

Abstract
Endothelial cell injury and vascular function strongly correlate with cardiac function following ischemia/reperfusion injury. Several studies indicate that endothelial cells are more sensitive to ischemia/reperfusion compared to cardiomyocytes and are critical mediators of cardiac ischemia/reperfusion injury. H2S is involved in the regulation of cardiovascular system homeostasis and can act as a cytoprotectant during ischemia/reperfusion. Activation of ERK1/2 in endothelial cells after H2S stimulation exerts an enhancement of angiogenesis while its inhibition significantly decreases H2S cardioprotective effects. In this work, we investigated how H2S pretreatment for 24 hours prevents the ischemia/reperfusion injury and promotes angiogenesis on microvascular endothelial cells following an ischemia/reperfusion protocol in vitro, using a hypoxic chamber and ischemic buffer to simulate the ischemic event. H2S preconditioning positively affected cell viability and significantly increased endothelial cell migration when treated with 1 μM H2S. Furthermore, mitochondrial function was preserved when cells were preconditioned. Since ERK1/2 phosphorylation was extremely enhanced in ischemia/reperfusion condition, we inhibited ERK both directly and indirectly to verify how H2S triggers this pathway in endothelial cells. Taken together, our data suggest that H2S treatment 24 hours before the ischemic insult protects endothelial cells from ischemia/reperfusion injury and eventually decreases myocardial injury.
1. Introduction
Cardiovascular diseases represent the leading cause of death and disabilities in the industrialized countries, and ischemic heart disease (IHD) majorly represents them. IHD causes 46% of cardiovascular deaths in men and 38% in women worldwide [1]. One of the major challenges in treating the ischemic heart is to avoid ischemia reperfusion injury (IRI); the limitation of which can result of pivotal importance in case of programmed ischemia/hypoxia such as open-heart surgery, transplantation, or primary percutaneous coronary intervention [2–4].
IRI is the trigger leading to cell death by apoptosis and necrosis, and the causes of the activation of these phenomena are mainly oxidative stress, inflammation, and intracellular Ca2+ overload [5, 6]. In addition, reperfusion can also lead to severe ventricular arrhythmia and, as a consequence, death [7]. Low cardiac output, perioperative myocardial infarction, and arrhythmias can, in turn, be the causes of I/R injury following cardiac surgery [8].
Paradoxically, the restoration of blood flow in the ischemic tissue can exacerbate the damage more than the ischemic event itself, causing a phenomenon known as reperfusion injury [2, 9]. Indeed, the restoration of coronary blood flow, although necessary after the ischemic episode, leads to the death of cardiac myocytes that were potentially viable at the onset of reperfusion [10].
Since 1980, many important contributions shed light on the importance of the endothelium in the cardiovascular system, redirecting the investigating approach to the cardiovascular system [5, 6, 11–16]. In the last 40 years, many advances have been made toward a deeper comprehension of the role of different cell types participating in the ischemic scenario and the attention on ECs proportionally increased.
In support to these findings, several studies indicate that endothelial cells are as sensitive to ischemia/reperfusion as cardiomyocytes and therefore are critical mediators of cardiac IRI [17–22].
Furthermore, reoxygenation of the ischemic tissue causes oxidative stress: during reperfusion, xanthine oxidase, NADPH oxidase, and the mitochondrial electron transport chain (mETC) generate reactive oxygen species (ROS), mediating an increased myocardial injury [23, 24]. The abovementioned enzymes also provide the greatest amount of superoxide anion in ECs [6].
During IRI, there is a deficiency of vasodilator molecules such as endothelin [25], angiotensin, prostacyclin, and nitric oxide, which directly influence metabolic and contractile function in the adult heart [22, 26]. After the IR insult, ECs undergo changes in cytoskeletal architecture and expression of adhesion molecules (e.g., CAMs and E- and P-selectins). The activation of quiescent ECs induces the recruitment of neutrophils which initiate the inflammatory response. In parallel, ECs permeability increase resulting in the loss of barrier function and capillary leakage [3, 5, 27].
The process of angiogenesis, which is de novo formation of micro vessels, in pathological conditions is not only essential to prevent heart failure in the long term but also has the potential to support the recovery of the ischemic myocardium in the days following myocardial infarction. Impairment of myocardial angiogenesis causes a reduction in myocardial perfusion and fatal ischemic cardiomyopathy [28–30]. Moreover, EC injury and vascular function strongly correlate with the cardiac performance following IRI [31].
To prevent pathological remodeling in the structure of blood vessels, ECs produce and release two endogenous gasotransmitters, hydrogen sulfide (H2S) and nitric oxide (NO). In particular, H2S has been known for many years for being a toxic agent. Nowadays, it is recognized as being a key factor in the regulation of inflammatory and immune response, cardiovascular and nervous systems, and gastrointestinal tract function [32–35]. The cardiac tissue is capable of producing H2S endogenously, due to the presence of a discreet quantity of cystathionine-γ-lyase (CSE), one of the enzymes responsible for the production of this gasotransmitter [36]. H2S can act as a cytoprotectant against oxidative stress and as an antiapoptotic agent by preserving mitochondrial function during ischemia/reperfusion [37, 38].
The identification of the reperfusion injury salvage kinase (RISK) pathway led the way to understand how to improve the clinical outcomes of acute myocardial infarction by mediating a programmed cell survival [9, 39, 40] when activated specifically at the time of reperfusion [41]. The RISK pathway is linked to two signaling cascades: PI3K-Akt and MEK1-ERK1/2. Hu et al. observed not only that preconditioning rat myocytes with NaHS (1–100 μM) increased cell viability but also that if ERK1/2 or Akt were blocked during preconditioning or ischemia there was a significant decrease of the H2S cardioprotective effect [42].
Studies on isolated rat liver mitochondria showed a biphasic effect of H2S on mETC. Concentrations ranging from 100 nM to 1 μM stimulated electron transport, whereas 10 μM or higher provoked its inhibition [43]. The same bimodal effect has been shown by Pupo et al., where H2S induced no effect at the lowest and highest concentrations (0.5 and 100 μM) and a significant cellular response at 1 μM in terms of cell migration and proliferation [44].
Both PI3K-Akt and MEK-ERK1/2 pathways have been demonstrated to be downstream effectors of H2S signaling cascade also in ECs, thus promoting migration, proliferation, and angiogenesis [35, 45–48].
Despite the growing literature in the field, it is still not clear whether H2S could be more effective as a preconditioning or postconditioning agent [7, 19, 40, 42, 49, 50].
In our study, we decided to investigate the role of H2S as a preconditioning trigger molecule, by using different micromolar concentrations of an inorganic H2S donor, namely, sodium hydrosulfide (NaHS). Specifically, we investigated whether a 24-hour treatment could trigger a cascade that would enhance the endothelial response after an ischemic insult in an in vitro model of human microvascular endothelial cells (HMEC-1). The in vitro model is particularly feasible to address functional recovery of ECs since it allows exposure to hypoxia and drugs while maintaining the ability to proliferate and migrate and, therefore, performing experimental assays.
2. Materials and Methods
2.1. Cell Culture
Human microvascular endothelial cells 1 (HMEC-1) (ATCC® CRL3243™) were cultured in EndoGRO™-MV Complete Culture Media Kit (Millipore) and 1% penicillin/streptomycin (Pan Biotech) and passaged 80-90% confluence.
Dulbecco's Modified Eagle's Medium (DMEM) with phenol red (Sigma-Aldrich) 2% FBS (Microgem), 1% penicillin/streptomycin, and 1% L-glutamine (Microgem) was used for preconditioning and reoxygenation processes.
2.2. Hydrogen Sulfide Preconditioning and Ischemia/Reperfusion Protocol
Sodium hydrosulfide hydrate (NaHS) (Sigma-Aldrich) was used as a saline hydrogen sulfide donor. NaHS was freshly prepared in phenol red DMEM 2% FBS on the day of the experiment at a concentration of 5 mM; the stock solution was used for dilutions to reach our working conditions (1-10-100 μM).
In vitro simulation of ischemia/reperfusion injury was induced using an ischemic buffer (pH 6.2) as described in literature [25], and severe hypoxic conditions were reached through incubation in a sealed chamber (see Scheme 1) endowed with a regulator of gas mixture (InvivO2 200, Ruskinn, United Kingdom).
Scheme 1.
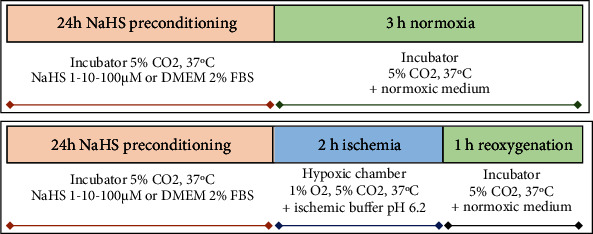
Schematic representation of the experimental design used. On the upper side is represented the “normoxia” setting, whereas on the lower panel is the setting used to simulate the in vitro “ischemia-reperfusion” condition.
A minimum level of 1% O2 can be achieved by this system, and no complete anoxia is guaranteed from the manufacturer. Moreover, to the best of our knowledge, 1% O2 at atmospheric pressure matches the oxygen partial pressure of ischemic tissues, including the myocardium [51]. The buffer was equilibrated overnight in a hypoxic chamber (1% O2, 5% CO2, 37°C). CO2 levels were set to 5% to buffer pH in medium, matching normoxic incubation. Cells were put in the hypoxic chamber, and the medium was changed with the equilibrated ischemic buffer. Ischemic protocol lasted for 2 hours in every experiment; cells were then reoxygenated with fresh medium for 1 hour in a normoxic incubator (37°C, 5% CO2).
2.3. Solutions and Reagents
2.3.1. Ischemic Buffer
The buffer was freshly prepared before each experiment. The following formulation was used: NaCl 137 mM, KCl 12 mM, MgCl2 0.49 mM, CaCl2 0.9 mM, HEPES 4 mM, and Na L-lactate 20 mM (all purchased from Sigma-Aldrich). The ingredients were dissolved into double-distilled water, and pH was adjusted to reach 6.2 before bringing the solution to the required volume (100 mL).
2.3.2. AZD6244
Also called Selumetinib, it is a selective MEK1/2 inhibitor. The inhibitor was solubilized in DMSO, as 10 mM stock solutions at -20°C. The stock was diluted to obtain a final concentration of 1 μM.
2.3.3. SCH772984 (Aurogene)
Selective ERK1/2 inhibitor was dissolved in DMSO, as 5 mM stock solutions at -80°C. The stock was diluted to reach a working concentration of 1 μM.
2.3.4. Antibodies
Primary antibodies anti-Heat-shock protein 90 (HSP90), p44/42 MAPK (ERK1/2), and phospho-p44/42 MAPK (p-ERK1/2) were all purchased from Cell Signaling Technology (The Netherlands). Secondary anti-rabbit antibody was purchased from ImmunoReagents, Inc. (North Carolina, USA). All antibodies were used according to the manufacturer's instructions.
2.4. Viability Assay
In order to address how cell viability could be influenced by preconditioning, an MTT assay was performed at 24 hours from the end of reoxygenation. Cells were plated in a 96-well plate at a density of 0.5 × 104 cells/well in growth medium. On the next day, cells were preconditioned as previously described. After 24 hours of preconditioning, cells would undergo normoxic or IR condition following the ischemia/reperfusion protocol mentioned earlier. At the end of reperfusion, media was changed for all cells to DMEM 2% FBS, in order to keep cells alive for the next 24 hours but avoiding cell proliferation. After one day, MTT solution was added to each well (10 μL/well), and plates were kept in the dark in an incubator for 3 hours. Then, media was removed and 100 μL DMSO was added to each well, and absorbance was detected at 570 nm using a microplate reader. At least eight wells for each condition were analyzed.
2.5. Cell Migration Assay
To assess cell migration, cells were plated into three-chamber silicone-culture inserts (Ibidi) in a 12-well plate at a density of 4 × 105 cells/mL in growth medium. This density was chosen as the most appropriate to have cells at 90-100% confluence overnight. On the next day, cells were treated with NaHS as described before, by removing the medium from each chamber and adding the desired treatment or just control medium, being careful not to scratch cells away. After 24 hours, all culture inserts were removed, and cells were gently washed with warm PBS with Ca/Mg. At this point, cells underwent IR protocol for 2 hours. At the end of the ischemia, cells were gently washed with warm PBS and growth medium was added to each well. After 6 hours of migration, cells were fixed in 4% PAF. Images were then acquired using a Nikon Eclipse Ti-E microscope with a ×10 lens. The MetaMorph software was used to both acquire and analyze all images. Cell motility was expressed as percentage of wound closure. At least three fields for each condition were analyzed.
2.6. In Vitro Angiogenesis Assay
The formation of capillary-like structures in vitro was studied on growth factor-reduced Matrigel (Corning, USA). Matrigel was used according to the manufacturer's instructions. ECs were plated into Petri dishes and treated according to our protocols on the following day. After in vitro-simulated IR injury, cells were seeded at 2.5 × 104 cells per well onto Matrigel-precoated 24-well plate in growth medium. After 16 hours, cell organization was observed using a 5x lens. Images were acquired using the Infinity Analyze software (Lumenera Corporation). A minimum of three fields was analyzed for each condition.
Image analysis was performed using ImageJ's plugin Angiogenesis Analyzer, which works on phase contrast (RGB colors, 24 bit) or fluorescence images (8 or 16 bit). In order to avoid artifacts, some background noise had to be removed from the images using the “blurred mask tool.” Once the image has been modified, the program can be run and the images are automatically analyzed. The images are returned with different paths traced in a color-code mode and a table with different automatically measured parameters. We choose to include in our analysis three parameters: (I) number of master segments, (II) total master segment length, and (III) number of isolated segments.
2.7. Quantification of Mitochondrial Mass and Membrane Depolarization
To measure the depolarization of mitochondrial membrane in relationship with mitochondrial mass, endothelial cells were plated at a density of 2 × 104 cells/well on 1.5 mm ø cover slips in growth medium. Cells were treated as previously described (see Hydrogen Sulfide Preconditioning and Ischemia/Reperfusion Protocol) with 1 μM NaHS. After reperfusion, all specimens were incubated in DMEM 2% FBS with both MitoTracker™ Green FM (Thermo Fisher) 200 nM and MitoTracker™ Red CMXROS (Thermo Fisher) 50 nM in the dark for 30 minutes at 37°C. After staining, cells were washed with PBS and fixed with 4% PAF. After fixation, coverslips were mounted on microscope slides and observed at a confocal microscope (Zeiss LSM800) with a 63x oil-immersion objective. GMT has an absorption/emission spectrum of 490 nm/516 nm, while the RMT one is 579 nm/599 nm. Images were acquired with ZEN System software with a resolution of 512 × 512 pixels.
2.8. Western Blot
Cells were seeded into Petri dishes and were allowed to reach confluence before being treated with NaHS (1-10-100 μM) and/or AZD6244 1 μM and then underwent IR simulation protocol. At the end of the protocol, cells were scraped and lysed with RIPA buffer (Sigma-Aldrich), containing protease and phosphatase inhibitors (Complete protease inhibitor tablets and PhosStop, Roche). Whole cell lysates were separated by SDS-PAGE and wet-transferred on PVDF membranes, according to the manufacturer's protocol (Bio-Rad). After transfer, membranes were blocked in 4% nonfat dried milk or 4% BSA (depending on the primary antibody), for 1 hour at room temperature. Membranes were washed once in TBST and incubated with antibodies against HSP90 (1 : 1,000), ERK1/2 (1 : 1,000), and phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (1 : 1,000) overnight at 4°C. Membranes were washed with TBST three times for 10 min. Anti-rabbit secondary antibody incubation (1 : 5,000) lasted for 1 hour. After another 30′ of washing with TBST 1x, chemiluminescence was detected using the ECL system (Bio-Rad) using a ChemiDoc Touch instrument (Bio-Rad).
2.9. Image Analysis
2.9.1. Tubulogenesis Assay
During the analysis, we kept into consideration the following parameters: number of master segments, number of master junctions, total master segment length, and number of isolated segments. A well-constructed network should have more master elements (segments and junctions), thus creating a more physiological and functional structure. The images were analyzed with ImageJ's Angiogenesis Analyzer plugin, and only the aforementioned parameters were kept into consideration. All images were converted to RGB format if needed.
2.9.2. Mitochondrial Staining
Multichannel images were split to separate the two channels. Then, the corrected total cell fluorescence (CTCF) was calculated for each cell in both channels as described in literature [52]. For each cell, the ratio between mitochondrial depolarization and mass was calculated and normalized on the control condition.
2.9.3. Image Lab Quantification
Using Image Lab software, bands were detected automatically and then subtracting the background to obtain a better quantification. Density of bands was computed keeping into consideration the value of the integrated density adjusted by subtracting the background. All proteins were quantified by normalizing the density over the loading control (HSP90).
2.10. Statistical and Computer Analysis
Results are expressed as mean ± SEM. Differences between groups were analyzed by one-way ANOVA or Kruskal-Wallis or Mann–Whitney t-test. Significance was established at p value < 0.05. All raw data were analyzed using Microsoft Excel and Prism 6. All experiments were repeated at least three times.
3. Results
3.1. Biological Effects of NaHS Preconditioning
During acute myocardial infarction, endothelial cells can go towards death through apoptosis, necrosis, or autophagy [53–55]. To assess endothelial cell response after IR injury in terms of viability, an MTT assay was performed after 24 hours from the injury.
As shown in Figure 1, there was a decrease in the IR condition compared to normoxia, but H2S did not significantly increase cell viability in the post-IR condition.
Figure 1.
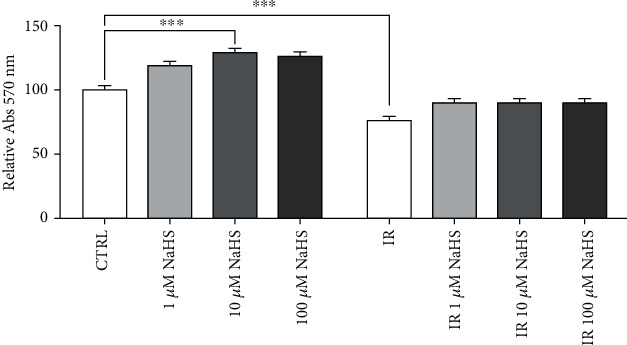
Normalized viability on the normoxic control condition. Statistical significance was set at p value < 0.05 (∗∗∗p ≤ 0.001).
The highest viability was observed at 10 μM, but only in normoxic conditions.
Cell migration is one of the essential processes during angiogenesis. In fact, cells have to migrate to create new vessels. The creation of new vessels is important after IR injury in order to allow an adequate blood supply to the myocardium and, eventually, to replace arteries that have been disrupted by the ischemic event and subsequent reperfusion. To study this process, we observed cell migration following IR simulation. After 6 hours, the percentage of wound closure was calculated and compared to the normoxic control condition (Figure 2(b)).
Figure 2.
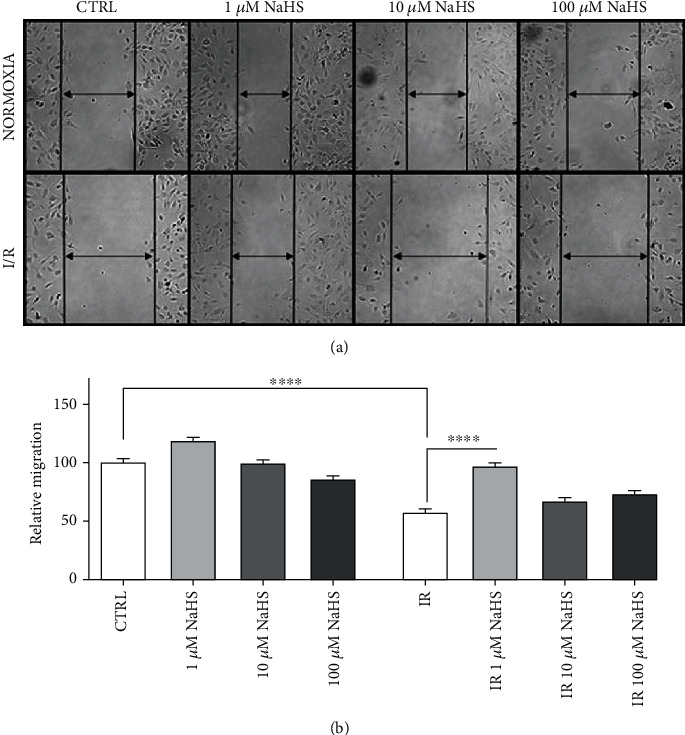
(a) Representative images of cells after 6 hours in the different experimental conditions. (b) Relative migration, all data sets were normalized on the normoxic control condition (CTRL). Statistical significance: ∗∗∗∗p < 0.0001.
We observed a significant improvement of cell migration in cells treated with NaHS, with the strongest effect at 1 μM NaHS, both in normoxia and post-IR.
To study whether NaHS treatment had a proangiogenic effect on cells that underwent IR injury simulation, we studied their ability to form capillary-like structures in vitro focusing on the most promising concentration of 1 μM NaHS.
After IR, cells were detached and plated in Matrigel-precoated 24-well multiwell in EndoGRO 10% FBS for 16 hours. As shown in Figure 3, there is a positive trend in terms of capillary formation in both normoxia and IR, upon treatment of cells with 1 μM NaHS, but no statistical significance was observed.
Figure 3.
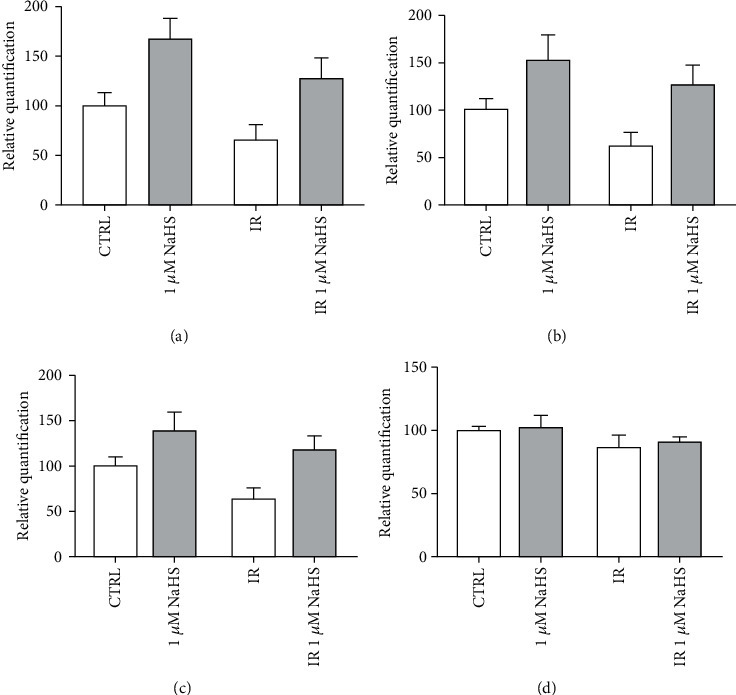
Quantification of main elements of the in vitro capillary network: (a) total master segment length; (b) number of master segments; (c) number of junctions; (d) number of isolated elements.
We decided to perform a double MitoTracker™ staining on all samples to investigate whether mitochondria were affected by NaHS preconditioning. MitoTracker™ Green FM stains all mitochondria, whereas MitoTracker™ Red CMXROS stains only viable mitochondria. Red fluorescence is directly proportional to membrane depolarization.
By calculating the ratio between red and green fluorescence, we determined the influence of NaHS and IR on mitochondria in endothelial cells.
NaHS treatment decreased the mitochondrial mass with a similar fashion in normoxia and IR (Figure 4(b)). However, the mitochondrial function did not change much between the two IR experimental conditions. On the other hand, NaHS treatment caused an improvement in mitochondria viability, observed as an increased ratio between vital (red) and total (green) mitochondria, both in IR and normoxic conditions (Figures 4(a)–4(d)).
Figure 4.
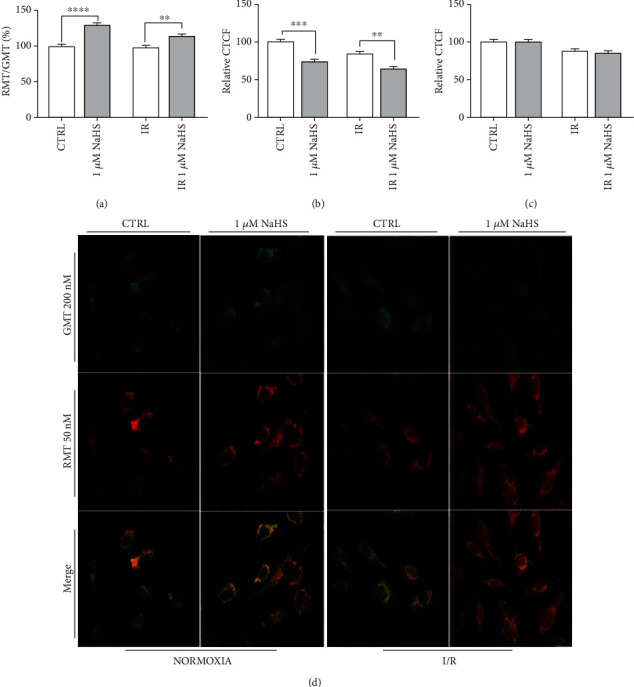
(a) Ratio between red (RMT) and green (GMT) MitoTracker™; (b, c) GMT and RMT fluorescence, respectively, calculated for each experimental condition as a percentage of CTRL; (d) representative fields of stained cells. All data sets were normalized on the normoxic control condition (CTRL). Statistical significance: ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
3.2. NaHS Preconditioning and IR Injury Influence ERK1/2 Phosphorylation
Since ERK1/2 is deeply implicated in both cell migration and angiogenesis, but also in inducing apoptosis, we evaluated whether IR and/or NaHS treatment could influence its level of phosphorylation. Protein expression and phosphorylation levels were assessed through western blot.
ERK appeared to be strongly phosphorylated in IR condition, whereas NaHS treatment attenuated the p-ERK/ERK ratio, bringing it towards a more physiological condition (Figure 5).
Figure 5.
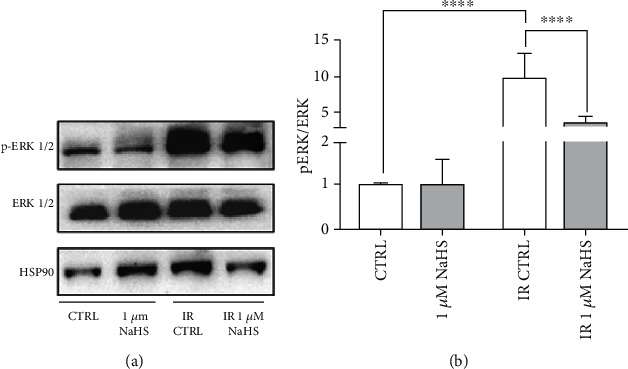
(a) Representative experiment of ERK1/2 and p-ERK1/2; (b) ratio of p-ERK on total ERK (n = 4). Both total and phosphorylated proteins were first normalized on the loading control (HSP90), and then, the ratio was calculated. All data sets were normalized on the normoxic control condition (CTRL). Statistical significance: ∗∗∗∗p < 0.0001.
3.3. Effects of MEK-ERK Pathway Inhibition
After assessing the levels of ERK phosphorylation, we decided to investigate if NaHS directly modulated the ERK pathway.
At first, we targeted its upstream modulator, MEK, using its specific inhibitor AZD6244 before preconditioning. Both viability and cell migration were tested.
Considering cell viability, there was no relevant difference with or without inhibitor and/or 1 μM NaHS, confirming what we had already observed (see Figure 1).
When we tested the effects on cell migration, we confirmed the effect of preconditioning with 1 μM NaHS in both normoxia and IR. Moreover, we observed that the treatment with MEK inhibitor only slightly decreased the rate of cell migration compared to the control condition, especially in normoxia. An even slighter effect was observed when we used both the inhibitor and NaHS (Figure 6(b)).
Figure 6.
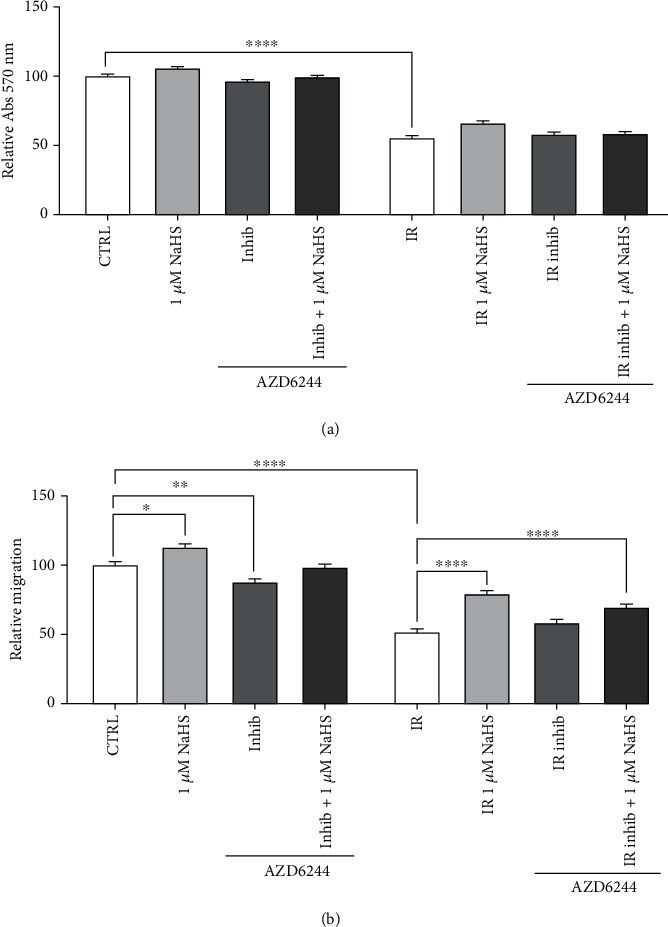
(a) Cell viability after MEK inhibition (1 μM AZD6244) and 1 μM NaHS preconditioning. (b) Cell migration after MEK inhibition (1 μM AZD6244) and 1 μM NaHS preconditioning. Statistical significance: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001.
After evaluating the effects on upstream inhibition, we decided to investigate whether NaHS preconditioning could rescue the biological effects after direct ERK1/2 inhibition.
Again, we assessed the inhibition on both cell viability and cell migration, which are two key pathways in which ERK1/2 is strongly involved.
No relevant changes were observed in IR condition, but a significant decrease in cell viability was observed in normoxia when cells were treated with both ERK inhibitor and NaHS (Figure 7(a)).
Figure 7.
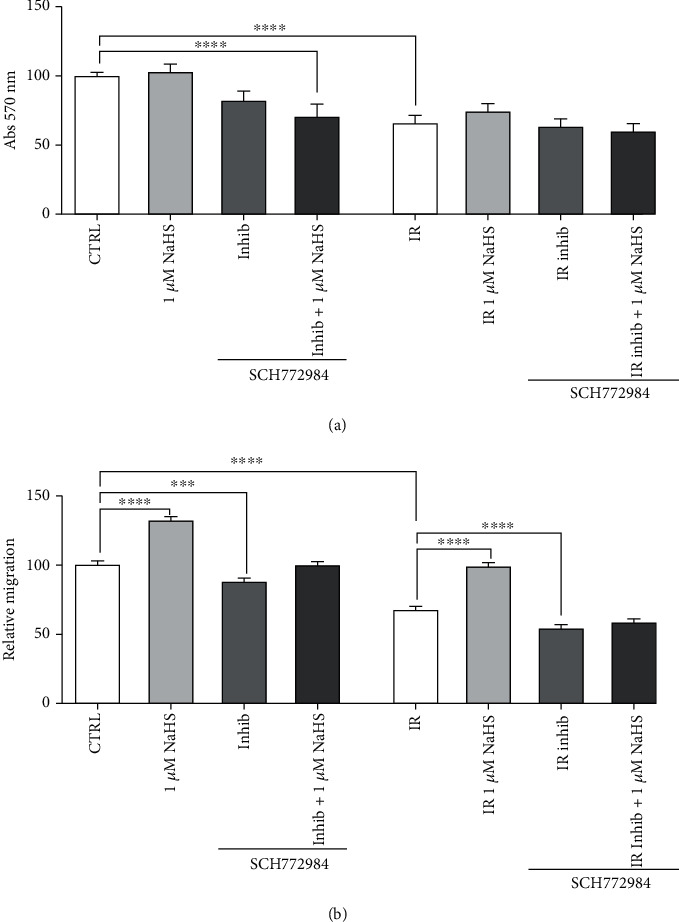
(a) Normalized viability after direct ERK1/2 inhibition (1 μM SCH772984) and 1 μM NaHS preconditioning. (b) Cell migration on 1 μM NaHS preconditioned cells after ERK1/2 inhibition (1 μM SCH772984). Statistical significance: ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Focusing on cell migration, in normoxia, ERK inhibition caused a significant decrease in cell migration, but NaHS treatment was able to rescue this inhibition, restoring the migration (Figure 7(b)). However, in IR, the direct inhibition of ERK actually caused a very strong decrease in cell migration when compared to the IR untreated cells, which could not be rescued by NaHS treatment (Figure 7(b)).
4. Discussion
Cardiovascular diseases are the leading cause of death worldwide. In 2016, they represented up to 30% of all global deaths [56, 57]. CVDs include different clinical conditions, most of which are associated to a pathological condition known as ischemia/reperfusion injury, such as myocardial infarction and stroke [58–61]. Whether this phenomenon affects the clinical outcome, it depends on the site, the duration, and the severity of the ischemic event itself. Nevertheless, postischemic reperfusion could exacerbate the damage both locally and systemically. This paradox relies on an increase in inflammatory response and, later on, to a profound tissue injury [2]. During an ischemic insult, moreover, there are several biochemical and cellular changes caused by the rapid depletion of oxygen and nutrients that cells cannot bear altogether with exacerbated effects during the reperfusion phase [62–64].
Endothelial integrity is pivotal for a proper vascularization which is, in turn, a key step for restoration of the physiological state as demonstrated by the several complications for patients undergoing incomplete cardiac revascularization [65, 66]. In this regard, it is reported that endothelial cells are very sensitive to IRI, even more than other cell types [20, 22, 27]. In addition, they are critical mediators for the onset of the inflammatory response, the generation of ROS, and the rapid restoration of the physiological pH.
In the ischemic myocardium, these events negatively affect the surrounding tissue, exacerbating the reperfusion injury and worsening the outcomes [2, 67].
A considerable part of literature about IRI has its focus on cardiac cells, while a small amount of data is available about endothelial cells, despite their importance in this context. Considering the role of ECs in both injury and recovery after the ischemic insult, the role of endothelial function in the hypoxic/ischemic scenario is of scientific relevance to ameliorate tissue recovery.
To this end, the action of hydrogen sulfide as a protective agent on endothelial cells against hypoxic/ischemic stress is promising because of the recent description of its beneficial effects in similar settings [68, 69]. H2S is known to act as a secondary messenger [70, 71], and there is evidence that it can be beneficial in IRI in several experimental models [72, 73]; hence, its potential as endogenous and exogenous therapeutic agent is gaining scientific attention. In order to study the effects solely on the endothelium, we decided to use an in vitro model of human microvascular endothelial cells to test the direct effects of hydrogen sulfide as a preconditioning agent, by using the inorganic donor NaHS. The aim was to recover the physiological state after the ischemic event. In particular, we expected to trigger a response in endothelial cells that would last until the end of the ischemic event, although NaHS is known to be rapidly hydrolyzed in water, establishing equilibrium between H2S and S2−+HS− species [74].
The preconditioning phenomenon can play an important role in the attenuation of ischemic consequences as demonstrated by the modulation of angiogenic activity of ECs. We verified this hypothesis by addressing the effects of H2S on cell migration and the ability to create capillary-like structures in vitro. Data showed that 1 μM NaHS pretreatment in cells that underwent IR protocol was able to rescue the defect in the migration rate, restoring the properties observed in the control-normoxic condition. This finding suggests a potential role of the gasotransmitter in promoting tissue repair following IRI.
There is still open debate about the actual in vivo levels of endogenous hydrogen sulfide. In order to test different conditions, the comparison of 2 putative concentrations (1 and 10 μM) can help decipherer the biological effects of constitutive deficiency. Low levels, 1 μM NaHS, restored the physiological condition in cell migration, whereas 10 μM gave a less intense increase in consistence with previously described bimodal activities shown in other cell types [44]. These data contribute to the hypothesis that endogenous and exogenous H2S, and their respective levels, might have opposite effects on some important cellular functions.
Goubern et al. suggest that exogenous H2S could function as an electron donor and as a potential inorganic energy source in mammalian cells at low micromolar concentrations [75]. The described mitoprotectant behavior could involve mitochondrial biogenesis and dynamics, as demonstrated in an endothelial model of rat aortic endothelial cells (RAEC) in which exogenous H2S exerts antifission and promitophagy effects when compared to the control in which endogenous H2S is modulated [76]. An enhanced mitophagy could explain diminished mitochondrial content in preconditioned I/R cells compared to the untreated control (Figure 4).
Despite the decrease in the mitochondrial mass, the ratio of functional mitochondria was enhanced in cells pretreated with NaHS. These data could be explained by an H2S-mediated enhancement of mitochondrial function and its suggested role of ROS scavenger.
The next step was to understand whether a 24-hour preconditioning could trigger a modulation of ERK phosphorylation, because of the role that the MEK/ERK pathway has in endothelial cell migration, other than apoptosis. Most interestingly, IR alone significantly enhanced ERK1/2 phosphorylation, while 1 μM NaHS treatment managed to drastically reduce this effect, going towards a more physiological (CTRL) phosphorylation state. On the other hand, the treatment did not affect ERK1/2 phosphorylation in normoxic cells.
Last, since it appeared that ERK was modulated by the NaHS treatment, we inhibited both ERK1/2 and its upstream activator, MEK, with the selective inhibitors AZD6244 and SCH772984. After testing the effects on cell viability and cell migration, we observed that the direct inhibition of ERK, but not MEK, decreased cell migration and could not be rescued by H2S treatment, thus indicating that H2S modulates the ERK pathway acting at the level of ERK phosphorylation.
Further experiments are needed to better describe the involvement of this pathway activated 24 hours ahead of the ischemic insult and how it is sustained over time. Moreover, it might be interesting to see whether the same concentrations could trigger similar effects in the postconditioning window and what similarities the pre- and postconditioning scenarios might have in common.
5. Conclusion
In our study, we showed that, despite its rapid metabolization, H2S could be used as a long-term preconditioning agent to ameliorate the effects of IRI. Moreover, this work is aimed at investigating the role of H2S as a modulator of an ERK1/2-dependent pathway, thus suggesting an important role for microvascular endothelial cells to participate in the response to ischemia/reperfusion injury. A deeper comprehension of endothelial cells in this context can contribute to design better strategies to preserve and repair tissues after a hypoxic/ischemic insult.
Acknowledgments
The authors would like to thank the constant support given by Dr. Tullio Genova and the Laboratory of Cellular and Molecular Angiogenesis, led by Prof. Luca Munaron (University of Turin). We would also like to thank Prof. Luca Primo and his group (IRCCS Institute, Candiolo—University of Turin) for their support. This research was funded by “Fondo Finanziamento delle Attività Base di Ricerca” and “Fondo Ricerca Locale” 2018, 2019, and 2020.
Data Availability
All the data used to support the findings of this study are included within the article.
Conflicts of Interest
The authors declare no conflict of interest.
Authors' Contributions
E.Z. was responsible for conceptualization; E.Z. was responsible for methodology; E.Z. and E.A. were responsible for software; E.Z. and E.A. were responsible for validation; E.Z. and E.A. were responsible for formal analysis; E.Z. and D.M. were responsible for investigation; E.Z. and E.A. were responsible for data curation; E.Z. and E.A. were responsible for writing—original draft preparation; E.Z., E.A., and D.M. were responsible for writing—review and editing; D.M. was responsible for supervision; D.M. was responsible for funding acquisition. All authors have read and agreed to the published version of the manuscript.
References
- 1.Virani S. S., Alonso A., Benjamin E. J., et al. Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation. 2020;141(9):e139–e596. doi: 10.1161/CIR.0000000000000757. [DOI] [PubMed] [Google Scholar]
- 2.Yellon D. M., Hausenloy D. J. Myocardial reperfusion injury. The New England Journal of Medicine. 2007;357(11):1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 3.Yang Q., He G. W., Underwood M. J., Yu C. M. Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: perspectives and implications for postischemic myocardial protection. American Journal of Translational Research. 2016;8(2):765–777. [PMC free article] [PubMed] [Google Scholar]
- 4.Kiss A., Heber S., Kramer A.-M., et al. MicroRNA expression profile changes after cardiopulmonary bypass and ischemia/reperfusion-injury in a porcine model of cardioplegic arrest. Diagnostics. 2020;10(4):p. 240. doi: 10.3390/diagnostics10040240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bassenge E. Endothelial function in different organs. Progress in Cardiovascular Diseases. 1996;39(3):209–228. doi: 10.1016/S0033-0620(96)80002-8. [DOI] [PubMed] [Google Scholar]
- 6.Davies P. F. Flow-mediated endothelial mechanotransduction. Physiological Reviews. 1995;75(3):519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakamura Y., Saito S., Miyagawa S., et al. Perioperative ischaemic reperfusion injury and allograft function in the early post-transplantation period. Interactive Cardiovascular and Thoracic Surgery. 2019;29(2):230–236. doi: 10.1093/ICVTS/IVZ086. [DOI] [PubMed] [Google Scholar]
- 8.Turer A. T., Hill J. A. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. The American Journal of Cardiology. 2010;106(3):360–368. doi: 10.1016/j.amjcard.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soares R. O. S., Losada D. M., Jordani M. C., Évora P., Castro-E-Silva O. Ischemia/reperfusion injury revisited: an overview of the latest pharmacological strategies. International Journal of Molecular Sciences. 2019;20(20):p. 5034. doi: 10.3390/ijms20205034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piper H. M., García-Dorado D., Ovize M. A fresh look at reperfusion injury. Cardiovascular Research. 1998;38(2):291–300. doi: 10.1016/S0008-6363(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 11.Griffith T. M., Edwards D. H., Lewis M. J., Newby A. C., Henderson A. H. The nature of endothelium-derived vascular relaxant factor. Nature. 1984;308(5960):645–647. doi: 10.1038/308645a0. [DOI] [PubMed] [Google Scholar]
- 12.Ignarro L. J. Biological actions and properties of endothelium-derived nitric oxide formed and released from artery and vein. Circulation Research. 1989;65(1):1–21. doi: 10.1161/01.RES.65.1.1. [DOI] [PubMed] [Google Scholar]
- 13.Moncada S., Palmer R. M., Higgs E. A. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacological Reviews. 1991;43(2):109–142. [PubMed] [Google Scholar]
- 14.Pober J. S., Cotran R. S. Cytokines and endothelial cell biology. Physiological Reviews. 1990;70(2):427–451. doi: 10.1152/physrev.1990.70.2.427. [DOI] [PubMed] [Google Scholar]
- 15.Vane J. R. The Croonian Lecture, 1993. The endothelium: maestro of the blood circulation. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences. 1994;343(1304):225–246. doi: 10.1098/rstb.1994.0023. [DOI] [PubMed] [Google Scholar]
- 16.Yanagisawa M., Kurihara H., Kimura S., et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332(6163):411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 17.Tsao P. S., Aoki N., Lefer D. J., Johnson G., Lefer A. M. Time course of endothelial dysfunction and myocardial injury during myocardial ischemia and reperfusion in the cat. Circulation. 1990;82(4):1402–1412. doi: 10.1161/01.CIR.82.4.1402. [DOI] [PubMed] [Google Scholar]
- 18.Lefer A. M., Tsao P. S., Lefer D. J., Ma X. L. Role of endothelial dysfunction in the pathogenesis of reperfusion injury after myocardial ischemia. The FASEB Journal. 1991;5(7):2029–2034. doi: 10.1096/fasebj.5.7.2010056. [DOI] [PubMed] [Google Scholar]
- 19.Richard V., Kaeffer N., Tron C., Thuillez C. Ischemic preconditioning protects against coronary endothelial dysfunction induced by ischemia and reperfusion. Circulation. 1994;89(3):1254–1261. doi: 10.1161/01.CIR.89.3.1254. [DOI] [PubMed] [Google Scholar]
- 20.Singhal A. K., Symons J. D., Boudina S., Jaishy B., Shiu Y.-T. Role of endothelial cells in myocardial ischemia-reperfusion injury. Vascular Disease Prevention. 2010;7(1):1–14. doi: 10.2174/1874120701007010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hausenloy D. J., Yellon D. M. Ischaemic conditioning and reperfusion injury. Nature Reviews. Cardiology. 2016;13(4):193–209. doi: 10.1038/nrcardio.2016.5. [DOI] [PubMed] [Google Scholar]
- 22.Hernández-reséndiz S., Muñoz-vega M., Contreras W. E., Crespo G. E. Responses of endothelial cells towards ischemic conditioning following acute myocardial infarction. Conditioning Medicine. 2018;1:247–258. [PMC free article] [PubMed] [Google Scholar]
- 23.Hofmann J., Otarashvili G., Meszaros A., et al. Restoring mitochondrial function while avoiding redox stress: the key to preventing ischemia/reperfusion injury in machine perfused liver grafts? International Journal of Molecular Sciences. 2020;21(9):p. 3132. doi: 10.3390/ijms21093132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yadav M., Kumari P., Yadav V., Kumar S. Pharmacological preconditioning with phosphodiestrase inhibitor: an answer to stem cell survival against ischemic injury through JAK/STAT signaling. Heart Failure Reviews. 2020;25(2):355–366. doi: 10.1007/s10741-019-09822-0. [DOI] [PubMed] [Google Scholar]
- 25.Rocca C., Femminò S., Aquila G., et al. Notch1 mediates preconditioning protection induced by GPER in normotensive and hypertensive female rat hearts. Frontiers in Physiology. 2018;9:p. 521. doi: 10.3389/fphys.2018.00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brutsaert D. L. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiological Reviews. 2003;83(1):59–115. doi: 10.1152/physrev.00017.2002. [DOI] [PubMed] [Google Scholar]
- 27.Yu H., Kalogeris T., Korthuis R. J. Reactive species-induced microvascular dysfunction in ischemia/reperfusion. Free Radical Biology & Medicine. 2019;135:182–197. doi: 10.1016/j.freeradbiomed.2019.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Laan A. M., Piek J. J., van Royen N. Targeting angiogenesis to restore the microcirculation after reperfused MI. Nature Reviews. Cardiology. 2009;6(8):515–523. doi: 10.1038/nrcardio.2009.103. [DOI] [PubMed] [Google Scholar]
- 29.Shah A. M., Mann D. L. In search of new therapeutic targets and strategies for heart failure: recent advances in basic science. The Lancet. 2011;378(9792):704–712. doi: 10.1016/S0140-6736(11)60894-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carmeliet P., Ng Y.-S., Nuyens D., et al. Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nature Medicine. 1999;5(5):495–502. doi: 10.1038/8379. [DOI] [PubMed] [Google Scholar]
- 31.Di Napoli P., Di Crecchio A., Contegiacomo G., et al. Endothelial protective effect of verapamil against acute myocardial contractile dysfunction in isolated working rat hearts subjected to global ischemia. Annals of the New York Academy of Sciences. 1998;853(1 CARDIAC SARCO):311–315. doi: 10.1111/j.1749-6632.1998.tb08287.x. [DOI] [PubMed] [Google Scholar]
- 32.Zhang X., Bian J.-S. Hydrogen sulfide: a neuromodulator and neuroprotectant in the central nervous system. ACS Chemical Neuroscience. 2014;5(10):876–883. doi: 10.1021/cn500185g. [DOI] [PubMed] [Google Scholar]
- 33.Kimura H. Hydrogen sulfide: its production, release and functions. Vol. 41. Springer Vienna; 2011. [DOI] [PubMed] [Google Scholar]
- 34.Lobov G. I., Sokolova I. B., Gorshkova O. P., Shvetsova M. E., Dvoretskii D. P. Contribution of hydrogen sulfide to dilation of rat cerebral arteries after ischemia/reperfusion injury. Bulletin of Experimental Biology and Medicine. 2020;168(5):597–601. doi: 10.1007/s10517-020-04759-z. [DOI] [PubMed] [Google Scholar]
- 35.Zhang L., Wang Y., Li Y., et al. Hydrogen sulfide (H2S)-releasing compounds: therapeutic potential in cardiovascular diseases. Frontiers in Pharmacology. 2018;9 doi: 10.3389/fphar.2018.01066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geng B., Yang J., Qi Y., et al. H2S generated by heart in rat and its effects on cardiac function. Biochemical and Biophysical Research Communications. 2004;313(2):362–368. doi: 10.1016/J.BBRC.2003.11.130. [DOI] [PubMed] [Google Scholar]
- 37.Elrod J. W., Calvert J. W., Morrison J., et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proceedings of the National Academy of Sciences. 2007;104(39):15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao L.-L., Huang X.-W., Wang Y.-G., Cao Y.-X., Zhang C.-C., Zhu Y.-C. Hydrogen sulfide protects cardiomyocytes from hypoxia/reoxygenation-induced apoptosis by preventing GSK-3β-dependent opening of mPTP. American Journal of Physiology-Heart and Circulatory Physiology. 2010;298(5):H1310–H1319. doi: 10.1152/ajpheart.00339.2009. [DOI] [PubMed] [Google Scholar]
- 39.Hausenloy D., Yellon D. M. New directions for protecting the heart against ischaemia–reperfusion injury: targeting the reperfusion injury salvage kinase (RISK)-pathway. Cardiovascular Research. 2004;61(3):448–460. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 40.Yellon D. M., Hausenloy D. J. Realizing the clinical potential of ischemic preconditioning and postconditioning. Nature Clinical Practice. Cardiovascular Medicine. 2005;2(11):568–575. doi: 10.1038/ncpcardio0346. [DOI] [PubMed] [Google Scholar]
- 41.Rossello X., Yellon D. M. The RISK pathway and beyond. Basic Research in Cardiology. 2018;113(1):p. 2. doi: 10.1007/s00395-017-0662-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu Y., Chen X., Pan T. T., et al. Cardioprotection induced by hydrogen sulfide preconditioning involves activation of ERK and PI3K/Akt pathways. Pflügers Archiv - European Journal of Physiology. 2007;455(4):607–616. doi: 10.1007/s00424-007-0321-4. [DOI] [PubMed] [Google Scholar]
- 43.Módis K., Coletta C., Erdélyi K., Papapetropoulos A., Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. The FASEB Journal. 2013;27(2):601–611. doi: 10.1096/fj.12-216507. [DOI] [PubMed] [Google Scholar]
- 44.Pupo E., Fiorio Pla A., Avanzato D., et al. Hydrogen sulfide promotes calcium signals and migration in tumor-derived endothelial cells. Free Radical Biology & Medicine. 2011;51(9):1765–1773. doi: 10.1016/j.freeradbiomed.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 45.Kolluru G. K., Shen X., Kevil C. G. A tale of two gases: NO and H2S, foes or friends for life? Redox Biology. 2013;1(1):313–318. doi: 10.1016/j.redox.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun H. J., Wu Z. Y., Nie X. W., Bian J. S. Role of endothelial dysfunction in cardiovascular diseases: the link between inflammation and hydrogen sulfide. Frontiers in Pharmacology. 2020;10 doi: 10.3389/fphar.2019.01568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katsouda A., Bibli S. I., Pyriochou A., Szabo C., Papapetropoulos A. Regulation and role of endogenously produced hydrogen sulfide in angiogenesis. Pharmacological Research. 2016;113(Part A):175–185. doi: 10.1016/j.phrs.2016.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin F., Yang Y., Wei S., et al. Hydrogen sulfide protects against high glucose-induced human umbilical vein endothelial cell injury through activating PI3K/Akt/eNOS pathway. Drug Design, Development and Therapy. 2020;14:621–633. doi: 10.2147/DDDT.S242521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banu S. A., Ravindran S., Kurian G. A. Hydrogen sulfide post-conditioning preserves interfibrillar mitochondria of rat heart during ischemia reperfusion injury. Cell Stress & Chaperones. 2016;21(4):571–582. doi: 10.1007/s12192-016-0682-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia N. A., Moncayo-Arlandi J., Vazquez A., et al. Hydrogen sulfide improves cardiomyocyte function in a cardiac arrest model. Annals of Transplantation. 2017;22:285–295. doi: 10.12659/aot.901410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boyman L., Williams G. S. B., Wescott A. P., Leach J. B., Kao J. P. Y., Lederer W. J. Real-time local oxygen measurements for high resolution cellular imaging. Journal of Molecular and Cellular Cardiology. 2019;127:97–104. doi: 10.1016/j.yjmcc.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burgess A., Vigneron S., Brioudes E., Labbé J. C., Lorca T., Castro A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(28):12564–12569. doi: 10.1073/pnas.0914191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Penna C., Mancardi D., Raimondo S., Geuna S., Pagliaro P. The paradigm of postconditioning to protect the heart. Journal of Cellular and Molecular Medicine. 2008;12(2):435–458. doi: 10.1111/j.1582-4934.2007.00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takagi H., Matsui Y., Sadoshima J. The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxidants & Redox Signaling. 2007;9(9):1373–1382. doi: 10.1089/ars.2007.1689. [DOI] [PubMed] [Google Scholar]
- 55.Vinten-Johansen J., Granfeldt A., Mykytenko J., Undyala V. V., Dong Y., Przyklenk K. The multidimensional physiological responses to postconditioning. Antioxidants & Redox Signaling. 2011;14(5):791–810. doi: 10.1089/ars.2010.3396. [DOI] [PubMed] [Google Scholar]
- 56.Hausenloy D. J., Boston-Griffiths E., Yellon D. M. Cardioprotection during cardiac surgery. Cardiovascular Research. 2012;94(2):253–265. doi: 10.1093/cvr/cvs131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nowbar A. N., Gitto M., Howard J. P., Francis D. P., Al-Lamee R. Mortality from ischemic heart disease. Circulation. Cardiovascular Quality and Outcomes. 2019;12(6):p. e005375. doi: 10.1161/CIRCOUTCOMES.118.005375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carden D. L., Granger D. N. Pathophysiology of ischaemia-reperfusion injury. The Journal of Pathology. 2000;190(3):255–266. doi: 10.1002/(SICI)1096-9896(200002)190:3<255::AID-PATH526>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 59.Perrelli M.-G. Ischemia/reperfusion injury and cardioprotective mechanisms: role of mitochondria and reactive oxygen species. World Journal of Cardiology. 2011;3(6):186–200. doi: 10.4330/wjc.v3.i6.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang D., Xie P., Liu Z. Ischemia/reperfusion-induced MKP-3 impairs endothelial NO formation via inactivation of ERK1/2 pathway. PLoS One. 2012;7(7):p. e42076. doi: 10.1371/journal.pone.0042076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu M.-Y., Yiang G.-T., Liao W.-T., et al. Current mechanistic concepts in ischemia and reperfusion injury. Cellular Physiology and Biochemistry. 2018;46(4):1650–1667. doi: 10.1159/000489241. [DOI] [PubMed] [Google Scholar]
- 62.Szocs K. Endothelial dysfunction and reactive oxygen species production in ischemia/reperfusion and nitrate tolerance. General Physiology and Biophysics. 2004;23(3):265–295. [PubMed] [Google Scholar]
- 63.Dymkowska D., Drabarek B., Podszywałow-Bartnicka P., Szczepanowska J., Zabłocki K. Hyperglycaemia modifies energy metabolism and reactive oxygen species formation in endothelial cells in vitro. Archives of Biochemistry and Biophysics. 2014;542:7–13. doi: 10.1016/J.ABB.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 64.Essick E. E., Sam F. Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxidative Medicine and Cellular Longevity. 2010;3(3):p. 177. doi: 10.4161/oxim.3.3.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamazaki S., Fujibayashi Y., Rajagopalan R. E., Meerbaum S., Corday E. Effects of staged versus sudden reperfusion after acute coronary occlusion in the dog. Journal of the American College of Cardiology. 1986;7(3):564–572. doi: 10.1016/S0735-1097(86)80466-1. [DOI] [PubMed] [Google Scholar]
- 66.Tong G., Zhang B., Zhou X., et al. Kappa-opioid agonist U50,488H-mediated protection against heart failure following myocardial ischemia/reperfusion: dual roles of heme oxygenase-1. Cellular Physiology and Biochemistry. 2016;39(6):2158–2172. doi: 10.1159/000447911. [DOI] [PubMed] [Google Scholar]
- 67.Hausenloy D. J., Yellon D. M. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. Journal of Clinical Investigation. 2013;123(1):92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Papapetropoulos A., Pyriochou A., Altaany Z., et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(51):21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang M.-J., Cai W.-J., Li N., Ding Y.-J., Chen Y., Zhu Y.-C. The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxidants & Redox Signaling. 2010;12(9):1065–1077. doi: 10.1089/ars.2009.2945. [DOI] [PubMed] [Google Scholar]
- 70.Wang R. Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? The FASEB Journal. 2002;16(13):1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 71.Andreadou I., Iliodromitis E. K., Rassaf T., Schulz R., Papapetropoulos A., Ferdinandy P. The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. British Journal of Pharmacology. 2015;172(6):1587–1606. doi: 10.1111/bph.12811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mancardi D., Florio Pla A., Moccia F., Tanzi F., Munaron L. Old and new gasotransmitters in the cardiovascular system: focus on the role of nitric oxide and hydrogen sulfide in endothelial cells and cardiomyocytes. Current Pharmaceutical Biotechnology. 2011;12(9):1406–1415. doi: 10.2174/138920111798281090. [DOI] [PubMed] [Google Scholar]
- 73.Mancardi D., Penna C., Merlino A., Del Soldato P., Wink D. A., Pagliaro P. Physiological and pharmacological features of the novel gasotransmitter: hydrogen sulfide. Biochimica et Biophysica Acta. 2009;1787(7):864–872. doi: 10.1016/j.bbabio.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hughes M. N., Centelles M. N., Moore K. P. Making and working with hydrogen sulfide: Free Radical Biology and Medicine. 2009;47(10):1346–1353. doi: 10.1016/j.freeradbiomed.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 75.Goubern M., Andriamihaja M., Nübel T., Blachier F., Bouillaud F. Sulfide, the first inorganic substrate for human cells. The FASEB Journal. 2007;21(8):1699–1706. doi: 10.1096/fj.06-7407com. [DOI] [PubMed] [Google Scholar]
- 76.Liu N., Wu J., Zhang L., et al. Hydrogen sulphide modulating mitochondrial morphology to promote mitophagy in endothelial cells under high-glucose and high-palmitate. Journal of Cellular and Molecular Medicine. 2017;21(12):3190–3203. doi: 10.1111/jcmm.13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data used to support the findings of this study are included within the article.