

Abstract
Stalled mRNA translation results in the production of incompletely synthesized proteins that are targeted for degradation by ribosome-associated quality control (RQC). Here we investigated the fate of defective proteins translated from stall-inducing, nonstop mRNA that escape ubiquitylation by the RQC protein LTN1. We found that nonstop protein products accumulated in nucleoli and this localization was driven by polylysine tracts produced by translation of the poly(A) tails of nonstop mRNA. Nucleolar sequestration increased the solubility of invading proteins but disrupted nucleoli, altering their dynamics, morphology, and resistance to stress in cell culture and intact flies. Our work elucidates how stalled translation may affect distal cellular processes and may inform studies on the pathology of diseases caused by failures in RQC and characterized by nucleolar stress.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12192-021-01200-w.
Keywords: Nonstop mRNA, Nucleolus, Ribosome-associated quality control (RQC), LTN1, Phase separation, Protein quality control
Introduction
Defects in mRNA can cause ribosomes to stall and abort translation (Frischmeyer et al. 2002a; Doma and Parker 2006; Joazeiro 2017). The most common mRNA defect is premature polyadenylation, in which the polyadenylation machinery utilizes cryptic signals upstream of the stop codon, placing a poly(A) tail within the open reading frame and truncating the mRNA (Frischmeyer et al. 2002a; van Hoof et al. 2002; Tian et al. 2007; Klauer and van Hoof 2012). Premature adenylation upstream of the stop codon occurs in ~1% of all mRNAs (Graber et al. 1999; Frischmeyer et al. 2002a; Klauer and van Hoof 2012; Zhou et al. 2018). Translation of these “nonstop” mRNAs proceeds into the poly(A) tail, where ribosomes translate several lysine-encoding AAA codons before stalling (Frischmeyer et al. 2002b; van Hoof et al. 2002; Ito-Harashima et al. 2007b; Bengtson and Joazeiro 2010a; Brandman et al. 2012a; Verma et al. 2013; Defenouillère et al. 2013a; Juszkiewicz and Hegde 2017; Sundaramoorthy et al. 2017). Premature polyadenylation therefore produces truncated nascent polypeptide chains appended with polylysine. To protect cells from these defective proteins, a process called ribosome-associated quality control (RQC) detects stalled translation events and ubiquitylates stalled nascent chains via the E3 ubiquitin ligase Listerin (LTN1), targeting them for destruction by the proteasome (Ito-Harashima et al. 2007a; Bengtson and Joazeiro 2010b; Brandman et al. 2012b; Defenouillère et al. 2013b; Shao et al. 2013) (Fig. 1a).
Fig. 1.

PolyK tracts drive the protein products of nonstop mRNA into nucleoli. a Schematic of RQC pathway acting on GFP nonstop substrate. Translation proceeds through the UTR into the poly(A) tail, creating a polyK tract. Ribosome stalling then leads to dissociation of the 60S subunit, ubiquitination of the nascent chain by LTN1, and subsequent degradation. b Localization of GFP-nonstop and GFP-stop. HeLa cells were transfected with GFP reporter constructs, then fixed and immunostained with nucleolin (NCL). White dashed lines mark transfected cell outlines. Scale bar = 10 μm. c Effect of C-terminal cleavage on nucleolar localization. HeLa cells were transfected with GFP-TEV-containing constructs (shown in diagram) with or without a plasmid encoding the Tobacco Etch Virus (TEV) protease. Scale bar = 10 μm. d Localization of GFP reporters in HeLa cells immunostained with NCL. Scale bar = 10 μm
Events that trigger excessive ribosome stalling or compromise RQC have deleterious consequences. On the cellular level, loss of LTN1 function results in proteotoxic stress (Yonashiro et al. 2016; Choe et al. 2016; Defenouillère et al. 2016). On an organismal level, LTN1 hypomorphic mice develop a neurodegenerative disease characterized by progressive impairment of motor and neuronal function (Chu et al. 2009). Similarly, mutation of a tRNA along with loss of GTP Binding Protein 2 (GTPBP2), a protein important for resolving stalled ribosomes, causes neurodegeneration (Ishimura et al. 2014). Finally, in both mice and humans, mutations to the RQC gene nuclear export mediator factor (NEMF) are associated with neuromuscular disease (Martin et al. 2020). How the diverse complement of defective proteins that accumulate in these conditions affects and is tolerated by cells is poorly understood.
To understand the effects of defective proteins produced by stalled translation on cells, we studied the fate of escaped RQC substrates arising from the translation of nonstop mRNA. We found that protein produced from nonstop mRNA accumulated in nucleoli, driven by polybasic tracts from translated poly(A) tails, which served as nucleolar targeting sequences. Nucleolar localization increased the solubility of these defective proteins, potentially providing a protective effect to cells, yet also altered nucleoli, with loss of LTN1 leading to disruption of nucleolar integrity in cell culture and in intact flies. Our study demonstrates how failures in co-translational protein quality control can disrupt distal cellular processes and may help define the consequences of defective RQC.
Materials and methods
Plasmid construction
The GFP nonstop and stop reporters were constructed by Gibson cloning (Gibson Assembly Master Mix, NEB) eGFP with or without a terminal stop codon into the pCMV AAV backbone which was used as an expression vector and used to make AAV. Stbl3 E. coli (Invitrogen) was used for all pCMV AAV vector cloning. The in-frame stop codon in the BGH 3′ UTR was removed to allow for readthrough into the poly(A) tail. The GFP polyK (AAA), polyK (AAG), and GFP-TEV constructs were constructed by inserting the new sequence with C-terminal stop codons onto the reverse primer, amplifying the GFP nonstop and reinserting into the pCMV AAV backbone with the stop-removed 3′ UTR. Note that if not specified in the text, GFP-polyK is encoded by “AAA” codons. GFP UTR was constructed by amplifying the sequence from GFP-nonstop and using Gateway cloning to insert the sequence with a stop codon before the polyadenylation signal sequence into the pCMV AAV backbone with the no-stop 3′ UTR. The GFP-UTR-polyK construct was made by adding the new sequence to a reverse primer, amplifying from the GFP UTR construct, and inserting into the pCMV AAV backbone with the no-stop 3′ UTR. All constructs were confirmed by sequencing. pEGFP-C1-Fibrillarin was a gift from Sui Huang (Addgene plasmid #26673; http://n2t.net/addgene:26673; RRID: Addgene_26673) (Chen and Huang 2001).
Cell culture and transfections
HeLa cells (ATCC) were maintained in Dulbecco’s modified Eagle medium (DMEM; Invitrogen) supplemented with 10% FBS (Sigma) and penicillin-streptomycin (Gemini Bio-Products). Prior to plating cells on a cell culture plate, the individual wells were treated with 3 mg/ml collagen solution (Advanced BioMatrix, 5005-B) diluted 1:100 in PBS for at least 12 h for glass or at least 1 h for plastic. The HUVEC human umbilical vein endothelial cell line (ATCC) was grown on collagen-coated vessels and maintained in EGM media supplemented with 2% FBS, hydrocortisone, hFGF-B, VEGF, R3-IGF-1, ascorbic acid, hEGF, and heparin (EGM-2 BulletKit; Lonza). Following the manufacturer’s protocol, Polyjet (SignaGen Laboratories) was used to transfect HeLa cells.
In vivo TEV protease digestion
Constructs containing the TEV cut site were co-transfected with pcDNA3.1 TEV protease into HeLa cells. Cells were fixed and immunostained 24 h post-transfection. pcDNA3.1 TEV (full-length) was a gift from Xiaokun Shu (Addgene plasmid #64276; http://n2t.net/addgene:64276; RRID: Addgene_64276) (To et al. 2015).
siRNA knockdowns
Lipofectamine RNAiMAX (Invitrogen) was used for siRNA transfection of HUVEC cells according to the manufacturer’s protocol. siRNA transfections were done every third day with assays performed on day 5. Knockdown was confirmed by qPCR and/or by flow cytometry (assaying for siLTN1 stabilization of the RFP-T2A-GFP nonstop reporter).
Amylo-glo staining
HeLa cells seeded on non-coated glass coverslips were transfected with cDNAs encoding GFP-stop, GFP-nonstop, GFP-UTR, GFP-polyK, and GFP-UTR-polyK using Lipofectamine 2000 (Thermo Scientific) according to the manufacturer’s instructions. Twenty-four hours later, cells were washed with cold PBS, fixed for 9 min at room temperature using 3.7% formaldehyde in PBS, and permeabilized with 0.2% Triton X-100 in PBS using a quick wash without agitation. Cells were then stained for 15 min at room temperature with Amylo-glo (1×) in 0.9% NaCl and washed for 5 min with 0.9% NaCl. Cells were immediately analyzed by confocal microscopy.
For experiments in LTN1-depleted cells, HeLa cells seeded on non-coated glass coverslips were lipofected with either siGENOME Non-Targeting siRNA (siRNA CTL) or SMARTpool ON-TARGETplus LTN1 siRNA (L-006968-00-0005, Dharmacon). Forty-eight hours post-transfection, cells were either left untreated or treated with 10 μM MG132 for 13 h, followed by recovery in drug-free medium for 5 h. Cells were then processed for Amylo-glo staining as described above.
Immunostaining
On the fifth day following the start of knockdown, the day after reporter transfection, or the second day after AAV reporter infection, HUVEC cells were washed once with PBS (Invitrogen) and fixed for 15 min with 4% formaldehyde (Ted Pella). After two PBS washes, cells were permeabilized and blocked with 10% FBS, 1% BSA, and 0.1% Triton X-100 (Sigma-Aldrich) for 30 min. Then, mouse α-nucleolin (C23) antibody (clone MS-3; Santa Cruz Biotechnologies, cat. #: SC-8031) diluted in permeabilization/block buffer (1:5000) was added for 2–4 h at room temperature. Following three 5-min PBS washes, goat α-mouse IgG secondary antibody (DyLight 650; Thermo Fisher Scientific, cat. #: 62265) diluted in permeabilization/block buffer (1:1000) was incubated for 1–2 h at room temperature. After three PBS washes, nuclei were stained with Hoechst. Other antibodies used for immunostaining and any procedural modifications are listed below: mouse α-UBF (1:100, Santa Cruz Biotechnologies, cat. #: SC-13125 )—cells were fixed with ice-cold methanol for 5 min, then blocked and stained with primary and secondary antibodies in 10% FBS and 1% BSA in PBS; rabbit α-FBL (1:500, One World Labs, cat. #: MCA-38F3); and rabbit α-NPM1 (1:1000, Santa Cruz Biotechnologies, cat. #: SC-6013-R).
Fractionation of NP-40 soluble and insoluble proteins
Twenty-four hours after transfection, cells were harvested in a buffer containing 1% NP-40 (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, and 1 mM EDTA) and passed through a 26G needle 3 times. Cells were lysed on ice for 10 min, then spun at 10,000×g at 4°C for 10 min. The supernatant was collected as NP-40 soluble fraction, while the pellet (NP-40 insoluble fraction) was resuspended with 2% SDS Laemmli buffer. β-Mercaptoethanol was added to the protein fractions, which were then boiled for 3 min at 100°C . Protein fractions were then separated by SDS-PAGE, followed by western blot using an anti-GFP antibody (632381, Clontech).
High-throughput nucleolar morphology microscopy
HUVEC cells were infected with AAV (MOI of 10) expressing reporter constructs for 2 days or siRNA knockdown was performed for 5 days. Cells were then fixed and immunostained as described in the “Immunostaining” section. Images were acquired with MicroManager v1.4 software package (Edelstein et al. 2010, 2014) on a Nikon Eclipse TiE inverted fluorescence microscope with a ×40 (NA 0.95) air objective (Nikon Instruments) equipped with a CCD camera (Andor Technologies). Data were collected from a minimum of three independent experiments. For each experiment, at least 500 cells were analyzed for each condition. The Matlab software platform (MathWorks) was used for high-throughput image analysis. First, a mask covering the nucleus was constructed from the Hoechst image. Very large (cells touching) or very small (debris or dividing cells) nuclei were not included in the analysis. Next, the program looked within the nuclear area defined by the Hoechst mask in the nucleolin channel to identify nucleoli. From here, parameters such as area, perimeter, extent (roundness), and eccentricity (circularity) were calculated for the different conditions.
Other fixed cell microscopy
Images were acquired on a CoolSnap HQ CCD camera (Photometrics) with a DeltaVision Core deconvolution microscope using 0.2-μm steps. Pictures shown in Figs. 2a, d and 3a were acquired using a Leica SP8 confocal microscope equipped with a 405-nm and white light lasers using a ×63 oil immersion objective. The Amylo-glo and GFP intensities were calculated in specific ROI using ImageJ (https://imagej.nih.gov/ij) and the mean ratio intensity of Amylo-glo was divided by the mean ratio intensity of GFP. One hundred cells in three independent experiments were analyzed.
Fig. 2.

Nucleolar targeting increases solubility of amyloidogenic sequences. a, b 24 h post-transfection HeLa cells expressing GFP-UTR were assessed for nucleolar and perinuclear amyloid by Amylo-glo staining. Microscopy is quantified in b. The mean Amylo-glo and GFP intensities were measured in 100 ROIs inside nucleoli and 100 perinuclear aggregates, in three independent experiments. Scale bar = 10 μm. c Fluorescence recovery after photobleaching (FRAP) was performed on the nucleolar (orange) and perinuclear (gray) compartments of HeLa cells expressing GFP-UTR. d Cells expressing GFP constructs for 24 h were assessed for amyloid by Amylo-glo staining. Scale bar = 10 μm. e FRAP was performed on the nucleoli of cells expressing the indicated constructs. f Anti-GFP western blot was performed on solubility-fractionated HeLa cell lysates. Lysate loading volumes were normalized so the soluble and insoluble fractions represent equivalent numbers of cells
Fig. 3.

Accumulating nonstop proteins disrupt nucleoli. a, b siRNA-treated HeLa cells were treated with 10 μM MG132 for 13 h and allowed to recover for 5 h. Cells were assessed for amyloid by Amylo-glo staining. Scale bar = 10 μm. Microscopy is quantified in b: p = 0.003 for three independent experiments, ± SEM. c, d FRAP was performed using GFP-FBL in HeLa cells treated with siLTN1 and siSCR (c) or RFP-nonstop and RFP-stop (d). Data is presented as means ± the standard error (SE) of three independent experiments (5–20 cells each). p values are of the mobile fractions. e–g siLTN1-treated and GFP nonstop-expressing HUVEC cells have smaller, rounder nucleoli. Results of automated image analysis quantifying changes in nucleolar morphology of siLTN1 (f) and GFP-nonstop (g) HUVEC cells. Data are presented as means −1 ± SE of the control distribution (siSCR (f) and GFP-stop (g)) for >three independent experiments (>500 cells each). Red dots represent replicates. *p < 0.05; **p < 0.054. n.s., not significant. h–j Muscle cells from flies expressing one or two copies of an RNAi transgene targeting Ltn1 were immunostained for FBL without (h) or with (i) 37°C heat shock. Scale bar = 5 μm. Data are quantified in j. *p < 0.05. n.s., not significant
Fluorescence recovery after photobleaching
Cells were plated on glass-bottom 96-well plates (Cell-Vis). After 18–24 h, GFP FBL was transfected, and cells were imaged 24 h later. Fluorescence recovery after photobleaching (FRAP) was performed with an inverted Zeiss LSM 880 laser scanning confocal microscope using a ×63 oil objective. A small region of interest was bleached by scanning with the 405-nm laser at 100% power. A normalizing, unbleached area as well as a non-fluorescent background region was also collected. Images were captured every 0.663 s for a total of 100 images. Data was exported from FRAP movies using Fiji (Schindelin et al. 2012) with the Bio-Formats plugin (Linkert et al. 2010). Data was analyzed using the easyFRAP-web single-page application (Koulouras et al. 2018). Full scale normalized data was plotted against time and fit with a double exponential.
FRAP measurements on HeLa cells transfected with cDNAs encoding GFP-nonstop, GFP-UTR, GFP-polyK, and GFP-UTR-PolyK were performed on a Leica SP8 confocal microscope equipped with a 405-nm and white light lasers using a ×63 oil immersion objective. A nucleolus was bleached for 1 s using a laser intensity of 100% at 405nm. Recovery was recorded for 100–120 time points after bleaching (60–300 s). Analysis of the recovery curves was carried out using a custom written FIJI/ImageJ routine. The equation used for FRAP analysis is as follows ((Ibleach − Ibackground)/(Ibleach(t0) − Ibackground(to)))/((Itotal − Ibackground)/(Itotal(t0) − Ibackground(to))), where Itotal is the fluorescence intensity of the entire cellular structure, Ibleach represents the fluorescence intensity in the bleach area, and Ibackground the background of the camera offset. Fluorescent density analysis was performed using FIJI/ImageJ and selecting specific region of interest (ROI). When necessary, image drift correction was applied using StackReg plug-in function of the FIJI software suite prior to FRAP analysis. FRAP curves were averaged to obtain the mean and standard deviation. p values were calculated by comparing the means of the plateau values using a paired t-test.
Flow cytometry
HeLa cells were trypsinized and resuspended in serum-free media supplemented with 5 mM EDTA prior to measurement on an Accuri C6 plus cytometer equipped with 488- and 640-nm lasers (BD Biosciences). Untransfected cells were gated out during analysis.
AAV production and infections
293T cells were transfected with adenovirus helper, AAV helper, and pAAV CMV GFP constructs using HBS-calcium chloride transfection. Adherent and floating cells were collected after 3 days and freeze-thawed three times, treated with benzonase for 30 min at 37°C, and supernatants were collected after spinning for 13,500 rpm for 30 min at 4°C.
Hydropathicity assessment
The translated sequence of the GFP-nonstop 3′UTR was run through the ExPASy ProtScale amino acid calculator for hydrophobicity (Gasteiger et al. 2005) using the Kyte and Doolittle scale (Kyte and Doolittle 1982). The average hydrophobicity value of the translated human proteome was calculated based on calculated amino acid abundances (Nakamura et al. 2000).
Simulations
Simulations were performed using a coarse-grained model in which proteins and nucleolar material are represented as one or more beads on a cubic lattice. A nearest-neighbor lattice interaction potential was used, and simulations were run under conditions in which a single phase-separated droplet is formed and stable at equilibrium. Further details can be found in the Supplemental Methods.
Drosophila genetics
Flies were raised in fly food at 25°C according to standard procedures. Fly food consisted of the following: water, 17 l; agar, 93 g; cornmeal, 1716 g; brewer’s yeast extract, 310 g; sucrose, 517 g; dextrose, 1033 g. Drosophila stock used: Ltn1 (CG32210); RNAi: V41866 (VDRC); MHC-Gal4 (lab stock). Homozygous Ltn-RI indicates two copies of the Ltn1-RNAi transgene driven by MHC-Gal4 while heterozygous Ltn1-RI signifies one copy of the Ltn1-RNAi transgene driven by MHC-Gal4.
Drosophila immunostaining
Drosophila larval body wall muscle dissection was performed according to the standard neuromuscular junction (NMJ) protocol. For the heat shock condition, 3rd instar larvae were heat-shocked at 37°C water bath for 1 h and recovered at room temperature for 1 h before dissection. Briefly, 3rd instar larvae were picked out of vials and washed with PBS. On a dissecting Petri dish, the flies were fixed at anterior and posterior ends with two dissection pins. A cut was made from the anterior to the posterior along the midline using a dissecting scissor. The larval body wall was straightened at the edges using four more pins. Internal organs were removed using forceps. Larval body wall was then washed with PBS 3 times. Bouin’s solution (Sigma) was added to completely immerse the tissue for 5 min of fixation. Larval tissue was then transferred into 500-μl Eppendorf tubes and washed 3 times for 15 min each in 0.3% Triton X-100 with PBS with shaking. Rabbit α-FBL (1:200, One World Labs, cat. #: MCA-38F3) primary antibody was incubated at 4°C overnight with gentle shaking. DAPI (1 μg/ml, cat. #: Sigma-D9542) and secondary antibody (1:300) were added and incubated for 2 h at room temperature. After washing 3 times for 15 min each with 0.3% Triton X-100 in PBS, larval tissues were mounted in SlowFade Antifade medium (Invitrogen). Images were taken using a Leica SP8 confocal microscope and processed in Photoshop. Sample numbers are n=18, 3 biological repeats per genotype.
FBL foci inside nuclei were quantified using ScanR. Briefly, nuclei and nucleoli were assessed by DAPI and FBL staining, respectively. Nuclei and FBL-positive nucleoli were segmented using the intensity and the edge module, respectively. Next, FBL foci were segmented using the edge module and automatically counted in both nuclei and nucleoli. The number of FBL foci dispersed in the nucleus was obtained by subtracting the number of foci inside nucleoli from the number of FBL foci inside nuclei.
Results
Translation of the poly(A) tail localizes nonstop substrates to nucleoli
To track the fate of proteins produced during stalled translation, we exogenously expressed GFP from a cistron lacking a stop codon (GFP-nonstop) in HeLa cells (Fig. S1a, b). This reporter was engineered with the commonly used bGH 3′ UTR. Mapping the 3′ end of the nonstop transcript confirmed the absence of in-frame stop codons and confirmed that polyadenylation occurred at the expected site on the reporter mRNA (Fig. S1c). GFP-nonstop protein was stabilized by siRNA knockdown of LTN1, confirming that ribosomes translating GFP-nonstop stalled and triggered RQC (Fig. S1d). Some RQC substrates have been observed to aggregate in yeast (Yonashiro et al. 2016; Choe et al. 2016; Defenouillère et al. 2016). Aggregated proteins can be pathological correlates and drivers of disease processes (Ross and Poirier 2004; Aguzzi and O’Connor 2010) and thus may underlie stalling and RQC-related neurodegenerative phenotypes. Unexpectedly, we found that GFP-nonstop did not form cytoplasmic inclusions, but instead localized to discrete structures inside the nucleus of all cells (Fig. 1b, top). Staining for nucleolar components revealed that GFP-nonstop localized to the nucleolus and was distributed throughout all of the nucleolar subcompartments (Fig. 1b, S1e, f), contrasting with the diffuse distribution of GFP-stop (Fig. 1b, bottom).
To verify that the extra sequence produced from the GFP-nonstop construct (vs. GFP-stop) was required for its nucleolar localization, we removed the C-terminal end of the protein in vivo using an engineered TEV cleavage site (GFP-TEV-nonstop) and coexpression of TEV protease. As predicted, this resulted in loss of nucleolar localization (Fig. 1c, top) that was specific to the nonstop construct, as the localization of the diffuse GFP-TEV-stop was unchanged by TEV coexpression (Fig. 1c, bottom). We sought to determine if an isolated feature of the nonstop protein was sufficient for nucleolar localization. GFP-nonstop translates two additional mRNA regions that differentiate it from GFP-stop: the 3′ UTR and the poly(A) tail. While the UTR can encode many different amino acids, the AAA codons in the poly(A) tail encode for lysine and thus translation of poly(A) tails produces polylysine (polyK) tracts. Previous work has shown that polybasic sequences can act as both nuclear (Lang et al. 2018) and nucleolar (Scott et al. 2010) targeting sequences. Consistent with this, adding a templated, C-terminal polyK tract to GFP was sufficient to cause nucleolar localization (Fig. 1d, top). This nucleolar localization was not codon-specific as polyK tracts encoded by both “AAA” and “AAG” codons yielded comparable localization (Fig. 1d, top and middle). Expression of the UTR sequence without polyK (GFP-UTR) was not sufficient to cause nucleolar-specific localization of the reporter, resulting in localization throughout the cell and the formation of cytoplasmic inclusions, in line with our original expectation (Fig. 1d, bottom). Thus, we conclude that the polyK tracts of nonstop proteins are necessary and sufficient to localize them to nucleoli, and that nucleolar sequestration prevents cytoplasmic inclusions
Nucleolar targeting increases solubility of amyloidogenic proteins
Nucleolar targeting has been proposed to solubilize misfolded proteins (Frottin et al. 2019; Mediani et al. 2019). To understand the effect of the local environment on the extent of aggregation of our reporters, we took advantage of our observation that GFP-UTR localized to both nucleoli and the cytosol. This allowed us to use an amyloid-specific dye (Amylo-glo) to determine if GFP-UTR forms amyloids in different compartments of the same cell. Strikingly, GFP-UTR stained for Amylo-glo in the cytosol (suggesting the presence of amyloid) but not in the nucleolus (suggesting soluble protein) (Fig. 2a, b). Proteins trapped within aggregates can be less able to exchange with other proteins and thus demonstrate decreased mobility by fluorescent recovery after photobleaching (FRAP) analysis. We found that the perinuclear aggregate fraction of GFP-UTR had dramatically reduced mobility relative to the nucleolar fraction of the protein (Fig. 2c). Collectively, these data suggest that the nucleolar environment solubilized protein that would otherwise have aggregated in the cytosol.
To understand the effect of nucleolar localization of nonstop proteins, HeLa cells were transfected with fluorescent reporters with differing C-termini and stained for amyloid (Fig. 2d). GFP-nonstop-expressing cells stained negatively for nucleolar amyloid, similar to GFP-stop (Fig. 2d, top two panels). However, GFP-nonstop is expressed at relatively low levels due to its mRNA being a constitutive target of nonstop mRNA degradation (Klauer and van Hoof 2012). In addition, GFP-nonstop is actively cleared from the nucleolus as demonstrated by the loss of nucleolar GFP signal after treatment with the translation inhibitor cycloheximide (Fig. S2a). This nucleolar clearance is proteasome dependent, as the loss of nucleolar GFP upon cycloheximide treatment was prevented by the proteasome inhibitor MG132 (Fig. S2a, b).
To increase the levels of the GFP-nonstop protein product, we encoded an equivalent sequence to GFP-nonstop on a gene containing a terminal stop codon, GFP-UTR-polyK. GFP-UTR-polyK was expressed at much higher levels and, in contrast to GFP-nonstop, caused strong Amylo-glo staining within nucleoli (Fig. 2d, second to bottom panel). The UTR sequence was essential for this effect, as expression of GFP-polyK resulted in no Amylo-glo staining (Fig. 2d, bottom panel), while GFP-UTR produced Amylo-glo positive cytoplasmic aggregates (Fig. 2d, middle panel).These results suggest that the amount and the nature of invading proteins determine if the nucleolar environment will be able to solubilize them. Measurements of reporter mobility by FRAP supported this model; GFP-polyK had the highest mobility, GFP-nonstop had a reduced mobile fraction and a somewhat delayed recovery time, and the high-expressing GFP-UTR-polyK had reduced mobile fraction and a further delayed recovery time (Fig. 2e). Using centrifugation to separate soluble and insoluble proteins revealed the same hierarchy of effect; the GFP-stop and most of the GFP-nonstop were found in the soluble fraction (Fig. 2f). By contrast, the majority of the GFP-UTR was insoluble. The GFP-UTR-polyK is proportionately more soluble than GFP-UTR, consistent with an increased solubility in nucleoli versus the cytosol (we note that the presence of polylysine also likely contributes to the increased solubility of GFP-UTR-polyK vs. GFP-UTR independent of nucleolar localization). Taken together, these data support a model of enhanced solubility being conferred to escaped RQC substrates through localization in the nucleolar compartment and interaction with resident nucleolar proteins before eventual proteasomal degradation.
LTN1 depletion disrupts nucleoli
We sought to probe the consequences of the influx of endogenous nonstop proteins into nucleoli that is predicted to occur with higher substrate load due to impaired RQC. We blocked RQC by knocking down LTN1, the E3 ligase that marks nonstop proteins for degradation. After 72 h of treatment with siRNA targeting LTN1, LTN1 mRNA levels were efficiently decreased. The effect was specific as the mRNA level of another important RQC component, NEMF, was not decreased (Fig. S2a, b). In cells depleted of LTN1, endogenous nonstop substrates are predicted to escape degradation and build up in the nucleolus. To assay nucleolar integrity, we measured the ability of nucleoli to recover from stress induced by the proteasome inhibitor MG132. MG132 induces nucleolar amyloid body formation (an indicator of nucleolar stress) that can dissolve following MG132 removal (Mediani et al. 2019) (Fig. 3a, b). MG132 also blocks degradation of nonstop proteins that accumulate in the nucleolus (Fig. S2c, d), and is thus predicted to increase their levels in nucleoli when RQC is impaired. Cells lacking LTN1 had defects dissolving nucleolar amyloid bodies after MG132 removal, suggesting that in the absence of LTN1, nucleoli were more vulnerable to stressors. This is consistent with our model that impaired RQC causes increased levels of nonstop proteins that then invade the nucleolus, interact with resident proteins, and alter the state of nucleoli.
We next explored if escaped RQC substrates alter nucleolar dynamics. The nucleolus is an archetypal membraneless organelle that is formed at least in part through liquid-liquid phase separation (Mitrea et al. 2016; Feric et al. 2016). Nucleoli are self-organizing and serve to concentrate RNA and proteins into a constrained space, thus enhancing reactions (Lam and Trinkle-Mulcahy 2015). We hypothesized that an influx of new proteins into nucleoli may impact the dynamics of nucleolar-resident proteins. To test this, we used FRAP to examine the mobility of the nucleolar protein fibrillarin (FBL) in cells under two conditions in which endogenous nonstop proteins are predicted to accumulate in the nucleolus due to RQC capacity being exceeded. Both knockdown of LTN1 (lowering RQC capacity, Fig. S1d) and expressing high levels of an exogenous nonstop protein (saturating RQC capacity, Fig. S3a) significantly increased the mobility of GFP-FBL, quantified as the average mobile fraction after 50 s (Fig. 3c, d). The observed difference in FBL mobility was most apparent in the mobile fraction of later timepoints, as t1/2 values between conditions were not significantly different. Taken together, these results suggest that when LTN1 cannot efficiently ubiquitylate the pool of polyK-containing nonstop substrates, these substrates enter the nucleolus, interact with resident proteins, and alter nucleolar dynamics.
We next looked to assess changes in nucleolar morphology, as it provides a visible readout of the underlying biophysical state of the nucleolus (Brangwynne et al. 2011). Because HeLa are a transformed cell line with heterogeneous nucleoli, we instead used human umbilical vein endothelial cells (HUVEC), which are primary cells that can be cultured in vitro and are amenable to high-throughput imaging due to their flat, uniform appearance. Cells were immunostained with the nucleolar marker nucleolin and qualitative differences could be observed between nucleolar morphologies, with siLTN1 nucleoli appearing smaller and rounder than siSCR nucleoli (Fig. S3b). To quantify these differences, we systematically analyzed cells (>1500 cells/condition) for nucleolar area, extent (~roundness), eccentricity (~circularity), and the size of the largest nucleolus per cell. Quantitative analysis confirmed that siLTN1 nucleoli had lower 2D area as measured by a single image, were rounder, and were more circular, and the largest area nucleolus per cell was smaller than observed in siSCR cells (Fig. 3e, f). Next, we sought to determine if we could recapitulate the altered nucleolar morphology with GFP-nonstop expression alone, as with the nucleolar mobility phenotype (Fig. 3c, d). Indeed, expression of the GFP-nonstop substrate had a weaker but similar trend of effects on nucleolar morphology, with a reduced area, greater extent, less eccentricity, and reduced size of the largest nucleolus per cell relative to a GFP-stop control (Fig. 3g). By contrast, expression of GFP-polyK had no significant impact on nucleolar morphology (Fig. S3c), suggesting that specific features of RQC substrates affect nucleoli. Remarkably, despite altered nucleolar morphology, LTN1-deficient HUVEC cells were able to synthesize normal levels of rRNA and support normal growth rates (Fig. S4a–d). This suggests that nucleolar state can serve as a sentinel of cellular stress below toxic levels.
To measure the effect of impaired RQC in an intact animal, LTN1 was selectively knocked down in the muscle cells of Drosophila melanogaster and nucleoli were assessed by FBL staining (Fig. 3h, j). Strikingly, LTN1 knockdown caused dramatic defects in nucleolar morphology, with smaller contiguous nucleolar staining and dispersion of FBL throughout the nucleus. This phenotype mimicked the effect of 37°C heat shock (Fig. 3i, j) and was amplified when two copies of LTN1 RNAi transgene were expressed to enhance LTN1 RNAi effect or when LTN1 knockdown was combined with heat shock. Thus, loss of LTN1 causes severe nucleolar stress (equivalent to heat shock) and loss of resistance to stressors in animals. Collectively, our experiments suggest that nucleoli are stressed by specific features of nonstop proteins when a cell’s RQC capacity is limited.
Discussion
In this work, we use cultured cells and Drosophila melanogaster to examine how cells are affected by stalled proteins that escape co-translational quality control. We find that the protein products of nonstop mRNAs accumulate in nucleoli via polyK tracts produced by the translation of poly(A) tails. We conclude that the exposed hydrophobic residues of misfolded regions of defective proteins interact with resident nucleolar proteins, increasing the solubility of nonstop proteins but disrupting nucleolar dynamics and morphology (Fig. 4).
Fig. 4.
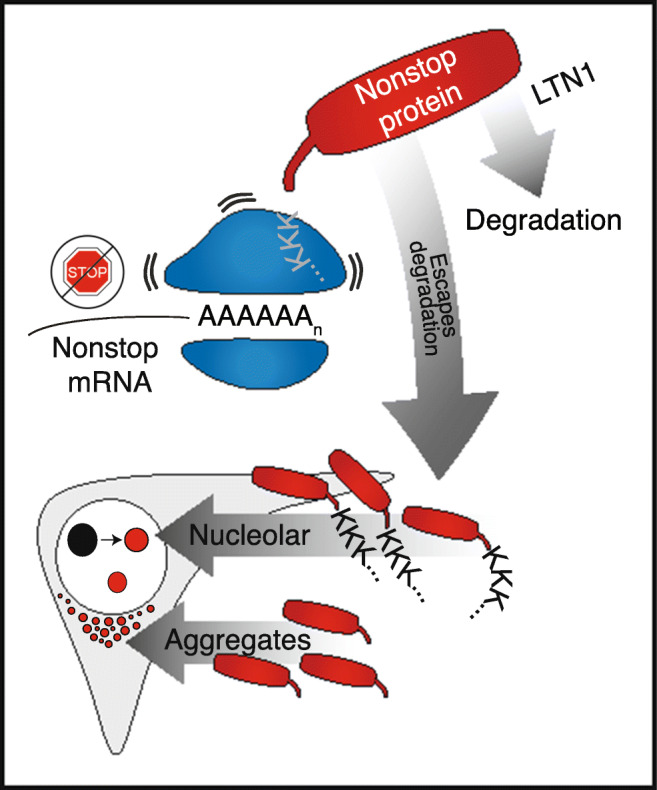
Invading nucleolar proteins experience greater solubility but can also disrupt nucleoli. Model for the effect of nonstop proteins on cells: translation of the poly(A) tail of nonstop mRNAs produces a polyK tract. This causes the ribosome to stall, and in the case of insufficient LTN1, cells accumulate nonstop proteins. The polyK tail localizes the nonstop protein to nucleoli, bringing the aggregation-prone, hydrophobic translated 3′ UTR with it. Interactions between nucleolar components and the infiltrating aggregation-prone protein increase solubility of defective protein but alter nucleolar dynamics and morphology, and interfere with the ability to recover from stress (depicted in the model with the transition from black to red nucleoli)
Because 3′ UTRs did not evolve to be translated, they may not have been subjected to negative selection disfavoring aggregation-prone, pathologic sequences. As with our reporter UTR, translation of the 3′ UTR of some disease-associated nonstop transcripts have been shown to unmask aggregation-prone, hydrophobic sequences (Rebelo et al. 2016; Bock et al. 2018). Thus, translated 3′ UTRs arising from stop codon readthrough (Dunn et al. 2013) may generally pose a danger to the cell such that increasing their solubility by nucleolar localization may be an important quality control strategy. Similarly, misfolded proteins generated by translation of prematurely polyadenylated mRNA may be solubilized and less toxic to cells when sequestered within nucleoli. Recent work has demonstrated that lysine-rich dipeptides are more strongly recruited to nucleoli compared to arginine-rich dipeptides, supporting a model in which polylysine provides explicit nucleolar targeting (Greig et al. 2019).
It was counterintuitive to observe that both knockdown of LTN1 and expression of an exogenous nonstop protein—perturbations that increase the nucleolar load of defective proteins—led to rounder nucleoli with smaller cross sections. By contrast, GFP-polyK partitions into the nucleolus yet do not lead to a change in nucleolar morphology (Fig. S3c). This suggests that the observed nucleolar changes with GFP-nonstop may not simply be due to the interactions that facilitate partitioning into the nucleolus, but that the translated 3′ UTR, which drives aggregation (Fig. 2) and is enriched in hydrophobic residues (Fig. S5a), contributes attractive intermolecular interactions that underlie altered nucleoli behavior. The observed morphological changes are interpretable under a simple biophysical model of phase separation in which the partitioning of hydrophobic residues in nonstop proteins into nucleoli strengthens the driving force for phase separation (Fig. S5b–d). In the spirit of simple models that are able to capture key features of nucleolar organization (Feric et al. 2016), we modeled this using a simple single-bead-per-protein lattice-based model (Fig. S5e). In this model, GFP-nonstop or GFP-polyK proteins partition into a phase-separated droplet consisting of “nucleolar material.” Our experimental results suggest that unlike GFP-polyK, GFP-nonstop engages in non-specific interactions with the nucleolar material driven by hydrophobic residues from the translated 3′ UTR. To test if this simple interpretation would yield the observed morphological changes, our computational model defines stronger GFP-nonstop interactions with nucleolar material than GFP-polyK. In agreement with our experimental results, GFP-polyK robustly partitions into the droplet but there are no concentration-dependent changes to droplet morphology (Fig. S5f, g). In contrast, GFP-nonstop protein displays enhanced partitioning and concentration-dependent morphological changes, with droplets becoming smaller and rounder with increasing concentrations of GFP-nonstop (Fig. S5f, g). This simple model recapitulates how an increase in defective proteins might affect morphology of the nucleolus through altering its physical properties, but is consistent with a general model in which disruption to intra-nucleolar interactions can have complex and non-monotonic effects on nucleolar size, dynamics, and function (Fig. S5h).
The interaction of escaped quality control substrates with nucleoli is supported by the finding that model substrates are more soluble in nucleoli than in the cytoplasm. This observation dovetails with recent work showing that importins recruited by nuclear localization sequences act as disaggregases in addition to facilitating transport into the nucleus (Guo et al. 2018). Our data suggest that insoluble substrates containing nucleolar localization sequences are kept from aggregating by virtue of being in the nucleolar environment. In some cases, nucleolar sequestration of defective proteins may have a protective effect. For example, a truncated, nucleolar localized version of the ALS-associated TDP-43 protein decreases cell death by assuming a non-toxic oligomeric state in nucleoli (Kitamura et al. 2016). In addition, nucleolar sequestration of aggregation-prone proteins is emerging as a general response to proteotoxic stress, since it has been recently reported to occur upon stress conditions such as heat shock, acidosis, transcriptional stress, and proteasome failure. Importantly, misfolded proteins in nucleoli, including nonstop proteins (Fig. S2a, b), are cleared over time by the cell’s protein quality control systems. As such, nucleoli act as reservoirs to prevent irreversible protein aggregation upon stress (Audas et al. 2016; Frottin et al. 2019; Mediani et al. 2019). Both protein-protein interactions and protein-RNA interactions are involved in the transient detention of aggregation-prone substrates inside nucleolar subcompartments that then acquire transient amyloidogenic properties. In particular, ribosomal intergenic spacer noncoding RNAs that are selectively upregulated upon stress conditions have been shown to interact with a variety of proteins, retaining them inside nucleolar subcompartments upon stress (Audas et al. 2012; Wang et al. 2018).
However, nucleoli have a limited buffering capacity and nucleolar sequestration of aggregation-prone proteins is not without consequences, as shown here in cells and flies lacking LTN1. We propose that the accumulation of misfolded proteins in nucleoli leads to enhanced intra-nucleolar interactions driven by aggregation-prone multivalent misfolded proteins, exhausting nucleolar material and reducing the nucleolar capacity to solubilize further nucleolar aggregation-prone proteins. These interactions alter nucleolar morphology, nucleolar size, and nucleolar dynamics. Furthermore, under extreme proteostatic stress, nucleolar fragmentation occurs (Fig. 3h–j), an observation consistent with a model in which nucleolar misfolded proteins compete for “native” nucleolar interaction sites, impacting nucleolar integrity. Essentially, our results are consistent with a general model in which the nucleolus plays a critical but finite role in buffering proteostatic stress.
While a causative link between the nucleolar abnormalities we observe and disease has not yet been shown, previous work has indicated that disruption of nucleoli may have deleterious consequences (Boulon et al. 2010). Defective clearance of misfolded proteins from nucleoli due to protein quality control impairment leads to irreversible amyloidogenesis and decreases cell viability (Audas et al. 2016; Frottin et al. 2019; Mediani et al. 2019). This has important implications in human disease, as the accumulation of aberrant disease-linked proteins such as arginine-rich C9orf72 dipeptide repeats disrupts nucleoli and is causative for amyotrophic lateral sclerosis (ALS) and frontotemporal degeneration (FTD) and has been associated with neurodegeneration (Parlato and Kreiner 2013; Hernández-Ortega et al. 2016; Lee et al. 2016; Frottin et al. 2019; Mediani et al. 2019). Future work will need to address to what extent nucleolar accumulation of nonstop mRNA protein products contributes to nucleolar dysfunction in ALS/FTD and its role in neurodegeneration caused by a hypomorphic LTN1 allele (Chu et al. 2009). Beyond neurodegeneration, enlarged nucleoli are a classical hallmark in many transformed cells, an observation ascribed to enhanced ribogenesis as required by rapid cell division (Derenzini et al. 1998). Our results suggest enlarged nucleoli will also enhance the proteostatic capacity of a cell, improving fitness under both acute and chronic stress and providing an additional broad-spectrum survival adaptation for transformed cells. Our study elucidates how failures in co-translational protein quality control can disrupt distal cellular processes and may help understand the pathological drivers of neurodegenerative disease.
Supplementary information
(PDF 11755 kb)
(DOCX 8 kb)
Acknowledgments
We thank the members of the Brandman laboratory for helpful discussions and commentary on the manuscript. Additionally, we acknowledge the members of the Julie Theriot Lab (Stanford/University of Washington) and Aaron Straight (Stanford) labs for use of microscopes, reagents, and expertise. We also thank people from the CIGS microscopy facility (University of Modena and Reggio Emilia) for technical support. This work was performed while ASH was a postdoctoral fellow in the laboratory of Dr. Rohit V. Pappu (RVP) at Washington University in St. Louis and supported by the Human Frontiers Science Program (grant RGP0034/2017 to RVP and S.A.). S.A. was supported by the European Researchn Council (PhaseAge, No. 725836). S.C. acknowledges funding from AriSLA Foundation (MLOpathy); Cariplo Foundation (Rif. 2014-0703); MAECI (Dissolve_ALS); MIUR (Departments of excellence 2018-2022; E91I18001480001). S.C. and S.A. are grateful to EU Joint Programme - Neurodegenerative Disease Research (JPND) project. The project is supported through funding organizations under the aegis of JPND (http://www.neurodegenerationresearch.eu/). This project has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under grant agreement no. 643417. A preprint of this work was uploaded to BioRxiv (Davis et al., 2019)
Funding
This research was supported by a Stanford Dean’s Postdoctoral Fellowship to ZHD as well as a grant (R01GM115968) from the National Institutes of Health to OB. Work in BL’s lab was supported by grants (R01NS084412, R01NS083417 and R01AR0748750) from the National Institutes of Health.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Serena Carra, Email: serena.carra@unimore.it.
Onn Brandman, Email: onn@stanford.edu.
References
- Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9:237–248. doi: 10.1038/nrd3050. [DOI] [PubMed] [Google Scholar]
- Audas TE, Jacob MD, Lee S. Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Mol Cell. 2012;45:147–157. doi: 10.1016/j.molcel.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Audas TE, Audas DE, Jacob MD, Ho JJD, Khacho M, Wang M, Perera JK, Gardiner C, Bennett CA, Head T, Kryvenko ON, Jorda M, Daunert S, Malhotra A, Trinkle-Mulcahy L, Gonzalgo ML, Lee S. Adaptation to Stressors by Systemic Protein Amyloidogenesis. Dev Cell. 2016;39:155–168. doi: 10.1016/j.devcel.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson MH, Joazeiro CAP. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature. 2010;467:470–473. doi: 10.1038/nature09371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson MH, Joazeiro CAP. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature. 2010;467:470–473. doi: 10.1038/nature09371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock AS, Günther S, Mohr J, Goldberg LV, Jahic A, Klisch C, Hübner CA, Biskup S, Beetz C. A nonstop variant in REEP1 causes peripheral neuropathy by unmasking a 3′ UTR-encoded, aggregation-inducing motif. Hum Mutat. 2018;39:193–196. doi: 10.1002/humu.23369. [DOI] [PubMed] [Google Scholar]
- Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. The nucleolus under stress. Mol Cell. 2010;40:216–227. doi: 10.1016/j.molcel.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li GW, Zhou S, King D, Shen PS, Weibezahn J, Dunn JG, Rouskin S, Inada T, Frost A, Weissman JS. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell. 2012;151:1042–1054. doi: 10.1016/j.cell.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li GW, Zhou S, King D, Shen PS, Weibezahn J, Dunn JG, Rouskin S, Inada T, Frost A, Weissman JS. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell. 2012;151:1042–1054. doi: 10.1016/j.cell.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Mitchison TJ, Hyman AA. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc Natl Acad Sci U S A. 2011;108:4334–4339. doi: 10.1073/pnas.1017150108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Huang S. Nucleolar components involved in ribosome biogenesis cycle between the nucleolus and nucleoplasm in interphase cells. J Cell Biol. 2001;153:169–176. doi: 10.1083/jcb.153.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe Y-J, Park S-H, Hassemer T, Körner R, Vincenz-Donnelly L, Hayer-Hartl M, Hartl FU. Failure of RQC machinery causes protein aggregation and proteotoxic stress. Nature. 2016;531:191–195. doi: 10.1038/nature16973. [DOI] [PubMed] [Google Scholar]
- Chu J, Hong NA, Masuda CA, Jenkins BV, Nelms KA, Goodnow CC, Glynne RJ, Wu H, Masliah E, Joazeiro CAP, Kay SA. A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proc Natl Acad Sci U S A. 2009;106:2097–2103. doi: 10.1073/pnas.0812819106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis ZH, Mediani L, Vinet J et al (2019) Protein products of non-stop mRNA disrupt nucleolar homeostasis. Preprint at. 10.1101/851741
- Defenouillère Q, Yao Y, Mouaikel J, et al. Cdc48-associated complex bound to 60S particles is required for the clearance of aberrant translation products. Proc Natl Acad Sci U S A. 2013;110:5046–5051. doi: 10.1073/pnas.1221724110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defenouillère Q, Yao Y, Mouaikel J, et al. Cdc48-associated complex bound to 60S particles is required for the clearance of aberrant translation products. Proc Natl Acad Sci U S A. 2013;110:5046–5051. doi: 10.1073/pnas.1221724110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defenouillère Q, Zhang E, Namane A, Mouaikel J, Jacquier A, Fromont-Racine M. Rqc1 and Ltn1 Prevent C-terminal Alanine-Threonine Tail (CAT-tail)-induced Protein Aggregation by Efficient Recruitment of Cdc48 on Stalled 60S Subunits. J Biol Chem. 2016;291:12245–12253. doi: 10.1074/jbc.M116.722264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derenzini M, Trerè D, Pession A, Montanaro L, Sirri V, Ochs RL. Nucleolar function and size in cancer cells. Am J Pathol. 1998;152:1291–1297. [PMC free article] [PubMed] [Google Scholar]
- Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–564. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JG, Foo CK, Belletier NG, Gavis ER, Weissman JS. Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. Elife. 2013;2:e01179. doi: 10.7554/eLife.01179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, et al. Computer control of microscopes using μManager. Curr Protoc Mol Biol Chapter. 2010;14(Unit14):20. doi: 10.1002/0471142727.mb1420s92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N et al (2014) Advanced methods of microscope control using μManager software. J Biol Methods 1. 10.14440/jbm.2014.36 [DOI] [PMC free article] [PubMed]
- Feric M, Vaidya N, Harmon TS, Mitrea DM, Zhu L, Richardson TM, Kriwacki RW, Pappu RV, Brangwynne CP. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell. 2016;165:1686–1697. doi: 10.1016/j.cell.2016.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischmeyer PA, van Hoof A, O’Donnell K, et al. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295:2258–2261. doi: 10.1126/science.1067338. [DOI] [PubMed] [Google Scholar]
- Frischmeyer PA, van Hoof A, O’Donnell K, et al. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295:2258–2261. doi: 10.1126/science.1067338. [DOI] [PubMed] [Google Scholar]
- Frottin F, Schueder F, Tiwary S, Gupta R, Körner R, Schlichthaerle T, Cox J, Jungmann R, Hartl FU, Hipp MS. The nucleolus functions as a phase-separated protein quality control compartment. Science. 2019;365:342–347. doi: 10.1126/science.aaw9157. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S', Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005. pp. 571–607. [Google Scholar]
- Graber JH, Cantor CR, Mohr SC, Smith TF. In silico detection of control signals: mRNA 3’-end-processing sequences in diverse species. Proc Natl Acad Sci U S A. 1999;96:14055–14060. doi: 10.1073/pnas.96.24.14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig JA, Nguyen TA, Lee M, et al (2019) Arginine-enriched mixed-charge domains provide cohesion for nuclear speckle condensation. bioRxiv 771592 [DOI] [PMC free article] [PubMed]
- Guo L, Kim HJ, Wang H, et al. Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell. 2018;173:677–692.e20. doi: 10.1016/j.cell.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Ortega K, Garcia-Esparcia P, Gil L, Lucas JJ, Ferrer I. Altered machinery of protein synthesis in Alzheimer’s: from the nucleolus to the ribosome. Brain Pathol. 2016;26:593–605. doi: 10.1111/bpa.12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Zhou H, Yang XL, Schimmel P, Senju S, Nishimura Y, Chuang JH, Ackerman SL. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014;345:455–459. doi: 10.1126/science.1249749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito-Harashima S, Kuroha K, Tatematsu T (2007a) Translation of the poly (A) tail plays crucial roles in nonstop mRNA surveillance via translation repression and protein destabilization by proteasome in yeast. Genes, Translation of the poly(A) tail plays crucial roles in nonstop mRNA surveillance via translation repression and protein destabilization by proteasome in yeast [DOI] [PMC free article] [PubMed]
- Ito-Harashima S, Kuroha K, Tatematsu T, Inada T. Translation of the poly (A) tail plays crucial roles in nonstop mRNA surveillance via translation repression and protein destabilization by proteasome in yeast. Genes Dev. 2007;21:519–524. doi: 10.1101/gad.1490207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joazeiro CAP. Ribosomal Stalling During Translation: Providing Substrates for Ribosome-Associated Protein Quality Control. Annu Rev Cell Dev Biol. 2017;33:343–368. doi: 10.1146/annurev-cellbio-111315-125249. [DOI] [PubMed] [Google Scholar]
- Juszkiewicz S, Hegde RS. Initiation of Quality Control during Poly(A) Translation Requires Site-Specific Ribosome Ubiquitination. Mol Cell. 2017;65:743–750.e4. doi: 10.1016/j.molcel.2016.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura A, Nakayama Y, Shibasaki A, Taki A, Yuno S, Takeda K, Yahara M, Tanabe N, Kinjo M. Interaction of RNA with a C-terminal fragment of the amyotrophic lateral sclerosis-associated TDP43 reduces cytotoxicity. Sci Rep. 2016;6:19230. doi: 10.1038/srep19230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauer AA, van Hoof A. Degradation of mRNAs that lack a stop codon: a decade of nonstop progress. WIREs RNA. 2012;3:649–660. doi: 10.1002/wrna.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulouras G, Panagopoulos A, Rapsomaniki MA, Giakoumakis NN, Taraviras S, Lygerou Z. EasyFRAP-web: a web-based tool for the analysis of fluorescence recovery after photobleaching data. Nucleic Acids Res. 2018;46:W467–W472. doi: 10.1093/nar/gky508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lam YW, Trinkle-Mulcahy L (2015) New insights into nucleolar structure and function. F1000Prime Rep 7:48 [DOI] [PMC free article] [PubMed]
- Lang W-H, Calloni G, Vabulas RM. Polylysine is a Proteostasis Network-Engaging Structural Determinant. J Proteome Res. 2018;17:1967–1977. doi: 10.1021/acs.jproteome.8b00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K-H, Zhang P, Kim HJ, et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell. 2016;167:774–788.e17. doi: 10.1016/j.cell.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkert M, Rueden CT, Allan C, Burel JM, Moore W, Patterson A, Loranger B, Moore J, Neves C, MacDonald D, Tarkowska A, Sticco C, Hill E, Rossner M, Eliceiri KW, Swedlow JR. Metadata matters: access to image data in the real world. J Cell Biol. 2010;189:777–782. doi: 10.1083/jcb.201004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PB, Kigoshi-Tansho Y, Sher RB, Ravenscroft G, Stauffer JE, Kumar R, Yonashiro R, Müller T, Griffith C, Allen W, Pehlivan D, Harel T, Zenker M, Howting D, Schanze D, Faqeih EA, Almontashiri NAM, Maroofian R, Houlden H, Mazaheri N, Galehdari H, Douglas G, Posey JE, Ryan M, Lupski JR, Laing NG, Joazeiro CAP, Cox GA. NEMF mutations that impair ribosome-associated quality control are associated with neuromuscular disease. Nat Commun. 2020;11:4625. doi: 10.1038/s41467-020-18327-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mediani L, Guillén-Boixet J, Vinet J, et al. Defective ribosomal products challenge nuclear function by impairing nuclear condensate dynamics and immobilizing ubiquitin. EMBO J. 2019;38:e101341. doi: 10.15252/embj.2018101341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrea DM, Cika JA, Guy CS, Ban D, Banerjee PR, Stanley CB, Nourse A, Deniz AA, Kriwacki RW (2016) Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. Elife 5. 10.7554/eLife.13571 [DOI] [PMC free article] [PubMed]
- Nakamura Y, Gojobori T, Ikemura T. Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 2000;28:292. doi: 10.1093/nar/28.1.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlato R, Kreiner G. Nucleolar activity in neurodegenerative diseases: a missing piece of the puzzle? J Mol Med. 2013;91:541–547. doi: 10.1007/s00109-012-0981-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebelo AP, Abrams AJ, Cottenie E, Horga A, Gonzalez M, Bis DM, Sanchez-Mejias A, Pinto M, Buglo E, Markel K, Prince J, Laura M, Houlden H, Blake J, Woodward C, Sweeney MG, Holton JL, Hanna M, Dallman JE, Auer-Grumbach M, Reilly MM, Zuchner S. Cryptic Amyloidogenic Elements in the 3′ UTRs of Neurofilament Genes Trigger Axonal Neuropathy. Am J Hum Genet. 2016;98:597–614. doi: 10.1016/j.ajhg.2016.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MS, Boisvert F-M, McDowall MD, et al. Characterization and prediction of protein nucleolar localization sequences. Nucleic Acids Res. 2010;38:7388–7399. doi: 10.1093/nar/gkq653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, von der Malsburg K, Hegde RS. Listerin-dependent nascent protein ubiquitination relies on ribosome subunit dissociation. Mol Cell. 2013;50:637–648. doi: 10.1016/j.molcel.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaramoorthy E, Leonard M, Mak R, et al. ZNF598 and RACK1 Regulate Mammalian Ribosome-Associated Quality Control Function by Mediating Regulatory 40S Ribosomal Ubiquitylation. Mol Cell. 2017;65:751–760.e4. doi: 10.1016/j.molcel.2016.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Pan Z, Lee JY. Widespread mRNA polyadenylation events in introns indicate dynamic interplay between polyadenylation and splicing. Genome Res. 2007;17:156–165. doi: 10.1101/gr.5532707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To T-L. Piggott BJ, Makhijani K, et al. Rationally designed fluorogenic protease reporter visualizes spatiotemporal dynamics of apoptosis in vivo. Proc Natl Acad Sci U S A. 2015;112:3338–3343. doi: 10.1073/pnas.1502857112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoof A, Frischmeyer PA, Dietz HC, Parker R. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science. 2002;295:2262–2264. doi: 10.1126/science.1067272. [DOI] [PubMed] [Google Scholar]
- Verma R, Oania RS, Kolawa NJ, Deshaies RJ. Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. Elife. 2013;2:e00308. doi: 10.7554/eLife.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Tao X, Jacob MD, et al. Stress-Induced Low Complexity RNA Activates Physiological Amyloidogenesis. Cell Rep. 2018;24:1713–1721.e4. doi: 10.1016/j.celrep.2018.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonashiro R, Tahara EB, Bengtson MH, Khokhrina M, Lorenz H, Chen KC, Kigoshi-Tansho Y, Savas JN, Yates JR, III, Kay SA, Craig EA, Mogk A, Bukau B, Joazeiro CAP. The Rqc2/Tae2 subunit of the ribosome-associated quality control (RQC) complex marks ribosome-stalled nascent polypeptide chains for aggregation. Elife. 2016;5:e11794. doi: 10.7554/eLife.11794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Dang Y, Zhou M, Yuan H, Liu Y (2018) Codon usage biases co-evolve with transcription termination machinery to suppress premature cleavage and polyadenylation. Elife 7. 10.7554/eLife.33569 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 11755 kb)
(DOCX 8 kb)