

There is a Blood Commentary on this article in this issue.
Key Points
Low ABRs were maintained with long-term emicizumab prophylaxis; bleeding into target joints decreased substantially and no new ones formed.
Emicizumab had a favorable long-term safety profile, with no unexpected safety signals or treatment-related deaths observed.
Abstract
Prophylaxis with emicizumab, a subcutaneously administered bispecific humanized monoclonal antibody, promotes effective hemostasis in persons with hemophilia A (PwHAs). The primary efficacy, safety, and pharmacokinetics of emicizumab were reported previously, but long-term data were limited. Here, data from 401 pediatric and adult PwHAs with/without factor VIII (FVIII) inhibitors who were enrolled in the phase 3 HAVEN 1, HAVEN 2, HAVEN 3, and HAVEN 4 studies (NCT02622321, NCT02795767, NCT02847637, NCT03020160) have been pooled to establish a long-term efficacy, safety, and pharmacokinetics profile. Across a median efficacy period of 120.4 weeks (interquartile range, 89.0-164.4) (data cutoff 15 May 2020), the model-based treated annualized bleed rate (ABR) was 1.4 (95% confidence interval [CI], 1.1-1.7). ABRs declined and then stabilized at <1 in an analysis of 24-week treatment intervals; at weeks 121 to 144 (n = 170), the mean treated ABR was 0.7 (95% CI, 0-5.0). During weeks 121 to 144, 82.4% of participants had 0 treated bleeds, 97.6% had ≤3 treated bleeds, and 94.1% reported no treated target joint bleeds. Bleeding into target joints decreased substantially. Emicizumab was well tolerated, and no participant discontinued because of adverse events beyond the 5 previously described. This data cutoff includes the previously reported 3 thrombotic microangiopathies (one in the PwHA with fatal rectal hemorrhage) and 2 thromboembolic events, all associated with activated prothrombin complex concentrate use, as well as a myocardial infarction and a venous device occlusion. With 970.3 patient-years of exposure, emicizumab prophylaxis maintained low bleed rates in PwHAs of all ages with/without FVIII inhibitors and remains well tolerated, with no new safety concerns identified.
Visual Abstract

Introduction
Persons with hemophilia A (PwHAs) have a deficiency in coagulation factor VIII (FVIII), resulting in spontaneous and traumatic bleeding, most commonly into joints, muscles, and soft tissues; intracranial bleeding can be life-threatening.1,2 Without adequate prophylaxis, recurrent joint bleeding results in hemophilic arthropathy, which is the most serious long-term complication.3 FVIII prophylactic infusions have high treatment burden (typically requiring ≥2 time-consuming infusions every week and often hindered by poor venous access) and can contribute to reduced adherence.4 For 25% to 30% of previously untreated PwHAs with a severe phenotype, treatment is further complicated by the development of FVIII inhibitors, which render FVIII replacement therapies ineffective.5
Emicizumab improves hemostasis by bridging activated FIX and FX to substitute for the function of missing activated FVIII.6,7 It has been demonstrated to be effective for the prevention of bleeds in PwHAs with or without FVIII inhibitors when administered subcutaneously once weekly, once every 2 weeks, or once every 4 weeks.8-11 The efficacy and safety of emicizumab across a broad population of children and adults with hemophilia A, with or without FVIII inhibitors, have been demonstrated in the HAVEN clinical trials (HAVEN 1 [NCT02622321], HAVEN 2 [NCT02795767], HAVEN 3 [NCT02847637], HAVEN 4 [NCT03020160]).8-11 During the primary analyses of HAVEN 1-4 (supplemental Table 1, available on the Blood Web site), emicizumab prophylaxis resulted in marked reductions in annualized bleeding rates (ABRs) of treated bleeds, irrespective of FVIII inhibitor status, and the majority of participants (54% to 90%) receiving emicizumab in each study did not report any treated bleeds.8-11
Emicizumab was well tolerated in the HAVEN trials, with a favorable overall safety profile. The most common related adverse events (AEs) were injection-site reactions (ISRs; observed in 15% to 31% of participants across studies); no ISR required treatment modification.8-11 The development of antidrug antibodies (ADAs) with neutralizing potential was rare (<1%); 3 participants developed ADAs with neutralizing potential (HAVEN 1, n = 1; HAVEN 2, n = 2) and displayed a decline in pharmacokinetics (PK). Of these, 1 participant discontinued treatment because of a loss of efficacy.12 In HAVEN 1, 3 participants experienced thrombotic microangiopathies (TMAs), and 2 experienced thromboembolic events (TEs); all TMAs and TEs occurred in participants who received, on average, a cumulative dose >100 U/kg per 24 hours of activated prothrombin complex concentrate (aPCC) for ≥24 hours.8 One of these participants experienced a fatal rectal hemorrhage that was considered unrelated to emicizumab; the TMA was resolving at the time of death.13 At the time of primary analysis, no fatality, TMA, or TE was reported in HAVEN 2, HAVEN 3, or HAVEN 4.9-11
Because emicizumab is the first nonfactor therapy with a novel mechanism of action approved for prophylaxis in hemophilia A,14 documentation of its long-term safety and efficacy is especially important. Here, we report the long-term safety, efficacy, and PK of emicizumab in PwHAs across HAVEN 1-4. The cutoff date for the current analysis (15 May 2020) expands upon the original cutoff dates for the primary publications of HAVEN 1 (25 October 2016), HAVEN 2 (30 April 2018), HAVEN 3 (15 September 2017), and HAVEN 4 (15 December 2017).8-11
Methods
Study design, participants, and data collection
Pooled data from long-term follow-up of HAVEN 1-4 are used to provide an aggregated analysis of efficacy and safety. For all participants to remain on study to obtain long-term data, HAVEN 3 and HAVEN 4 studies were extended from their original anticipated completion dates of 30 September 2019 and 31 December 2019, respectively. All HAVEN studies are ongoing for participants who are unable to access emicizumab in their country.
HAVEN 1-4 are phase 3 multicenter open-label studies. Individual study designs, locations, inclusion and exclusion criteria, methods, and types of data collected have been reported previously8-11 and are briefly outlined in Figure 1.
Figure 1.
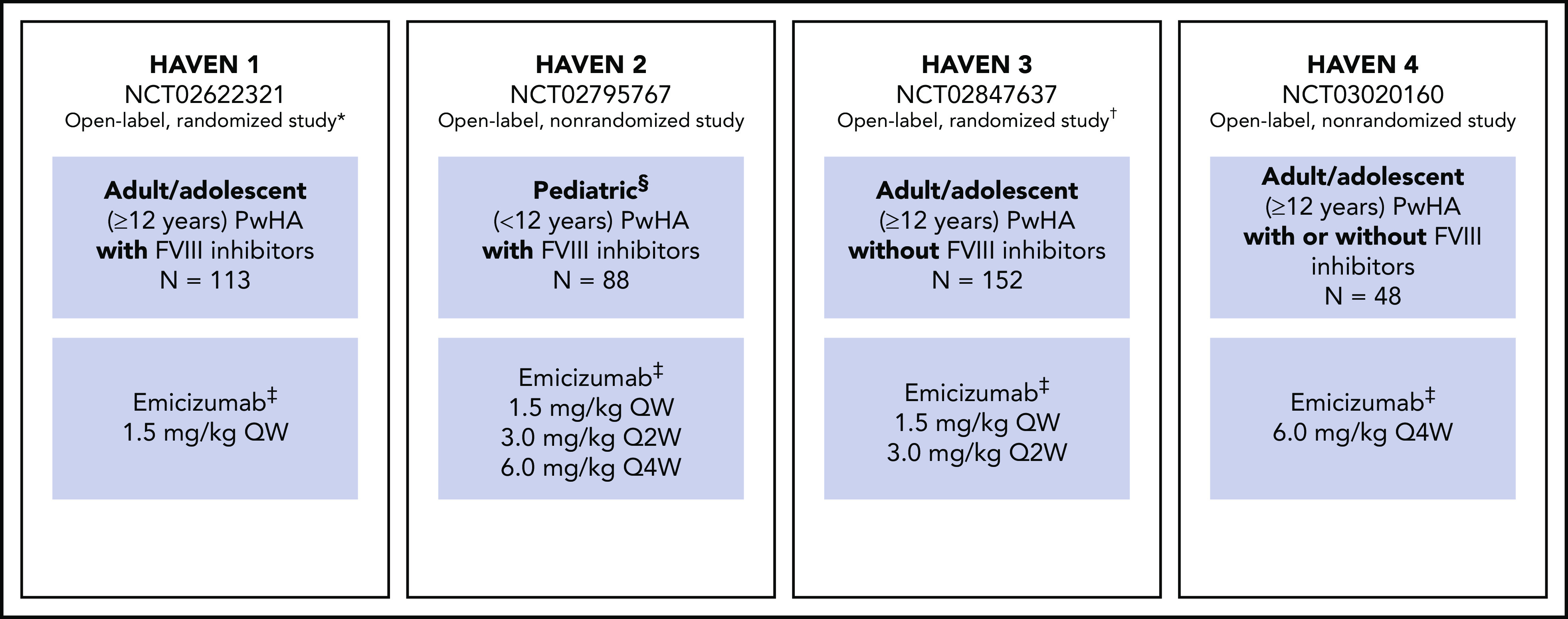
Study overview for HAVEN 1, HAVEN 2, HAVEN 3, and HAVEN 4.8-11 *Participants receiving episodic bypassing agents prior to study entry were randomized to emicizumab prophylaxis (arm A) or no emicizumab (arm B, control), and those receiving prophylactic bypassing agents prior to study entry received emicizumab prophylaxis (arm C). After completing the first 24 weeks of the trial, participants in the control arm (arm B) could receive emicizumab prophylaxis. A fourth arm also receiving emicizumab prophylaxis (arm D) consisted of participants enrolled after arms A through C closed. †Participants receiving episodic FVIII prior to study entry were randomized (2:2:1) to emicizumab once weekly (arm A), emicizumab every 2 weeks (arm B) or no prophylaxis (arm C, control), and those receiving prophylactic FVIII prior to study entry received emicizumab once weekly (arm D). ‡Maintenance doses. With the exception of the HAVEN 4 PK run-in cohort (n = 7), all maintenance doses were preceded by loading doses of 3.0 mg/kg once weekly for 4 weeks. §Adolescents aged 12 to 17 years were also eligible to enroll in HAVEN 2 if they weighed <40 kg; 3 participants were aged 12 to 17 years. QW, once weekly; Q2W, once every 2 weeks; Q4W, once every 4 weeks.
All adult participants provided written informed consent prior to study entry. For participants <18 years of age, informed consent was provided by a parent or legally authorized representative, along with informed assent in those ages 8 to 17 years. Each study protocol was approved by the relevant independent ethics committee/institutional review board at each participating institution, and the study was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice.15
Data were collected consistently across all 4 studies via the Bleed and Medication Questionnaire.
End points
End points across the HAVEN program were aligned during study conception,8-11 which allowed for pooling in this analysis. Efficacy end points included treated bleeds as the primary end point, and all bleeds, treated spontaneous bleeds, treated joint bleeds, and treated target joint bleeds were the secondary end points. The percentages of participants with 0 treated bleeds or 0 to 3 treated bleeds, as well as the proportion of resolved target joints, were also evaluated. Target joint resolution was defined as ≤2 spontaneous or traumatic bleeding events in 52 weeks in a joint previously defined as a target joint (eg, had ≥3 bleeds over 24 weeks; supplemental Methods).16
In addition, this pooled analysis evaluates the annualized infusion rate (AIR) for PwHAs without FVIII inhibitors, which included infusions of extended or standard half-life FVIII used to treat a bleed; this was also calculated for PwHAs with FVIII inhibitors taking aPCC and/or recombinant activated FVII (rFVIIa). Safety end points included the incidence of AEs, serious AEs (SAEs), TEs, and TMAs. PK end points included the mean trough concentration of emicizumab over time. Long-term immunogenicity (ie, development of ADAs with or without neutralizing potential) was evaluated but will be reported separately.12
Statistical analysis
In this descriptive analysis, data (beyond the primary analysis) from HAVEN 1-4 (data cutoff date: 15 May 2020) were pooled to establish a comprehensive efficacy and safety profile of emicizumab. Only data from participants with ≥1 postbaseline assessment were included in the analysis.
Treated bleeds, all bleeds, treated spontaneous bleeds, treated joint bleeds, and treated target joint bleeds are presented as medians, calculated mean ABRs, and model-based ABRs via a negative-binomial regression model, as in the primary analyses.8 In this long-term analysis, mean ABRs were calculated from data pooled across HAVEN 1-4 in discrete consecutive 24-week time intervals, whereas model-based ABRs were used for variable time periods (ie, when considering bleeds across the entire study period), because this method takes into account the varying time periods for each individual. For each 24-week interval, only participants exposed to emicizumab (through uptitration for those who were uptitrated) during the entire time interval were available for inclusion in the relevant ABR calculations. Each 24-week interval required available data from ≥10 participants for the calculation of ABR; data for time intervals with <10 participants are not reported. Confidence intervals (CIs) were obtained via exact Poisson and Clopper-Pearson methods, where indicated. AIR or annualized consumption was calculated by dividing the FVIII, aPCC, or rFVIIa amount administered within the time interval or efficacy period by the duration of that period in days, multiplied by 365.25. Participants who received FVIII prophylaxis prior to enrollment in HAVEN 3 (arm D) and HAVEN 4 were allowed to continue it for 1 week after the start of emicizumab. Therefore, their FVIII consumption data for the first week of emicizumab prophylaxis were excluded. Also, for participants randomized to the control arms in HAVEN 1 and HAVEN 3, only the data after initiation of prophylaxis were used in the analysis.
Consistent with the International Society on Thrombosis and Haemostasis definition, target joints were defined as major joints (eg, hip, elbow, wrist, shoulder, knee, and ankle) in which ≥3 bleeding events occurred over a 24-week period.16 Because assessment of target joint resolution is across ≥52 weeks, participants eligible for this analysis had to have received ≥52 weeks of emicizumab (through uptitration, if applicable).
The efficacy period for each individual started on the first day of emicizumab prophylaxis (at the beginning of the study for participants randomized to an active arm and later on for those randomized to a control arm). The efficacy period ended at the date of clinical cutoff, the day of study withdrawal, or the day before uptitration, whichever occurred first. For participants randomized to an active arm who withdrew before dosing, the efficacy period started and ended on the day of enrollment (ie, an efficacy period of 1 day).
Data analyses were conducted by study statisticians and clinical pharmacologists (employed by the sponsor) who vouch for the completeness and accuracy of the statistical analyses. Data were made available to all authors, who confirm adherence to the protocol and statistical analyses plans.
Results
Participant demographics
In total, 401 participants were enrolled in HAVEN 1-4 (Figure 2). Of these, 400 participants were included in the efficacy analyses; 1 participant in HAVEN 3, who was randomized to the no prophylaxis arm (arm C), was lost to follow-up, received no emicizumab treatment, and is not included. One additional participant in HAVEN 1 who was randomized to an active arm (arm A) discontinued prior to emicizumab treatment. This participant is included in the efficacy population (n = 400) but was excluded from the safety population (n = 399).8,10 To date, 244 participants have completed the studies, of whom 242 continue to receive emicizumab in an alternate setting (eg, commercial or posttrial access program.)
Figure 2.
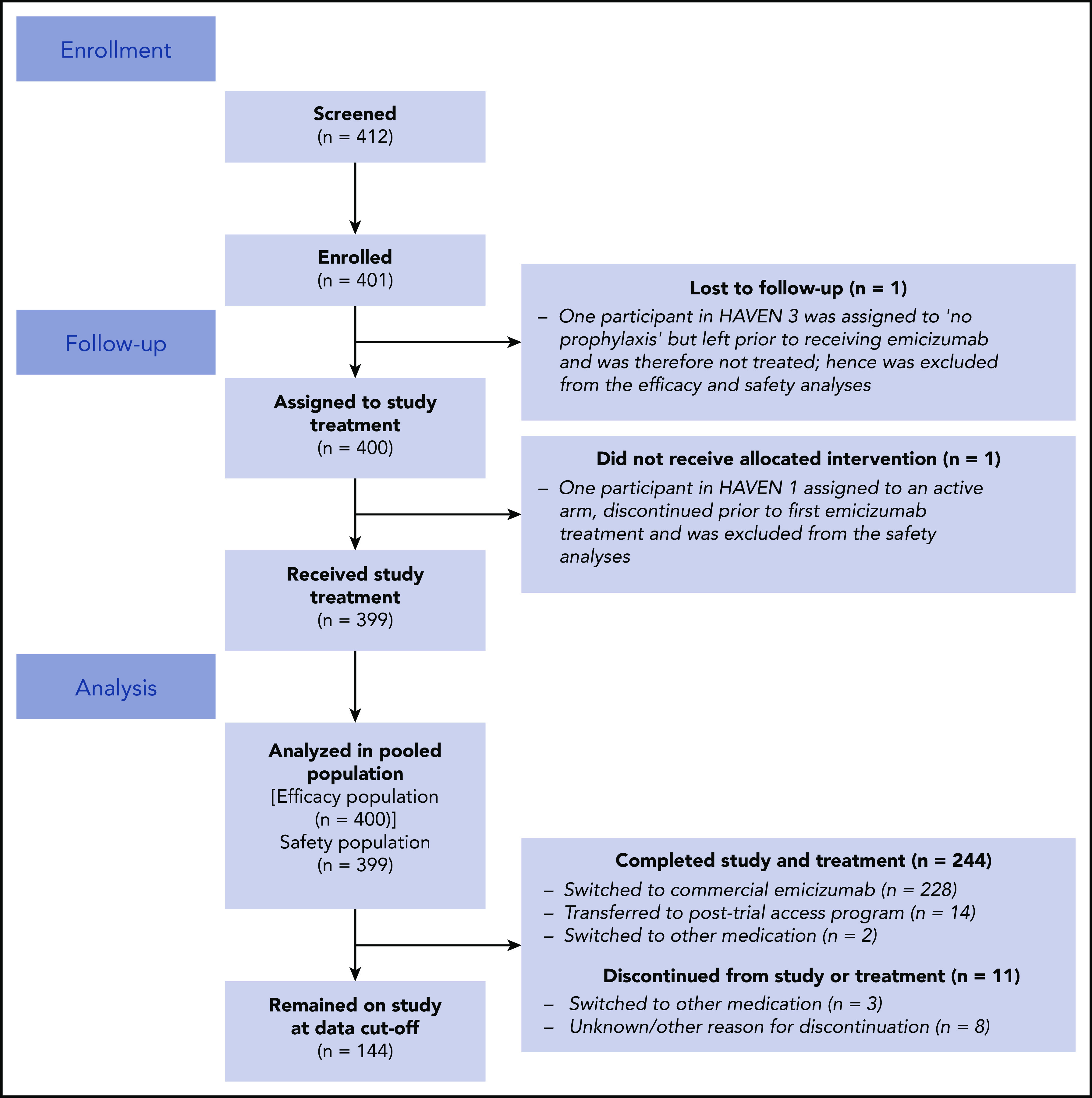
Participant disposition (total population).
Baseline demographics and clinical characteristics are described for the 401 enrolled participants (Table 1). The population includes adults (≥18 years; n = 269; 67.1%) and children and adolescents (n = 132; 32.9%). Also included are those with FVIII inhibitors (n = 209; 52.1%) and those without (n = 192; 47.9%).8-11 The median duration of the efficacy period was 120.4 weeks (interquartile range [IQR], 89.0-164.4), and a total of 970.3 patient-years of emicizumab exposure was included. Across HAVEN 1-4, 61.0% of participants had target joints before study entry, and there was a median of 8.0 (IQR, 5.0-15.0) bleeding events in the 24 weeks prior to study entry (Table 1).
Table 1.
Baseline demographics and disease characteristics across HAVEN 1-4 clinical trials
| HAVEN 1 | HAVEN 2 | HAVEN 3 | HAVEN 4 | Total | |
|---|---|---|---|---|---|
| Patients enrolled, n* | 113 | 88 | 152 | 48 | 401 |
| Participants in ITT group (efficacy population), n | 113 | 88 | 151† | 48 | 400 |
| Participants treated with emicizumab (safety population), n | 112‡ | 88 | 151† | 48 | 399 |
| Duration of exposure, median (IQR), wk | 109.3 (92.1-167.1) | 92.1 (68.3-124.4) | 163.4 (108.1-170.4) | 150.6 (84.4-153.0) | 120.4 (89.0-164.4) |
| Total patient-years of emicizumab exposure | 270.5 | 166.1 | 417.5 | 116.2 | 970.3 |
| Age | |||||
| Median (range), y | 29.0 (12-75) | 7.0 (1-15) | 38.0 (13-77) | 38.0 (14-68) | 28.0 (1-77) |
| <18 y, n (%) | 32 (28.3) | 88 (100) | 8 (5.3) | 4 (8.3) | 132 (32.90) |
| ≥65 y, n (%) | 5 (4.4) | 0 (0) | 5 (3.3) | 3 (6.3) | 13 (3.2) |
| Race, n (%) | |||||
| White | 75 (66.4) | 54 (61.4) | 102 (67.1) | 36 (75.0) | 267 (66.6) |
| Asian | 21 (18.6) | 13 (14.8) | 32 (21.0) | 10 (20.8) | 76 (19.0) |
| African American | 11 (9.7) | 12 (13.6) | 8 (5.3) | 1 (2.1) | 32 (8.0) |
| Other or unknown | 6 (5.3) | 9 (10.2) | 10 (6.6) | 1 (2.1) | 26 (6.4) |
| Previous treatment regimen, n (%) | |||||
| Episodic | 64 (56.6) | 22 (25.0) | 88 (58.3) | 18 (37.5) | 192 (48.0) |
| Prophylactic | 49 (43.4) | 66 (75.0) | 63 (41.7) | 30 (62.5) | 208 (52.0) |
| FVIII inhibitors at baseline, n (%) | |||||
| Yes | 113 (100) | 88 (100) | 0 (0) | 8 (16.7) | 209 (52.1) |
| No | 0 (0) | 0 (0) | 152 (100) | 40 (83.3) | 192 (47.9) |
| Previously underwent immune tolerance induction therapy, n (%) | 58 (51.3) | 63 (71.6) | 0 (0)§ | 6 (12.5) | 127 (31.7) |
| Bleeds in 24 wk prior to study entry, median (IQR), n | 10.0 (6.0-17.0) | 6.0 (3.5-9.0) | 9.0 (3.0-17.0) | 5.0 (2.0-10.5) | 8.0 (5.0-15.0) |
| Presence of target joints¶ at baseline, n (%) | 77 (68.8) | 34 (38.6) | 102 (67.1) | 31 (64.6) | 244 (61.0) |
ITT, intent-to-treat.
The demographics table is based on those who enrolled (N = 401). Participants included in the efficacy analysis only (N = 400).
One participant in HAVEN 3 assigned to no prophylaxis was lost to follow-up prior to the switch to emicizumab and, therefore, was not treated. This patient was excluded from the efficacy and safety analyses.
One participant in HAVEN 1 assigned to an active arm discontinued prior to first emicizumab treatment and was excluded from the safety analyses.
Historic immune tolerance induction therapy information was not recorded explicitly in the case report form for this noninhibitor study. However, 1 HAVEN 3 participant is known to have undergone immune tolerance induction therapy in 1987, because this information was listed as previous/concomitant medications for the participant.
In line with International Society on Thrombosis and Haemostasis definitions,16 target joints were defined as major joints (eg, hip, elbow, wrist, shoulder, knee, and ankle) in which ≥3 spontaneous bleeding events occurred over a 24-week treatment period.
Overall, 11 participants discontinued treatment: 4 because of AEs, 1 because of lack of efficacy alongside an SAE, and 6 because of other reasons (eg, lost to follow-up, personal reasons, physician decision). The 4 who discontinued because of AEs (alopecia/pruritus/skin necrosis, headache/lethargy, depressed mood/insomnia/nightmare, and superficial thrombophlebitis) were described in the primary analyses.8-11 The participant who discontinued because of lack of efficacy was also described previously; the SAE that contributed to withdrawal from treatment was presence of neutralizing ADAs.9 During the long-term follow-up period, no other participants withdrew because of AEs.
Efficacy
Compliance
Long-term compliance in reporting bleeds and medications was >90% across all 4 studies. Bleed and Medication Questionnaires were completed on 94.1% of 95 788 available days in HAVEN 1, 90.9% of 60 676 available days in HAVEN 2, 94.8% of 147 493 available days in HAVEN 3, and 93.4% of 40 830 available days in HAVEN 4.
Treated bleeds
Across HAVEN 1-4, the model-based ABR for treated bleeds over the entire study period was 1.4 (95% CI, 1.1-1.7). In all 4 studies, the calculated mean ABR for treated bleeds generally decreased over successive 24-week treatment intervals (Figure 3; supplemental Table 2). The median ABR for treated bleeds was maintained at 0 throughout the study period. Weeks 121 to 144 is the latest 24-week time interval with pooled data robustly representing HAVEN 1-4. During this period, 97.6% (n = 170) of participants taking emicizumab had ≤3 treated bleeds, with 82.4% of participants reporting 0 treated bleeding events (Figure 4; supplemental Table 2). ABRs for treated bleeds in PwHAs with FVIII inhibitors (HAVEN 1 and HAVEN 2), PwHAs without FVIII inhibitors (HAVEN 3), or a mixture of PwHAs with or without inhibitors (HAVEN 4) showed similar emicizumab efficacy, irrespective of FVIII inhibitor status (Figure 3). Although ABRs in HAVEN 4 appeared to be higher in later time intervals, in general, ABRs remained well <2, regardless of study and time interval, and the overall model-based ABR in HAVEN 4 (1.8 [negative binomial model-based ABR]; 95% CI, 1.2-2.9) is comparable with that in HAVEN 3 (1.2 [negative binomial model-based ABR]; 95% CI, 0.9-1.6) and HAVEN 1 (2.4 [negative binomial model-based ABR]; 95% CI, 1.5-3.9).
Figure 3.
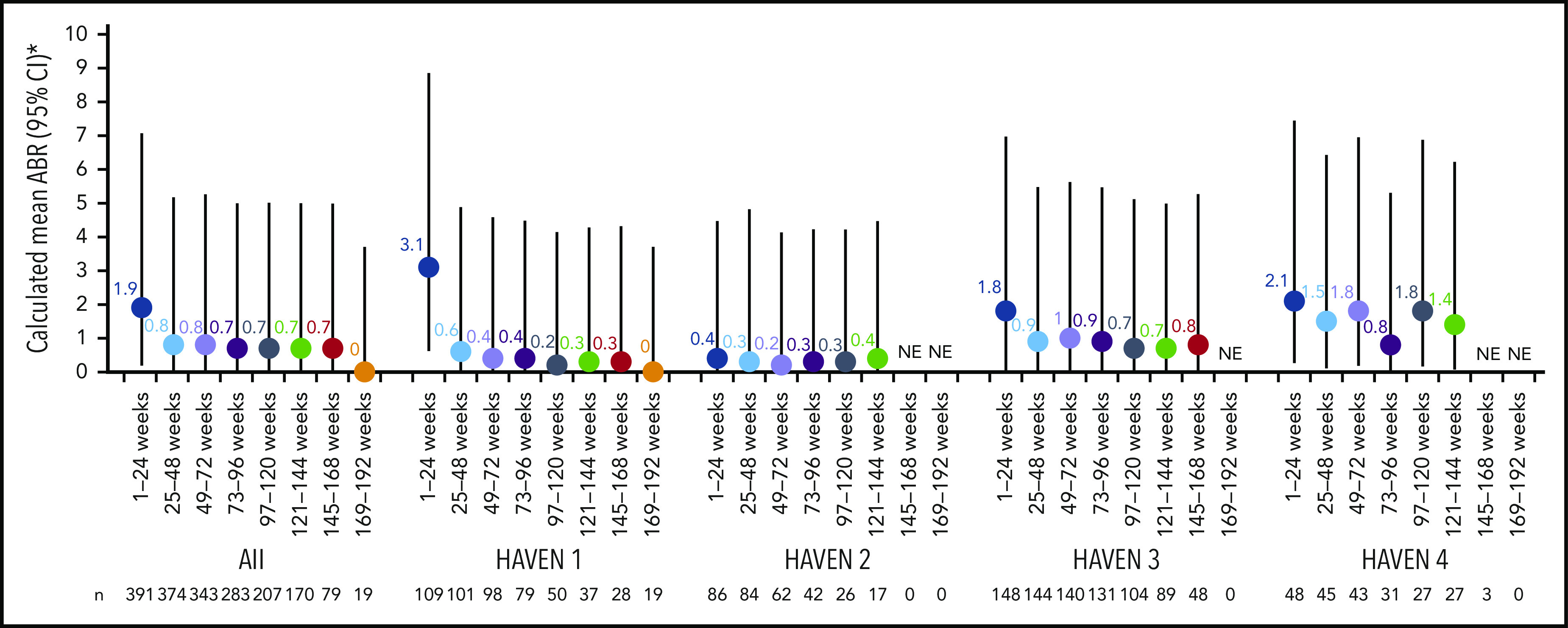
Pooled treated ABR and by HAVEN study across 24-week treatment intervals. 95% CI for the mean were calculated via the exact Poisson method. A treated bleed was defined as a bleed followed by treatment of a bleed; bleeds due to surgery/procedures were excluded. Only data for time intervals with ≥10 participants are reported; for inclusion within a time window, participants needed to have completed the entire 24-week interval (without uptitration). *Based on the calculated ABR for bleeds treated with coagulation factors. NE, not estimated.
Figure 4.
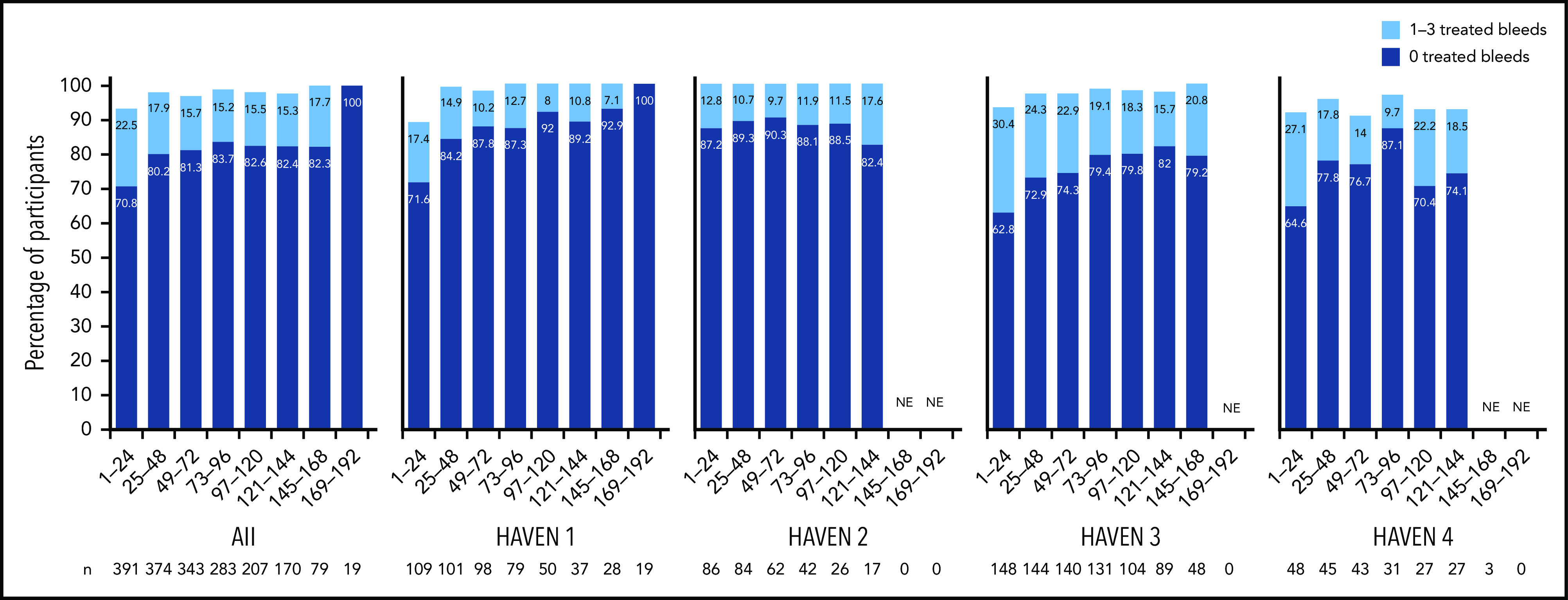
Proportion of participants with 0 or 1 to 3 treated bleeds by HAVEN study across 24-week treatment intervals. A treated bleed was defined as a bleed followed by treatment of a bleed; bleeds due to surgery/procedures were excluded. Only data for time intervals with ≥10 participants are reported; for inclusion within a time window, participants needed to have completed the entire 24-week interval (without uptitration). NE, not estimated.
All bleeds
All bleeds included untreated bleeds and those treated with coagulation factor products. Over the entire study duration, the model-based ABR for all bleeds across HAVEN 1-4 was 2.6 (negative binomial model-based ABR) (95% CI, 2.2-3.1). The calculated mean ABR for all bleeds decreased with each consecutive 24-week treatment interval (Table 2), and the calculated median ABR for all bleeds was 0 across all 24-week time intervals. The proportion of participants with 0 all bleeds increased over each treatment interval. At weeks 121 to 144, 74.1% of participants had 0 all bleeds, and 97.6% had ≤3 all bleeds.
Table 2.
ABRs and proportion of participants with 0 and 0 to 3 bleeds over consecutive 24-week intervals among participants of HAVEN 1-4 treated with emicizumab
| Weeks 1-24 (n = 391) | Weeks 25-48 (n = 374) | Weeks 49-72 (n = 343) | Weeks 73-96 (n = 283) | Weeks 97-120 (n = 207) | Weeks 121-144 (n = 170) | |
|---|---|---|---|---|---|---|
| Treated bleeds | ||||||
| ABR calculated, mean (95% CI) | 1.9 (0.2-7.1) | 0.8 (0-5.2) | 0.8 (0-5.2) | 0.7 (0-5.0) | 0.7 (0-5.0) | 0.7 (0-5.0) |
| Participants with 0 bleeds n, % (95% CI*) | 277, 70.8 (66.1-75.3) | 300, 80.2 (75.8-84.1) | 279, 81.3 (76.8-85.3) | 237, 83.7 (78.9-87.9) | 171, 82.6 (76.8-87.5) | 140, 82.4 (75.8-87.8) |
| Participants with 1-3 bleeds n, % (95% CI*) | 88, 22.5 (18.5–27.0) | 67, 17.9 (14.2-22.2) | 54, 15.7 (12.1-20.0) | 43, 15.2 (11.2-19.9) | 32, 15.5 (10.8-21.1) | 26, 15.3 (10.2-21.6) |
| Participants with 0-3 bleeds n, % (95% CI*) | 365, 93.4 (90.4-95.6) | 367, 98.1 (96.2-99.2) | 333, 97.1 (94.7-98.6) | 280, 98.9 (96.9-99.8) | 203, 98.1 (95.1-99.5) | 166, 97.6 (94.1-99.4) |
| All bleeds | ||||||
| Mean ABR calculated (95% CI) | 3.8 (1.0-9.9) | 2.0 (0.2-7.2) | 1.6 (0.1-6.6) | 1.3 (0.1-6.1) | 1.2 (0.1-5.9) | 1.0 (0-5.5) |
| Participants with 0 bleeds n, % (95% CI*) | 203, 51.9 (46.8–57.0) | 232, 62.0 (56.9-67.0) | 228, 66.5 (61.2-71.5) | 198, 70.0 (64.3-75.3) | 147, 71.0 (64.3-77.1) | 126, 74.1 (66.9-80.5) |
| Participants with 0-3 bleeds n, % (95% CI*) | 332, 84.9 (81.0–88.3) | 349, 93.3 (90.3-95.6) | 323, 94.2 (91.1-96.4) | 275, 97.2 (94.5-98.8) | 201, 97.1 (93.8-98.9) | 166, 97.6 (94.1-99.4) |
| Treated spontaneous bleeds | ||||||
| ABR calculated, mean (95% CI) | 0.8 (0-5.3) | 0.2 (0-4.2) | 0.2 (0-4.2) | 0.2 (0-4.0) | 0.2 (0-4.1) | 0.3 (0-4.2) |
| Participants with 0 bleeds n, % (95% CI*) | 329, 84.1 (80.1-87.6) | 346, 92.5 (89.4-95.0) | 314, 91.5 (88.1-94.3) | 272, 96.1 (93.2-98.0) | 192, 92.8 (88.3-95.9) | 156, 91.8 (86.6-95.4) |
| Participants with 0-3 bleeds n, % (95% CI*) | 383, 98.0 (96.0-99.1) | 372, 99.5 (98.1-99.9) | 343, 100 (98.9-100.0) | 282, 99.6 (98.1-100.0) | 207, 100 (98.2-100.0) | 169, 99.4 (96.8-100.0) |
| Treated joint bleeds | ||||||
| ABR calculated, mean (95% CI) | 1.4 (0.1-6.2) | 0.4 (0-4.6) | 0.5 (0-4.8) | 0.4 (0-4.5) | 0.5 (0-4.6) | 0.4 (0-4.5) |
| Participants with 0 bleeds n, % (95% CI*) | 304, 77.7 (73.3-81.8) | 328, 87.7 (83.9-90.9) | 301, 87.8 (83.8-91.0) | 251, 88.7 (84.4-92.1) | 184, 88.9 (83.8-92.8) | 153, 90.0 (84.5-94.1) |
| Participants with 0-3 bleeds n, % (95% CI*) | 372, 95.1 (92.5-97.1) | 371, 99.2 (97.7-99.8) | 338, 98.5 (96.6-99.5) | 280, 98.9 (96.9-99.8) | 204, 98.6 (95.8-99.7) | 167, 98.2 (94.9-99.6) |
| Treated target joint bleeds | ||||||
| ABR calculated, mean (95% CI) | 0.8 (0-5.2) | 0.3 (0-4.2) | 0.3 (0-4.4) | 0.3 (0-4.2) | 0.2 (0-4.2) | 0.2 (0-4.1) |
| Participants with 0 bleeds n, % (95% CI*) | 336, 85.9 (82.1-89.2) | 350, 93.6 (90.6-95.9) | 320, 93.3 (90.1-95.7) | 264, 93.3 (89.7-95.9) | 193, 93.2 (88.9-96.3) | 160, 94.1 (89.5-97.1) |
| Participants with 0-3 bleeds n, % (95% CI*) | 382, 97.7 (95.7-98.9) | 371, 99.2 (97.7-99.8) | 339, 98.8 (97.0-99.7) | 280, 98.9 (96.9-99.8) | 206, 99.5 (97.3-100.0) | 169, 99.4 (96.8-100.0) |
95% CIs were calculated using the Clopper-Pearson method.
95% CIs of proportion are indicated.
Treated spontaneous bleeds
Across HAVEN 1-4, the model-based ABR for treated spontaneous bleeds was 0.6 (negative binomial model-based ABR) (95% CI, 0.4-0.8) over the entire study period, and the calculated mean ABR showed an initial reduction that was maintained over time (Table 2). The calculated median ABR for treated spontaneous bleeds was 0 across all 24-week time intervals. Across all studies, the proportion of participants with 0 treated spontaneous bleeds increased at the start and was maintained at >91% in subsequent treatment intervals. At weeks 121 to 144, 91.8% of participants had 0 treated spontaneous bleeds, and 99.4% reported ≤3 treated spontaneous bleeds.
Treated joint bleeds
The model-based ABR for treated joint bleeds was 0.9 (negative binomial model-based ABR) (95% CI, 0.7-1.2) across all studies over the entire study period, and the mean calculated ABR for treated joint bleeds was 1.4 (95% CI, 0.1-6.2) from weeks 1 to 24, decreasing to 0.4 (95% CI, 0-4.5) by weeks 121 to 144 (Table 2). The calculated median ABR for treated joint bleeds was 0 across all 24-week time intervals. Across all studies, the proportion of participants with 0 treated joint bleeds reached 90.0% by weeks 121 to 144. A total of 98.2% of participants (167/170) at weeks 121 to 144 reported ≤3 treated joint bleeds.
Treated target joint bleeds
The model-based ABR for treated target joint bleeds was 0.5 (negative binomial model-based ABR) (95% CI, 0.4-0.7) across all studies over the entire study period, and the mean calculated ABR for treated target joint bleeds was 0.8 (95% CI, 0-5.2) from weeks 1 to 24, decreasing to 0.2 (95% CI, 0-4.1) by weeks 121 to 144 (Table 2). The calculated median ABR for treated target joint bleeds was 0 across all 24-week time intervals.
At baseline, 244 of 401 enrolled participants (61.0%) had target joints (Table 1). Of those evaluable (ie, with ≥52 weeks of emicizumab prophylaxis up to any uptitration), 202 of 226 (89.4%) participants did not have any spontaneous or traumatic bleeding into a target joint while receiving emicizumab (Table 3). Indeed, during weeks 121 to 144, 160 of 170 (94.1%) participants reported 0 treated target joint bleeds, and 169 of 170 (99.4%) had ≤3 treated target joint bleeds. At the clinical cut off, 95.1% of the 530 target joints observed at baseline in the evaluable population had resolved across all 4 studies (Table 3).
Table 3.
Target joint resolution with emicizumab prophylaxis by study
| HAVEN 1 (n = 113) | HAVEN 2 (n = 88) | HAVEN 3 (n = 151) | HAVEN 4 (n = 48) | Total (N = 400) | |
|---|---|---|---|---|---|
| Evaluable participants with target joints at baseline* | 68 (60.2) | 33 (37.5) | 96 (63.6) | 29 (60.4) | 226 (56.5) |
| Target joints at baseline among evaluable participants, n | 159 | 57 | 237 | 77 | 530 |
| Proportion of evaluable participants with no spontaneous or traumatic bleeds in target joints | 63 (92.6) | 32 (97.0) | 81 (84.4) | 26 (89.7) | 202 (89.4) |
| Target joints with 0 spontaneous or traumatic bleeds among target joints from evaluable participants | 154 (96.8) | 56 (98.2) | 218 (92.0) | 70 (90.9) | 498 (94.0) |
| Target joints resolved among target joints from evaluable participants† | 155 (97.5) | 56 (98.2) | 223 (94.1) | 70 (90.9) | 504 (95.1) |
| Target joints resolved among target joints from evaluable participants‡ | 156 (98.1) | 57 (100) | 234 (98.7) | 76 (98.7) | 524 (98.9) |
Unless otherwise stated, data are n (%).
Evaluable participants are those with ≥52 weeks of emicizumab prophylaxis (up to uptitration, if applicable). Of those, participants with target joints at baseline are shown.
Target joint resolution was defined as ≤2 spontaneous or traumatic bleeding events in a 52-week period in a joint previously defined as a target joint.
Alternative target joint resolution definition: ≤2 spontaneous bleeding events in a 52-week period in a joint previously defined as a target joint.
FVIII use
The mean AIR for FVIII treatment of bleeds among participants without FVIII inhibitors in the efficacy analysis was 3.7 (95% CI, 0.9-9.7). Although AIR fluctuated, a general and modest decline was seen over time, resulting in an AIR for treatment of bleeds of 2.5 (95% CI, 0.4-8.0) at weeks 121 to 144 (Table 4).
Table 4.
Pooled AIR and cumulative amount of FVIII in PwHAs without FVIII inhibitors across 24-week treatment intervals in the HAVEN 3 and 4 studies
| Treatment for bleed | Weeks 1-24 | Weeks 25-48 | Weeks 49-72 | Weeks 73-96 | Weeks 97-120 | Weeks 121-144 |
|---|---|---|---|---|---|---|
| Evaluable participants, n | 186 | 182 | 175 | 154 | 127 | 112 |
| FVIII AIR | 3.3 (0.8-9.2) | 3.7 (0.9-9.8) | 3.4 (0.8-9.4) | 2.1 (0.3-7.4) | 2.0 (0.2-7.2) | 2.5 (0.4-8.0) |
| FVIII, U/kg | 101.6 (82.8-123.4) | 107.6 (88.3-130.0) | 100.4 (81.7-122.1) | 62.9 (48.3-80.5) | 56.7 (42.9-73.5) | 71.5 (55.9-90.2) |
Unless otherwise noted, data are mean (95% CI).
A total of 191 PwHAs without FVIII inhibitors were included in the efficacy analysis, because 1 participant in HAVEN 3 assigned to no prophylaxis was lost to follow-up and not treated. This patient was excluded from the efficacy analysis.
Examination of FVIII annualized consumption in PwHAs without FVIII inhibitors revealed that the use of FVIII to treat bleeds generally decreased over time, from a mean of 101.6 (95% CI, 82.8-123.4) U/kg at weeks 1 to 24 to a mean of 71.5 (95% CI, 55.9-90.2) U/kg at weeks 121 to 144, with some fluctuations in between.
rFVIIa and aPCC use
The mean AIR for rFVIIa treatment of bleeds in PwHAs with FVIII inhibitors was 2.7 (95% CI, 0.5-8.4). Examination of rFVIIa annualized consumption per patient revealed that usage to treat bleeds tended to decrease over time from a mean of 657.9 μg/kg (95% CI, 608.5-710.1) at weeks 1 to 24 to 93.5 μg/kg (95% CI, 75.5-114.5) at weeks 121 to 144, with some fluctuations (Table 5).
Table 5.
Pooled annualized consumption of rFVIIa and aPCC in PwHAs with FVIII inhibitors across 24-week treatment intervals in the HAVEN 1, HAVEN 2, and HAVEN 4 studies
| Treatment for bleed | Weeks 1-24 | Weeks 25-48 | Weeks 49-72 | Weeks 73-96 | Weeks 97-120 | Weeks 121-44 |
|---|---|---|---|---|---|---|
| Evaluable participants, n | 203 | 192 | 167 | 125 | 79 | 57 |
| rFVIIa AIR | 5.1 (1.7-11.9) | 1.2 (0.1-5.9) | 2.0 (0.3-7.3) | 2.3 (0.4-7.8) | 2.2 (0.3-7.6) | 1.4 (0.1-6.3) |
| rFVIIa consumption, μg/kg | 657.9 (608.5-710.1) | 114.3 (94.3-137.3) | 202.2 (175.3-232.1) | 147.3 (124.5-173.1) | 171.4 (146.7-199.1) | 93.5 (75.5-114.5) |
| aPCC AIR | 1.2 (0.1-6.0) | 0.5 (0-4.7) | 0.1 (0-3.9) | 0 (0-3.8) | 0 (0-3.8) | 0* |
| aPCC consumption, U/kg | 87.8 (70.4-108.2) | 11.6 (5.9-20.5) | 2.9 (0.6-8.7) | 0.5 (0-4.7) | 0.3 (0-4.3) | 0* |
Unless otherwise noted, data are mean (95% CI).
95% CIs are not reported when all participants had a value of 0.
The mean AIR for aPCC used to treat bleeds in PwHAs with FVIII inhibitors was 0.4 (95% CI, 0-4.6), and it also generally decreased over time. Annualized mean consumption of aPCC per patient decreased considerably from 87.8 U/kg (95% CI, 70.4-108.2) during weeks 1 to 24 to 11.6 (95% CI, 5.9-20.5) U/kg during weeks 25 to 48, and then to 2.9 (95% CI, 0.6-8.7) U/kg during weeks 49 to 72, with no aPCC consumed during weeks 121 to 144 (Table 5). This reflects amendments to recommendations regarding the concomitant use of aPCC with emicizumab after TMA and TEs were noted in HAVEN 1; therefore, the drop in mean AIR for aPCC by a 24-week period cannot be interpreted as a measure of emicizumab efficacy. Of note, at the time of the release of these recommendations (17 October 2016), the median exposure of the 113 HAVEN 1 participants who were dosed with emicizumab was 20 weeks (IQR, 7-31). In addition, 20 participants were already enrolled in HAVEN 2 (median exposure, 12 weeks). In HAVEN 4, no participants with FVIII inhibitors were exposed at that time.
Safety
Overall, 381 of the 399 participants (95.5%) in the safety population reported ≥1 AE during a median exposure of 130.3 (range, 3.4-221.1) weeks (Table 6). The most common AE was nasopharyngitis (n = 126; 31.6%; supplemental Table 3). The most common treatment-related AE was ISR (n = 107; 26.8%); the majority of all ISRs (treatment related or not) were mild (104/111; 93.7%) and occurred during the first 24 weeks (93/111; 83.8%). The proportion of participants with ISRs declined over time to <1% of the total number in the safety population, and no participants discontinued emicizumab because of an ISR. The only other treatment-related AEs that occurred in ≥1% of participants were headache (n = 4; 1%) and indeterminable ABO blood type (n = 6; 1.5%).
Table 6.
AEs in participants treated with emicizumab across HAVEN 1-4 over a median exposure of 130.3 weeks (range, 3.4-221.1)
| Total (N = 399)* | |
|---|---|
| Participants with ≥1 AE | 381 (95.5) |
| AE with fatal outcome | 1 (0.3) |
| SAE | 93 (23.3) |
| AE leading to withdrawal from treatment | 5 (1.3) |
| AE leading to dose modification/interruption | 9 (2.3) |
| Grade ≥3 AE | 87 (21.8) |
| ISR† | 111 (27.8) |
| AEs of special interest | |
| Systemic hypersensitivity/anaphylactic/anaphylactoid reaction‡ | 1 (0.3) |
| TMA associated with concomitant aPCC and emicizumab | 3 (0.8) |
| Other TMA | 0 |
| TE associated with concomitant aPCC and emicizumab | 2 (0.5) |
| Other TE | 2 (0.5) |
All data are n (%). Multiple occurrences of the same AE in each participant are counted only once.
The safety population included only those participants who received emicizumab. This excludes 1 participant in HAVEN 1 who discontinued prior to emicizumab treatment and 1 additional participant from HAVEN 3 who was assigned to no prophylaxis and was lost to follow-up and not treated.
All ISRs, regardless of relatedness to treatment. The majority of local ISRs were mild (104/111; 93.7%).
Assessed using Sampson criteria and includes all participants who experienced indicative symptoms. One participant was identified through algorithmic analysis as potentially having a systemic hypersensitivity/anaphylactic/anaphylactoid reaction (he had experienced symptoms of abdominal pain and cough); however, medical review of the case showed that Sampson criteria were not met.
In total, 144 SAEs occurred in 93 participants. Bleeding-related SAEs that triggered hospitalization were reported in 32 participants and included the preferred term “hemorrhage,” “hemarthrosis,” “hematoma,” or “hematuria” (and variations thereof specifying location), according to investigator description. Six participants (1.5%) had treatment-related SAEs: cavernous sinus thrombosis, neutralizing antibodies, skin necrosis, and superficial thrombophlebitis (n = 1 each) and TMAs (n = 3). All 3 TMAs and 2 of 4 TEs were associated with concomitant aPCC use (Table 6), and all were reported previously in HAVEN 1.8 The 2 TEs not associated with concomitant aPCC use (device occlusion of a peripherally inserted central catheter and acute myocardial infarction [MI]) were reported on day 196 (HAVEN 1) and on day 1017 (HAVEN 3), respectively. Both were assessed as unrelated to emicizumab by the investigators, both resolved, and each individual continued emicizumab. The person with the nonserious device occlusion had a history of device-related thrombosis before receiving emicizumab. The person with MI was older than 65 years, had previously undiagnosed coronary artery disease, was treated for the event, and recovered with reduced heart function. No deaths were observed beyond the one caused by rectal hemorrhage that was described in the HAVEN 1 primary analysis.8,13
Pharmacokinetics
As reported in the primary analyses, mean emicizumab trough concentrations ∼50 µg/mL were achieved by week 5 following loading dosing across all studies. With maintenance doses of 1.5 mg/kg once weekly, 3 mg/kg once every 2 weeks, or 6 mg/kg once every 4 weeks, efficacious plasma trough concentrations >30 µg/mL were sustained for the duration of exposure (supplemental Figure 1). Although the mean plasma trough concentration with emicizumab, 6 mg/kg once every 4 weeks, remained below those observed with more frequent dosing regimens, 95% CIs generally overlapped, and bleed protection was sustained throughout the follow-up period.
Discussion
In the primary analyses of the HAVEN studies, emicizumab prophylaxis resulted in statistically significant and clinically meaningful reductions in ABRs across bleed-related end points, regardless of age, FVIII inhibitor status, or dosing regimen.8-10 In this pooled analysis of HAVEN 1-4, ABRs continued to decrease with ongoing emicizumab treatment and were maintained at <1 after 24 weeks of prophylaxis. During weeks 121 to 144, the vast majority (99.4%) of participants had ≤3 treated target joint bleeds, and 82.4% (140/170) of participants did not report any treated bleeds. Furthermore, the proportion of participants with ≤3 treated bleeds (spontaneous, joint, and all) within successive 24-week intervals approached 100% within 1 year from emicizumab initiation and was maintained for >2 years; this may reflect improved joint health achieved through the reduction in recurrent hemarthroses.17 Across all 4 studies, bleeding into target joints decreased substantially (Table 3), and 85.9% to 94.1% of participants did not have any target joint bleeds across 24-week time intervals up to 144 weeks (Table 2), suggesting improved bleed control and resolution of joint morbidity as observed in HAVEN 3.17 It is important to note that compliance in bleeds/medications reporting remained >90% throughout participation in the studies.
Across each 24-week treatment interval, the ABR for treated bleeds was similar among PwHAs with and without FVIII inhibitors (Figure 3). Likewise, ABRs in adolescents and adults in HAVEN 1 and HAVEN 3 stabilized around the level reported in pediatric participants in HAVEN 2 by the treatment interval containing weeks 73 to 96; this trend was seen as early as 24 weeks following initiation of emicizumab prophylaxis. ABRs in HAVEN 4, which evaluated a once-every-4-weeks regimen, were slightly higher across the 24-week treatment intervals compared with the other HAVEN studies (Figure 3). This may be due to an outlier with 18 bleeds (as previously reported)11 combined with the relatively small number of participants in the study. Indeed, smaller sample sizes are subject to variability. The overall conclusion from the primary analysis of HAVEN 4 was that emicizumab administered once every 4 weeks provided sustained clinically meaningful bleed prevention11; this is consistent with 2 other phase 3 studies (HAVEN 5 and HOHOEMI) evaluating the once-every-4-weeks regimen, with ABRs of 1.0 (95% CI, 0.5-1.8) and 0.7 (95% CI, 0.2-2.6), respectively.18,19
We provide exploratory observations regarding FVIII use, with the caveat that change in factor VIII consumption is dependent on the ways in which the data are analyzed. Our pooled analysis of nearly 200 participants without FVIII inhibitors suggests that annualized consumption of FVIII tended to decrease over time. Similarly, the consumption of rFVIIa also generally decreased over time. As expected, in line with recommendations on the concomitant use of aPCC with emicizumab, aPCC consumption was ∼0 for the latter half of this long-term analysis.
Emicizumab exhibited a consistently favorable long-term safety profile, with no unexpected or new safety signals. No fatalities or TMAs were reported across HAVEN 1-4, beyond those described in the HAVEN 1 primary analysis.8 Of 4 TEs reported, 2 were associated with concomitant aPCC use (cavernous sinus thrombosis and skin necrosis–superficial thrombophlebitis) during HAVEN 1. Of the 2 not associated with concomitant aPCC, device occlusion was reported in HAVEN 1 during weeks 25 to 48, and acute MI was reported in HAVEN 3 during weeks 145 to 168. Following the identification of TMA and TEs in association with the administration of high doses of aPCC during HAVEN 1, study protocols for HAVEN 1-4 were amended to recommend avoiding coadministration of emicizumab and aPCC.
The limitations of this analysis are the same as those outlined in the respective primary article for each study.8-11 Additional limitations include those associated with pooling of study data, which, for example, had slightly different definitions for joint bleeds. As noted in "Methods," each of the studies had different population characteristics (eg, age, FVIII inhibitor status) and, therefore, different joint disease. Also, there are limitations associated with long-term analyses, such as reporting fatigue and attrition resulting from participants switching to commercial product.
In summary, long-term safety and efficacy data for emicizumab are consistent with the findings of the primary analyses and indicate that PwHAs treated with emicizumab continue to experience improvements in the control of bleeding events and target joint resolution. Emicizumab continued to demonstrate a favorable safety profile, with no discontinuations due to AEs, and no deaths or TMAs beyond those reported in the primary analyses. This, coupled with the marked reduction in treatment burden afforded by subcutaneous administration once weekly, once every 2 weeks, or once every 4 weeks, makes emicizumab prophylaxis an efficacsious and well-tolerated option for improving the care of PwHAs.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
Medical writing assistance, under the direction of the authors, was provided by Sophie Nobes, Andrea Bothwell, and Rebecca A. Bachmann (Gardiner-Caldwell Communications) and funded by F. Hoffmann-La Roche Ltd. The HAVEN studies were funded by F. Hoffmann-La Roche Ltd and Chugai Pharmaceutical Co, Ltd.
Footnotes
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents see https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: All authors conceived and designed the study, analyzed and interpreted data, revised the manuscript critically, provided final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Conflict-of-interest disclosure: M.U.C. has received honoraria and consulting fees from Genentech, Inc/F. Hoffmann-La Roche Ltd, Takeda, Sanofi, Pfizer, Bayer, Global Blood Therapeutics, bluebird bio, uniQure, Spark Therapeutics, Inc, BioMarin, Kedrion, Grifols, and HEMA Biologics; has received speakers’ bureau fees from Genentech, Inc/F. Hoffmann-La Roche Ltd, Takeda, Bayer, Global Blood Therapeutics, BioMarin, and Novo Nordisk; has had travel expenses paid by Genentech, Inc/F. Hoffmann-La Roche Ltd, Takeda, Sanofi, Pfizer, Bayer, Global Blood Therapeutics, uniQure, Spark Therapeutics, Inc, BioMarin, Kedrion, and HEMA Biologics; has received expert testimony fees from Genentech, Inc/F. Hoffmann-La Roche Ltd; has received research funding from Pfizer; and has stock ownership in Alnylam Pharmaceuticals. C.N. has received consultancy fees from Bayer, BioMarin, CSL Behring, Freeline, LFB Biopharmaceuticals, Novo Nordisk, Octapharma, Pfizer, F. Hoffmann-La Roche Ltd, Sanofi, Shire/Takeda, Sobi, and Spark Therapeutics, Inc; has received research funding from CSL Behring, Octapharma, Shire/Takeda, and Sobi; as has had travel expenses and accommodations paid by CSL Behring, F. Hoffmann-La Roche Ltd, and Sobi. I.P.-P. is an employee of Genentech, Inc. T.C. is an employee of, and has stock ownership in, Genentech, Inc/F. Hoffmann-La Roche Ltd. S.C. and M.L. are employees of, and have stock ownership in, F. Hoffmann-La Roche Ltd. J.M. is president of the College of Pathologists and is an employee of the University of the Witwatersrand and National Health Laboratory Service and has received honoraria, consulting fees, speakers’ bureau fees, and research funding from CSL Behring, Catalyst Biosciences, Freeline Therapeutics, Novo Nordisk, F. Hoffmann-La Roche Ltd, Sanofi, Spark Therapeutics, Inc, and Takeda. G.Y. has received honoraria from BioMarin, Genentech, Inc/F. Hoffmann-La Roche Ltd, Grifols, Kedrion, Novo Nordisk, Sanofi Genzyme, Spark Therapeutics, Inc, Takeda; has acted as a consultant for and had travel expenses paid by BioMarin, Freeline, Genentech, Inc/F. Hoffmann-La Roche Ltd, Grifols, Kedrion, Novo Nordisk, Sanofi Genzyme, Spark Therapeutics, Inc, Takeda, and UniQure; has provided expert testimony for Bayer, CSL Behring, and Genentech, Inc; and has received research funding from Grifols, Takeda, and Genentech, Inc. R.K.-J. has received honoraria and consultancy fees from Takeda, Chugai Pharmaceutical Co, Genentech, Inc/F. Hoffmann-La Roche Ltd, CSL Behring, BioMarin, and CRISPR; has been a member of the speakers’ bureau for Genentech, Inc/F. Hoffmann-La Roche Ltd. and Sanofi; and has received research funding from Pfizer, CSL Behring, and Genentech, Inc. M.E.M. has acted as a consultant and is a member of the speakers’ bureau for Bayer, BioMarin, Catalyst, CSL Behring, Grifols, Kedrion, Novo Nordisk, Octapharma, Pfizer, F. Hoffmann-La Roche Ltd, Sobi, and Takeda. M.N. and N.S.B. are employees of F. Hoffmann-La Roche Ltd. M.H. is an employee of Roche Canada; has had travel expenses paid by, and holds stock ownership in, F. Hoffmann-La Roche Ltd; and was recently employed by PRO Unlimited. M.S. has received honoraria from Chugai Pharmaceutical Co, CSL Behring, Bayer, Sanofi, Novo Nordisk, Takeda, Pfizer, Sysmex, and KM Biologics; has received research funding from Chugai Pharmaceutical Co, Bayer, Novo Nordisk, Sanofi, KM Biologics, Asahi KASEI, Sysmex, and Takeda; and holds patents with Chugai Pharmaceutical Co and Sysmex. V.J.-Y. has received reimbursement for attending symposia/congresses and/or honoraria for speaking or consulting and/or funds for research from Takeda, Bayer, CSL Behring, Grifols, BioMarin, Novo Nordisk, Sobi, F. Hoffmann-La Roche Ltd, Octapharma, and Pfizer. C.S. and E.A. are employees of, and hold stock ownership in, F. Hoffmann-La Roche Ltd. G.G.L. is a previous employee of Genentech, Inc and a current employee of Spark Therapeutics, Inc (leadership role); reports stock ownership in F. Hoffmann-La Roche Ltd; and holds patents with Baxalta, Inc. S.W.P. is an employee of the University of Michigan and is Chair of the Medical and Scientific Advisory Council to the National Hemophilia Foundation; has acted as a consultant for ApcinteX, Bayer, BioMarin, Catalyst Biosciences, CSL Behring, HEMA Biologics, Freeline, Novo Nordisk, Pfizer, Genentech, Inc/F. Hoffmann-La Roche Ltd, Sangamo Therapeutics, Sanofi, Takeda, Spark Therapeutics, Inc, and uniQure; and has received research funding from Siemens. J.O. has received honoraria, consultancy fees, and speakers’ bureau fees from Bayer, Biogen Idec, Biotest, CSL Behring, Chugai, Grifols, Novo Nordisk, Octapharma, Pfizer, F. Hoffmann-La Roche Ltd, Takeda, and Swedish Orphan Biovitrum and has received research funding from Bayer, Biotest, CSL Behring, and Takeda.
The current affiliation for M.E.M. is Center for Thrombosis and Hemorrhagic Diseases, Humanitas Clinical and Research Center–Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Rozzano, Milan, Italy.
The current affiliation for E.A. is Finnish Health Authority, Helsinki, Finland.
The current affiliation for G.G.L. is Spark Therapeutics, Inc, Philadelphia, PA.
Correspondence: Michael U. Callaghan, Department of Pediatrics, Central Michigan University School of Medicine, 1937 Wakerobin Rd, Detroit, MI 48301; e-mail: michael.callaghan@cmich.edu.
REFERENCES
- 1.Mannucci PM, Tuddenham EG. The hemophilias–from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773-1779. [DOI] [PubMed] [Google Scholar]
- 2.Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Treatment Guidelines Working Group on Behalf of The World Federation Of Hemophilia. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1-e47. [DOI] [PubMed] [Google Scholar]
- 3.Knobe K, Berntorp E. Haemophilia and joint disease: pathophysiology, evaluation, and management. J Comorb. 2011;1(1):51-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thornburg CD, Duncan NA. Treatment adherence in hemophilia. Patient Prefer Adherence. 2017;11:1677-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arshad S, Singh A, Awasthi NP, Kumari S, Husain N. Clinicopathological parameters influencing inhibitor development in patients with hemophilia A receiving on-demand therapy. Ther Adv Hematol. 2018;9(8):213-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitazawa T, Igawa T, Sampei Z, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18(10):1570-1574. [DOI] [PubMed] [Google Scholar]
- 7.Shima M, Hanabusa H, Taki M, et al. Factor VIII-mimetic function of humanized bispecific antibody in hemophilia A. N Engl J Med. 2016;374(21):2044-2053. [DOI] [PubMed] [Google Scholar]
- 8.Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377(9):809-818. [DOI] [PubMed] [Google Scholar]
- 9.Young G, Liesner R, Chang T, et al. A multicenter, open-label phase 3 study of emicizumab prophylaxis in children with hemophilia A with inhibitors. Blood. 2019;134(24):2127-2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahlangu J, Oldenburg J, Paz-Priel I, et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N Engl J Med. 2018;379(9):811-822. [DOI] [PubMed] [Google Scholar]
- 11.Pipe SW, Shima M, Lehle M, et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open-label, non-randomised phase 3 study. Lancet Haematol. 2019;6(6):e295-e305. [DOI] [PubMed] [Google Scholar]
- 12.Paz-Priel I, Chang T, Asikanius E, et al. Immunogenicity of emicizumab in people with hemophilia A (PwHA): results from the HAVEN 1-4 studies [abstract]. Blood. 2018;132(suppl1). Abstract 633. [Google Scholar]
- 13.Khoo L, Matthews S, Kershaw G, et al. Case report of a fatal rectal haemorrhage in a person with severe haemophilia A receiving emicizumab and high-dose bypassing agents in the HAVEN 1 study. Haemophilia. 2020;26(6):e340-e342. [DOI] [PubMed] [Google Scholar]
- 14.Mannucci PM. Hemophilia therapy: the future has begun. Haematologica. 2020;105(3):545-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.World Medical Association . World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191-2194. [DOI] [PubMed] [Google Scholar]
- 16.Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A; Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Hemostasis . Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935-1939. [DOI] [PubMed] [Google Scholar]
- 17.Kiialainen A, Niggli M, Kempton C, et al. Bone and joint health markers in persons with hemophilia A (PwHA) treated with emicizumab in HAVEN 3 [abstract]. Blood. 2019;134(suppl 1). Abstract 626.31262783 [Google Scholar]
- 18.Wang S, Zhao X, Wang X, et al. A randomized, multicenter, open-label, Phase III clinical trial to evaluate the efficacy, safety, and pharmacokinetics of prophylactic emicizumab versus no prophylaxis in persons with hemophilia A in the Asia-Pacific region (HAVEN 5) [abstract]. Res Pract Thromb Haemost. 2020;4(suppl1). Abstract PB0957. [Google Scholar]
- 19.Shima M, Nogami K, Nagami S, et al. A multicentre, open-label study of emicizumab given every 2 or 4 weeks in children with severe haemophilia A without inhibitors. Haemophilia. 2019;25(6):979-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.