

SUMMARY
Synapses exhibit multiple forms of short-term plasticities, which have been attributed to the heterogeneity of neurotransmitter release probability. However, the molecular mechanisms that underlie the differential release states remain to be fully elucidated. The Unc-13 proteins appear to have key roles in synaptic function through multiple regulatory domains. Here, we report that deleting the M domain in Caenorhabditis elegans UNC-13MR leads to a significant increase in release probability, revealing an inhibitory function of this domain. The inhibitory effect of this domain is eliminated when the C1 and C2B domains are absent or activated, suggesting that the M domain inhibits release probability by suppressing the activity of C1 and C2B domains. When fused directly to the MUNC2C fragment of UNC-13, the M domain greatly enhances release probability. Thus, our findings reveal a mechanism by which the UNC-13 M domain regulates synaptic transmission and provides molecular insights into the regulation of release probability.
In brief
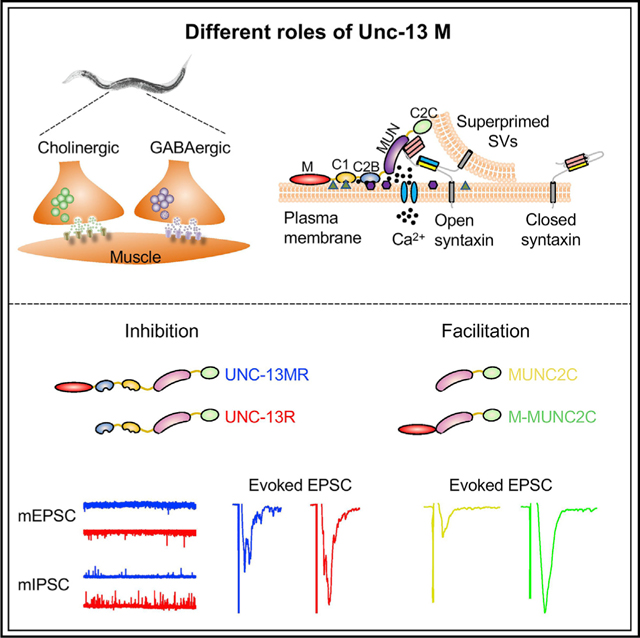
Liu et al. demonstrate that the M domain in Unc-13 has two opposite roles in synaptic transmission. It inhibits neurotransmitter release by suppressing the activity of the adjacent C1 and C2B domains and promotes neurotransmitter release when fused to the N-terminal of the MUNC2C fragment.
Graphical Abstract
INTRODUCTION
Synaptic strength is altered in response to changes in neuronal activity, thereby producing multiple forms of short-term plasticity (Atwood and Karunanithi, 2002). Presynaptic mechanisms that affect the probability of synaptic vesicle (SV) release are believed to have important roles in regulating synaptic strength. The presynaptic fusion machinery, including the SNARE complex, Munc13s, synaptotagmin RIMs, and RIM-binding proteins (RIM-BPs), are involved in the regulation of release probability in many synapse types (Südhof and Rizo, 2011; Jahn and Fasshauer, 2012). Synaptic release probability can be downregulated or upregulated by synaptic proteins, which leads to differential changes in plasticity, such as synaptic depression and facilitation (Dobrunz and Stevens, 1997; Fioravante and Regehr, 2011; Regehr, 2012). However, the mechanisms by which synaptic proteins function to control release probability remain to be fully elucidated.
Of the known synaptic proteins that form the fusion machinery, the Unc13 family of proteins are particularly important because they are involved in almost all steps of the exocytic process, including docking, priming, and fusion (Aravamudan et al., 1999; Augustin et al., 1999a; Richmond et al., 1999; Madison et al., 2005; Hammarlund et al., 2007), as well as short-term plasticity (Junge et al., 2004; Chen et al., 2013; Calloway et al., 2015; Xu et al., 2017). Four Munc13 isoforms have been identified in the mammalian nervous system, Munc13–1, ubMunc13–2, bMunc13–2, and Munc13–3. Both Munc13–1 and ubMunc13–2 possess an N-terminal C2A domain, which physically binds to the active-zone protein RIM and localizes those proteins close to the Ca2+ entry sites (Brose et al., 1995; Song et al., 1998; Betz et al., 2001; Deng et al., 2011). At many synapses, Munc13–1 has been shown to be the prominent isoform that mediates fast SV release (Betz et al., 1998; Augustin et al., 1999a). In contrast, bMunc13–2 and Munc13–3 lack an N-terminal C2A domain, leading to more diffuse expression relative to active-zone proteins (Chen et al., 2013). Synaptic transmission is completely arrested in Munc13–1/2 double-knockout hippocampal neurons, in which only these two Munc13 isoforms are expressed (Rosenmund et al., 2002; Varoqueaux et al., 2002). Munc13–3 is primarily expressed in the cerebellum, where its deletion also causes a synaptic transmission defect with a decreased release probability (Augustin et al., 1999b, 2001; Ishiyama et al., 2014). In addition, expression of Munc13–3 has been found in perforant path terminals targeting the dendrites of granule cells in the hippocampus (Netrakanti et al., 2015), and in calyx of Held synapses (Chen et al., 2013).
The importance of Unc13 proteins in synaptic transmission was first demonstrated in the fruit fly Drosophila melanogaster and the soil nematode Caenorhabditis elegans (Aravamudan et al., 1999; Richmond et al., 1999; Madison et al., 2005). C. elegans expresses two UNC-13 isoforms: a long isoform UNC-13L, which contains an N-terminal C2A domain, and a short isoform UNC-13MR (also called UNC-13S; Figure 1A), which has a unique N-terminal M domain that lacks the C2A motif. At the C. elegans neuromuscular junction (NMJ), UNC-13L colocalizes with the active-zone protein UNC-10/RIM and mediates a fast SV release component, analogous to Munc13–1 (Hu et al., 2013). In contrast, UNC-13MR exhibits a more diffuse synaptic distribution and mediates a slow component of SV release, indicating the two UNC-13 isoforms have differential roles in synaptic transmission. We have shown that UNC-13L regulates release probability through several distinct functional domains within the N-terminal (Michelassi et al., 2017; Li et al., 2019; Liu et al., 2019a). The C2A domain promotes SV release by binding to the active-zone protein unc-10/RIM, and the X, C1, and C2B domains inhibit SV release by multiple mechanisms, revealing two opposing functions of UNC-13L in regulating synaptic transmission. Similar effects have also been observed in studies of Munc13–1 (Basu et al., 2007; Lou et al., 2008; Shin et al., 2010; Liu et al., 2016; Camacho et al., 2017), demonstrating a conserved functionality of this protein. These results provide significant evidence that the C2A-containing UNC-13/Munc13 functions as one of the major substrates that regulates the heterogeneity of synaptic release probability and short-term plasticity.
Figure 1. The M domain in UNC-13MR inhibits spontaneous and evoked neurotransmitter release.

Miniature EPSCs/IPSCs and stimulus-evoked EPSCs were recorded from body wall muscle of adult worms in various Ca2+ levels.
(A) The domain structures of UNC-13MR and UNC-13R are illustrated. Ligands for each domain are indicated.
(B) Representative locomotory trajectories of 10 animals for wild type, unc-13(s69), and unc-13 rescued by single-copy insertion of UNC-13MR and UNC-13R under the pan-neuronal promoter. The starting points of each trajectory have been aligned for clarity.
(C) Quantification of the average locomotion speed for the indicated genotypes or transgenes.
(D) Representative mEPSC and mIPSC traces (recorded at 0-mM and 1-mM Ca2+) from the indicated genotypes. (E and F) Quantification of the frequency and amplitude of the mEPSCs and mIPSCs under all tested Ca2+ levels (0, 0.25, 0.5, and 1 mM) from the same genotypes as in (D).
(G) Example traces of stimulus-evoked EPSCs recorded in various Ca2+ levels (0.25, 0.5, and 1 mM) from wild-type (black), UNC-13MR-rescued (blue), and UNC-13R-rescued (red) animals.
(H–J) Quantification of the evoked EPSC amplitude and charge transfer from the same genotypes and Ca2+ levels as in (G).
(K) Ratio of the evoked EPSC charge transfer in UNC-13R-rescued worms to that in UNC-13MR-rescued animals in different Ca2+ levels.
(L–N) Comparison of the concentration effect for mEPSC frequency, mIPSC frequency, and evoked charge transfer versus extracellular Ca2+ concentration between the UNC-13MR- and UNC-13R-rescued animals.
(O) Quantification of the 10%–90% rise time and decay of the evoked EPSCs from wild-type, UNC-13MR, and UNC-13R-rescued animals (in 1-mM Ca2+).
(P) Representative traces of averaged mEPSCs (in 1-mM Ca2+) and quantification of mEPSC decay from wild-type, UNC-13MR, and UNC-13R-rescued animals. Data are means ± SEM. ##p < 0.01, ###p < 0.001 when compared with the wild type. *p < 0.05, **p < 0.01, ***p < 0.001 when compared with UNC-13MR-rescued animals; n.s., non-significant when compared with UNC-13MR-rescued animals. One-way ANOVA test for data in (C), (I), (J), and (P); one-way ANOVA after the Kruskal-Wallis test for data in (E) and (H). The number of worms analyzed for each genotype is indicated under the bar graphs.
The role of the N-terminal region of Unc-13 proteins that lack the C2A domain, however, remains unclear. Prior studies have shown that the coiled-coil motif in the N-terminal of bMunc13–2 binds to the active-zone protein ELKS1 and that interaction is critical for proper localization of bMunc13–2 and is required for SV priming in hippocampal synapses (Kawabe et al., 2017). Moreover, at Drosophila olfactory synapses, the active-zone protein Syd-1 clusters UNC-13B, an isoform without C2A in its N-terminal, at specific positions of the release site (Fulterer et al., 2018). These observations suggest various mechanisms by which the C2A-lacking Unc-13s are recruited to active zones. However, the M domain in worm UNC-13 does not appear to share similarities with the N-terminal of rat bMunc13–2 or Drosophila UNC-13B, suggesting that the M domains may have differential functions.
In this study, we report functions of the N-terminal M domain in the worm UNC-13MR isoform. Our data suggest that the M domain decreases release probability by regulating C1 and C2B. We further demonstrate that the M domain promotes release when fused directly to the C-terminal MUNC2C fragment in UNC-13. Furthermore, we demonstrate that the N-terminal sequence of rat bMunc13–2 exhibits similar regulatory functions as the worm M domain, despite poor sequence homology. Thus, our findings indicate that the M domain has important and conserved functions in controlling UNC-13MR-mediated synaptic transmission, thereby providing significant mechanistic advances in our understanding of how UNC-13s/Munc13s regulate synaptic release probability and plasticity.
RESULTS
The M domain inhibits spontaneous and evoked neurotransmitter release
To determine the role of the M domain in UNC-13MR, we generated a construct lacking the M domain (termed UNC-13R) (Figure 1A). Single-copy expression of UNC-13MR or UNC-13R in the nervous system in unc-13-null mutants (s69) partially restored locomotion (Figures 1B and 1C), indicating both proteins could support neuronal function. To directly assess the role of the M domain in synaptic transmission, we first examined spontaneous neurotransmitter release at the NMJs of UNC-13MR- or UNC-13R-rescued animals. Because C. elegans NMJs are innervated by both cholinergic excitatory neurons and GABAergic inhibitory neurons, the frequencies of miniature excitatory currents (mEPSCs) and miniature inhibitory postsynaptic currents (mIPSCs) were analyzed separately. We consider these events as miniature currents based on their discrete appearance and amplitude distributions (Figures S1A–S1C). The frequencies of both mEPSCs and mIPSCs were significantly higher in UNC-13R-rescued animals compared with those of UNC-13MR over a range of Ca2+ concentrations (0, 0.25, 0.5, and 1mM). These data indicate that the M domain inhibits spontaneous release in UNC-13MR, even in nominally Ca2+-free medium (Figures 1D–1F). The enhanced spontaneous release frequency in UNC-13R relative to UNC-13MR was more pronounced at inhibitory synapses, particularly in 0Ca2+ saline, in which the UNC-13R-rescued animal mIPSCs were increased 15-fold but only 2-fold in 1-mM Ca2+. In previous studies, we have shown that the acetylcholine (Ach) current amplitude in body-wall muscles is 50% reduced in the absence of Ca2+; therefore, we speculate that the lower mEPSC frequencies in 0Ca2+ saline may partially reflect a detection limitation. In addition to the lower mEPSC frequency, we also observed smaller mEPSC amplitude in UNC-13MR-rescued animals (Figures 1E and S1D), consistent with our previous results (Hu et al., 2013).
To determine whether the M domain also regulates evoked release, ventral nerve cord stimulation anterior to the voltage-clamped muscle was recorded. UNC-13R-rescued animals exhibited significantly greater amplitude-evoked EPSCs and evoked charge integrals compared with UNC-13MR-rescued animals at all Ca2+ concentrations tested (0.25, 0.5, and 1 mM) (Figures 1G–1J). These results reveal inhibitory function of the M domain in both Ca2+-independent and Ca2+-dependent neurotransmitter release mediated by UNC-13MR.
The inhibitory effects of the M domain were also observed when multicopy UNC-13MR and UNC-13R extrachromosomal arrays were examined (Figures S1E–S1I; Tables S1 and S2), indicating that the difference between UNC-13MR and UNC-13R is unlikely to reflect differences in expression levels.
Compared with the UNC-13MR-rescued animals, the charge transfer of the evoked EPSCs was increased 6-fold in 0.25-mM Ca2+ and only 1.5-fold in 1-mM Ca2+ (Figure 1K). Both spontaneous and evoked EPSCs rescued by UNC-13MR and UNC-13R displayed an obvious Ca2+ concentration dependence, whereas the steepness of the concentration-effect relationship in UNC-13MR-rescued animals was less than that in UNC-13R-rescued animals (Figures 1L–1N), indicating that the M domain regulates the Ca2+ dependence of SV release.
It has previously been reported that the evoked EPSCs mediated by UNC-13MR display slower kinetics relative to UNC-13L (Hu et al., 2013). To determine whether the M domain of UNC-13MR regulates release kinetics, we analyzed the rise time and the decay of the evoked EPSCs (in 1-mM Ca2+). These parameters were indistinguishable between UNC-13MR and UNC-13R-rescued animals (Figure 1O). Moreover, the averaged mEPSCs in UNC-13MR and UNC-13R-rescued animals also exhibited comparable decay (Figure 1P). These results indicate that the M domain does not affect release kinetics. Taken together, these findings demonstrate an inhibitory function of the M domain in the short unc-13 isoform in the regulation of synaptic transmission.
The probability of SV release is enhanced by the removal of the M domain
The increase in Ca2+-triggered neurotransmitter release in the UNC-13R-rescued animals may arise from a larger primed vesicle pool. To test that, we assessed SV priming in UNC-13MR and UNC-13R-rescued animals. SV priming was measured by the pulsed application of hypertonic sucrose solution (1 M, 2 s) onto the ventral nerve cord. The size of the readily releasable vesicle pool (RRP) was calculated by integrating the sucrose-evoked current (Rosenmund and Stevens, 1996; Li et al., 2019; Liu et al., 2019a). As shown in Figure 2A, the sucrose current was nearly eliminated in unc-13 mutants but was fully restored by single-copy expression of UNC-13MR, indicating that UNC-13MR is sufficient to maintain SV priming. Compared with UNC-13MR, the charge integral of UNC-13R-rescued animals was slightly increased (Figure 2B). However, the mEPSC charge integral of UNC-13R-rescued animals was also increased (Figure 2C); therefore the quantal content calculated by dividing the hyperosmotic charge integral by that of the mEPSCs was unchanged in UNC-13R relative to UNC-13MR-rescued animals (Figure 2D). These data suggest that the primed vesicle pool is unaltered in UNC-13R.
Figure 2. Deleting the M domain increases the fusogenicity of synaptic vesicles.

(A) Hypertonic sucrose-evoked current (recorded in 1-mM Ca2+) from wild-type (black), unc-13-null mutant (gray), UNC-13MR-rescued (blue), and UNC-13R-rescued (red) animals.
(B) Averaged charge transfer from the sucrose-evoked currents in (A).
(C and D) Quantification of the charge transfer of the mEPSCs (recorded in 1-mM Ca2+) and averaged quantal content in UNC-13MR- and UNC-13R-rescued animals.
(E) Presynaptic ultrastructure of cholinergic motor neuron synapses in wild-type, unc-13-null mutant, UNC-13MR-rescued, and UNC-13R-rescued animals. Asterisks indicate dense projection (DP); arrowheads indicate synaptic vesicles docked at the plasma membrane. Scale bar, 100 nm.
(F) Quantification of the number of synaptic vesicles docked at the plasma membrane per synaptic profile.
(G) Summary of the spontaneous vesicular release rate (mEPSC frequency divided by the number of vesicles in the RRP) in tested Ca2+ levels (0.25, 0.5, and 1 mM).
(H) Quantification of the probability of synaptic vesicle release (Pvr) in various Ca2+ levels (0.25, 0.5, and 1 mM) from the indicated genotypes.
Data are means ± SEM. ###p < 0.001 when compared with wild types, one-way ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001 when compared with UNC-13MR-rescued animals, Student’s t test; n.s., non-significant when compared with UNC-13MR-rescued animals, Student’s t test. The number of worms analyzed for each genotype is indicated under the bar graphs.
Previous data have shown that unc-13 mutant defects in SV priming correlate with reduced morphologically docked SVs at C. elegans NMJs in specimens prepared by high-pressure freeze fixation and freeze substitution (HPF/FS) (Weimer et al., 2006). Based on the hyperosmotic data, we would expect a similar number of docked SVs in UNC-13MR and UNC-13R-rescued animals. Morphometric analysis of synaptic profiles imaged by serial-section electron microscopy (EM) confirmed that the number of docked SVs was significantly reduced in unc-13-null mutants compared with the wild type (Figures 2E and 2F). Both UNC-13MR and UNC-13R restored the SV-docking defect of unc-13 mutants to the same extent (Figures 2E and 2F), indicating that the M domain does not affect overall SV docking, a result that is consistent with the similar RRP sizes determined electrophysiologically. Other parameters, such as the total number of SVs, the terminal area, and the SV distribution in each synaptic profile, were also indistinguishable between UNC-13MR and UNC-13R rescued animals (Figures S2B–S2H). Taken together, these results suggest that the M domain inhibits neurotransmitter release through a post-priming regulatory mechanism.
We next analyzed the probability of SV release in UNC-13MR and UNC-13R-rescued worms. Release probability was first assessed at the single-SV level by calculating the ratio of the mEPSC frequency to the quantal content of the RRP (termed “spontaneous release rate”). The spontaneous release rate was markedly increased by UNC-13R compared with that of UNC-13MR (Figure 2G). Similarly, at the whole-synapse level, the probability of release (Pvr), which was determined as the ratio of the evoked charge transfer to the RRP. Pvr (Gerber et al., 2008) was also significantly enhanced in UNC-13R relative to that of UNC-13MR at all Ca2+ concentrations (0.25, 0.5, and 1 mM Ca2+) as shown in Figure 2H. These results suggest that the M domain of UNC-13MR decreases the overall probability of SV release through molecular mechanisms that act downstream of vesicle priming.
M domain inhibition of SV release acts through the C1 and C2B domains
How might the M domain regulate release probability? Recent studies have shown that the neighboring diacylglycerol-activated C1 domain and the Ca2+/phospholipid-activated C2B domain common to all UNC-13/Munc13 isoforms are involved in the regulation of synaptic-release probability and plasticity (Basu et al., 2007; Shin et al., 2010). Evidence suggests that C1 and C2B are autoinhibitory because disinhibition upon Ca2+ or diacylglycerol (DAG) binding allows a superpriming function of UNC-13/Munc13. Consistent with that model, deletion of the C1 or C2B domain in C. elegans UNC-13L causes an increase in both synaptic transmission and release probability (Li et al., 2019).
Given that the M-domain-mediated inhibition of UNC-13MR is relieved in high-Ca2+ conditions, which can directly or indirectly activate C1 (possibly by producing more DAG) and C2B, we hypothesized that the M domain exerts an inhibitory effect on release probability by regulating the activity of the C1 and C2B domains. If true, we would expect to see a change in M domain inhibition in the absence of the C1 and C2B domains.
Because both the C1 and C2B domains inhibit synaptic transmission mediated by UNC-13L, we first examined whether these two domains have similar functions in UNC-13MR. As shown in Figure S3, deleting the C1 or the C2B domain in UNC-13MR significantly enhanced the frequency of mEPSCs and mIPSCs in the absence of Ca2+, consistent with the results observed in UNC-13L (Li et al., 2019). These results indicate that these two domains are functionally conserved in both UNC-13 isoforms. We next investigated M domain inhibition in the absence of C1, C2B, or both (Figure 3A). Our results revealed that deleting either the C1 or the C2B domain almost abolished the inhibitory effect of the M domain on the mEPSCs (MRΔC1 versus RΔC1, MR-ΔC2B versus RΔC2B; Figures 3B–3E). The inhibition of mIPSCs was decreased in MRΔC1 and MRΔC2B in the absence of Ca2+ but was unaltered in 1-mM Ca2+ (Figures 3B–3E). Furthermore, removing C1 and C2B simultaneously in UNC-13MR completely eliminated the M domain inhibition (MRΔC1ΔC2B versus RΔC1-ΔC2B) of spontaneous release (Figures 3B–3E). This disinhibition was confirmed over a range of Ca2+ concentrations (0.25, 0.5, and 1 mM) (Figures 3F and 3G). These results indicate that the inhibitory effects of the M domain on spontaneous release rely on the presence of the C1 and C2B domains. Moreover, deleting the C1 or the C2B domain or both also abolished the effects of the M domain on mEPSC amplitude (Figures 1E and S5). Similarly, the inhibitory effect of the M domain on stimulus-evoked release was removed in the absence of the C1, C2B, or C1 and C2B domains (Figures 3H and 3I), a finding that was confirmed by comparing the evoked EPSCs in UNC-13MRΔC1-ΔC2B and UNC-13RΔC1ΔC2B at various Ca2+ concentrations (0.25, 0.5, and 1 mM) (Figure 3J). Strikingly, removal of both C1 and C2B increased the evoked EPSC, suggesting that C1 and C2B inhibitory regulation in the short UNC-13MR isoform is additive. Collectively, these results demonstrate that the M domain in UNC-13MR inhibits SV release by regulating the neighboring C1 and C2B domains.
Figure 3. The inhibitory effects of the M domain are eliminated in the absence of the C1 and C2B domains.

(A) Cartoon depicting the domain structure of UNC-13MR and UNC-13R lacking their C1, C2B, or C1 and C2B domains. (B and D) Representative mEPSC and mIPSC traces (recorded at 0-mM and 1-mM Ca2+) from the indicated genotypes.
(C and E) Quantification of the frequency and amplitude of the mEPSCs and mIPSCs from the same genotypes as in (B) and (D).
(F and G) Comparison of the mEPSC and mIPSC frequency between MRΔC1ΔC2B- and RΔC1ΔC2B-rescued animals in various Ca2+ levels (0.25, 0.5, and 1 mM).
(H) Example traces of stimulus-evoked EPSCs from the indicated genotypes.
(I) Quantification of the evoked EPSC amplitude and charge transfer from the same genotypes as in (H).
(J) Comparison of the amplitude and charge transfer of the evoked EPSCs between MRΔC1ΔC2B- and RΔC1ΔC2B-rescued animals in various Ca2+ levels (0.25, 0.5, and 1 mM).
Data are means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, n.s., non-significant when compared with the same C1 or C2B or C1 and C2B deletion in UNC-13MR; one-way ANOVA test for data in (C), (E), and (I), Student’s t test for data in (F), (G), and (J). The number of worms analyzed for each genotype is indicated under the bar graphs.
C1 and C2B domain activation eliminates M domain inhibition
How does the M domain regulate the C1 and the C2B domains to inhibit SV release? It is known that the C1 domain binds phorbol ester/DAG and that the C2B domain interacts with the plasma membrane in the presence of Ca2+ (Maruyama and Brenner, 1991; Shin et al., 2010). Functional studies in UNC-13L have shown mutations in the C1 (H696K) and C2B (D3,4N) domains that mimic DAG and Ca2+ binding, respectively, increase synaptic transmission and release probability by relieving autoinhibition of the MUN domain (Michelassi et al., 2017; Li et al., 2019). This raises the question of whether M-domain-mediated inhibition is still observed in UNC-13MR when C1 and C2B are activated.
To address that question, we first examined whether those C1- and C2B-activating mutations increase synaptic transmission in UNC-13MR, as found in UNC-13L. Our results showed that UNC-13MR carrying the H348K mutation (MRH348K) or the D3,4N mutations (MRD3,4N) produced significantly higher mEPSC and mIPSC frequencies than observed with wild-type UNC-13MR (Figure S3). These results suggest that the C1 and C2B domains are activated by these mutations in UNC-13MR, consistent with previous findings (Basu et al., 2007; Lou et al., 2008; Michelassi et al., 2017; Li et al., 2019) and indicate that the C1 and C2B domains function is conserved in the two UNC-13 isoforms.
We next examined the effect of introducing HK and DN mutations into UNC-13MR and UNC-13R (termed “MRHK,DN” and “RHK,DN”) on M domain functions (Figure 4A). We found that the mEPSC and mIPSC frequencies (in 0- and 1-mM Ca2+), as well as the evoked EPSC amplitude and charge transfer (in 1-mM Ca2+), were indistinguishable between MRHK,DN- and RHK,DN-rescued worms (Figures 4B–4E), demonstrating that the inhibitory effects of the M domain are diminished by the activity of the C1 and C2B domains and are eliminated upon the activation of both domains, simultaneously.
Figure 4. Activation of the C1 and C2B domains eliminates the M-domain inhibition.

(A) Cartoon depicting the HK and DN mutations in UNC-13MR and UNC-13R.
(B) Representative mEPSC and mIPSC traces (recorded in 0-mM and 1-mM Ca2+) from the MRHK,DN and RHK,DN transgenic worms.
(C) Quantification of the mEPSC and mIPSC frequencies from the same genotypes as in (B).
(D) Example traces of stimulus-evoked EPSCs from the MRHK,DN and RHK,DN transgenic worms (recorded in 1-mM Ca2+).
(E) Quantification of the evoked EPSC amplitude and charge transfer from the same genotypes as in (D).
Data are means ± SEM. n.s., non-significant when compared with UNC-13MRHK,DN-rescued animals, Student’s t test. The number of worms analyzed for each genotype is indicated under the bar graphs.
Prior studies have demonstrated that the C2B domain exhibits Ca2+-dependent interaction with the plasma membrane (Shin et al., 2010). However, the Ca2+ dependency of the C1 domain binding to DAG remains controversial (Maruyama and Brenner, 1991; Betz et al., 1998; Shen et al., 2005). Nevertheless, Ca2+ is required for PLCβ to hydrolyze PIP2 to produce DAG (Ellis et al., 1998), which subsequently activates the C1 domain. It is, therefore, possible that both the C1 and C2B domains are more active at higher Ca2+ levels and that the M domain should produce relatively weak inhibition in high Ca2+ and strong inhibition in low Ca2+, a notion which is well supported by our findings. Compared with UNC-13MR-rescued animals, the ratio of increase in evoked charge in UNC-13R-rescued animals was markedly decreased in high Ca2+ (Figure 1K). Taken together, our results indicate that the M domain inhibits synaptic transmission by suppressing the activity of the C1 and C2B domains and that Ca2+-induced activation of the C1 and C2B domains weakens M domain inhibition.
The M domain facilitates SV release when fused to the MUNC2C fragment
Compared to UNC-13MR, the evoked EPSCs rescued by UNC-13MRΔC1ΔC2B were dramatically increased (charge transfer, 13pC versus 60pC; Figures 1H and 3I). The N terminus of MR-ΔC1ΔC2B contains only the M domain and three linkers (Figure 4A), a sequential structure that is similar to that of a previously studied hyperactive truncated form of UNC-13L (i.e., super UNC-13 or sUNC-13, produced by deleting the X, C1, and C2B domains of UNC-13L), leaving the C2A domain and the same three linkers in its N terminus (Li et al., 2019). The evoked EPSCs rescued by sUNC-13 were increased 6-fold compared with UNC-13L (20pC versus 120pC) (Li et al., 2019). Moreover, the C2A domain in sUNC-13 produced strong facilitation of SV release.
Prompted by these results, we hypothesized that the M domain in MRΔC1ΔC2B might also facilitate SV release. However, such effect may be masked by the presence of the linker domains (Figure 4A), which can enhance synaptic transmission via an unknown mechanism (Figures 3H and 3I; see Discussion) (Li et al., 2019). Given that the MUNC2C fragment has been proposed to be the minimal region for SV-priming and Ca2+-triggered fusion (Basu et al., 2005; Madison et al., 2005; Liu et al., 2016), we tested the possibility that the M domain promotes SV release by directly fusing it to the MUNC2C fragment (termed “M-MUNC2C”; Figure 5A). We first assessed the role of the MUNC2C fragment in rescuing spontaneous and evoked release in unc-13 mutants. Overexpression of MUNC2C in unc-13 mutants partially restored mEPSCs and almost fully restored mIPSCs (Figures 5B–5E), indicating that this fragment is still functional for spontaneous release. The evoked EPSCs, however, were only slightly restored by MUNC2C overexpression (Figures 5F and 5G), consistent with previous findings (Madison et al., 2005; Liu et al., 2016). These results indicate that MUNC2C is likely to be the minimal fragment responsible for spontaneous and evoked release. Notably, MUNC2C exhibited better rescue in spontaneous release than it did evoked release, possibly reflecting previous observations that these two types of release modes are differentially regulated by the fusion machinery (Varoqueaux et al., 2005; Liu et al., 2019b). Moreover, the MUNC2C fragment appeared to better rescue inhibitory neurotransmitter release, consistent with prior studies that found that bMunc13–2 is almost fully functional in inhibitory hippocampal synapses but not in excitatory synapses (Augustin et al., 1999a). We did not see a further increase of mEPSCs or mIPSCs in M-MUNC2C-rescued animals (Figures 5B–5E), suggesting that the M domain does not facilitate spontaneous release. However, compared with MUNC2C-rescued animals, the evoked EPSCs in the M-MUNC2C chimera were dramatically increased (Figures 5F and 5G). The increase in evoked EPSCs was also observed at a lesser Ca2+ concentration (Figure 5H). These results suggest that the M domain has an intrinsic facilitatory capability that promotes evoked neurotransmitter release in this chimeric construct. The sucrose-evoked currents rescued by MUNC2C and M-MUNC2C were comparable (Figures 5I and 5J), suggesting that the M domain does not affect SV priming. Instead, the low-release probability of MUNC2C was significantly enhanced by the inclusion of the M domain (Pvr; Figure 5J). Taken together, these results reveal a facilitatory function of the M domain in evoked neurotransmitter release.
Figure 5. The M domain promotes evoked neurotransmitter release and enhances release probability.

(A) Cartoon depicting the domain structure of UNC-13 MUNC2C and M-MUNC2C.
(B and C) Representative mEPSC and mIPSC traces (recorded in 0-mM and 1-mM Ca2+) from wild-type (black), unc-13 (gray), MUNC2C overexpression rescued (yellow), and M-MUNC2C overexpression rescued (green) animals.
(D and E) Quantification of the frequency of the mEPSCs and mIPSCs from the same genotypes as in (B) and (C).
(F) Example traces of stimulus-evoked EPSCs from wild-type (black), unc-13 (gray), MUNC2C overexpression rescued (yellow)m and M-MUNC2C overexpression rescued (green) animals (recorded in 1-mM Ca2+).
(G) Quantification of the evoked EPSC amplitude and charge transfer from the same genotypes as in (F).
(H) Quantification of the evoked EPSCs amplitude in various Ca2+ concentrations (0.25, 0.5, and 1 mM).
(I) Hypertonic sucrose-evoked current recorded from wild-type (black), unc-13 (gray), MUNC2C overexpression rescued (blue), and M-MUNC2C overexpression rescued (red) animals.
(J) Averaged charge transfer from the sucrose-evoked currents in (I), and quantification of the probability of synaptic vesicle release (Pvr) from the indicated genotypes. Data are means ± SEM. ###p < 0.001 when compared with the wild type; **p < 0.01, ***p < 0.001 when compared with the MUNC2C overexpression rescued animals; n.s., non-significant when compared with the MUNC2C overexpression rescued animals; one-way ANOVA test for data in (D), (E), and (H); one-way ANOVA following the Kruskal-Wallis test for data in (G) and (H).
(K) Example traces of mEPSCs and mIPSCs from MUNC2C SCI rescue and M-MUNC2C SCI rescue animals (in 1 mM Ca2+).
(L) Quantification of the mEPSC and mIPSC frequencies in (K).
(M and N) Representative traces of evoked EPSCs and quantification of the amplitude and charge transfer from the indicated genotypes. Data are means ± SEM.
**p < 0.01, ***p < 0.001 when compared with the MUNC2C SCI rescued animals; Student’s t test. The number of worms analyzed for each genotype is indicated under the bar graphs.
Because the inhibitory effects of the M domain do not rely on the expression level of UNC-13MR (Figures 1 and S1), we next examined whether the facilitatory function of the M domain depends on the expression level of M-MUNC2C. To do that, we constructed MUNC2C and M-MUNC2C single-copy insertion (SCI) rescue in unc-13 mutants. Unlike MUNC2C overexpression, we found that MUNC2C SCI failed to restore locomotion (data not shown) and synaptic transmission (Figures 5K–5N). Both spontaneous and evoked EPSCs (in 1-mM Ca2+) in the MUNC2C SCI-rescued animals were nearly the same as those in the unc-13 mutants. These results indicate that the rescue ability of MUNC2C is dependent on expression level. Similarly, M-MUNC2C SCI did not restore synaptic transmission to the same level as that obtained with M-MUNC2C overexpression. However, the rescue of both spontaneous and evoked release by M-MUNC2C SCI was significantly increased compared with that observed after rescue of MUNC2C SCI (Figures 5K–5N). These results further demonstrate that the M domain is capable of enhancing Ca2+-dependent neurotransmitter release and release probability, despite the expression level of the UNC-13 fragment.
Expression level of the UNC-13 proteins is not regulated by the M domain
The inhibitory and facilitatory functions of the M domain may arise from its roles in regulating the abundance of UNC-13 proteins. To investigate that, we made fusion proteins of UNC-13MR, UNC-13R, MUNC2C, and M-MUNC2C, tagged with mApple in their C terminus. The expression levels of UNC-13 proteins were measured based on the mApple fluorescence intensity. As shown in Figure 6, UNC-13MR::mApple and UNC-13R::mApple displayed comparable fluorescence intensity, suggesting that they were expressed at similar levels. Similarly, the fluorescence intensities of MUNC2C::mApple and M-MUNC2C::mApple were also indistinguishable. These results demonstrate that the inhibition and facilitation in synaptic transmission and release probability mediated by the M domain do not result from changes in the abundance of UNC-13 proteins.
Figure 6. The M domain does not regulate UNC-13 protein level and localization.

(A) Representative confocal z stack images for mApple-tagged UNC-13MR, UNC-13R, MUNC2C, and M-MUNC2C (all driven by the unc-129 promoter). Scale bar, 5 μm.
(B) Quantification of the fluorescence intensity from the lines in (A).
(C) Representative confocal z stack images for UNC-13MR and UNC-13R (tagged with mApple), and their colocalization with UNC-10/RIM (tagged with GFP). Scale bar, 5 μm.
(D) Quantification of the Pearson correlation coefficient between UNC-13MR/UNC-13R and UNC-10.
(E) Representative confocal z stack images for UNC-13MR::mApple, MRΔC1ΔC2B::mApple, UNC-13R::mApple, and RΔC1ΔC2B::mApple.
(F) Quantification of the fluorescence intensity from the lines in (E).
Data are means ± SEM. *p < 0.05, n.s., non-significant, one-way ANOVA. The number of worms analyzed for each genotype is indicated in the bar graphs.
To determine whether the M domain is required for the localization of Unc-13 proteins, we assessed the position of UNC-13MR and UNC-13R relative to UNC-10/RIM. We have previously demonstrated that compared with UNC-13L, which displays strong colocalization with UNC-10/RIM, UNC-13MR localizes further away from UNC-10/RIM, leading to a lower Pearson correlation coefficient (Hu et al., 2013). Here, we found that the Pearson correlation coefficient between UNC-13R and UNC-10 was indistinguishable from that between UNC-13MR and UNC-10, indicating that the subcellular localization of the Unc-13 proteins is not altered by the lack of the M domain (Figures 6C and 6D).
We next asked whether the dramatically increased synaptic transmission in MRΔC1ΔC2B and RΔC1ΔC2B are caused by enhanced Unc-13 protein levels. To assess that, we made fusion proteins of MRΔC1ΔC2B and RΔC1ΔC2B, tagged with mApple. By measuring the mApple fluorescence, we found that the RΔC1ΔC2B::mApple was not significantly different from that of UNC-13R::mApple, whereas the MRΔC1ΔC2B::mApple was slightly decreased compared with UNC-13MR::mApple (Figures 6E and 6F). These results indicate that the increased synaptic transmission in MRΔC1ΔC2B and RΔC1ΔC2B-rescued animals arise from functional changes in Unc-13 proteins but not in their expression levels.
The bMunc13–2 N terminus inhibits synaptic transmission
These experiments revealed functions of the N-terminal M domain in UNC-13MR in regulating release probability. Both bMunc13–2 and Munc13–3 also lack a C2A domain in their N terminus and have been found to regulate the kinetics of the priming pool in some synapses (Chen et al., 2013). UNC-13MR in the worm also has essential roles in regulating the kinetics of SV fusion (Hu et al., 2013), suggesting that the UNC-13 and Munc13 proteins lacking a C2A domain may function similarly. However, sequence analysis of the N terminus in bMunc13–2 and Munc13–3, revealed no similarity with that of the M domain in UNC-13MR (Figure S4). In fact, these N-terminal sequences have no homology to any known proteins (Augustin et al., 1999b; Kohn et al., 2000). Despite the lack of sequence similarity, we examined whether mammalian N-terminal domain could exert similar functions to the UNC-13 M domain in regulating synaptic transmission.
We focused on bMunc13–2 because it is the primary Munc13 isoform to be coexpressed with Munc13–1 in some synapses, such as those in the hippocampus (Varoqueaux et al., 2002), although Munc13–3 has also been found in a subpopulation of hippocampal synapses (Netrakanti et al., 2015). Moreover, bMunc13–2 can functionally replace Munc13–1 to mediate SV release in inhibitory hippocampal neurons (Varoqueaux et al., 2002), demonstrating the importance of this isoform. We, therefore, constructed chimeric UNC-13 proteins containing the N terminus of bMunc13–2 (0–765aa) fused to the worm UNC-13R (termed “NbMunc13−2R”) to examine whether it inhibits SV release and to the worm MUNC2C (termed “NbMunc13−2MUNC2C”) to determine whether it is capable of facilitating C. elegans MUNC2C function (Figure 7A). The bMunc13–2 N terminus did not appear to inhibit spontaneous release because both mEPSCs and mIPSCs were unaltered in NbMunc13−2R-rescued animals compared with those in UNC-13R-rescued animals (Figures 7B and 7C; Table S1). However, we found strong inhibition by the bMunc13–2 N terminus on evoked EPSCs in NbMunc13−2R-rescued animals (Figures 7D and 7E; Table S2), consistent with the results in UNC-13MR-rescued worms. These results indicate that the bMunc13–2 N terminus also inhibits Ca2+-dependent neurotransmitter release, similar to the M domain of UNC-13MR. Compared with MUNC2C, evoked EPSCs were significantly increased in NbMunc13−2MUNC2C-rescued animals (Figures 7D and 7E; Tables S1 and S2), whereas spontaneous EPSCs were unchanged (Figures 7B and 7C; Table S1), demonstrating that the bMunc13–2 N terminus is able to facilitate SV release. Taken together, these results revealed that the N terminus of bMunc13–2 exhibits similar inhibitory and facilitatory functions to that involving the M domain of UNC-13MR in regulating Ca2+-dependent neurotransmitter release, likely via conserved mechanisms.
Figure 7. The N terminus in bMunc13–2 exhibits both inhibition and facilitation in synaptic transmission.

(A) Cartoon depicting the domain structure of UNC-13R, NbMunc13−2-R, MUNC2C, and NbMunc13−2-MUNC2C.
(B) Representative mEPSC and mIPSC traces (recorded in 1-mM Ca2+) from the indicated genotypes (UNC-13R, NbMunc13−2-R, MUNC2C, and NbMunc13−2-UNC2C rescue animals).
(C) Quantification of the frequency and amplitude of the mEPSCs and mIPSCs from the same genotypes as in (B).
(D) Example traces of stimulus-evoked EPSCs (recorded in 1mM Ca2+) from indicated genotypes.
(E) Quantification of the evoked EPSC amplitude and charge transfer from the same genotypes as in (D).
Data are means ± SEM. #p < 0.05 when compared with MUNC2C rescue; **p < 0.01 when compared with UNC-13R rescue; n.s., non-significant, one-way ANOVA. The number of worms analyzed for each genotype is indicated under the bar graphs.
DISCUSSION
Although the functional importance of UNC-13/Munc13 proteins lacking a C2A domain has been reported, the detailed mechanisms by which they regulate neurotransmitter release and release probability are poorly understood. By conducting a structure-function analysis of the worm UNC-13MR isoform, we show that the N-terminal M domain strongly inhibits neurotransmitter release and release probability. This inhibitory function appears to be conserved because the N-terminal sequence of bMunc13–2 can substitute for the M domain of UNC-13MR. We further demonstrate that M domain-mediated inhibition requires the presence of the C1 and the C2B domains, with activation of both C1 and C2B mitigating M-domain inhibition. Furthermore, we demonstrate that the M domain has an intrinsic facilitatory ability and strongly promotes neurotransmitter release and release probability when fused to the N terminus of the MUNC2C fragment, suggesting multiple effects of the M domain in UNC-13MR. This study reveals interesting and important regulatory functions of the M domain, thereby providing mechanistic insights by which UNC-13/Munc13 regulates synaptic transmission and release probability.
Sequences and functions of the N terminus in UNC-13s/Munc13s
Based on their N-terminal sequences, UNC-13/Munc13 isoforms can be divided into two groups based on the presence or absence of the C2A domain. The C-terminal sequences (spanning from the C1 to the C2C domain) in all isoforms share high sequence similarity in domain structures. Among the UNC-13/Munc13 isoforms that have an N-terminal C2A domain, sequence similarity in this region is also apparent. In contrast, the M domain of worm UNC-13MR has no sequence homology to other proteins (Kohn et al., 2000), including the N-terminal sequences of bMunc13–2 and Munc13–3 (Figure S4), which also lack a C2A domain. Furthermore, the N-terminal sequences of bMunc13–2 and Munc13–3 only share a small stretch of similarity, spanning 54 residues (Figure S4, blue) (Augustin et al., 1999b) that does not exist in the worm UNC-13 M domain. Both bMunc13–2 and Munc13–3 have been shown to be important in synaptic transmission (Augustin et al., 2001; Rosenmund et al., 2002; Varoqueaux et al., 2002). Moreover, recent studies have demonstrated that the N-terminal of bMunc13–2 contains a coiled-coil motif, which binds to the active zone protein ELKS1 and regulates synaptic localization and function of bMunc13–2 (Kawabe et al., 2017). The lack of sequence conservation between the N-terminal of UNC-13 and bMunc13–2/Munc13–3 prompted us to address the function of the M domain in C. elegans. Does this region regulate synaptic transmission? Do the N termini of these three UNC-13/Mun13 proteins have conserved functions and are they similar to the C2A domain? What is their physiological relevance? These questions motivated us to investigate the functional roles of the M domain in C. elegans.
Prior studies have shown that the N-terminal C2A domain in both UNC-13L and Munc13–1 form inhibitory homodimers that undergo a molecular switch upon C2A binding to the zinc finger domain of UNC-10/RIM forming UNC-10/UNC-13 heterodimers that promote release (Camacho et al., 2017; Liu et al., 2019a). By contrast, little is known about the biochemical characteristics of the N-terminal domains in UNC-13MR, bMunc13–2, and Munc13–3. Nevertheless, our data show that the N-terminal domains in UNC-13MR and bMunc13–2 exhibit conserved functions, inhibiting synaptic transmission mediated by UNC-13R. Furthermore, attaching the UNC-13 M domain or bMunc13–2 N-terminal is able to facilitate synaptic transmission mediated by the minimal fusion domain encompassing MUNC2C, indicating that the N termini exhibit functional conservation.
Given that UNC-13/Munc13 proteins have been implicated in multiple steps of the SV cycle, including docking, priming, and fusion (Aravamudan et al., 1999; Augustin et al., 1999a; Richmond et al., 1999; Varoqueaux et al., 2002; Madison et al., 2005; Weimer et al., 2006), we assessed the involvement of the M domain in each of these stages. Our EM data revealed that the number of docked vesicles in UNC-13R-rescued animals is indistinguishable from that in UNC-13MR (Figure 2). Similarly, the SV numbers in the primed vesicle pool derived from hyperosmotic responses were also comparable between UNC-13MR and UNC-13R-rescued animals. Furthermore, the sucrose-evoked currents in the M-MUNC2C chimera were unchanged compared with those of MUNC2C. Together, these results suggest that the M domain functions downstream of docking and priming to regulate release probability.
Possible model for the M-domain inhibition
Both spontaneous and evoked neurotransmitter release were significantly increased in UNC-13R-rescued animals lacking the M domain, revealing an inhibitory function of that domain (Figure 1). Notably, this M-domain inhibition only occurred in the presence of the neighboring C1 and C2B domains. Deletion of C1, C2B, or both exhibited similar release properties in UNC-13MR and UNC-13R. Mutations that mimic the activation of C1 and C2B also abolished M-domain inhibition (Figure 4). These results indicate that the M domain exerts an inhibitory function by suppressing the activation of the C1 and C2B domains. However, it is still unclear how that suppression occurs. Because the activity of C1 and C2B is Ca2+ dependent (Maruyama and Brenner, 1991; Shin et al., 2010), it is possible that the M domain suppresses Ca2+ entry, thereby inhibiting Ca2+-triggered neurotransmitter release. However, that would not explain why M-domain inhibition was strongest in 0-mM Ca2+.
Analysis of the crystal structure of Munc13 C1C2BMUN suggests a “bridge model,” in which the C2C domain interacts with the vesicle membrane and the C1-C2B act as a module interacting with both the plasma membrane and the MUN domain (Liu et al., 2016; Xu et al., 2017). The interaction between C2B and the MUN domain is not thought to interfere with the ability of the MUN domain to bind syntaxin, correctly orient SNARE complex assembly, and prime vesicles for fusion (Lai et al., 2017; Michelassi et al., 2017; Palfreyman and Jorgensen, 2017). Instead, evidence suggests that activation of the C1-C2B module by Ca2+ or DAG causes a conformational change at the membrane interface, which also alleviates autoinhibition of the MUN domain by C2B, allowing the MUN domain to increase release probability (superpriming) through an unspecified post-priming function (Basu et al., 2007; Michelassi et al., 2017). It is, therefore, possible that the M domain stabilizes (or locks) the C1 and C2B in their inactive membrane-bound conformation. This would allow vesicle priming to occur but lead to lower release probabilities (Figure S6A). Sufficient Ca2+ influx may relieve M-domain inhibition (unlocking) of C1 and C2B, allowing them to undergo conformational changes that allow the MUN domain to enhance release probability (Figure S6B). This would explain why M-domain inhibition is strong in low Ca2+ but weak in high Ca2+. In the complete absence of Ca2+, the M domain stabilization or lock would be even tighter, and UNC-13MR would be unable to trigger Ca2+-independent neurotransmitter release.
It should be noted that changes in the number of release sites at active zones also affect release probability. Prior studies have reported that Munc13 proteins are involved in the assembly of multiple release sites at a single synapse (Böhme et al., 2016; Sakamoto et al., 2018). Thus, although the total docked and primed SVs are unaltered, the greater release probability in UNC-13R-rescued animals may arise from more release sites. A future study will focus on this possibility to uncover more potential roles of the M domain.
Facilitatory function of the M domain
The facilitatory effects of the N-terminal C2A domain in UNC-13L can also be observed in a chimera consisting of C2AMUNC2C, which markedly increases spontaneous and evoked release and release probabilities compared with MUNC2C alone (Li et al., 2019). Moreover, the C2A domain also increased spontaneous release in the absence of Ca2+, indicating that its facilitatory function is Ca2+ independent. By contrast, the M domain facilitated SV release in a Ca2+-dependent manner. Increases in evoked EPSCs by the M-MUNC2C chimera were observed in 1-mM and 0.5-mM Ca2+, but not in 0.25-mM Ca2+ (Figure 5H), and no increase in spontaneous release was found in the presence or absence of Ca2+. These results demonstrate that, unlike the C2A domain, the facilitation of synaptic transmission by the M domain is Ca2+ dependent.
How does the M domain promote release of SVs? Prior studies have shown that the MUNC2C fragment is the minimal domain responsible for UNC-13/Munc13 activity. Overexpression of MUNC2C restored only 20% of the evoked EPSCs in unc-13 mutants, demonstrating that this fragment is inefficient in triggering synaptic transmission, possibly because this domain is not stable under physiological conditions. Another possibility is that the MUNC2C fragment is unable to efficiently connect the vesicle and the plasma membranes to establish the UNC-13 bridge. It is, therefore, likely that the M domain directly or indirectly interacts with the plasma membrane or other unknown membrane protein(s), thereby bridging the two membranes (Figure S6C). In this way, the M domain could stabilize the MUN domain in a conformation and position to catalyze SNARE complex assembly and promote fusion.
The N-terminal of bMunc13–2 also exhibited inhibitory and facilitatory regulation when fused to the UNC-13R or the UNC-13 MUNC2C fragment (Figure 7), similar to the UNC-13 M domain. These results suggest that the N termini of UNC-13MR and bMunc13–2 use similarly conserved mechanisms to regulate synaptic transmission. Prior studies have shown that a short stretch of sequence (Figure S4) in the N-terminal of bMunc13–2 forms a coiled-coil structure, and that motif binds to the active-zone protein ELKS1 to localize bMunc13–2 at active zones and regulate SV priming (Kawabe et al., 2017). Thus, the facilitatory function of the M domain may also arise from a direct interaction with ELKS1. However, the UNC-13M domain does not contain a similar binding motif, and no coiled-coil structure is predicted in the M domain, suggesting that the mechanism whereby ELKS1 recruits bMunc13–2 to the active zone may not exist in worm UNC-13MR. Nevertheless, our results cannot rule out the possibility that the UNC-13 M domain interacts with other active zone proteins to facilitate synaptic transmission. Interestingly, one recent study has found that Syd-1 clusters UNC-13B at specific positions of the release site at Drosophila synapses (Fulterer et al., 2018), indicating that multiple active-zone proteins may be involved in the recruitment of UNC-13/Munc13 proteins.
Functional diversity of UNC-13s/Munc13s in regulating release probability
As important synaptic-hub proteins, UNC-13s/Munc13s contain multiple functional domains, which have various binding partners, such as UNC-10/RIM, DAG, Ca2+/phospholipids, and the SNARE proteins. Because of their binding properties, UNC-13/Munc13 proteins are involved in several cellular pathways by acting as essential substrates that regulate synaptic release (Betz et al., 1998; Lackner et al., 1999; Hu et al., 2015). Moreover, modulation in the function of UNC-13s/Munc13s alters superpriming in several synapse types (Lee et al., 2013; Taschenberger et al., 2016). Previous studies, together with our recent work, have shown that distinct domains in UNC-13L/Munc13–1 have opposing regulatory functions in synaptic transmission and release probability. The C2A domain promotes SV release and enhances release probability (Camacho et al., 2017; Liu et al., 2019a), whereas the X, C1, and C2B domains inhibit synaptic transmission and release probability (Michelassi et al., 2017; Li et al., 2019). Thus, the net regulatory effect of UNC-13L/Munc13–1 on synaptic transmission reflects a balance of facilitation and inhibition. In this study, we further demonstrate that the C1 and C2B domains inhibit synaptic transmission in UNC-13MR, analogous to their functions in UNC-13L. The M domain also inhibits synaptic transmission and release probability through a mechanism that involves C1 and C2B. These findings, therefore, provide significant insights into the mechanisms that govern the function of UNC-13/Munc13 isoforms that lack C2A domains in synaptic release probability and short-term plasticity. Future studies will focus on the biochemical characteristics and crystal structure of the M domain to further elucidate mechanistic details by which this occurs.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zhitao Hu (z.hu1@uq.edu.au).
Materials availability
All new strains and plasmids created in this study will be provided on request with no restrictions.
Data and code availability
This study did not generate/analyze datasets/code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strain maintenance and genetic manipulation were performed as previously described (Brenner, 1974). Animals were cultivated at room temperature on nematode growth medium (NGM) agar plates seeded with OP50 bacteria. On the day before experiments L4 larval stage animals were transferred to fresh plates seeded with OP50 bacteria for all the electrophysiological recordings. The following strains were used:
Wild-type, N2 bristol KP6901 unc-13(s69)
ZTH456 hztSi1 [Psnb-1::UNC-13MR]; unc-13(s69)
CZ15872 juSi67 [Prgef-1::UNC-13R]; unc-13(s69) (Zhou et al., 2013)
ZTH11 hztEx11 [Psnb-1::UNC-13MR]; unc-13(s69)
ZTH99 hztEx20 [Psnb-1::UNC-13R]; unc-13(s69)
ZTH498 hztEx53 [Psnb-1::UNC-13MRΔC1]; unc-13(s69)
ZTH521 hztEx68 [Psnb-1::UNC-13RΔC1]; unc-13(s69)
ZTH517 hztEx55 [Psnb-1::MRΔC2B]; unc-13(s69)
ZTH507 hztEx69 [Psnb-1::UNC-13RΔC2B]; unc-13(s69)
ZTH495 hztEx70 [Psnb-1::MRΔC1ΔC2B]; unc-13(s69)
ZTH696 hztEx109 [Psnb-1::UNC-13RΔC1ΔC2B]; unc-13(s69)
ZTH421 hztEx54 [Psnb-1::UNC-13MRH348K]; unc-13(s69)
ZTH447 hztEx56 [Psnb-1::UNC-13MRD3,4N]; unc-13(s69)
ZTH451 hztEx72 [Psnb-1::UNC-13MRHK, D3,4N]; unc-13(s69)
ZTH448 hztEx71 [Psnb-1::UNC-13RHK, D3,4N]; unc-13(s69)
ZTH858 hztSi7 [Prab-3::UNC-13MUNC2C]; unc-13(s69)
ZTH752 hztSi5 [Prab-3::UNC-13M-MUNC2C]; unc-13(s69)
ZTH442 hztEx107 [Psnb-1::UNC-13MUNC2C]; unc-13(s69)
ZTH577 hztEx108 [Psnb-1::UNC-13M-MUNC2C]; unc-13(s69)
ZTH738 hztEx111 [Psnb-1:: NbMunc13−2::UNC-13R]; unc-13(s69)
ZTH740 hztEx112 [Psnb-1::NbMunc13−2:: UNC-13(MUNC2C)]; unc-13(s69)
ZTH686 hztEx79 [Punc-129::UNC-13MR::mApple]; NuIs165 [Punc-129::UNC-10::GFP]
ZTH711 hztEx80 [Punc-129::UNC-13R::mApple]; NuIs165 [Punc-129::UNC-10::GFP]
ZTH693 hztEx82 [Punc-129::UNC-13MUNC2C::mApple]; NuIs165 [Punc-129::UNC-10::GFP]
ZTH692 hztEx81 [Punc-129::UNC-13(M-MUNC2C)::mApple]; NuIs165 [Punc-129::UNC-10::GFP]
ZTH924 hztEx113 [Psnb-1::NbMunc13−2(Δ145−187)::UNC-13R]; unc-13(s69)
ZTH921 hztEx95 [Punc-129::UNC-13MRΔC1ΔC2B::mApple]; NuIs165 [Punc-129::UNC-10::GFP]
ZTH702 hztEx66 [Punc-129::UNC-13RΔC1ΔC2B::mApple]; NuIs165 [Punc-129::UNC-10::GFP]
METHOD DETAILS
Constructs, transgenes and germline transformation
The snb-1 promoter (3kb) and unc-129 promoter (2.6kb) were inserted into JB6 vector between the SphI and BamHI sites, all the UNC-13 expression constructs were amplified by PCR and inserted into JB6 vector between the KpnI and NotI sites. Red fluorescent protein mApple was inserted between NotI and MluI sites in-frame with unc-13 genes for imaging experiments. The miniMos-based single-copy insertion plasmid KP3313 (modified from pCFJ910) was gift from Josh Kaplan Lab. The rab-3 promoter (1.2kb) was inserted into KP3313 between SbfI and XmaI, unc-13 genes were constructed using AscI and NotI.
Transgenic strains were isolated by microinjection of various plasmids using either Pmyo-2::NLS-GFP (KP#1106) or Pmyo-2::NLS-mCherry (KP#1480) as the co-injection marker.
Single-copy insertions were generated using miniMos-based single copy insertion (minimosSCI) method, as previously described (Frøkjaer-Jensen et al., 2014).
Electrophysiology
Electrophysiology was conducted on dissected C. elegans as previously described (Li et al., 2019; Liu et al., 2019a). Worms were superfused in an extracellular solution containing 127 mM NaCl, 5 mM KCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 20 mM glucose, 1 mM CaCl2, and 4 mM MgCl2, bubbled with 5% CO2, 95% O2 at 22°C. The 1mM CaCl2 was replaced by 1mM MgCl2 to record mEPSCs and mIPSCs in 0mM of Ca2+. Whole-cell recordings were carried out at −60mV for all EPSCs, including mEPSCs, evoked EPSCs, and sucrose-evoked responses. The holding potential was switched to 0mV to record mIPSCs. The internal solution contained 105 mM CH3O3SCs, 10 mM CsCl, 15 mM CsF, 4mM MgCl2, 5mM EGTA, 0.25mM CaCl2, 10mM HEPES, and 4mM Na2ATP, adjusted to pH 7.2 using CsOH. Stimulus-evoked EPSCs were stimulated by placing a borosilicate pipette (5–10 μm) near the ventral nerve cord (one muscle distance from the recording pipette) and applying a 0.4 ms, 85 μA square pulse using a stimulus current generator (WPI).
For estimating the readily releasable pool size, a pipette containing 1M sucrose solution was placed at the end of the patched muscle cell (around half muscle distance from the recording pipette), and a 20 psi, 2 s pressure pulse was applied by Picospritzer (Parker) to create a rapid jump in osmolarity at the neuromuscular junction (< 200 msec latency on average). The integrated charge transfer was computed as a function of time throughout the sucrose delivery, and the charge accumulation was corrected for the baseline holding current and spontaneous fusion events prior to sucrose application.
Fluorescence imaging
Animals were immobilized on 2% agarose pads with 30 mM levamisole. Fluorescence imaging was performed on a spinning-disk confocal system (3i Yokogawa W1 SDC) controlled by Slidebook 6.0 software. Animals were imaged with an Olympus 100× 1.4 NA Plan-Apochromat objective. Z series of optical sections were acquired at 0.11 μm steps. Images were deconvolved with Huygens Professional version 16.10 (Scientific Volume Imaging, the Netherlands) and then processed to yield maximum intensity projections using ImageJ 1.51n (Wayne Rasband, National Institutes of Health) (Schneider et al., 2012).
Electron microscopy
Samples were prepared using high-pressure freeze fixation (Weimer et al., 2006). ~30 young adult hermaphrodites were placed in each specimen chamber containing E. coli and were rapidly frozen at −180°C under high pressure (Leica HPM 100). Frozen specimens then underwent freeze substitution (Leica Reichert AFS) during which samples were held at −90°C for 107 hr in 0.1% tannic acid and 2% OsO4 in anhydrous acetone. The temperature was then increased at 5°C/hr to −20°C, kept at −20°C for 14 hr, and increased by 10°C/hr to 20°C. After fixation, samples were infiltrated with 50% Epon/acetone for 4 hr, 90% Epon/acetone for 18 hr, and 100% Epon for 5 hr. Finally, samples were embedded in Epon and incubated for 48 hr at 65°C. Ultrathin (40 nm) serial sections were cut using a Leica Ultracut 6 and collected on formvar-covered, carbon-coated copper grids (EMS, FCF2010-Cu). Poststaining was performed using 2.5% aqueous uranyl acetate for 4 min, followed by Reynolds lead citrate for 2 min. Images were acquired using a JEOL JEM-1220 transmission electron microscope operating at 80 kV using a Gatan digital camera at a magnification of 100k (1.8587 pixels/nm). Images were collected from the ventral nerve cord region anterior to the vulva for all genotypes. Serial micrographs were manually annotated using NIH ImageJ/Fiji software to quantify the number of SVs, number of plasma membrane docked SVs, and distance of docked SVs from the dense projection (DP) for each synaptic profile. Specimens were encrypted to ensure unbiased analysis. Cholinergic synapses were identified on the basis of their typical morphology (White et al., 1986). A synapse was defined as a series of sections (profiles) containing a dense projection as well as two flanking sections on both sides without dense projections. SVs were identified as spherical, light gray structures with an average diameter of ~30 nm. A docked synaptic vesicle was defined as a synaptic vesicle whose membrane was morphologically contacting the plasma membrane.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data acquisition and analysis
All electrophysiological data were obtained using a HEKA EPC10 double amplifier (HEKA Elektronik) filtered at 2 kHz, and analyzed with open-source scripts developed by Eugene Mosharov (http://sulzerlab.org/Quanta_Analysis_8_20.ipf) in Igor Pro 7 (Wavemetrics). All imaging data were analyzed in ImageJ software. Each set of data represents the mean ± SEM of an indicated number (n) of animals. To analyze mEPSCs and mIPSCs, a 4pA peak threshold was preset, above which release events are clearly distinguished from background noise. The analyzed results were re-checked by eye to ensure that the release events were accurately selected.
Statistical analysis
All data were statistically analyzed in Prism 8 software. Normality distribution of the data was determined by the D’Agostino-Pearson normality test. When the data followed a normal distribution, an unpaired Student’s t test (two-tailed) or one-way ANOVA was used to evaluate the statistical significance. In other cases a Mann-Whitney test or one-way ANOVA following Kruskal-Wallis test was used. A summary of all electrophysiological data is provided in Tables S1 and S2, with the results presented as mean ± SEM (* p < 0.05, ** p < 0.01, ***p < 0.001).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli OP50 | Caenorhabditis Genetics Center (CGC) | OP50 |
| Chemicals, peptides, and recombinant proteins | ||
| CaCl2 | Fluka Analytical | Cat#21114 |
| MgCl2 | Fluka Analytical | Cat#63020 |
| NaCl | Sigma-Aldrich | Cat#S9888 |
| NaHCO3 | Sigma-Aldrich | Cat#S6014 |
| Sucrose | Sigma | Cat#S9378 |
| KCl | Sigma | Cat#P5405 |
| NaH2PO4 | Sigma-Aldrich | Cat#S0751 |
| Glucose | Sigma | Cat#G8270 |
| CsCl | Sigma | Cat#C3309 |
| CsF | Aldrich | Cat#289345 |
| EGTA | Sigma | Cat#E3889 |
| HEPES | Sigma | Cat#H4034 |
| Na2ATP | Sigma-Aldrich | Cat#2383 |
| CsOH | Aldrich | Cat#C8518 |
| Agarose | Sigma | Lot #SLBR6299V |
| All trans-Retinal | Sigma-Aldrich | CAS #116–31-4 |
| Experimental models: organisms/strains | ||
| C. elegans: N2 | Caenorhabditis Genetics Center | N2 |
| C. elegans: unc-13(s69) | Caenorhabditis Genetics Center | BC168 |
| C. elegans: hztSi1 [Psnb-1::UNC-13MR]; unc-13(s69) | This paper | ZTH456 |
| C. elegans: juSi67 [Prgef-1::UNC-13R]; unc-13(s69) | Zhou et al., 2013 | CZ15872 |
| C. elegans: hztEx11 [Psnb-1::UNC-13MR]; unc-13(s69) | This paper | ZTH11 |
| C. elegans: hztEx20 [Psnb-1::UNC-13R]; unc-13(s69) | This paper | ZTH99 |
| C. elegans: hztEx53 [Psnb-1::UNC-13MRΔC1]; unc-13(s69) | This paper | ZTH498 |
| C. elegans: hztEx68 [Psnb-1::UNC-13RΔC1]; unc-13(s69) | This paper | ZTH521 |
| C. elegans: hztEx55 [Psnb-1::MRΔC2B]; unc-13(s69) | This paper | ZTH517 |
| C. elegans: hztEx69 [Psnb-1::UNC-13RΔC2B]; unc-13(s69) | This paper | ZTH507 |
| C. elegans: hztEx70 [Psnb-1::MRΔC1ΔC2B]; unc-13(s69) | This paper | ZTH495 |
| C. elegans: hztEx109 [Psnb-1::UNC-13RΔC1ΔC2B]; unc-13(s69) | This paper | ZTH696 |
| C. elegans: hztEx54 [Psnb-1::UNC-13MRH348K]; unc-13(s69) | This paper | ZTH421 |
| C. elegans: hztEx56 [Psnb-1::UNC-13MRD3,4N]; unc-13(s69) | This paper | ZTH447 |
| C. elegans: hztEx72 [Psnb-1::UNC-13MRHK, D3,4N]; unc-13(s69) | This paper | ZTH451 |
| C. elegans: hztEx71 [Psnb-1::UNC-13RHK, D3,4N]; unc-13(s69) | This paper | ZTH448 |
| C. elegans: hztSi7 [Prab-3::UNC-13MUNC2C]; unc-13(s69) | This paper | ZTH858 |
| C. elegans: hztSi5 [Prab-3::UNC-13M-MUNC2C]; unc-13(s69) | This paper | ZTH752 |
| C. elegans: hztEx107 [Psnb-1::UNC-13MUNC2C]; unc-13(s69) | This paper | ZTH442 |
| C. elegans: hztEx108 [Psnb-1::UNC-13M-MUNC2C]; unc-13(s69) | This paper | ZTH577 |
| C. elegans: hztEx111 [Psnb-1:: NbMunc13−2-UNC-13R]; unc-13(s69) | This paper | ZTH738 |
| C. elegans: hztEx112 [Psnb-1::NbMunc13−2-UNC-13(MUNC2C)]; unc-13(s69) | This paper | ZTH740 |
| C. elegans: hztEx79 [Punc-129::UNC-13MR::mApple]; NuIs165 [Punc-129::UNC-10::GFP] | This paper | ZTH686 |
| C. elegans: hztEx80 [Punc-129::UNC-13R::mApple]; NuIs165 [Punc-129::UNC-10::GFP] | This paper | ZTH711 |
| C. elegans: hztEx82 [Punc-129::UNC-13MUNC2C::mApple]; NuIs165 [Punc-129::UNC-10::GFP] | This paper | ZTH693 |
| C. elegans: hztEx81 [Punc-129::UNC-13(M-MUNC2C)::mApple]; NuIs165 [Punc-129::UNC-10::GFP] | This paper | ZTH692 |
| C. elegans: hztEx113 [Psnb-1::NbMunc13−2(D145−187)-UNC-13R]; unc-13(s69) | This paper | ZTH924 |
| C. elegans: hztEx95 [Punc-129::UNC-13MRΔC1ΔC2B::mApple]; NuIs165 [Punc-129::UNC-10::GFP] | This paper | ZTH921 |
| C. elegans: hztEx66 [Punc-129::UNC-13RΔC1ΔC2B::mApple]; NuIs165 [Punc-129::UNC-10::GFP] | This paper | ZTH702 |
| Oligonucleotides | ||
| Unc-13 ΔC1 Forward: CAACTCAAAGAGCAGCCGAGAAGTCG | IDT | N/A |
| Unc-13 ΔC1 Reverse: TGCTCTTTGAGTTGTTGCACTGATCGGGTAGA | IDT | N/A |
| Unc-13 ΔC2B Forward: CAAATGGAAGGGAGAAGAGAAACTTGCACC | IDT | N/A |
| Unc-13 ΔC2B Reverse: CTCCCTTCCATTTGCTACTTCCTTCCAAAATAG | IDT | N/A |
| Unc-13 C2B D3,4N Forward: aagttcgagtttggaacgagaataatgatttgaaatc | IDT | N/A |
| Unc-13 C2B D3,4N Reverse: gatttcaaatcattattctcgttccaaactcgaactt | IDT | N/A |
| Unc-13 C1 HK Forward: caactcctaagaacttcgcaactacaacattc | IDT | N/A |
| Unc-13 C1 HK Reverse: gttgcgaagttcttaggagttgttgcac | IDT | N/A |
| Recombinant DNA | ||
| Psnb-1::UNC-13MR | This paper | Phzt11 |
| Psnb-1::UNC-13R | This paper | Phzt90 |
| Psnb-1::UNC-13MRΔC1 | This paper | Phzt387 |
| Psnb-1::UNC-13RΔC1 | This paper | Phzt523 |
| Psnb-1::UNC-13MRΔC2B | This paper | Phzt229 |
| Psnb-1::UNC-13RΔC2B | This paper | Phzt103 |
| Psnb-1::UNC-13MRΔC1ΔC2B | This paper | Phzt409 |
| Psnb-1::UNC-13RΔC1ΔC2B | This paper | Phzt501 |
| Psnb-1::UNC-13MRH348K | This paper | Phzt92 |
| Psnb-1::UNC-13MRD3,4N | This paper | Phzt356 |
| Psnb-1::UNC-13MRHK, D3,4N | This paper | Phzt367 |
| Psnb-1::UNC-13RHK, D3,4N | This paper | Phzt369 |
| Prab-3::UNC-13MUNC2C | This paper | Phzt746 |
| Prab-3::UNC-13M-MUNC2C | This paper | Phzt934 |
| Psnb-1::UNC-13MUNC2C | This paper | Phzt352 |
| Psnb-1::UNC-13M-MUNC2C | This paper | Phzt652 |
| Psnb-1:: NbMunc13−2-UNC-13R | This paper | Phzt943 |
| Psnb-1::NbMunc13−2-UNC-13(MUNC2C) | This paper | Phzt935 |
| Punc-129::UNC-13MR::mApple | This paper | Phzt1152 |
| Punc-129::UNC-13R::mApple | This paper | Phzt217 |
| Punc-129::UNC-13MUNC2C::mApple | This paper | Phzt925 |
| Punc-129::UNC-13(M-MUNC2C)::mApple | This paper | Phzt926 |
| Psnb-1::NbMunc13−2(D145−187)-UNC-13R | This paper | Phzt1071 |
| Punc-129::UNC-13MRΔC1ΔC2B::mApple | This paper | Phzt1173 |
| Punc-129::UNC-13RΔC1ΔC2B::mApple | This paper | Phzt1018 |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| PatchMaster | HEKA Elektronik | V2×73.2 |
| Igor Pro | Wavemetrics | Version 7 |
| SigmaPlot | Systat Software Inc. | Version 13.0 |
| Other | ||
| Microscope Cover Glass | Fisher Scientific | Lot#060214–9 |
| Glass Capillaries | World Precision Instruments, Inc. | Lot#2009330 |
Highlights.
The M domain in UNC-13MR inhibits the probability of neurotransmitter release
Removal or activation of C1 and C2B eliminates the inhibitory effects of the M domain
The M domain promotes neurotransmitter release when fused with the MUNC2C fragment
The N terminus of bMunc13–2 has a similar function in the M domain
ACKNOWLEDGMENTS
We thank the C. elegans Genetics Stock Center for strains. We thank members of the Hu laboratory. We thank Rowan Tweedale for critically reading the manuscript. We thank Professor Yishi Jin for providing the CZ15872 strain. We thank Professor Nils Brose for providing the bMunc13–2 plasmid. This work was supported by an Australia Research Council Discovery Project grant (DP160100849 to Z.H.), a National Health and Medical Research Council Project grant (APP1122351 to Z.H.), a NIH/NEI R21 grant (1R21EY029450–01 to P.B., J. L., and Z.H.)and a NARSAD Young Investigator grant (24980 to Z.H.). This work made use of the BioCryo facility of Northwestern University’s NUANCE Center, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the MRSEC program (NSF DMR-1720139) at the Materials Research Center; the International Institute for Nanotechnology (IIN); and the State of Illinois, through the IIN. Further, this work used instrumentation provided by the University of Illinois Research Resources Center, Electron Microscopy Core. Some strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440)
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests. Received: May 28, 2020
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108828.
REFERENCES
- Aravamudan B, Fergestad T, Davis WS, Rodesch CK, and Broadie K. (1999). Drosophila UNC-13 is essential for synaptic transmission. Nat. Neurosci 2, 965–971. [DOI] [PubMed] [Google Scholar]
- Atwood HL, and Karunanithi S. (2002). Diversification of synaptic strength: presynaptic elements. Nat. Rev. Neurosci 3, 497–516. [DOI] [PubMed] [Google Scholar]
- Augustin I, Rosenmund C, Südhof TC, and Brose N. (1999a). Munc13–1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400, 457–461. [DOI] [PubMed] [Google Scholar]
- Augustin I, Betz A, Herrmann C, Jo T, and Brose N. (1999b). Differential expression of two novel Munc13 proteins in rat brain. Biochem. J 337, 363–371. [PMC free article] [PubMed] [Google Scholar]
- Augustin I, Korte S, Rickmann M, Kretzschmar HA, Südhof TC, Herms JW, and Brose N. (2001). The cerebellum-specific Munc13 isoform Munc13–3 regulates cerebellar synaptic transmission and motor learning in mice. J. Neurosci 21, 10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu J, Shen N, Dulubova I, Lu J, Guan R, Guryev O, Grishin NV, Rosenmund C, and Rizo J. (2005). A minimal domain responsible for Munc13 activity. Nat. Struct. Mol. Biol 12, 1017–1018. [DOI] [PubMed] [Google Scholar]
- Basu J, Betz A, Brose N, and Rosenmund C. (2007). Munc13–1 C1 domain activation lowers the energy barrier for synaptic vesicle fusion. J. Neurosci 27, 1200–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Südhof TC, Rettig J, and Brose N. (1998). Munc13–1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron 21, 123–136. [DOI] [PubMed] [Google Scholar]
- Betz A, Thakur P, Junge HJ, Ashery U, Rhee JS, Scheuss V, Rosenmund C, Rettig J, and Brose N. (2001). Functional interaction of the active zone proteins Munc13–1 and RIM1 in synaptic vesicle priming. Neuron 30, 183–196. [DOI] [PubMed] [Google Scholar]
- Böhme MA, Beis C, Reddy-Alla S, Reynolds E, Mampell MM, Grasskamp AT, Lützkendorf J, Bergeron DD, Driller JH, Babikir H, et al. (2016). Active zone scaffolds differentially accumulate Unc13 isoforms to tune Ca(2+) channel-vesicle coupling. Nat. Neurosci 19, 1311–1320. [DOI] [PubMed] [Google Scholar]
- Brenner S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, Hofmann K, Hata Y, and Südhof TC (1995). Mammalian homologues of Caenorhabditis elegans unc-13 gene define novel family of C2-domain proteins. J. Biol. Chem 270, 25273–25280. [DOI] [PubMed] [Google Scholar]
- Calloway N, Gouzer G, Xue M, and Ryan TA (2015). The active-zone protein Munc13 controls the use-dependence of presynaptic voltage-gated calcium channels. eLife 4, e07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho M, Basu J, Trimbuch T, Chang S, Pulido-Lozano C, Chang SS, Duluvova I, Abo-Rady M, Rizo J, and Rosenmund C. (2017). Heterodimerization of Munc13 C2A domain with RIM regulates synaptic vesicle docking and priming. Nat. Commun 8, 15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Cooper B, Kalla S, Varoqueaux F, and Young SM Jr. (2013). The Munc13 proteins differentially regulate readily releasable pool dynamics and calcium-dependent recovery at a central synapse. J. Neurosci 33, 8336–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Kaeser PS, Xu W, and Südhof TC (2011). RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron 69, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, and Stevens CF (1997). Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron 18, 995–1008. [DOI] [PubMed] [Google Scholar]
- Ellis MV, James SR, Perisic O, Downes CP, Williams RL, and Katan M. (1998). Catalytic domain of phosphoinositide-specific phospholipase C (PLC). Mutational analysis of residues within the active site and hydrophobic ridge of plcdelta1. J. Biol. Chem 273, 11650–11659. [DOI] [PubMed] [Google Scholar]
- Fioravante D, and Regehr WG (2011). Short-term forms of presynaptic plasticity. Curr. Opin. Neurobiol 21, 269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjær-Jensen C, Davis MW, Sarov M, Taylor J, Flibotte S, LaBella M, Pozniakovsky A, Moerman DG, and Jorgensen EM (2014). Random and targeted transgene insertion in Caenorhabditis elegans using a modified Mos1 transposon. Nat. Methods 11, 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulterer A, Andlauer TFM, Ender A, Maglione M, Eyring K, Woitkuhn J, Lehmann M, Matkovic-Rachid T, Geiger JRP, Walter AM, et al. (2018). Active zone scaffold protein ratios tune functional diversity across brain synapses. Cell Rep. 23, 1259–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber SH, Rah JC, Min SW, Liu X, de Wit H, Dulubova I, Meyer AC, Rizo J, Arancillo M, Hammer RE, et al. (2008). Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science 321, 1507–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Palfreyman MT, Watanabe S, Olsen S, and Jorgensen EM (2007). Open syntaxin docks synaptic vesicles. PLoS Biol. 5, e198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Tong XJ, and Kaplan JM (2013). UNC-13L, UNC-13S, and Tomosyn form a protein code for fast and slow neurotransmitter release in Caenorhabditis elegans. eLife 2, e00967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Vashlishan-Murray AB, and Kaplan JM (2015). NLP-12 engages different UNC-13 proteins to potentiate tonic and evoked release. J. Neurosci 35, 1038–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama S, Schmidt H, Cooper BH, Brose N, and Eilers J. (2014). Munc13–3 superprimes synaptic vesicles at granule cell-to-basket cell synapses in the mouse cerebellum. J. Neurosci 34, 14687–14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, and Fasshauer D. (2012). Molecular machines governing exocytosis of synaptic vesicles. Nature 490, 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge HJ, Rhee JS, Jahn O, Varoqueaux F, Spiess J, Waxham MN, Rosenmund C, and Brose N. (2004). Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell 118, 389–401. [DOI] [PubMed] [Google Scholar]
- Kawabe H, Mitkovski M, Kaeser PS, Hirrlinger J, Opazo F, Nestvogel D, Kalla S, Fejtova A, Verrier SE, Bungers SR, et al. (2017). ELKS1 localizes the synaptic vesicle priming protein bMunc13–2 to a specific subset of active zones. J. Cell Biol 216, 1143–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn RE, Duerr JS, McManus JR, Duke A, Rakow TL, Maruyama H, Moulder G, Maruyama IN, Barstead RJ, and Rand JB (2000). Expression of multiple UNC-13 proteins in the Caenorhabditis elegans nervous system. Mol. Biol. Cell 11, 3441–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner MR, Nurrish SJ, and Kaplan JM (1999). Facilitation of synaptic transmission by EGL-30 Gqa and EGL-8 PLCb: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron 24, 335–346. [DOI] [PubMed] [Google Scholar]
- Lai Y, Choi UB, Leitz J, Rhee HJ, Lee C, Altas B, Zhao M, Pfuetzner RA, Wang AL, Brose N, et al. (2017). Molecular mechanisms of synaptic vesicle priming by Munc13 and Munc18. Neuron 95, 591–607.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Ho WK, Neher E, and Lee SH (2013). Superpriming of synaptic vesicles after their recruitment to the readily releasable pool. Proc. Natl. Acad. Sci. USA 110, 15079–15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Liu H, Hall Q, Wang W, Yu Y, Kaplan JM, and Hu Z. (2019). A hyperactive form of unc-13 enhances Ca2+ sensitivity and synaptic vesicle release probability in C. elegans. Cell Rep. 28, 2979–2995.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Seven AB, Camacho M, Esser V, Xu J, Trimbuch T, Quade B, Su L, Ma C, Rosenmund C, and Rizo J. (2016). Functional synergy between the Munc13 C-terminal C1 and C2 domains. eLife 5, e13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Li L, Nedelcu D, Hall Q, Zhou L, Wang W, Yu Y, Kaplan JM, and Hu Z. (2019a). Heterodimerization of UNC-13/RIM regulates synaptic vesicle release probability but not priming in C. elegans. eLife 8, e40585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sugiura Y, Südhof TC, and Lin W. (2019. b). Ablation of all synaptobrevin vSNAREs blocks evoked but not spontaneous neurotransmitter release at neuromuscular synapses. J. Neurosci 39, 6049–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Korogod N, Brose N, and Schneggenburger R. (2008). Phorbol esters modulate spontaneous and Ca2+-evoked transmitter release via acting on both Munc13 and protein kinase C. J. Neurosci 28, 8257–8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison JM, Nurrish S, and Kaplan JM (2005). UNC-13 interaction with syntaxin is required for synaptic transmission. Curr. Biol 15, 2236–2242. [DOI] [PubMed] [Google Scholar]
- Maruyama IN, and Brenner S. (1991). A phorbol ester/diacylglycerol-binding protein encoded by the unc-13 gene of Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 88, 5729–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelassi F, Liu H, Hu Z, and Dittman JS (2017). A C1-C2 module in Munc13 inhibits calcium-dependent neurotransmitter release. Neuron 95, 577–590.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netrakanti PR, Cooper BH, Dere E, Poggi G, Winkler D, Brose N, and Ehrenreich H. (2015). Fast cerebellar reflex circuitry requires synaptic vesicle priming by munc13–3. Cerebellum 14, 264–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfreyman MT, and Jorgensen EM (2017). Unc13 Aligns SNAREs and superprimes synaptic vesicles. Neuron 95, 473–475. [DOI] [PubMed] [Google Scholar]
- Regehr WG (2012). Short-term presynaptic plasticity. Cold Spring Harb. Perspect. Biol 4, a005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, and Jorgensen EM (1999). UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat. Neurosci 2, 959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, and Stevens CF (1996). Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16, 1197–1207. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Sigler A, Augustin I, Reim K, Brose N, and Rhee JS (2002). Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron 33, 411–424. [DOI] [PubMed] [Google Scholar]
- Sakamoto H, Ariyoshi T, Kimpara N, Sugao K, Taiko I, Takikawa K, Asanuma D, Namiki S, and Hirose K. (2018). Synaptic weight set by Munc13–1 supramolecular assemblies. Nat. Neurosci 21, 41–49. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen N, Guryev O, and Rizo J. (2005). Intramolecular occlusion of the diacylglycerol-binding site in the C1 domain of munc13–1. Biochemistry 44, 1089–1096. [DOI] [PubMed] [Google Scholar]
- Shin OH, Lu J, Rhee JS, Tomchick DR, Pang ZP, Wojcik SM, Camacho-Perez M, Brose N, Machius M, Rizo J, et al. (2010). Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat. Struct. Mol. Biol 17, 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Ailenberg M, and Silverman M. (1998). Cloning of a novel gene in the human kidney homologous to rat munc13s: its potential role in diabetic nephropathy. Kidney Int. 53, 1689–1695. [DOI] [PubMed] [Google Scholar]
- Südhof TC, and Rizo J. (2011). Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol 3, a005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, Woehler A, and Neher E. (2016). Superpriming of synaptic vesicles as a common basis for intersynapse variability and modulation of synaptic strength. Proc. Natl. Acad. Sci. USA 113, E4548–E4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, and Rosenmund C. (2002). Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc. Natl. Acad. Sci. USA 99, 9037–9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Sons MS, Plomp JJ, and Brose N. (2005). Aberrant morphology and residual transmitter release at the Munc13-deficient mouse neuromuscular synapse. Mol. Cell. Biol 25, 5973–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer RM, Gracheva EO, Meyrignac O, Miller KG, Richmond JE, and Bessereau JL (2006). UNC-13 and UNC-10/rim localize synaptic vesicles to specific membrane domains. J. Neurosci 26, 8040–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, and Brenner S. (1986). The structure of the nervous system of the nematode caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci 314, 1–340. 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Xu J, Camacho M, Xu Y, Esser V, Liu X, Trimbuch T, Pan YZ, Ma C, Tomchick DR, Rosenmund C, and Rizo J. (2017). Mechanistic insights into neurotransmitter release and presynaptic plasticity from the crystal structure of Munc13–1 C1C2BMUN. eLife 6, e22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Stawicki TM, Goncharov A, and Jin Y. (2013). Position of UNC-13 in the active zone regulates synaptic vesicle release probability and release kinetics. eLife 2, e01180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.