

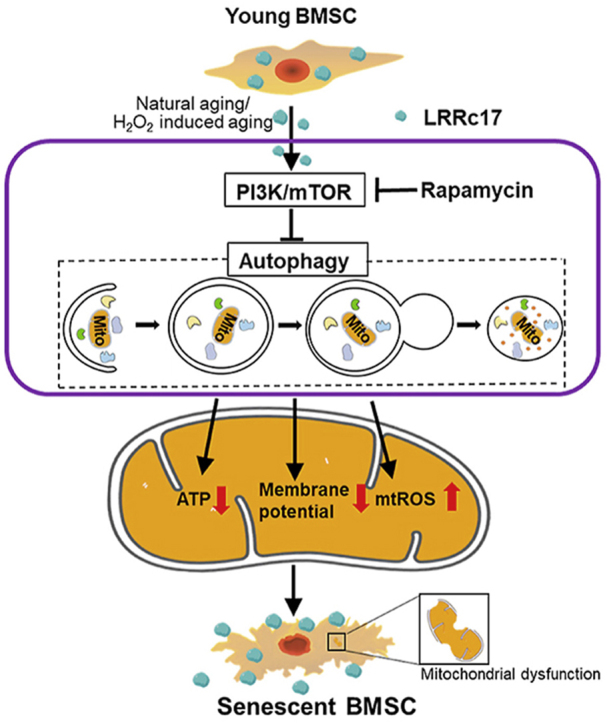
Abstract
Senescence of bone marrow-derived mesenchymal stem cells (BMSCs) has been widely reported to be closely correlated with aging-related diseases, including osteoporosis (OP). Moreover, the beneficial functions of BMSCs decline with age, limiting their therapeutic efficacy in OP. In the present study, using RNA sequencing (RNA-Seq), we found that leucine-rich repeat containing 17 (LRRc17) expression in BMSCs was highly positively correlated with age. Therefore, we investigated whether LRRc17 knockdown could rejuvenate aged MSCs and increase their therapeutic efficacy in OP. Consistent with the RNA-Seq results, the protein expression of LRRc17 in senescent BMSCs was significantly increased, whereas LRRc17 knockdown inhibited cell apoptosis and reduced the expression of age-related proteins and G2 and S phase quiescence. Furthermore, LRRc17 knockdown shifted BMSCs from adipogenic to osteogenic differentiation, indicating the critical role of LRRc17 in BMSC senescence and differentiation. Additionally, similar to rapamycin (RAPA) treatment, LRRc17 knockdown activated mitophagy via inhibition of the mTOR/PI3K pathway, which consequently reduced mitochondrial dysfunction and inhibited BMSC senescence. However, the effects of LRRc17 knockdown were significantly blocked by the autophagy inhibitor hydroxychloroquine (HCQ), demonstrating that LRRc17 knockdown prevented BMSC senescence by activating mitophagy. In vivo, compared with untransfected aged mouse-derived BMSCs (O-BMSCs), O-BMSCs transfected with sh-LRRc17 showed effective amelioration of ovariectomy (OVX)-induced bone loss. Collectively, these results indicated that LRRc17 knockdown rejuvenated senescent BMSCs and thus enhanced their therapeutic efficacy in OP by activating autophagy.
Keywords: BMSCs, LRRc17, Aging, Mitophagy, Osteoporosis
Graphical abstract
Abbreviations
- BMSCs
bone marrow-derived mesenchymal stem cells
- O-BMSC
Aged mouse-derived BMSC
- Y-BMSC
Young mouse-derived BMSC
- OP
Osteoporosis
- LRRc17
leucine-rich repeat containing 17
- RNA-Seq
RNA sequencing
- RAPA
Rapamycin
- HCQ
Hydroxychloroquine
- OVX
Ovariectomy
- ROS
Reactive oxygen species
- PBS
Phosphate-buffered saline
- α-MEM
Alpha minimum essential medium
- FBS
Fetal bovine serum
- OC
Osteoclast
- OB
Osteoblast
- BMMs
Bone marrow-derived macrophages
- M-CSF
Macrophage colony stimulating factor
- RANKL
Receptor activator for nuclear factor-κB ligand
- TRAP
Tartrate resistant acid phosphatase
- MC3T3-E1
Mouse embryo osteoblast precursor cells
- ELISA
Enzyme linked immunosorbent assay
- PI
Propidium iodide
- HEK293T
Human embryonic kidney 293T
- JC-1
Tetrechloro-tetraethylbenzimidazol carbocyanine iodide
- mtROS
Mitochondria ROS
- ATP
Adenosine-triphosphate
- OCR
Oxygen consumption rate
- RT-PCR
Real-time polymerase chain reaction
- DAPI
4’,6-diamidino-2-phenylindole
- micro-CT
Microcomputed tomography
- BMD
Bone mineral density
- Tb.Th
Trabecular bone thickness
- Tb.N
Trabecular bone number
- BV/TV
Bone volume per tissue volume
- Tb.Sp
Trabecular separation
- Ct.Th
Cortical bone thickness
- BFR
Bone formation rate
- MAR
Mineral deposition rate
1. Introduction
Mesenchymal stem cells (MSCs) possess proliferative capacity and differentiation potential, and their pluripotency makes them an attractive resource for regenerative cell therapy [1]. MSCs are present in various tissues, including dental pulp, adipose tissue, placenta and bone marrow (BM), among which bone marrow is one of the most common sources used in preclinical and clinical studies due to its high cell number and proliferation activity [2]. Numerous studies have also demonstrated the potential of BM-derived MSCs to treat age-related diseases, including osteoporosis (OP), diabetes mellitus (DM), osteoarthritis (OA), myocardial infarction (MI) and Crohn's disease (CD) [3,4].
Aging is a complex, progressive and inevitable physiological process accompanied by the accumulation of damaged macromolecules and contributes to the development of organ dysfunction. Stem cell senescence results in stem cell exhaustion and leads to tissue failure and pathological and physiological aging [5]. For example, OP is triggered not only by increased bone resorptive activity but also by dysfunction of MSCs that exhibit a distinct shift from osteogenic to adipogenic differentiation and an associated reduction in self-renewal capacity [6]. In addition, the senescence of MSCs may have a profound impact on their therapeutic function. Khan et al. demonstrated that young mouse-derived MSC (Y-BMSC) treatment significantly improved left ventricle systolic and diastolic function in a myocardial infarction mouse model, while the therapeutic effect of MSCs decreased as the age of the donor mice increased [7]. Thus, investigating the mechanism involved in MSC senescence and exploring novel strategies to rejuvenate aged MSCs are essential for their therapeutic applications [8].
The molecular mechanism of MSC senescence is partially related to the p21/p53 and PI3K/AKT pathways [9,10]. In the presence of intracellular or extracellular stimuli, including reactive oxygen species (ROS) and severe inflammation or trauma, the upstream regulator of the p21/p53 pathway is activated, accompanied by diminished telomerase activity and dysfunctional organelle accumulation, especially mitochondria. Moreover, cellular quiescence and changes in differentiation potential are mainly attributed to impaired mitochondria due to decreased autophagy activity [11]. Autophagy is essential for cell metabolism, function and homeostasis and is defined as a lysosomal degradation pathway [12]. Recent evidence has suggested that autophagy deficiency is correlated with impaired osteogenic ability of senescent BMSCs in osteoporosis [13]. In addition, LRRc17, a vital regulator of osteoclast development, is involved in the interactions between osteoclasts and osteoblasts, which are essential to ensure the proper regulation of orthotropic factors in bone metabolism [14]. Both autophagy and LRRc17 are undoubtedly indispensable for bone homeostasis. However, whether there is a correlation between them and the specific mechanism underlying such a correlation have not yet been reported. In this study, we demonstrated that LRRc17 controls BMSC senescence by altering autophagy activity and mitochondrial bioenergetics. These findings indicated that LRRc17 could be a potential therapeutic target for rejuvenating senescent MSCs and treating age-related bone loss.
2. Materials and methods
2.1. Animals
All animal experiments were conducted according to the requirements of the Animal Welfare Act. All experimental procedures were performed according to the Ethics Committee of Sichuan University. C57BL/6 mice were divided into three age groups, a young group (<8 weeks), a middle-aged group (5–8 months), and an aged group (18 months), as previously reported [15].
2.2. Isolation and identification of BMSCs
BMSCs were harvested from the bone marrow of C57BL/6 mice as previously described [16,17]. Briefly, bone marrow cells were flushed out and collected from the femur and tibia of mice, plated in T25 flasks, and cultured overnight in a 37 °C incubator with 5% CO2. Then, nonadherent cells were removed by rinsing twice with phosphate-buffered saline (PBS). The adherent cells were maintained in alpha minimum essential medium (α-MEM) containing 20% fetal bovine serum (FBS), 2 mM Glutamax, and 1% penicillin and streptomycin (PS).
For flow cytometric analysis of surface immunophenotypic markers, BMSCs were incubated with PE-conjugated anti-CD29 (BD Biosciences, USA), SCA-1 (eBiosciences, USA), Alexa Fluor-conjugated anti-CD45 (BD Biosciences, USA), CD11b (BD Biosciences, USA), and FITC-conjugated anti-CD29 (BioLegend, USA) at room temperature for 25 min and then analyzed via flow cytometry (BD Biosciences, USA).
2.3. RNA-sequencing (RNA-seq)
Total RNA of Y-BMSCs and O-BMSCs at passage 3 were isolated using TRIzol (Life Technologies, USA). RNA-Seq library construction and RNA high-throughput sequencing were performed by Beijing Genomic Institution (BGI, China) on a BGISEQ-500 high-throughput sequencer. Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, Gene Set Enrichment Analysis (GSEA), Weighted correlation network analysis (WGCNA) and time course analysis were analyzed according to a previously described method [18].
2.4. Differentiation potential
For osteogenesis and adipogenesis induction of BMSCs, several specific factors were used as previously reported [19]. In brief, for adipogenesis, BMSCs at passage3 (5 × 105 cells/cm2) were cultured α-MEM plus 10% FBS added with 50 μM isobutylmethylxanthine (IBMX), 0.5 μM dexamethasone, 50 μM indomethacin and 10 μg/ml insulin. After 21 d of culture, the cells were fixed with 10% formalin and stained with 0.5% fresh Oil-red O solution (Cyagen, USA) for 30 min. For osteogenesis, BMSCs (4 × 105 cells/cm2) were cultured in α-MEM plus 10% FBS added with 50 μg/ml ascorbate-2, 10 mM β-glycerol phosphate and 0.1 μM dexamethasone. After 14 d of culture, the cells were fixed with 10% formalin, and stained with 0.1% Alizarin red solution (Cyagen, USA) for 30 min.
2.5. Osteoclast (OC) and MC3T3-E1 culture
For in vitro osteoclast differentiation, bone marrow cells were harvested from the femora and tibiae of 8-week-old mice. Then, the bone marrow cells (0.5 × 106) were differentiated into bone marrow-derived macrophages (BMMs) in α-MEM supplemented with 50 ng/mL recombination macrophage colony stimulating factor (M-CSF), Glutamax, 10% heat-inactivated FBS, and 1% PS in a 12-well plate for 16 h in a 37 °C incubator with 5% CO2. The non-adherent cells were then maintained in the presence of 50 ng/mL receptor activator for nuclear factor-κB ligand (RANKL) and 50 ng/mL M-CSF for an additional 5 days. For the coculture assay, osteoclasts were cocultured with BMSCs at a 1:3 ratio for 5 d in a Transwell system. Tartrate resistant acid phosphatase (TRAP) staining was performed using a kit (Solarbio, China) following the manufacturer's protocols. The mouse osteoblast cell line Mouse embryo osteoblast precursor cells (MC3T3-E1) (subclone 4) was purchased from the American Type Culture Collection (ATCC, USA) and cultured in α-MEM containing 10% FBS and 1% PS in a 37 °C incubator with 5% CO2. For the coculture assay, osteoblasts were cocultured with BMSCs at a 1:3 ratio for 10 d. Alizarin red staining was conducted using a kit (Cyagen, USA) following the manufacturer's instructions.
2.6. Bone resorption assay
To examine osteoclast function, BMMs were seeded on bovine bone slices at a density of 2.5 × 104 cells/cm2 in triplicate. After adhering, cells were stimulated with 50 ng/ml M-CSF and 50 ng/ml RANKL for 7 days. Then, the cells were subsequently washed by mechanical agitation and sonication. Resorption pits were visualized by scanning electron microscope (ZEISS, Germany) and Image J software was used to quantify the area of bone resorption [20].
2.7. Proliferation assay
A proliferation assay was used to determine cell growth kinetics. In brief, 1 × 105 BMSCs were cultured in 6-well plates and counted daily for 9 successive days using a hemocytometer. The EdU assay was carried out as previously described [21].
2.8. CFU analysis
BMSCs were rinsed twice with PBS and fixed with 4% formaldehyde for 20 min at room temperature. Then, 0.5% crystal violet was used to stain the BMSCs for 30 min, and the cells were washed with PBS [22]. Colonies composed of 50 or more cells were considered for counting.
2.9. SA-β-gal staining
SA-β-gal activity was measured with a staining kit (Beyotime, China) following the manufacturer's protocols. The number of blue SA-β-gal-positive cells was calculated as previously reported [23].
2.10. Cell death evaluation
A cell death detection enzyme linked immunosorbent assay (ELISA) kit (Roche, USA) was applied to measure cell apoptosis. The relative apoptosis ratio was detected at an absorption wavelength of 405 nm and a reference wavelength of 490 nm according to the manufacturer's protocol.
2.11. Cell cycle and apoptosis analysis
For the cell cycle assay, BMSCs were harvested and fixed with 70% ethanol overnight at 4 °C. Then, the fixed BMSCs were incubated with 100 μg/mL RNase A (Sigma-Aldrich, USA) and 20 μg/mL propidium iodide (PI) (Sigma-Aldrich, USA) for 30 min. The samples were analyzed by flow cytometry, and the data were analyzed with ModFit LT (version 5.0). For the apoptosis analysis, cells were collected and analyzed with the Annexin V-FITC/PI Apoptosis Detection Kit (BD, USA).
2.12. Telomerase activity
BMSCs were lysed to detect telomerase activity. Next, a TeloTAGGG Telomerase PCR ELISA Kit (Roche, USA) was used to perform sequential reaction steps in accordance with the manufacturer’s protocol. The absorbance of the PCR amplification product at 450 nm was determined using a microplate reader within 30 min.
2.13. Lentiviral transfection of BMSCs
Construction and transfection of the lentiviral vectors were performed according to previously described methods [24]. In brief, lentiviral sh-LRRc17 or overexpression vectors were cotransfected into human embryonic kidney 293T (HEK293T) cells, and the three packaging plasmids included pHBLVTM, psPAX2 and pMD2.G. Lentiviral particles were obtained after 48 h of transfection via cell harvest and concentration with ultracentrifugation at 72,000 g for 2 h. Following titer determination, BMSCs were transfected at a multiplicity of infection (MOI) of 50 for 24 h.
2.14. Mitochondrial morphological analyses
Mitochondrial morphometric analyses were performed as previously reported [25]. BMSCs were labeled with MitoTracker Green (50 nM, Invitrogen, USA) at 37 °C for 30 min, and representative images were acquired with a laser scanning confocal microscope. Mitochondrial length and complexity were reflected by the aspect ratio (AR, major axis/minor axis) and form factor (FF, 4π × (area/perimeter2)), respectively, which were calculated with ImageJ software (Wayne; USA).
2.15. Mitochondrial membrane potential (ΔΨ) and mtROS determination
Tetrechloro-tetraethylbenzimidazol carbocyanine iodide (JC-1, 10 μg/ml; Beyotime, China) staining was performed at 37 °C for 20 min to determine BMSC mitochondrial membrane potential. For mitochondria ROS (mtROS) detection, BMSCs were stained with 4 μM MitoSOX (Invitrogen, USA) at 37 °C for 20 min, and mtROS quantification was performed using flow cytometry.
2.16. Adenosine-triphosphate (ATP) measurement
Intracellular ATP was determined by an ATP measurement kit (Beyotime, China) according to the manufacturer's protocol. The ATP luminescence signal of supernatants mixed with luciferase assay buffer was determined by a luminescence microplate reader and then normalized to the protein concentration.
2.17. Mitochondrial oxygen consumption rate (OCR) measurement
The mitochondrial OCR of BMSCs was conducted with a Seahorse XF Cell Mito Stress Test Kit (Agilent, USA) in accordance with the manufacturer's protocol as previously reported [26].
2.18. Real-time polymerase chain reaction (RT-PCR)
Total RNA was isolated using TRIzol (Life Technologies, USA). Then, a high-capacity cDNA reverse transcription kit (Vazyme, China) was used to reverse-transcribe 1 μg of mRNA into cDNA in accordance with the manufacturer’s protocol. RT-PCRs were performed on an ABI7900 PCR system (Applied Biosystems, USA) using SYBR Green MasterMix (Vazyme, China). GAPDH was used as a reference gene. The primers are shown in Supplementary Table 1.
2.19. Immunofluorescence
To observe the colocalization of LC3B with mitochondria in BMSCs, BMSCs were incubated with MitoTracker Deep Red (200 nM; Invitrogen, USA) at 37 °C for 10 min, fixed with methanol and then treated with 0.2% Triton X-100 (Sigma-Aldrich, USA) to increase cell membrane penetrability. Cells were incubated with LC3B (1:200 dilution; CST, USA) at 4 °C overnight and then incubated with fluorescently labeled secondary antibodies (1:250 dilution; ABclonal, China) for 1 h at room temperature. Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, USA) for 5 min. Immunofluorescence images were acquired by confocal laser scanning microscopy.
2.20. Western blot
Protein expression in BMSCs was determined by immunoblotting as previously described [27]. Anti-LRRc17 (1:1000, Proteintech), anti-p21 (1:1000, Affinity), anti-p16 (1:1000, CST), anti-p53 (1:1000, CST), anti-mTOR (1:500, Affinity), anti-phospho-mTOR (1:500, CST), anti-PI3K (1:1000, CST), anti-phospho-PI3K (1:1000, Affinity), anti-P62 (1:5000, ABclonal), anti-LC3B (1:1000, CST), anti-Beclin1 (1:1000, CST), anti-OPA1 (1:1000, CST), anti-Drp1 (1:1000, CST), anti-JNK (1:1000, Abclonal) abti-phosphor-JNK (1:1000, Abclonal) and anti-Calpain1 (1:1000, Abclonal) antibodies were used to analyze protein expression. The signal was visualized using an Immobilon Western Chemiluminescent HRP Substrate Kit (Millipore, USA). Quantification of the gray values of the bands was performed with ImageJ software.
2.21. OVX-induced osteoporosis and BMSC transplantation
Six-month-old female C57BL/6 mice were divided into six groups: sham (n = 7), OVX (n = 7), OVX + Y-BMSCs (n = 7), OVX + O-BMSCs (n = 7), OVX + O-BMSCsLV-GFP (n = 7) and OVX + O-BMSCssh-LRRc17 (n = 7). Four month after ovariectomy, ovariectomized mice were anesthetized and immediately received local transplantation of BMSCs (1 × 106 cells suspended in 100 μl normal saline) as previously proposed [28]. Briefly, 1 × 106 BMSCs in 100 μl of normal saline were delivered with a 22-gauge needle into the marrow cavity through the metaphysis and during needle removal to evenly distribute the cells. One month after transplantation, the mice were euthanized, and samples were collected for subsequent testing.
2.22. Micro-CT and analysis
Microcomputed tomography (micro-CT) was applied to determine bone mass. After anesthesia with 1% pentobarbital, the mice were secured on the table in the prone position. Then, femurs were imaged by microcomputed tomography (PerkinElmer, USA) at 55 kV at an 8 μm voxel size. After the image was reconstructed, the area of interest (ROI) of trabecular bone was analyzed within the 0.3-0.8 mm distal metaphysis from the growth plate. Analyze V12.0 software was used to analyze the data and quantify the parameters, including bone mineral density (BMD), trabecular bone thickness (Tb.Th), trabecular bone number (Tb.N), bone volume per tissue volume (BV/TV), and trabecular separation (Tb.Sp).
2.23. Calcein labeling assay
C57/BL6 mice were intraperitoneally injected with 20 mg/kg calcein (Sigma-Aldrich, USA), dissolved in PBS at a concentration of 2 mg/ml with 1 mg/ml NaHCO3 (Sigma-Aldrich, USA) at 16 d and 2 d before sacrifice. After sacrifice, the left femur was obtained, fixed in 4% formaldehyde, and embedded in methyl methacrylate. A hard tissue slicing machine (SP1600; Leica, Germany) was used to sagittally section specimens into 30-mm sections away from light. Then, cortical endosteum surfaces were evaluated using a fluorescence microscope (STP6000; Leica, Germany) with an excitation wavelength of 488 nm. The bone formation rate (BFR), mineral deposition rate (MAR) and the recommended relevant calculations were used for quantitative analysis using ImageJ 1.47 software [29].
2.24. Histology and immunohistochemistry
The femur was decalcified by 10% EDTA and then embedded in paraffin. Five-micrometer sagittal sections of the metaphysis were prepared, stained with Toluidine blue, TRAP, hematoxylin-eosin (HE) and Masson, and observed by light microscopy (Zeiss, Germany) [30].
For NFATc1 and RUNX2 immunohistochemical staining, after quenching with endogenous peroxidase, achieving antigen retrieval, and blocking nonspecific binding sites, the femur section were incubated with anti-NFATc1 (1:100; Abclonal, China), anti-RUNX2 (1:100; Huabio, China) at 4 °C overnight, followed by incubation with HRP-conjugated goat anti-rabbit secondary antibodies (Invitrogen, USA) at room temperature for 30 min. Finally, the sections were exposed to diaminobenzidine peroxidase substrate for 5 min and counterstained with Mayer’s hematoxylin.
2.25. Statistics
A two-tailed Student's t-test was applied when two groups were compared. One-way ANOVA with Bonferroni adjustment was performed to determine the differences among multiple groups. Data are shown as the mean ± SD. For all analyses, P values < 0.05 were considered statistically significant.
3. Results
3.1. LRRc17 expressed by BMSCs was positively correlated with age in mice
BMSCs were isolated from young, middle-aged and aged C57/BL6 mice and separately cultured to passage three (Figs. S1a and b). Cell death analysis showed that the number of dead cells increased with age in both adherent BMSCs and the culture medium supernatant (Fig. 1a). Furthermore, detection of telomerase activity revealed that telomerase activity gradually decreased, and the reduction was more obvious in female mice (Fig. 1b). In addition, SA-β-gal staining revealed that the ratio of senescent cells in O-BMSCs was notably higher than that in BMSCs isolated from young and middle-aged mice (Fig. 1c). Additionally, the expression of p16, p21 and p53 was also elevated with age (Fig. 1d–g). To reveal the underlying causes of these phenomena, we further analyzed Y-BMSCs and O-BMSCs by RNA sequencing. KEGG pathway analysis showed that the cellular senescence signaling pathway was highly enriched (Fig. 1h). Gene Set Enrichment Analysis (GSEA) showed that pathways were mainly associated with autophagy and p53 signaling (Fig. S2 a, b). Furthermore, weighted gene coexpression network analysis (WGCNA) showed that PTGFR, MX1, IFIT1B, LRRc17, CH3I2, VILL, PENK, GBJ2 and GJB2 might interact with each other (Fig. 1i). Among these genes, LRRc17 (level 1: organismal systems, level 2: development) has been widely reported to be correlated with bone homeostasis regulation, and the log2 (Old/Young) ratio was 2.50. Furthermore, time course analysis showed that LRRc17 was positively correlated with age (Fig. 1j), suggesting that LRRc17 may be a critical factor for regulating senescence.
Fig. 1.

Senescent BMSCs show high expression of LRRc17. BMSCs were separately isolated from young, middle-aged and aged mice and then cultured to passage 3. (a) A photometric enzyme immunoassay was performed to assess the BMSC death ratio via quantitative assessment of cytoplasmic histone-associated DNA fragments (n = 3). (b) Telomerase activity was determined with a commercial TeloTAGGG Telomerase PCR ELISA kit (n = 3). (c) Quantitative analysis of SA-β-gal-positive BMSCs. Five fields from each section were randomly selected to calculate the positive SA-β-gal cell ratio (n = 3). Scale bar, 10 μm. (d–g) BMSCs were lysed and prepared to measure the expression levels of p16, p21 and p53 by Western blotting (n = 3). Aged, middle-aged, and young BMSCs were subjected to RNA-seq, and differentially expressed genes were enriched and subjected to (h) KEGG pathway enrichment analysis, (i) weighted gene coexpression network analysis (WGCNA), (j) and time course analysis (n = 3). All data are shown as the mean ± SD. **P < 0.01, *P < 0.05; NS: not significant (P > 0.05).
3.2. LRRc17 controlled BMSC senescence and differentiation
To determine whether LRRc17 is involved in cellular senescence, we knocked down LRRc17 in O-BMSCs via lentiviral transfection and found that LRRc17 silencing significantly decreased the expression levels of p16, p21 and p53 in O-BMSCs compared with control cells, but there were no notable changes in the GFP-transfected cells (Fig. 2a). Furthermore, Y-BMSCs were induced to senescence, characterized by increased expression of p16, p21, and p53, via H2O2 treatment. However, senescence was significantly reversed when LRRc17 was downregulated (Fig. 2b, Figs. S3a–d). In addition, numerous reports have found that in metabolic disorders, senescent cells produce more cytotoxic components, which leads to abnormal cell proliferation and a bias toward apoptosis. Thus, ROS detection, apoptosis and the cell cycle were assessed, and we found that LRRc17 knockdown significantly decelerated ROS accumulation after H2O2 treatment (Fig. S3e). Moreover, apoptosis analysis revealed that the proportion of apoptotic cells declined notably (Fig. S3f), and the number of cells in the quiescent phase (G0/G1) was also much lower in the LRRc17 knockdown group than in the H2O2-treated group (Fig. S3g). Our data demonstrated that LRRc17 knockdown effectively alleviated BMSC senescence.
Fig. 2.

Silencing LRRc17 alleviates BMSC senescence and alters differentiation potential. O-BMSCs at passage 3 were treated with 100 nM RAPA, followed by transfection of LVsh-LRRc17 or LVGFP for 72 h. (a) Quantitative analysis of the protein expression levels of p16, p21, and p53 in 4 different groups (O-BMSCs, RAPA-treated O-BMSCs, LVsh-LRRc17-transfected O-BMSCs, and LVGFP-transfected O-BMSCs) by Western blotting (n = 3). Senescence was induced in Y-BMSCs at passage 3 with 400 μM H2O2, followed by treatment with 100 nM RAPA or lentivirus transfection for 72 h. (b) Quantitative analysis of the levels of p16, p21, and p53 in 4 different groups (Y-BMSCs, H2O2-treated young BMSCs, H2O2- and RAPA-treated young BMSCs, H2O2- and LVsh-LRRc17-transfected young BMSCs) by Western blotting (n = 3). (c) Quantitative analysis of the protein levels of p16, p21, and p53 in 3 different groups (Y-BMSCs, LVGFP-transfected Y-BMSCs, and LVov-LRRc17-transfected Y-BMSCs) by Western blotting (n = 3). Young, middle-aged, old, youngov-LRRc17, and oldsh-LRRc17 BMSCs at passage 3 were subjected to osteogenic and adipogenic induction. (d, e) BMSCs (3 × 105) were subjected to osteogenic induction for 14 d, and the mineralized nodules were quantified by calculating the ratio of red mineralization area to total area after alizarin red staining (n = 3). Scale bar, 200 μm. (f, g) BMSCs (3 × 105) were subjected to adipogenic induction for 21 days. Then, lipid droplets were quantified by calculating the ratio of red lipid droplet area to total area after Oil Red O staining (n = 3). Scale bar, 200 μm. (h, i, j) The expression levels of ALP and RUNX2 were detected by Western blotting after osteogenic induction (n = 3). (k) The expression levels of PPAR-γ and LPL were detected and quantified by Western blotting after adipogenic induction. β-Actin was used as an internal control (n = 3). All data are shown as the mean ± SD. **P < 0.01, *P < 0.05; NS, not significant (P > 0.05). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Senescence of BMSCs leads to changes in their differentiation potential; thus, we next investigated whether LRRc17 is involved in the aging process and multidifferentiation potential of BMSCs. Here, the results showed that overexpression of LRRc17 accelerated the senescence of Y-BMSCs, as indicated by the noticeably increased protein levels of p16, p21 and p53 in the LRRc17 overexpression group compared with the GFP transfection group (Fig. 2c). In addition, similar to O-BMSCs, after overexpression of LRRc17, the osteogenic capacity of Y-BMSCs declined, while the adipogenic capacity increased (Fig. 2d–g). More interestingly, after LRRc17 was knocked down, the osteogenic differentiation ability of O-BMSCs was increased, and their adipogenic differentiation ability was decreased. Furthermore, consistent with the above results, the expression of key osteogenesis genes, including ALP and RUNX2 (Fig. 2h, i, j), and the adipogenic genes PPARγ and LPL in BMSCs was increased (Fig. 2k). These findings suggested that overexpression of LRRc17 accelerated the aging process in BMSCs, which led to preferential adipogenic differentiation of MSCs rather than osteogenic differentiation.
3.3. Mitochondrial impairment in senescent BMSCs was alleviated by LRRc17 knockdown
Mitochondrial dysfunction leads to dysfunctional reactive oxygen species metabolism, resulting in DNA impairment and DNA damage response activation, which accelerates the aging process. MitoTracker Green staining showed that compared with that of Y-BMSCs, the mitochondrial morphology of O-BMSCs was discontinuous and showed obvious fragmentation (Fig. 3a). Similar to the O-BMSCs, H2O2-induced BMSCs also exhibited mitochondrial fragmentation, while the mitochondrial impairment was significantly improved after transfection of sh-LRRc17 (Fig. 3b and c). Moreover, the expression of the mitochondrial inner membrane fusion protein OPA1 was also significantly increased, while the expression of the mitochondrial fission protein Drp1 was reduced (Fig. 3d). Taken together, these results revealed that LRRc17 knockdown alleviated H2O2-induced mitochondrial fission in BMSCs.
Fig. 3.

Knockdown of LRRc17 reverses mitochondrial dysfunction in senescent BMSCs. (a) Representative images of MitoTracker Green fluorescence in BMSCs from young and aged mouse BMSCs. (b) Representative images of Y-BMSCs with different treatments visualized with MitoTracker Green staining. The groups are described in Fig. 2b. (c) Computer-assisted morphometric analyses of mitochondrial morphology (n = 3). (d) Western blotting analysis with quantification of Drp1 and OPA1 proteins in BMSCs (n = 3). (e) Morphology and mtROS of BMSCs were detected with MitoSOX (red) and MitoTracker (green), respectively. Scale bar, 5 μm. (f) mtROS staining was conducted with MitoSOX Red and is shown as the relative mean fluorescence intensity as measured by flow cytometry (n = 3). (g) Detection of JC-1 aggregates (red) and monomers (green) in BMSCs by confocal fluorescence microscopy. Scale bar, 10 μm. (h, i) Flow cytometry assessment of mitochondrial membrane potential (aggregate fluorescence/monomer fluorescence) of BMSCs (n = 3). (j) Quantification of ATP content by microplate reader (n = 3). (k, l) BMSCs were harvested and seeded in a Seahorse × 24 microplate. OCR was detected with a Cell Mito Stress Test Kit, and basal respiration and maximal respiration were analyzed with Wave Software (n = 3). All data are shown as the mean ± SD. **P < 0.01, *P < 0.05; NS, not significant (P > 0.05). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Subsequently, the function of mitochondria was further evaluated. MitoSOX staining showed that mtROS increased sharply after H2O2 induction, indicating that mitochondria undergo noticeable oxidative stress damage (Fig. 3e). However, LVsh-LRRc17 transfection effectively decreased the accumulation of mtROS (Fig. 3f). Furthermore, under H2O2 stimulation, the red fluorescence of JC-1 increased, while the green fluorescence decreased in LRRc17 knockdown MSCs, revealing that LVsh-LRRc17 transfection prevented mitochondrial membrane potential disruption (Fig. 3g). In line with the fluorescence images, flow cytometry analysis of JC-1 also verified these results (Fig. 3h and i). As a consequence, we also found that LVsh-LRRc17 transfection reversed the decrease in ATP production induced by H2O2 stimulation (Fig. 3j). Finally, OXPHOS characteristics were obtained by calculating the differences in oxygen consumption rate (OCR) upon the injection of Oligo, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), and rotenone (ROT) + antimycin A (AA), and we found that the basal and maximal mitochondrial respiration of BMSCs in the LVsh-LRRc17-transfected group were significantly higher than those in the H2O2-induced senescence group (Fig. 3k and l). Together, these data suggested that knockdown of LRRc17 not only restored the morphology of mitochondria but also effectively improved mitochondrial bioenergetics.
3.4. LRRc17 knockdown activated mitophagy through the PI3K/mTOR pathway
Autophagy, an evolutionarily conserved process involving the degradation of unnecessary or dysfunctional cell components by the lysosomal machinery, plays an essential role in mitochondrial quality control via clearance of damaged mitochondria. Here, we found that the expression of both LC3 II and Beclin1 was decreased in H2O2-induced senescent Y-BMSCs, suggesting that autophagy activity was inhibited in senescent BMSCs (Fig. 4a and b). To further investigate the role of autophagy in MSC senescence, H2O2-treated BMSCs and O-BMSCs were treated with RAPA (an inhibitor of mTOR), and autophagy was activated. The aging phenotype (characterized by increased expression of p16, p53, and p21, increased apoptosis, and decreased cell proliferation) (Fig. 2a, b, Figs. S3a–d) and mitochondrial damage, including mitochondrial fragmentation (Fig. 3b and c) and mtROS production (Fig. 3e and f), the decline of OCR (Fig. 3k, l), ATP production (Fig. 3j) and mitochondrial membrane potential (Fig. 3g–i), were significantly alleviated, indicating that autophagy protected BMSCs against senescence. Interestingly, we found that the protein expression of LC3II and Beclin1 was increased, while the protein expression of P62 was decreased in the sh-LRRc17 group, indicating that LRRc17 knockdown activated autophagy (Fig. 4a–d).
Fig. 4.

LRRc17 knockdown maintains autophagic flux and mitophagy via mTOR/PI3K inhibition in senescent BMSCs. (a, b) Western blot analysis and quantification of the expression of LRRc17, p-mTOR/mTOR, p-PI3K/PI3K, Beclin1, P62, and LC3Ⅱ in 4 different groups as described in Fig. 2b (n = 3). (c, d) Western blot analysis and quantification of the protein levels of LRRc17, mTOR, p-mTOR, PI3K, p-PI3K, Beclin1, P62, and LC3Ⅱ in the 4 different groups as described in Fig. 2a (n = 3). Y-BMSCs were treated with H2O2 or transfected with LVsh-LRRc17 for 72 h, followed by treatment with 10 μM HCQ for 8 h before cells were collected and lysed for Western blotting. (e) Representative images and quantitative analysis of the Western blotting results for P62 and LC3B in BMSCs. β-Actin was used as an internal control (n = 3). (f) Colocalization of LC3B (green) and mitochondria (red) was assessed to evaluate mitophagy in the different groups, and nuclear fluorescence was visualized with DAPI staining (blue) (n = 3). Scale bar, 5 μm. All data are shown as the mean ± SD. **P < 0.01, *P < 0.05; NS, not significant (P > 0.05). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Autophagy is regulated by mTOR dependent pathway (PI3K/AKT/mTOR signaling) and mTOR independent pathways including MAPK/JNK, Calpain1/Atg5 signaling. Here, we found that the expressions of p-JNK and calpain1 significantly increased in H2O2-induced senescent BMSCs, but there were no significant changes after LRRC17 knockdown, suggesting mTOR independent pathways are not regulated by LRRc17 (Fig. S4). Moreover, the H2O2-induced increase in the expression of p-PI3K and p-mTOR, which negatively regulate the autophagy process, was significantly reduced in the sh-LRRc17 group (Fig. 4a–d). Based on these results, we speculated that sh-LRRc17-mediated prevention of BMSC senescence may rely on the activation of autophagy.
To determine the role of autophagy in LRRc17-mediated BMSC senescence, the autophagy-lysosomal inhibitor HCQ was employed to block the degradation of autophagic vacuoles. After treatment with HCQ, LC3Ⅱ accumulated significantly, and P62 levels also showed obvious enhancement (Fig. 4e). Consistent with these results, the colocalization of LC3B and mitochondria in Y-BMSCs increased greatly after HCQ treatment (Fig. 4f). These results revealed that autophagic flux was blocked. Next, we explored the effects of autophagic flux blockade on BMSC senescence. As shown in Fig. 5, HCQ promoted the accumulation of JC-1 monomers in mitochondria, indicating their low membrane potential, and this effect was accompanied by an increase in mtROS in control BMSCs (Fig. 5a and b). In addition, in H2O2-induced senescent BMSCs, the sh-LRRC17-mediated improvement of mitochondrial function was significantly blocked after HCQ treatment. Consequently, the SA-β-gal-positive BMSC ratio also increased markedly in the presence of HCQ (Fig. 5c). Similarly, p16, p21, and p53 expression significantly increased in HCQ-treated BMSCs (Fig. 5d and e). These results appeared to prove that the blockade of autophagic vacuole degradation led to cellular senescence, and the alleviation of senescence mediated by sh-LRRc17 relied on the activation of autophagy and the improvement of mitochondrial function.
Fig. 5.

LRRc17 downregulation prevents H2O2-induced BMSC senescence by enhancing mitophagy. O-BMSCs transfected with LVsh-LRRc17 were treated with 10 μM HCQ for 24 h before the experiment, and young BMSCs were used as a control group. (a) The mitochondrial membrane potential of BMSCs was detected by measuring JC-1 fluorescence with flow cytometry (n = 3). (b) mtROS were stained with MitoSOX Red and are shown as the relative mean fluorescence intensity as measured by flow cytometry (n = 3). (c) Quantitative analysis of SA-β-gal-positive cells. Five fields from each section were randomly selected to calculate the positive SA-β-gal cell ratio (n = 3). Scale bar, 10 μm. (d) Western blotting results for p16, p21 and p53 expression in BMSCs. β-Actin was used as an internal control. (e) The bar charts show the quantitative results of the indicated proteins (n = 3). All data are shown as the mean ± SD. **P < 0.01, *P < 0.05; NS, not significant (P > 0.05). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.5. LRRc17 expressed by BMSCs modulated osteoclastogenesis and osteogenesis by inhibiting the differentiation of OCs and MC3T3-E1 cells
To further substantiate the correlation between LRRc17 expression in BMSCs and bone homeostasis in vitro, MSCs were cocultured with OCs induced by primary bone marrow-derived mononuclear cells for 5 days, and then OCs were stained with TRAP. After coculture, the number of TRAP-positive cells in the OC + BMSC group was less than that in the OC group. Moreover, the inhibitory effect of BMSCs on OC differentiation was further weakened when BMSCs were transfected with LVov-LRRc17 (Figs. S5a–c). Moreover, the expression of transcription factors involved in osteoclast differentiation, including Nfatc1, Fos, Src, Acp5 and Ctsk, also declined considerably (Fig. S5d). Similarly, after BMSCs and OBs were cocultured for 10 days, alizarin red staining showed that LRRc17 overexpression in BMSCs significantly reduced the osteogenic differentiation potential of MC3T3-E1 cells (Figs. S5e and f). Furthermore, the expression of the key osteogenic proteins ALP and RUNX2 was consistent with the results of alizarin red staining (Figs. S5g–i). In addition, the gene expression levels of the osteogenesis-related transcription factors Bsp, Sp7/Osterix and RUNX2 were also inhibited in LRRc17-overexpressing BMSCs (Fig. S5j-l). These data showed that LRRc17 inhibited both OC and MC3T3-E1 differentiation via paracrine signaling to contribute to bone homeostasis regulation in vitro.
3.6. Knockdown of LRRc17 in O-BMSCs alleviated bone loss in OVX mice
To investigate whether knockdown of LRRc17 affects O-BMSCs in the treatment of osteoporosis in vivo, OVX-induced osteoporotic mice were euthanized and evaluated one month after local BMSC transplantation through the metaphysis. Micro-CT analysis showed that O-BMSCs possessed a decreased ability to alleviate bone loss in osteoporotic mice compared with Y-BMSCs, but LRRc17 knockdown in O-BMSCs reversed this effect, as shown by increased BMD, BV/TV, Tb.Th, Tb.N and decreased Tb.Sp (Fig. 6a–c). Moreover, calcitonin double labeling analysis found that O-BMSC transplantation increased new bone formation in osteoporotic mice after LRRc17 knockdown (Fig. 6d and e). Furthermore, pathological staining of OBs and OCs showed that in the O-BMSC transplantation group with LRRc17 knockdown, the number of toluidine blue-labeled osteoblasts was notably greater than that in the O-BMSC transplantation group (Fig. 6f and g). Conversely, there was a significant decrease in TRAP-labeled osteoclasts (Fig. 6h and i). Furthermore, derangement distribution and expanded bone marrow cavity were restored by knockdown of LRRc17 in O-MSCs (Fig. 7a and b), and the beneficial effects of O-MSCs on bone loss were remarkably improved when the LRRc17 was knocked down (Fig. 7c and d). These results suggested that LRRc17 knockdown improved the ability of BMSCs to alleviate bone loss in osteoporotic mice.
Fig. 6.

Transplantation of O-BMSCs transfected with sh-LRRc17 alleviates the osteoporotic phenotype and reverses bone loss in OVX mice. Five groups of mice were sacrificed 1 month after BMSC transplantation (n = 7 per group). (a) Representative micro-CT images of trabecular bone sections. Scale bars: 500 mm (top) and 100 mm (bottom). (b, c) Quantitative analysis of the corresponding parameters, including BMD, BV/TV, Tb.Th, Tb.Sp, and Tb.N. (d) Representative fluorescence images of double calcein labeling and (e) quantitative analysis of MAR, MS/BS, and BFR. Scale bar, 25 μm. (f) Representative images of OB staining with toluidine blue (red arrows). Scale bars, 50 μm. Quantitative analysis of (g) the number of osteoblasts per bone surface (N.Ob/BS) and osteoblast surface over bone surface (Ob.S/BS). (h) Representative images of OC staining with TRAP (red arrows). Scale bar, 50 μm. Quantitative analysis of (i) the number of osteoclasts per bone surface (N.Oc/BS) and osteoclast surface over bone surface (Oc.S/BS). All data are shown as the mean ± SD. **P < 0.01, *P < 0.05; NS, not significant (P > 0.05). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Fig. 7.

LRRc17 knockdown of O-BMSCs improves the defect area in OVX mice. The histological assessment of the defect area by HE staining (a) and Masson staining (b). Images of immunohistochemical staining of osteoclast factor NFATc1 (c) and osteogenic factor RUNX2 (d). Scale bar, 50 μm. (e, f) The qutification of NFATc1 and RUNX2 in mice from different groups. All data were shown as mean ± SD. **P < 0.01, *P < 0.05.
4. Discussion
The correlation between stem cell senescence and degenerative diseases and the underlying molecular mechanism have generated tremendous interest over the years. In our study, we revealed for the first time that overexpression of the key soluble cytokine LRRc17 was positively correlated with the aging process of BMSCs. LRRc17 accumulation led to a shift towards adipogenic differentiation and cellular senescence of Y-BMSCs. Moreover, LRRc17 downregulation reversed H2O2-induced or natural aging-induced senescence and mitochondrial dysfunction and increased autophagy by inhibiting the PI3K/mTOR pathway. Furthermore, LVsh-LRRc17-transfected O-BMSC transplantation ameliorated OP-induced bone loss via activation of autophagy in BMSCs and inhibition of osteoclastogenesis in vivo. Taken together, our results uncovered a notable role for LRRc17 in the BMSC aging process.
In vitro prolonged culture, previous studies have demonstrated that several bioactive factors secreted by nonsenescent MSCs can postpone replicative senescence by improving osteogenic differentiation potential and cell proliferation [31]. We confirmed the effect of aging on BMSCs, which showed clear signs of senescence between young, middle-aged and old C57/BL6 mice, consistent with prior studies reported by Li Yang et al. The hallmarks of aging include augmentation of SA‐β‐gal, increased cell death ratio, and decreased telomerase activity. More importantly, senescence appears to alter BMSC differentiation potential. O-BMSCs exhibited impaired pluripotency; in addition, the dynamic balance between osteogenesis and adipogenesis was disrupted [32].
A growing number of studies have suggested a negative regulatory role of LRRc17 in blocking NF-kB ligand (RANKL) receptor activator-mediated osteoclastogenesis in bone remodeling. Furthermore, LRRc17 is highly expressed in OBs and is markedly inhibited by 1,25(OH)2D3, an active metabolite of VD and a pro-osteoclastogenic factor. In addition, LRRc17 inhibits RANKL-induced osteoclast differentiation [14,33,34]. Here, we unexpectedly found through transcriptome sequencing that LRRc17 was highly expressed in aged mouse-derived BMSCs. To investigate this hypothesis, analysis of the differentiation ability of BMSCs was performed. Senescent BMSCs in bone marrow exhibited a shift in multilineage differentiation potential from osteogenic to adipogenic as well as a marked decrease in regenerative capacity [35]. We found that LRRc17 overexpression in Y-BMSCs had a negative effect on osteogenic ability but was beneficial to adipogenesis. LRRc17 knockdown in O-BMSCs, however, resulted in decreased adipogenesis and elevated osteogenesis as well as decreased expression of age-related proteins. Thus, our study demonstrated for the first time that aging disrupted the dynamic balance between the adipogenic and osteogenic potential of BMSCs by modulating the expression of LRRc17.
Oxidative stress-induced mitochondrial impairment is one of the major injuries that accelerates cellular senescence in vitro and in vivo [36]. Continuous oxidation contributes to DNA impairment and disrupted DNA repair, protein structural instability and mitochondrial dysfunction, which lead to mutual transformation that accelerates the aging process. Several studies have also noted that restoring mitochondrial dysfunction prevents senescence in musculoskeletal tissue and MSCs [11,[37], [38], [39]]. In our study, similar results were observed in O-BMSCs, and we found that the mitochondria showed decreased integrity, maturity and abundance in senescent BMSCs. Moreover, LRRc17 downregulation reversed BMSC senescence via stabilization of mitochondrial function. Our results further indicated that knockdown of LRRc17 improved the morphological integrity of mitochondria. The crucial mitochondrial characteristic involved in the senescence of BMSCs is mitochondrial dynamism (i.e., cycles of fission and fusion), which is involved in the quality control mechanisms that determine mitochondrial mass and morphology [40]. In accordance with previous reports, our data demonstrated enhanced mitochondrial fusion, as evidenced by elongated mitochondria, with a decrease in Drp1 protein expression and an increase in OPA1 protein expression in LVsh-LRRc17-transfected BMSCs compared with H2O2-induced senescent BMSCs. Based on these findings, we conclude that LRRc17 mediates BMSC senescence by regulating mitochondrial dynamics.
Autophagy is an intracellular degradation system with multiple pathophysiological and physiological functions [41]. Mounting evidence has revealed that mitochondria-specific autophagy performs a variety of interrelated functions involved in maintaining cell homeostasis, especially in regulating cell metabolism and lifespan and participating in cell differentiation [42]. Our data also showed that the increased oxidative stress damage in senescent BMSCs was correlated with a decrease in mitophagy. Furthermore, age-related mitochondrial dysfunction has been recognized to promote and strengthen the aging response by stimulating the p16 and p53/p21 pathways [43], which is consistent with our result that after transfection with sh-LRRc17, senescence-associated proteins such as p53, p16 and p21 were also effectively inhibited. Moreover, recent studies have reported that autophagy enhancement in senescent MSCs through a genetic approach or pharmacological stimulation by the mTOR inhibitor RAPA restored the proliferative ability and osteogenic differentiation of MSCs in vitro [6]. Laura García-Prat et al. reported that overexpressing Atg7 (a key regulator of autophagosome formation) restored autophagy and consequently prevented stem cell senescence [11]. Nonetheless, the role of LRRc17 in BMSC autophagy and the consequent effects of improving mitochondrial function by removing damaged mitochondria have not been reported. As shown in Fig. 3, Fig. 4, Fig. 5, we first revealed that LRRc17 knockdown activated autophagy and enhanced the colocalization between LC3B and mitochondria, thereby inhibiting mitochondrial impairment and cell senescence. Further experiments indicated that the LRRc17 knockdown-induced inhibition of BMSC senescence was counteracted by HCQ treatment. All the results illustrated that mitophagy enhancement induced by LRRc17 knockdown maintains the regenerative abilities of MSCs.
Autophagy has been reported to alleviate estrogen-induced osteoporosis by maintaining the stemness and cellular function of MSCs [44]. Some reports have also shown that autophagy enhances the osteogenic capacity of hMSCs isolated from the vertebrae of senile osteoporosis patients [13]. Moreover, activation of autophagy in MSCs has been demonstrated to significantly attenuate the osteoporotic phenotype in OVX mice compared with sham-operated mice [44,45]. Here, we found that after silencing LRRc17, autophagy activity in O-BMSCs was elevated. Consequently, the therapeutic effect of O-BMSCs on osteoporosis was also significantly improved, as shown by an increased number and volume of trabecular bone in the femur, a faster formation ratio of new bone, a decreased number of TRAP-positive osteoclasts, and an increased number of osteoclasts. Therefore, our data suggest that sh-LRRc17-mediated BMSC improvement in osteoporosis is correlated with bone homeostasis by activating autophagy.
The mTOR signaling pathway plays a pivotal role in BMSC senescence [46]. Additionally, it has been widely reported that the protective effect of autophagy in senescent BMSCs is regulated by the Akt/mTOR signaling pathway. In addition, inhibition of PI3K/AKT/mTOR signaling maintains MSCs in their immature undifferentiated state by providing a stem cell niche during long-term culture expansion [47]. mTOR is hyperactivated, thus blocking autophagy induction and downregulating autophagosome formation-related proteins, including FOXO1 and ATG7, accompanied by a decline in mitophagy activity [48]. In our study, we found that LRRc17 downregulation blocked the PI3K/mTOR pathway and thus activated autophagy. However, the underlying mechanisms of how LRRC17 modulates PI3K/mTOR remain unclear. RANKL-induced NFATc1 expression and osteoclastogenesis were inhibited by LRRc17-mediated blockade of PLCγ signaling [14]. PLCγ has been implicated in cell proliferation and differentiation via its activation of downstream target genes such as PKC and PI3K, which are highly associated with the progression of brain diseases, hematopoietic function and diabetes [49]. Whether the excessive secretion of LRRc17 in O-BMSCs inhibits the autophagic capacity of BMSCs by activating PLCγ-mediated PI3K/mTOR signaling should be further confirmed.
In addition to mitophagy, mitochondrial fusion/fission and mitochondrial biogenesis also play crucial roles in maintaining mitochondrial homeostasis. Alterations in mitophagy, biogenesis, and dynamic fusion/fission are common responses of mitochondria to oxidative stresses and stimuli [50]. mTOR signaling is also involved in mitochondrial fusion and fission as well as the regulation of mitochondrial biosynthesis. A recent study found that the AMPK/SIRT1/PGC1-α pathway blocked the upregulation of age-related mTOR and thus led to increased mtDNA integrity and decreased mitochondrial fragmentation and maintained mitochondrial biogenesis [51]. Moreover, mTORC1 controls mitochondrial respiration and energy metabolism by modulating the transport of the nucleus-encoded gene TFAM to mitochondria, which is predominantly mediated by 4E-BPs [52]. In our study, we found that compared with mitochondrial fragmentation in O-BMSCs, mitochondrial fission was notably suppressed after LRRc17 knockdown. Therefore, we deduced that the protective effect of LRRc17 knockdown on mitochondrial bioenergetics may occur not only through PI3K/mTOR pathway-regulated mitophagy but also through mTOR-related mitochondrial biogenesis and metabolism.
Collectively, our findings revealed that LRRc17 knockdown inhibited the senescence of O-BMSCs via activation of mitophagy, which improved the therapeutic effect of O-BMSCs on osteoporosis. It should also be noted that there were no obvious differences in FPKM between middle-aged and young mouse-derived BMSCs, which may be due to an insufficient age gap between young and middle-aged mice. Furthermore, we still do not fully understand how LRRc17 modulates the PI3K/mTOR pathway. In addition, the C57/BL6 osteoporosis model used in our research consisted of only female mice and thus did not fully represent natural aging in males. More studies need to be conducted on other aging mouse models, including the senescence-accelerated mouse model, which was generated by selective inbreeding of the AKR/J strain.
In conclusion, our current study revealed for the first time the negative role of LRRc17 in BMSC senescence through blocking mitophagy capacity, and O-BMSCs with LRRc17 knockdown alleviated aging-related bone loss in an OVX mouse model. These findings indicate that targeting LRRc17 could be a promising therapeutic approach to rejuvenate senescent BMSCs and attenuate degenerative disorders.
Declaration of competing interest
The authors declare no potential conflicts of interest.
Acknowledgments
We are grateful to Li Zhou, Bo Su, Zhen Yang, Di Wang, Linqiao Tang and Yan Liang from Core Facility of West China Hospital, Sichuan University for their support. This research was funded by grants from the Chengdu Giant Panda Breeding Research Foundation (CPF2015-08), the National Natural Science Foundation of China (81900666), and 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University, China (ZYGD18014).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.101963.
Contributor Information
Jingqiu Cheng, Email: jqcheng@scu.edu.cn.
Jie Zhang, Email: zjie@scu.edu.cn.
Author contributions
Fei Liu and Yujia Yuan designed the study and wrote the paper; Fei Liu Lin Bai performed the experiment; Longhui Yuan and Lan Li analyzed the data; Jingping Liu, Younan Chen helped perform the analysis with constructive discussions, Yanrong Lu, Jingqiu Cheng and Jie Zhang critically revised the paper; All authors have read and approve the final manuscript.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Hwang E.S. Senescence suppressors: their practical importance in replicative lifespan extension in stem cells. Cell. Mol. Life Sci. 2014;71:4207–4219. doi: 10.1007/s00018-014-1685-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramakrishnan A., Torok-Storb B., Pillai M.M. Primary marrow-derived stromal cells: isolation and manipulation. Methods Mol. Biol. 2013;1035:75–101. doi: 10.1007/978-1-62703-508-8_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiernan J., Hu S., Grynpas M.D., Davies J.E., Stanford W.L. Systemic mesenchymal stromal cell transplantation prevents functional bone loss in a mouse model of age-related osteoporosis. Stem Cells Transl Med. 2016;5:683–693. doi: 10.5966/sctm.2015-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y., Wu Q., Wang Y., Li L., Bu H., Bao J. Senescence of mesenchymal stem cells (Review) Int. J. Mol. Med. 2017;39:775–782. doi: 10.3892/ijmm.2017.2912. [DOI] [PubMed] [Google Scholar]
- 5.Ren X., Hu B., Song M., Ding Z., Dang Y., Liu Z. Maintenance of nucleolar homeostasis by CBX4 alleviates senescence and osteoarthritis. Cell Rep. 2019;26:3643–3656 e3647. doi: 10.1016/j.celrep.2019.02.088. [DOI] [PubMed] [Google Scholar]
- 6.Infante A., Rodriguez C.I. Osteogenesis and aging: lessons from mesenchymal stem cells. Stem Cell Res. Ther. 2018;9:244. doi: 10.1186/s13287-018-0995-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan M., Mohsin S., Khan S.N., Riazuddin S. Repair of senescent myocardium by mesenchymal stem cells is dependent on the age of donor mice. J. Cell Mol. Med. 2011;15:1515–1527. doi: 10.1111/j.1582-4934.2009.00998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rubin H. Promise and problems in relating cellular senescence in vitro to aging in vivo. Arch. Gerontol. Geriatr. 2002;34:275–286. doi: 10.1016/s0167-4943(01)00221-7. [DOI] [PubMed] [Google Scholar]
- 9.Chen H.F., Shi B.Y., Feng X.B., Kong W., Chen W.W., Geng L.Y. Leptin and neutrophil-activating peptide 2 promote mesenchymal stem cell senescence through activation of the phosphatidylinositol 3-kinase/akt pathway in patients with systemic lupus erythematosus. Arthritis & Rheumatology. 2015;67:2383–2393. doi: 10.1002/art.39196. [DOI] [PubMed] [Google Scholar]
- 10.Fujimaki S., Wakabayashi T., Takemasa T., Asashima M., Kuwabara T. The regulation of stem cell aging by Wnt signaling. Histol. Histopathol. 2015;30:1411–1430. doi: 10.14670/HH-11-657. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Prat L., Martinez-Vicente M., Perdiguero E., Ortet L., Rodriguez-Ubreva J., Rebollo E. Autophagy maintains stemness by preventing senescence. Nature. 2016;529:37–42. doi: 10.1038/nature16187. [DOI] [PubMed] [Google Scholar]
- 12.Han Y., Zhang L., Xing Y., Zhang L., Chen X., Tang P. Autophagy relieves the function inhibition and apoptosispromoting effects on osteoblast induced by glucocorticoid. Int. J. Mol. Med. 2018;41:800–808. doi: 10.3892/ijmm.2017.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wan Y., Zhuo N., Li Y., Zhao W., Jiang D. Autophagy promotes osteogenic differentiation of human bone marrow mesenchymal stem cell derived from osteoporotic vertebrae. Biochem. Biophys. Res. Commun. 2017;488:46–52. doi: 10.1016/j.bbrc.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Kim T., Kim K., Lee S.H., So H.S., Lee J., Kim N. Identification of LRRc17 as a negative regulator of receptor activator of NF-kappaB ligand (RANKL)-induced osteoclast differentiation. J. Biol. Chem. 2009;284:15308–15316. doi: 10.1074/jbc.M807722200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tyrrell D.J., Blin M.G., Song J., Wood S.C., Zhang M., Beard D.A. Age-associated mitochondrial dysfunction accelerates atherogenesis. Circ. Res. 2020;126:298–314. doi: 10.1161/CIRCRESAHA.119.315644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao L., Yang X., Su X., Hu C., Zhu X., Yang N. Redundant miR-3077-5p and miR-705 mediate the shift of mesenchymal stem cell lineage commitment to adipocyte in osteoporosis bone marrow. Cell Death Dis. 2013;4 doi: 10.1038/cddis.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sui B.D., Hu C.H., Zheng C.X., Shuai Y., He X.N., Gao P.P. Recipient glycemic micro-environments govern therapeutic effects of mesenchymal stem cell infusion on osteopenia. Theranostics. 2017;7:1225–1244. doi: 10.7150/thno.18181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ning L.J., Zhang Y.J., Zhang Y., Qing Q., Jiang Y.L., Yang J.L. The utilization of decellularized tendon slices to provide an inductive microenvironment for the proliferation and tenogenic differentiation of stem cells. Biomaterials. 2015;52:539–550. doi: 10.1016/j.biomaterials.2015.02.061. [DOI] [PubMed] [Google Scholar]
- 20.Wu H., Hu B., Zhou X., Zhou C., Meng J., Yang Y. Artemether attenuates LPS-induced inflammatory bone loss by inhibiting osteoclastogenesis and bone resorption via suppression of MAPK signaling pathway. Cell Death Dis. 2018;9 doi: 10.1038/s41419-018-0540-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shu Y.J., Weng H., Ye Y.Y., Hu Y.P., Bao R.F., Cao Y. SPOCK1 as a potential cancer prognostic marker promotes the proliferation and metastasis of gallbladder cancer cells by activating the PI3K/AKT pathway. Mol. Canc. 2015;14:12. doi: 10.1186/s12943-014-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuo R., Liu M., Wang Y., Li J., Wang W., Wu J. BM-MSC-derived exosomes alleviate radiation-induced bone loss by restoring the function of recipient BM-MSCs and activating Wnt/beta-catenin signaling. Stem Cell Res. Ther. 2019;10:30. doi: 10.1186/s13287-018-1121-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma Y., Qi M., An Y., Zhang L., Yang R., Doro D.H. Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging Cell. 2018;17 doi: 10.1111/acel.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao L., Su X., Yang X., Hu C., Li B., Lv Y. TNF-α inhibits FoxO1 by upregulating miR-705 to aggravate oxidative damage in bone marrow-derived mesenchymal stem cells during osteoporosis. Stem Cell. 2016;34:1054–1067. doi: 10.1002/stem.2274. [DOI] [PubMed] [Google Scholar]
- 25.Yu T., Jhun B.S., Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxidants Redox Signal. 2011;14:425–437. doi: 10.1089/ars.2010.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yau W.W., Singh B.K., Lesmana R., Zhou J., Sinha R.A., Wong K.A. Thyroid hormone (T3) stimulates brown adipose tissue activation via mitochondrial biogenesis and MTOR-mediated mitophagy. Autophagy. 2019;15:131–150. doi: 10.1080/15548627.2018.1511263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu D., Wu H., Wang C., Li Y., Tian H., Siraj S. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 2018;26:1735–1749. doi: 10.1038/s41418-018-0251-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinder B.P., Novak S., Wee N.K.Y., Basile M., Maye P., Matthews B.G. Engraftment of skeletal progenitor cells by bone-directed transplantation improves osteogenesis imperfecta murine bone phenotype. Stem Cell. 2020;38:530–541. doi: 10.1002/stem.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dempster D.W., Compston J.E., Drezner M.K., Glorieux F.H., Kanis J.A., Malluche H. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 2013;28:2–17. doi: 10.1002/jbmr.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sui B., Hu C., Zhang X., Zhao P., He T., Zhou C. Allogeneic mesenchymal stem cell therapy promotes osteoblastogenesis and prevents glucocorticoid-induced osteoporosis. Stem Cells Transl Med. 2016;5:1238–1246. doi: 10.5966/sctm.2015-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang B., Lee W.Y., Huang B., Zhang J.F., Wu T., Jiang X. Secretome of human fetal mesenchymal stem cell ameliorates replicative senescen. Stem Cell. Dev. 2016;25:1755–1766. doi: 10.1089/scd.2016.0079. [DOI] [PubMed] [Google Scholar]
- 32.Turinetto V., Vitale E., Giachino C. Senescence in human mesenchymal stem cells: functional changes and implications in stem cell-based therapy. Int. J. Mol. Sci. 2016;17 doi: 10.3390/ijms17071164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong N., Kim B.J., Kim C.H., Baek K.H., Min Y.K., Kim D.Y. Low plasma level of leucine-rich repeat-containing 17 (LRRc17) is an independent and additive risk factor for osteoporotic fractures in postmenopausal women. J. Bone Miner. Res. 2016;31:2106–2114. doi: 10.1002/jbmr.2902. [DOI] [PubMed] [Google Scholar]
- 34.Baumhoer D., Zillmer S., Unger K., Rosemann M., Atkinson M.J., Irmler M. MicroRNA profiling with correlation to gene expression revealed the oncogenic miR-17-92 cluster to be up-regulated in osteosarcoma. Cancer Genet. 2012;205:212–219. doi: 10.1016/j.cancergen.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 35.Macias I., Alcorta-Sevillano N., Rodriguez C.I., Infante A. Osteoporosis and the potential of cell-based therapeutic strategies. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21051653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campisi J. From cells to organisms: can we learn about aging from cells in culture? Exp. Gerontol. 2001;36:607–618. doi: 10.1016/s0531-5565(00)00230-8. [DOI] [PubMed] [Google Scholar]
- 37.Habiballa L., Salmonowicz H., Passos J.F. Mitochondria and cellular senescence: implications for musculoskeletal ageing. Free Radic. Biol. Med. 2019;132:3–10. doi: 10.1016/j.freeradbiomed.2018.10.417. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y., Liu Y., Chen E., Pan Z. The role of mitochondrial dysfunction in mesenchymal stem cell senescence. Cell Tissue Res. 2020;382:457–462. doi: 10.1007/s00441-020-03272-z. [DOI] [PubMed] [Google Scholar]
- 39.Shen G., Ren H., Shang Q., Qiu T., Yu X., Zhang Z. Autophagy as a target for glucocorticoid-induced osteoporosis therapy. Cell. Mol. Life Sci. 2018;75:2683–2693. doi: 10.1007/s00018-018-2776-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmitt K., Grimm A., Dallmann R., Oettinghaus B., Restelli L.M., Witzig M. Circadian control of DRP1 activity regulates mitochondrial dynamics and bioenergetics. Cell Metabol. 2018;27:657–666 e655. doi: 10.1016/j.cmet.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 41.Kang C., Elledge S.J. How autophagy both activates and inhibits cellular senescence. Autophagy. 2016;12:898–899. doi: 10.1080/15548627.2015.1121361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sui B., Hu C., Jin Y. Mitochondrial metabolic failure in telomere attrition-provoked aging of bone marrow mesenchymal stem cells. Biogerontology. 2016;17:267–279. doi: 10.1007/s10522-015-9609-5. [DOI] [PubMed] [Google Scholar]
- 43.Muñoz-Espín D., Serrano M. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014;15:482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 44.Qi M., Zhang L., Ma Y., Shuai Y., Li L., Luo K. Autophagy maintains the function of bone marrow mesenchymal stem cells to prevent estrogen deficiency-induced osteoporosis. Theranostics. 2017;7:4498–4516. doi: 10.7150/thno.17949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang X., Hou Z., Xie Y., Yan F., Li S., Zhu X. Icariin promotes osteogenic differentiation of bone marrow stromal cells and prevents bone loss in OVX mice via activating autophagy. J. Cell. Biochem. 2019;120:13121–13132. doi: 10.1002/jcb.28585. [DOI] [PubMed] [Google Scholar]
- 46.Gu Z., Tan W., Ji J., Feng G., Meng Y., Da Z. Rapamycin reverses the senescent phenotype and improves immunoregulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling pathway. Aging (Albany NY) 2016;8:1102–1114. doi: 10.18632/aging.100925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gharibi B., Farzadi S., Ghuman M., Hughes F.J. Inhibition of Akt/mTOR attenuates age-related changes in mesenchymal stem cells. Stem Cell. 2014;32:2256–2266. doi: 10.1002/stem.1709. [DOI] [PubMed] [Google Scholar]
- 48.Bordi M., Darji S., Sato Y., Mellen M., Berg M.J., Kumar A. mTOR hyperactivation in Down Syndrome underlies deficits in autophagy induction, autophagosome formation, and mitophagy. Cell Death Dis. 2019;10:563. doi: 10.1038/s41419-019-1752-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Follo M.Y., Manzoli L., Poli A., McCubrey J.A., Cocco L. PLC and PI3K/Akt/mTOR signalling in disease and cancer. Adv Biol Regul. 2015;57:10–16. doi: 10.1016/j.jbior.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 50.Yu B., Ma J., Li J., Wang D., Wang Z., Wang S. Mitochondrial phosphatase PGAM5 modulates cellular senescence by regulating mitochondrial dynamics. Nat. Commun. 2020;11:2549. doi: 10.1038/s41467-020-16312-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang N., Luo Z., Jin M., Sheng W., Wang H.T., Long X. Exploration of age-related mitochondrial dysfunction and the anti-aging effects of resveratrol in zebrafish retina. Aging (Albany NY) 2019;11:3117–3137. doi: 10.18632/aging.101966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morita M., Gravel S.P., Chenard V., Sikstrom K., Zheng L., Alain T. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metabol. 2013;18:698–711. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.