

Abstract
The glucagon-like peptide 1 receptor (GLP-1R) is a class B G-protein coupled receptor (GPCR) and diabetes drug target expressed mainly in pancreatic β-cells that, when activated by its agonist glucagon-like peptide 1 (GLP-1) after a meal, stimulates insulin secretion and β-cell survival and proliferation. The N-terminal region of GLP-1 interacts with membrane-proximal residues of GLP-1R, stabilizing its active conformation to trigger intracellular signaling. The best-studied agonist peptides, GLP-1 and exendin-4, share sequence homology at their N-terminal region; however, modifications that can be tolerated here are not fully understood. In this work a functional screen of GLP-1 variants with randomized N-terminal domains reveals new GLP-1R agonists and uncovers a pattern whereby a negative charge is preferred at the third position in various sequence contexts. We further tested this sequence-structure-activity principle by synthesizing peptide analogues where this position was mutated to both canonical and non-canonical amino acids. We discovered a highly active GLP-1 analogue in which the native glutamate residue three positions from the N-terminus was replaced with the sulfo-containing amino acid cysteic acid (GLP-1-CYA). The receptor binding and downstream signaling properties elicited by GLP-1-CYA were similar to the wild type GLP-1 peptide. Computational modeling identified a likely mode of interaction of the negatively charged side chain in GLP-1-CYA with an arginine on GLP-1R. This work highlights a strategy of combinatorial peptide screening coupled with chemical exploration that could be used to generate novel agonists for other receptors with peptide ligands.
Graphical Abstract

INTRODUCTION:
The secretion of glucagon-like peptide-1 (GLP-1) from intestinal L-cells and its subsequent activation of glucagon-like peptide 1 receptor (GLP-1R) on β-cells triggers a host of functions including insulin secretion, β-cell proliferation, and β-cell survival to boost the body’s capacity to control blood glucose levels1–4. These activities help promote glucose metabolism and nutrient absorption after a meal, and thus GLP-1R agonists, including analogues of GLP-1, have been clinically approved as therapeutics for type 2 diabetes5–7. Additionally, GLP-1R agonists have been shown to offer neuroprotective properties in cell culture and animal models8–10 and are currently in Phase 2 clinical trials for the treatment of Parkinson’s Disease (Trial ID NCT04154072).
Given the importance of GLP-1R as a therapeutic target, understanding the GLP-1–GLP-1R interaction is of chief importance for studying metabolic physiology and further developing pharmacological agents that can leverage the therapeutic potential of GLP-1R activation. Like other class B GPCRs, the general mode of interaction between GLP-1R and GLP-1 can be understood according to the “two-domain model”11,12. In this model, GLP-1R contains a 130-amino-acid long extracellular domain (ECD) which binds the C-terminal helical portion of GLP-1, initiating the peptide-receptor interaction (Figure 1a). The N-terminal portion of GLP-1 interacts more closely with the transmembrane domains and extracellular loops of the receptor, forming contacts which stabilize the active conformation of GLP-1R to trigger activation of intracellular signaling.
FIGURE 1: GLP-1R peptide agonists.


a) Cryo-EM structure of the GLP-1 peptide agonist interaction with activated GLP-1R18 labeled to show how this interaction follows the “two-domain model” whereby the C-terminus of the peptide binds the extracellular domain (ECD) and the N-terminus binds to residues near the membrane to activate the receptor. b) Alignment of GLP-1R agonists GLP-1, exendin-4, and Ex4-P5 where the orange residues are those shared between GLP-1 and the other two peptides, the blue residues are residues shared only between exendin-4 and Ex4-P5, the gray residue is unique to exendin-4, and the green residues are those which are unique to Ex4-P5.
The precise determinants for agonism at the critical N-terminal region of the GLP-1 peptide have been investigated but details are still fully emerging. There is relatively high convergence (~90%) in the first nine amino acids between the two most well-studied GLP-1R agonist peptides, GLP-1 and exendin-4, a peptide isolated from Gila monster lizard saliva that has similar GLP-1R agonist activity as GLP-113. Alanine-scanning experiments and cryo-EM analysis of GLP-1 in complex with GLP-1R have highlighted several amino acid residues within the N-terminal region, including the first, third, fourth, and fifth residues, that contact the receptor and are necessary for full agonist activity of the wild type GLP-1 sequence14,15. However, the discovery of active GLP-1R agonists with sequences divergent from GLP-1 at the N-terminus challenges this understanding. The engineered peptide Ex4-P5, for example, shares no amino acids in common with its parent exendin-4 at the first six amino acids of its N-terminus (Figure 1b), but can activate its primary signaling axis of cAMP production to a similar extent as exendin-4 in GLP-1R-expressing mammalian (CHO-K1 and HEK293) cells16, pointing to more flexibility in the sequences that can trigger GLP-1R activation than previously understood. Recent structural data highlights features that can be used towards GLP-1R agonist design17–20, but understanding non-covalent interactions driving agonism within a variety of peptide sequence contexts remains a major goal of the field.
Peptide ligands drive class B GPCR activity, unlike many GPCRs which are triggered by small molecules, presenting a unique opportunity to discover new agonists and explore sequence-function relationships between ligands and receptors. In many small molecule and peptide screens, ligand variation is generated by dedicated chemical synthesis of library members21,22. In contrast, methods based on expression of peptide libraries in a host organism—for example, phage or yeast—rely on variation generated with recombinant DNA that links genotype to phenotype23. Combined with an appropriate functional screening assay, these approaches are attractive for discovering novel peptide ligands that induce receptor activation without the need to chemically synthesize each individual variant.
Here, the determinants of GLP-1R agonism were explored by screening a peptide library, secreted in yeast, of GLP-1 variants containing randomized N-terminal sequences. We uncovered novel GLP-1R agonists and determined that the N-terminal region of GLP-1 can tolerate a high level of sequence diversity. Moreover, we found that a negative charge at the third amino acid position of GLP-1 was a critical driver of agonism and confirmed this insight by creating a highly active GLP-1 peptide containing a non-canonical cysteic acid residue at this position.
RESULTS & DISCUSSION:
GLP-1 peptide library generation and screening using a GLP-1R activation assay
Key contacts between the N-terminal region of GLP-1 with the membrane-proximal region of GLP-1R stabilizes an activating conformational change of the receptor, leading to signaling primarily through the cyclic adenosine monophosphate (cAMP) secondary messenger1. To explore which N-terminal contacts are most important for receptor activation, we created a library of GLP-1 peptides where DNA encoding for the first 5 amino acids was randomized by saturation mutagenesis. A mutagenic oligonucleotide pool was synthesized using a mix of trimer phosphoramidites representing the three-nucleotide codons for nineteen amino acids excluding cysteine to avoid disulfide bonds (ELLA Biotech)24. This pool of oligonucleotides was incorporated into the GLP-1 peptide sequence by polymerase chain reaction (PCR) and the resulting library of DNA inserts was introduced into the yeast strain Saccharomyces cerevisiae by homologous recombination. Individual yeast clones, which each contained a unique GLP-1 variant, were analyzed to confirm that the sequence composition was as expected after saturation mutagenesis of the targeted region (Supplemental Information 3.1). Individual transformed yeast colonies were inoculated into microtiter plates and cultured such that the yeast in each well secreted a different GLP-1 peptide variant.
To measure agonist activity, we created a mammalian reporter cell line in the parental line CHO-K1 by transducing both the receptor and a gene expression reporter element comprised of green fluorescent protein (GFP) under the control of the cAMP Response Element (CRE), which promotes gene expression upon activation of cAMP signaling, a system that is commonly used for detection of GLP-1R activity16,25. A screening workflow was then established to enable detection of yeast-secreted active GLP-1 peptide variants in a microtiter plate format and assessing their agonist activity through treatment and subsequent flow cytometry analysis of the CHO-K1-CRE-GFP-GLP-1R cells (referred to as GLP-1R reporter cells) (Figure 2a). Using this plate-based functional screening workflow, approximately 1700 individual peptide variants were screened.
FIGURE 2: GLP-1 peptide library screening.
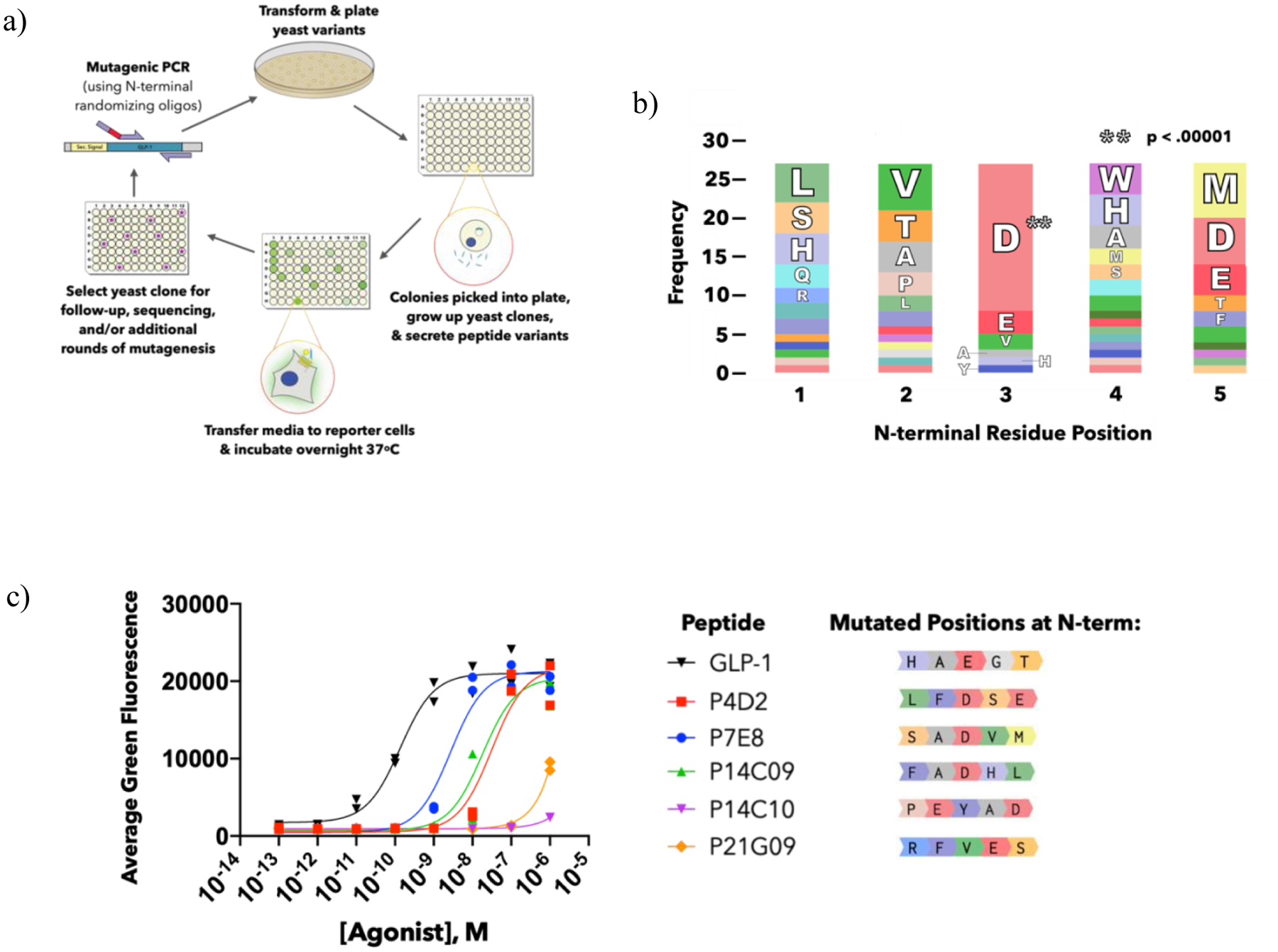
a) Schematic of peptide mutagenesis and library screening workflow. b) Summary of GLP-1 library screening hits via amino acid frequencies of the five randomized residues. The amino acid frequency out of the total 27 hits identified is represented by the its share of the bar at each position, colored by amino acid identity. For full list of hit sequences, see Supp Table 3.3–1. The p-value was assigned based on a chi-square test comparing observed frequencies to expected frequencies of amino acids at each position. c) CRE reporter cell activities of selected hits identified from the saturation mutagenesis screen as compared to wild-type GLP-1. The first five N-terminal residues are shown (residues 6–31 shared with GLP-1, see Figure 1b). Data points represent duplicate measurements.
GLP-1 variants with a modified N-terminal region function as GLP-1R agonists
Despite randomizing five amino acids at the N-terminal region of GLP-1, after screening over a thousand library variants, we identified dozens of active peptides as determined by statistical analysis of how significantly they exceeded the average fluorescence of the other variants on the screening plate (Supp Info 3.2). An analysis of the sequences that comprise the first five amino acids in these active GLP-1 variants reveals a significant amount of variability in the N-terminal residues that yield active GLP-1R agonists (Figure 2b, Supp Table 3.3–1). Five of these screening hits were randomly chosen for solid phase peptide synthesis and purified by solid phase extraction or by HPLC, along with wild-type GLP-1. The GLP-1R reporter cells were then used to compare their activity (Figure 2c). We identified clone P7E8 as one of the top hits from the peptides tested, although its EC50 was approximately 2 logs less active than GLP-1, and we chose to use this variant for follow-up screening and study.
To further improve its activity, clone P7E8 was subjected to a second round of mutagenesis where we further diversified the five amino acid N-terminal region with single point mutations. Using a Python script, we generated mutagenic oligonucleotide sequences to code for all single amino acid mutants (expect for cysteine) This oligonucleotide pool was synthesized (IDT Technologies), incorporated into the GLP-1 sequence using PCR to create a library of inserts, and transformed into yeast. We then repeated the yeast secretion and mammalian cell screen workflow under higher stringency by diluting the peptide-containing media an additional 25x prior to analysis (Supp Info 3.4.1). We screened roughly 200 yeast clones, focusing on any wells whose fluorescence exceeded that of the parent P7E8 clone. Using this strategy, we identified two putatively improved variants of P7E8, containing V4A or V4M point mutations (Figure 3a). We then chemically synthesized, purified, and tested the activity of these variants in the GLP-1R reporter cell assay (Figure 3b). We found that the activity of P7E8-V4M was significantly improved compared to the P7E8 parent (EC50 = 150 ± 100 pM versus 1.5 ± 1 nM, p= 0.019) and was similar to the activity of wild-type GLP-1 (EC50 = 90 pM ± 70 pM, p= 0.12). P7E8-V4A had an EC50 value of 290 ± 200 pM, which was significantly improved compared to P7E8 (p= 0.024) but was less active than wild-type GLP1 (p= 0.017). These studies confirm that GLP-1 variants with disparate N-terminal sequences as compared to GLP-1 can function as GLP-1R agonists.
FIGURE 3: Sequence-activity screening of GLP-1 variant P7E8.

a) Sequences and EC50 values of P7E8 (blue) and variants identified from a second round of mutagenesis and screening (V4A, V4M), and P7E8 variants with glutamate or asparagine substituted at the third position (D3E, D3N). Sequence differences from GLP-1 (black) are underlined and differences between P7E8 and its variants are highlighted in bold. Range describes the top and bottom bounds of the approximate 95% confidence interval of the estimated EC50. b,c) Dose response curves of modified peptides tested for cAMP activity in CHO-K1-CRE-GFP reporter cells. b) GLP-1, P7E8, P7E8 V4A, P7E8 V4M and c) GLP-1, P7E8, P7E8 D3N, P7E8 D3E. Data points represent duplicate measurements.
Analysis of the frequencies at which amino acids appeared in our screening hits revealed a striking prevalence of aspartate appearing at the third position, with this occurrence appearing in 70% of the hit sequences (Figure 2b). Glutamate, another negatively charged amino acid that is present in wild type GLP-1, was the second highest frequency residue present at this position, and together, over 80% of our hits contained a negatively charged amino acid at the third position. Since the GLP-1 library was generated using a saturation mutagenesis technique that randomized the sequences at the codon level to minimize library bias, this pattern suggests that maintaining this chemical preference in the context of a number of different N-terminal sequences confers some level of peptide activity. In Figure 2c, two variants that lacked a negative charge at the third position (P14C10, P21G09), originally identified as hits in the yeast secretion format, were only weakly active upon testing as synthetic peptides. This likely resulted from high peptide expression levels in the non-diluted yeast supernatant used for the first round of screening. These results also reveal quantitative limitations of the yeast secretion and screening platform, and highlight the importance of validating hits using synthetic, purified, and quantified peptides as we have done in Figures 2–4.
FIGURE 4: Non-canonical amino acid substitutions at the third position of GLP-1.


a) Structures of the glutamate side chain and negatively charged non-canonical amino acid side-chain substitutions at the third position of GLP-1. b) Activity of non-canonically substituted GLP-1 variants and GLP-1. Black: GLP-1; Red: GLP-1-CYA; Green: GLP-1-PS; Blue: GLP-1-NPA. c) Putative interaction site of cysteic acid residue (cyan) with arginine 190 of GLP-1R (yellow) in lowest energy computational model of GLP-1-CYA peptide bound to activated GLP-1R. The interaction of the glutamate residue of wild-type GLP-1 (magenta) with arginine 190 of GLP-1R (orange) is also shown for comparison18. d) Competition binding assay results of GLP-1 and GLP-1-CYA binding to GLP-1R expressed on CHO-K1 cells. e) Dose-response of GLP-1 and GLP-1-CYA measuring peptide-mediated activation of ERK1/2 in CHO-K1-GLP-1R cells. Black: GLP-1; Red: GLP-1-CYA. Error bars of (e) represent standard deviation of measurements from three separate peptide treatments at the concentration with two separate TR-FRET instrument readings each.
A negatively charged amino acid at position 3 of P7E8 is a key driver of GLP-1R activity
Previous alanine scanning experiments confirm the importance of several N-terminal residues for full GLP-1 activity, including the negatively charged glutamate residue at the third position14,26. In addition, an alignment of GLP-1, Ex4, and Ex4-P5 as shown in Figure 1b reveals that all three of these peptides contain either an aspartate or a glutamate at this position. To similarly demonstrate the importance of this residue in the context of the P7E8 variant, we synthesized point mutants with the third position aspartate changed to either another negatively charged residue (glutamate) or to its closest structural counterpart asparagine, which shares its side chain geometry and shape but lacks its negative charge. For each of these mutants, as well as P7E8, we measured cAMP activity in a dose-response manner using the GLP-1R reporter cell assay (Figure 3c). We found that the original P7E8 variant containing aspartate at position 3 retained the highest agonist activity (EC50 = 1.5 ± 1 nM), while mutation to glutamate (P7E8 D3E) reduced activity (EC50 = 7.5 ± 3 nM). Mutation to asparagine (P7E8 D3N) further reduced activity (EC50 = 65 ± 30 nM) as compared to P7E8. These results show that aspartate is important for conferring full GLP-1R activity in the context of the P7E8 peptide, further highlighting that a charged amino acid side chain at position 3 is a key property driving GLP-1R activity.
Introduction of cysteic acid at position 3 of GLP-1 further validates the importance of negative charge for GLP-1R activity
The co-complex cryo-EM structure of GLP-1/GLP-1R reveals a potential electrostatic interaction of glutamate 3 of the GLP-1 peptide with arginine 190 of the GLP-1R, providing a rationale for our mutagenesis results18. We used this information for structure-activity-based chemical design to further validate the relevance of this interaction, introducing several non-canonical amino acids at the third position of GLP-1 and determining their effects on GLP-1R agonism.
GLP-1 variants were synthesized with non-canonical residues at the third position that differed in the chemistry of their R groups but maintained at least −1 formal charge at the distal end of on their R-group (Figure 4a). The first was a phosphoserine residue where there is a permanent negative charge on the phosphate entity separating the negative moiety from the peptide backbone (GLP-1-PS). We also synthesized a mutant containing cysteic acid, the oxidized and permanently charged version of cysteine (GLP-1-CYA). Lastly, we incorporated the amino acid 4-nitrophenylalanine, which has an intervening benzene ring but a distal negatively charged nitrate moiety (GLP-1-NPA).
The activity of each purified peptide was measured in a dose dependent manner using the GLP-1R reporter cells (Figure 4b). We found that the 4-nitrophenylalanine substitution dramatically decreased peptide activity (EC50 = 160 ± 50 nM) compared to wild-type GLP-1, likely because of the disruption in N-terminal binding from the structurally rigid aromatic R-group. The phosphoserine substitution yielded a peptide that was still fairly active but whose altered R-group reduced peptide activity ~5 fold compared to GLP-1 (EC50 = 2 ± 2 nM vs 0.4 ± 0.5 nM for GLP-1). The cysteic acid substitution, however, maintained a high level of activity (EC50 = 0.2 ± 0.1 nM), indicating it is at least equally able to engage the receptor and activate cAMP signaling as the natural GLP-1 glutamate residue (p= 0.068), highlighting an approach for identifying novel GLP-1 variants containing non-canonical residues.
Computational Modeling of GLP-1-CYA
Since it proved to be a highly active GLP-1R agonist, we investigated how the cysteic acid residue in GLP-1-CYA could be engaging with GLP-1R using computational modeling. To visualize the mode of interaction, cysteic acid was modeled in place of glutamate using Rosetta Remodel27, starting from the cryo-EM structure of activated GLP-1R bound to wild-type GLP-1. We began by generating a rotamer library with the cysteic acid residue that would allow Remodel to stochastically sample various bond angles and thus various spatial orientations of this non-canonical R-group during the process of minimizing the energy of the co-complexed structure of GLP-1R and GLP-1-CYA. The resulting lowest energy conformation reveals a possible interaction mode of GLP-1-CYA with GLP-1R (Figure 4c). Oxygen atoms on the negatively charged sulfo moiety of the cysteate R-group come within close enough proximity (~2 Å) to engage in hydrogen bonding with the hydrogens on the positively charged guanidinium R-group of the receptor’s arginine residue at position 190, forming a non-covalent salt bridge interaction. This modeling and the functional activity elicited by a cysteic acid substituted GLP-1 variant further corroborates the importance of a charged residue at the third position of GLP-1, and provides a visual guide for how this new chemistry might be interacting with a key residue on the receptor.
Further characterization of GLP-1 and GLP-1-CYA
To further characterize the pharmacological properties of the non-canonically substituted GLP-1-CYA variant we measured relative receptor binding and ERK1/2 activity, another signaling axis triggered by activation of GLP-1R. To measure binding, a competition assay was performed with CHO-K1 cells expressing exogenous GLP-1R in which either unlabeled GLP-1 or GLP-1-CYA was used to compete off GLP-1 labeled at the C-terminus with 6-carboxyfluorescein (GLP-1-FAM). IC50 values of GLP-1-CYA (20 ± 4 nM) and wild-type GLP1 (30 ± 10 nM) to GLP-1R expressed on mammalian cells were found to be similar between these peptides (Figure 4d). We next measured the phosphorylation of ERK1/2 (Thr 202 and Tyr 204) in response to GLP-1 or GLP-1-CYA over a range of peptide concentrations (Figure 4e). Both peptides activated ERK1/2 phosphorylation in GLP-1R reporter cells, with the GLP-1-CYA peptide variant appearing to show a slight improvement over GLP-1 (EC50 = 1.6 ± 1.8 nM vs. 14 ± 21 nM); however, the results are not statistically significant (p= 0.07). Collectively, these studies demonstrate that a GLP-1 peptide containing a non-canonical amino acid can mirror GLP-1 activity in three of its key pharmacological effects: cAMP pathways activation, binding affinity, and ERK1/2 pathway activation.
CONCLUSIONS:
Our study explores the sequence space of the N-terminal region of the GLP-1 peptide ligand in the context of GLP-1R activity. Using a saturation mutagenesis and library screening approach, we found that the first five amino acids of GLP-1 can contain disparate, but not completely random, sequences to achieve competency in stimulating GLP-1R activity similar to the naturally occurring GLP-1 peptide. We also highlighted a pattern among the library hits in which the third position from the N-terminal of active peptides favored a negatively charged aspartate or glutamate in a variety of sequence contexts. We further validated this finding by showing that mutation to asparagine at this position resulted in significantly reduced GLP-1R activity. This information was leveraged to create a novel GLP-1R agonist containing a non-canonical cysteic acid in place of this glutamate residue in GLP-1 that maintains GLP-1R binding and pharmacological activity. We also used computational modeling based on the cryo-EM structure of GLP-1/GLP-1R to propose a hypothesized interaction between this negatively charged non-canonical amino acid and a positively charged arginine residue on the receptor. We are interested in exploring the pharmacokinetics and plasma stability of GLP-1 variants containing non-canonical amino acids in a future study.
In addition to findings observed with GLP-1-CYA, the charge-charge interaction between GLP-1/GLP-1R could potentially be exploited in other ways by those developing small molecule antagonists or agonists of this receptor. Moreover, other class B GPCRs share some of GLP-1R’s structural properties, including its “two domain” peptide binding model, and have a high level of conservation in the “polar interaction network” made up of residues in the core of the transmembrane helices that are thought to help transmit ligand binding to the cytoplasmic face of the receptor28. Thus, the approach used here could be applied to reveal sequence-function patterns that generate active agonists for other GPCRs with peptide ligands. In particular, combinatorial peptide screening coupled with chemical exploration represents a strategy to reveal and then exploit important ligand-receptor contacts that is complementary to the use of structural data alone.
EXPERIMENTAL/METHODS SECTION:
GLP-1R cAMP response element reporter cell line
The GLP-1R cAMP response element reporter cell line was created by first transducing CHO-K1 cells (ATCC) with the CRE-GFP Cignal Lentiviral Assay prepared lentivirus reporter (Qiagen) followed by puromycin selection and confirmation of activity with forskolin to create CHO-K1-CRE-GFP cells29. The GLP-1R open reading frame sequence was obtained from the Harvard Center for Cancer Systems biology Human ORFeome (version 5.1) and cloned with its native signal sequence into the pLV-Neomycin vector, a generous gift from the Meyer Lab, to make pLV-GLP1R (Supp Info 2.3)30,31. pLV-GLP1R along with third-generation packaging plasmids pMDLg, pRSV-rev, and pCMV-VSVG were transfected in HEK293T using Lipofectamine 2000 in T75 flask format for lentiviral packaging. Transfected cells were then cultured at 37°C and 5% CO2 for 3 days in serum-free OptiMEM media (Gibco), with virus-containing supernatant collected daily. Supernatant was passed through a 0.45 μm filter (Millipore) and concentrated to 180 μl using an Amicon Ultra-15 centrifugal protein concentration device with a 100,000 Da cut-off membrane (Millipore) and aliquoted into 30 μl portions for transduction. To create CHO-K1-CRE-GFP-GLP-1R reporter cells, CHO-K1-CRE-GFP cells were transduced with the GLP-1R lentivirus and after selection with geneticin (neomycin) and puromycin for both genetic components (Gibco), reporter cell activity was confirmed by treatment with GLP-1R agonists and flow cytometry to confirm dose-responsive production of GFP upon agonist treatment. Additionally, we used this GLP-1R lentivirus to generate a CHO-K1-GLP-1R cell line without reporter activity for performing mammalian cell binding and pERK1/2 activation assays. Both cell lines were maintained for all subsequent experiments in Ham’s F12K media supplemented with 10% fetal bovine serum (Gibco).
Yeast secretion strain and library creation
GLP-1 variants for the library screen were expressed in Saccharomyces cerevisiae strain YVH10 (ATCC MYA-4940) in a plasmid derived from the yeast secretion vector pD1214-AK (ATUM) called pCKL1 (Supp Info 2.1) whereby expression is constitutive under the control of TEF promoter and secretion is controlled by the α-factor secretion signal and a Kex protease site that directs cleavage just before the first residue of the peptide sequence32. Wild type GLP-1 was cloned into pCKL1 and propagated in DH10B Escherichia coli. Isolated pCKL1-GLP1 plasmid was transformed into YVH10 yeast by electroporation.
The saturation mutagenesis library was created using mutagenic oligonucleotides, randomized at the first five N-terminal codon positions into any of the 20 amino acids except cysteine by trimer phosphoramidite synthesis, with the codon used for each amino acid specified in Supp. Info 2.2.3 (ELLA Biotech)33. The P7E8 library, containing single point mutations within the first 5 N-terminal amino acid residues, was generated using a computational script to list all 90 possible single amino acid mutations that could be generated from its peptide sequence (except for cysteine) and synthesizing and pooling the corresponding oligonucleotide sequences also encoding each amino acid as indicated in Supp. Info 2.2.3 (Integrated DNA Technologies). For both libraries, the pCKL1-GLP1 plasmid DNA was used as a polymerase chain reaction template when the mutagenic primers were used to incorporate changes. The mutated DNA inserts were then extended with flanking primers to generate inserts with a ~50bp overlap on each end with the pCKL1 vector, and the resulting inserts were transformed into YVH10 along with linearized pCKL1 by homologous recombination34. Transformed yeast were plated on SD-SCAA selective media as previously described35. Yeast colonies expressing individual GLP-1 variants were inoculated into deep-well 96-well plates in 1 ml of liquid SD-SCAA media and grown for 3 days, during which peptides were constitutively secreted into the media of each well. Yeast were pelleted to the bottom of the plate and supernatant containing peptide variants was harvested for treatment of CHO-K1-CRE-GFP-GLP-1R reporter cells in the screening experiments.
GLP-1 variant activity screen
To screen the GLP-1 N-terminal peptide variants for their GLP-1R agonist activity, 20 μl of harvested YVH10 yeast secretion supernatant was added to CHO-K1-CRE-GFP-GLP-1R reporter cells grown overnight at 37°C/5% CO2 in a 96 well plate after seeding at 10,000 cells/well. Treated reporter cells were incubated overnight at 37°C/5% CO2 to allow for receptor activation and GFP production. The next day the cells were washed with phosphate buffered saline (Hyclone), dissociated from the culture plate with 2.5% trypsin (Gibco), and the trypsin’s action quenched in complete media (Ham’s F12K media, 10% fetal bovine serum, Gibco). Cell suspensions were transferred to V-bottom plates for automatic sampling and measurement of each well’s reporter cells by a flow cytometer by collecting 500 cells and calculating the average green fluorescence for the well (Accuri C6). Wells that were considered hits had a well average that passed a statistical threshold of three standard deviations above the average green fluorescence of all the measured wells on the plate (Supp Info 3.2). Plasmid DNA from selected yeast clones was isolated using a Zymoprep yeast plasmid isolation kit followed by transformation and amplification in DH10B E. coli and plasmid purification. DNA was sequenced by single primer extension using an ABI 3730xl DNA Analyzer and the Thermo Fisher Big Dye Terminator kit (Sequetech).
Synthesis of GLP-1 variants
Solid phase peptide synthesis (SPPS) was used to generate peptide variants for follow-up testing. The first 26 amino acids, which were identical in all GLP-1 derivatives, were synthesized using automated fast-flow peptide synthesis (AFPS) in less than 1 h of total synthesis time36,37. We then split the resin and manually coupled the remaining five residues, including the non-canonical amino acid building blocks. Peptides were synthesized at 90° C in the reactor on 100 mg of HMPB-ChemMatrix® resin (0.44 mmol/g) with manually coupled first amino acid (glycine). Cleavage yields a C-terminal carboxylic acid. The coupling agent for the synthesis was 0.38M HATU & 0.38M PyAOP in amine-free DMF; HATU except S&A with HATU; N, Q, R, V, & T with PyAOP. Each amino acid was deprotected with 40% Piperidine, 2% formic acid in amine-free DMF. Amino acids for synthesis were obtained from Novabiochem and prepared to 0.4 M amino acids in amine-free DMF. Peptides were deprotected and cleaved from resin for 2 hr at room temperature in TFA/TIPS/EDT/H2O (94%/1%/2.5%/2.5%), triturated with ice-cold ether (3x). Samples were reconstituted in 50:50 Water:Acetonitrile + 0.1% TFA solution and resin was removed with a 0.2 μm nylon filter. The filtered samples were then lyophilized. Using this strategy, we obtained multiple GLP-1 derivatives per day with an average crude purity of 63%, which were subjected to additional testing after high-performance liquid chromatography (HPLC) purification.
Solid phase extraction and/or mass-directed reversed phased RP-HPLC was used to purify peptide preparations. See Supplementary Information sections 4 and 5 for detailed preparation information on each peptide. Crude and purified peptide samples were analyzed by analytical HPLC and liquid chromatography with mass spectrometry (LCMS). Analytical HPLC was performed on an Agilent 1200 Series instrument using a method of 5%–65% Solvent B at 1% B/min (Solvent A: Water + 0.1% TFA, Solvent B: Acetonitrile + 0.08% TFA) using an Agilent Zorbax 300SB-C3 column (5μm, 2.1×150 mm). LCMS) was performed on an Agilent 6520 Accurate mass Q-TOF LC/MS using a gradient of 1–91% Solvent B at 6% B/min (Solvent A: Water + 0.1% FA, Solvent B: Acetonitrile + 0.1% FA) using an Agilent Zorbax 300SB-C3 column (5μm 300 Å).
GLP-1R agonist activity & binding assays
CHO-K1-CRE-GFP-GLP-1R reporter cells were seeded at 10,000 cells/well in a 96-well plate and grown overnight at 37°C and 5% CO2. The next day, dilutions of purified synthetic peptides or commercially sourced control GLP-1 peptide (Anaspec) were made at 20X in phosphate buffered saline and used to treat plated reporter cells overnight. Treated adherent cells were dissociated in 0.25% trypsin-EDTA (Gibco), quenched in complete media, and green fluorescence of reporter cells was measured on an Accuri C6 flow cytometer. Data was exported for analysis in FlowJo software, and the mean fluorescence of the mammalian cell population for each sample was analyzed in Graph Pad Prism. EC50 curves were plotted using a three-parameter dose-response curve fit with all replicate data and 95% confidence intervals for EC50 values were determined. Analyses were repeated, treating each replicate series separately and p-values were calculated by a one-sided t-test of the EC50 values of the replicate series for the samples being compared.
For GLP-1R competition binding assays, GLP-1 conjugated at the C-terminal end with 6-carboxyfluorescein (GLP1-FAM) was used as the labeled competitor (Anaspec). CHO-K1 GLP1R cells were incubated with increasing concentrations of unlabeled test peptides and 30 nM of GLP1-FAM in complete media at 4°C for 2 hr in the dark38. Then, three washes in phosphate buffered saline with 0.1% bovine serum albumin (B-PBS) were performed on ice with cold centrifuging. Cells were resuspended in 100 μl of B-PBS and analyzed on an Accuri C6 flow cytometer to measure the fluorescence associated with the cells and an average fluorescence of the cells collected was calculated for each sample. Data were analyzed in Graph Pad Prism with a three-parameter sigmoidal binding curve.
To evaluate ERK1/2 phosphorylation, the Advanced phospho-ERK (Thr202/Tyr204) HTRF (homogenous time-resolved fluorescence) cellular kit was used (Cisbio)39. The assay measures phosphorylated ERK1/2 in cellular lysates through increased FRET due to bridging of antibodies in the presence of pERK1/2. CHO-K1-GLP-1R cells were treated for 8 min with agonist peptides to induce receptor activation and ERK1/2 signaling and were then immediately lysed with the provided buffer. After incubation with HTRF-labeled antibodies, phosphorylated ERK1/2 was measured by time-resolved fluorescence resonance energy transfer using a Tecan Infinite M1000 PRO HTRF plate reader and expressed as a ratio of fluorescence at 665nm/620nm, multiplied by 1000 as recommended by the manufacturer. These results were analyzed and plotted in GraphPad Prism using a three-parameter non-linear dose-response curve using biological triplicates with two HTRF measurements each and 95% confidence intervals for EC50 values were determined. Analyses were repeated, treating each triplicate series separately and p-values were determined by a one-sided t-test of the EC50 values of the replicate series for the samples being compared.
GLP-1R computational structural modeling with Rosetta Remodel
To model how the cysteic acid residue would interact with GLP-1R, we started by creating a rotamer library for the cysteic acid residue. To do so, we implemented the rotamer library creation protocol described in Renfrew et al40. In brief, atomic connections for CYA, including an acetyl cap at the N-terminus and n-methyl cap at the C-terminus, were built using PyMOL41, with energy minimization performed in Avogadro42 using UFF (universal force field). Two chi angles were chosen for generating rotamer angles, formed by rotating along the bonds between the alpha carbon and the beta carbon and the bond between the beta carbon and the sulfur atom of the sulfo group in cysteic acid. We sampled in 5-degree intervals for each of the two chi angles, and 10-degree intervals for each of the backbone phi and psi angles, generating 5184 (72×72) conformations for each 1296 (36×36) phi/psi bin. Rotamer libraries were generated through the MakeRotLib protocol in Rosetta, with cluster centroids for K-means chosen for each chi angle at 60, 180, and 300 degrees based on the side chain chemistry.
Next, this non-canonical CYA amino acid was modeled using Rosetta Remodel27 to introduce the desired point-mutation. For this simulation, we wanted an unbiased local sampling of the cysteic acid conformations while allowing the surrounding residues to respond. It is possible to use either RosettaRelax and FixBB to accomplish this, but RosettaRemodel offers a simpler setup. According to our setup, the protocol relaxed the backbone of a local section of the structure (as designated “movable” in the blueprint file) while automatically setting up full (coordinate-based) constraints for all other residues. The protocol also automatically discovered neighboring side chains through the “-find_neighbors” flag and allowed their conformations to update in response to the glutamate to cysteic acid mutation (Supp Info 6). We prepared the cryo-EM structure of activated GLP-1R bound to wild-type GLP-1 (ID 5VAI) by relaxing with all-heavy atom constraints. The CYA residue was then modeled into the EM structure using Rosetta Remodel, bypassing fragment insertion in 1000 parallel trajectories. The two flanking residues on each side of CYA, as well as neighboring residues (6A), were allowed to be repacked without altering their amino acid identity. Models were scored by total energy, and the top scoring model was chosen as the representative model (Figure 4c). The top scoring model ranked by total CYA residue energy likewise displays the same interaction with R190 on GLP-1R.
Supplementary Material
WHY ACS CHEM BIO & CONTENTS OF SI:
In this work chemistry and biology critically enable discovery of novel bioactive peptides, which are used as tools to study sequence-structure-activity principles in a physiologically-relevant ligand-receptor system. These efforts combine mutagenesis, screening, and chemical design of novel peptide ligands with advanced rapid solid phase peptide synthesis techniques and biochemical characterization. In the Supporting Information, we provide supplemental details on peptide screening methodology, identification of DNA and peptide sequences used in this work, and additional details on peptide synthesis and characterization.
ACKNOWLEDGEMENTS:
The authors would like to thank A. Bisaria for graciously providing the lentivirus 3rd generation system used to engineer GLP-1R into cell lines. We would also like to thank the Qi lab at Stanford for use of their tissue culture facilities and for providing the HEK293T cells for lentivirus production. We would like to thank J. Yeung and B. Atsavapranee for technical assistance on screening experiments. We are grateful to past and current members of the Cochran lab who contributed to scientific discussions, particularly A. Mitchell for assisting with vectors used for yeast peptide secretion.
FUNDING SOURCES: This work was supported by a pilot grant from the Stanford Diabetes Research Center awarded to C.K.L. and J.R.C. C.K.L. is supported by an NIH Cell and Molecular Biology Training Grant (T32 GM007276). Novo Nordisk and Bristol-Myers Squibb Unrestricted Grant in Synthetic Organic Chemistry was awarded to B.L.P. R.A.P.S is partially funded by a fellowship from the National Institute of Standards and Technology (NIST).
ABBREVIATIONS:
- GLP-1R
glucagon-like peptide-1 receptor
- GLP-1
glucagon-like peptide-1
- ECD
extracellular domain
- cAMP
cyclic adenosine monophosphate
- CRE
cAMP Response Element
- ERK
extracellular-signal-regulated kinase
- PBS
phosphate buffered saline
- EM
electron microscopy
- CYA
cysteic acid
- NPA
4-nitrophenylalanine
- PS
phosphoserine
- AFPS
automated fast flow peptide synthesis
- FAM
6-carboxyfluorescein
- HMPB
4-(4-Hydroxymethyl-3-methoxyphenoxy)-butyric acid
- PyAOP
(7-Azabenzotriazol-1-yloxy)trispyrrolidinophosphonium hexafluorophosphate
- TFA
trifluoroacetic acid
- TIPS
Triisopropylsilyl ether
- EDT
1,2-Ethanedithiol
- HATU
Hexafluorophosphate Azabenzotriazole Tetramethyl Uronium
- LCMS
liquid chromatography mass spectrometry
- HPLC
high performance liquid chromatography
- FRET
fluorescence resonance energy transfer
- HTRF
homogenous time-resolved fluorescence
Footnotes
COMPETING INTERESTS:
Authors are inventors on intellectual property related to scientific findings described in this manuscript. J.R.C. is a co-founder of xCella Biosciences and Combangio, Inc., and a co-founder and Director of Trapeze Therapeutics. B.L.P. is a co-founder of Amide Technologies and Resolute Bio. Both companies focus on the development of protein and peptide therapeutics.
REFERENCES:
- 1.Campbell JE & Drucker DJ Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 17, 819–837 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Meloni AR, Deyoung MB, Lowe C & Parkes DG GLP-1 receptor activated insulin secretion from pancreatic β-cells: Mechanism and glucose dependence. Diabetes, Obes. Metab 15, 15–27 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yusta B et al. GLP-1 receptor activation improves β cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 4, 391–406 (2006). [DOI] [PubMed] [Google Scholar]
- 4.MacDonald PE et al. The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes 51, (2002). [DOI] [PubMed] [Google Scholar]
- 5.Drucker DJ Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 27, 740–756 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Mathieu C et al. Efficacy and safety of liraglutide added to insulin treatment in type 1 diabetes: The adjunct one treat-to-target randomized trial. Diabetes Care 39, 1702–1710 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Bucheit JD et al. Oral Semaglutide: A Review of the First Oral Glucagon-Like Peptide 1 Receptor Agonist. Diabetes Technol. Ther 22, 10–18 (2020). [DOI] [PubMed] [Google Scholar]
- 8.During MJ et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med 9, 1173–1179 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Hunter K & Hölscher C Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci. 13, 4–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yun SP et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med 24, 931–938 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pal K, Melcher K & Xu HE Structure and mechanism for recognition of peptide hormones by Class B G-protein-coupled receptors. Acta Pharmacol. Sin 33, 300–11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilmen A, Göke B & Göke R The isolated N-terminal extracellular domain of the glucagon-like peptide-1 (GLP)-1 receptor has intrinsic binding activity. FEBS Lett. 398, 43–47 (1996). [DOI] [PubMed] [Google Scholar]
- 13.Eng J, Kleinman WA, Singh L, Singh G & Raufman JP Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom: Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J. Biol. Chem 267, 7402–7405 (1992). [PubMed] [Google Scholar]
- 14.Adelhorst K, Hedegaard BB, Knudsen LB & Kirk O Structure-activity studies of glucagon-like peptide-1. J. Biol. Chem 269, 6275–6278 (1994). [PubMed] [Google Scholar]
- 15.Xiao Q et al. Biological activities of glucagon-like peptide-1 analogues in vitro and in vivo. Biochemistry 40, 2860–2869 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Zhang H et al. Autocrine selection of a GLP-1R G-protein biased agonist with potent antidiabetic effects. Nat. Commun 6, 8918 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang YL et al. Phase-plate cryo-EM structure of a biased agonist bound human GLP-1 receptor-Gs complex. Nature 555, 121–125 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546, 248–253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wootten D et al. The Extracellular Surface of the GLP-1 Receptor Is a Molecular Trigger for Biased Agonism. Cell 165, 1632–1643 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu F et al. Full-length human GLP-1 receptor structure without orthosteric ligands. Nat. Commun 11, 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swinney DC & Anthony J How were new medicines discovered? Nat. Rev. Drug Discov 10, 507–519 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Klabunde T & Hessler G Drug Design Strategies for Targeting G-Protein-Coupled Receptors. ChemBioChem 3, 928–944 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Kariolis MS, Kapur S & Cochran JR Beyond antibodies: Using biological principles to guide the development of next-generation protein therapeutics. Curr. Opin. Biotechnol 24, 1072–1077 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Kayushin A, Korosteleva M & Miroshnikov A Large-scale solid-phase preparation of 3′-unprotected trinucleotide phosphotriesters - Precursors for synthesis of trinucleotide phosphoramidites. Nucleosides, Nucleotides and Nucleic Acids 19, 1967–1976 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Chen J et al. Identifying glucagon-like peptide-1 mimetics using a novel functional reporter gene high-throughput screening assay. Peptides 28, 928–934 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Xiao Q, Jeng W, Wheeler MB, Medicine D & Ms C Characterization of glucagon-like peptide-1 receptor-binding determinants. J. Mol. Endocrinol 8, 321–335 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Huang P-S et al. RosettaRemodel: A Generalized Framework for Flexible Backbone Protein Design. PLoS One 6, e24109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Graaf C et al. Extending the Structural View of Class B GPCRs. Trends Biochem. Sci 42, 946–960 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Totsuka Y, Ferdows MS, Nielsen TB & Field JB Effects of forskolin on adenylate cyclase, cyclic AMP, protein kinase and intermediary metabolism of the thyroid gland. Biochim. Biophys. Acta - Gen. Subj 756, 319–327 (1983). [DOI] [PubMed] [Google Scholar]
- 30.Lamesch P et al. hORFeome v3.1: A resource of human open reading frames representing over 10,000 human genes. Genomics 89, 307–315 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayer A et al. Engulfed cadherin fingers are polarized junctional structures between collectively migrating endothelial cells. Nat. Cell Biol 18, 1311–1323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilcox CA & Fuller RS Posttranslational Processing of the Prohormone-cleaving Kex2 Protease in the. J. Cell Biol 115, 297–307 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Randolph J, Yagodkin A, Lamaitre M, Azhayev A & Mackie H Codon based mutagenesis using trimer phosphoramidites. Nucleic Acids Symp. Ser 52, 479–479 (2008). [Google Scholar]
- 34.Hua S, Qiu M, Chan E, Zhu L & Luo Y Minimum Length of Sequence Homology Required for in vivo Cloning by Homologous Recombination in Yeast. Plasmid 38, 91–96 (1997). [DOI] [PubMed] [Google Scholar]
- 35.Robinson AS, Hines V & Wittrup KD Protein Disulfide lsomerase Overexpression Increases Secretion of Foreign Proteins in Saccharomyces cerevisiae. 12, 381–384 (1994). [DOI] [PubMed] [Google Scholar]
- 36.Hartrampf N et al. Synthesis of proteins by automated flow chemistry. Science 368, 980–987 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Mijalis AJ et al. A fully automated flow-based approach for accelerated peptide synthesis. Nat. Chem. Biol 13, 464–466 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Hunter SA & Cochran JR Cell-Binding Assays for Determining the Affinity of Protein–Protein Interactions: Technologies and Considerations. Methods in Enzymology 580, (Elsevier Inc., 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eglen RM et al. The Use of AlphaScreen Technology in HTS: Current Status. Curr. Chem. Genomics 1, 2–10 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Renfrew PD, Choi EJ & Kuhlman B Using Non-canonical Amino Acids in Computational Protein-Peptide Interface Design. PLoS One (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeLano WL The PyMOL Molecular Graphics System, Version 1.8. Schrödinger LLC http://www.pymol.org (2014). doi: 10.1038/hr.2014.17 [DOI] [Google Scholar]
- 42.Hanwell MD et al. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform 4, 17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.