

Summary
Mesenchymal-derived osteoblasts play a key role in bone formation via synthesis and mineralization of the bone and bone remodeling. Osteoclasts are multinucleated cells of hematopoietic origin with a role in bone resorption. Here, we describe a protocol for generating primary cultures of these two cell types from bone tissue including the femur, tibia, and humerus of young mice. We describe methods for addressing their activity and/or differentiation, enabling studying the effects of various treatments during or following differentiation ex vivo.
For further practical example of using these protocols, please refer to Chevalier et al. (2020).
Subject areas: Cell Biology, Cell culture, Cell isolation, Metabolism, Cell Differentiation
Graphical Abstract
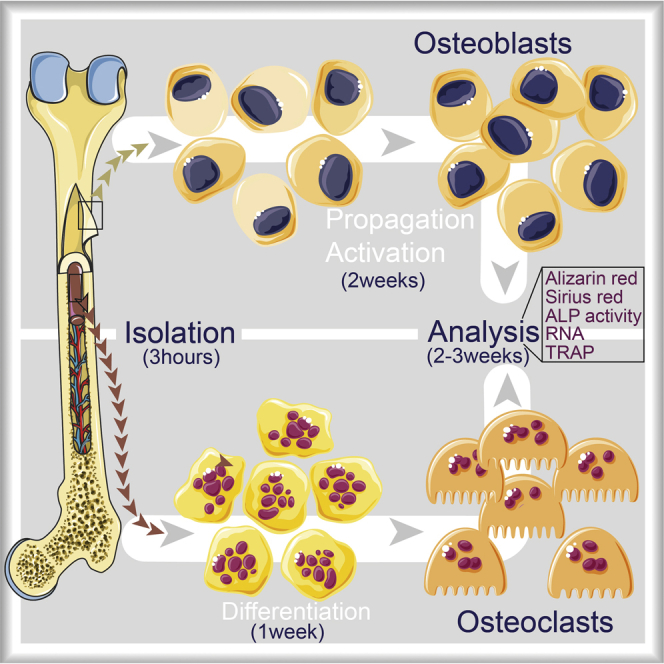
Highlights
-
•
Protocols for primary cell culture of osteoblasts and osteoclasts from young mice
-
•
Assess osteoblasts through ALP activity, calcium/collagen deposition, and RNA expression
-
•
Assessment of activity and differentiation of osteoclasts using TRAP and RNA expression
Mesenchymal-derived osteoblasts play a key role in bone formation via synthesis and mineralization of the bone and bone remodeling. Osteoclasts are multinucleated cells of hematopoietic origin with a role in bone resorption. Here, we describe a protocol for generating primary cultures of these two cell types from bone tissue including the femur, tibia, and humerus of young mice. We describe methods for addressing their activity and/or differentiation, enabling studying the effects of various treatments during or following differentiation ex vivo.
Before you begin
Experimental design
This protocol provides different experimental conditions and final assessments that might not all be required by the experimental design pertinent to the addressed question. Please refer to the flowchart (Figure 1) to define what experimental design will be suitable for your specific question. For a practical example of using these protocols, please refer to (Chevalier et al., 2020).
-
1.For osteoblasts, different validation tests are proposed in this protocol to assess different functions, each requiring a certain number of cells. These include:
-
a.RNA extraction (optimized for 6-well plate) (see step 30)
-
b.ALP (alkaline phosphatase) quantification (optimized for 6-well plate) (see step 31)
-
c.Mineralization production (Alizarin Red staining) (optimized for 12-well plate) (see step 32)
-
d.Collagen production (Sirius red staining) (optimized for 12-well plate) (see step 33)
-
a.
-
2.For osteoclasts, the treatment can be applied either during the differentiation process, or after the differentiation. Ultimately this protocol proposes various validation tests for osteoclasts that can assess the treatment effect in both conditions:
-
a.RNA isolation: optimized for 6-well plate (see step 34)
-
b.TRAP staining: optimized for 12-well plate (see step 35)
-
a.
-
3.
Please refer to each specific section (see step 23e for osteoblasts, step 29c for osteoclasts) to calculate the number of cells and plates required for the final design of your experimental setup. It is recommended that each treatment condition is performed at least in triplicates, where pooling of three animals is considered as one isolation, and then the triplicate of this is considered as a technical replicate. The full protocol should be repeated at least in three isolations, here named as “runs”.
Figure 1.

Flow chart with the experimental setup
General laboratory preparations
Timing: 30–60 min
-
4.
Prepare all stock solutions.
-
5.
Prepare media.
-
6.
Set water bath to 37°C.
-
7.
Warm media to 37°C.
-
8.
Sterilize all the tools (scissors, tweezers) for dissecting the mice and collecting bone by soaking them in 70% EtOH.
-
9.
All procedures are performed in a Class II biological hood with standard aseptic technique and primary cells are cultured in a humidified 37°C incubator with 5% CO2.
Note: none of the culture flasks or plates were pre-coated before experiment.
Key resource table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| L-Ascorbic acid-2-phosphate | Sigma-Aldrich | 49752-10G |
| Beta-glycerophosphate | Sigma-Aldrich | G9422-50G |
| Recombinant murine M-CSF | PeproTech | 31502 |
| Recombinant murine sRANK ligand | PeproTech | 315-11C |
| αMEM medium | BioConcept | 1-23P10-M |
| 100× Penicillin streptomycin | Gibco | 15140 |
| L-Glutamine 200 mM | Gibco | 25030 |
| MEM Amino Acids solution | BioConcept | 5-12K01-H |
| Sodium bicarbonate | Sigma-Aldrich | S5761-500G |
| Fetal bovine serum | Sigma-Aldrich | 12133C-1000ML |
| Collagenase I | Thermo Fisher Scientific | 17100-017 |
| Collagenase II | Sigma-Aldrich | C6885-1G |
| NaCl | Sigma-Aldrich | 71376-1KG |
| PBS without CaCl2 and Mg2 | Life Technologies | 10010-15 |
| Trizol | Thermo Fisher Scientific | 15596018 |
| p-Nitrophenylphosphate (4-nitrophenyl phosphate disodium salt hexahydrate, p-NPP) | Sigma-Aldrich | 71768-25G |
| 2-Amino-2-methyl-1-propanol | Sigma-Aldrich | A 9199 |
| MgCl2 | Sigma-Aldrich | M4880-100G |
| NaOH | Merck | 6498.1000 |
| Alizarin Red S | Sigma-Aldrich | A5533 |
| Hexadecyl pyridinium chloride | Sigma-Aldrich | C9002-100G |
| 37% Formaldehyde (Formol) | Sigma-Aldrich | 47608-0250 |
| Direct Red 80 | Sigma-Aldrich | P744 |
| Saturated picric acid | Sigma-Aldrich | P6744-1GA |
| HCl 37% | Acros Oganics | 124630010 |
| Fast Violet B Salt | ChemCruz | sc-215029B |
| Naphthol AS-TR phosphate disodium salt | Sigma-Aldrich | N6125 |
| Sodicum acetate | Sigma-Aldrich | A1045,1000 |
| Acetic acid glacial | Carlo Erba | 401391 |
| Sodium fluoride | Sigma-Aldrich | S7920-100G |
| Sirius red dye reagent: Direct red 80 | Sigma-Aldrich | P744 |
| Critical commercial assays | ||
| Pierce BCA Protein Assay Kit | Thermo Scientific | 23225 |
| Experimental models: organisms/strains | ||
| C57BL/6J mice | Janvier Labs | SC-C57J-M |
| Others | ||
| Cell incubator | Heraeus | HERAcell240 |
| Light microscope | Life Technologies | EVOS FL |
| Spectrophotometer plate reader | Molecular Devices | SpectraMax Paradigm |
| Sonicator | Branson Ultrasonics | Sonifier 250 |
| Centrifuge | Thermo Scientific | Multifuge3SR+ |
| Centrifuge | Eppendorf | 5427 R |
| Thermoblock | Eppendorf | ThermoMixer F1.5 |
| Orbital shaker | Heidolph | Rotamax120 |
| Magnetic stirrer | Heidolph | MR 2002 |
| 96-well cell culture plates | Corning | 353072 |
| 6-well cell culture plates | Corning | 353046 |
| 12-well cell culture plates | Corning | 353043 |
| 25 m2 Cell culture flasks | Thermo Fisher Scientific | 156367 |
| 75 m2 Cell culture flasks | Thermo Fisher Scientific | 156499 |
| Vacuum filter systems, 500 mL | Corning | CLS430769-12EA |
| 0.45 μm Pore filter | MillexGV | SLHV033RS |
| Disposable syringes (1 mL) | Becton Dickinson | 300300 |
| 25-gauge needle | Becton Dickinson | 300600 |
| Cell scraper S | Techno Plastic Products | 99002 |
Note: General laboratory consumables like serological pipettes (1 mL, 5 mL, 10 mL, 25 mL, 50 mL), p10, p20, p200 and p1000 pipet tips, aspirating pipettes, centrifuge tubes (15 mL, 50 mL) and Eppendorf tubes (1.5 mL and 2 mL), and haemocytometer (for cell counting) are also required.
Materials and equipment
| Reagent | Stock Concentration |
|---|---|
| L-ascorbic acid-2-phosphate | 50 mg/mL in 2 mL MilliQ water |
| Beta-Glycerophosphate | 1 M in 2 mL MilliQ water |
| Recombinant Murine M-CSF | 10 μg/mL in 1 mL MilliQ water |
| Recombinant Murine s-RANK Ligand | 100 μg/mL in 1 mL MilliQ water |
Growth and differentiation factor stock solutions
Sterilize L-ascorbic acid-2-phosphate and Beta-Glycerophosphate via filtering through 0.22 μm filter.
Storage conditions: The powder of the recombinant M-CSF and RANK ligand can be kept at −20°C until the expiration date or at 4°C for 6 months. All stock solutions can be stored at −20°C up to 3 months. However, the working dilutions from these stock solutions can only be kept at 4°C up to 1 week.
CRITICAL: Prepare sterile aliquots of stock solutions according to your experimental setting. Avoid repeated freeze-thaw cycles.
Basal medium preparation
| Reagent | Final Concentration | Amount in 500 mL Milli-Q water |
|---|---|---|
| α-MEM powder | 10 g/L | 5 g |
| Sodium bicarbonate (NaHCO3) | 2.2 g/L | 1.1 g |
| Penicillin Streptomycin solution | 1× | 5 mL (from 100× stock) |
| L-Glutamine | 2 mM | 5 mL (from 200 mM stock) |
| MEM amino acids solution | 0.375× | 3.75 mL (from 50× stock) |
| Fetal Bovine Serum (FBS) (heat-inactivated) | 10% | 50 mL |
| Milli-Q water | - | Up to 500 mL |
Mix all the ingredients and then filter through 0.22 μm pore medium filter.
Note: This medium can be stored up to 1 week at 4°C.
Preparation of digestion medium
Prepare adequate volume by dissolving collagenase II into basal medium at a final concentration of 1 mg/mL. Filter through 0.22 μm pore medium filter.
Note: This medium should be prepared fresh and used immediately.
Preparation of osteoblast stimulation medium
| Reagent | Final Concentration | Amount |
|---|---|---|
| L-ascorbic acid-2-phosphate (50 mg/mL stock) | 50 μg/mL | 0.25 mL (from 50 mg/mL stock |
| Beta-Glycerophosphate (1 M stock) | 10 mM | 2.5 mL (from 1 M stock) |
| Basal medium | - | 250 mL |
Note: Further diluted stock solutions may be used for easier pipetting of accurate volumes.
Storage conditions: Storage of this medium can be up to 1 week at 4°C.
Preparation of osteoclast differentiation medium
| Reagent | Final Concentration | Amount |
|---|---|---|
| M-CSF (10 μg/mL stock) | 30 ng/mL | 0.75 mL (from 10 μg/mL stock) |
| RANKL (100 μg/mL stock) | 100 ng/mL | 0.25 mL (from 100 μg/mL stock) |
| Basal Medium | - | 250 mL |
Note: Further diluted stock solutions may be used for easier pipetting of accurate volumes.
Storage conditions: Storage of this medium can be up to 1 week at 4 oC.
Tissue non-specific alkaline phosphatase activity measurement stock solutions
| Solution | Stock Concentration |
|---|---|
| p-nitrophenylphosphate(4 Nitrophenyl phosphate disodium salt hexahydrate, p-NPP) | 100 mM in 10 mL MilliQ water |
| 2-amino-2-methyl-1-propanol | 0.7 M in 1 L MilliQ water |
| MgCl2 | 100 mM in 500 mL MilliQ water |
| NaOH | 1 M in 500 mL MilliQ water |
Storage Conditions: Powder p-NPP can be stored at −20°C for longer periods. 2-amino-2-methyl-1-propanol solution should be stored at 4°C in a dark setting.
TRAP staining stock solutions
| Acetate-Tartrate buffer (pH:5.0) | Final Concentration | Amount |
|---|---|---|
| Sodium Acetate trihydrate | 19 mg/mL | 9.5 g |
| Glacial Acetic Acid (100%) | 0.45% | 2.25 mL |
| Sodium Tartrate trihydrate | 150 μg/mL | 75 mg |
| Milli-Q water | Up to 500 mL, adjust pH to 5 with NaOH |
| Sodium Fluoride solution | Final Concentration/Amount |
|---|---|
| Sodium Fluoride | 4.2 g/L in 250 mL MilliQ water |
Note: The solutions can be kept at 4°C for several months.
Step-by-step method details
Part 1: Collecting bones and bone marrow
-
1.
Prepare 12-well cell culture plate containing 1 mL basal medium (3 wells for each bone).
-
2.
Prepare 1.5 mL Eppendorf tube for each collected bone, filled with 1 mL of basal medium.
-
3.
Sacrifice 3-weeks-old male mice and immediately dip the whole body into 70% EtOH.
-
4.
Bring it to the primary cell culture hood and place it in a petri dish.
-
5.
Extract the femurs, tibias, and humeri with sterile tools and remove the skin, tendons, and muscles with a sterile gauze (Figures 2A–2E).
Note: The cleaned bones should not contain any residue of muscle, tendons, or skin.
-
6.
Put each cleaned bone in individual well of the 12 well plate.
-
7.
Repeat the process for each mouse.
Note: 3 mice would provide enough cells to plan an experiment with multiple conditions. Pooling 3 different animals increases reproducibility between experiments.
-
8.
Once all bones are collected and cleaned, cut the epiphyses of the bones with a small scissors.
-
9.
Flush the bone marrow several times with a 1 mL syringe with 25G needle in a 1.5-mL Eppendorf tube with 1 mL basal medium, until the bone becomes completely devoid of bone marrow (the bone becomes white) (Figures 2F and 2G).
Note: For each bone, use 1 mL of medium for collecting the bone marrow in different Eppendorf tubes.
-
10.
Place the bone back in a clean well of the 12-well plate containing basal medium and follow the osteoblast culture protocol (see Part 2).
-
11.
Keep the bone marrow-containing Eppendorf tubes for osteoclast culture protocol (see Part 3).
Figure 2.

Representative images of collecting bone and bone marrow
(A and B) Mouse dissection and collection of arm and leg.
(C and D) Removal of skin from leg (C) and arm (D).
(E) Cleaned bones after the removal of muscles and tendons.
(F and G) Collection of bone marrow (from tibia) after cutting the epiphyses. The collection of the bone marrow should be in 1.5 mL tube. Shown here is a microplate for visualization purpose.
Part 2: Osteoblast culture protocol
-
12.
Flush the bones at least four times with basal medium in the 12-well plate to clean the bone from other cells. The bone should look clear from any residual bone marrow or attached muscle cells. Move the bone to a new clean well filled with basal medium and flush one last time to remove any unwanted residual cells.
Note: Using several wells of the 12-well plate ensures sterility and decreases the risks of other cell type contamination. This is an important step to avoid contamination of other cell types in the osteoblast culture.
-
13.
Prepare Digestion medium freshly as described in material and equipment section.
-
14.
Cut each bone with sterile scissors into small pieces with a length of 1 mm and put them into a 25 cm2 flask containing 6 mL digestion medium. All the bones from one mouse can be pooled in one flask, leaving 3 flasks at the end of this step.
Note: Too big bone pieces might reduce the migration out of the osteoblasts.
-
15.
Incubate the flask for 90 min at 37°C with mild shaking in vertical position.
Note: Using a flask is required to ensure sterility of the preparation.
-
16.
Aspirate the digestion medium containing collagenase, keeping the bone pieces in the flask. Discard the medium containing unwanted digested cells.
-
17.
Rinse the flask with 10 mL basal medium and discard the medium, keeping the bone pieces in the flask. Repeat this step 2 times.
-
18.
Add 6 mL basal medium to the flask with bone pieces and incubate the cells to spread and proliferate at 37°C 5% CO2 for 3–4 days. The cells will migrate from the bone pieces to attach to the flask and start proliferating (Figure 3A).
-
19.
Check the flasks once to see if the cells spread from the bone particles (Figure 3A).
-
20.
After 3–4 days, change the medium if the cells have not exceeded 70%–80% confluence.
Note: The time to reach 70%–80% confluence might be variable but should not exceed 15 days.
-
21.
Once the cells reach 80% confluence, follow the passaging protocol below (see step 22).
Note: It is possible that the 3 flasks (issued from different animals) proliferate at different rates. If this is the case, wait for each flask to reach 70%–80% confluence and passage them independently.
-
22.Osteoblast passage protocol:Timing: 30 min –1 h for preparation of passaging preparation. See the timing in section for the whole experiment duration.
For the first passage: (Passage 1)
-
a.For a 25 cm2 flask, add 2 mL of collagenase I solution (1 mg/mL in 0.9% NaCl)Note: Ensure that the 2 mL covers the surface of the flask.CRITICAL: Always prepare fresh collagenase solution from powder and pass through a 0.22 μm filter to have a sterile preparation.
-
b.Incubate 8 min at 37°C.Note: Cells will not detach (or only partially) at this step. It will mainly help to weaken the extracellular matrix.
-
c.Add 5 mL basal medium to stop the digestion and collect the medium completely in a 50 mL falcon tube. Keep at 15°C–25°C until the next step.
-
d.To the 25 cm2 flask from which the medium has been taken out, add 2 mL trypsin and incubate 3 min at 37°C to detach the rest of the cells.
-
e.Check under the microscope to confirm that most of the cells are well detached form the flask and add 5 mL basal medium to stop the digestion. Flush the flask several times to detach the cells and collect the medium in a 50 mL falcon-tube that is used in step 22c.
-
f.Note: It is normal that some cells will stay attached to the flask (strongly), they are mostly dendritic cells and should not be taken.
-
g.Centrifuge at 178 × g for 8 min at 4°C.
-
h.During the centrifugation, use a 5 mL serological pipette with a cut tip to enlarge the aspirating end, and collect the remaining pieces of bones with 5 mL basal medium. Transfer it all to a new 75 cm2 flask.
-
i.At the end of the centrifugation, aspirate and discard the supernatant from the cells and resuspend them in 15 mL basal medium.
-
j.Transfer all the cells to the respective new 75 cm2 flasks. Be sure that the bone pieces are evenly spread through the flasks before incubating at 37°C.
-
k.Let the cells to grow (Figure 3B), until they reach 90% confluence. In the meantime, change the medium with basal medium every 3–4 days.
-
l.When cells reach 90% confluence (Figure 3C), proceed with new passage using the protocol from steps 22 m–22v.For passage 2 to 5:
-
m.For each 75 cm2 flasks, add 6 mL of collagenase I solution (1 mg/mL in 0.9% NaCl).Note: Ensure that the solution covers the entire surface of the flask.CRITICAL: Always prepare fresh collagenase solution from powder and pass through a 0.22 μm filter to have a sterile preparation.
-
n.Incubate 8 min at 37°C.Note: Cells will not detach (or only partially) at this step. It will mainly help to weaken the extracellular matrix.
-
o.Add 10 mL basal medium to stop the digestion and collect the medium in a 50 mL falcon tube. Keep at 15°C–25°C until the next step.
-
p.To the 75 cm2 flask from which the medium has been taken out, add 6 mL trypsin and incubate 3 min at 37°C.
-
q.Check under the microscope to confirm that >90% of the cells are well detached form the flask and add 10 mL basal medium to stop the digestion. Flush the flask several times to detach the cells and collect the medium in a 50 mL falcon-tube that is used in step 22o.Note: It is normal that some cells will stay attached to the flask (strongly), they are mostly dendritic cells and should not be taken.
-
r.Centrifuge at 178 × g or 8 min at 4°C. The 75 cm2 flasks containing the remaining bone pieces can be discarded.
-
s.At the end of the centrifugation, aspirate and discard the supernatant from the cells and resuspend them in 15 mL basal medium.
-
t.Count the cells and seed approximately 9 × 106 cells in 15 mL basal medium (6 × 105 cells/mL or 1.2 × 105/cm2) in a new 75 m2 culture flask (for each of the 3 original flasks)Note: This is an estimation that should be optimized in each experimental setting. Ideally cells should cover approximately 60% of the flask surface. This seeding is optimal to promote growth of the osteoblasts.
-
u.Let the cells to grow (Figure 3B), until they reach 90% confluence. In the meantime, change the medium with basal medium every 3–4 days.
-
v.When cells reach 90% confluence (Figure 3C), passage them again (repeat the process from steps 22 m–22v.
-
a.
Figure 3.

Spreading of osteoblasts
(A–D) Light microscopy images (10×) of:
(A) Osteoblasts spreading from the bone pieces (step 18).
(B) Osteoblasts spreading and proliferation after passage.
(C) Osteoblast with 90% confluence.
(D) Confluent Osteoblasts.
Scale is 400 μm.
Important: Osteoblasts will be ready to be used for experiments between 3 to 5 passages. The 3 passages will improve the purity of the osteoblasts culture from other cell types contamination. Once all three T75 flasks (from three different animals) reach between 70%–90% confluence after being passaged at least 3 times, they can be pooled, and the experimental design for treatments can start.
Note: In case of any bacterial, fungal or mycoplasma contamination, trash the flask immediately and continue with the remaining flasks only.
-
23.Seeding osteoblasts according to your experimental designCRITICAL: According to your experimental setup, estimate the cell number needed for a particular experiment (quantitative PCR, ALP measurement, mineralization, collagen production), for each condition (controls, treatments), and for replicates. Please check the flow chart to plan your experiment and check the number of cells required per well of each plate in step 23e.CRITICAL: Calculate the number of plates that will be needed in your experimental setting and freshly prepare the Osteoblast stimulation medium accordingly (see materials and equipment section).
-
a.Once all three T75 flasks reach approximately 90% confluence after being passaged at least 3 times, they can be pooled, and the experimental design can start.
-
b.At the end of the step 22 s, pool the three 15 mL basal media containing the cells of the 3 animals in one 50-mL tube.
-
c.Centrifuge 8 min at 178 × g at 4°C.
-
d.Resuspend in 10 mL of basal medium and count the live cells using Trypan blue.Note: The cell quantity can vary but 3 × 107 cells can be expected per isolation.
-
e.Seed the cells into different plates in accordance with the concentrations below (Figure 3A):For a 6-well plate: 1.4 × 105 cells/well in 2 mL medium (7 × 104cells/mL or 1.5 × 104cells/cm2)For a 12-well plate: 7 × 104 cells/well in 1 mL medium (7 × 104 cells/mL or 1.8 × 104cells/cm2)
-
f.Check your cells every day until they reach 100% confluence (Figure 3C).
-
g.As soon as 100% confluence is reached, change the medium to osteoblast stimulation medium and your treatment of interest.
-
h.Change the medium every 3–4 days with osteoblast stimulation medium and your treatment of interest.Note: If the half-life of your molecule is very short, you might consider changing the medium more often, but we would not recommend more than every 2 days as the cell stimulation might be impacted.
-
i.Stop the experiment and perform the different protocol according to their recommended time.Note: For characterization of osteoblasts, RNA collection (step 30) and alkaline phosphatase activity measurements (step 31) can be done after 7 days of culture.Note: Measurement of mineralization with Alizarin red staining (step 32) and measurement of collagen production with Sirius red staining (step 33) can be done after 14–21 days of culture (Figure 3D).
-
a.
Part 3: Osteoclast culture protocol
-
24.
After flushing the bone marrow from each bone in 1.5-mL Eppendorf tubes (see step 9), resuspend the bone marrow by gently flushing up and down through a needle (25G) several times. Avoid making bubbles.
-
25.
Pool all the bone marrow from one mouse (coming from tibia, femur, and humeri from the same mouse) and pass the cells through a 70-μm filter to collect them in a 50-mL falcon- tube.
-
26.
Put these cells in a 75 cm2 flask by topping the volume up to 15 mL with basal medium containing 10 ng/mL M-CFS (one flask per animal).
-
27.
Incubate for 24 h at 37°C in a 5% CO2 incubator.
-
28.
Next day, confirm there is no bacterial or fungal contamination in the flasks by checking the medium coloration and cell growth and collect the supernatant that contains the cells that were not adhering to the flask. These are typically the non- adherent hematopoietic progenitors. The supernatant of the flasks can be pooled in one 50 mL tube, and the flasks discarded. Proceed immediately with your experimental setup.
Note: Mycoplasma contamination is also possible if your cells still don`t grow. You can check the mycoplasma contamination using commercially available PCR kits.
-
29.
Seeding hematopoietic progenitors for osteoclast differentiation according to your experimental design
Important: According to your the experimental setup, estimate the cell number needed for a particular experiment (quantitative PCR, TRAP staining), for each condition (controls, treatments), and for replicates. Calculate the number of plates that will be needed in your experimental setting and prepare fresh basal medium with 30 ng/mL M-CFS. Please check flow chart to plan your experiment and check the number of cells required per well of each plate in step 29c.Note: Effects of a treatment can be analyzed during the differentiation time from hematopoietic progenitors to osteoclasts, or on mature osteoclasts, or both. Plan the number of plates accordingly.-
a.Centrifuge cells collected from the 3 animals at step 28 in 50 mL tube (8 min at 178 × g-4°C) and resuspend in 5 mL basal medium containing 30 ng/mL M-CFS.
-
b.Count the live cells using trypan blue.Note: The cell quantity can vary but up to 1.5 × 107 cells can be expected from isolation.
-
c.Seed the cells in accordance with the suggested concentrations below in basal medium containing 30 ng/mL M-CFS (Figure 4A).For a 6-well plate: 4 × 105 cells/well in 2 mL medium (2 × 105cells/mL or 4.2 × 104 cells/cm2).For a 12-well plate: 1 × 105 cells/well in 1 mL medium (2.6 × 104cells/cm2).
-
d.After 48 h, change the medium and initiate the differentiation using the osteoclast differentiation medium (See material and equipment section).Important: If you want to check the effect of treatment on osteoclast differentiation, start supplementing the differentiation medium with the appropriate treatment.
-
e.Check the cells every day to follow the differentiation process (Figures 4B–4D)CRITICAL: It is important to follow the differentiation process every day. Indeed, differentiated osteoclasts (big multinucleated cells) cannot be cultured very long, therefore after the differentiation happened, no more than 1 or 2 days of culture can be extended; otherwise, the cells will burst and die. (see Figure 3E as an example of this process).
-
f.Change the osteoclast differentiation medium every 4 days.Note: If the medium is changed more frequently, the differentiation may not occur.
-
g.If the effect of the treatment needs to be assessed on differentiated osteoclasts, wait until the osteoclasts are differentiated (big multinucleated cells), and add your treatment for 1 or 2 days before stopping the cell culture.CRITICAL: Be sure you started your treatment at the correct timing as the differentiated osteoclasts could die very fast. Therefore, after the differentiation happened, no more than 1 or 2 days of treatment can be applied. Stop the experiment and perform the different protocols to assess the osteoclasts differentiation.Note: For characterization of osteoclasts, RNA collection (see step 34) and TRAP staining (see step 35) can be performed.
-
a.
Figure 4.

Differentiation stages of osteoclasts
(A–E) Light Microscopy Images (10×) of:
(A) Myeloid progenitors before differentiation medium is added to the cells.
(B) Early osteoclast differentiation (2 to 4 days after differentiation medium started) indicated with white arrows.
(C) Intermediary osteoclast differentiation, cells are bigger and start to be multinucleated (5 to 7 days after differentiation medium is added).
(D) Full osteoclasts differentiation, cells contain numerous nucleus and are very big (7 to 10 days after differentiation medium added).
(E) Dead Osteoclast (burst), one day after (D).
Star indicates burst osteoclasts. Scale is 400 μm. Arrows indicate multinucleation of the osteoclasts.
Part 4: Analysis of osteoblasts
-
30.RNA Extraction (6-well plate)
-
a.Aspirate the medium from the cells when they are at day 7 of differentiation. Wash the cells with 2 mL cold PBS.Note: You could keep the medium supernatant by freezing it at −80°C for further analysis (see expected outcomes for possibilities).
-
b.Aspirate the PBS and add 1 mL of Trizol onto the wells under a chemical hood.Note: You can use different RNA isolation methods according to your preference.
-
c.Pipette up and down several times and collect all the cells in a 1.5 mL Eppendorf tube.
 Pause point: At this step, samples can be stored at −20°C for a week prior to the isolation.
Pause point: At this step, samples can be stored at −20°C for a week prior to the isolation. -
d.Proceed with RNA extraction as per the Trizol isolation protocol according to the manufacturer’s guidelines (https://assets.thermofisher.com/TFS-Assets/LSG/manuals/trizol_reagent.pdf).
-
a.
-
31.Tissue Non-Specific Alkaline Phosphatase (TNAP) Activity Measurement (6-well plate)
-
a.Prepare required materials:
-
i.Cell scraper
-
ii.Timer
-
iii.Sonicator
-
iv.Centrifuge
-
v.96-well plate
-
vi.Spectrophotometer
-
vii.Thermoblock set to 37°C
-
i.
-
b.Rinse cell cultures at day 7 of the protocol with 2 mL of sterile PBS under cell culture hood after aspirating the media.
-
c.Discard the PBS and scrape the cells in 0.5 mL Milli-Q H2O with a cell scraper.
-
d.Collect the 0.5 mL of scraped cells in water in Eppendorf tubes.Note: You can store these samples at −20°C.
-
e.Sonicate the samples three times with 5 seconds of pausing with 30% duty cycle (intermittent pulsing) and output control as "2" that adjusts the amplitude of power 20%.Note: Please check the key resources table about the sonicator used in this study and check the compatibility with the one you will use.Note: At this step, keeping samples on ice is important for avoid heating and deterioration of proteins.
-
f.Centrifuge at 11292 × g for 15 min at 4°C
-
g.Meanwhile, prepare the reactive solution for measurement and incubate at 37°C.10 mL of reactive solution preparation (pH=10.5):
-
i.1 mL of 100 mM p-NPP
-
ii.8 mL of 0.7 M 2-amino-2-methyl-1-propanol
-
iii.0.1 mL of 100 mM MgCl2
-
iv.0.9 mL of Milli-Q Water
-
i.
-
h.From the centrifuged sample transfer 100 μL of the supernatant to a 1.5 mL tube.
-
i.Take 200 μl of supernatant in a new collection tube and store at −20°C to measure the protein concentration later, which will be used for final calculation of the ALP activity.Note: Do not forget to collect the same amount of Milli-Q water as a blank from both steps 31h and 31i.
-
j.Incubate the tubes at 37°C for 10 min.
-
k.Add 900 μL of warm reactive solution to the 100 μL warm samples (37°C) and start a timer.
-
l.Wait for the color of the solution to turn yellow.Note: p-NPP (colorless) becomes yellow when hydrolyzed by ALP into para-nitrophenol.Note: This should take about 5 min, but incubation time depends on differentiation status of cell cultures.
-
m.When the solution is yellow, stop the reaction by adding 200 μL of NaOH 1N and stop the timer: Record the time.
-
n.Vortex samples briefly.
-
o.Transfer 200 μL from each tube in a 96-well plate.
-
p.Read the optical density (OD) at 405 nm with a spectrophotometer.Note: This is recommended to be performed in duplicates.
-
q.Measure the protein concentration from step 31i with a standard Bradford or BCA protocol (by putting 10 μL of protein in duplicates in 96-well plate and with generating a std curve 500 ng - 5 μg/mL).
-
r.The ALP activity can be calculated as follows:
-
a.
| ALP activity (Units/mg of proteins)) = [((ODs-ODb) × Rvol (mL)) / ((time(min) × ε (in mM) × Svol (mL))] / Sprotein (mg/mL). |
ODs = Average optical density at 405 nm of the sample
ODb = Average optical density at 405 nm of the blank
Rvol = Reaction volume = 1 mL
Svol = Sample volume = 0.1 mL
ε = Extinction coefficient: 18.3 mmol/L for p-nitrophenol at 405 nm
Time = time recorded in step 31m
Sprotein = protein concentration of the sample
-
32.Alizarin Red-S Staining for Measuring Mineralization (for 12-well plate)Note: After 14–21 days of differentiation, depending on the confluency, this staining can be done. Cells should not be overgrown and start detaching from the surface.Preparation of working solutions
Solution Working Concentration Alizarin Red-S 2% (w/v) in 50 mL MilliQ water Hexadecyl pyridinium chloride 10% (w/v) in 100 mL MilliQ water CRITICAL: Preparation of solutions and every step of this experiment should be done in a fume hood due to toxicity of the hexadecyl pyridinium chloride and the formol.Note: Hexadecyl pyridinium chloride will dissolve in water upon heating and agitation of the solution-
a.Rinse cell cultures with 2 mL of sterile PBS under cell culture hood.
-
b.Fix cells with 2 mL of 3.7% formol per well for 1–2 h at 15°C–25°C.Note: This step can also be done for 24 h at 4°C.
-
c.Discard formol and rinse with 2 mL of Milli-Q water per well.Pause point: Plates can be kept at 4°C for one week at this step by keeping cells in water.
-
d.Rinse cells with 2 mL of Milli-Q water per well.
-
e.Stain with 1 mL of 2% (w/v) Alizarin Red-S for a maximum of 1 h at 15°C–25°C under gentle agitation.
-
f.To remove excess unbound dye, wash the plate several times with Milli-Q water by filling the wells to the top and aspirating it. Repeat the wash until the water becomes clear. (Might require multiple washes.)Note: In this step, being gentle to the cells is important to prevent dissociation cells from the plate.
-
g.Let the plates dry under the hood or gently tap the plate to a tissue paper to dry it.Note: There can be some dye remnants in the wells of the plate, to remove them as much as possible, one can use cotton swabs to clean the dye from the sides of the plate.
-
h.Take pictures of the cells under the microscope (Figure 5). This can be either used as illustration, or to identify the regions of calcification. For quantitative analysis of the staining, see the next two steps.
-
i.Destain the cells with 2 mL of 10% (w/v) hexadecyl pyridinium chloride for 1 h at 15°C–25°C under gentle agitation. The solution becomes violet after this incubation.Note: Hexadecyl pyridinium chloride could form precipitates at 15°C–25°C, one can pre-heat the solution to dissolve the precipitates.
-
j.Collect 200 μL of each well in the 96-well plate and measure the absorbance at 550 nm with spectrophotometer.Note: Do not forget to add 200 μL hexadecyl pyridinium chloride as blank control.CRITICAL: Sample solution must be diluted with hexadecyl pyridinium chloride solution if sample absorption is superior to 1.2 value.
-
k.Alizarin Red staining quantification can be then estimated using the following formula:
Where Abs sample= Optical density of the sample measured at 550 nmAbs blank= Optical density of the blank measured at 550 nmAlizarin red staining (AU)= Abs sample – Abs blank Note: It is recommended to normalize the results with the control group.
-
a.
-
33.Sirius Red Staining for Collagen Deposition (12-well plate)Note: After 14–21 days of differentiation, depending on the confluency. This staining can be done at the same time together with Alizarin red staining.Preparation of working solutions:
Solution Working Concentration Formaldehyde (Formol) 3.7% in 100 mL MilliQ water Saturated aqueous picric acid pH:3.5 1.3% in 50 mL MilliQ water Direct Red 80 0.1% (w/v) in 50 mL Saturated aqueous picric acid HCl 0.01N in 250 mL MilliQ water NaOH 0.1N in 50 mL MilliQ water CRITICAL: Picric acid and HCl are toxic; work under a chemical hood.-
a.Prepare required material:
-
i.37°C incubator
-
ii.Orbital shaker
-
iii.Spectrophotometer plate reader
-
iv.96-well plate
-
i.
-
b.Rinse cell cultures with 2 mL of sterile PBS under cell culture hood after aspirating the media.
-
c.Fix cells with 2 mL of 3.7% formol per well for 1–2 h at 15°C–25°C.Note: This step can also be done for 24 h at 4°C.
-
d.Discard formol and rinse with 2 mL of Milli-Q water per well.Note: Plates can be kept at 4°C for 1 week at this step by keeping cells in water. Rinsing can be done also with deionized water.
-
e.Wash the plate 3 times with deionized water.
-
f.Dry the culture plates at 15°C–25°C.Note: Drying can be done either in 37°C incubator, or by placing the plates inversely on a tissue paper at 15°C–25°C.
-
g.Add 1 mL of Sirius red dye reagent per well for 1 h with low shaking at 15°C–25°C
-
h.Wash extensively with 0.01 N HCl to remove all unbound dye and aspirate the solution.Note: This step should be repeated as many times as needed to remove all the excess dye, while being careful not to detach the cells from sides of the plate when adding the hydrochloric acid solution.
-
i.Remove the excess of wash solution and let the plates dry under the hood, or gently tap the plate to a tissue paper to dry.
-
j.Take pictures of the cells under the microscope (Figure 6). This can either be used as illustration, or to identify the region of collagen deposition. For quantitative analysis of the staining, see next two steps.
-
k.Dissolve Sirius red the dye by adding 0.2 mL 0.1 N sodium hydroxide and with low shaking for 30 min at 15°C–25°C.
-
l.Collect 200 μL of each well in 96 well plate and measure the absorbance at 550 nm with spectrophotometer.Note: Do not forget to add 200 μL sodium hydroxide solution as blank control.CRITICAL: sample solution must be diluted with sodium hydroxide solution if sample absorption is superior to 1.2.
-
m.Sirius Red staining quantification can be then estimated using the following formula:
Where Abs sample= Optical density of the sample measured at 550 nmSirius red staining (AU)= Abs sample – Abs blank Abs blank= Optical density of the blank measured at 550 nmNote: It is recommended to normalize the results with the control group.
-
a.
Figure 5.

Alizarin red staining
Representative image of osteoblasts after Alizarin Red-S staining 17 days after seeding the cells visualized using light microscopy (10×, scale: 200 μm). Arrows indicate the mineralized plaque.
Figure 6.
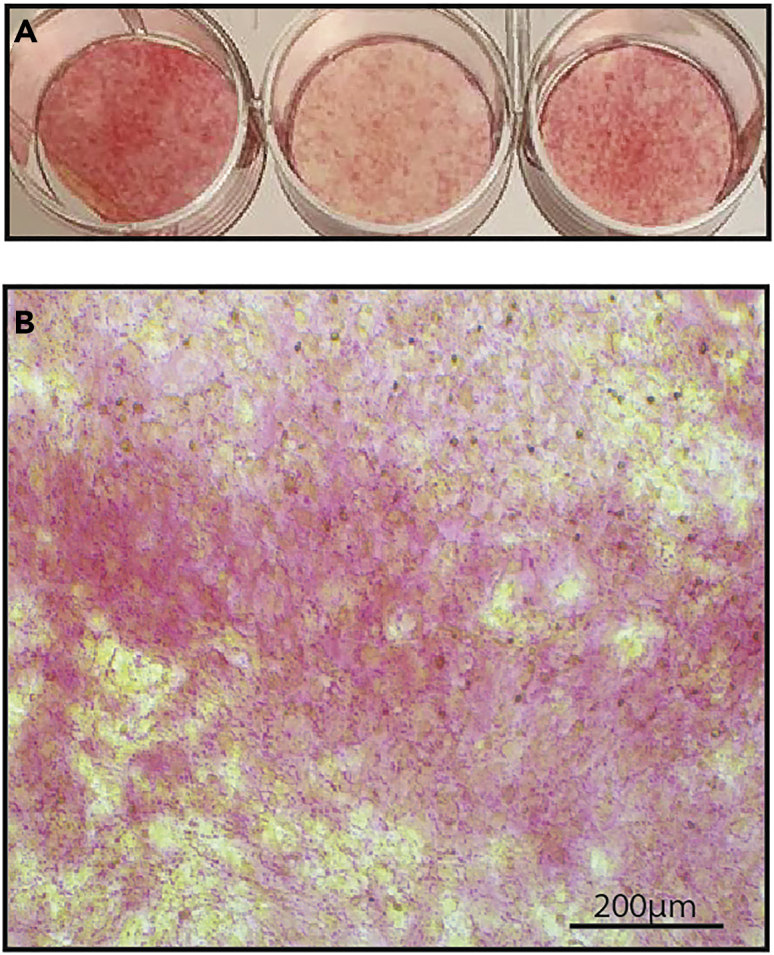
Sirius red staining
Representative image of osteoblasts after Sirius red 17 days after seeding the cells visualized using a standard camera (A) or under light microscope (B; 10×, Scale: 200 μm).
Part 5: Analysis of osteoclasts
-
34.
RNA Extraction (6-well plate):
This can be done as described in step 30.
-
35.
TRAP Staining (12-well plate)
Preparation of staining solutions, to be prepared fresh every time:Solution Working Concentration Fast violet B salt 7 mg/mL in 100 mL Acetate-Tartrate buffer Naphthol AS-TR phosphate disodium salt 2 mg/mL in 100 mL Acetate-Tartrate buffer Note: For Fast violet B dye, after dissolving let it agitate for 30 min. It is normal to still have few undissolved particles. Filter the solution through a 45-μm filter (prepare 1 mL in excess for the filtering volume). Before filtering the solution is orange and becomes yellow after filtration.Storage Conditions: Fast violet B powder should be kept at 4°C. Naphthol powder should be kept at −20°C.-
a.Prepare required materials:
-
i.Magnetic stirrer
-
ii.0.45 μm filter
-
i.
-
b.Fix and wash the cells as in the Alizarin red staining (Steps 32a–32d).
-
c.Mix fast violet solution and naphthol solution at a 1:1 ratio in a small beaker by pouring both solutions at the same time. Meanwhile, gently agitate with magnetic stirrer for approximately 1 min: small precipitates should appear.
-
d.Add 500 μl of this solution per well.Note: It is important that solution is homogenous when displayed in the different wells (including precipitates).
-
e.Incubate 14–16 h at 4°C.
-
f.Next day, discard the solution and wash with 2 mL water.
-
g.Add 500 μl of sodium fluoride solution and incubate for 30 min at 15°C–25°C.
-
h.Discard the solution and add 2 mL of water.
-
i.Proceed with imaging (Figure 7) of the plate and count the TRAP-positive cells (osteoclasts) by counting the multinucleated, violet stained cells.Note: Outcomes can be measured as number of differentiated osteoclasts per number of cells (in %) or the number of differentiated osteoclasts per surface area of the well.
-
a.
Figure 7.

Differentiated osteoclasts after TRAP
Representative image of differentiated osteoclasts after TRAP staining visualized using light microscopy (10×, Scale: 200 μm).
Expected outcomes
This is protocol for primary cell cultures of the 2 main cell types involved in the bone remodeling: osteoblasts and osteoclasts. Using this protocol, you can test your treatment of interest either during the differentiation process, or when the cells are fully differentiated.
Starting with the sacrifice of 3 mice, you can expect to have approximately 3 × 107 cells (from pooling of the 3 animals and after 3 passages) to start treatment for primary osteoblast and 1.5 × 107 cells(from pooling of 3 animals) to start treatment for primary osteoclast differentiation. Depending on the number of conditions you want to test, you should be able to analyze different outcomes (see the appropriate protocol to calculate the number of plates you can expect). Pooling 3 is with a goal of excluding the inter-mouse variability. Alternatively, using one mouse at a time can be considered (e.g., in case of transgenic mice), but that would require sailing down the amount of reagents and plate sizes, and would need to be additionally optimized.
As an example, we propose different tests that can be performed on the cultured cells to assess the differentiation capacity and activity of both osteoclasts and osteoblasts. However, many other technics exists that are not referenced here. As an example, you may want to investigate osteoclast apoptosis, and use a TUNEL assay procedure (Penolazzi et al., 2008, 2003), or the osteoclasts activity using a resorption pit assay (Vesprey and Yang, 2016).
For the osteoblasts, we exemplify assessing their activity by measuring RNA expression of some key markers. A list of targets can be found in Chevalier et al. 2020, and these include Collagen type 1 (Col1a), Osteopontin (Opn) or Osteocalcin (Ocn). The bone formation capacity can also be measured by the activity of the ALP which is critical for the bone mineralization process. We also describe analysis of the mineralization process by measuring the deposition of calcium in form of hydroxyapatite using the chelating property of Alizarin Red on calcium cations and its birefringent product. Finally, we describe analysis of the collagen deposition during the bone formation process using the chemical properties of Sirius red compound. Our isolation and differentiation protocols are suitable with other experimental setups that can be used and determined by the researchers to assess parameters pertinent to their experimental question.
For the osteoclasts, we describe assessing their activity and differentiation by measuring RNA expression of several key markers. The list of potential targets can be found in Chevalier et al. 2020, and these include Matrix metalloprotease 9 (Mmp9), Cathepsin K (Ctsk) and Acid phosphatase 5, tartrate resistant (Trap5b). We also provide a protocol for measuring the Tartrate-resistant acid phosphatase (TRAP), which is used as a marker to identify osteoclasts. Again, here we describe the basic analysis of the osteoclast differentiation, but the experimenters should define what setting would be appropriate to answer their experimental question.
The collected cell medium (e.g., before the RNA isolation), can be used for measuring several secreted proteins, e.g., osteocalcin as marker for osteoblasts activity.
Limitations
Here we propose a protocol that isolates osteoclasts on one side and osteoblast on the other. Although this is beneficial for assessing the individual aspect of each cell types, the strong interconnection and communication normally happening in bone tissue cannot be reflected here. Both activation of osteoclasts and osteoblasts in bone tissue are triggered by complex signaling cascade where both osteoblast and osteoclasts act on each other. Here we use a differentiating cocktail that simplifies this natural complexity.
This protocol cannot be used to assess structural alteration of the bone. It reflects the individual activity and differentiation of osteoclasts and osteoblasts, and should be used, analyzed, and interpreted as such.
As for any cell culture experiment, the use of different batches of Fetal Bovine Serum (FBS), can influence the cell growth and differentiation. It is therefore important to test different batches and select ones that will be maintained for the whole experimental process (Fang et al., 2017; Thermo Fisher, 2010; van der Valk et al., 2018).
Troubleshooting
There could be several pitfalls that one may envision, some of which being obvious and similar to other cell culture experiments, while other being specific for the bone-derived cultures and experiments. Since this protocol is aimed for researchers with different scientific backgrounds and training, below we outline possible solutions for both.
Problem 1
Fungi or bacterial contamination of the cell culture.
Potential solution
Before pooling cells from the 3 animals, it is important to verify under the microscope that no contamination occurred. If contamination is found once the animals have been pooled, the whole experiment should be discarded. As for many cell culture experiments, the contamination can be uncovered by a color changing of the medium, and confirmed under the microscope. The contamination can occur during the cell isolation step and should be noticeable within couple of days. If only one cell flasks (originating from one animal) is contaminated, it should be immediately discarded and the experiment can be continued with the 2 remaining ones. If more than one flask is contaminated, the experiment should be repeated and more caution would be needed during sterilization of the instruments, and strict adherence to the good practices of working under laminar flow sterile hood.
Problem 2
Uneven osteoblast growth on the flask (steps 18–22)
Potential solution
To avoid osteoblast overgrowth localized around the bone pieces, it is important to evenly spread the bone pieces in the flask and regularly gently shake the flask to circulate the bone pieces evenly in the flask. This should enable a more homogenous colonization of the flask and avoid local over-confluence that would be detrimental to the cells.
Problem 3
The osteoblasts from the three T75 flasks (originating from three animals) are not growing at the same speed, and the passaging cannot be performed at the same time (step 21). Note that the growing time of the culture can vary mainly depending on the time required for the preceding steps to isolate the cells, a longer isolation can increase cell death and consequently reduce the cell growth to the required confluence.
Potential solution
This phenomenon is not unexpected and different factors can influence the cell growth rate. If the isolation process time was not even between animals, and cells stay variable periods without sufficient nutrients during the isolation, the rates of the cell growth may differ. Also, the interindividual diversity between animals may interfere in the process. This is one of the main reasons why we recommend using 3 different animal that would be pooled together for the experimental process to minimize these initial differences.
Consequently, it may happen that some flasks will need one or 2 more passages in addition, until the remaining ones reach the 3 necessary passages. However, if more than 2 passages differences occur to have the 3 flasks ready, the higher passage line should be excluded. Indeed, comparing cells at different passages is not recommended as the cell differentiation might be affected differently. By pooling 3 different cells extraction from 3 different animals we minimize this effect and increase the reproducibility between the different runs, but the number of passages should be kept as similar as possible. If the 3 flasks show very different growth rates, and the above recommendations can’t be followed, you should consider repeating the whole experiment (run). If after 15 days the cells do not reach confluence, it is likely they will not reach the required confluency, and the experiment can be discarded.
Problem 4
Osteoclasts are not differentiating (Part 3)
Potential solution
It is possible that the osteoclasts do not differentiate and start dying instead. This may be due to an excessive change of the medium. The differentiation process needs several secreted factors that would be washed out by changing the medium too frequently. Please refer to the step 29f for the optimal time to change the medium. The FBS can also interfere in the differentiating process and using a different batch might cause problems. The growth hormones and the metabolites present in different batches of FBS can interfere in the differentiation, longevity, and metabolism of the osteoclast. Different concentration of FBS could be tested as well, as suggested by Xiong et al (Xiong et al., 2015). Finally, make sure that the differentiating molecules (M-CSF and RANKL) are fresh and at the correct concentration.
Problem 5
Osteoclasts burst (see Figures 4D and 4E)
Potential solution
The osteoclast differentiation is a very sensitive process. The differentiated osteoclasts survive in cell culture for a maximum of only 1 or 2 days. It is therefore important to check the cell culture every day to identify completion of differentiation (when cells get bigger and multinucleated as shown in Figure 4C) and stop the experiment immediately to analyze the outcome. If the cells had already started bursting, the experiment needs to be started over. Again the differentiating molecules (RANKL and M-CSF) can interfere with the differentiation speed, they should be added fresh and at the correct concentration to minimize this problem.
Problem 6
Alizarin red staining does not show chunks of staining in mineralized plaques as expected (step 32 h).
Potential solution
The full activity of the osteoblasts can be seen by the deposition of mineralized plaques and collagen (Alizarin red and Sirius red staining). These processes accumulate with time and it is necessary to maintain the cells in culture for minimum 14 days before being able to observe those depositions. Therefore, it is very important that the cells reach 100% confluence and are well spread across the well surface when the stimulation medium is added to potentiate the osteoblast differentiation. Please note that the number of cells given by the protocol is an estimation, and should be optimized in the experimenters’ own settings. It is also possible that the number of passages performed in the initial phase of the protocol was not sufficient (step 22). Those steps are important for the purity of the osteoblast culture and insufficient passages might increase chances of residual contamination from other cell populations. The mineralization is also dependent of the full differentiation of the osteoblast and the stimulation medium is an important factor that can affect this. Please make sure that the L-ascorbic acid-2-phosphate and Beta-Glycerophosphate are still fresh and at the correct concentration. Optimizing their concentration in the experimenter’s own setting might also be considered, having in mind that 10 mM Beta-Glycerophosphate, concentration that despite being commonly used, is higher that the physiological concentrations of approximately 4 mM. Finally, using 6-well plates instead of 12 well plates in order to reduce surface tension effect and improve the mineralization process might be considered.
Problem 7
TRAP staining does not work, osteoclasts cells appeared multinucleated, but do not appear violet (step 35i).
Potential solution
The step 35c of the protocol is critical for the effectiveness of the reaction. It is necessary that the 2 solutions are poured at the same time and rate under agitation. As soon as a small precipitate appears, the solution should immediately be applied to the cells. Freshly preparing this solution is critical for the reaction to be efficient.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mirko Trajkovski (Mirko.Trajkovski@unige.ch).
Materials availability
This study did not generate new unique reagents. All reagents are available as specified in the Key Resource Table.
Data and code availability
This study did not generate/analyze [data sets/code].
Acknowledgments
We thank all members of Ferrari and Trajkovski laboratories for discussions and advice. This work is part of a project that has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (ERC Consolidator Grant agreement No. 815962, Healthybiota) and from the Clayton Foundation for biomedical research.
Author contributions
C.C., M.Ç., J.B., and C.T. optimized the protocol and performed the experiments. N.B., S.F., and M.T. supervised the work. C.C., M.Ç., and M.T. wrote the manuscript with input from all authors.
Declaration of interests
The authors declare no competing interests.
References
- Chevalier C., Kieser S., Çolakoğlu M., Hadadi N., Brun J., Rigo D., Suárez-Zamorano N., Spiljar M., Fabbiano S., Busse B. Warmth prevents bone loss through the gut microbiota. Cell Metab. 2020;32:575–590.e7. doi: 10.1016/j.cmet.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C.-Y., Wu C.-C., Fang C.-L., Chen W.-Y., Chen C.-L. Long-term growth comparison studies of FBS and FBS alternatives in six head and neck cell lines. PLoS One. 2017;12 doi: 10.1371/journal.pone.0178960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penolazzi L., Lambertini E., Borgatti M., Piva R., Cozzani M., Giovannini I., Naccari R., Siciliani G., Gambari R. Decoy oligodeoxynucleotides targeting NF-kappaB transcription factors: induction of apoptosis in human primary osteoclasts. Biochem. Pharmacol. 2003;66:1189–1198. doi: 10.1016/s0006-2952(03)00470-2. [DOI] [PubMed] [Google Scholar]
- Penolazzi L., Lampronti I., Borgatti M., Khan M.T.H., Zennaro M., Piva R., Gambari R. Induction of apoptosis of human primary osteoclasts treated with extracts from the medicinal plant Emblica officinalis. BMC Complement. Altern. Med. 2008;8:59. doi: 10.1186/1472-6882-8-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Valk J., Bieback K., Buta C., Cochrane B., Dirks W.G., Fu J., Hickman J.J., Hohensee C., Kolar R., Liebsch M. Fetal bovine serum (FBS): past - present - future. ALTEX. 2018 doi: 10.14573/altex.1705101. [DOI] [PubMed] [Google Scholar]
- Vesprey A., Yang W. Pit assay to measure the bone resorptive activity of bone marrow-derived osteoclasts. Bio Protoc. 2016;6 doi: 10.21769/BioProtoc.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Q., Zhang L., Xin L., Gao Y., Peng Y., Tang P., Ge W. Proteomic study of different culture medium serum volume fractions on RANKL-dependent RAW264.7 cells differentiating into osteoclasts. Proteome Sci. 2015;13:16. doi: 10.1186/s12953-015-0073-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermo Fisher Hyclone. (2010) Growth comparison studies between FBS and other serum products [Online]. Available at: http://apps.thermoscientific.com/media/BID/BPP/appnotes/serumgrowthcomparison/growth-comparison-fbs-vs-other-serum.pdf. (Accessed 01 February 2021).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze [data sets/code].