The American Journal of Pathology. 2010;176(6):2880–2890. doi: 10.2353/ajpath.2010.090853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.He Y., Leung K. W., Zhuo Y. H., Ge J. Pro370Leu mutant myocilin impairs mitochondrial functions in human trabecular meshwork cells. Molecular Vision. 2009;15:815–825. [PMC free article] [PubMed] [Google Scholar]
- 74.Ferreira S. M., Lerner S. F., Brunzini R., Reides C. G., Evelson P. A., Llesuy S. F. Time course changes of oxidative stress markers in a rat experimental glaucoma model. Investigative Opthalmology & Visual Science. 2010;51(9):4635–4640. doi: 10.1167/iovs.09-5044. [DOI] [PubMed] [Google Scholar]
- 75.Ko M.-L., Peng P.-H., Ma M.-C., Ritch R., Chen C.-F. Dynamic changes in reactive oxygen species and antioxidant levels in retinas in experimental glaucoma. Free Radical Biology and Medicine. 2005;39(3):365–373. doi: 10.1016/j.freeradbiomed.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 76.Kong Y. X. G., van Bergen N., Bui B. V., Chrysostomou V., Vingrys A. J., Trounce I. A. Impact of aging and diet restriction on retinal function during and after acute intraocular pressure injury. Neurobiology of Aging. 2012;33:p. 1126. doi: 10.1016/j.neurobiolaging.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 77.Kawasaki A., Otori Y., Barnstable C. J. Müller cell protection of rat retinal ganglion cells from glutamate and nitric oxide neurotoxicity. Investigative Ophthalmology & Visual Science. 2000;41:3444–3450. [PubMed] [Google Scholar]
- 78.Skytt D. M., Klawonn A. M., Stridh M. H., et al. siRNA knock down of glutamate dehydrogenase in astrocytes affects glutamate metabolism leading to extensive accumulation of the neuroactive amino acids glutamate and aspartate. Neurochemistry International. 2012;61(4):490–497. doi: 10.1016/j.neuint.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 79.Farkas R. H., Chowers I., Hackam A. S., Kageyama M., Nickells R. W., Otteson D. C. Increased expression of iron-regulating genes in monkey and human glaucoma. Investigative Ophthalmology & Visual Science. 2004;45(5):1410–1417. doi: 10.1167/iovs.03-0872. [DOI] [PubMed] [Google Scholar]
- 80.Benoist d’Azy C., Pereira B., Chiambaretta F., Dutheil F. Oxidative and anti-oxidative stress markers in chronic glaucoma: a systematic review and meta-analysis. PLoS One. 2016;11 doi: 10.1371/journal.pone.0166915.e0166915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yuki K., Murat D., Kimura I., Tsubota K. Increased serum total antioxidant status and decreased urinary 8-hydroxy-2′-deoxyguanosine levels in patients with normal-tension glaucoma. Acta Ophthalmologica. 2010;88(7):e259–e264. doi: 10.1111/j.1755-3768.2010.01997.x. [DOI] [PubMed] [Google Scholar]
- 82.Ramdas W. D., Wolfs R. C. W., Kiefte-de Jong J. C., et al. Nutrient intake and risk of open-angle glaucoma: the Rotterdam study. European Journal of Epidemiology. 2012;27(5):385–393. doi: 10.1007/s10654-012-9672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ko M.-L., Peng P.-H., Hsu S.-Y., Chen C.-F. Dietary deficiency of vitamin E aggravates retinal ganglion cell death in experimental glaucoma of rats. Current Eye Research. 2010;35(9):842–849. doi: 10.3109/02713683.2010.489728. [DOI] [PubMed] [Google Scholar]
- 84.Ozdemir G., Tolun F. I., Gul M., Imrek S. Retinal oxidative stress induced by intraocular hypertension in rats may be ameliorated by brimonidine treatment and N-acetyl cysteine supplementation. Journal of Glaucoma. 2009;18(9):662–665. doi: 10.1097/ijg.0b013e31819c46b1. [DOI] [PubMed] [Google Scholar]
- 85.Lee K. Y. C., Nakayama M., Aihara M., Chen Y.-N., Araie M. Brimonidine is neuroprotective against glutamate-induced neurotoxicity, oxidative stress, and hypoxia in purified rat retinal ganglion cells. Molecular Vision. 2010;16:246–251. [PMC free article] [PubMed] [Google Scholar]
- 86.Yoles E., Wheeler L. A., Schwartz M. Alpha2-adrenoreceptor agonists are neuroprotective in a rat model of optic nerve degeneration. Investigative Ophthalmology & Visual Science. 1999;40(1):65–73. [PubMed] [Google Scholar]
- 87.Lv B., Chen T., Xu Z., Huo F., Wei Y., Yang X. Crocin protects retinal ganglion cells against H2O2-induced damage through the mitochondrial pathway and activation of NF-κB. International Journal of Molecular Medicine. 2016;37(1):225–232. doi: 10.3892/ijmm.2015.2418. [DOI] [PubMed] [Google Scholar]
- 88.Russo R., Cavaliere F., Rombolà L., et al. Rational basis for the development of coenzyme Q10 as a neurotherapeutic agent for retinal protection. Progress in Brain Research. 2008;173:575–582. doi: 10.1016/s0079-6123(08)01139-4. [DOI] [PubMed] [Google Scholar]
- 89.Ahn H. R., Lee H. J., Kim K.-A., et al. Hydroxycinnamic acids in Crepidiastrum denticulatum protect oxidative stress-induced retinal damage. Journal of Agricultural and Food Chemistry. 2014;62(6):1310–1323. doi: 10.1021/jf4046232. [DOI] [PubMed] [Google Scholar]
- 90.Piotrowska-Nowak A., Kosior-Jarecka E., Schab A., et al. Investigation of whole mitochondrial genome variation in normal tension glaucoma. Experimental Eye Research. 2019;178:186–197. doi: 10.1016/j.exer.2018.10.004. [DOI] [PubMed] [Google Scholar]
- 91.Wiggs J. L., Pasquale L. R. Genetics of glaucoma. Human Molecular Genetics. 2017;26(R1):R21–R27. doi: 10.1093/hmg/ddx184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bailey J. N. C., Loomis S. J., Loomis S. J., et al. Genome-wide association analysis identifies TXNRD2, ATXN2 and FOXC1 as susceptibility loci for primary open-angle glaucoma. Nature Genetics. 2016;48(2):189–194. doi: 10.1038/ng.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Caprioli J., Munemasa Y., Kwong J. M., Piri N. Overexpression of thioredoxins 1 and 2 increases retinal ganglion cell survival after pharmacologically induced oxidative stress, optic nerve transection, and in experimental glaucoma. Transactions of the American Ophthalmological Society. 2009;107:161–165. [PMC free article] [PubMed] [Google Scholar]
- 94.Rezaie T., Child A., Hitchings R., Brice G., Miller L., Coca-Prados M. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295(5557):1077–1079. doi: 10.1126/science.1066901. [DOI] [PubMed] [Google Scholar]
- 95.Stone E. M., Fingert J. H., Alward W. L., Nguyen T. D., Polansky J. R., Sunden S. L. Identification of a gene that causes primary open angle glaucoma. Science. 1997;275(5300):668–670. doi: 10.1126/science.275.5300.668. [DOI] [PubMed] [Google Scholar]
- 96.Sheffield V. C., Stone E. M., Alward W. L. M., et al. Genetic linkage of familial open angle glaucoma to chromosome 1q21-q31. Nature Genetics. 1993;4(1):47–50. doi: 10.1038/ng0593-47. [DOI] [PubMed] [Google Scholar]
- 97.Charliat G., Jolly D., Blanchard F. Genetic risk factor in primary open-angle glaucoma: a case-control study. Ophthalmic Epidemiology. 1994;1(3):131–138. doi: 10.3109/09286589409047221. [DOI] [PubMed] [Google Scholar]
- 98.Fingert J. H., Robin A. L., Stone J. L., et al. Copy number variations on chromosome 12q14 in patients with normal tension glaucoma. Human Molecular Genetics. 2011;20(12):2482–2494. doi: 10.1093/hmg/ddr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sears N. C., Boese E. A., Miller M. A., Fingert J. H. Mendelian genes in primary open angle glaucoma. Experimental Eye Research. 2019;186 doi: 10.1016/j.exer.2019.107702.107702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Collins D. W., Gudiseva H. V., Trachtman B. T., Jerrehian M., Gorry T., Merritt W. T., III Mitochondrial sequence variation in African-American primary open-angle glaucoma patients. PLoS One. 2013;8 doi: 10.1371/journal.pone.0076627.e76627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jeoung J. W., Seong M.-W., Park S. S., Kim D. M., Kim S. H., Park K. H. Mitochondrial DNA variant discovery in normal-tension glaucoma patients by next-generation sequencing. Investigative Opthalmology & Visual Science. 2014;55(2):986–992. doi: 10.1167/iovs.13-12968. [DOI] [PubMed] [Google Scholar]
- 102.Sundaresan P., Simpson D. A., Sambare C., et al. Whole-mitochondrial genome sequencing in primary open-angle glaucoma using massively parallel sequencing identifies novel and known pathogenic variants. Genetics in Medicine. 2015;17(4):279–284. doi: 10.1038/gim.2014.121. [DOI] [PubMed] [Google Scholar]
- 103.Larsen N. B., Rasmussen M., Rasmussen L. J. Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion. 2005;5(2):89–108. doi: 10.1016/j.mito.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 104.Lascaratos G., Garway-Heath D. F., Willoughby C. E., Chau K.-Y., Schapira A. H. V. Mitochondrial dysfunction in glaucoma: understanding genetic influences. Mitochondrion. 2012;12(2):202–212. doi: 10.1016/j.mito.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 105.Khawaja A. P., Cooke Bailey J. N., Kang J. H., et al. Assessing the association of mitochondrial genetic variation with primary open-angle glaucoma using gene-set analyses. Investigative Opthalmology & Visual Science. 2016;57(11):5046–5052. doi: 10.1167/iovs.16-20017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Beal M. F., Flint Beal M. Mitochondria take center stage in aging and neurodegeneration. Annals of Neurology. 2005;58(4):495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- 107.Tsilis A. G., Tsilidis K. K., Pelidou S. H., Kitsos G. Systematic review of the association between Alzheimer’s disease and chronic glaucoma. Clinical Ophthalmology. 2014;8:2095–2104. doi: 10.2147/OPTH.S69534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Larsson N.-G. Somatic mitochondrial DNA mutations in mammalian aging. Annual Review of Biochemistry. 2010;79(1):683–706. doi: 10.1146/annurev-biochem-060408-093701. [DOI] [PubMed] [Google Scholar]
- 109.Bender A., Krishnan K. J., Morris C. M., et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nature Genetics. 2006;38(5):515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 110.Mathew A., Lindsley T. A., Sheridan A., et al. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. Journal of Alzheimer’s Disease. 2012;30(3):617–627. doi: 10.3233/jad-2012-120145. [DOI] [PubMed] [Google Scholar]
- 111.Abramova N. E., Davies K. J. A., Crawford D. R. Polynucleotide degradation during early stage response to oxidative stress is specific to mitochondria. Free Radical Biology and Medicine. 2000;28(2):281–288. doi: 10.1016/s0891-5849(99)00239-7. [DOI] [PubMed] [Google Scholar]
- 112.Matheoud D., Sugiura A., Bellemare-Pelletier A., Laplante A., Rondeau C., Chemali M. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell. 2016;166(2):314–327. doi: 10.1016/j.cell.2016.05.039. [DOI] [PubMed] [Google Scholar]
- 113.Wu W., Xu H., Wang Z., Mao Y., Yuan L., Luo W. PINK1-Parkin-Mediated mitophagy protects mitochondrial integrity and prevents metabolic stress-induced endothelial injury. PLoS One. 2015;10 doi: 10.1371/journal.pone.0132499.e0132499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cowan K., Anichtchik O., Luo S. Mitochondrial integrity in neurodegeneration. CNS Neuroscience & Therapeutics. 2019;25(7):825–836. doi: 10.1111/cns.13105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vives-Bauza C., Zhou C., Huang Y., et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proceedings of the National Academy of Sciences. 2010;107(1):378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang Y., Nartiss Y., Steipe B., McQuibban G. A., Kim P. K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy. 2012;8(10):1462–1476. doi: 10.4161/auto.21211. [DOI] [PubMed] [Google Scholar]
- 117.Chernyshova K., Inoue K., Yamashita S.-I., Fukuchi T., Kanki T. Glaucoma-associated mutations in the optineurin gene have limited impact on parkin-dependent mitophagy. Investigative Opthalmology & Visual Science. 2019;60(10):3625–3635. doi: 10.1167/iovs.19-27184. [DOI] [PubMed] [Google Scholar]
- 118.Shim M. S., Takihara Y., Kim K.-Y., Iwata T., Yue B. Y. J. T., Inatani M. Mitochondrial pathogenic mechanism and degradation in optineurin E50K mutation-mediated retinal ganglion cell degeneration. Scientific Reports. 2016;6:p. 33830. doi: 10.1038/srep33830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Morton S., Hesson L., Peggie M., Cohen P. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Letters. 2008;582(6):997–1002. doi: 10.1016/j.febslet.2008.02.047. [DOI] [PubMed] [Google Scholar]
- 120.Sirohi K., Kumari A., Radha V., Swarup G. A glaucoma-associated variant of optineurin, M98K, activates Tbk1 to enhance autophagosome formation and retinal cell death dependent on Ser177 phosphorylation of optineurin. PLoS One. 2015;10 doi: 10.1371/journal.pone.0138289.e0138289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Minegishi Y., Iejima D., Kobayashi H., et al. Enhanced optineurin E50K-TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Human Molecular Genetics. 2013;22(17):3559–3567. doi: 10.1093/hmg/ddt210. [DOI] [PubMed] [Google Scholar]
- 122.Fingert J. H., Miller K., Hedberg-Buenz A., et al. Transgenic TBK1 mice have features of normal tension glaucoma. Human Molecular Genetics. 2017;26(1):124–132. doi: 10.1093/hmg/ddw372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Matsumoto G., Shimogori T., Hattori N., Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Human Molecular Genetics. 2015;24(15):4429–4442. doi: 10.1093/hmg/ddv179. [DOI] [PubMed] [Google Scholar]
- 124.Li Y., Kang J., Horwitz M. S. Interaction of an adenovirus E3 14.7-kilodalton protein with a novel tumor necrosis factor alpha-inducible cellular protein containing leucine zipper domains. Molecular and Cellular Biology. 1998;18(3):1601–1610. doi: 10.1128/mcb.18.3.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Toth R. P., Atkin J. D. Dysfunction of optineurin in amyotrophic lateral sclerosis and glaucoma. Frontiers in Immunology. 2018;9:p. 1017. doi: 10.3389/fimmu.2018.01017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Swarup G., Sayyad Z. Altered functions and interactions of glaucoma-associated mutants of optineurin. Frontiers in Immunology. 2018;9:p. 1287. doi: 10.3389/fimmu.2018.01287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhu G., Wu C.-J., Zhao Y., Ashwell J. D. Optineurin negatively regulates TNFα- induced NF-κB activation by competing with NEMO for ubiquitinated RIP. Current Biology. 2007;17(16):1438–1443. doi: 10.1016/j.cub.2007.07.041. [DOI] [PubMed] [Google Scholar]
- 128.Tanishima M., Takashima S., Honda A., et al. Identification of optineurin as an interleukin-1 receptor-associated kinase 1-binding protein and its role in regulation of MyD88-dependent signaling. Journal of Biological Chemistry. 2017;292(42):17250–17257. doi: 10.1074/jbc.m117.813899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nagabhushana A., Bansal M., Swarup G. Optineurin is required for CYLD-dependent inhibition of TNFα-induced NF-κB activation. PLoS One. 2011;6 doi: 10.1371/journal.pone.0017477.e17477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sudhakar C., Nagabhushana A., Jain N., Swarup G. NF-κB mediates tumor necrosis factor α-induced expression of optineurin, a negative regulator of NF-κB. PLoS One. 2009;4(4):p. e5114. doi: 10.1371/journal.pone.0005114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Louis C., Burns C., Wicks I. TANK-binding kinase 1-dependent responses in health and autoimmunity. Frontiers in Immunology. 2018;9:p. 434. doi: 10.3389/fimmu.2018.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pomerantz J. L., Baltimore D. NF-kappa B activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. The EMBO Journal. 1999;18(23):6694–6704. doi: 10.1093/emboj/18.23.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liddelow S. A., Guttenplan K. A., Clarke L. E., et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vecino E., Rodriguez F. D., Ruzafa N., Pereiro X., Sharma S. C. Glia-neuron interactions in the mammalian retina. Progress in Retinal and Eye Research. 2016;51:1–40. doi: 10.1016/j.preteyeres.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 135.Iadecola C., Nedergaard M. Glial regulation of the cerebral microvasculature. Nature Neuroscience. 2007;10(11):1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- 136.Ullian E. M., Sapperstein S. K., Christopherson K. S., Barres B. A. Control of synapse number by glia. Science. 2001;291(5504):657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- 137.Vohra R., Tsai J. C., Kolko M. The role of inflammation in the pathogenesis of glaucoma. Survey of Ophthalmology. 2013;58(4):311–320. doi: 10.1016/j.survophthal.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 138.Soto I., Howell G. R. The complex role of neuroinflammation in glaucoma. Cold Spring Harbor Perspectives in Medicine. 2014;4(8) doi: 10.1101/cshperspect.a017269.a017269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang L., Cioffi G. A., Cull G., Dong J., Fortune B. Immunohistologic evidence for retinal glial cell changes in human glaucoma. Investigative Ophthalmology & Visual Science. 2002;43(4):1088–1094. [PubMed] [Google Scholar]
- 140.Inman D. M., Horner P. J. Reactive nonproliferative gliosis predominates in a chronic mouse model of glaucoma. Glia. 2007;55(9):942–953. doi: 10.1002/glia.20516. [DOI] [PubMed] [Google Scholar]
- 141.Tezel G. L. N., Chauhan B. C., LeBlanc R. P., Wax M. B. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Investigative Opthalmology & Visual Science. 2003;44(7):3025–3033. doi: 10.1167/iovs.02-1136. [DOI] [PubMed] [Google Scholar]
- 142.Fard M. A., Moghimi S., Sahraian A., Ritch R. Optic nerve head cupping in glaucomatous and non-glaucomatous optic neuropathy. British Journal of Ophthalmology. 2019;103(3):374–378. doi: 10.1136/bjophthalmol-2018-312161. [DOI] [PubMed] [Google Scholar]
- 143.Dai C., Khaw P. T., Yin Z. Q., Li D., Raisman G., Li Y. Structural basis of glaucoma: the fortified astrocytes of the optic nerve head are the target of raised intraocular pressure. Glia. 2012;60(1):13–28. doi: 10.1002/glia.21242. [DOI] [PubMed] [Google Scholar]
- 144.Sun D., Qu J., Jakobs T. C. Reversible reactivity by optic nerve astrocytes. Glia. 2013;61(8):1218–1235. doi: 10.1002/glia.22507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bariş M., Tezel G. Immunomodulation as a neuroprotective strategy for glaucoma treatment. Current Ophthalmology Reports. 2019;7(2):160–169. [PMC free article] [PubMed] [Google Scholar]
- 146.Nikolskaya T., Nikolsky Y., Serebryiskaya T., Zvereva S., Sviridov E., Dezso Z. Network analysis of human glaucomatous optic nerve head astrocytes. BMC Medical Genomics. 2009;2:p. 24. doi: 10.1186/1755-8794-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kompass K. S., Agapova O. A., Li W., Kaufman P. L., Rasmussen C. A., Hernandez M. R. Bioinformatic and statistical analysis of the optic nerve head in a primate model of ocular hypertension. BMC Neuroscience. 2008;9:p. 93. doi: 10.1186/1471-2202-9-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ginhoux F., Greter M., Leboeuf M., et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Neufeld A. H. Microglia in the optic nerve head and the region of parapapillary chorioretinal atrophy in glaucoma. Archives of Ophthalmology. 1999;117(8):1050–1056. doi: 10.1001/archopht.117.8.1050. [DOI] [PubMed] [Google Scholar]
- 150.Yuan L., Neufeld A. H. Activated microglia in the human glaucomatous optic nerve head. Journal of Neuroscience Research. 2001;64(5):523–532. doi: 10.1002/jnr.1104. [DOI] [PubMed] [Google Scholar]
- 151.Tezel G., Wax M. B. Increased production of tumor necrosis factor-α by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. The Journal of Neuroscience. 2000;20(23):8693–8700. doi: 10.1523/jneurosci.20-23-08693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bosco A., Romero C. O., Breen K. T., et al. Neurodegeneration severity can be predicted from early microglia alterations monitored in vivo in a mouse model of chronic glaucoma. Disease Models & Mechanisms. 2015;8(5):443–455. doi: 10.1242/dmm.018788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bosco A., Inman D. M., Steele M. R., et al. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Investigative Opthalmology & Visual Science. 2008;49(4):1437–1446. doi: 10.1167/iovs.07-1337. [DOI] [PubMed] [Google Scholar]
- 154.Jha M. K., Seo M., Kim J.-H., Kim B.-G., Cho J.-Y., Suk K. The secretome signature of reactive glial cells and its pathological implications. Biochimica et Biophysica Acta (BBA)—Proteins and Proteomics. 2013;1834(11):2418–2428. doi: 10.1016/j.bbapap.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 155.Neufeld A. H., Hernandez M. R., Gonzalez M. Nitric oxide synthase in the human glaucomatous optic nerve head. Archives of Ophthalmology. 1997;115(4):497–503. doi: 10.1001/archopht.1997.01100150499009. [DOI] [PubMed] [Google Scholar]
- 156.Cho K. J., Kim J. H., Park H.-Y. L., Park C. K. Glial cell response and iNOS expression in the optic nerve head and retina of the rat following acute high IOP ischemia-reperfusion. Brain Research. 2011;1403:67–77. doi: 10.1016/j.brainres.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 157.Vidal L., Díaz F., Villena A., Moreno M., Campos J. G., Vargas I. P. d. Nitric oxide synthase in retina and optic nerve head of rat with increased intraocular pressure and effect of timolol. Brain Research Bulletin. 2006;70(4–6):406–413. doi: 10.1016/j.brainresbull.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 158.Shareef S., Sawada A., Neufeld A. H. Isoforms of nitric oxide synthase in the optic nerves of rat eyes with chronic moderately elevated intraocular pressure. Investigative Ophthalmology & Visual Science. 1999;40(12):2884–2891. [PubMed] [Google Scholar]
- 159.Neufeld A. H., Sawada A., Becker B. Inhibition of nitric-oxide synthase 2 by aminoguanidine provides neuroprotection of retinal ganglion cells in a rat model of chronic glaucoma. Proceedings of the National Academy of Sciences. 1999;96(17):9944–9948. doi: 10.1073/pnas.96.17.9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yang X., Luo C., Cai J., et al. Neurodegenerative and inflammatory pathway components linked to TNF-α/TNFR1 signaling in the glaucomatous human retina. Investigative Opthalmology & Visual Science. 2011;52(11):8442–8454. doi: 10.1167/iovs.11-8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tezel G. L. N., Yang X., Luo C., Peng Y., Sun S. L., Sun D. Mechanisms of immune system activation in glaucoma: oxidative stress-stimulated antigen presentation by the retina and optic nerve head glia. Investigative Opthalmology & Visual Science. 2007;48(2):705–714. doi: 10.1167/iovs.06-0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tezel G., Thornton I. L., Tong M. G., et al. Immunoproteomic analysis of potential serum biomarker candidates in human glaucoma. Investigative Opthalmology & Visual Science. 2012;53(13):p. 8222. doi: 10.1167/iovs.12-10076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tezel G., Hernandez R., Wax M. B. Immunostaining of heat shock proteins in the retina and optic nerve head of normal and glaucomatous eyes. Archives of Ophthalmology. 2000;118(4):511–518. doi: 10.1001/archopht.118.4.511. [DOI] [PubMed] [Google Scholar]
- 164.Steele M. R., Inman D. M., Calkins D. J., Horner P. J., Vetter M. L. Microarray analysis of retinal gene expression in the DBA/2J model of glaucoma. Investigative Opthalmology & Visual Science. 2006;47(3):977–985. doi: 10.1167/iovs.05-0865. [DOI] [PubMed] [Google Scholar]
- 165.Panagis L., Zhao X., Ge Y., Ren L., Mittag T. W., Danias J. Gene expression changes in areas of focal loss of retinal ganglion cells in the retina of DBA/2J mice. Investigative Opthalmology & Visual Science. 2010;51(4):2024–2034. doi: 10.1167/iovs.09-3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Johnson E. C., Jia L., Cepurna W. O., Doser T. A., Morrison J. C. Global changes in optic nerve head gene expression after exposure to elevated intraocular pressure in a rat glaucoma model. Investigative Opthalmology & Visual Science. 2007;48(7):p. 3161. doi: 10.1167/iovs.06-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yang J., Yang P., Tezel G., Patil R. V., Hernandez M. R., Wax M. B. Induction of HLA-DR expression in human lamina cribrosa astrocytes by cytokines and simulated ischemia. Investigative Ophthalmology & Visual Science. 2001;42(2):365–371. [PubMed] [Google Scholar]
- 168.Chidlow G., Ebneter A., Wood J. P. M., Casson R. J. Evidence supporting an association between expression of major histocompatibility complex II by microglia and optic nerve degeneration during experimental glaucoma. Journal of Glaucoma. 2016;25(8):681–691. doi: 10.1097/ijg.0000000000000447. [DOI] [PubMed] [Google Scholar]
- 169.Duarte J. N., Cragnolini J. J., Swee L. K., et al. Generation of immunity against pathogens via single-domain antibody-antigen constructs. The Journal of Immunology. 2016;197(12):4838–4847. doi: 10.4049/jimmunol.1600692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.West A. P., Shadel G. S., Ghosh S., Ghosh S. Mitochondria in innate immune responses. Nature Reviews Immunology. 2011;11(6):389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kawasaki T., Kawai T. Toll-like receptor signaling pathways. Frontiers in Immunology. 2014;5:p. 461. doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Takeuchi O., Akira S. Innate immunity to virus infection. Immunological Reviews. 2009;227(1):75–86. doi: 10.1111/j.1600-065x.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tang D., Kang R., Coyne C. B., Zeh H. J., Lotze M. T. PAMPs and DAMPs: signal 0 s that spur autophagy and immunity. Immunological Reviews. 2012;249(1):158–175. doi: 10.1111/j.1600-065x.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.West A. P. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology. 2017;391:54–63. doi: 10.1016/j.tox.2017.07.016. [DOI] [PubMed] [Google Scholar]
- 175.Nakahira K., Hisata S., Choi A. M. K. The roles of mitochondrial damage-associated molecular patterns in diseases. Antioxidants & Redox Signaling. 2015;23(17):1329–1350. doi: 10.1089/ars.2015.6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tezel G., Yang J., Wax M. B. Heat shock proteins, immunity and glaucoma. Brain Research Bulletin. 2004;62(6):473–480. doi: 10.1016/s0361-9230(03)00074-1. [DOI] [PubMed] [Google Scholar]
- 177.Chen H., Cho K.-S., Vu T. H. K., Shen C.-H., Kaur M., Chen G. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nature Communications. 2018;9:p. 3209. doi: 10.1038/s41467-018-06428-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Johnson E. C., Morrison J. C. Friend or foe? Resolving the impact of glial responses in glaucoma. Journal of Glaucoma. 2009;18(5):341–353. doi: 10.1097/ijg.0b013e31818c6ef6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Cooper M. L., Crish S. D., Inman D. M., Horner P. J., Calkins D. J. Early astrocyte redistribution in the optic nerve precedes axonopathy in the DBA/2J mouse model of glaucoma. Experimental Eye Research. 2016;150:22–33. doi: 10.1016/j.exer.2015.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Son J. L., Soto I., Oglesby E., et al. Glaucomatous optic nerve injury involves early astrocyte reactivity and late oligodendrocyte loss. Glia. 2010;58(7):780–789. doi: 10.1002/glia.20962. [DOI] [PubMed] [Google Scholar]
- 181.Wang R., Seifert P., Jakobs T. C. Astrocytes in the optic nerve head of glaucomatous mice display a characteristic reactive phenotype. Investigative Opthalmology & Visual Science. 2017;58(2):924–932. doi: 10.1167/iovs.16-20571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Inman D. M., Harun-Or-Rashid M. Metabolic vulnerability in the neurodegenerative disease glaucoma. Frontiers in Neuroscience. 2017;11:p. 146. doi: 10.3389/fnins.2017.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Howell G. R., Macalinao D. G., Sousa G. L., et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. Journal of Clinical Investigation. 2011;121(4):1429–1444. doi: 10.1172/jci44646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Glass C. K., Saijo K., Winner B., Marchetto M. C., Gage F. H. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Heid M. E., Keyel P. A., Kamga C., Shiva S., Watkins S. C., Salter R. D. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. The Journal of Immunology. 2013;191(10):5230–5238. doi: 10.4049/jimmunol.1301490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhou R., Yazdi A. S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 187.Gurung P., Lukens J. R., Kanneganti T.-D. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends in Molecular Medicine. 2015;21(3):193–201. doi: 10.1016/j.molmed.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Liu Q., Zhang D., Hu D., Zhou X., Zhou Y. The role of mitochondria in NLRP3 inflammasome activation. Molecular Immunology. 2018;103:115–124. doi: 10.1016/j.molimm.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 189.Dela Cruz C. S., Kang M.-J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion. 2018;41:37–44. doi: 10.1016/j.mito.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yu J., Nagasu H., Murakami T., et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proceedings of the National Academy of Sciences. 2014;111(43):15514–15519. doi: 10.1073/pnas.1414859111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chi W., Li F., Chen H., et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1 production in acute glaucoma. Proceedings of the National Academy of Sciences. 2014;111(30):11181–11186. doi: 10.1073/pnas.1402819111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Puyang Z., Feng L., Chen H., Liang P., Troy J. B., Liu X. Retinal ganglion cell loss is delayed following optic nerve crush in NLRP3 knockout mice. Scientific Reports. 2016;6:p. 20998. doi: 10.1038/srep20998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yang X., Hondur G., Tezel G. Antioxidant treatment limits neuroinflammation in experimental glaucoma. Investigative Opthalmology & Visual Science. 2016;57(4):p. 2344. doi: 10.1167/iovs.16-19153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yan X., Tezel G., Wax M. B., Edward D. P. Matrix metalloproteinases and tumor necrosis factor α in glaucomatous optic nerve head. Archives of Ophthalmology. 2000;118(5):666–673. doi: 10.1001/archopht.118.5.666. [DOI] [PubMed] [Google Scholar]
- 195.Tezel G., Li L. Y., Patil R. V., Wax M. B. TNF-alpha and TNF-alpha receptor-1 in the retina of normal and glaucomatous eyes. Investigative Ophthalmology & Visual Science. 2001;42(8):1787–1794. [PubMed] [Google Scholar]
- 196.Bonifati D., Kishore U. Role of complement in neurodegeneration and neuroinflammation. Molecular Immunology. 2007;44(5):999–1010. doi: 10.1016/j.molimm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 197.Ricklin D., Hajishengallis G., Yang K., Lambris J. D. Complement: a key system for immune surveillance and homeostasis. Nature Immunology. 2010;11(9):785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Stevens B., Allen N. J., Vazquez L. E., et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 199.Storrs S. B., Kolb W. P., Olson M. S. C1q binding and C1 activation by various isolated cellular membranes. Journal of Immunology. 1983;131(1):416–422. [PubMed] [Google Scholar]
- 200.Kohl J. The role of complement in danger sensing and transmission. Immunologic Research. 2006;34(2):157–176. doi: 10.1385/ir:34:2:157. [DOI] [PubMed] [Google Scholar]
- 201.Reis E. S., Mastellos D. C., Hajishengallis G., Lambris J. D. New insights into the immune functions of complement. Nature Reviews Immunology. 2019;19(8):503–516. doi: 10.1038/s41577-019-0168-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kuehn M. H., Kim C. Y., Ostojic J., et al. Retinal synthesis and deposition of complement components induced by ocular hypertension. Experimental Eye Research. 2006;83(3):620–628. doi: 10.1016/j.exer.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 203.Stasi K., Nagel D., Yang X., et al. Complement component 1Q (C1Q) upregulation in retina of murine, primate, and human glaucomatous eyes. Investigative Opthalmology & Visual Science. 2006;47(3):1024–1029. doi: 10.1167/iovs.05-0830. [DOI] [PubMed] [Google Scholar]
- 204.Tezel G., Yang X., Luo C., et al. Oxidative stress and the regulation of complement activation in human glaucoma. Investigative Opthalmology & Visual Science. 2010;51(10):5071–5082. doi: 10.1167/iovs.10-5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Johnson E. C., Doser T. A., Cepurna W. O., et al. Cell proliferation and interleukin-6-type cytokine signaling are implicated by gene expression responses in early optic nerve head injury in rat glaucoma. Investigative Opthalmology & Visual Science. 2011;52(1):504–518. doi: 10.1167/iovs.10-5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Miyahara T., Kikuchi T., Akimoto M., Kurokawa T., Shibuki H., Yoshimura N. Gene microarray analysis of experimental glaucomatous retina from cynomologous monkey. Investigative Opthalmology & Visual Science. 2003;44(10):4347–4356. doi: 10.1167/iovs.02-1032. [DOI] [PubMed] [Google Scholar]
- 207.Bosco A., Anderson S. R., Breen K. T., et al. Complement C3-targeted gene therapy restricts onset and progression of neurodegeneration in chronic mouse glaucoma. Molecular Therapy. 2018;26(10):2379–2396. doi: 10.1016/j.ymthe.2018.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Jha P., Banda H., Tytarenko R., Bora P. S., Bora N. S. Complement mediated apoptosis leads to the loss of retinal ganglion cells in animal model of glaucoma. Molecular Immunology. 2011;48(15-16):2151–2158. doi: 10.1016/j.molimm.2011.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Howell G. R., Soto I., Ryan M., Graham L. C., Smith R. S., John S. W. M. Deficiency of complement component 5 ameliorates glaucoma in DBA/2J mice. Journal of Neuroinflammation. 2013;10:p. 76. doi: 10.1186/1742-2094-10-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Reinehr S., Gomes S. C., Gassel C. J., et al. Intravitreal therapy against the complement factor C5 prevents retinal degeneration in an experimental autoimmune glaucoma model. Frontiers in Pharmacology. 2019;10 doi: 10.3389/fphar.2019.01381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gasque P., Dean Y. D., McGreal E. P., VanBeek J., Morgan B. P. Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology. 2000;49(1-2):171–186. doi: 10.1016/s0162-3109(00)80302-1. [DOI] [PubMed] [Google Scholar]
- 212.Piccio L., Rossi B., Scarpini E., et al. Molecular mechanisms involved in lymphocyte recruitment in inflamed brain microvessels: critical roles for P-selectin glycoprotein ligand-1 and heterotrimeric gi-linked receptors. The Journal of Immunology. 2002;168(4):1940–1949. doi: 10.4049/jimmunol.168.4.1940. [DOI] [PubMed] [Google Scholar]
- 213.Charabati M., Rabanel J.-M., Ramassamy C., Prat A. Overcoming the brain barriers: from immune cells to nanoparticles. Trends in Pharmacological Sciences. 2020;41(1):42–54. doi: 10.1016/j.tips.2019.11.001. [DOI] [PubMed] [Google Scholar]
- 214.Flammer J., Mozaffarieh M. What is the present pathogenetic concept of glaucomatous optic neuropathy? Survey of Ophthalmology. 2007;52(Suppl 2):S162–S173. doi: 10.1016/j.survophthal.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 215.Uhler T. A., Piltz-Seymour J. Optic disc hemorrhages in glaucoma and ocular hypertension: implications and recommendations. Current Opinion in Ophthalmology. 2008;19(2):89–94. doi: 10.1097/icu.0b013e3282f3e6bc. [DOI] [PubMed] [Google Scholar]
- 216.Tezel G., Siegmund K. D., Trinkaus K., Wax M. B., Kass M. A., Kolker A. E. Clinical factors associated with progression of glaucomatous optic disc damage in treated patients. Archives of Ophthalmology. 2001;119(6):813–818. doi: 10.1001/archopht.119.6.813. [DOI] [PubMed] [Google Scholar]
- 217.Lorenz K., Beck S., Keilani M. M., Wasielica-Poslednik J., Pfeiffer N., Grus F. H. Longitudinal analysis of serum autoantibody-reactivities in patients with primary open angle glaucoma and optic disc hemorrhage. PLoS One. 2016;11 doi: 10.1371/journal.pone.0166813.e0166813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Guo C., Wu N., Niu X., Wu Y., Chen D., Guo W. Comparison of T Helper cell patterns in primary open-angle glaucoma and normal-pressure glaucoma. Medical Science Monitor. 2018;24:1988–1996. doi: 10.12659/msm.904923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Yang J., Patil R. V., Yu H., Gordon M., Wax M. B. T cell subsets and sIL-2R/IL-2 levels in patients with glaucoma. American Journal of Ophthalmology. 2001;131(4):421–426. doi: 10.1016/s0002-9394(00)00862-x. [DOI] [PubMed] [Google Scholar]
- 220.Wax M. B., Tezel G., Yang J., et al. Induced autoimmunity to heat shock proteins elicits glaucomatous loss of retinal ganglion cell neurons via activated T-cell-derived fas-ligand. Journal of Neuroscience. 2008;28(46):12085–12096. doi: 10.1523/jneurosci.3200-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Joachim S. C., Gramlich O. W., Laspas P., Schmid H., Beck S., von Pein H. D. Retinal ganglion cell loss is accompanied by antibody depositions and increased levels of microglia after immunization with retinal antigens. PLoS One. 2012;7 doi: 10.1371/journal.pone.0040616.e40616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Gramlich O. W., Beck S., von Thun und Hohenstein-Blaul N., et al. Enhanced insight into the autoimmune component of glaucoma: IgG autoantibody accumulation and pro-inflammatory conditions in human glaucomatous retina. PLoS One. 2013;8(2) doi: 10.1371/journal.pone.0057557.e57557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Gramlich O. W., Ding Q. J., Zhu W., Cook A., Anderson M. G., Kuehn M. H. Adoptive transfer of immune cells from glaucomatous mice provokes retinal ganglion cell loss in recipients. Acta Neuropathologica Communications. 2015;3:p. 56. doi: 10.1186/s40478-015-0234-y. [DOI] [PMC free article] [PubMed] [Google Scholar]