

Abstract
Background:
Early-life wheezing-associated respiratory infection with human rhinovirus (RV) is associated with asthma development. RV infection of six day-old immature mice causes mucous metaplasia and airway hyperresponsiveness which is associated with the expansion of IL-13-producing type 2 innate lymphoid cells (ILC2s) and dependent on IL-25 and IL-33. We examined regulation of this asthma-like phenotype by IL-1β.
Methods:
Six day-old wild type or NRLP3−/− mice were inoculated with sham or RV-A1B. Selected mice were treated with IL-1 receptor antagonist (IL-1RA), anti-IL-1β or recombinant IL-1β.
Results:
RV infection induced Il25, Il33, Il4, Il5, Il13, muc5ac and gob5 mRNA expression, ILC2 expansion, mucus metaplasia and airway hyperresponsiveness. RV also induced lung mRNA and protein expression of pro-IL-1β and NLRP3 as well as cleavage of caspase-1 and pro-IL-1β, indicating inflammasome priming and activation. Lung macrophages were a major source of IL-1β. Inhibition of IL-1β signaling with IL-1RA, anti-IL-1β or NLRP3 KO increased RV-induced type 2 cytokine immune responses, ILC2 number and mucus metaplasia, while decreasing IL-17 mRNA expression. Treatment with IL-1β had the opposite effect, decreasing IL-25, IL-33 and mucous metaplasia while increasing IL-17 expression. IL-1β and IL-17 each suppressed Il25, Il33 and muc5ac mRNA expression in cultured airway epithelial cells. Finally, RV-infected 6 day-old mice showed reduced IL-1β mRNA and protein expression compared to mature mice.
Conclusion:
Macrophage IL-1β limits type 2 inflammation and mucous metaplasia following RV infection by suppressing epithelial cell innate cytokine expression. Reduced IL-1β production in immature animals provides a mechanism permitting asthma development after early-life viral infection.
Keywords: asthma, IL-17, IL-25, IL-33, NLRP3, type 2 innate lymphoid cell
Introduction
Early-life wheezing-associated respiratory infection with human rhinovirus (RV) has been associated with asthma development 1–5. We have shown that RV infection of six-day old immature mice causes the development of a chronic asthma-like mucous metaplasia phenotype which requires expansion of IL-13-producing ILC2s 6–9. ILC2 expansion is driven by the epithelial-derived innate cytokines IL-25 and IL-33 10.
RV infection induces IL-1β secretion in cultured bronchial epithelial cells 11,12 and peripheral blood mononuclear cells 13. Experimental human RV infection increases nasal IL-1β 14–16. Bioactive IL-1β is a consequence of inflammasome activation, produced by progressive proteolytic cleavage of procaspase-1 and pro-IL-1β 17. In cultured bronchial epithelial cells, RV infection induces activation of the nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing 3 (NLRP3) inflammasome 18,19. We recently found that, following acute RV infection of adult mice, NLRP3 inflammasome activation is required for maximal IL-1β production, airway inflammation and airway hyperresponsiveness in vivo 20. Toll like receptor 2 was required for inflammasome priming and viral RNA was required for inflammasome activation. We therefore examined the roles of NLRP3 and IL-1β, a key regulator of the innate immune response, in our immature mouse model of asthma development.
In general, IL-1β creates a pro-inflammatory milieu with the production of IL-6, IL-17 and chemokines which attract neutrophils to the airways. In cultured human airway epithelial cells, IL-1β is required for RV-induced expression of IL-6 and the neutrophil chemoattractants CXCL2, CXCL5 and CXCL8 12. IL-1β, especially in synergy with IL-23, plays an essential role in the induction or expansion of murine and human Th17 cells 21–23. In addition, IL-1β promotes differentiation and function of IL-17-producing type 3 innate lymphoid cells (ILC3s) 24–26. On the other hand, under certain conditions, IL-1β may promote type 2 eosinophilic inflammation. Intranasal treatment with IL-1β, in combination with endotoxin-free ovalbumin, induces allergic sensitization in naïve mice, in contrast to treatment with ovalbumin alone which has no effect 27. IL-1β−/− mice show reduced expression of neutrophil chemoattractants, the type 2 cytokine IL-33 and Muc5ac in response to successive house dust mite and dsRNA treatment 28. We found that IL-1β was required for RV-induced neutrophilic inflammation in naïve mice and eosinophilic inflammation in house dust mite-challenged mice.20 Finally, recent studies have shown that type 2 innate lymphoid cells (ILC2s) cultured in the presence of IL-1β increase IL-5 and IL-13 production as well as mRNA expression of Il17rb and Il1rl1, which encode subunits of the IL-25 and IL-33 receptors, respectively 29,30. Based on the stimulation of type 2 cytokine production from ILC2s in vitro 29,30, we hypothesized that, in immature mice with ILC2-dependent mucous metaplasia, IL-1β is required for maximum RV-induced ILC2 expansion and development of the persistent asthma-like phenotype.
Material and Methods
Ethics statement.
Mouse work was approved by the University of Michigan Animal Care and Use Committee, protocol #PRO00006118, and performed in according to the 2011 Guide for the Care and Use of Laboratory Animals.
RV infection of mice.
RV-A1B (ATCC, Manassas, VA) was partially purified from infected HeLa cell lysates by ultrafiltration using a 100 kD cut-off filter 31,32 and titered by plaque assay 33. Similarly concentrated and purified HeLa cell lysates were used for sham infection. Six day-old C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME), NLRP3−/− and IL-1β−/− mice34, male or female, were inoculated through intranasal route under Forane anesthesia with RV-A1B (2 × 106 PFU per mouse) or sham HeLa cell lysates. Selected mice were treated with 1 ng or 10 ng of recombinant mouse IL-1β (R&D Systems, Minneapolis, MN) intranasally, or 1 or 2 μg/g body weight of human recombinant IL-1 receptor antagonist (IL-1RA, PeproTech, Rocky Hill, NJ) intraperitoneally one hour before RV infection, followed by a half dose of IL-1β or IL-1RA on day 1. IL-1RA is effective against mouse IL-1 receptor. Additional mice were treated with either 1 μg/g body weight of neutralizing antibody to IL-1β, IL-1α (R&D Systems, Minneapolis, MN) or isotype control (polyclonal goat IgG, R&D Systems) intraperitoneally 1 h prior to RV infection. (The same concentration of neutralizing antibody was sufficient to block RV-induced airway inflammation in adult mice20.) Lungs were harvested 1, 7, or 21 days after infection for analysis.
Histology and immunofluorescence microscopy.
Three weeks after RV infection, lungs were perfused through the pulmonary artery with phosphate-buffered saline containing 5 mM EDTA. Next, lungs were inflated and fixed with 4% paraformaldehyde overnight. Five-micrometer–thick paraffin sections and processed for histology or fluorescence microscopy as described 35. Lung sections were stained with periodic acid–Schiff (PAS) (Sigma-Aldrich, St Louis, MO) or Alexa Fluor 488–conjugated anti-Muc5ac at 1 μg/mL (Thermo Fisher Scientific, Rockford, IL) to visualize mucus. For IL-25 and IL-33 staining, lung sections were harvested two days post-RV infection and stained with Alexa Fluor 488–conjugated rabbit anti-mouse IL-25/IL-17E (Millipore, Billerica, MA), Alexa Fluor 555-congjugated goat anti-mouse IL-33 (R&D Systems), and Alexa Fluor 555-congjugated rabbit anti-enterovirus D68 VP3 (interacting with RVA1B VP3; GeneTex, Irvine, CA). Levels of PAS, Muc5ac, IL-25 or IL-33 staining in the airway epithelium were quantified by NIH ImageJ software (Bethesda, MD). PAS, Muc5ac expression was represented as the fraction of PAS+ or Muc5ac+ epithelium compared with the total basement membrane length. One section from each of four lungs per group was analyzed. Sections contained 6–26 individual airways (average, 14). Lung sections were also incubated with Alexa Fluor 488-conjugated anti-mouse IL-1β (R&D Systems), Alexa Fluor 488-conjugated anti-mouse NLRP3 (Cell Signaling Technology, Danvers, MA) and Alexa Fluor 647-conjugated anti-mouse F4/80 (Biolegend, San Diego, CA).
Macrophage depletion.
Depletion of alveolar macrophages was accomplished by intranasal administration of liposomes containing clodronate (dichloromethylenediphosphonic acid, disodium salt, Millipore Sigma, Burlington, MA), as previously described 35. PBS-containing liposomes were used for control experiments. Liposomes were kept at 4°C under N2 until use. Depletion was performed 24 h before sham or RV infection by introducing 50 μl of clodronate- or PBS-containing liposomes intranasally under Forane anesthesia.
Flow cytometric analysis.
Lungs from sham- and RV-treated immature C57BL/6J or IL-1β−/− mice were harvested one or seven days post-infection, perfused with PBS containing EDTA, minced, and digested in collagenase IV. Cells were filtered and washed with RBC lysis buffer, and dead cells were stained with PacBlue (Thermo Fisher Scientific). To identify the cellular source of IL-1β, lung cells were harvested one day post-infection and stained with fluorescent-tagged anti-CD45, anti-F4/80, and anti-CD11b (all from BioLegend). Cells were subsequently treated with permeabilization buffer (eBioscience) and stained with anti-IL-1β (eBioscience). To identify ILC2s, cells were then stained with fluorescent-tagged antibodies for lineage markers (CD3ε, TCRb, B220/CD45R,Ter-119, Gr-1/Ly-6G/Ly-6C, CD11b, CD11c, F4/80, and FcεRIa; all from BioLegend), anti-CD25 (BioLegend), and anti-CD127 (eBioscience), as described9. Cells were fixed, subjected to flow cytometry, and analyzed on an LSR Fortessa (BD Biosciences, San Jose, CA). Data were collected using FACSDiva software (BD Biosciences) and analyzed using FlowJo software (TreeStar, Ashland, OR).
ILCs culture.
Lungs from sham- and RV-treated immature C57BL/6J or IL-1β−/− mice were harvested seven days post-infection for ILCs isolation by flow cytometry (Sony MA900 Cell Sorter). Lung cells were processed as described above and stained with fluorescent-tagged antibodies for lineage markers, CD45 and CD127. Lineage-negative CD45 and CD127 ILCs were plated on round-bottom 96-well plates at 104 cells per well and cultured in RPMI 1640 supplemented with 10% FBS, IL-2, and IL-7 (20 ng/ml each) (R&D Systems). Twenty-four hours later, cells were stimulated with IL-1β (10 ng/ml) + IL-12 (50 ng/ml), IL-25 (50 ng/ml) + IL-33 (50 ng/ml), or IL-1β (10 ng/ml) + IL-23 (50 ng/ml, all from R&D Systems). After 24 h, cell pellet RNA was extracted for quantitative real-time PCR, as described below.
Western blot assay.
Lungs were harvested one day post-infection, dissolved in lysis buffer and homogenized for Western blot assay using anti-mouse IL-1β (R&D Systems), anti-mouse caspase-1 (Abcam, Cambridge, MA), anti-mouse NLRP3 (Cell Signaling Technology), and anti-β actin (Millipore Sigma, Burlington, MA).
Quantitative real-time polymerase chain reaction (qPCR).
After solubilization with Trizol (Invitrogen), RNA was extracted from cells and tissue according to manufacturer’s recommendations. Purified RNA was processed for first strand cDNA and qPCR using reverse transcriptase and SYBR green qPCR reagents (ThermoFisher Scientific). For in vivo experiments, mRNA Il1b, Il18, Aim2, Nlrp1, Nlrp3, Nlrc5, Il1rn, Il1r1, Tnf, Cxcl1, Cxcl10 and Il33 were measured 1 day post infection; mRNA Il12b, Il25, Il13, Muc5ac, Muc5b and Gob5 were measured 7 days post infection10; mRNA Ifng and Il17 were measured 1 and 7 days post infection. Expression levels were normalized to GAPDH using the ΔΔCt method. Primers used are described in Supplemental Table S1. To quantify virus particles, qPCR for positive-strand viral RNA was conducted using RV-specific primers and probes (forward primer: 5’-GTGAAGAGCCSCRTGTGCT-3’; reverse primer: 5’-GCTSCAGGGTTAAGGTTAGCC-3’; probe: 5’-FAM-TGAGTCCTCCGGCCCCTGAATG-TAMRA-3’) 36.
Measurement of IL-1β, IL-25 and IL-33 protein levels.
Lung IL-1β (R&D Systems), IL-25 and IL-33 (Thermo Fisher Scientific) were measured by ELISA. ELISA data were analyzed by BioTek Gen5 software (Winooski, VT). Total lung protein concentration was measured by BCA protein assay (Thermo Fisher Scientific).
Human bronchial epithelial cell culture.
Airway epithelial cells were isolated from tracheobronchial trimmings of unused healthy donor lungs under a protocol approved by the University of Michigan Investigational Review Board (protocol number HUM00000230). Primary airway epithelial cells were cultured in Transwells at air-liquid interface as described previously, with some modifications37. Briefly, airway epithelial cells were cultured under submerged conditions in complete PneumaCult-Ex Plus medium (Stemcell Technologies, Vancouver, CA) for 1 week. Cells were transferred to Transwells and cultured with complete medium in both basal and apical wells until confluence was reached. Cells were then maintained at air-liquid interface for three weeks in PneumaCult-ALI maintenance medium. Cells were infected with sham or RV-A1B at an MOI of 10 for 12 hours. Selected wells were treated with human recombinant IL-1β and IL-17 at concentrations 10 ng/mL or 30 ng/mL.
Quantification and statistical analysis.
Data are represented as mean ± standard error. Statistical significance was assessed by unpaired t-test, one-way ANOVA or two-way ANOVA, as appropriate. Group differences were pinpointed by a Tukey multiple comparison test.
Results
RV infection activates the inflammasome in vivo in six day-old mice.
Our recent study showed that RV infection of mature mice induces lung inflammasome priming and activation 20. To examine developmental differences, we collected lungs from RV-infected six-day-old and eight week-old mice and measured mRNA and protein expression of IL-1β and IL-1 receptor antagonist (IL-RA). IL-1β and IL-1RA mRNA and protein expression was increased in RV-infected 6-day-old mice one day post-infection (Fig. 1A), but expression was significantly lower in immature mice compared to eight week-old mice. Il1b, Il1rn and Il33 mRNA as well as IL-1β protein peaked at day 1 post-infection in RV-infected 6-day-old mice (Fig. 1B). Il25 mRNA was elevated on day 2 after infection and peaked on day 7, consistent with our previous study10. In addition, mRNA expression of Nlrp1, Nlrp3 and Nlrc5 but not Il18 was increased (Fig. 1C). RV increased protein expression of NLRP3 and pro-IL-1β (Fig. 1D, 1E), indicative of the RV-induced priming step. RV also triggered cleavage of pro-IL-1β and caspase-1 and subsequent production of IL-1β and caspase-1 p12 (Fig. 1D, 1E), demonstrating inflammasome activation in the lungs of RV-infected immature mice.
FIG 1. RV activates inflammasome in vivo in immature mice.
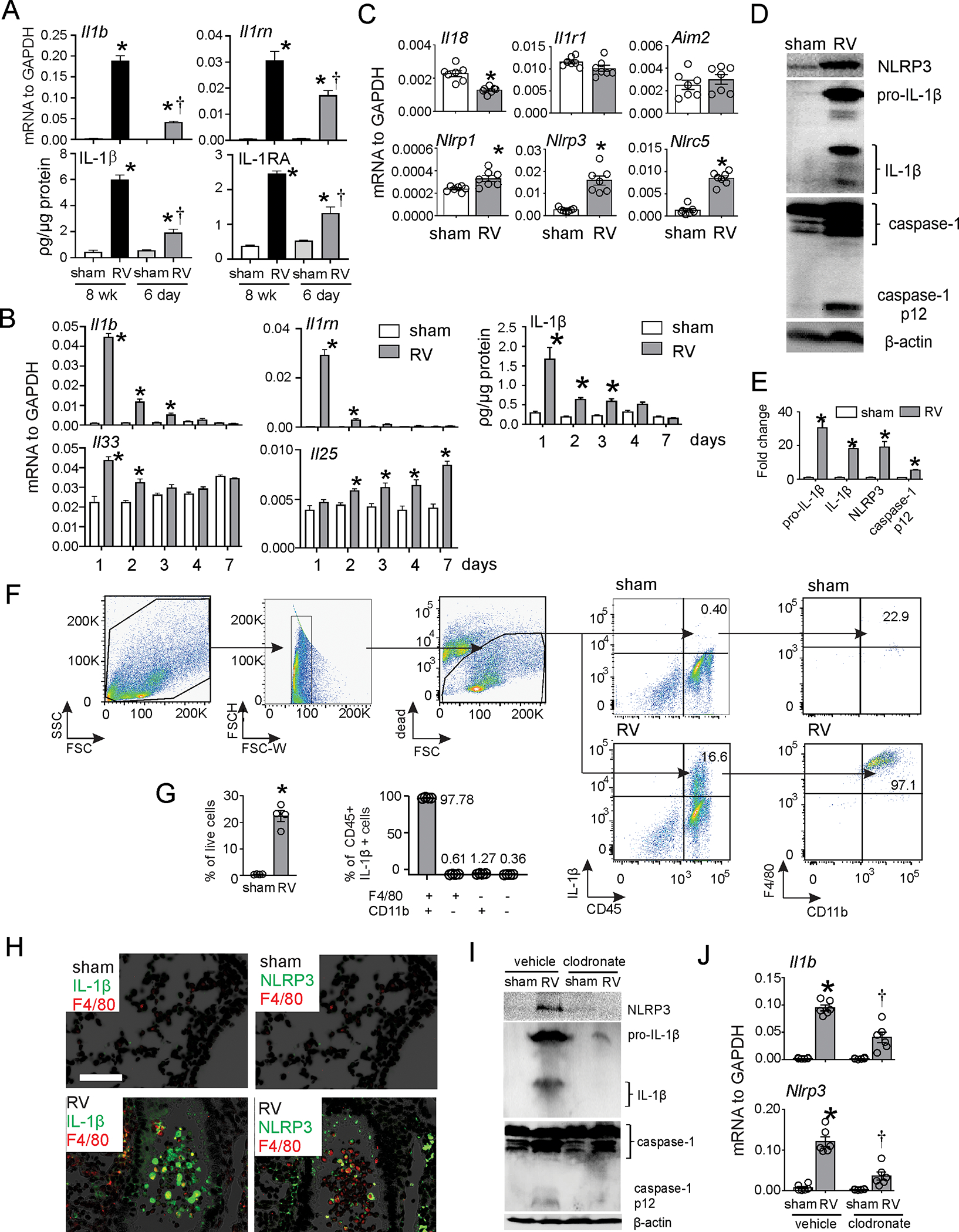
A. Eight week-old (adult) or 6 day-old (immature) C57BL/6 mice were inoculated with sham or RV. Lung mRNA and protein expression were measured 1 day later. (N=4, mean±SEM, *different from sham, †different from adult RV, p<0.05, one-way ANOVA with Tukey’s multiple comparisons test.) B. Six day-old immature C57BL/6 mice were inoculated with sham or RV. Lung mRNA and protein expression were measured 1, 2, 3, 4 or 7 days later. (N=4–7, mean±SEM, *different from sham, p<0.05, one-way ANOVA with Tukey’s multiple comparisons test. C. Six day-old immature C57BL/6 mice were inoculated with sham or RV. Lung mRNA was measured 1 day after infection. (N=6, mean±SEM, *different from sham, p<0.05, unpaired t test. D. One day after infection, whole lungs were homogenized within the lysis buffer and subjected to Western blot. Anti-mouse-IL-1β recognizes pro-IL-1β and its bioactive form IL-1β. Anti-mouse-caspase-1 detects both caspase-1 and its cleaved form, caspase-1 p12. E. Group mean relative expression levels were normalized to β-actin. (N=6, mean±SEM, *different from sham, p<0.05, one-way ANOVA.) F and G. Lung IL-1β+ cells in RV-infected six-day-old mice. IL-1β+ cells were identified 1 d after infection. IL-1β+ cells were analyzed as a percentage of live cells (left panel) and F4/80+ and CD11b+ cells were analyzed as a percentage of CD45+IL-1β+ cells (right panel) (n = 4, mean±SEM, *different from WT sham, p<0.05, unpaired t-test). H. Lungs were stained for IL-1β (green) NLRP3 (green), F4/80 (red) and nuclei (DAPI, black; bar, 50 μm). I. and J. Clodronate- or PBS-containing liposomes were delivered to mice intranasally 24 hours before sham or RV infection. One day after infection, lungs were harvested for mRNA and Western blot (N=6, mean±SEM, *different from sham, p<0.05, one-way ANOVA).
We performed flow cytometry to determine the cellular source of IL-1β. RV–infected 6-day-old mice showed a greater percentage of CD45+ IL-1β+ lung cells (Fig. 1F), and almost all of them were F4/80+ CD11b+ exudative macrophages (Fig. 1G). Wes also examined airway IL-1β and NLRP3 deposition by immunofluorescence. Infection with RV increased airway IL-1β and NLRP3 expression, with the strongest signal found in F4/80-positive cells, indicative of airway macrophages (Fig. 1H). There was less IL-1β and NLRP3 staining in the airway epithelium.
Next, we delivered clodronate- or PBS-containing liposomes to mice intranasally to deplete macrophages as previously described 35. Twenty-four hours later, mice were inoculated with sham or RV. RV-induced protein expression of NLRP3 and pro-IL-1β as well as production of mature IL-1β and caspase-1 p12 were reduced in clodronate-treated mice (Fig. 1I). Clodronate treatment also significantly reduced whole lung IL-1β mRNA in RV-infected mice (Fig. 1J). Together, these data confirm the macrophage to be a major cellular source of inflammasome activation.
Inhibition of IL-1β signaling prior to RV infection amplifies ILC2 expansion and development of the asthma-like phenotype in immature mice.
To further investigate the role of IL-1β, we employed an antagonist of IL-1 receptor (IL-1RA) and a neutralizing antibody against IL-1β. Again, early-life RV infection induced a mucous metaplasia phenotype, as evidenced by periodic acid–Schiff (PAS) staining and Muc5ac protein deposition in the airway epithelium 21 days after infection (Fig. 2A and 2B). RV infection also expanded the population of lineage-negative CD25+ CD127+ ILC2s seven days after infection (Fig. 2C and 2D). We have previous shown that ILC2 expansion peak at seven days and is maintained 21 days after infection10. In contrast to RV-infected mature mice, IL-1RA treatment augmented RV-induced PAS staining, Muc5ac protein accumulation (Fig. 2A, 2B) and ILC2 expansion (Fig. 2C, 2D). mRNA expression of the ILC2 products IL-5 and IL-13 and the mucus-related genes Muc5ac and Gob5 were also significantly augmented in RV-infected, IL-1RA-treated mice (Fig. 2E). mRNA expression was increased in a dose-dependent manner. In addition, IL-25 and IL-33 mRNA and protein expression were induced by RV infection and further increased in the presence of IL-1RA (Fig 2F, 2G). (Levels of IL-25 and IL-33 were measured at days 7 and 1 after infection, respectively, when their production is maximal10.) These results are consistent with the notion that IL-1β limits development of the mucous metaplasia phenotype via regulation of innate cytokine expression and ILC2 expansion. IL-1RA did not block RV-induced mRNA expression of Tnf, Cxcl1, Cxcl10 or Ifng. However, IL-1RA decreased Il17 mRNA expression (Fig. 2H). IL-1RA treatment was associated with a slight increase (0.2 log) in viral copy number (Fig 2I).
FIG 2. IL-1RA increased RV-induced ILC2 expansion and mucus metaplasia in 6-day old wild type mice.
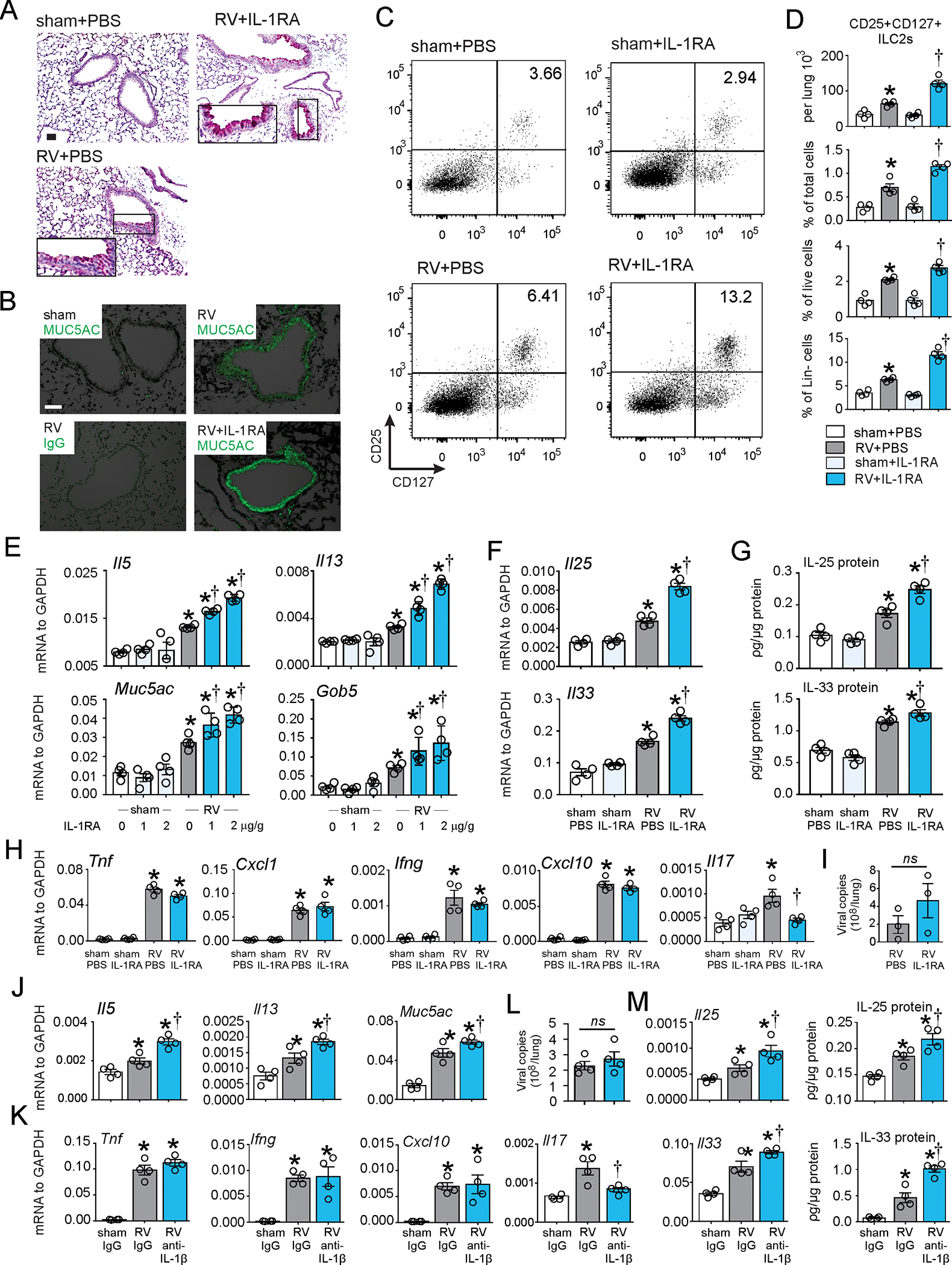
Six day-old C57BL/6 mice were inoculated with sham or RV intranasally. IL-1RA or vehicle was given intraperitoneally one hour before RV infection, followed by a half dose of IL-1RA on day 1. The concentration of IL-1RA was 2 μg/g unless otherwise noted. PAS staining (A; bar=50 μm), Muc5ac immunofluorescence (B; bar=50 μm), lung lineage-negative CD25+ CD127+ ILC2s (C and D) and whole-lung mRNA and protein expression (E-H) were examined. Il33, Il17, Ifng, Tnf, Cxcl1 and Cxcl10 mRNA and IL-11 protein were examined at one day post-infection. Il25, Il5, Il13, Muc5ac and Gob5 mRNA IL-25 protein, PAS staining, Muc5ac immunofluorescence and lung ILC2s were examined 7 days post-infection. I. RV positive-strand RNA was assessed 24 h after infection, and presented as viral copy number in total lung. (N =3–4, mean±SEM, *different from sham, †different from RV, p<0.05, p<0.05, one-way ANOVA). J-M, 6-day old wild type C57BL/6 mice were inoculated with sham, RV, in combination with either isotype IgG control or anti-IL-1β Ab. Whole-lung mRNA (J, K) and vRNA (L) were measured using quantitative PCR, and whole lung IL-25 and IL-33 protein (M) were examined by ELISA. (N =4, mean±SEM, *different from WT sham, †different from WT RV, p<0.05, one-way ANOVA).
Since IL-1RA is a competitive inhibitor of both IL-1α and IL-1β, we employed neutralizing antibodies against IL-1β and IL-1α to specify their individual roles. Consistent with the effects of IL-1RA, anti-IL-1β increased RV-induced mRNA expression of Il5, Il13 and Muc5ac (Fig. 2J). Anti-IL-1β had no significant effect on mRNA expression of Tnf, Ifng, or Cxcl10. However, anti-IL-1β decreased Il17 mRNA expression (Fig. 2K). Anti-IL-1β also increased mRNA and protein expression of IL-25 and IL-33 (Fig. 2L). In contrast, anti-IL-1α had no significant effect on Il25, Il33, Il13, Il5, Muc5ac, Gob5 or Il17 mRNA expression (Supplemental Figure 1).
NLRP3 KO increases RV-induced type 2 immune responses in vivo.
NLRP3 is required for the RV-induced inflammasome activation but not the priming 20. We therefore employed NLRP3−/− mice to examine the requirement of NLRP3 for type 2 immune responses and development of mucous metaplasia. Compared to wild type mice, RV infection of six day-old NLRP3−/− mice induced a similar level of pro-IL-1β protein (Fig. 3A and 3B) and Il1b mRNA (Fig. 3C), indicating intact inflammasome priming. However, caspase-1 p12 and IL-1β levels were significantly decreased, indicative of impaired IL-1β maturation and secretion (Fig. 3A and 3B). Twenty-one days after RV infection, NLRP3−/− mice showed increased PAS staining and Muc5ac protein accumulation compared to wild-type mice (Fig. 3D and 3E). mRNA expression of Il5 and Il13 as well as the mucus-related genes Muc5ac and Gob5 was significantly increased in NLRP3−/− mice compared to wild type mice (Fig. 3F). IL-25 and IL-33 mRNA and protein expression were also significantly higher (Fig. 3G). NLRP3 KO did not block RV-induced mRNA expression of Cxcl1, Cxcl10 or Ifng (Fig 3H). NLRP3 KO mice showed a modest but statistically insignificant increase in viral copy number (Fig 3I). These results further demonstrate that inhibition of IL-1β during RV infection of immature mice increases innate cytokine expression and development of a mucous metaplasia phenotype.
FIG 3. NLRP3 KO increases RV-induced innate cytokine expression, type 2 immune responses and mucus metaplasia in 6-day old wild type mice.
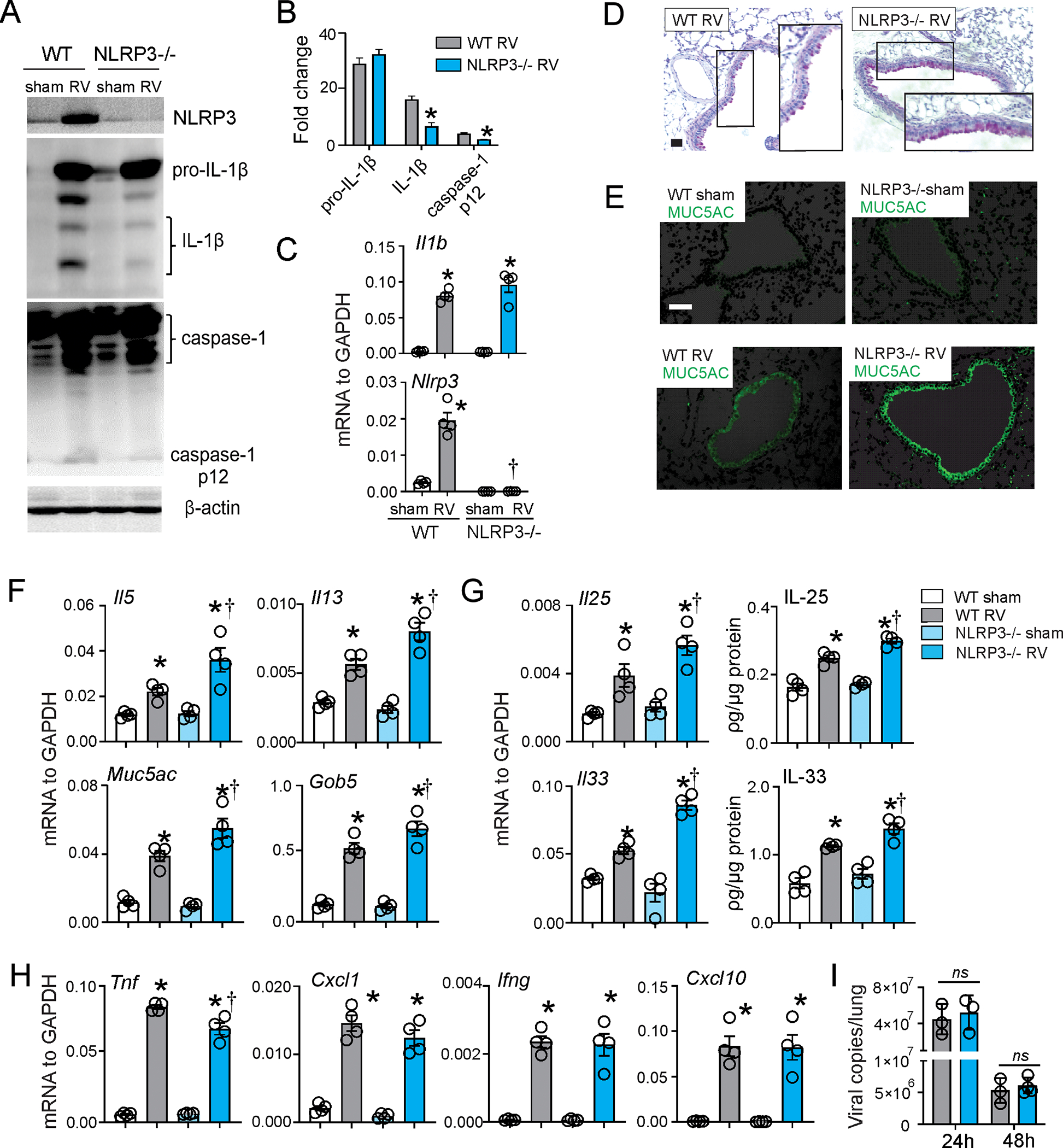
Six day-old wild type C57BL/6 and NLRP3−/− mice were inoculated with sham or RV. A, B. One day after infection, whole lungs were homogenized in lysis buffer and subjected to Western blot. Anti-mouse-IL-1β recognizes pro-IL-1β and its bioactive form IL-1β. Anti-mouse-caspase-1 detects both caspase-1 and its cleaved form, caspase-1 p12. Group mean relative expression levels were normalized to β-actin. (N=3, mean±SEM, *different from wild type RV, p<0.05, one-way ANOVA.) C. mRNA expression of Il1b and Nlrp3 in wild type and NLRP3−/− mice. (N=4, mean±SEM, *different from WT sham, †different from WT RV, p<0.05, one-way ANOVA). D and E. PAS staining and Muc5ac immunofluorescence were examined 21 d post-infection (bar=50 μm). Whole-lung mRNA and protein expression were examined one or seven days post-infection. F-G Il33, Il1b, Nlrp3, Ifng, Tnf, Cxcl1 and Cxcl10 mRNA and IL-33 protein were examined one day post-infection; Il25, Il5, Il13, Muc5ac and Gob5 mRNA and IL-25 expression were examined 7 days post-infection (n =4, mean±SEM, *different from WT sham, †different from WT RV, p<0.05, one-way ANOVA). I. RV positive-strand RNA was assessed 24 h and 48 h after infection, and presented as viral copy number in total lung. (N =3–4, mean±SEM, *different from sham, †different from RV, p<0.05, one-way ANOVA).
Effect of IL-1β KO on ILC2 maturation.
To further investigate the role of IL-1β in ILC2 expansion and development of mucous metaplasia and airway hyperresponsiveness in RV-infected immature mice, we first infected 6 day-old wild type and IL-1β−/− mice with RV-A1B. In contrast to our results with IL-1RA and anti-IL-1β, IL-1β deficiency blocked RV-induced mRNA expression of Il5, Il13, Muc5ac and Gob5 (Fig. 4A), PAS staining and Muc5ac protein accumulation compared to wild-type mice (Fig. 4B and 4C). Similarly, airway hyperresponsiveness to RV was blocked in IL-1β−/− mice (Fig. 4D). IL-1β deficiency blocked RV-induced IL-25 and IL-33 mRNA and protein expression (Fig. 4E, 4F). Compared to wild type mice, IL-1β−/− mice showed reduced mRNA expression of Tnf, Ifng, Cxcl10, Il17 and Il1rl1, which encodes the IL-33 receptor ST2 (Fig. 4G). Viral copy number was significantly higher in IL-1β−/− mice (Fig 4H).
FIG 4. IL-1β-KO blocked RV-induced development of an asthma-like phenotype and innate cytokine expression in immature mice.
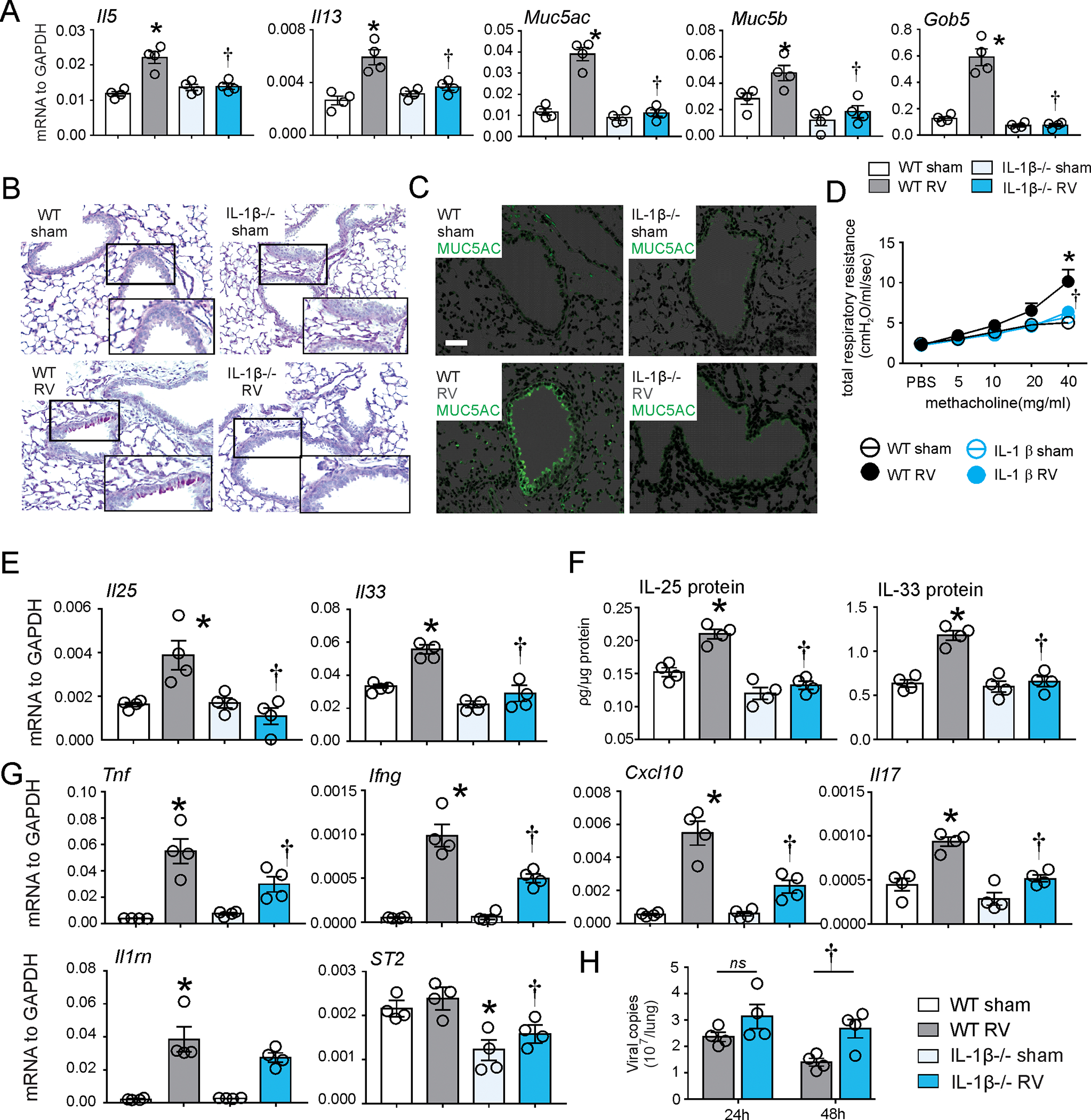
6-day old wild type C57BL/6 and IL-1β−/− mice were inoculated with sham or RV. A. Lung mRNA expression was measured 7 d post infection. (n = 4, mean±SEM, *different from WT sham, †different from WT RV, p<0.05, one-way ANOVA). Mucous metaplasia was assessed by PAS staining (B) and Muc5ac immunofluorescence (C). Lung sections prepared 3 wk after treatment of 6-d-old mice. D, Airway responsiveness of four wk-old baby mice, 21 d after sham and RV infection (n = 4, mean±SEM, *different from WT sham, †different from WT RV, p<0.05, two-way ANOVA). E, F. Whole-lung IL-25 and IL-33 mRNA and protein expression were examined seven and one day post-infection, respectively. G. Il1rl1, Il1rn, Ifng, Tnf, Il17 and Cxcl10 mRNA were examined one day post-infection. H. RV positive-strand RNA was assessed 24 h and 48 h after infection, and presented as viral copy number in total lung (n =4, mean±SEM, *different from WT sham, †different from WT RV, p<0.05, one-way ANOVA).
The significant reduction in Il1rl1 in IL-1β−/− mice, with or without RV infection, suggested a developmental deficiency of type 2 immunity in IL-1β−/− mice. To test this, we sorted Lin-CD45+CD127+ ILCs 38 from both wild type and IL-1β−/− mice, and then cultured and stimulated them ex vivo (Fig 5A). In the absence of cytokine stimulation, expression of mRNAs encoding the type 2 cytokines IL-5 and IL-13, the innate cytokine receptors IL-17RB and ST2 and the ILC2 transcription factor GATA3 was significantly reduced in the sorted cells from the IL-1β−/− mice (Fig. 5B). No significant difference in Ifn and Il17 mRNA was observed. Treatment with the ILC2 agonists IL-25 and IL-33 increased Il5, Il13 and Il1rl1, in wild type mice but not in IL-1β-/−. In contrast, there was no defect in the Ifn mRNA response to the ILC1 stimuli IL-1β and IL-12. There was no induction of Il17 mRNA. Taken together, these results demonstrate that the absence of IL-1β during development leads to a defect in ILC2 maturation which makes the cells unresponsive to IL-25 and IL-33. This physiologic state contrasts to the absence of IL-1β signaling after RV infection, which promotes ILC2 responses.
FIG 5. IL-1β−/− mice demonstrate deficient ILC2 maturation.
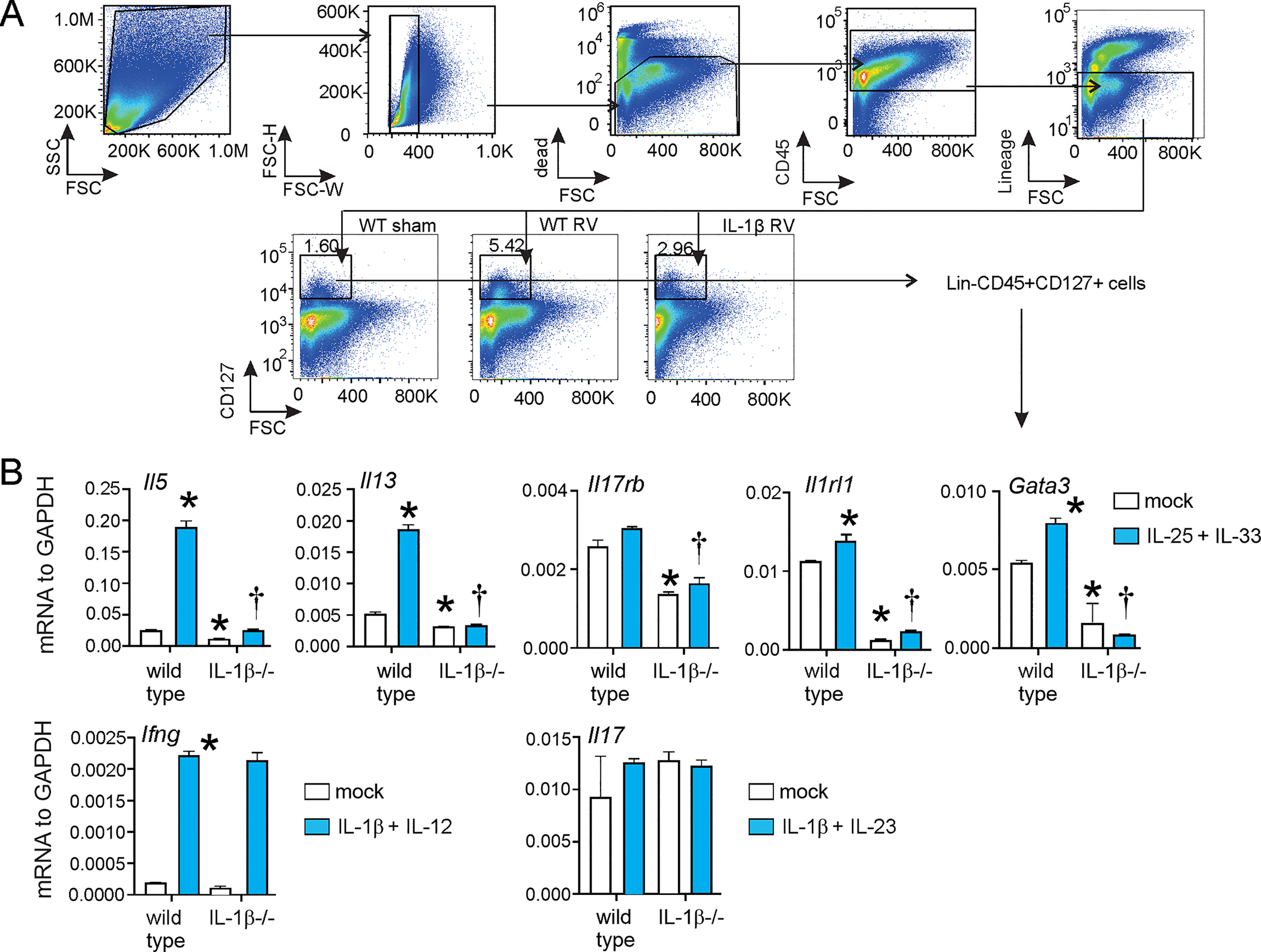
Six day-old wild type C57BL/6 and IL-1β−/− mice were inoculated with sham or RV. (A) Lungs were collected from sham or RV-infected wild type or immature mice, and cell suspensions were sorted for Lin- CD45+ CD127+ ILCs. Sorted ILCs were stimulated with combinations of type 1 (IL-1β + IL-12), type 2 (IL-25 + IL-33) and type 3 (IL-1β + IL-23) stimuli. (B) The cell pellet was collected for mRNA expression by quantitative PCR (N=3/group). (*different from wild type mock, †different from stimulated wild type, p<0.05 one-way ANOVA).
IL-1β protects against RV-induced type 2 immune responses in vivo.
We next examined the effects of exogenous IL-1β on RV-induced type 2 immune responses. Two doses of recombinant IL-1β were given intranasally to RV-infected 6 day-old mice; the first dose was given 1 h prior to infection and the second dose was given 24 h after infection. One group of mice received 1 ng per dose and a second group of mice received 10 ng per dose. Seven days after infection, mice treated with exogenous IL-1β showed decreased RV-induced mRNA expression of Il5 and Il13 as well as mucus-related genes Muc5ac and Gob5 (Fig. 6A). On the other hand, exogenous IL-1β increased RV-induced Ifng and Il17 mRNA, and there was no effect of IL-1β on Tnfa, Cxcl1 or Cxcl2 (Fig. 6B). IL-1β treatment had no significant effect on viral copy number (Fig 6C). In addition, IL-1β inhibited lung IL-25 and IL-33 mRNA and protein expression (Fig. 6D). IL-1β also attenuated IL-25 and IL-33 deposition but not RV immunoreactivity in the airway epithelium (Fig. 6E). Twenty-one days after infection, IL-1β-treated, RV-infected mice showed significantly reduced PAS staining and Muc5ac expression in the airways (Fig. 6F).
FIG 6. IL-1β treatment is protective against RV-induced type 2 inflammation.
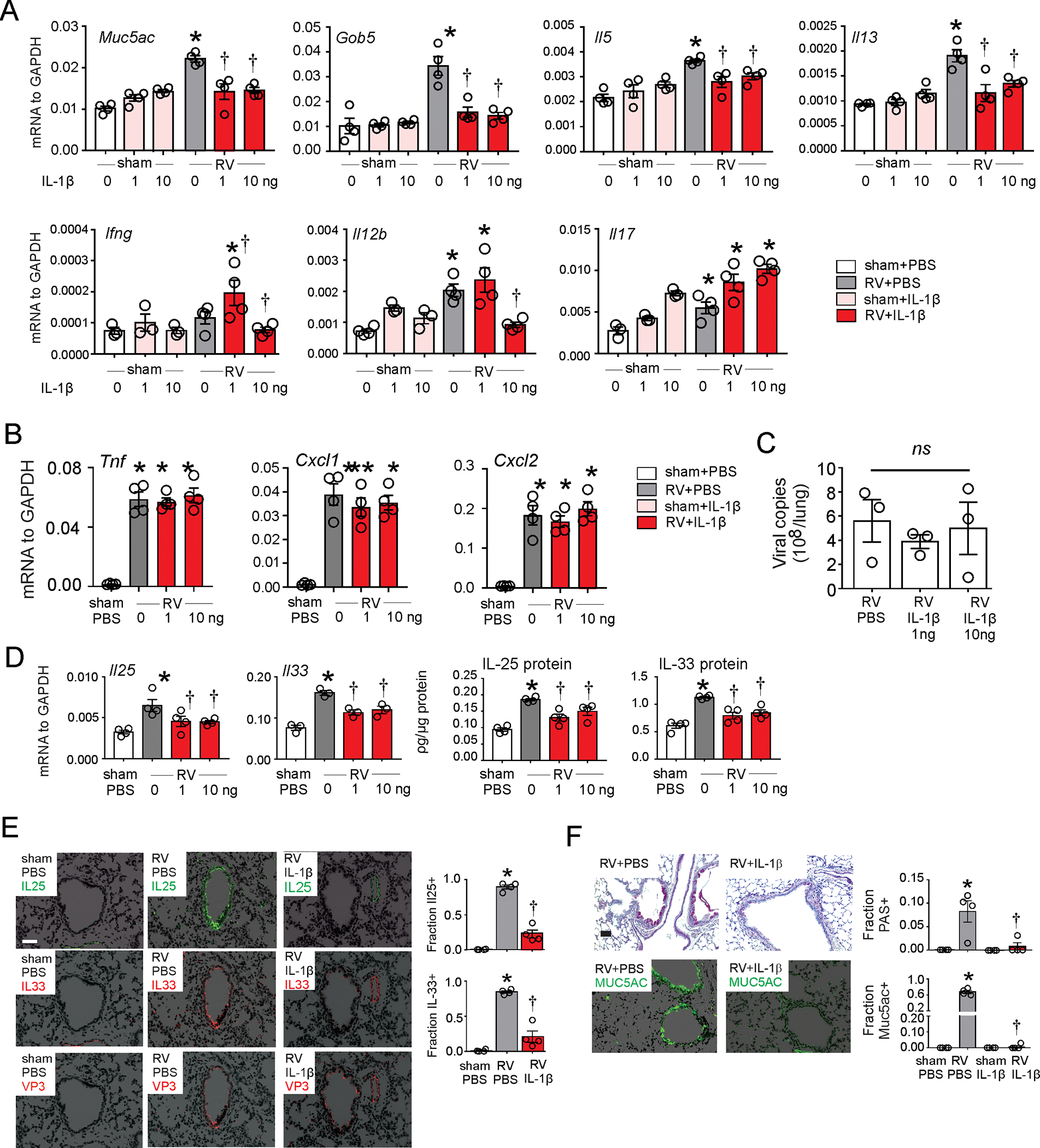
Six day-old wild type C57BL/6 mice were inoculated with sham or RV in combination with recombinant mouse IL-1β. A-C. Whole-lung mRNA and protein were assessed 1 day (Cxcl 1, Cxcl2, Tnfα and Il33) or 7 days (Il5, Il13, Il17, Il25, Ifng, Muc5ac and Gob5) post infection. D. RV positive strand RNA was assessed 24 h after infection, and presented as viral copy number in total lung. (N =3–4, mean±SEM, *different from sham, p<0.05; † different from RV, p<0.05, one-way ANOVA). E. Two days post-infection, lungs were stained for IL-33 (red), IL-25 (green), RV VP3 protein (red), and nuclei (DAPI, black). Scale bar, 50 μm. IL-25 and IL-33 were quantified as the fraction of epithelium that was positively stained, measured by NIH ImageJ software (N =4, mean±SEM, *different from sham, p<0.05; † different from RV, p<0.05, one-way ANOVA). F. PAS staining and Muc5ac immunofluorescence were examined 21 d post infection (bar=50 μm). PAS and Muc5ac were quantified as the fraction of epithelium that was positively stained, measured by NIH ImageJ software (N =4, mean±SEM, *different from sham, p<0.05; † different from RV, p<0.05, one-way ANOVA).
IL-1β inhibits RV induced human epithelial-derived innate cytokine expression.
Innate cytokines are produced by the airway epithelium in response to allergens, pathogens, pollutants, and toxic compounds. To determine the effects of IL-1β on the epithelial IL-25 and IL-33 expression, we infected human bronchial epithelial cells with RV in combination with human recombinant IL-1β or IL-17. RV infection increased mRNA expression of Il25, Il33 and Muc5ac but not Cxcl1, Cxcl8 or Cxcl10 (Fig. 7). Both IL-1β and IL-17 suppressed RV-induced mRNA expression of Il25, Il33, and Muc5ac. IL-1β and IL-17 had no effect in sham-infected cells (data not shown). IL-1β and IL-17 treatment significantly decreased viral copy number (Fig. 7). Together, these data suggest that IL-1β prevents type 2 inflammation and asthma development following early-life viral infection by suppressing epithelial cell innate cytokine expression.
FIG 7. IL-1β inhibits RV induced human epithelial-derived innate cytokine expression.
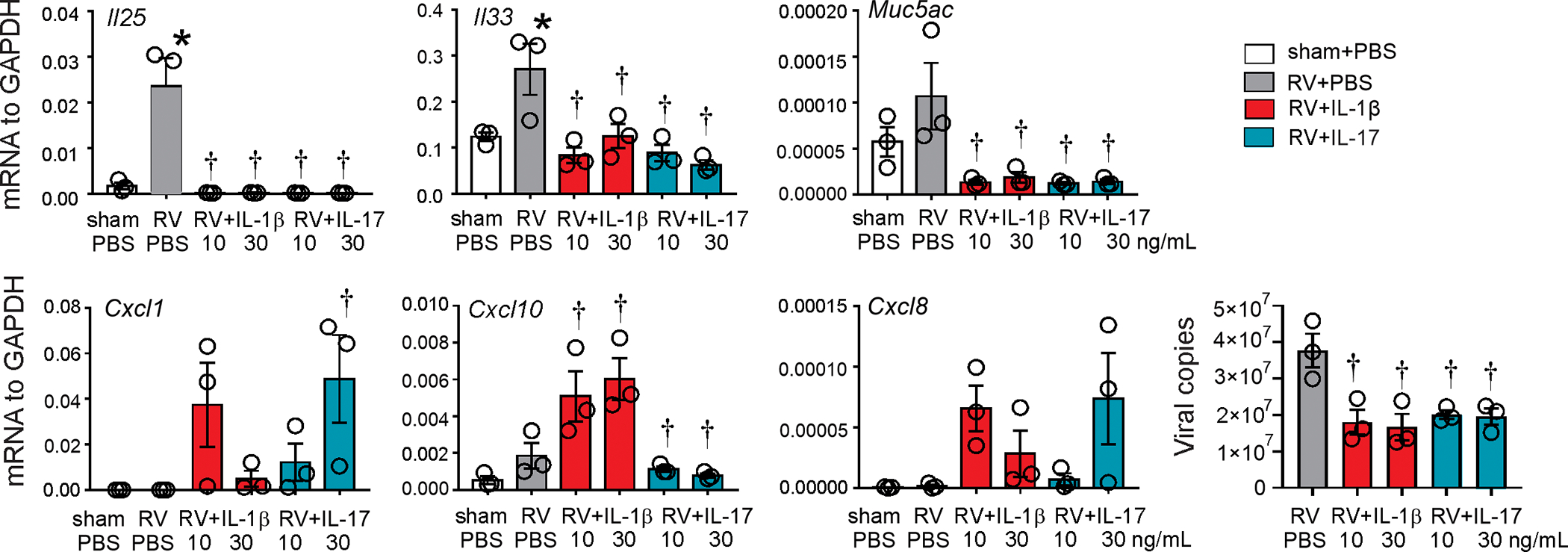
Human bronchial derived epithelial cells were infected with RV at an MOI of 20 in combination with recombinant human IL-1β and IL-17. mRNA expression and RV positive-strand RNA were measured 12 hours post infection. (n =3, mean±SEM, *different from sham, p<0.05; † different from RV, p<0.05, one-way ANOVA).
Discussion
Early-life respiratory viral infection has been associated with asthma development. In a prospective study of Finnish infants hospitalized for wheezing, RV was the most common virus isolated and was significantly associated with a diagnosis of asthma 6 years after hospitalization 39. In the University of Wisconsin Childhood Origins of Asthma Study, infants with a family history of allergy and wheezing-associated illnesses with RV were more likely to develop asthma than infants with allergen sensitization or infection with respiratory syncytial virus 40,41. The association between asthma and wheezing illnesses with RV was still present at age 13 years 1. Similarly, infants in the Netherlands Generation R study with bronchitis, bronchiolitis and pneumonia before three years of age were more likely to have lower lung function and asthma at 10 years of age 2. In the latter study, allergic sensitization did not factor into the associations seen. These data are consistent with the notion that early-life RV infections induce a non-allergic asthma phenotype 3,42. It has also been suggested that early life RV infections could drive the development of atopic sensitization and subsequent allergic airways disease 4.
RV infection of six day-old immature mice causes the development of a chronic asthma-like mucous metaplasia phenotype which is associated with expansion of IL-13-producing ILC2s and dependent on IL-25 and IL-33 8–10,36. We recently found that RV-induced inflammasome activation is required for maximal airway inflammation and hyperresponsiveness in naive and house dust mite-exposed mature mice with allergic airways disease 20. We therefore hypothesized that IL-1β is required for maximum RV-induced ILC2 expansion and the development of mucous metaplasia. However, we found that inhibition of IL-1β signaling with IL-1RA or anti-IL-1β administered prior to RV infection increased type 2 immune responses, ILC2 number and mucus metaplasia. Knockout of NLRP3, which is required for the RV-induced inflammasome activation 20, also increased type 2 cytokine responses. Treatment with IL-1β attenuated the asthma-like phenotype, including deposition of IL-25 and IL-33 in the airway epithelium. Finally, IL-1β suppressed Il25, Il33 and muc5ac mRNA expression in cultured airway epithelial cells. Together, our data suggest that macrophage IL-1β limits type 2 inflammation and mucous metaplasia following early life viral infection by suppressing epithelial cell innate cytokine expression. These results are consistent with a previous study showing that Heligmosomoides polygyrus bakeri-induced IL-1β expression suppresses intestinal epithelial cell IL-25 and IL-33 production.43 In the latter case, suppression of epithelial IL-25 and IL-33 production attenuates parasite expulsion, allowing pathogen chronicity. In the case of early-life viral infection, IL-1β appears to play a protective role, limiting expression of innate cytokines, type 2 cytokines and mucus-related genes and blocking the development of mucous metaplasia. Finally, we found that, compared to mature mice, immature mice show reduced IL-1β production in response to RV infection, consistent with the notion that a limited IL-1β response permits development of the mucous metaplasia phenotype.
In our study, IL-1RA reduced, and IL-1β enhanced, Il17 transcription. IL-17 also attenuated epithelial cell innate cytokine expression. It is therefore possible that the effect of IL-1β was at least partially mediated by IL-17. IL-17 treatment attenuates ovalbumin-induced Th2-mediated allergic airways disease 44. Among the innate immune cells, γδ T cells and type 3 innate lymphoid cells produce IL-17 in response to IL-1β 24,45. We have found that, in adult mice, RV infection expands these cell populations, though to a lesser extent than enterovirus-D68 infection 46. A subpopulation of ILC2s that can convert into IL-17-producing NKp44- ILC3-like cells has recently been identified 38. However, in our study lung ILCs did not produce IL-17 in response to IL-1β and IL-23 stimulation. IL-1β also increased expression of the canonical type 1 cytokine IFN-γ, which we have shown to directly suppress ILC2 function. 7 In contrast to IL-1β, IFN-γ had no effect on lung IL-25 or IL-33 production.
While in vitro studies of RV-induced inflammasome activation have focused on airway epithelial cells 18,19, we found that CD11b+ F4/80+ macrophages in the airway lumen and subepithelium produce IL-1β in response to RV infection in vivo. CD11b+ exudative macrophages are recruited to the lung following RV infection 47, influenza infection 48 and LPS administration 49. These data are consistent with previous work showing that caspase-1 inflammasome activation in the hematopoietic, but not stromal, compartment was required to induce protective antiviral immunity in influenza-infected mice 50. These data provide support to the concept that airway macrophages 35,51, ILC2s 8–10 and other innate immune cells are an important source of pro-inflammatory cytokines following RV infection, interacting with airway epithelial cells to determine the final response to RV infection.
We found that IL-1β tended to reduce viral copy number whereas IL-1β blockade tended to increase vRNA. It is therefore conceivable that IL-1β inhibits the RV-induced mucous metaplasia phenotype by decreasing viral load. However, IL-1β administration increased IL-17 mRNA and IFN-γ expression, demonstrating that the inhibitory effect of IL-1β on type 2 gene expression and mucous metaplasia was not due to a general suppression of viral-induced responses. In addition, changes in vRNA were small, rarely reaching statistical significance and reaching at most 0.3 log (in IL-1β KO mice). Finally, IL-1β did not appear to reduce RV immunoreactivity in the airway epithelium (Figure 6E).
One unexpected aspect of our study is the contradictory effect of IL-1β KO mice compared to inhibition or activation of IL-1β signaling prior to RV infection. Studies of cultured ILCs from immature mouse lungs showed that IL-1β KO block ILC2 maturation, as evidenced by reduced mRNA expression of Il17rb, Il1rl1 and Gata3 and insensitivity to IL-25 and IL-33 stimulation. Recent studies have shown that ILC2s cultured in the presence of IL-1β show increased IL-5 and IL-13 production as well as increased mRNA expression of Il17rb and Il1rl1, which encode unique subunits of the IL-25 and IL-33 receptors, respectively 29,30.
The immature immune system is qualitatively different from that of adult, refractory to type 1 and permissive to type 2 responses. Infection of mice with RV induces an age-dependent immune response in the airways. Early-life RV infection, but not adult infection, increases expression of IL-4, IL-5, IL-13, IL-25 and IL-33 7–10,36. In contrast, induction of the type 1 cytokines IFN-γ, IL-12 p40 and TNF-α is diminished in neonates compared to adults. In this context, upregulation of the macrophage IL-1β response pulls the immune response towards a mature antiviral response and away from a pro-asthmatic phenotype. Further insight into this pathway may lead to therapeutic interventions against asthma development.
Supplementary Material
Acknowledgments:
The authors thank Dr. Gabriel Núñez (University of Michigan, Ann Arbor, MI) for his gift of IL1β −/− and NLRP3−/− mice. This work was supported NIH grant R01 AI120526 (to M.B.H.).
Footnotes
Declaration of Interests:
The authors declare no competing interests
References
- 1.Rubner FJ, Jackson DJ, Evans MD, et al. Early life rhinovirus wheezing, allergic sensitization, and asthma risk at adolescence. J Allergy Clin Immunol 2017;139(2):501–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Meel ER, den Dekker HT, Elbert NJ, et al. A population-based prospective cohort study examining the influence of early-life respiratory tract infections on school-age lung function and asthma. Thorax. 2018;73(2):167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moraes TJ, Sears MR. Lower respiratory infections in early life are linked to later asthma. Thorax. 2018;73(2):105–106. [DOI] [PubMed] [Google Scholar]
- 4.Martorano LM, Grayson MH. Respiratory viral infections and atopic development: From possible mechanisms to advances in treatment. Eur J Immunol. 2018;48(3):407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hasegawa K, Hoptay CE, Harmon B, et al. Association of type 2 cytokines in severe rhinovirus bronchiolitis during infancy with risk of developing asthma: A multicenter prospective study. Allergy. 2019;74(7):1374–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneider D, Hong JY, Popova AP, et al. Neonatal rhinovirus infection induces persistent mucous metaplasia and airways hyperresponsiveness J Immunol. 2012;188:2894–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han M, Hong JY, Jaipalli S, et al. IFN-γ Blocks Development of an Asthma Phenotype in Rhinovirus-Infected Baby Mice by Inhibiting Type 2 Innate Lymphoid Cells. Am J Respir Cell Mol Biol. 2017;56(2):242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajput C, Cui T, Han MY, et al. ROR alpha-dependent type 2 innate lymphoid cells are required and sufficient for mucous metaplasia in immature mice. Am J Physiol-Lung Cell Mol Physiol. 2017;312(6):L983–L993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong JY, Bentley JK, Chung Y, et al. Neonatal rhinovirus induces mucous metaplasia and airways hyperresponsiveness through IL-25 and type 2 innate lymphoid cells. J Allergy Clin Immunol 2014;134(2):429–439.e428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han M, Rajput C, Hong JY, et al. The Innate Cytokines IL-25, IL-33, and TSLP Cooperate in the Induction of Type 2 Innate Lymphoid Cell Expansion and Mucous Metaplasia in Rhinovirus-Infected Immature Mice. J Immunol. 2017;199(4):1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Terajima M, Yamaya M, Sekizawa K, et al. Rhinovirus infection of primary cultures of human tracheal epithelium: role of ICAM-1 and IL-1beta. Am J Physiol. 1997;273(4 Pt 1):749–759. [DOI] [PubMed] [Google Scholar]
- 12.Piper SC, Ferguson J, Kay L, et al. The Role of Interleukin-1 and Interleukin-18 in Pro-Inflammatory and Anti-Viral Responses to Rhinovirus in Primary Bronchial Epithelial Cells. PLoS One 2013;8(5):e63365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simpson JL, Carroll M, Yang IA, et al. Reduced Antiviral Interferon Production in Poorly Controlled Asthma Is Associated With Neutrophilic Inflammation and High-Dose Inhaled Corticosteroids. Chest. 2016;149(3):704–713. [DOI] [PubMed] [Google Scholar]
- 14.Proud D, Gwaltney JM, Hendley JO, Dinarello CA, Gillis S, Schleimer RP. Increased Levels of Interleukin-1 Are Detected in Nasal Secretions of Volunteers during Experimental Rhinovirus Colds. J Infect Dis. 1994;169(5):1007–1013. [DOI] [PubMed] [Google Scholar]
- 15.Yoon HJ, Zhu Z, Gwaltney JM, Elias JA. Rhinovirus Regulation of IL-1 Receptor Antagonist In Vivo and In Vitro: A Potential Mechanism of Symptom Resolution. J Immunol. 1999;162(12):7461–7469. [PubMed] [Google Scholar]
- 16.Kluijver JD, Grünberg K, Pons D, et al. Interleukin‐1β and interleukin‐1ra levels in nasal lavages during experimental rhinovirus infection in asthmatic and non‐asthmatic subjects. Clin Exp Allergy. 2003;33(10):1415–1418. [DOI] [PubMed] [Google Scholar]
- 17.He Y, Hara H, Núñez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci. 2016;41(12):1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Triantafilou K, Kar S, Kuppeveld FJMv, Triantafilou M. Rhinovirus-Induced Calcium Flux Triggers NLRP3 and NLRC5 Activation in Bronchial Cells. Am J Respir Cell Mol Biol. 2013;49(6):923–934. [DOI] [PubMed] [Google Scholar]
- 19.Shi L, Manthei DM, Guadarrama AG, Lenertz LY, Denlinger LC. Rhinovirus-Induced IL-1β Release from Bronchial Epithelial Cells Is Independent of Functional P2X7. Am J Respir Cell Mol Biol. 2012;47(3):363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han MY, Bentley JK, Rajput C, et al. Inflammasome activation is required for human rhinovirus-induced airway inflammation in naive and allergen-sensitized mice. Mucosal Immunol. 2019;12(4):958–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17–producing helper T cells. Nat Immunol 2007;8:950. [DOI] [PubMed] [Google Scholar]
- 22.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17–producing human T helper cells. Nature Immunol 2007;8:942. [DOI] [PubMed] [Google Scholar]
- 23.Sutton C, Brereton C, Keogh B, Mills KHG, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17–producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203(7):1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HY, Lee HJ, Chang Y-J, et al. Interleukin-17–producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nature Med. 2013;20:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mortha A, Chudnovskiy A, Hashimoto D, et al. Microbiota-Dependent Crosstalk Between Macrophages and ILC3 Promotes Intestinal Homeostasis. Science. 2014;343(6178):1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seo S-U, Kuffa P, Kitamoto S, et al. Intestinal macrophages arising from CCR2+ monocytes control pathogen infection by activating innate lymphoid cells. Nature Commun. 2015;6:8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi T, Iijima K, Checkel JL, Kita H. IL-1 Family Cytokines Drive Th2 and Th17 Cells to Innocuous Airborne Antigens. Am J Respir Cell Mol Biol. 2013;49(6):989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahmutovic Persson I, Menzel M, Ramu S, Cerps S, Akbarshahi H, Uller L. IL-1β mediates lung neutrophilia and IL-33 expression in a mouse model of viral-induced asthma exacerbation. Respir Res. 2018;19(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohne Y, Silver JS, Thompson-Snipes L, et al. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nature Immunol. 2016;17:646. [DOI] [PubMed] [Google Scholar]
- 30.Bal SM, Bernink JH, Nagasawa M, et al. IL-1β, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nature Immunol. 2016;17:636. [DOI] [PubMed] [Google Scholar]
- 31.Newcomb DC, Sajjan U, Nanua S, et al. Phosphatidylinositol 3-Kinase Is Required for Rhinovirus-induced Airway Epithelial Cell Interleukin-8 Expression. J Biol Chem. 2005;280(44):36952–36961. [DOI] [PubMed] [Google Scholar]
- 32.Newcomb DC, Sajjan US, Nagarkar DR, et al. Human rhinovirus 1B exposure induces phosphatidylinositol 3-kinase-dependent airway inflammation in mice. Am J Respir Crit Care Med. 2008;177(10):1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fiala M, Kenny GE. Enhancement of Rhinovirus Plaque Formation in Human Heteroploid Cell Cultures by Magnesium and Calcium. J Bacteriol. 1966;92(6):1710–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seo SU, Kamada N, Munoz-Planillo R, et al. Distinct Commensals Induce Interleukin-1 beta via NLRP3 Inflammasome in Inflammatory Monocytes to Promote Intestinal Inflammation in Response to Injury. Immunity. 2015;42(4):744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagarkar DR, Bowman ER, Schneider D, et al. Rhinovirus infection of allergen-sensitized and -challenged mice induces eotaxin release from functionally polarized macrophages. J Immunol. 2010;185:2525–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider D, Hong JY, Popova AP, et al. Neonatal rhinovirus infection induces mucous metaplasia and airways hyperresponsiveness. J Immunol 2012;188(6):2894–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider D, Ganesan S, Comstock AT, et al. Increased Cytokine Response of Rhinovirus-infected Airway Epithelial Cells in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2010;182:332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernink JH, Ohne Y, Teunissen MBM, et al. c-Kit-positive ILC2s exhibit an ILC3-like signature that may contribute to IL-17-mediated pathologies. Nature Immunol. 2019;20(8):992–1003. [DOI] [PubMed] [Google Scholar]
- 39.Kotaniemi-Syrjanen A, Vainionpaa R, Reijonen TM, Waris M, Korhonen K, Korppi M. Rhinovirus-induced wheezing in infancy--the first sign of childhood asthma? J Allergy Clin Immunol. 2003;111(1):66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemanske RF Jr, Jackson DJ, Gangnon RE, et al. Rhinovirus illnesses during infancy predict subsequent childhood wheezing. J Allergy Clin Immunol 2005;116(3):571–577. [DOI] [PubMed] [Google Scholar]
- 41.Jackson DJ, Gangnon RE, Evans MD, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med. 2008;178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taussig LM, Wright AL, Holberg CJ, Halonen M, Morgan WJ, Martinez FD. Tucson children’s respiratory study: 1980 to present. J Allergy Clin Immunol 2003;111(4):661–675. [DOI] [PubMed] [Google Scholar]
- 43.Zaiss MM, Maslowski KM, Mosconi I, Guenat N, Marsland BJ, Harris NL. IL-1β Suppresses Innate IL-25 and IL-33 Production and Maintains Helminth Chronicity. PLoS Pathogen. 2013;9(8):e1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schnyder-Candrian S, Togbe D, Couillin I, et al. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med. 2006;203(12):2715–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KHG. Interleukin-1 and IL-23 Induce Innate IL-17 Production from γδ T Cells, Amplifying Th17 Responses and Autoimmunity. Immunity. 2009;31(2):331–341. [DOI] [PubMed] [Google Scholar]
- 46.Rajput C, Han M, Bentley JK, et al. Enterovirus D68 infection induces IL-17-dependent neutrophilic airway inflammation and hyperresponsiveness. JCI Insight. 2018;3(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung Y, Hong JY, Lei J, Chen Q, Bentley JK, Hershenson MB. Rhinovirus Infection Induces Interleukin-13 Production from CD11b-Positive, M2-Polarized Exudative Macrophages. Am J Respir Cell Mol Biol 2015;52(2):205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ Monocyte-Derived Dendritic Cells and Exudate Macrophages Produce Influenza-Induced Pulmonary Immune Pathology and Mortality. J Immunol. 2008;180(4):2562–2572. [DOI] [PubMed] [Google Scholar]
- 49.Duan M, Li WC, Vlahos R, Maxwell MJ, Anderson GP, Hibbs ML. Distinct Macrophage Subpopulations Characterize Acute Infection and Chronic Inflammatory Lung Disease. J Immunol. 2012;189(2):946–955. [DOI] [PubMed] [Google Scholar]
- 50.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206(1):79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bentley JK, Sajjan US, Dzaman MB, et al. Rhinovirus colocalizes with CD68- and CD11b-positive macrophages following experimental infection in humans. J Allergy Clin Immunol. 2013;132(3):758–761 e753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.