

Abstract
Background:
Alzheimer’s disease (AD) is a neurodegenerative disorder that causes cognitive dysfunction. Previous studies have suggested that amyloid plaques, mainly comprising of amyloid-beta peptides, play a pivotal role in AD pathophysiology. This study focuses on the evaluation of the effects of amyloid precursor protein (APP) overexpression on NF-κB, Rho-GTPase and Bcl-2 mediated pro-apoptotic pathways in neuronal cells.
Methods:
A lentiviral transduction system was used to generate SH-SY5Y cells overexpressing APP. Immunoblotting was conducted to determine expression levels of NF-κB, Rho-GTPase, and Bcl-2 family proteins in the APP overexpressed cells.
Results:
In the NF-κB signaling pathway, APP-overexpressing SH-SY5Y cells showed that there was a reduction of p-NF-κB (p< 0.05) and IKKα. Subsequently, there was upregulation of protein expression of NF-Κb, IKKβ and IκBα. On the other hand, protein expression of RhoC (p< 0.05) and Rac1/2/3 was upregulated as compared to the control group. Meanwhile, a decrease in RhoA, Cdc42 (p< 0.05) and p-Rac1/cdc42 protein levels was observed in the APP-overexpressed group. Lastly, in the pro-apoptotic pathway, the expression of Bcl-2, Bid, Bok and Puma (p< 0.05) was up regulated in the APP-overexpressed group. Downregulation of Bad and Bim expression was observed in the APP-overexpressed as compared to the control group, and Bax expression remained unchanged in the APP-overexpressed group.
Conclusion:
APP overexpression regulated signaling in the NF-κB, Rho-GTPase and Bcl-2 family pathways in neuronal cells, suggesting that these are involved in promoting neuronal survival and modulating synaptic plasticity in AD. However, further studies are essential to elucidate the APP-mediated mechanism of action.
Key Words: Alzheimer’s disease, Amyloid precursor protein, Bcl-2 family proteins, NF-κB, Rho-GTPase
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease affecting the hippocampal and entorhinal cortical regions of the brain, with the progression of cognitive deterioration correlating with age (1). Apart from aging, biological factors such as genetic mutations and polymorphisms, atypical inflammatory or immune responses, oxidative stress, traumatic injury, drugs and hormone therapy have been implicated in the pathogenesis of AD. There are two main neurological features involving abnormal protein deposition in AD pathophysiology: hyperphosphorylated tau proteins and beta-amyloid (Aβ) peptide accumulation that result in neurofibrillary tangles and amyloid plaque formation (1, 2). Based on statistics from the Alzheimer’s Association, AD prevalence is estimated to grow to 65 million by the year 2030. At present, there is no specific treatment to cure AD completely, and the available drugs such as donepezil and rivastigmine are for symptomatic relief. Therefore, the race to understand the complexity of AD pathogenesis amongst neurosurgeons and scientists aims to develop alternative therapies to improve AD patients’ and their caretakers’ quality of life. Numerous studies support that excessive neurotoxic Aβ peptides derived from amyloid precursor protein (APP) play an important role in the pathological event of AD, eventually leading to neuronal cell death and ultimately dementia (3).
APP is a transmembrane protein that is processed by three enzymes, namely α-, β-, and γ-secretase. APP undergoes proteolytic processing via two pathways; the amyloidogenic and non-amyloidogenic pathways, of which the latter is more predominant in AD patients (4, 5). In the amyloidogenic pathway, β-secretase cleaves APP to soluble APP protein beta (sAPPβ) and leaves the 99-amino-acids C-terminal fragment (C99) within the membrane, which is consequently processed by γ-secretase to release Aβ and APP intracellular C-terminal domain (AICD) (6). On the other hand, in the non-amyloidogenic pathway, α-secretase cleaves APP to release soluble APP protein alpha (sAPPα) and C83, which are further processed by γ-secretase to produce soluble fragments p3 and AICD (7).
As aforementioned, the pathways underlying the pathogenesis of AD is a subject of intensive study by the neuroscience community. Previous studies show that the transcriptional activity of Nuclear Factor Kappa Beta (NF-κB) is increased in a variety of tissues with aging, and is associated with numerous age-related degenerative diseases including AD (8). NF-κB represents a family of inducible transcription factors comprising of NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB and c-Rel (9). In unstimulated cells, NF-κB is sequestered in the cytoplasm by the inhibitor of NF-κB proteins (IκB), which are IκBα, IκBβ, IκBε,, and Bcl-3 respectively (10). Upon activation, the NF-κB pathway is initiated by the signal-induced degradation of IκB proteins. This occurs primarily via the activation of a kinase called the IκB kinase (IKK) which is composed of a heterodimer of the catalytic subunits IKKα, IKKβ, and IKKγ (10). When activated, the IκB kinase phosphorylates two serine residues located in an IκB regulatory domain. This causes the IκB proteins to undergo ubiquitination, which is followed by proteasomal degradation. With the degradation of IκB, the NF-κB complex is then freed to enter the nucleus to regulate gene expression. The activation of these genes by NF-κB then leads to the associated physiological response, for example, an inflammatory or immune response, a cell survival response, or cellular proliferation (11). Chen et al. showed that the NF-κB pathway is involved in the activation of β-secretase transcription that leads to enhanced production of Aβ which further promotes amyloid dysregulation in AD (12). In addition, transcription factors of the NF-κB pathway, including insulin-like growth factor (IGF-1), Forkhead transcription factors (FOXO), sirtuin (SIRT), and mammalian target of rapamycin (mTOR) play a critical role in regulating lifespan (13). Another component contributing to increased NF-κB activity associated with AD is the altered transcriptional phenotype which is a phenomenon in the aging cells. A specific senescence-associated secretory phenotype (SASP) elevates the expression of cytokines IL-6, IL-8, IL-7, increasing the risk of succumbing to age-related disorders (14).
Recently, numerous researchers have proposed that Rho-GTPase pathway proteins may correlate with Aβ production and thereby contribute to the loss of actin polymerization and dendritic spines, which results in the alteration of neural connectivity (15, 16). Rho-GTPases are guanine nucleotide-binding proteins that act as mediators for many cellular activities that influence cell migration, motility, adhesion, apoptosis, polarity and differentiation (17). In response to external and internal signals, Rho-GTPases are able to control signal transduction by oscillating between the GDP-bound inactive state and GTP-bound active state(18). Once Rho-GTPases are activated, they react with a spectrum of effectors to stimulate downstream signaling pathways. Among the members of the Rho-GTPases family, Rho, Rac and Cdc42 are the most prominent subfamilies whereas the others remain poorly understood (17). Over the years, studies have shown that Rho-GTPases contribute to various aspects of neuronal growth, including the formation of neurites, development and repair of dendritic spines, as well as axon pathfinding (18). The critical functions of Rho-GTPase family proteins in regulating nervous system development indicate that their dysregulation may be a causative factor in AD pathogenesis. Although the roles of Rho-GTPases in neural development have been well-documented, their contribution to neurodegeneration has been far less characterized.
Previous studies also showed that the mechanism of neuronal cell death is likely mediated via the intrinsic mitochondrial pathway, which subsequently leads to apoptosis. This pathway can be activated by highly toxic intracellular conditions, including oxidative stress (19, 20). Thus, it is possible that APP, an oxidative stress-inducing agent, may increase intracellular calcium levels, in turn causing excitotoxicity (19). These events are possibly regulated by the Bcl-2 family proteins through elevating proteins with pro-apoptotic characteristics and suppressing the anti-apoptotic proteins (21, 22).
Based on the literature, we surmise that AD is an aging-associated disease caused by the dysregulation of pathways involved in neuronal growth and development, eventually leading to neuronal cell death. Nevertheless, there are limited studies investigating the roles of APP in NF-κB, Rho-GTPase and Bcl-2 mediated pro-apoptotic pathways in neuronal cells. Thus, the present study aimed to provide a fundamental evaluation of the above-mentioned pathways in mediating AD pathogenesis.
Materials and Methods
Cell Culture
SH-SY5Y cell line was purchased from American Type Culture Collection (ATCC). The cell line was maintained in Dulbecco’s modified Eagle’s medium (Hyclone Laboratories, USA) supplemented with 10% foetal bovine serum (Biosera, South America) and 1% penicillin/ streptomycin (i-DNA Biotechnology, Singapore) at 37 °C with 5% CO2.
Lentivirus production and transduction
Lentiviral control and expression constructs targeting human APP were purchased from GeneCopoeia (USA). Briefly, high-titer lentiviruses were produced by co-transfection with packaging plasmids psPAX2 (Addgene plasmid #12260) and envelope plasmids pMD2.G (Addgene plasmid #12259) into HEK-293T using CalPhos Transfection Kits (Clontech, Mountain View, CA, USA). Supernatants containing lentiviruses were supplemented with polybrene (Sigma, USA) and used for the transduction of SH-SY5Y cells.
Protein extraction and immunoblotting
Protein lysates were extracted using ice-cold lysis buffer (1% NP-40, 1mM DTT, supplemented with protease and phosphatase inhibitors in PBS). Total protein (50 μg) was subjected to SDS-PAGE followed by immunoblotting. Primary antibodies against APP (803001; 1:1000) and GAPDH (sc-365062; 1:1,000) were obtained from Biolegend (San Diego, CA, USA) and Santa Cruz Biotechnology (Dallas, Texas, USA) respectively. NF-κB (#8242; 1:1000), p-NF-κB (#3033; 1:1000), IKKα (#2682; 1:1000), IKKβ (#2370; 1:1000), Iκ-B-α (#4814; 1:1000), RhoA (#2117; 1:1000), RhoC (#3430; 1:1000), Cdc42(#2466; 1:1000), Rac1/2/3 (#2465; 1:1000), p-Rac1/cdc42 (#2461; 1:1000), Bad (#9239;1:1000), Bid (#2002;1:1000), Bim (#2819; 1:1000), Puma (#4976; 1:1000), Bok (#4521; 1:1000), Bax (#2772; 1:1000) and Bcl-2 (#2870; 1:1000) were purchased from Cell Signaling Technology (Danvers, MA, USA). The protein expression level was then determined by densitometry with the aid of ImageLab™ 5.2.1 Software (Bio-rad Laboratories, USA).
Statistical Analysis
Immunoblot data were quantified by densitometry and presented in means ± standard deviation of at least two replicates. Statistical analysis was accomplished using Student’s t-test in Microsoft Excel, with p-values less than 0.05 considered as statistically significant.
Results
In order to generate the AD-like in vitro model, SH-SY5Y cells were transduced with lentiviral-constructs containing APP. Minimal morphological changes were observed in APP-overexpressed compared to control cells (Fig. 1A.).
Fig. 1.
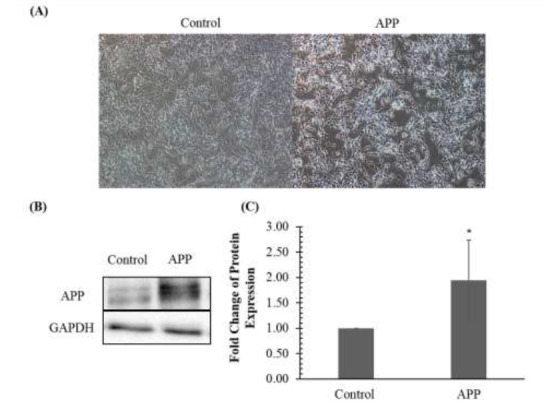
Amyloid precursor protein (APP) expression in SH-SY5Y cells following transduction. (A). Micrographs of SHSY5Y cells transduced with control and APP overexpression vector. Magnification, ×100. (B) APP expression validation in SH-SY5Y cells transduced with the control vector and APP by immunoblotting. (C) Fold change of protein expression in SH-SY5Y cells transduced with control vector and APP vector determined by densitometry. The bar chart shows the mean values of protein expression with ± standard deviation. * denotes that the P-value is less than 0.05 as compared to the control.
Next, the overexpression of APP was validated by immunoblotting. As shown in Fig. 1B., there was an approximately 2-fold increment of APP levels as compared to control (p< 0.05). The validated protein lysates were subsequently subjected to evaluation of signaling pathways.
To evaluate the effects of APP overexpression on proteins involved in the NF-κB pathway, immunoblotting was performed. Protein expression in the APP-overexpressed group was compared against control with GAPDH as the reference protein. Fig. 2. demonstrates that the protein expression of p-NF-κB and IKKα were down-regulated in the APP-overexpressed group. However, only the down-regulation of p-NF-κB was statistically significant. Meanwhile, there was upregulation of protein expression of NF-κB, IKKβ and IκBα. However, these observations were not statistically significant.
Fig. 2.
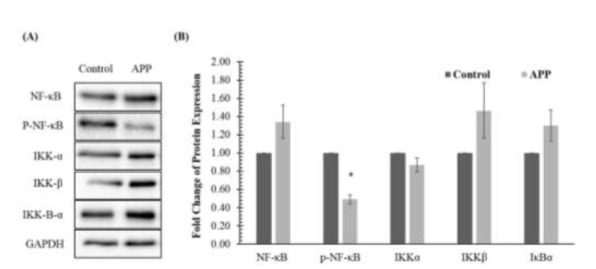
Effects of amyloid precursor protein (APP) overexpression on NF-κB signaling pathway in SH-SY5Y cells determined by (A) immunoblotting and (B) densitometry. Data is presented as mean ± standard deviations. * denotes that the p-value is less than 0.05 as compared to the control.
With reference to Figure 3, the expression of RhoC and Rac1/2/3 proteins showed up-regulation when compared to the control group. However, only the increment of RhoC was statistically significant. On the other hand, reduction in the protein levels of RhoA, Cdc42 and p-Rac1/cdc42 was observed in the APP-overexpressed group. Reduction of Cdc42 was statistically significant.
Fig. 3.
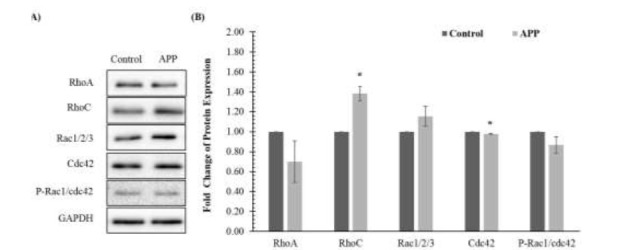
Effects of amyloid precursor protein (APP) overexpression on the Rho-GTPase signaling pathway in SH-SY5Y cells determined by (A) immunoblotting and (B) densitometry. Data is presented as mean ± standard deviations. * denotes that the p-value is less than 0.05 as compared to the control.
In the pro-apoptotic pathway, the APP-overexpressed group was up-regulated in the expression of Bcl-2, Bid, Bok and Puma (Fig. 4.). Meanwhile, down-regulation of Bad and Bim expression was observed in the APP-overexpressed group as compared to the control group. However, only the change in Puma expression was statistically significant. In present study, the expression of Bax remained unchanged in the APP-overexpressed group.
Fig. 4.
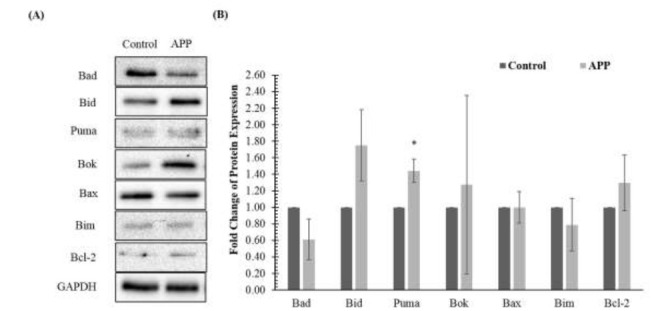
Effects of amyloid precursor protein (APP) overexpression on pro-apoptotic proteins in SH-SY5Y cells determined by (A) immunoblotting and (B) densitometry. Data is presented as mean ± standard deviations. * denotes that the p-value is less than 0.05 as compared to the control.
Discussion
In this study, the protein expression of NF-κB was not significantly upregulated, however, the activated NF-κB (p-NF-κB) was significantly down-regulated in the APP overexpressed group. A relevant study showed that inhibition of NF-κB activation reduced the size and complexity of the neurite arbors of sensory neurons (23). In agreement with this finding, pharmacological inhibition of NF-κB phosphorylation affects the formation of memory in the crab (24). A recent study suggested that NF-κB and Cdh-1, a cell-cycle regulator, are involved in the regenerative processes of post-mitotic neurons (25). However, the underlying molecular pathways that involve the stimulation of adult axoneogenesis are still poorly understood. Therefore, we postulate that the downregulation of p-NF-κB may be a contributing factor to AD pathophysiology. Inhibitory kappa-B-alpha (IκBα) protein is a member of the inhibitory NF-κB proteins (IκB), responsible for regulating the activity of p65 and other NF-κB proteins by sequestering the latter in the cytoplasm when in an inactive state (10, 26). In accordance with previous findings, the up-regulation of IκBα expression was observed in this study and the outcome also explained the basis for p-NF-κB downregulation.
We demonstrated that the expression of RhoA protein was down-regulated (not statistically significant) in APP-overexpressing SH-SY5Y cells. A similar phenomenon was also observed by Huesa et al., whereby the expression of RhoA was decreased in synapses and increased in the dystrophic neurites of AbetaPP mice (27). Moreover, total RhoA protein was decreased in the hippocampus of AD patients’ brains (27). Unlike RhoA, the function of RhoC in neural development still remains poorly understood. In cancer biology, RhoC is associated with cell proliferation and conversion of benign to malignant tumours (28), as it causes degradation and reconstruction of the extracellular matrix and remodeling of the cytoskeleton (29). Similarly, RhoC has been reported to regulate actin remodeling that is required for phagosome formation during FcγR-mediated phagocytosis (30). In the present study, the expression of RhoC was increased significantly in the cells overexpressing APP. Perhaps the observation plays a role in enhancing or synergizing the effect of decreased RhoA expression, thereby stimulating axonal growth and neurite branching. As mentioned, RhoA activation has an inhibitory effect on neurite growth and branching, whilst Rac activation has the opposite effect (31). This observation correlated with the present study whereby Rac1/2/3 expression was slightly increased.
Cdc42 and Rac function as regulatory molecules involved in cytoskeletal reformation, membrane trafficking, transcriptional regulation, cell growth and development (32). Based on the findings of several studies, up-regulated Cdc42 promoted neurite formation in Drosophila and rat cortical neurons (33, 34). Moreover, it was observed that expression of a dominant negative Cdc42 caused defects in neurite outgrowth, whereas Cdc42 reconstitution resulted in actin polymerization (35, 36). Similar findings were proposed by a few research groups, stating that the expression of Cdc42 and its downstream effector, p21-activated kinase, declined after treating cells with oligomeric Aβ25–35 and Aβ1–42, respectively (37-39). Therefore, the down-regulation of p-Rac1/cdc42 and Cdc42, as observed in the present study, may cause the inhibition of neurite outgrowth and favor dystrophic neurite formation.
In the present study, APP overexpression slightly upregulated Bok and Bid and significantly upregulated Puma, which in turn initiated the apoptotic pathway, particularly the intrinsic mitochondrial pathway. Based on the study by Certo et al., these activators first stimulate the pro-apoptotic pathway by contributing to mitochondrial outer membrane permeabilization (40). These proteins then recruit multimeric pores resulting in the opening of mitochondrial permeability transition pores followed by the release of cytochrome c into the cytoplasm (41-43). Consequently, cytochrome c serves as a scaffold to bind with apoptotic protease activating factor 1 and the subgroup family of cysteine proteases, caspase 9, to form a complex called apoptosome. This holoenzyme then activates caspase 3, resulting in neuronal cell death (44, 45). As a result, APP overexpression which upregulated the pro-apoptotic proteins would result in the activation of apoptosis in neuronal cells.
In conclusion, the current study sheds light on the fundamental pathways involved in the pathogenesis of AD, and demonstrated that these pathways potentially promote neuronal survival and modulate synaptic plasticity during the early stages AD pathogenesis. Nevertheless, much remains to be discovered about the pathogenesis of AD, for example, neurite outgrowth and mechanisms which associate the functional interactions between Bcl-2 family proteins in apoptosis stimulation and the pro-survival pathway activation.
Acknowledgements
The work was supported by grants FRGS/1/2016/SKK08/IMU/03/3 from the Ministry of Higher Education, Malaysia and BMSc I-2018 (10) from the International Medical University, Malaysia.
References
- 1.Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430(7000):631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. . 2000;33(1):95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 3.Nalivaeva NN, Turner AJ. The amyloid precursor protein: a biochemical enigma in brain development, function and disease. . FEBS Lett. 2013;587(13):2046–54. doi: 10.1016/j.febslet.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Dong S, Duan Y, Hu Y, Zhao Z. Advances in the pathogenesis of Alzheimer's disease: a re-evaluation of amyloid cascade hypothesis. . Transl Neurodegener. 2012;1:18. doi: 10.1186/2047-9158-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alva G, Potkin SG. Alzheimer disease and other dementias. . Clinics in geriatric medicine. 2003;19(4):763–776. doi: 10.1016/s0749-0690(03)00028-4. [DOI] [PubMed] [Google Scholar]
- 6.Selkoe DJ. Normal and abnormal biology of the beta-amyloid precursor protein. . Annu Rev Neurosci. . 1994;17:489–517. doi: 10.1146/annurev.ne.17.030194.002421. [DOI] [PubMed] [Google Scholar]
- 7.Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. . Neuromolecular Med. . 2010;12(1):1–12. doi: 10.1007/s12017-009-8104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tilstra JS, Clauson CL, Niedernhofer LJ, Robbins PD. NF-kappaB in Aging and Disease. . Aging Dis. . 2011;2(6):449–65. [PMC free article] [PubMed] [Google Scholar]
- 9.Sun SC, Chang JH, Jin J. Regulation of nuclear factor-kappaB in autoimmunity. Trends Immunol. . 2013;342(6):282–9. doi: 10.1016/j.it.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. . Cold Spring Harb Perspect Biol. . 2009;1(4):a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinz M, Scheidereit C. The IkappaB kinase complex in NF-kappaB regulation and beyond. . EMBO reports. . 2014;15(1):46–61. doi: 10.1002/embr.201337983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen CH, Zhou W, Liu S, Deng Y, Cai F, Tone M, et al. Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer's disease. . Int J Neuropsychopharmacol. . 2012;15(1):77–90. doi: 10.1017/S1461145711000149. [DOI] [PubMed] [Google Scholar]
- 13.Niedernhofer LJ, Robbins PD. Signaling mechanisms involved in the response to genotoxic stress and regulating lifespan. The international journal of biochemistry & cell biology. . 2008;40(2):176–180. doi: 10.1016/j.biocel.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coppe JP, Patil CK, Rodier F, Krtolica A, Beausejour CM, Parrinello S, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PloS one. 2010;5(2):e9188. doi: 10.1371/journal.pone.0009188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Musilli M, Nicolia V, Borrelli S, Scarpa S, Diana G. Behavioral effects of Rho GTPase modulation in a model of Alzheimer's disease. . Behavioural brain research. 2013;237:223–229. doi: 10.1016/j.bbr.2012.09.043. [DOI] [PubMed] [Google Scholar]
- 16.Bolognin S, Lorenzetto E, Diana G, Buffelli M. The potential role of rho GTPases in Alzheimer's disease pathogenesis. . Mol Neurobiol. 2014;50(2):406–22. doi: 10.1007/s12035-014-8637-5. [DOI] [PubMed] [Google Scholar]
- 17.Boureux A, Vignal E, Faure S, Fort P. Evolution of the Rho family of ras-like GTPases in eukaryotes. . Mol Biol Evol. 2007;24(1):203–16. doi: 10.1093/molbev/msl145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. . Genes Dev. . 2005;19(1):1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- 19.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. . 1992;12(2):376–89. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanamaru T, Kamimura N, Yokota T, Iuchi K, Nishimaki K, Takami S, et al. Oxidative stress accelerates amyloid deposition and memory impairment in a double-transgenic mouse model of Alzheimer's disease. Neuroscience letters. . 2015;587:126–131. doi: 10.1016/j.neulet.2014.12.033. [DOI] [PubMed] [Google Scholar]
- 21.Willis S, Day CL, Hinds MG, Huang DC. The Bcl-2-regulated apoptotic pathway. . J Cell Sci. 2003;116(Pt 20):4053–6. doi: 10.1242/jcs.00754. [DOI] [PubMed] [Google Scholar]
- 22.Akhtar RS, Ness JM, Roth KA. Bcl-2 family regulation of neuronal development and neurodegeneration. Biochim Biophys Acta. . 2004;1644(2-3):189–203. doi: 10.1016/j.bbamcr.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 23.Gutierrez H, Hale VA, Dolcet X, Davies A. NF-kappaB signalling regulates the growth of neural processes in the developing PNS and CNS. . Development. 2005;132(7):1713–26. doi: 10.1242/dev.01702. [DOI] [PubMed] [Google Scholar]
- 24.Merlo E, Freudenthal R, Romano A. The IkappaB kinase inhibitor sulfasalazine impairs long-term memory in the crab Chasmagnathus. . Neuroscience. 2002;112(1):161–72. doi: 10.1016/s0306-4522(02)00049-0. [DOI] [PubMed] [Google Scholar]
- 25.Haenold R, Weih F, Herrmann KH, Schmidt KF, Krempler K, Engelmann C, et al. NF-kappaB controls axonal regeneration and degeneration through cell-specific balance of RelA and p50 in the adult CNS. J Cell Sci. 2014;127(Pt 14):3052–65. doi: 10.1242/jcs.140731. [DOI] [PubMed] [Google Scholar]
- 26.Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR. Distinct roles of the Ikappa B kinase alpha and beta subunits in liberating nuclear factor kappa B (NF-kappa B) from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B. . J Biol Chem. 2002;227(6):3863–9. doi: 10.1074/jbc.M110572200. [DOI] [PubMed] [Google Scholar]
- 27.Huesa G, Baltrons MA, Gomez-Ramos P, Moran A, Garcia A, Hidalgo J, et al. Altered distribution of RhoA in Alzheimer's disease and AbetaPP overexpressing mice. J Alzheimers Dis. . 2010;19(1):37–56. doi: 10.3233/JAD-2010-1203. [DOI] [PubMed] [Google Scholar]
- 28.Horiuchi A, Imai T, Wang C, Ohira S, Feng Y, Nikaido T, et al. Up-regulation of small GTPases, RhoA and RhoC, is associated with tumor progression in ovarian carcinoma. . Laboratory investigation. . 2003;83(6):861–70. doi: 10.1097/01.lab.0000073128.16098.31. [DOI] [PubMed] [Google Scholar]
- 29.Ikoma T, Takahashi T, Nagano S, Li YM, Ohno Y, Ando K, et al. A definitive role of RhoC in metastasis of orthotopic lung cancer in mice. . Clin Cancer Res. . 2004;10(3):1192–200. doi: 10.1158/1078-0432.ccr-03-0275. [DOI] [PubMed] [Google Scholar]
- 30.Egami Y, Kawai K, Araki N. RhoC regulates the actin remodeling required for phagosome formation during FcgammaR-mediated phagocytosis. . Journal of cell science. . 2017;130(24):4168–4179. doi: 10.1242/jcs.202739. [DOI] [PubMed] [Google Scholar]
- 31.Watabe-Uchida M, Govek EE, Van Aelst L. Regulators of Rho GTPases in neuronal development. The Journal of neuroscience: the official journal of the Society for Neuroscience. . 2006;26(42):10633–5. doi: 10.1523/JNEUROSCI.4084-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wennerberg K, Der CJ. Rho-family GTPases: it's not only Rac and Rho (and I like it). . Journal of cell science. 2004;117(Pt 8):1301–12. doi: 10.1242/jcs.01118. [DOI] [PubMed] [Google Scholar]
- 33.Threadgill R, Bobb K, Ghosh A. Regulation of dendritic growth and remodeling by Rho, Rac, and Cdc42. . Neuron. 1997;19(3):625–634. doi: 10.1016/s0896-6273(00)80376-1. [DOI] [PubMed] [Google Scholar]
- 34.Luo L, Liao YJ, Jan LY, Jan YN. Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. . Genes & development. 1994;8(15):1787–802. doi: 10.1101/gad.8.15.1787. [DOI] [PubMed] [Google Scholar]
- 35.Moreau V, Way M. Cdc42 is required for membrane dependent actin polymerization in vitro. FEBS letters. 1998;427(3):353–356. doi: 10.1016/s0014-5793(98)00443-8. [DOI] [PubMed] [Google Scholar]
- 36.Ruchhoeft ML, Ohnuma S, McNeill L, Holt CE, Harris WA. The neuronal architecture of Xenopus retinal ganglion cells is sculpted by rho-family GTPases in vivo. . The Journal of neuroscience: the official journal of the Society for Neuroscience. 1999;19(19):8454–63. doi: 10.1523/JNEUROSCI.19-19-08454.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma QL, Yang F, Calon F, Ubeda OJ, Hansen JE, Weisbart RH, et al. p21-activated kinase-aberrant activation and translocation in Alzheimer disease pathogenesis. . The Journal of biological chemistry. . 2008;283(20):14132–43. doi: 10.1074/jbc.M708034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao L, Ma QL, Calon F, Harris-White ME, Yang F, Lim GP, et al. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nature neuroscience. . 2006;9(2):234–42. doi: 10.1038/nn1630. [DOI] [PubMed] [Google Scholar]
- 39.Shen JN, Wang DS, Wang R. The protection of acetylcholinesterase inhibitor on beta-amyloid-induced the injury of neurite outgrowth via regulating axon guidance related genes expressionin neuronal cells. . International journal of clinical and experimental pathology. . 2012;5(9):900–13. [PMC free article] [PubMed] [Google Scholar]
- 40.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. . Cancer cell. . 2006;9(5):351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 41.Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. PNAS. . 1998;95(9):4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, et al. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. PNAS. 1998;95(25):14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116(2):205–19. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 44.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. . Cell. 1997;91(4):479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 45.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. . Cell. 1997;90(3):405–13. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]