

Abstract
Background
Smooth muscle cells (SMC) play a critical role in atherosclerosis. The Aryl hydrocarbon receptor (AHR) is an environment-sensing transcription factor that contributes to vascular development, and has been implicated in coronary artery disease (CAD) risk. We hypothesized that AHR can affect atherosclerosis by regulating phenotypic modulation of SMC.
Methods
We combined RNA-Seq, ChIP-Seq, ATAC-Seq and in-vitro assays in human coronary artery SMC (HCASMC), with single-cell RNA-Seq (scRNA-Seq), histology, and RNAscope in an SMC-specific lineage-tracing Ahr knockout mouse model of atherosclerosis to better understand the role of AHR in vascular disease.
Results
Genomic studies coupled with functional assays in cultured HCASMC revealed that AHR modulates HCASMC phenotype and suppresses ossification in these cells. Lineage tracing and activity tracing studies in the mouse aortic sinus showed that the Ahr pathway is active in modulated SMC in the atherosclerotic lesion cap. Furthermore, scRNA-Seq studies of the SMC-specific Ahr knockout mice showed a significant increase in the proportion of modulated SMC expressing chondrocyte markers such as Col2a1 and Alpl, which localized to the lesion neointima. These cells, which we term “chondromyocytes” (CMC), were also identified in the neointima of human coronary arteries. In histological analyses, these changes manifested as larger lesion size, increased lineage-traced SMC participation in the lesion, decreased lineage-traced SMC in the lesion cap, and increased alkaline phosphatase activity in lesions in the Ahr knockout compared to wild-type mice. We propose that AHR is likely protective based on these data and inference from human genetic analyses.
Conclusions
Overall, we conclude that AHR promotes maintenance of lesion cap integrity and diminishes the disease related SMC-to-CMC transition in atherosclerotic tissues.
Keywords: Atherosclerosis; Smooth Muscle Cell Differentiation; Genetics, Animal Models; Coronary Calcification
Introduction
Coronary artery disease (CAD) is driven by both genetic and environmental risk. The contribution of genes and the environment is estimated to be ~50% each based on large human genetic studies.1, 2 Furthermore, genes can modify the disease process promoted by environmental exposures.3 Such interplay between genes and the environment has been difficult to study due to lack of power and lack of well-documented individual level measurement of exposure dosage.
The aryl hydrocarbon receptor (AHR) is a ligand activated transcription factor that is induced by both endogenous and exogenous compounds including environmental pollutants such as dioxin, polycyclic aromatic hydrocarbons (PAH), tobacco smoke, and organic waste products which have been linked to cardiovascular mortality.4, 5 AHR has established roles in the activation of detoxifying enzymes in response to pollutant exposure, but also emerging roles in vascular development and homeostasis. Specifically, disruption of the AHR pathway resulted in reduced coronary artery formation, and increased cardiovascular birth defects in animal models.6–8 It is yet unclear whether AHR plays a protective role in the vasculature in response to environmental toxins or if AHR is the driver pathway that is responsible for pathological changes in the context of exposure to these atherogenic toxins.
In our previous work, we have shown that TCF21, a highly replicated gene associated with CAD, interacts closely with AHR.9 We also found that TCF21 plays a central role in regulating SMC phenotype and in modulating SMC phenotypic modulation during atherosclerosis.10–12 However, the role of the AHR pathway in regulating SMC phenotypic modulation in atherosclerotic disease is not well understood.
In this study, we sought to further investigate the specific effects of AHR on the vascular smooth muscle cell phenotype in atherosclerotic disease. We show that AHR regulates the SMC phenotype by globally affecting the chromatin accessibility landscape of SMC and thus impacting expression of genes regulating development, cell migration, proliferation, as well as extracellular matrix organization and endochondral ossification. We further characterize the effect of AHR in a SMC-specific AHR knockout mouse model of atherosclerosis using single-cell sequencing and histological analysis, and conclude that AHR is a critical regulator of SMC contribution to atherosclerotic lesions.
Methods
All methods and materials used in this study are described in detail in the Materials and Methods section of the online-only Data Supplement. Below please see a brief summary of the most relevant details. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Culture of human coronary artery smooth muscle cell (HCASMC) -
Primary human coronary artery smooth muscle cells (HCASMC) were purchased from Cell Applications, Inc (San Diego, CA) and experiments were performed with HCASMC between passages 5–8.
Knockdown and overexpression -
The siRNA for AHR and TCF21 were purchased from Origene (SR300136, SR321985), and siRNA for AC003075.4 was custom designed (Ambion/Life Technologies Corp, Table I in the Supplement). For the overexpression study, HCASMC were transduced with 2nd generation lentivirus with AHR cDNA (HG10456-CY, Sino Biological) and AC003075.4 cDNA (custom gBlock fragment, IDT) cloned into pWPI (Addgene #12254).
qRT-PCR –
Gene expression levels were measured using Taqman probes (Invitrogen) (AHR, TNFRSF11B, HAPLN1) and custom probes (see Table I in the Supplement) for SYBR Green assay.
RNA-sequencing –
Four replicates were used for each sample. The RNA were processed and analyzed as described previously. The script for RNA-Seq analysis can be found in github (https://github.com/milospjanic/rnaSeqFPro). Additionally, web-based tools iDEP.90 (http://bioinformatics.sdstate.edu/idep/) was used for analysis and visualization using the counts data generated from FeatureCounts.13
ChIP-Seq and ATAC-Seq –
ChIP-seq was performed using AHR antibody (Santa Cruz, sc-5579) and TCF21 antibody (HPA013189, Sigma) according to previous published protocols.14 For ATAC-Seq, we followed the published Omni-ATAC-seq protocol.15 The script for analysis can be found in github (https://github.com/zhaoshuoxp/Pipelines-Wrappers/).
Mouse Models -
The animal study protocol was approved by the Administrative Panel on Laboratory Animal Care (APLAC) at Stanford University. The DRE-LacZ strain was re-derived from cryopreserved embryos at the Jackson Laboratories (B6.Cg-Tg(DRE-lacZ)2Gswz/J strain, JAX# 006229)16 then bred to ApoE−/− background. Final genotypes of SMC lineage-tracing (SMCLin) mice were: TgMyh11-CreERT2, ROSAtdT/tdT, ApoE−/− as previously described.12 For the SMC lineage-tracing, Ahr knockout (SMCLin-KO), Ahrflox/flox mice were obtained from Jackson Labs (JAX#006203) and bred with the SMCLin mice. As the Cre-expressing BAC was integrated into the Y chromosome, all lineage tracing mice in the study were male. Tamoxifen was administered by oral gavage at 8 weeks of age to induce the lineage marker and Ahr knockout, followed by high fat diet (HFD) initiation (Dyets #101511). The Cre-mediated excision was confirmed in the DNA obtained from vascular tissues (Figure I in the Supplement, Table I in the Supplement for primers).
Single-cell RNA-Seq -
For both SMCLin and SMCLin-KO genotype, three mice were used at 16 weeks of high fat diet. Mouse aortic root dissociation and processing of cells was described previously.12 Briefly, dissociated cells were sorted for live/dead signal and tdTomato+ signal. tdTomato+ cells were captured and library was prepared using the 10X Genomics platform. Fastq files from each experimental group were aligned to the reference genome using CellRanger software (10X Genomics), then analyzed using the R package Seurat.17 The gene expression values underwent normalization using the sctransform function followed by principal component analysis and UMAP (Uniform Manifold and Approximation and Projection) clustering.18 Raw data from the single-cell RNA-Seq experiments will be deposited in the GEO database. Primary accession codes are pending.
Histology –
The mouse aortic root was fixed in 0.4% PFA and embedded in OCT, and slides were prepared and processed as described previously.12 Immunohistochemistry was performed with anti-SM22α antibody (Abcam #ab14106), and CD68 antibody (Abcam #ab125212). For the alkaline phosphatase enzymatic assay, Ferangi Blue Chromogen kit (SKU#FB813H, Biocare Medical) was used as instructed. Areas of interest were quantified using ImageJ (NIH) software. The lesion cap was defined as 30μm segment from the luminal surface as previously described.12, 19 Researchers were blinded to the genotype of the animals until completion of the analysis.
HCASMC Phenotypic Assays -
The Radius Cell Migration Assay kit (Cell Biolabs) was used for the gap closure assay. For the proliferation assay, EdU was introduced into the cell culture 3 hours before assay for uptake. The protocol for Click-iT Plus EdU proliferation kit (Thermo Fisher) was followed as instructed. For the apoptosis assay, HCASMC was treated with Doxorubicin (1uM) for 24 hours to induce apoptosis. The RealTime-Glo Annexin V Apoptosis kit (Promega) was used to quantify the degree of apoptosis in HCASMC. To assay for calcification, HCASMC was exposed to calcification media with 10mM beta-glycerophophate and 100ug/ml ascorbic acid as described previously.20 The HCASMC were exposed to calcification media with 1% FBS for 10–14 days, then treated with 0.6N hydrochloric acid for 24 hours. The supernatant was collected for calcium assay using Calcium Colorimetric Assay kit (Sigma-Aldrich), and the cell layer was collected for protein quantification.
Statistics -
R or GraphPad Prism 7.0 was used for statistical analysis. For motif and gene enrichment analyses, binomial test was used in HOMER and GREAT.21, 22 For overlapping of genomic regions or gene sets, we used Fisher’s exact test to test for enrichment. For comparisons between two groups of equal variance, an unpaired two-tailed Student’s t-test was performed. For multiple comparison testing, one-way analysis of variance (ANOVA) accompanied by Tukey’s post hoc test were used as appropriate. All error bars represent standard error of the mean (SE). Number of stars for the P values in the graphs: ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05.
Results
AHR modulation in HCASMC results in transcriptional changes relevant to vascular function and disease
In order to examine the transcriptional pathways regulated by AHR, we treated human coronary artery smooth muscle cells (HCASMC) with AHR siRNA or control siRNA (Figure Ib in the Supplement), and performed bulk RNA sequencing (RNA-Seq). Overall, at an adjusted p-value less than 0.00001 and fold change greater than 1.3, there were 1004 up-regulated genes, and 814 down-regulated genes (Figure 1a, Table II in the Supplement). The differentially regulated genes were enriched in curated biological processes including vascular development, response to organic substance, and regulation of cell migration (Figure 1b, Table II in the Supplement). To further validate these findings, we also transduced AHR overexpressing lentivirus into HCASMC and performed RNA-seq. We again found that vascular development, and response to chemical stimulus were among the top enriched pathways, in addition to cell cycle/cell division (Figure IIa,b in the Supplement, Table II in the Supplement).
Figure 1. AHR modulates pathways relevant to vascular disease and induces chromatin accessibility in HCASMC.
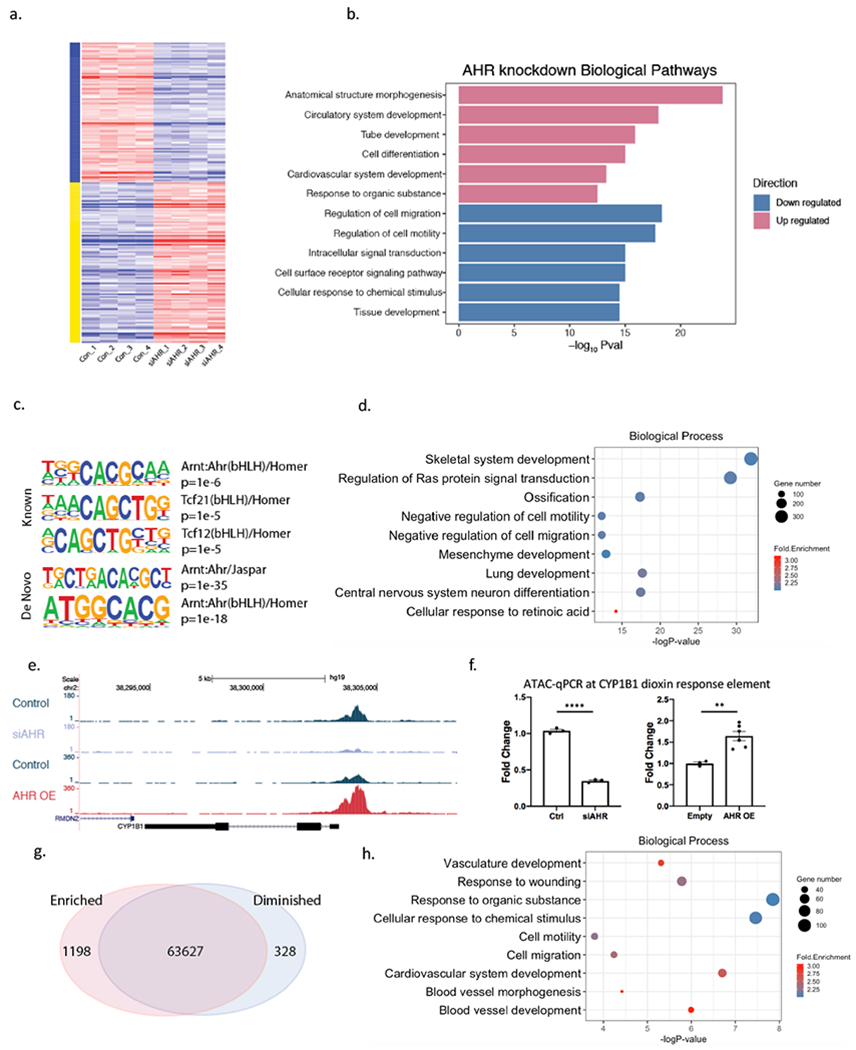
(a) Heatmap of differentially expressed genes in AHR knockdown RNA-Seq data in HCASMC show a significant impact of AHR modulation on the transcriptome. (b) Top enriched biological pathways from differentially expressed genes of AHR knockdown. (c) AHR ChIP-Seq peaks localize to AHR and TCF21 motifs with HOMER known and de novo analyses. (d) DAVID Gene Ontology analysis of AHR target genes identified by GREAT. (e) An ATAC-seq peak is visualized at the promoter region of CYP1B1, where the peak is diminished with knockdown and increased with AHR overexpression and (f) was confirmed with ATAC-qPCR (AHR knockdown by siRNA p<0.0001 (left), AHR overexpression p=0.0048 (right)). ****P < 0.0001, **P < 0.01. (g) AHR overexpression leads to an overall increase in open-chromatin regions. (h) DAVID Gene Ontology analysis of genes in the regions of increased chromatin accessibility signatures from AHR overexpression.
To determine which of the differentially expressed genes are directly regulated by AHR, we investigated the global pattern of AHR protein binding to chromatin by performing ChIP-Seq in HCASMC. We identified a total of ~17000 peaks with the AHR ChIP-Seq study in this cell type. AHR and TCF21 motifs were both significantly enriched within the peaks (Figure 1c, Table III in the Supplement). Genes identified by GREAT22 at AHR target loci were found by ontology analysis to be associated with terms related to development, ossification, response to retinoic acid, and cell migration (Figure 1d).
In addition to direct transcriptional regulation, we investigated whether AHR can affect gene expression in target loci through altering chromatin states. To examine this possibility, we performed the assay for transposase accessible chromatin (ATAC) followed by sequencing in HCASMC (ATAC-Seq). We found that AHR overexpression increased the chromatin accessibility in the promoter region of CYP1B1, and decreased accessibility when AHR was knocked down, as confirmed with ATAC-qPCR (Figure 1e, 1f). We then compared each treatment group to look for differentially enriched peaks. We found that AHR modulation had a significant impact on the global landscape of chromatin accessibility. When AHR was overexpressed, there were 1198 up-regulated peaks, and 328 down-regulated peaks at FDR<0.05 (Figure 1g). Globally, the open chromatin regions that enriched with AHR overexpression were clustered near genes related to response to organic substance, cardiovascular development and cellular migration and motility, mirroring those of the AHR ChIP-Seq enrichment results (Figure 1h, Table III in the Supplement).
The AHR downstream pathway overlaps with the TCF21 pathway and is enriched in regions of open chromatin in HCASMC
TCF21 is a causal CAD associated gene that is activated in the setting of vascular stress to promote modulation of SMC to develop a fibroblast-like phenotype, i.e., as a fibromyocyte (FMC).12 To extend our previous studies that indicated a regulatory interaction between AHR and TCF21,9 we compared their downstream pathways as determined by combined RNA-seq and ChIP-Seq analyses. There was significant overlap in both up- and down-regulated genes with AHR and TCF21 knockdown (Figure 2a). The pathways that were co-regulated in these experiments were enriched for regulation of cell migration, development, ECM organization, and cell adhesion by GO analysis with DAVID (Figure 2b). There was also enrichment for cytokine-cytokine receptor interaction, TGF-β pathway and ECM-receptor interaction KEGG pathways (Figure 2c, Table IV in the Supplement). We also considered the genes that were specifically regulated by AHR but not by TCF21. These genes were enriched for GO terms similar to the those identified for the overlap gene list including migration, but also specifically identified ossification/skeletal system development, collagen fibril organization, and SMC proliferation (Figure 2d, Table IV in the Supplement).
Figure 2. The AHR pathway overlaps TCF21 downstream pathways and is enriched in open chromatin regions in HCASMC.
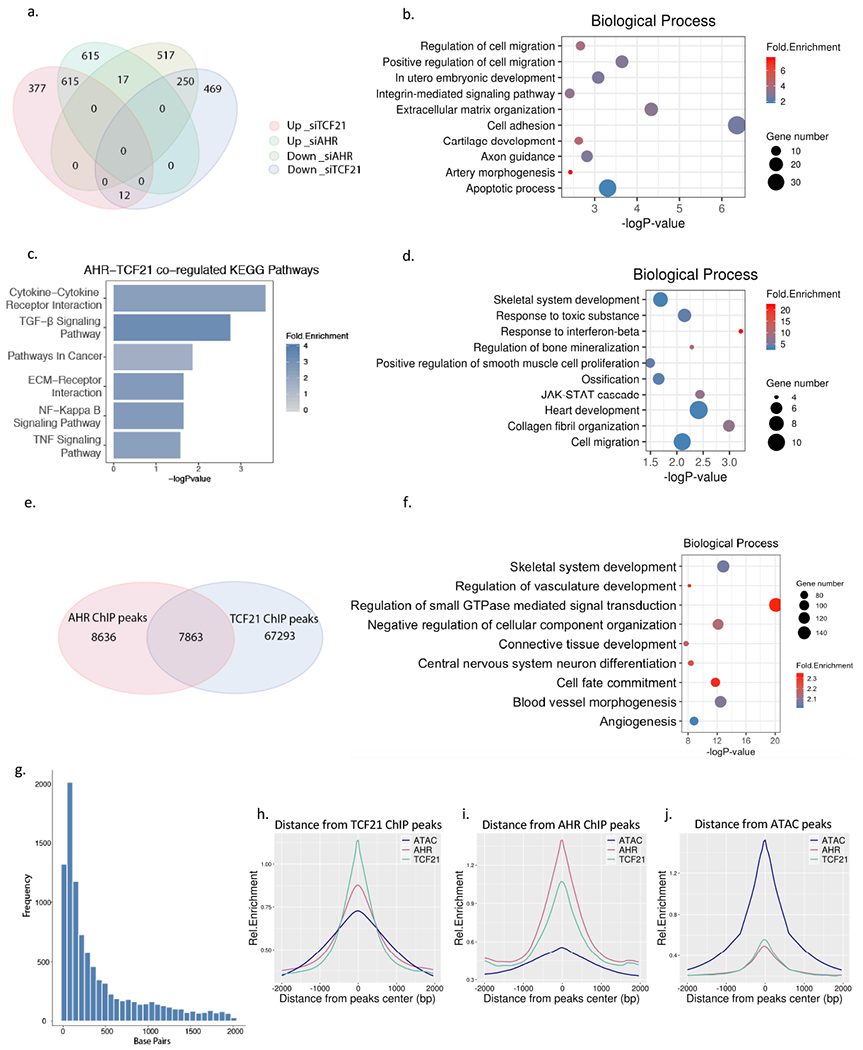
(a) Comparison of AHR knockdown RNA-Seq and TCF21 knockdown RNA-Seq results from HCASMC. A total of 1004 genes were up-reguated and 814 genes down-regulated in AHR KD, 1247 genes up-regulated, and 731 genes down-regulated in TCF21 KD (FDR <0.00001, Fold Change >1.3). Among these 615 up-regulated and 250 down-regulated genes overlapped between the AHR KD and TCF21 KD groups, respectively (Hypergeometric test p =1e-4642) (b) Top enriched biological pathways from up-regulated genes that are common to AHR KD and TCF21 KD (c) The top KEGG pathways enriched from genes regulated by both TCF21 and AHR. (d) Top enriched biological pathways unique to AHR KD. (e) AHR ChIP-Seq and TCF21 ChIP-Seq target loci share a significant number of intersected peaks. (f) DAVID Gene Ontology analysis of overlapping AHR and TCF21 binding peak genes identified by GREAT. (g) The distance between the AHR and TCF21 binding sites are on average 50-100 bp apart. (h-j) The peaks of TCF21 ChIP-Seq (green line), AHR ChIP-Seq (red line) and AHR overexpression ATAC-seq peaks (blue line) co-localize genome-wide, as shown by overlapping relative enrichment centered around each respective coordinates.
We then compared the AHR and TCF21 ChIP-Seq peaks to see which of these gene expression similarities were driven directly by joint binding at target loci. There was a significant overlap in the common peaks as identified by the two ChIP-seq studies (Figure 2e, p = 1e-4642, Fisher’s test), with 48% of the AHR peaks intersecting with TCF21 target loci. The common peaks were enriched for skeletal system development, cell motility and migration, and development (Figure 2f, Table IV in the Supplement). The distance between the summits of the peaks were compared, and the most common overlap was seen within 50–100 bp, but extended up to 1000 bp, suggesting that these two factors may interact through regulation of the local epigenome that mediates binding of AHR and TCF21 at these sites (Figure 2g). We further analyzed the chromatin accessibility at these ChIP-Seq peaks and found that AHR binding and TCF21 binding sites globally co-localized with open chromatin regions in HCASMC, suggesting that AHR and TCF21 affect the binding and function of each other (Figure 2h–j). Although co-immunoprecipitation studies failed to identify a direct interaction of these two transcription factors in solution (Figure IIIa in the Supplement), we found evidence of interaction when these proteins were cross-linked to chromatin by performing ChIP followed by western blotting, further supporting the hypothesis that the interaction of AHR and TCF21 is at least partially based on their joint chromatin scaffolding (Figure IIIb in the Supplement).
Single-cell RNA sequencing and in situ studies of mouse atherosclerotic lesions identify AHR pathway expression in FMC in the fibrous cap
Since AHR shares downstream molecular pathways with TCF21, we investigated whether AHR was also a regulator of phenotypic modulation in vivo. For these experiments, we performed single-cell RNA-seq using the SMC lineage-tracing mouse model SMCLin as described previously12. Briefly, we digested aortic root tissues and isolated tdTomato + cells by FACS and performed single cell RNA sequencing (scRNA-Seq). We found 7 clusters within the tdTomato+ cells, and identified the markers that characterized each cluster (Figure 3a, Figure 5 in the Supplement). Genes that distinguished clusters 0, 3, 4, and 5 included mature SMC markers such as Cnn1, indicating that they represent closely related mature SMC populations (Figure 3b). On the other hand, downregulation of mature SMC markers and upregulation of genes such as Lum and other markers established through our previous work identified clusters 1 and 2 as representing the fibroblast-like FMC (Figure 3c).12 Cluster 6 expressed markers for pericytes, and the small number of cells in cluster 7 expressed markers for macrophages, which were likely contaminants. Ahr expression was found predominantly in FMC, and also found to co-localize with tdTomato+ cells in the intima of the lesion and lesion cap using RNAscope and immunofluorescence (Figures IVa–c in the Supplement).
Figure 3. Single-cell and in situ analysis of atherosclerotic mouse aorta identifies AHR as a critical upstream regulator of phenotypic modulation and atherosclerosis.
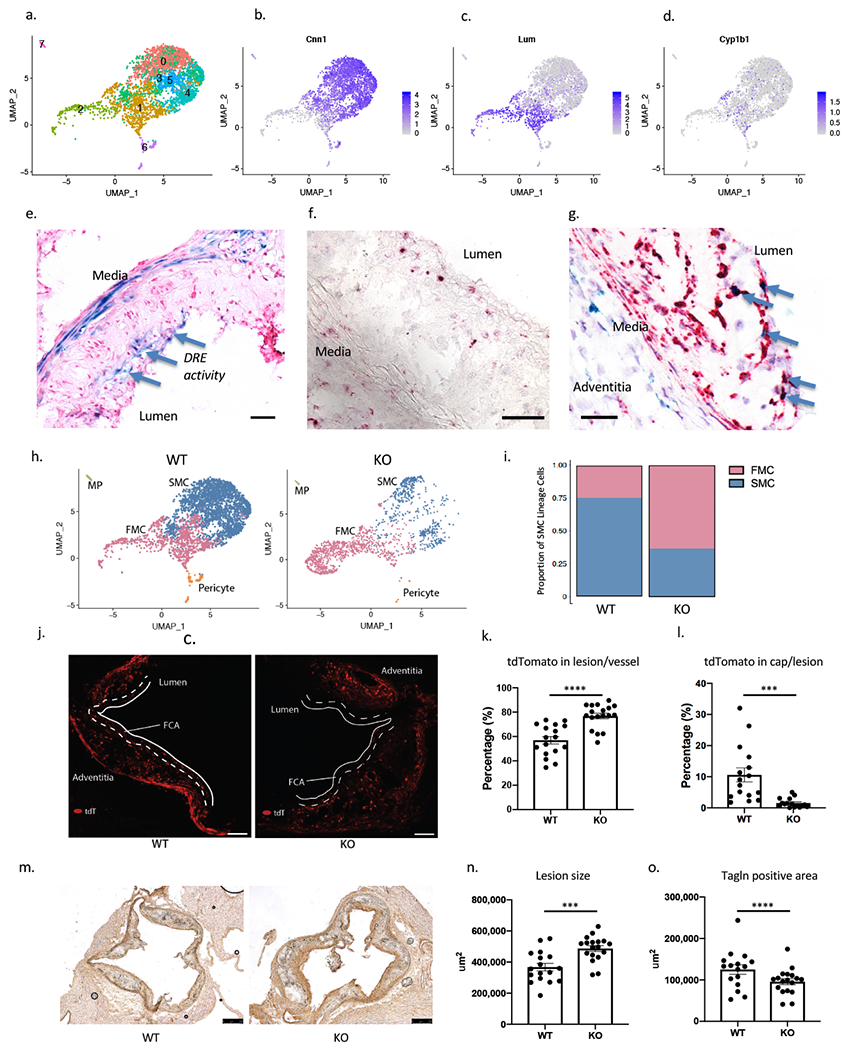
(a) UMAP plot of tdTomato+ SMC-lineage cells from SMCLin mice (n=3327 cells). (b, c) Featureplots of SMC marker Cnn1 and fibroblast marker Lum show delineation of SMC and FMC. (d) Cyp1b1 is expressed both in FMC and SMC, but expressed at higher levels in FMC (p=1.3e-9, Figure IV in the Supplement). (e) A dioxin reporter element (DRE) lacZ reporter transgene in ApoE null atherosclerosis model shows X-gal staining in media and the lesion cap (blue arrows) at 16 weeks of HFD (bar = 50μm). (f) RNAscope of vascular lesion of SMCLin mouse showed expression of Cyp1b1(red) at the fibrous cap and in the adventitia (bar = 50μm). (g) RNAscope of lesions for Cyp1b1 (green) and tdTomato (red) showed colocalization in the lesion cap (blue arrows). Cyp1b1 staining is also seen in the media and adventitia (bar = 50μm). (h) UMAP clusters of tdTomato+ cells from WT mice (left, n=3177), assigned as SMC and FMC based on their expression of SMC and fibroblast markers. KO tdTomato+ cells (right, n=1534) show shifting of transcriptional identity towards FMC compared to WT mice. (i) There is greater proportion of FMC in KO compared to the WT (WT 25% vs. KO 63%, Chi-square p=2.7e-145). (j) The tdTomato+ signal in lesion cap of WT (left) vs. KO (right) lesion after 16 weeks of high fat diet (bar = 50μm). (k) There is an increase in the tdTomato+ area in the lesion of KO animals compared to the WT mice (p<0.0001) (l) There is a significant decrease in the tdTomato+ area within the lesion cap, defined as 30um area from the luminal surface (p=0.0004). (m) Immunohistology was performed on WT (left) and KO mice (right) for Tagln (bar = 250μm). (n, o) The lesion size was increased (p=0.0005), but the Tagln+ area was reduced in the KO (p<0.0001). ****P < 0.0001, ***P < 0.001.
To assess the activity of the Ahr pathway in SMC lineage cells, we surveyed expression of the prototypical downstream gene Cyp1b1 in the single-cell analysis. We found that while Cyp1b1 was expressed in both SMC and FMC clusters, the average expression was greater in FMC (Figure 3d, Figure IVd in the Supplement). To identify the areas of the atherosclerotic lesion with AHR pathway activity, we then introduced a previously described transgene carrying the dioxin response element (DRE) lacZ reporter onto the ApoE−/− model of atherosclerosis.16 This transgene carries tandem repeats of the AHR binding motifs with minimal promoter driving lacZ expression. After 16 weeks of high fat diet, X-gal staining of the aortic sinus showed the presence of Ahr pathway activity in the media as well as the lesion cap (Figure 3e). We further assessed activity of the Ahr pathway by identifying the location of Cyp1b1 gene expression using RNAscope in-situ hybridization. We again found that Cyp1b1 gene expression was present in the media and the lesion cap, and that this signal in the lesion cap co-localized with tdTomato expression, confirming that the Ahr pathway activity in the lesion is present in the FMC population (Figures 3f, 3g).
SMC-specific Ahr KO increases the proportion of modulated SMC, producing larger disease lesions with fibrous caps deficient in SMC lineage cells
In order to understand how Ahr affects the process of phenotypic modulation in vivo in the disease setting, we introduced Ahr−flox/flox alleles onto the SMCLin line, producing mice with the genotype Myh11CreERT2, Ahr ΔSMC/ΔSMC, RosatdT/+, ApoE−/− (SMCLin-KO). We then compared the wild-type mice SMCLin (WT) and the SMCLin-KO (KO) mice on 16 weeks of HFD to assess the effect of AHR deletion on lesion characteristics. We performed scRNA-Seq on the tdTomato+ cells of WT and KO mice, and identified clustered cells as FMC, SMC, or pericytes (Figure 3h, WT left, KO right). There was a significant reduction of Ahr expression in the KO mice (Figure IVe in the Supplement). Overall, we found that the KO mice had a significantly larger proportion of FMC compared to the WT group (WT 25% vs. KO 63%, 4711 total cells from 3 mice/group, Chi-square p=2.7e-145, Figure 3i). In order to confirm these findings in a larger cohort of mice, we compared the proportion of tdTomato+ cell populations in histology sections in 15 WT and KO mice per group (Figure 3j). There was no difference in the overall tdTomato+ area in the vessel, however, the proportion of tdTomato+ area in the intimal lesion was higher in the KO group compared to the WT group (WT 57.0±3.1% vs. KO 76.8±2.3%, p<0.0001, Figure 3k) Additionally, the tdTomato+ FMC were significantly absent from the fibrous cap area (area extending 30μm from the lumen), suggesting either failure of cells to reach the cap or their exit from the cap and ingress back into the plaque (WT 10.6±2.2% vs. KO 1.5±0.4% by area, p=0.0004, Figure 3l). Consistent with these findings, we also found the number of DAPI+/tdTomato+ cells increased in the intima, and decreased in the lesion cap of the KO group (Figure Va–c in the Supplement).
We performed Tagln immunohistology of the aortic root to characterize the atherosclerotic lesions in the WT and KO mice (Figure 3m). We found the lesion size to be increased in the KO cohort compared to the wild type (367580±24295 vs. 486949±19086 μm2, p=0.0005, Figure 3n). The Tagln staining revealed that the overall SMC area in the lesion was decreased in the KO mice compared to the WT (WT 125017±11600 vs. KO 95748±7265 μm2, p<0.0001, Figure 3o), consistent with findings with the scRNA-Seq data. We also found the proportion of Tagln+ cells among tdTomato+ cells decreased significantly in the KO (Figure Vd in the Supplement). tdTomato+ media also decreased significantly in the KO compared to WT suggesting increased migration of the lineage traced cells (Figure Ve in the Supplement). We did not observe any significant difference in the prevalence of Cd68+ macrophages in the lesion (Figure Vf in the Supplement).
SMC-specific Ahr KO promotes the conversion of lineage traced SMC to a chondrogenic phenotype
In order to further characterize the phenotypically modulated SMC in the Ahr KO mice, we further divided the modulated population to FMC1 and FMC2 cells (Figure 4a), where FMC2 represents the population that was significantly increased in the KO mice compared to the WT (47.2% vs. 5.4%, chi-square p = 4.9e-262, Figure 4b). The phenotype of these cells was characterized by expression of genes such as Col2a1, Spp1, Alpl and Enpp1 (Table V in the Supplement), i.e. enriched for biological pathways related to ossification, collagen fibril organization and skeletal system development (Figure 4c). We then determined the upstream regulators driving transcriptional changes (top 100 upregulated genes) between FMC2 and FMC1 using IPA (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis).23 When these upstream regulators were queried using STRING-db,24 we found again that ossification and cartilage development, as well as TGF-β pathways were key biological processes responsible for promoting their phenotype and possibly regulating this transition from FMC1 to FMC2 (Figure 4d, 4e). As expected, Ahr was reported as the top down-regulated transcription factor, while the top up-regulated factors formed a closely linked network involved in bone formation, including Bmp2, Sox9, Tgfb1, Wnt3a, and Pth (Figure 4f). Transcription factors specific for chrondrocytes such as Sox9 and Runx2 were upregulated in the FMC2 population (Figure VIa in the Supplement). A differential expression analysis between the FMC2 cells from WT vs. KO also found enrichment of KO group for extracellular matrix organization, and chondrocyte differentiation (Figure VIb in the Supplement, Table V in the Supplement). Based on the strong chondrocyte-like transcriptional phenotype of the FMC2 population, and by comparison to the term fibromyocyte, we termed these cells “chondromyocytes (CMC)” to reflect their SMC origin and their chondrogenic phenotype.
Figure 4. SMC-specific Ahr KO produces modulated cells with a chondrogenic phenotype.
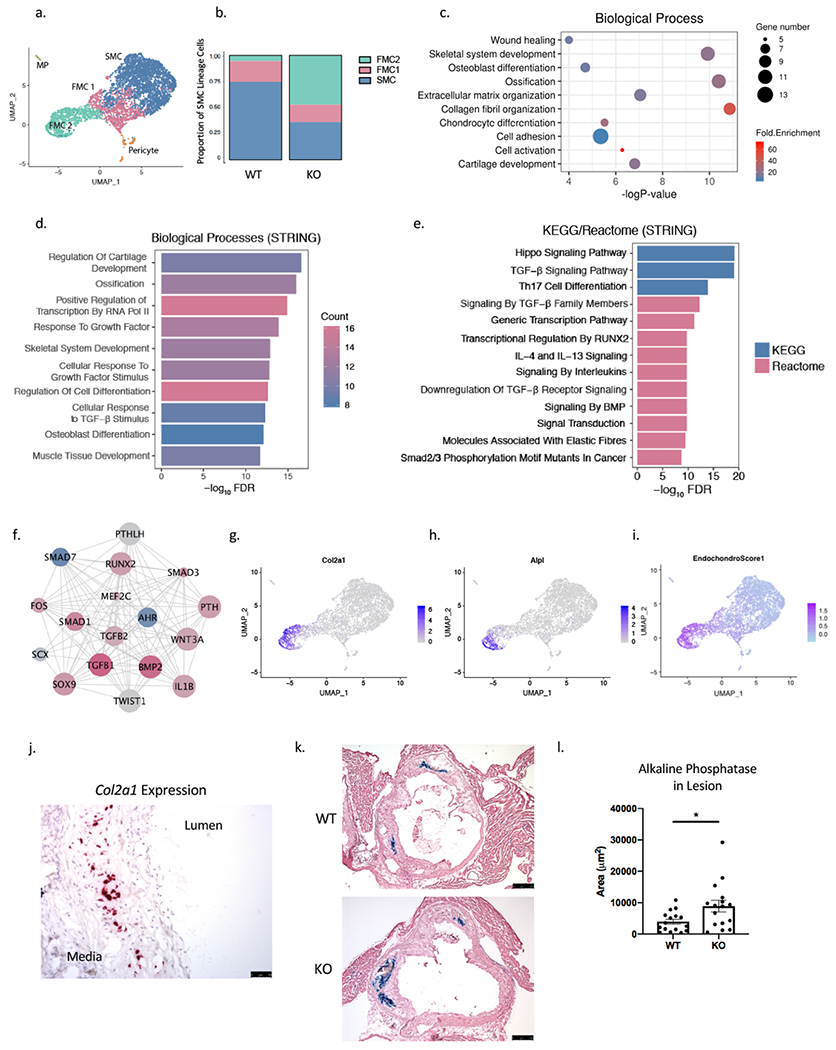
(a) On the combined UMAP of tdTomato+ lineage traced cells from WT and KO mice, the FMC population was further clustered to FMC1 and FMC2 population based on graph-based clustering with PCA. (b) The proportion of FMC2 cells was significantly greater for the KO compared to the WT group (Chi-square p = 4.9e-262). (c) DAVID Gene Ontology analysis of the top 100 differentially regulated genes in FMC2 compared to FMC1 population. (d) Enriched biological pathways and (e) KEGG/Reactome from the top upstream regulators of FMC1 to FMC2 transcriptional change. (f) A network of top upstream regulators of FMC2 vs. FMC1 transcriptional phenotype show AHR as top inhibited upstream factor (blue = inhibited, red = activated, size of circle = Z-score; nodes from Ingenuity Pathway Analysis, edges from STRING-db). (g) The FMC2 population was enriched for expression of chondrocyte/osteoblast markers Col2a1 (h) and Alpl, and (i) an endochondral score built from the average expression of 13 genes show a strong signal in the FMC2 population. (j) RNAscope of Col2a1 expression, which is localized to the neointimal layer of the atherosclerotic lesion in mouse aortic sinus. (k) Alkaline phosphatase activity is detected in the neointima of atherosclerotic lesion at 16 weeks of HFD, in both WT and KO (bar = 250μm). (l) The average lesion area with alkaline phosphatase activity is larger in KO group compared to the WT group (3936±794 vs. 8893±1850 μm2, p=0.02. *P < 0.05.
Localization and quantification of disease associated chondromyocytes
To characterize the location of CMC in transcriptomic space in relation to FMC and SMC, we visualized expression of Col2a1, and Alpl and noted that these genes identified CMC as being maximally separated from SMC, and closer to FMC, suggesting that they derive from FMC as part of a linear trajectory (Figures 4g, 4h). We then constructed an “Endochondral Score” based on the average expression of 13 representative genes from the gene ontology “Endochondral Ossification” (Extended Methods in Supplemental Material), and visualized this on a UMAP showing that the CMC population had a clear signal for endochondral ossification (Figure 4i). To define the anatomical location of CMC, we performed RNAscope as well as histochemical analysis of the atherosclerotic lesions. We found the representative CMC gene Col2a1 to be expressed in intimal cells of the lesion but not in the lesion cap, unlike the localization that we had observed for Cyp1b1 (Figure 4j). Alcian blue stain also showed presence of the proteoglycan components of the extracellular matrix associated with chondrocytes in the intima (Figure VIc in the Supplement). Furthermore, alkaline phosphatase (AP) activity was also localized using a chromogenic assay in the atherosclerotic lesions of mice (Figure 4k), and AP activity was found exclusively in the intima of the lesions. Consistent with our finding from the scRNA-Seq data showing greater CMC number in the KO mice, we found the relative AP-stained area to be larger in the KO cohort compared to the WT (Figure 4l).
AHR regulates HCASMC phenotype
We used cultured HCASMC to examine the effect of AHR on cell state phenotypes as predicted by the RNA-Seq and ChIP-Seq experiments. First, using a gap closure assay, we measured the effect of AHR knockdown on cell migration, and found an increase in the migration and cell coverage with silencing of AHR (p=0.048, Figure 5a). We also looked for any change in the proliferative capacity of the HCASMC with AHR modulation using an EdU proliferation assay. We found that AHR knockdown resulted in increased uptake of EdU, and the opposite when AHR was overexpressed (p<0.0001 for both, Figure 5b). We also assayed for apoptotic activity in HCASMC subjected to apoptotic stress induced by doxorubicin. AHR knockdown resulted in reduced apoptotic activity based on the Annexin V assay (p=0.016, two-way ANOVA, Figure 5c). We also investigated the rate of calcification of HCASMC grown in calcification media. The total calcification increased in cells where AHR was silenced, and decreased when AHR was overexpressed (Figure 5d). Overall, we found that AHR had an anti-migratory, anti-proliferative, anti-calcifying, and pro-apoptotic effect on HCASMC in vitro. These findings in human cells were congruent with the scRNA-Seq data in mouse.
Figure 5. AHR regulates HCASMC phenotype in vitro.
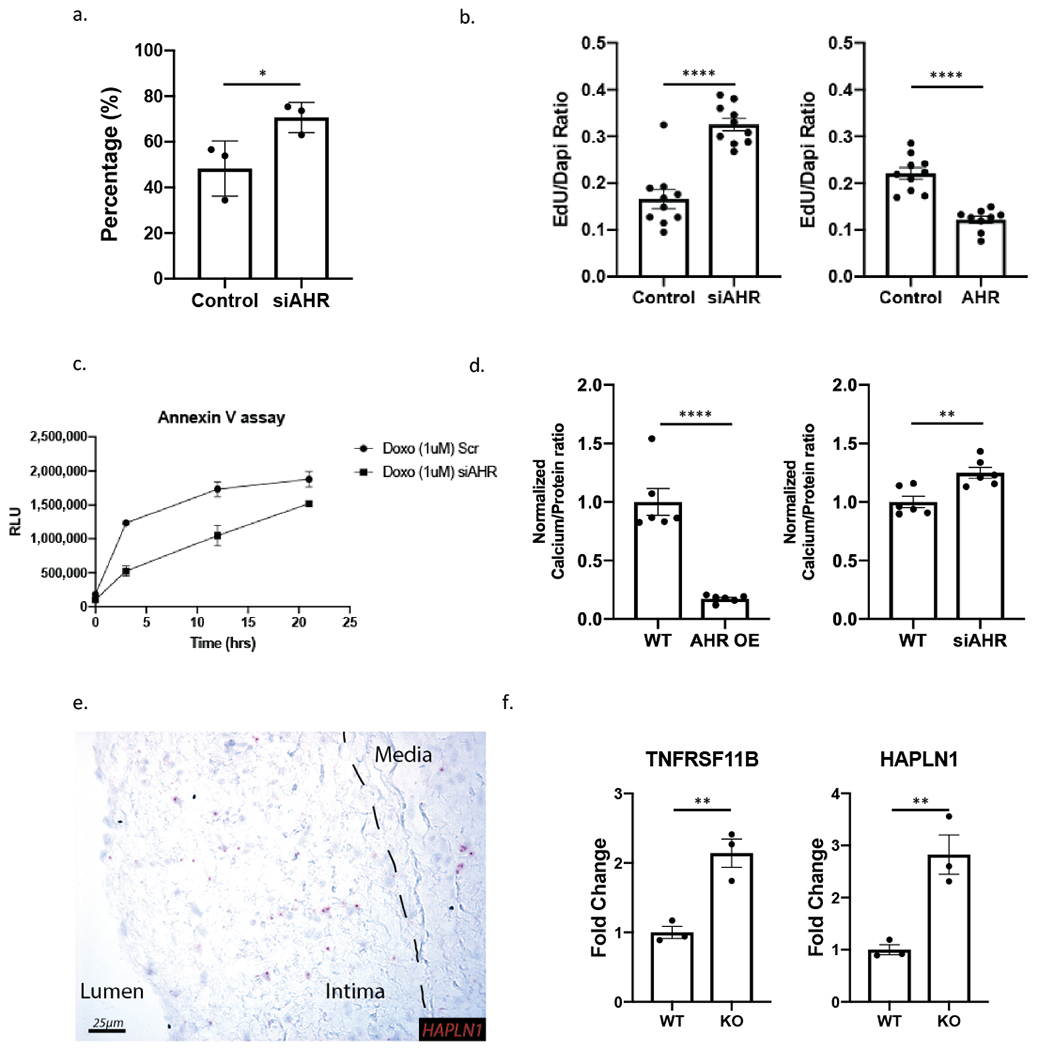
(a) Radial migration assay of HCASMC with scrambled control and AHR siRNA show increased migration with AHR knockdown (* p=0.049). (b) EdU uptake assay used to measure proliferation of HCASMC showed an increase with AHR knockdown and decrease with AHR overexpression (p<0.0001 for both). (c) Apoptosis quantification by Annexin V assay shows AHR knockdown to decrease apoptosis (p=0.016, 2-way ANOVA). (d) Calcification assay shows a decrease in calcification with AHR overexpression in HCASMC and an increase with AHR knockdown (p<0.0001 for AHR OE, p= 0.004 for AHR knockdown). (e) Chondrocyte marker HAPLN1 is also expressed in the diseased human coronary artery as determined by RNAscope, and concentrated in the intimal layer. (f) Chondrocyte markers identified in FMC2 are increased with AHR knockdown in HCASMC (p=0.0068 for TNFRSF11B, p=0.0094 for HAPLN1). ****P < 0.0001, **P < 0.01, *P < 0.05.
Chondromyocyte markers are expressed in human coronary artery lesion cells
We next investigated whether genes upregulated in CMC of Ahr KO mice were also expressed in human coronary artery cells of similar phenotype using our previously published scRNA-Seq dataset.12 We used CNN1 expression to identify mature contractile SMC, and LUM expression to identify fibroblasts, FMC and CMC (Figure VIIa–c in the Supplement). We found AHR expression present in the FMC/CMC and fibroblast populations (Figure VIId in the Supplement). Human cells expressing genes specific for the CMC population, TNFRSF11B and HAPLN1, were positioned in cluster 4 between the SMC and fibroblast clusters (Figure VIIe–h in the Supplement). We then applied the endochondral score on the human data, and showed that this cluster of cells had higher scores than others within the SMC-Fibroblast continuum (Figure VIIi in the Supplement). We probed for HAPLN1 gene expression in the human coronary artery tissues with RNAscope, and identified staining primarily in the intima, with little staining in either the lesion cap or media (Figure 5e). Further, there was a significant increase in the expression of both markers when AHR was knocked down with siRNA in HCASMC (Figure 5f). Additionally, ALPL was also negatively regulated by AHR overexpression (Figure VIIj in the Supplement). Taken together, these data suggest that CMC are present in the human plaque, with CMC similarly localized in the neointima of the coronary artery plaque.
Antisense long non-coding RNA AC003075.4 is associated by GWAS with CAD at 7p.21 where it regulates AHR expression
In the genomic locus at 7p.21, an LD block represented by lead SNP rs6968554 was found to be associated with coronary artery disease with a look-up in the CARDIOGRAM+C4D GWAS (p=1e-4, Figure 6a). Furthermore, this variant is a cis-eQTL for AC003075.4, a lncRNA that is anti-sense to AHR, in the GTEX database in tibial artery (p=2.5e-4, Figure 6b). We found that knocking down AC003075.4 resulted in the down-regulation of AHR expression, while overexpression resulted in the up-regulation of AHR (Figure 6c,d). The overexpression of AC003075.4 also resulted in decreased calcification of HCASMC in-vitro (Figure VIIIa in the Supplement). When AHR was knocked down, there was up-regulation of AC003075.4, and when AHR was overexpressed, there was down-regulation of AC003075.4, suggesting a negative feedback mechanism (Figure VIIIb in the Supplement). Furthermore, when TCF21 was overexpressed, AC003075.4 expression increased, and expression decreased with TCF21 knockdown (Figure 6e). These finding suggest that AHR expression is regulated by TCF21 and its antisense lncRNA, which are both associated with CAD phenotype by common variants.
Figure 6. Antisense long non-coding AC003075.4 is correlated with CAD in GWAS and regulates AHR expression.
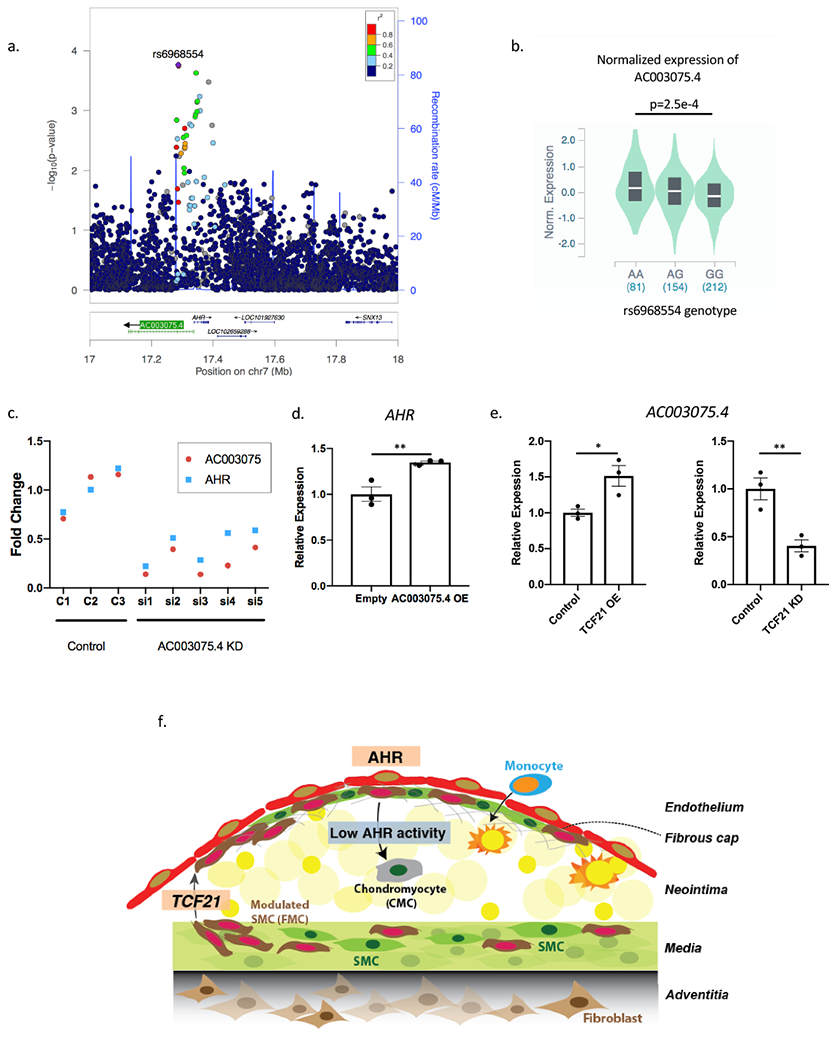
(a) rs6968554, a lead-SNP upstream of the AHR gene, is correlated to CAD on CARDIOGRAM+C4D (p=1e-4). (b) In GTEx vascular tissue (tibial artery), rs6968554 is an eQTL for AC003075.4 expression, a lncRNA that is anti-sense to AHR (ANOVA p=2.5e-4). (c-d) AC003075.4 and AHR expression are highly correlated (c) AC003075.4 knockdown decreases AHR expression (p = 0.0062), and (d) AC003075.4 overexpression increases AHR expression (p=0.012). (e) TCF21 positively regulates AC003075.4 expression in HCASMC (p=0.027 for overexpression, p=0.01 for knockdown). (f) We propose a working model of AHR function in the development of atherosclerosis. TCF21 is initially up-regulated and promotes SMC to dedifferentiate and participate in lesion and fibrous cap development. AHR is activated in the lesion cap and plays a role in maintaining the FMC in this region, with AHR loss promoting FMC to migrate into the neointima and assume a chondrocyte-like phenotype, i.e., transition to chondromyoctes (CMC). **P < 0.01, *P < 0.05.
Discussion
Environmental factors activate gene regulatory programs to contribute to the risk for coronary artery disease (CAD) through gene-environment interactions, and we have investigated the aryl hydrocarbon receptor (AHR) as a critical mediator of such effects. AHR is an inducible transcription factor that was first recognized for its ability to mediate the effect of a number of xenobiotic ligands such dioxin and PAH, which are established components of tobacco smoke, and its functional role in this regard considered harmful.4, 25 However, AHR is now recognized as an important developmental factor and physiological regulator in the vascular, immune and reproductive systems.6, 26, 27 AHR regulates cell cycle and cellular differentiation through interaction with numerous other signaling pathways, including those regulated by TGF-β and Notch.28, 29 Many of these actions are likely mediated by endogenous ligands, and for the most part considered beneficial. Thus, experiments described in this work have focused on the function of AHR in vascular smooth muscle cells in vascular disease, in the absence of known external environmental ligand activation.
AHR was identified as a downstream target of the CAD associated factor TCF21, which we have recently shown to promote phenotypic modulation as a mechanism of risk prevention.9, 12 In studies reported here, we gained further insight into the regulatory interaction between AHR and TCF21, and found a significant overlap between the transcriptional changes induced by AHR and TCF21 modulation that point to critical pathways involved in SMC modulation. Importantly, AHR showed a unique gene expression signature suggesting inhibition of ossification. And while AHR was found to regulate genes that modulate SMC migration and proliferation, the direction of effect as identified by in vitro assays was opposite to that of TCF21.10 These data thus suggested a distinct functional profile compared to TCF21.
We have previously used lineage tracing and scRNA-Seq to show that Tcf21 promotes the modulation of SMC to fibroblast-like cells that we termed fibromyocytes (FMC),12 and were thus interested to determine how Ahr might affect SMC phenotype in the disease setting. In contrast to the reduced FMC in lesion and lesion cap seen in the SMC-specific knockout of Tcf21, we found that there was an increase in SMC modulation with SMC-specific knockout of Ahr. Interestingly, loss of Ahr was found to promote further modulation of FMC to a chondrocyte-like cell phenotype. This is consistent with findings in model systems where AHR has been shown to suppress expression of pro-chondrocyte genes Sox9, and Runx2.30, 31 Also, by analogy to the de-differentiated FMC, previous reports suggest that AHR may maintain the “stemness” of progenitor cells. Watson et al. reported that AHR mediates inhibition of osteoblast differentiation in human mesenchymal stem cells32, and another recent report found that AHR pathway activation by an endogenous ligand can maintain the undifferentiated state of embryonic stem cells.33 Hence, the FMC may in fact be a form of multi-potent cell population that is derived from de-differentiated SMC population, ready to differentiate to a mesenchymal lineage cell-type, including the CMC and also back to a differentiated, quiescent SMC population. The chondrogenic cells described here were previously identified as the most de-differentiated portion of the FMC, but not characterized specifically as a discrete cell lineage.12 Given evidence from these studies showing that these cells are independently regulated with respect to cellular phenotype, we have elected to recognize them as a discrete group of modulated SMC, and use the term chondromyocyte to distinguish them from their mature SMC origin, and their related and presumably intermediate phenotypic FMC precursors.
Thus, findings reported here contribute to our understanding of the SMC response to stress in the setting of vascular disease, but suggest that the process of phenotypic modulation is more complex than previously appreciated.12, 19 Although it is not currently possible to integrate all available information, we propose the following model (Figure 6f). TCF21 expression is activated early in the disease process to promote SMC de-differentiation, proliferation and migration out of the media. Subsequently, the AHR pathway is upregulated in FMC and serves to maintain the FMC phenotype as these cells migrate to the fibrous cap. Loss of AHR results in FMC with greater migratory capacity reaching the cap and then moving into the plaque where they adopt a chrondrogenic phenotype, consistent with a recently proposed mechanism of cellular disease construction,34 or simply adopting a chondrogenic phenotype and expanding in the neointima where they contribute to lesion size and detract from cells contributing to the fibrous cap. Undoubtedly, additional genes such as KLF4 and OCT4 also control various aspects of the phenotypic modulation process,19, 35 and their roles will be better understood as scRNA-Seq data becomes available for knockout models of these and other relevant genes.
Given the upregulation of AHR by TCF21 and the overlap of regulated genes downstream, we expected that AHR would have a similar function to TCF21 in SMC. However, we found that its functional profile with in vitro assays was distinct to TCF21, and that its loss led to an increase in the number of modulated SMC. While this profile would be expected to decrease disease risk based on our previous studies of TCF21, the phenotype of the increased SMC-derived modulated cells in the knockout mice was quite different from the FMC we characterized previously, and more consistent with a chondrogenic type cell. Further, the causal relationship of the human data is complex, but suggests a protective role of AHR as the major disease allele “G” for lead SNP rs6968554 is associated with lower expression of regulatory lncRNA AC003075.4 and thus AHR. This direction is consistent with literature that higher level of coronary calcification is associated with increased incidence of myocardial infarction.36, 37 The exact mechanism of how osteochondrogenesis affects the stability of lesion remains inconclusive as the biomechanical alteration appears dependent on the type and location of calcification.38–40 The directionality of AHR may also be due to its role in maintaining modulated SMC in the lesion cap. Additional studies are required to better understand the complex relationship between the AHR gene, SMC phenotypic modulation associated with variance of this gene, and the directionality of disease risk.
Conclusion
In this study, we provide evidence that the AHR pathway, mediated by an environment-sensing transcription factor, is active in modulated SMC, and can inhibit the phenotypic modulation of SMC to chondrocyte-like cells during development of atherosclerosis.
Supplementary Material
Expanded Methods
Supplemental Table (Excel) Legends
Supplemental Figure Legends
Supplemental Figures I-VIII
Supplemental Table (Excel) Files I-V
Clinical Perspective.
What is new?
This study identifies a novel population of cells derived from smooth muscle cells (SMC), termed chondromyocytes, which have gene expression features of cartilage and bone formation within the atherosclerotic lesion.
The environment-sensing transcription factor aryl-hydrocarbon receptor (AHR) plays an important role in smooth muscle cell differentiation, ossification, and maintains the SMC-derived fibrous cap structure.
What are the clinical implications?
Human genetics suggest a protective role of AHR in the SMC during atherosclerosis, and therapies targeted to increase AHR activity in SMC may confer protection against adverse calcific remodeling of the atherosclerotic plaque.
Further characterization of the pathways that direct the modulation of SMC during atherosclerosis at the single-cell level can identify potential therapeutic targets to mitigate the risk of atherosclerosis.
Sources of Funding
This work was supported by National Institutes of Health grants K08HL133375 (J.B.K.), F32HL143847 (P.C.), R01HL109512 (T.Q.), R01HL134817 (T.Q.), R33HL120757 (T.Q.), R01DK107437 (T.Q.), R01HL139478 (T.Q.), American Heart Association 18CDA34110206 (R.W.), a grant from the Chan Zuckerberg Foundation – Human Cell Atlas Initiative (T.Q.), and a grant from the California Tobacco-Related Disease Research Program (T30IP0999) (J.B.K and T.Q.). The sequencing data were generated on an Illumina HiSeq 4000 that was purchased with funds from NIH under award number S10OD018220. Cell sorting was performed on an instrument in the Shared FACS Facility obtained using NIH S10 Shared Instrument Grant S10RR025518-01.
Non-standard Abbreviations and Acronyms
- AHR
Aryl-hydrocarbon Receptor
- HCASMC
Human Coronary Artery Smooth Muscle Cells
- CMC
Chondromyocytes
- FMC
Fibromyocytes
- DRE
Dioxin Response Element
Footnotes
Disclosures
Nothing to Disclose
References
- 1.Lusis AJ. Genetics of atherosclerosis. Trends Genet. 2012;28:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marenberg ME, Risch N, Berkman LF, Floderus B and de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med. 1994;330:1041–1046. [DOI] [PubMed] [Google Scholar]
- 3.Khera AV and Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet. 2017;18:331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beischlag TV, Luis Morales J, Hollingshead BD and Perdew GH. The aryl hydrocarbon receptor complex and the control of gene expression. Crit rev eukaryot gene expr. 2008;18:207–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Humblet O, Birnbaum L, Rimm E, Mittleman MA and Hauser R. Dioxins and cardiovascular disease mortality. Environ Health Perspect. 2008;116:1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ivnitski I, Elmaoued R and Walker MK. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) inhibition of coronary development is preceded by a decrease in myocyte proliferation and an increase in cardiac apoptosis. Teratology. 2001;64:201–212. [DOI] [PubMed] [Google Scholar]
- 7.Walisser JA, Bunger MK, Glover E and Bradfield CA. Gestational exposure of Ahr and Arnt hypomorphs to dioxin rescues vascular development. Proc Natl Acad Sci U S A. 2004;101:16677–16682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carreira VS, Fan Y, Wang Q, Zhang X, Kurita H, Ko CI, Naticchioni M, Jiang M, Koch S, Medvedovic M, et al. Ah Receptor Signaling Controls the Expression of Cardiac Development and Homeostasis Genes. Toxicol Sci. 2015;147:425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim JB, Pjanic M, Nguyen T, Miller CL, Iyer D, Liu B, Wang T, Sazonova O, Carcamo-Orive I, Matic LP, et al. TCF21 and the environmental sensor aryl-hydrocarbon receptor cooperate to activate a pro-inflammatory gene expression program in coronary artery smooth muscle cells. PLoS Genet. 2017;13:e1006750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nurnberg S, Cheng K, Raiesdana A, Kundu R, Miller C, Kim J, Arora K, Carcamo-Oribe I, Xiong Y, Tellakula N, et al. Coronary Artery Disease Associated Transcription Factor TCF21 Regulates Smooth Muscle Precursor Cells That Contribute to the Fibrous Cap. PLoS Genet. 2015;11:e1005155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagao M, Lyu Q, Zhao Q, Wirka RC, Bagga J, Nguyen T, Cheng P, Kim JB, Pjanic M, Miano JM, et al. Coronary Disease Associated Gene TCF21 Inhibits Smooth Muscle Cell Differentiation by Blocking the Myocardin-Serum Response Factor Pathway. Circ Res. 2020;126:517–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, Kundu R, Nagao M, Coller J, Koyano TK, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. 2019;25:1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ge SX, Son EW and Yao R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics. 2018;19:534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Q, Wirka R, Nguyen T, Nagao M, Cheng P, Miller CL, Kim JB, Pjanic M and Quertermous T. TCF21 and AP-1 interact through epigenetic modifications to regulate coronary artery disease gene expression. Genome Med. 2019;11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14:959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bemis JC, Nazarenko DA and Gasiewicz TA. Coplanar polychlorinated biphenyls activate the aryl hydrocarbon receptor in developing tissues of two TCDD-responsive lacZ mouse lines. Toxicol Sci. 2005;87:529–536. [DOI] [PubMed] [Google Scholar]
- 17.Butler A, Hoffman P, Smibert P, Papalexi E and Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature biotechnology. 2018;36:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hafemeister C and Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. bioRxiv. 2019. doi: /10.1101/576827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shankman L, Gomez D, Cherepanova O, Salmon M, Alencar G, Haskins R, Swiatlowska P, Newman A, Greene E, Straub A, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shioi A, Nishizawa Y, Jono S, Koyama H, Hosoi M and Morii H. Beta-glycerophosphate accelerates calcification in cultured bovine vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1995;15:2003–2009. [DOI] [PubMed] [Google Scholar]
- 21.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H and Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell. 2010;38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM and Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nature biotechnology. 2010;28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kramer A, Green J, Pollard J Jr., and Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larigot L, Juricek L, Dairou J and Coumoul X. AhR signaling pathways and regulatory functions. Biochim Open. 2018;7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sorg O AhR signalling and dioxin toxicity. Toxicol Lett. 2014;230:225–233. [DOI] [PubMed] [Google Scholar]
- 27.Gialitakis M, Tolaini M, Li Y, Pardo M, Yu L, Toribio A, Choudhary JS, Niakan K, Papayannopoulos V and Stockinger B. Activation of the Aryl Hydrocarbon Receptor Interferes with Early Embryonic Development. Stem Cell Reports. 2017;9:1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo J, Sartor M, Karyala S, Medvedovic M, Kann S, Puga A, Ryan P and Tomlinson CR. Expression of genes in the TGF-beta signaling pathway is significantly deregulated in smooth muscle cells from aorta of aryl hydrocarbon receptor knockout mice. Toxicol Appl Pharmacol. 2004;194:79–89. [DOI] [PubMed] [Google Scholar]
- 29.Huang B, Butler R, Miao Y, Dai Y, Wu W, Su W, Fujii-Kuriyama Y, Warner M and Gustafsson JA. Dysregulation of Notch and ERalpha signaling in AhR−/− male mice. Proc Natl Acad Sci U S A. 2016;113:11883–11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiong KM, Peterson RE and Heideman W. Aryl hydrocarbon receptor-mediated down-regulation of sox9b causes jaw malformation in zebrafish embryos. Molecular pharmacology. 2008;74:1544–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tong Y, Niu M, Du Y, Mei W, Cao W, Dou Y, Yu H, Du X, Yuan H and Zhao W. Aryl hydrocarbon receptor suppresses the osteogenesis of mesenchymal stem cells in collagen-induced arthritic mice through the inhibition of beta-catenin. Exp Cell Res. 2017;350:349–357. [DOI] [PubMed] [Google Scholar]
- 32.Watson ATD, Nordberg RC, Loboa EG and Kullman SW. Evidence for Aryl hydrocarbon Receptor-Mediated Inhibition of Osteoblast Differentiation in Human Mesenchymal Stem Cells. Toxicol Sci. 2019;167:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamamoto T, Hatabayashi K, Arita M, Yajima N, Takenaka C, Suzuki T, Takahashi M, Oshima Y, Hara K, Kagawa K, et al. Kynurenine signaling through the aryl hydrocarbon receptor maintains the undifferentiated state of human embryonic stem cells. Sci Signal. 2019;12: pii: eaaw3306. doi: 10.1126/scisignal.aaw3306. [DOI] [PubMed] [Google Scholar]
- 34.Misra A, Feng Z, Chandran RR, Kabir I, Rotllan N, Aryal B, Sheikh AQ, Ding L, Qin L, Fernandez-Hernando C, et al. Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat Commun. 2018;9:2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cherepanova OA, Gomez D, Shankman LS, Swiatlowska P, Williams J, Sarmento OF, Alencar GF, Hess DL, Bevard MH, Greene ES, et al. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat Med. 2016;22:657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raggi P, Callister TQ, Cooil B, He ZX, Lippolis NJ, Russo DJ, Zelinger A and Mahmarian JJ. Identification of patients at increased risk of first unheralded acute myocardial infarction by electron-beam computed tomography. Circulation. 2000;101:850–855. [DOI] [PubMed] [Google Scholar]
- 37.Wayhs R, Zelinger A and Raggi P. High coronary artery calcium scores pose an extremely elevated risk for hard events. J Am Coll Cardiol. 2002;39:225–230. [DOI] [PubMed] [Google Scholar]
- 38.Otsuka F, Sakakura K, Yahagi K, Joner M and Virmani R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol. 2014;34:724–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang H, Virmani R, Younis H, Burke AP, Kamm RD and Lee RT. The impact of calcification on the biomechanical stability of atherosclerotic plaques. Circulation. 2001;103:1051–1056. [DOI] [PubMed] [Google Scholar]
- 40.Kolodgie FD, Burke AP, Farb A, Weber DK, Kutys R, Wight TN and Virmani R. Differential accumulation of proteoglycans and hyaluronan in culprit lesions: insights into plaque erosion. Arterioscler Thromb Vasc Biol. 2002;22:1642–1648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded Methods
Supplemental Table (Excel) Legends
Supplemental Figure Legends
Supplemental Figures I-VIII
Supplemental Table (Excel) Files I-V