

Abstract
Socioeconomic status (SES) and education (EDU) are phenotypically associated with psychiatric disorders and behaviors. It remains unclear how these associations influence genetic risk for psychopathology, psychosocial factors, and EDU/SES individually. Using information from >1 million individuals, we conditioned the genetic risk for psychiatric disorders, personality traits, brain imaging phenotypes, and externalizing behaviors with genome-wide data for EDU/SES. Accounting for EDU/SES significantly affected the observed heritability of psychiatric traits ranging from 2.44% h2 decrease for bipolar disorder to 14.2% h2 decrease for Tourette syndrome. Neuroticism h2 significantly increased by 20.23% after conditioning with SES. After EDU/SES conditioning, neuronal cell-types were identified for risky behavior (excitatory), major depression (inhibitory), schizophrenia (excitatory and GABAergic), and bipolar disorder (excitatory). Conditioning with EDU/SES also revealed unidirectional causality between brain morphology, psychopathology, and psychosocial factors. Our results indicate that genetic discoveries related to psychopathology and psychosocial factors may be limited by genetic overlap with EDU/SES.
Introduction
Education (EDU) and socioeconomic status (SES) are risk or protective factors for traits related to mental health and disease.1, 2 Social position has been repeatedly correlated with mood, anxiety, and substance use related disorders, while EDU phenotypes such as educational attainment, math ability, and fluid intelligence are overall protective factors for development of neurological and psychiatric conditions.2 Though highly correlated, the specific EDU and/or SES phenotypes used in epidemiological studies clearly account in part for the incidence of numerous health outcomes, including self-reported health, chronic conditions, and overall mortality.3 It is therefore imperative to understand how EDU and SES phenotypes influence what we understand about human health and disease.
Genome-wide association studies (GWAS) are powerful hypothesis-generating investigation for detecting risk loci with respect to phenotypes of interest. Their widespread use has led to risk locus discovery underlying thousands of phenotypes across the spectrum of human health, including mental and physical traits, personality, anthropometric measures, intelligence, and behaviors.4 An observation generated from large-scale GWAS is the widespread presence of pleiotropy; a single SNP (or a set of SNPs) may have a range of relatively small effects on multiple similar or disparate phenotypes. On a genome-wide scale, these pleiotropic effects, detected using GWAS summary data, may be used to determine genetic correlations between phenotypes to putatively identify genetic underpinnings of trait pairs.5
The EDU phenotypes educational attainment and cognitive performance have relatively high SNP-heritability: the phenotypic variance explained by genetic information was 40%6 and 21.5%,7 respectively. SES is defined as the social standing or class of an individual or group, often measured as a combination of education, income, and occupation.8 SES phenotypes such as household income and Townsend deprivation index (i.e., measure of SES based on whether individuals own their homes, their employment status, their access to a vehicle, and whether or not individuals share living accommodations with others) are significantly heritable and show strong genetic correlation with EDU traits.9 Additionally, there is pleiotropy of genetic risks between EDU/SES and a range of psychopathologies and psychosocial factors (e.g., psychiatric disorders, personality traits, internalizing and externalizing behaviors, social science outcomes, and brain imaging phenotypes).10, 11
The epidemiological observations of high genetic correlations between genetic risk for EDU/SES, psychopathology, and psychosocial factors1, 2 raise two critical questions: (1) how might genetic variants with strong effects on EDU/SES affect our understanding of the overall genetic risk for psychopathology and psychosocial factors? and (2) is there evidence that genetic variants associated with psychopathology and psychosocial factors affect our understanding of the overall genetic risk for EDU/SES? The goal of this study was to investigate how the shared genetic effects between the general categories of EDU, SES, psychopathology, and psychosocial factors influence genetic risk for individual phenotypes within each of these classes.
There are several ways to approach these questions such as polygenic risk scoring (PRS) or multi-trait analysis of GWAS (MTAG). PRS12 is an analytic approach by which a persons’ genetic information is used to derive a numerical description of their risk to develop a disorder or display a certain trait.13 PRS is a tempting approach to answer our question; but PRS using psychopathology and psychosocial factors to predict the same or different phenotypes from an independent dataset often explain very little variance in the outcome phenotype.14, 15, 16 MTAG analyzes the GWAS of several traits with the goal of boosting statistical power and the detection of genetic signal across those traits. MTAG adjusts per-SNP effect estimates and association p-values using the strength of the genetic correlation between phenotypes.17 Genetic correlations between EDU/SES and related phenotypes have, however, demonstrable biases from environmental confounders. If genetic correlations involving EDU and SES proxy phenotypes are significantly upwardly biased, an MTAG adjustment of summary statistics may inappropriately correct (i.e., bias) the summary statistics used for this study. To disentangle the complex genetic overlaps between EDU/SES, psychopathology, and psychosocial factors, we therefore used multi-trait conditioning and joint analysis (mtCOJO), which generates conditioned GWAS summary statistics for each phenotype of interest after correcting for the per-SNP effects of another phenotype.18 The mtCOJO approach is not based on genetic correlation, but leverages the causal relationship between trait pairs inferred by Mendelian randomization (MR). For our phenotypes of interest, mtCOJO is an advantageous approach, which, in theory, is independent of the effects of environmental confounders.19, 20 MR detects causal inferences between trait pairs using non-modifiable risk factors (SNPs) under the assumption that (1) SNPs are associated with an exposure variable, (2) SNPs are associated with an outcome variable only through the exposure, and (3) SNPs are not associated with confounders of the relationship between exposure and outcome. Because SNPs are non-modifiable, environmental confounders of the relationship between SNP, exposure, and outcome should not influence MR estimates.19, 20
We used the mtCOJO approach to condition psychopathology and psychosocial factors with the per-SNP effects of EDU and SES phenotypes and investigate their underlying biology of more than 125 GWAS at multiple levels: (1) risk locus detection, (2) heritability (h2), (3) gene-set enrichment, (4) tissue transcriptomic profile enrichment, (5) cell type transcriptomic profile enrichment, (6) phenotype relationships via structural equation modeling and genetic correlation, and (7) latent genetically causal relationships (see flow diagram Fig S1). Our findings identify several cell types and phenotype relationships that were masked by the shared genetic etiology between psychopathology, psychosocial factors, and EDU/SES. Furthermore, we demonstrate that the same multi-level analyses of EDU and SES are largely robust to the effects of shared genetic etiology with psychopathology and psychosocial factors.
Results
An overview of all analytic approaches and their results is shown in Fig. S1.
Trait Inclusion
The genetic correlations (rg) between EDU (educational attainment, cognitive performance, highest math class, and self-rated math ability), SES (household income and Townsend deprivation index), and psychopathology and psychosocial factors (i.e., psychiatric disorders, personality traits, externalizing behaviors, social science outcomes, and brain imaging phenotypes) were estimated using the Linkage Disequilibrium Score Regression (LDSC) method (Figs. 1 and S2, Tables S1–S4).21 Description of brain imaging phenotype selection is described in the Supplemental Material. Note that sample overlap between EDU/SES, psychopathology, and psychosocial factors was accounted for in conditioning experiments via incorporation of the sampling covariance estimated from GWAS summary statistics.18, 22
Fig. 1. Trait inclusion genetic correlations.
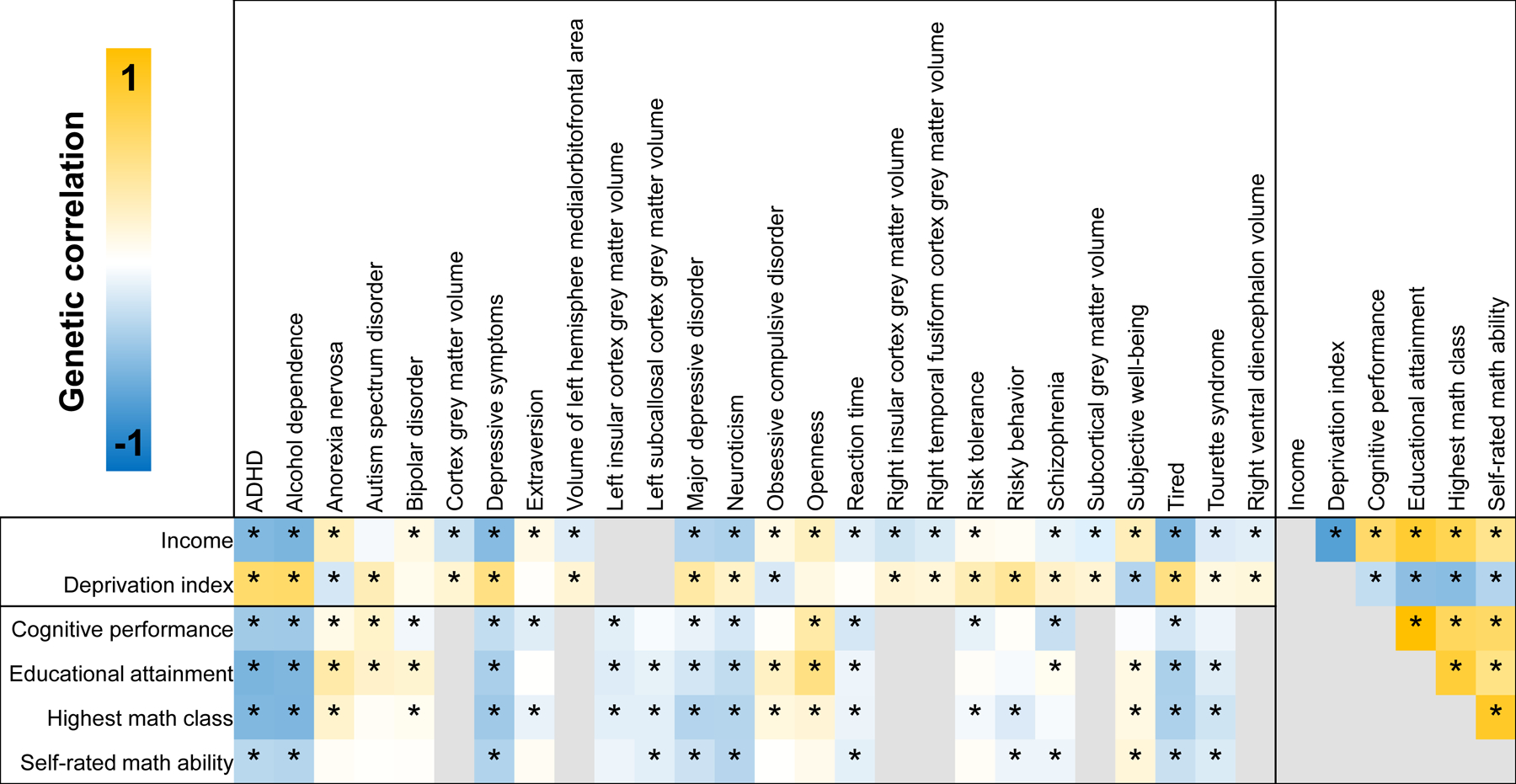
Genetic correlation between psychopathology, psychosocial factors, education phenotypes, and socioeconomic status phenotypes. Nominally significant genetic correlations were considered to select traits for inclusion in conditioning experiments (* = p<0.05).
Conditioning Heritability and Risk Locus Discovery
We tested the effects of conditioning on the observed-scale heritability estimates (h2) using LDSC.21 Except for major depressive disorder (MDD), anxiety, and posttraumatic stress disorder (PTSD), conditioning reduced the h2 for all psychiatric disorders relative to their original estimates. The decrease in h2 ranged from 2.44%±0.187 for bipolar disorder (original p=3.55×10−33, h2=4.39%, se=0.360; highest conditioned p=5.67×10−65, h2=2.22%, se=0.460; lowest conditioned p=4.05×10−80, h2=1.70%, se=0.440) to 29.0%±0.105 for Tourette syndrome (original p=6.56×10−98, h2=21.0%, se=2.52; highest conditioned p=2.61×10−18 h2=6.72%, se=0.770; lowest conditioned p=1.27×10−18, h2=6.43%, se=0.730; Figs. 2 and S3, Table S5). Tourette syndrome exhibited the largest decrease in h2 after conditioning with the effects of EDU/SES phenotypes (1.78×10−11<pdiff<3.02×10−11, mean h2 decrease compared to original Tourette syndrome = 0.144, se=0.001). Conversely, two phenotypes exhibited significant increases in h2 after conditioning with EDU/SES phenotypes: neuroticism (p=3.08×10−226, highest conditioned h2=20.2%, se=0.630; p=2.35×10−207, lowest conditioned h2=18.1%, se=0.590) and subjective well-being (p=8.11×10−62, highest conditioned h2=3.65%, se=0.220; p=4.67×10−52, lowest conditioned h2=3.34%, se=0.220).
Fig. 2. Heritability (h2) changes.
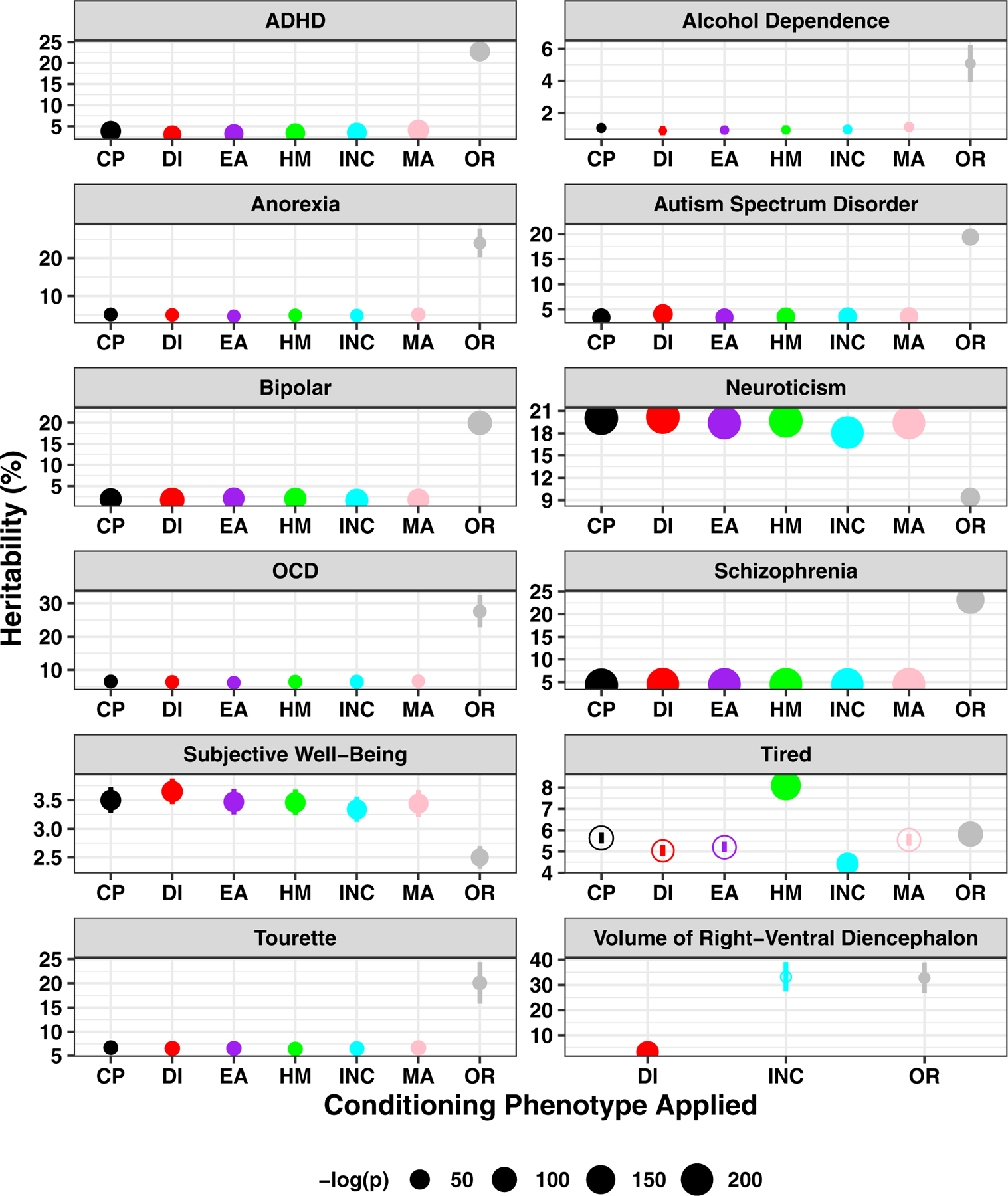
Each data point indicates an observed-scale h2 estimate and standard error of the original (OR; grey) indicated phenotype or that same phenotype after conditioning with education and socioeconomic status phenotypes (CP: cognitive performance, DI: deprivation index, EA: educational attainment, HM: highest math class, INC: income, MA: self-rated math ability). Two p-values are shown: (1) the h2 estimates of each conditioned phenotype was at least nominally significant (p<0.05) as demonstrated by the size of each data point, (2) solid data points indicate that the h2 estimate of a conditioned phenotype was significantly different from the original h2 estimate in grey. Phenotypes showing significant h2 changes are shown here while all phenotypes are presented in Fig S3.
Conditioning the neuroticism GWAS (p=3.88×10−46, original h2=9.41%, se=0.65) with EDU/SES phenotypes revealed several novel risk loci (ranging from 59 loci (neuroticism conditioned with income) to 100 loci (neuroticism conditioned with deprivation index; Fig. S4), increased h2 (1.94×10−32<p<5.27×10−28, mean h2 increase = 10.1%, se=0.747; Table S5), and confirmed known LD-independent risk loci. We observed an increase in the association signal in the neuroticism GWAS with the strongest effects observed after conditioning with SES phenotypes income (lambda GC=1.36; intercept=0.971, se=0.009) and deprivation index (lambda GC=1.75; intercept=0.967, se=0.009; Fig. S5 and Table S6). This increase was not related to an increase in the potential bias of population stratification (i.e., there was no significant change in the LDSC intercept, 0.884<pdiff<0.994), supporting that the observation was attributable to the increased detection of valid neuroticism polygenic signals. Using a physical proximity single-SNP-single-gene based annotation of conditioned neuroticism genomic risk loci (Table S7–S9), the top gene sets included Gene Ontology (GO) cellular component synapse (7.51×10−6<p<9.58×10−4, mean enrichment = 0.138, se=0.019), GO biological process long term synaptic potentiation (2.95×10−6<p<7.43×10−5, mean enrichment = 0.650, se=0.042), and GO cellular component synapse part (enrichment 2.46×10−5<p<0.001, mean enrichment = 0.144, se=0.017).
The significant increase in h2 for GWAS of subjective well-being (original p=7.47×10−36, h2=2.50%, se=0.20) uncovered a 5.7 kb genomic risk locus on chromosome 7 (minimum genome-wide significant p=1.45×10−8) which maps to the α2δ1 subunit of calcium voltage-gated channel (CACNA2D1, Tables S10–S12). The protein encoded by CACNA2D1 has been implicated in familial epilepsy and intellectual disability pedigrees but to our knowledge has not been implicated in genome-wide studies of these phenotypes.23, 24 The shared biology between neuroticism and subjective well-being and all other psychopathologies and psychosocial factors revealed similar results as those with EDU/SES phenotypes and are described in Supplementary Results (Figs. S3 and Tables S13–S17).
We next considered h2 estimates for psychopathology and psychosocial factors after conditioning in two additional experiments: (1) with latent factors representing EDU/SES phenotypes (excluding income, which failed to load on a latent factor; see Correlative, Latent, and Causal Relationships between Psychopathology and Psychosocial Factors) and (2) with all EDU/SES phenotypes simultaneously. All traits exhibited a reduction in SNP-h2 except for extraversion (p=3.47×10−41, h2=0.137, se=0.010 when conditioned with all EDU/SES phenotypes and p=6.32×10−4, h2=0.035, se=0.010 when conditioned with latent factors; Table S18). Though extraversion h2 increased, the conditioned GWAS resulted in no genome-wide significant findings.
Tissue-Type Transcriptomic Profile Enrichment Differences
After conditioning with GWAS of EDU/SES phenotypes (Table S19), schizophrenia was the only trait demonstrating significant changes in tissue-specific transcriptomic profile enrichment. Compared to the unconditioned schizophrenia GWAS, all conditioned schizophrenia brain tissue GTEx (Genotype-Tissue Expression)25 annotations, with the exception of c1 cervical spinal cord, had significantly decreased enrichments (Fig. S6 and Table S19). The maximum decrease was observed after conditioning schizophrenia with educational attainment (average beta decrease for all brain tissue annotations = 0.038, se=0.004).
After conditioning with EDU and SES phenotypes, the cerebellum and cerebellar hemisphere GTEx annotations remained the most enriched in the schizophrenia GWAS (original cerebellum p=1.76×10−22, enrichment=0.080, se=0.008; original cerebellar hemisphere p=1.28×10−22, enrichment=0.077, se=0.008; conditioned cerebellum 6.83×10−22<p<5.82×10−19, mean enrichment=0.047, se=0.001; conditioned cerebellar hemisphere 7.43×10−23<p<6.56×10−20, mean enrichment=0.047, se=0.001). After adjusting for the effects of cognitive performance and educational attainment, we uncovered enrichment of skeletal muscle tissue transcriptomic profiles in the schizophrenia GWAS (original skeletal muscle p=0.135, enrichment=0.010, se=0.009; skeletal muscle conditioned with educational attainment p=0.032, enrichment=0.010, se=0.006; skeletal muscle conditioned with cognitive performance p=0.024, enrichment=0.011, se=0.006).26 Though demonstrated in early studies of schizophrenia patients,27 contemporary studies are required to validate this enrichment.
Cell-Type Transcriptomic Profile Discoveries
Cell-type transcriptomic profile enrichments were evaluated in two ways: (1) assess differences in within-data-set cell-type enrichments before and after conditioning with EDU/SES (based on MAGMA cell-type enrichment Step 128) and (2) assess the effects of conditioning on the detection of conditionally independent proportional significance (PS) of the cell type enrichments (based on MAGMA cell-type enrichment Step 328). PS cell-types are those whose genetic signals could be differentiated from one another. PS values ≥ 0.80 indicate independent genetic signals relative to a second cell type. We then used genes whose expression profiles define the excitatory (Ex) and inhibitory (In) cell types of PsychENCODE29 to perform gene set enrichment analyses of GO and KEGG gene sets.
There were no statistically significant differences in cell-type transcriptomic profile enrichments for psychopathology and psychosocial factors (MAGMA cell-type Step 1) after conditioning with EDU/SES; however, we discovered several PS cell-type pairs not detected in the unconditioned GWAS for (1) risky behavior (Fig. 3 and Table S20), (2) MDD (Fig. 3 and Table S21), and (3) schizophrenia (Fig. S7 and Table S22; MAGMA cell-type Step 3).
Fig. 3. Cell-type transcriptomic profile enrichments underlying psychopathology and psychosocial factors.
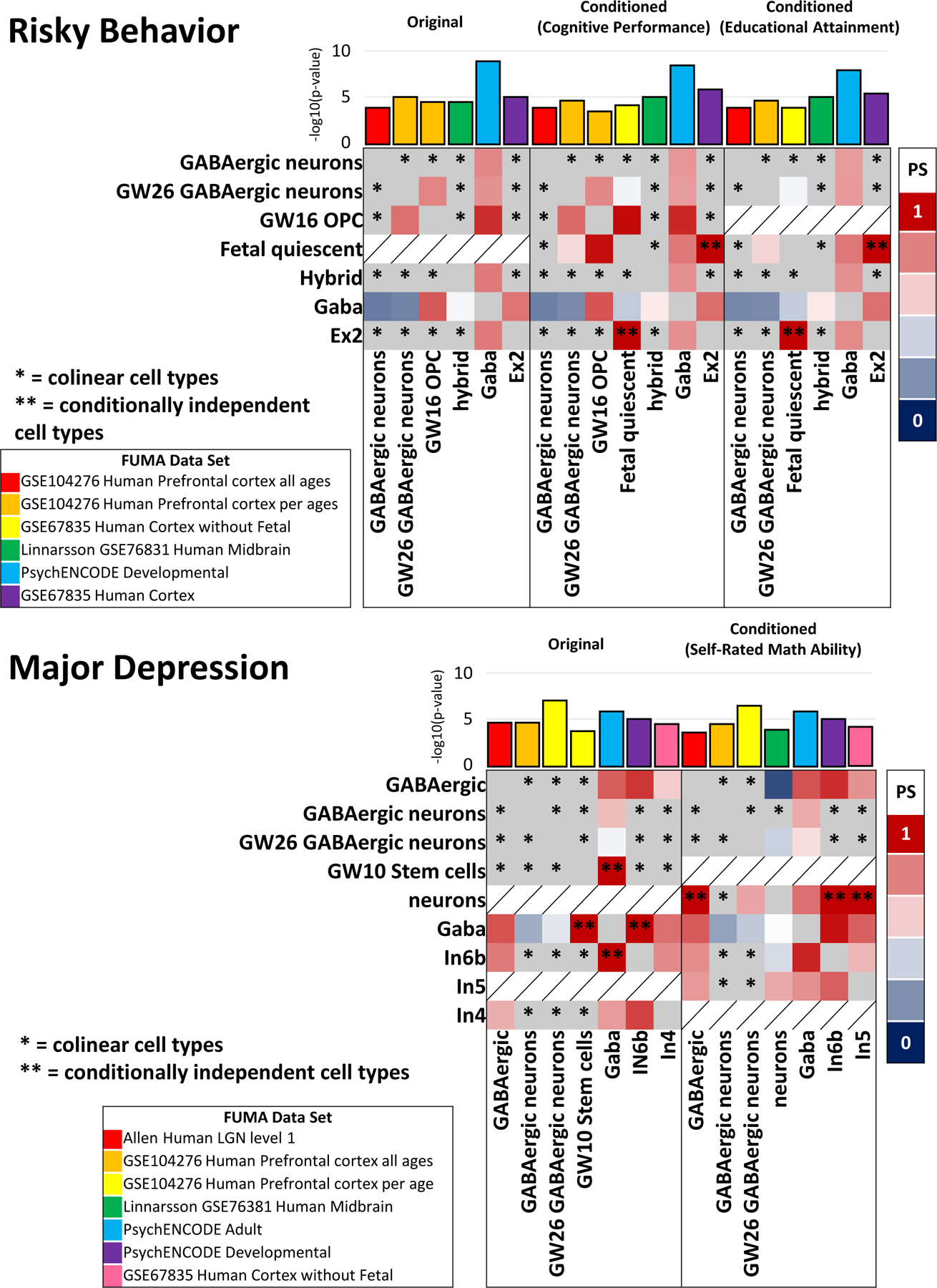
Cross-data-set proportional significance (PS) and conditional independent (i.e., genetic signatures of cell-type pairs are distinguishable) of cell-type transcriptomic profile enrichments underlying unconditioned and conditioned GWAS for (A) risky behavior and (B) major depression. The human cell-type data sets from FUMA are labeled individually for each panel using different colors; cell types in the x and y directions are conditionally independent signals from within-data-set analysis performed in FUMA (cell-type enrichment step 228). Genetic signals from colinear cell types labeled with a single asterisk could not be differentiated from one another in FUMA.
In unconditioned GWAS of risky behavior, there were no PS cell-type enrichments. After conditioning with EDU phenotypes, human cortex fetal quiescent and Ex2 were conditionally independent from one another (risky behavior conditioned with cognitive performance Ex2 p=7.48×10−4, β=0.035, se=0.011, PS=1.37; fetal quiescent p=0.032, β=0.023, se=0.012, PS=1.82; risky behavior conditioned with educational attainment Ex2 p=0.001, β=0.034, se=0.011, PS=1.38; fetal quiescent p=0.030, β=0.024, se=0.012, PS=1.77; (Fig. 3 and Table S20). The genes that define the Ex2 cell type were enriched in nervous system development (GO:0007399; enrichment FDR=3.70×10−4) and eye development (GO:001654; enrichment FDR=6.30×10−4) gene sets.
The unconditioned MDD GWAS exhibited cell-type transcriptomic profile enrichments between adult GABAergic neurons, In6b, and gestational week 10 (GW10) stem cells. After conditioning with self-rated math ability, the genetic signal from human midbrain neurons was conditionally independent from lateral geniculate nucleus (LGN) GABAergic neurons (p=0.002, β relative to midbrain neurons=0.041, se=0.014, PS=0.822; Fig 3), In6b neurons (p=6.59×10−6, β relative to midbrain neurons=0.517, se=0.11, PS=0.969), and In5 neurons (p=5.26×10−5, β relative to midbrain neurons=0.039, se=0.01, PS=0.813; (Fig. 3 and Table S21). The gene expression profiles of these cell types implicate the neurotransmitter transport (GO:0007269; enrichment FDR=0.003) and locomotory behavior (GO:0007626; enrichment FDR=0.015) gene sets in MDD psychopathology.
Correlative, Latent, and Causal Relationships between Psychopathology and Psychosocial Factors
We next evaluated relationships between phenotypes using three methods: genetic correlation, genomic structural equation modeling (genomic SEM), and latent causal variable (LCV) analysis.
Genetic correlations were assessed between all psychopathology and psychosocial factors after conditioning with individual EDU and SES phenotypes. Though small changes in genetic correlation magnitude were observed, psychopathology and psychosocial factor genetic correlations largely persisted (Fig. S8 and Table S24).
Genomic SEM30 was used to identify how unconditioned and conditioned psychopathology and psychosocial factors relate to a latent unobserved genetic factor connecting them (Fig. 4). In unconditioned models, exploratory factor analysis (EFA) identified a two-factor model as best suited to explain the relationships among psychopathology and psychosocial factors. In confirmatory factor analysis (CFA), these two latent factors generally highlight relationships between all psychiatric disorders and brain imaging phenotypes (F1) and anxiety, MDD, depressive symptoms, and neuroticism (F2). The correlation between unconditioned F1 and F2 was 0.14. After conditioning with highest math class, self-rated math ability, and deprivation index, the GWAS of neuroticism and MDD were no longer major contributors to the same factor. Conditioned F1 had major contributions from MDD (mean loading=0.611, se=0.005) and depressive symptoms (loading=0.538, se=0.098) while conditioned F2 had major contributions from neuroticism (loading=0.877, se=0.080) and anxiety (loading=0.658, se=0.009). Interestingly, after conditioning with the SES phenotype income, the SEM best-fit converged on a single common factor between all psychopathology and psychosocial factors with major contributions from MDD (loading=0.808, se=0.068) and depressive symptoms (loading=0.831, se=0.022).
Fig. 4. Trait loading onto latent factors.
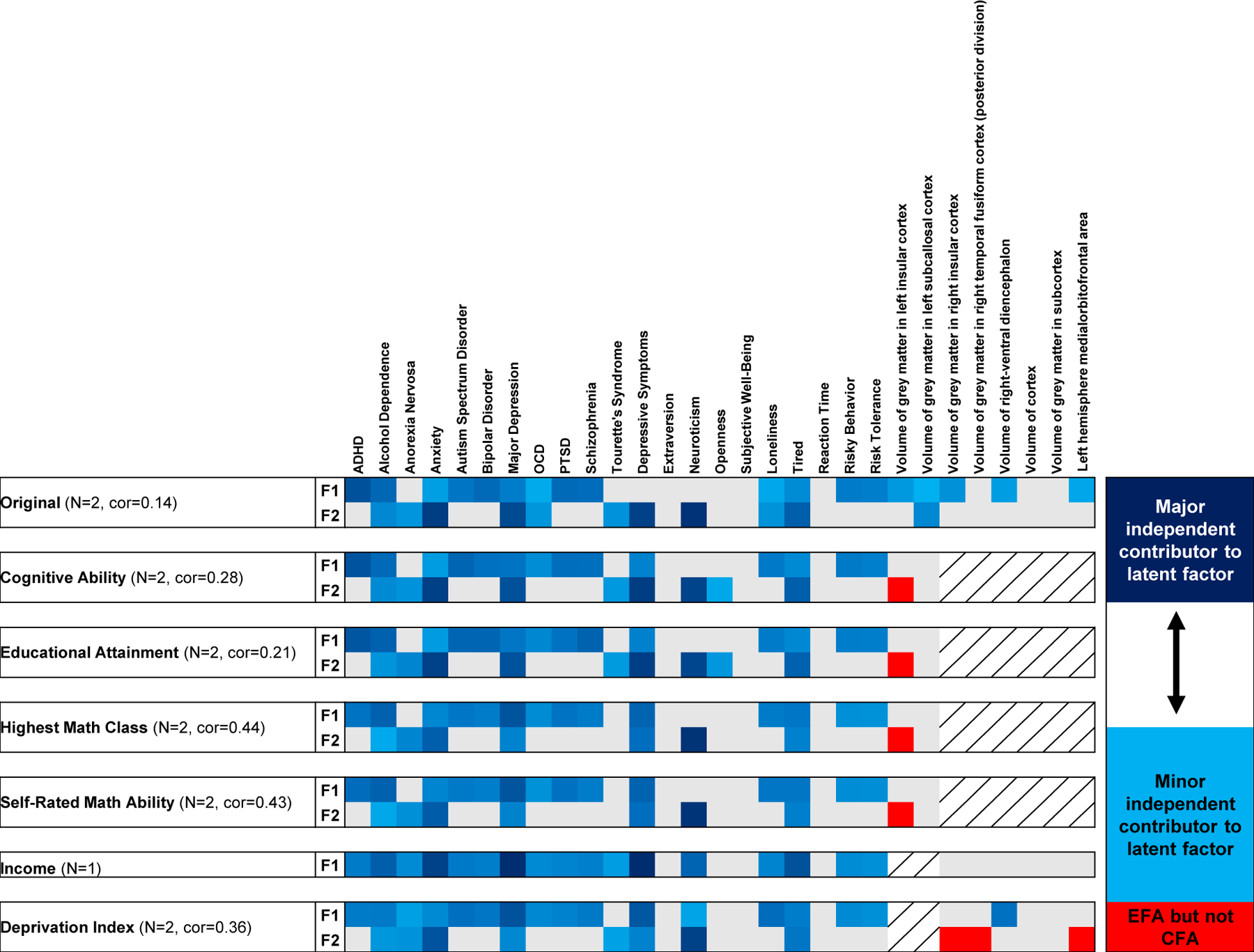
Genomic structural equation modeling of psychopathology and psychosocial factors before and after conditioning with education and socioeconomic status phenotypes. Each column shows the confirmatory factor analysis (CFA) loading value (blue shading indicating that a trait is a major contributor to the latent factor and blue tinting indicating that a trait is a minor independent contributor to the latent factor) for each psychopathology and psychosocial factors (in the x direction) into one of two factors (F1 and F2) from exploratory factor analysis (EFA). Grey boxes indicate that a given trait was not predicted to load onto a given factor column. Red boxes indicate that the trait was predicted by EFA to load onto a factor but did not independently load during CFA.
Latent Causal Variable (LCV) analyses were used to detect causal relationships between trait pairs that are independent of the genetic correlations between them.30 Considering only the unconditioned psychopathology and psychosocial factors, one trait pair exhibited significant genetic causality proportion (gĉp): left subcallosal cortex→obsessive compulsive disorder p=4.54×10−6, gĉp=0.167, se=0.047 (Tables 1 and S25 and Figs. 5 and 6). This partial causal relationship did not survive conditioning; however, thirteen unique trait pairs demonstrated significant gĉp after conditioning both traits with an EDU or SES phenotype (Table 1). Most notable were those causal relationships involving brain imaging phenotypes which became significant after conditioning with EDU phenotypes: (1) extraversion→left subcallosal cortex (1.23×10−13<p<1.83×10−6, mean gĉp=0.188, se=0.107) after conditioning with educational attainment, highest math class, and self-rated math ability, (2) left subcallosal cortex→subjective well-being (1.45×10−9<p<1.16×10−8, mean gĉp=0.745, se=0.009) after conditioning with cognitive performance, educational attainment, and highest math class, (3) openness→left insular cortex (2.54×10−23<p<3.63×10−8, mean gĉp=0.296 , se=0.050) after conditioning with cognitive performance, highest math class, and self-rated math ability. These average gĉp estimates represent only Bonferroni significant relationships; however, each trait pair listed was nominally significant after conditioning with all other EDU and SES phenotypes but not significant in the unconditioned experiment (Table 1).
Table 1. Causal inferences detected after multiple testing correction.
Significant causal relationships detected between psychopathology and psychosocial factors using latent causal variable analyses. For each significant causal estimate, all conditioned causal estimates between that phenotype pair are provided, highlighting at least nominally significant causal inferences after conditioning with education and socioeconomic status phenotypes that could not be detected in the original unconditioned trait pair.
| Trait 1 | Trait 2 | Conditioning | zscore | gcp.p | gcp.pm | gcp.pse | rho.est | rho.err | h2.zscore.1 | h2.zscore.2 | Notes |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Extraversion | Left Subcallosal Cortex | Original | 0.124 | 0.901 | −0.086 | 0.522 | −4.51E-05 | 0.038 | 7.772 | 12.423 | |
| Cognitive Performance | 3.626 | 4.57E-04 | −0.252 | 0.393 | 0.208 | 0.141 | 7.979 | 13.119 | |||
| Educational Attainment | 5.074 | 1.83E-06 | −0.165 | 0.378 | 0.201 | 0.141 | 7.742 | 13.207 | |||
| Highest Math Class | 8.599 | 1.23E-13 | −0.094 | 0.386 | 0.209 | 0.141 | 7.706 | 12.881 | |||
| Self-Rated Math Ability | 6.086 | 2.20E-08 | −0.304 | 0.326 | 0.19 | 0.13 | 6.92 | 11.845 | h2.zscore.1 too low | ||
| Left Subcallosal Cortex | Subjective Well-Being | Original | 0.336 | 0.737 | −0.004 | 0.434 | 0.036 | 0.029 | 6.894 | 20.441 | h2.zscore.1 too low |
| Cognitive Performance | 6.525 | 2.92E-09 | 0.746 | 0.168 | 0.28 | 0.091 | 7.365 | 25.776 | |||
| Educational Attainment | 6.227 | 1.16E-08 | 0.736 | 0.173 | 0.269 | 0.093 | 7.129 | 25.499 | |||
| Highest Math Class | 6.675 | 1.45E-09 | 0.753 | 0.164 | 0.265 | 0.09 | 7.04 | 25.116 | |||
| Self-Rated Math Ability | 7.699 | 1.05E-11 | 0.78 | 0.148 | 0.259 | 0.1 | 6.65 | 26.939 | h2.zscore.1 too low | ||
| Left Subcallosal Cortex | Alcohol Dependence | Original | 0.336 | 0.737 | −0.305 | 0.401 | −0.043 | 0.116 | 8.213 | 8.188 | |
| Cognitive Performance | 9.901 | 1.80E-16 | −0.293 | 0.254 | −0.146 | 0.202 | 7.244 | 8.203 | |||
| Educational Attainment | 1.256 | 0.212 | 0.042 | 0.164 | −0.099 | 0.216 | 6.467 | 8.001 | h2.zscore.1 too low | ||
| Highest Math Class | 1.008 | 0.316 | −0.005 | 0.21 | −0.087 | 0.214 | 6.484 | 8.148 | h2.zscore.1 too low | ||
| Self-Rated Math Ability | 3.422 | 9.05E-04 | −0.322 | 0.218 | −0.106 | 0.196 | 7.92 | 7.5 | |||
| Alcohol Dependence | Bipolar Disorder | Original | 0.093 | 0.926 | −0.023 | 0.56 | −0.001 | 0.022 | 7.986 | 35.945 | |
| Cognitive Performance | 6.884 | 5.37E-10 | 0.607 | 0.084 | 0.191 | 0.077 | 7.059 | 30.127 | |||
| Educational Attainment | 5.134 | 1.42E-06 | 0.843 | 0.252 | 0.282 | 0.084 | 6.346 | 30.211 | h2.zscore.1 too low | ||
| Highest Math Class | 6.201 | 1.30E-08 | 0.834 | 0.105 | 0.227 | 0.081 | 6.412 | 29.84 | h2.zscore.1 too low | ||
| Self-Rated Math Ability | 5.52 | 2.72E-07 | 0.197 | 0.051 | 0.187 | 0.073 | 8.028 | 27.169 | |||
| Income | 4.149 | 7.07E-05 | 0.563 | 0.168 | 0.262 | 0.08 | 6.556 | 31.5 | h2.zscore.1 too low | ||
| Deprivation Index | 0.789 | 0.431 | 0.433 | 0.333 | 0.207 | 0.078 | 5.75 | 32.739 | h2.zscore.1 too low | ||
| Bipolar Disorder | Major Depression | Original | 0.037 | 0.97 | 0.001 | 0.071 | 0.338 | 0.039 | 34.38 | 36.466 | |
| Cognitive Performance | 0.999 | 0.319 | 0.086 | 0.081 | 0.329 | 0.038 | 30.433 | 34.262 | |||
| Educational Attainment | 1.198 | 0.233 | 0.105 | 0.085 | 0.363 | 0.038 | 30.529 | 33.8 | |||
| Highest Math Class | 1.437 | 0.153 | 0.127 | 0.086 | 0.358 | 0.036 | 30.5 | 33.703 | |||
| Self-Rated Math Ability | 0.996 | 0.321 | 0.095 | 0.09 | 0.346 | 0.036 | 28.867 | 36.063 | |||
| Income | 1.777 | 0.078 | 0.139 | 0.078 | 0.362 | 0.036 | 31.788 | 34.07 | |||
| Deprivation Index | 4.844 | 4.71E-06 | 0.268 | 0.063 | 0.364 | 0.035 | 33.639 | 34.238 | |||
| Bipolar Disorder | Schizophrenia | Original | 0.182 | 0.856 | 0.024 | 0.133 | 0.711 | 0.026 | 32.985 | 34.422 | |
| Cognitive Performance | 0.649 | 0.518 | −0.121 | 0.173 | 0.701 | 0.023 | 30.263 | 40.071 | |||
| Educational Attainment | 0.619 | 0.537 | −0.107 | 0.164 | 0.697 | 0.023 | 30.312 | 39.843 | |||
| Highest Math Class | 0.755 | 0.452 | −0.138 | 0.175 | 0.707 | 0.023 | 29.911 | 40.022 | |||
| Self-Rated Math Ability | 0.757 | 0.451 | −0.133 | 0.17 | 0.704 | 0.024 | 28.396 | 36.96 | |||
| Income | 1.993 | 0.048 | 0.246 | 0.124 | 0.695 | 0.02 | 31.851 | 38.906 | |||
| Deprivation Index | 6.865 | 5.88E-10 | 0.68 | 0.11 | 0.674 | 0.021 | 33.358 | 40.103 | |||
| Bipolar Disorder | Volume of Right-Ventral Diencephalon | Original | 0.71 | 0.479 | 0.172 | 0.518 | −0.019 | 0.021 | 10.058 | 35.797 | |
| Income | 0.131 | 0.895 | 0.111 | 0.519 | 0.008 | 0.066 | 9.116 | 31.746 | |||
| Deprivation Index | 9.236 | 5.07E-15 | −0.673 | 0.171 | 0.184 | 0.04 | 28.882 | 33.264 | |||
| Left Insular Cortex | Depressive Symptoms | Original | 0.117 | 0.907 | 0.09 | 0.525 | −0.002 | 0.03 | 8.076 | 22.529 | |
| Cognitive Performance | 3.828 | 2.27E-04 | 0.683 | 0.204 | −0.283 | 0.093 | 7.767 | 23.319 | |||
| Educational Attainment | 2.828 | 0.005 | 0.612 | 0.241 | −0.264 | 0.095 | 7.547 | 22.23 | |||
| Highest Math Class | 4.04 | 1.06E-04 | 0.681 | 0.204 | −0.258 | 0.098 | 7.719 | 23.633 | |||
| Self-Rated Math Ability | 4.813 | 5.33E-06 | 0.736 | 0.175 | −0.281 | 0.104 | 8.284 | 23.571 | |||
| Obsessive Compulsive Disorder | Anorexia Nervosa | Original | 0.243 | 0.808 | 0.117 | 0.519 | −0.009 | 0.035 | 11.558 | 11.504 | |
| Cognitive Performance | 2.957 | 0.003 | −0.064 | 0.214 | 0.514 | 0.126 | 11.861 | 13.034 | |||
| Educational Attainment | 11.041 | 5.94E-19 | −0.805 | 0.375 | 0.479 | 0.135 | 10.921 | 12.701 | |||
| Highest Math Class | 4.252 | 4.81E-05 | 0.184 | 0.243 | 0.494 | 0.129 | 11.43 | 12.973 | |||
| Self-Rated Math Ability | 2.282 | 0.024 | −0.335 | 0.304 | 0.488 | 0.122 | 13.977 | 14.596 | |||
| Income | 6.585 | 2.20E-09 | −0.371 | 0.692 | 0.501 | 0.13 | 11.328 | 12.723 | |||
| Deprivation Index | 4.691 | 8.71E-06 | 0.144 | 0.46 | 0.495 | 0.129 | 11.647 | 12.78 | |||
| Openness | Autism Spectrum Disorder | Original | 0.274 | 0.784 | 0.097 | 0.544 | 0.315 | 0.111 | 14.625 | 8.038 | |
| Cognitive Performance | 6.147 | 1.67E-08 | 0.761 | 0.164 | 0.286 | 0.127 | 16.717 | 6.089 | h2.zscore.2 too low | ||
| Educational Attainment | 5.452 | 3.66E-07 | 0.741 | 0.174 | 0.26 | 0.138 | 16.176 | 4.898 | h2.zscore.2 too low | ||
| Highest Math Class | 5.865 | 5.96E-08 | 0.762 | 0.163 | 0.332 | 0.122 | 16.295 | 6.68 | h2.zscore.2 too low | ||
| Self-Rated Math Ability | 5.914 | 4.77E-08 | 0.777 | 0.157 | 0.351 | 0.116 | 15.955 | 7.782 | |||
| Income | 5.212 | 1.02E-06 | 0.742 | 0.174 | 0.343 | 0.122 | 16.291 | 6.466 | h2.zscore.2 too low | ||
| Deprivation Index | 5.219 | 9.93E-07 | 0.764 | 0.166 | 0.39 | 0.115 | 17.898 | 7.248 | |||
| Left Insular Cortex | Openness | Original | 0.498 | 0.619 | −0.225 | 0.314 | 0.041 | 0.057 | 7.151 | 8.094 | |
| Cognitive Performance | 5.967 | 3.77E-08 | 0.35 | 0.379 | 0.201 | 0.2 | 7.458 | 6.903 | h2.zscore.2 too low | ||
| Educational Attainment | 9.861 | 2.20E-16 | −0.092 | 0.381 | 0.187 | 0.221 | 7.311 | 5.574 | h2.zscore.2 too low | ||
| Highest Math Class | 8.221 | 8.04E-13 | 0.284 | 0.456 | 0.239 | 0.184 | 7.487 | 7.298 | |||
| Self-Rated Math Ability | 13.09 | 2.54E-23 | 0.252 | 0.384 | 0.198 | 0.17 | 7.583 | 7.598 | |||
| Right Insular Cortex | Volume of Right-Ventral Diencephalon | Original | 1.322 | 0.189 | 0.513 | 0.292 | −0.157 | 0.162 | 8.925 | 10.614 | |
| Income | 1.546 | 0.125 | 0.533 | 0.283 | −0.148 | 0.166 | 8.87 | 10.354 | |||
| Deprivation Index | 5.975 | 3.63E-08 | 0.478 | 0.178 | −0.131 | 0.096 | 8.735 | 30.666 | |||
| Volume of Right-Ventral Diencephalon | Openness | Original | 0.577 | 0.565 | −0.328 | 0.404 | −0.066 | 0.049 | 9.177 | 8.094 | |
| Income | 8.8 | 4.50E-14 | −0.203 | 0.442 | 0.27 | 0.096 | 28.449 | 8.041 | |||
| Deprivation Index | 0.042 | 0.966 | −0.061 | 0.302 | 0.153 | 0.17 | 9.327 | 7.409 |
Column Descriptions
zscore Z score for partial genetic causality
gcp p p; Significantly positive zscore implies trait 1 partially genetically causal for trait 2
gcp.pm posterior genetic causality proportion (positive = trait 1 > trait 2)
gcp.pse posterior standard error for genetic causality proportion
rho.est estimated genetic correlation
rho.err standard error for genetic correlation estimate
h2.zscore.1 z-score for trait 1 being significantly heritable
h2.zscore.2 z-score for trait 2 being significantly heritable
Fig. 5. Causal relationships masked by education and socioeconomic status effects.
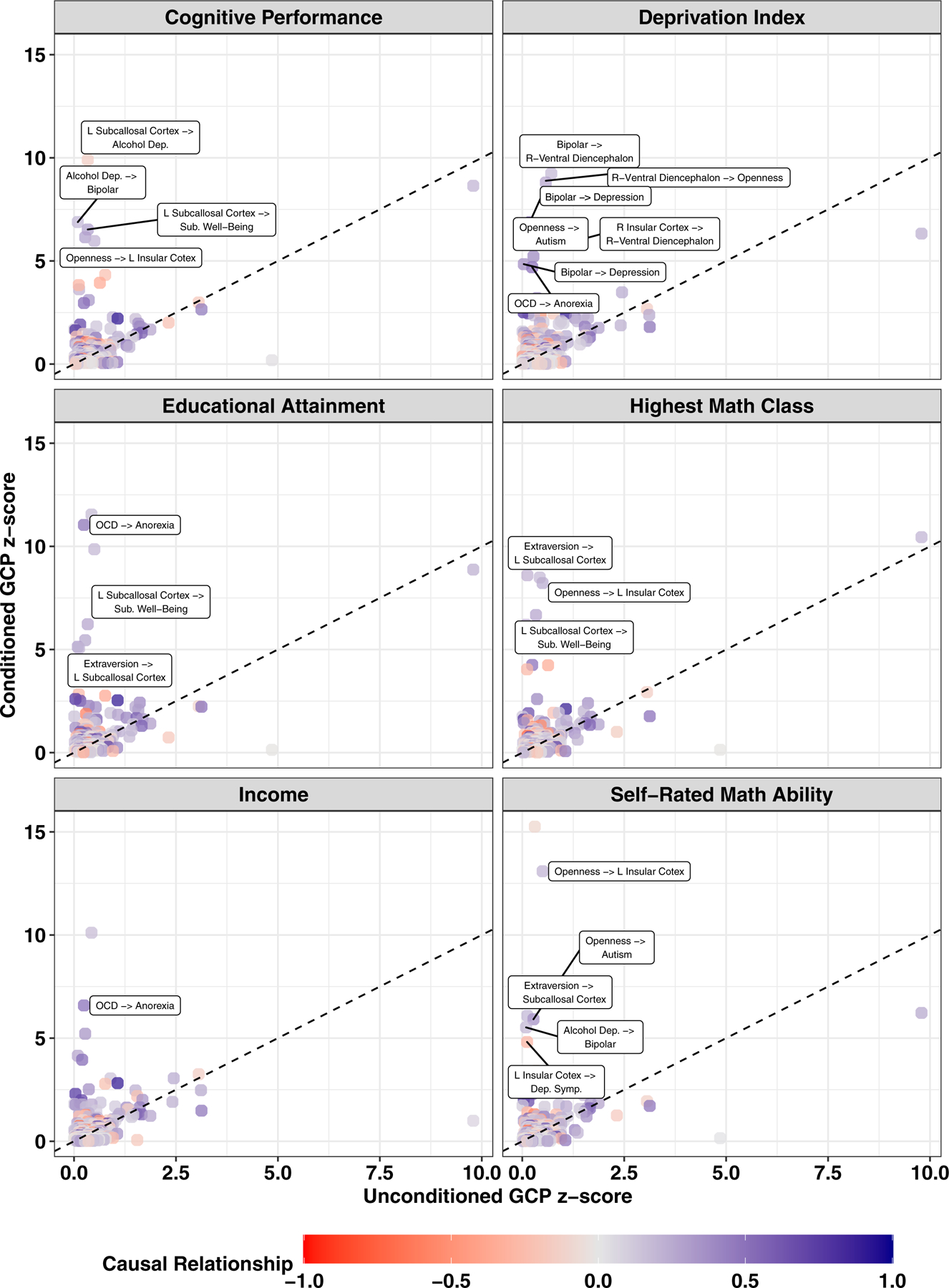
Latent Causal Variable (LCV) results detecting putative causal relationships between the genetic risk for two psychopathology and psychosocial factors. Each data point indicates a trait pair comparison in which the y-direction indicates a causal estimate whereby both traits have been conditioned on one education and socioeconomic status phenotype. The color of each data point indicates the magnitude of causal estimate (e.g., causal relationship = −1 indicates that trait 1 decreases trait 2) The dashed diagonal lines indicates a one-to-one relationship between x and y axes. Significant putative causal relationships are labeled and described in detail in Table 1.
Fig. 6. Latent causal variable (LCV) relationship network.
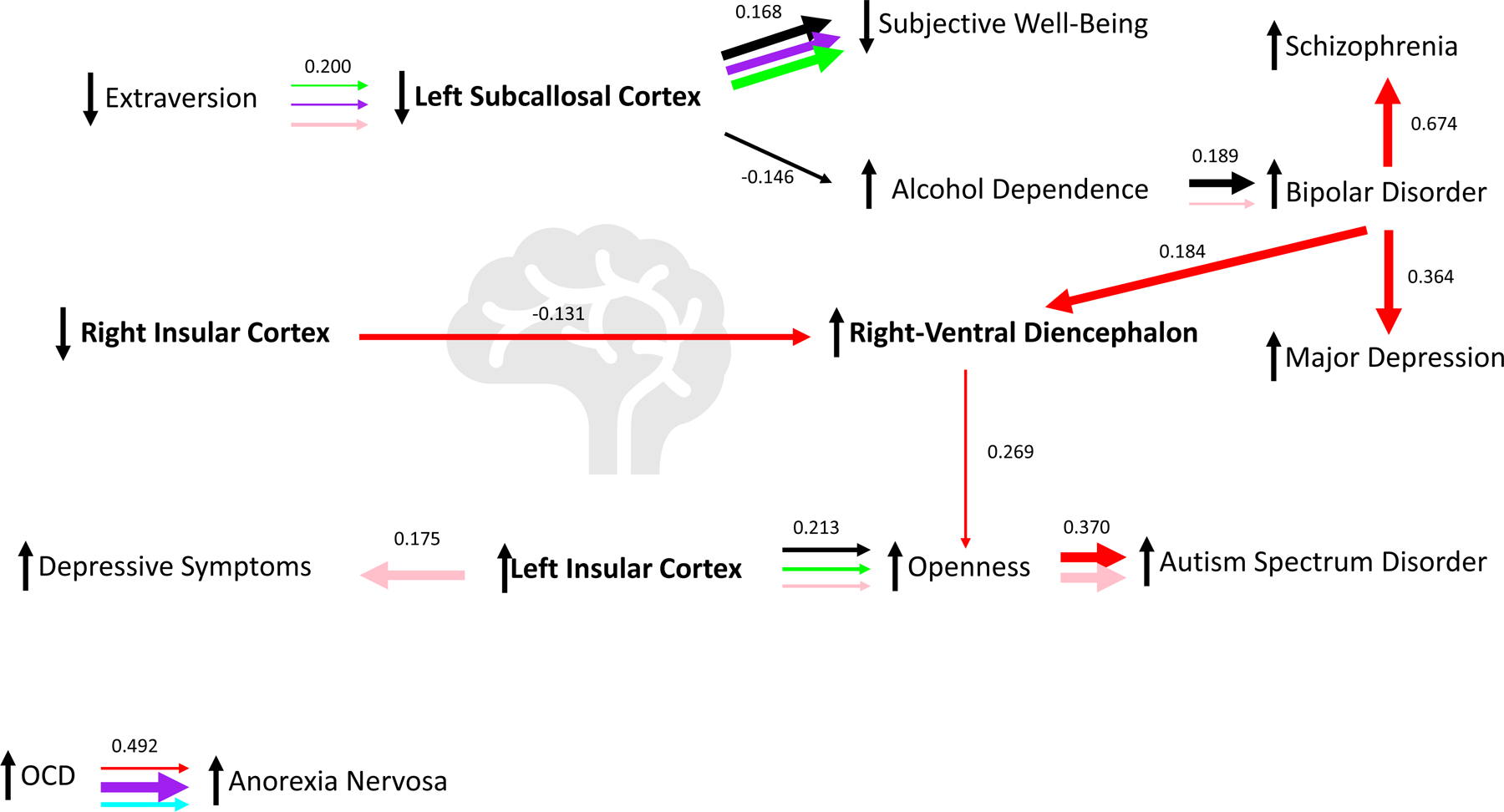
(A) Summary of the causal relationship (derived from Fig. 5) network originating from brain imaging phenotypes (bolded text). (B) Causal relationships with no evidence of brain imaging phenotype connection in the current study (derived from Fig. 5). Arrow thickness indicates the size of the estimated causal relationship between the two traits on either end of the arrow while triangles indicate the direction of causal effect; the color of each arrow indicates the education or socioeconomic status phenotype used to condition each trait of a trait pair (black = cognitive performance; purple = educational attainment; blue = income; red = deprivation index; green = highest math class; pink = self-rated math ability); mean genetic correlations from LCV are included above each set of horizontal arrows. Sample interpretation (from A): decreased extraversion causes lower left subcallosal cortex volume after removing the effects of highest math class, educational attainment, and self-rated math ability.
The EDU/SES phenotype that revealed the most latent causal relationships between psychopathology and psychosocial factors was Townsend deprivation index. Conditioning with this phenotype revealed 7/13 causal relationships, most of which involved bipolar disorder or the volume of the right-ventral diencephalon (Fig. 6 and Table S26).
Discussion
EDU and SES are important influences on numerous psychopathology, psychosocial, and mental disorders, but it has been difficult to determine the extent to which this is so, and the biological nature of the relationship. How much of the genetic risk for schizophrenia, for example, is caused by reduced educational attainment? Or how much of the risk for schizophrenia reflects a shared biology with the predisposition to educational attainment? These are important questions to answer if we are to understand the biology of both kinds of traits. To get at this question, we conditioned one on the other, and thereby statistically removed its effects, and then asked the question, “how much of the heritable risk for that trait remains?” In most cases, EDU/SES accounted for some of the genetic variance in the psychopathology and psychosocial phenotype and adjusting for EDU/SES reduced the strength of the association with the heritable risk for that disorder. However, in a few cases (depression, anxiety, neuroticism, PTSD, and subjective well-being) adjusting for EDU/SES either increased or did not change these associations. In the space below, we present a framework for interpreting the complexity of these findings.
The biology underlying psychiatric disorders was most affected by shared genetic etiology with EDU/SES proxies, as evidenced by significant decreases in h2 for all psychiatric disorder except MDD, anxiety, and PTSD when conditioned with EDU/SES. Conversely, conditioning the neuroticism and subjective well-being GWAS revealed additional risk loci that were not detected in their unconditioned GWAS. Using an independent method, Turley, et al. observed similar information gain;17 however, we demonstrated that this information gain is due to polygenicity rather than population substructure as evidenced by stable intercept estimates no different from the unconditioned neuroticism GWAS but increasing lambda GC. That is, we believe this demonstrated biological underpinnings, as opposed to the underlying population genetics phenomena. Unlike Turley, et al., we do not report comparable risk locus gain with subjective well-being.
In structural equation models of psychopathology and psychosocial factors, neuroticism and MDD originally loaded onto the same common factor. After conditioning with highest math class, self-rated math ability, and deprivation index, the loadings of neuroticism and MDD separate, suggesting that their unique biology may be distinguishable. This is consistent with our observation of ameliorated genetic correlation between these two phenotypes due to conditioning. Hill, et al., described two factors of neuroticism, one of which aligns more closely with anxiety and tension phenotypes and the second of which aligns more closely with worry and vulnerability phenotypes.31 With genomic SEM, we support these claims: neuroticism loads onto the same common factor as anxiety while MDD aligns with depressive symptoms and loneliness. We demonstrate here that neuroticism and MDD are highly positively genetically correlated in their unconditioned versions. Based on the present results, we hypothesize that conditioning these phenotypes with EDU and SES reveals their unique genetic architectures. We demonstrate that, after conditioning with EDU/SES, general neuroticism appears more similar to the Hill, et al. anxiety/tension phenotype. Lastly, our genomic SEM data mirror those genetic correlation results from Hill, et al., adding weight to our observed two-factor model.31
Cell type transcriptomic profile enrichments underlying the GWAS of psychopathology and psychosocial factors were robust to the effects of EDU and SES phenotypes, but we uncovered cell-type information for risky behavior, MDD, schizophrenia, and bipolar disorder, which highlight cell-specific processes. The cell-types discovered in the conditioned schizophrenia GWAS overlap with those in the conditioned bipolar disorder GWAS. These findings recapitulate common therapeutic targets for these disorders. For example, inhibitory and GABAergic neuron transcriptomic profile enrichments were detected in the conditioned MDD GWAS and these are common targets of emerging therapeutic options (e.g., scopolamine, an antidepressant which blocks the M1 receptor of GABAergic interneurons in the medial prefrontal cortex;32 ketamine blocking the activation of somatostatin interneurons in PFC33) for MDD and depressive symptoms.32 Detection of overlapping cell-type information may support drug repurposing efforts in psychiatric disorders and related mental health conditions, though the effect of these detected cell types as therapeutic targets requires experimental validation.
Using genome-wide information, we uncovered putatively causal relationships between many psychopathology, psychosocial factors, and brain measurements. These analyses detected well-known relationships between traits (e.g., bipolar disorder, schizophrenia, and MDD) but also elucidated several relationships involving brain imaging phenotypes. In particular we identified the volume of the left subcallosal cortex as a possible mediator of the relationships between several psychopathology and psychosocial factors (e.g., extraversion, subjective well-being, and alcohol dependence), which in turn demonstrate potential causal relationships with mood disorders, which are commonly comorbid with alcohol dependence.34 The brain structural convergence detected here may indicate common disease mechanisms; however, these commonalities may be confounded by fine-grained nuances of the relationship between brain microstructure and mental health and disease. The LCV method used to identify these causal relationships does not support multivariable analyses nor does it employ sensitivity tests to detect horizontal pleiotropy (i.e., a SNP is associated with both phenotypes through separate mechanisms) or effect-size outlier SNPs. These observations likely confound our causal inferences and require more robust testing with traditional Mendelian randomization methods suited to accommodate weak genetic instruments (i.e., those SNPs not strongly associated with either phenotypes of interest, typically with association p-values greater than 5×10−8).35, 36, 37 It should be recognized that LCV and other causal inference methods indeed have reduced power with respect to highly polygenic traits, such as those studied here, leading towards downwardly biased genetic causality proportion estimates. It is therefore unlikely that estimates with high genetic causality proportions are false positives.30
Certain relationships regarding psychopathology and psychosocial factor conditioning that might have been expected were not observed in our study. Intellectual abilities are genetically correlated with ASD and ADHD and disabilities therein often co-occur with ASD and/or ADHD diagnoses.38, 39 According to the Diagnostic and Statistical Manual of Mental Disorders (5th Edition, DSM-5), diagnosis of intellectual disability or global developmental delay must be eliminated as possible explanations of ASD symptoms prior to making a formal ASD diagnosis. We had hypothesized that after conditioning with the effects of EDU phenotypes, these psychiatric disorders might demonstrate notable changes in their genetically predicted underlying biology, but this was not the case. This lack may suggest that ADHD and ASD diagnosis criteria robustly capture elements unique to the disorders rather than those shared with EDU/SES phenotypes. In other words, ascertainment of cases at the extreme ends of spectrum disorders40, 41 reliably capture trait specific biology with minimal phenotype confounding from shared effects with EDU and SES.
Information derived from the GWAS of EDU and SES phenotypes was generally robust to conditioning with psychopathology and psychosocial factors. When conditioned with individual psychopathology and psychosocial factors, we detected relatively few changes to the genetically predicted biology of EDU/SES phenotypes. Only when EDU/SES phenotypes were conditioned with several psychopathology and psychosocial factors did we observed changes in h2 and genetically predicted biology. When assessing the relationship between unconditioned EDU and SES phenotypes by genomic structural equation modeling, we revealed cognitive performance and highest math class as driving factors linking EDU and SES phenotypes. When conditioned, we uncovered an independent contribution of income to a common factor with educational attainment and self-rated math ability. Based on recent work of Morris, et al. to uncover why EDU and SES phenotypes are related to one another, and the near perfect loading of educational attainment on the common factor, these observations point to educational attainment as a mediator of the genetic and phenotypic correlations between EDU and SES.
Tissue and cell-type transcriptomic profile analyses of EDU, SES, psychopathology, and psychosocial phenotypes highlighted differences in cortical and cerebellar tissue enrichment. Though not significantly decreased in all phenotypes after conditioning, the bidirectional changes in cerebellar and cortical tissue enrichment (i.e., EDU/SES conditioned with psychopathology and psychosocial factors and psychopathology and psychosocial factors conditioned with EDU/SES) highlight the importance of these brain regions and their shared transcriptional regulation in human mental health and disease.42 Furthermore, this observation of cerebellar and cortical tissue changes support the common psychopathology factor (a p-factor) studied extensively in recent mental health and disease research.43
Our study has three primary limitations. First, we did not select independent genetic correlates from the psychopathology and psychosocial factors tested with which to condition the EDU and SES phenotypes. Due to high genetic correlation between psychopathology and psychosocial factors, this approach may have introduced bias in our reporting of which EDU and SES phenotypes were robust to shared genetic etiology with all psychopathology and psychosocial factors. This potential over-conditioning likely drove our results towards the null (e.g., non-significant h2) and therefore, we have not reported gene set, tissue transcriptomic profile enrichment, cell-type transcriptomic profile enrichment, or genomic SEM loadings for EDU/SES traits where there might have been over-conditioning. For this reason, our results do not imply that, for example, educational attainment is a more powerful or specific EDU phenotype than cognitive ability. Second, it has recently been demonstrated that the origin of phenotypic and genetic correlations between EDU and SES phenotypes may be driven by dynastic effects and/or assortative mating acting independently or in concert6. Dynastic effects describe a condition where offspring inherit phenotype-associated genetic risks and phenotype-associated environmental risks. Assortative mating exists when mate pairs are non-randomly chosen based on certain attributes. We hypothesize that the dynastic and assortative mating events described between EDU and SES phenotypes6 may also appear in phenotypic and genetically correlated EDU, SES, and psychopathology and psychosocial factor pairs. Future studies will be required to describe how these evolutionary and social pressures influence the correlative and causal relationships uncovered here (e.g., OCD→anorexia nervosa after conditioning with the effects of educational attainment, income, and deprivation index). Third, the UK Biobank is considered a generally healthy cohort not enriched for any trait or disorder of interest. To our knowledge, the brain imaging GWAS (performed on a subset of UK Biobank participants) used here were not adjusted for variables related to blood pressure, height, weight, and bone mineral composition, substance-related (recreational or prescribed), or psychiatric variables. The presence of these variables in sufficient quantities among those UKB brain imaging participants could alter brain volumes affecting the results of the genetic analyses conducted.
By conditioning psychopathology and psychosocial factors for the shared genetic etiology with EDU and SES phenotypes, this study elucidates biological underpinnings and causal relationships between phenotypes. These biological mechanisms, cell-types, tissue-types, and causal trait pairs could not have been detected without adjusting the effects of EDU and SES. This study highlights how the pervasive effects of EDU and SES may mask genetically determined underlying biology of psychopathology and psychosocial factors in support of multi-trait analyses of GWAS to enable trait-specific discoveries.
Methods
This study was conducted using genome-wide association statistics generated by previous studies. Owing to the use of previously collected, deidentified, aggregated data, this study did not require institutional review board approval. Ethical approval had been obtained in all original studies (Table S1). An overview of all materials, methods, and key findings from this investigation of the genetic overlap between EDU, SES, and psychopathology and psychosocial factors is shown in a flow diagram in Fig. S1.
Trait Description and Selection
Four EDU (educational attainment, highest math class, self-rated math ability, and cognitive performance) and two SES phenotypes (average household income before tax and Townsend deprivation index) from the Social Science Genetic Association Consortium (SSGAC), UK Biobank (UKB), and 23andMe were used in this study to condition psychopathology and psychosocial factors. These unconditioned phenotypes were characterized on the level of heritability, tissue transcriptomic profile enrichment, and cell-type transcriptomic profile enrichment in Fig. S1. Briefly, the educational attainment phenotype describes the number of years of schooling completed per participant. Highest math class and self-rated math ability were derived from the 23andMe survey regarding participant mathematical background. For self-rated math ability, 23andMe participants were asked “how would you rate your mathematical ability” ranging from “very poor” to “excellent.” For highest math class, 23andMe participants were asked to indicate the most advanced mathematics course they have successfully completed (excluding statistics courses) ranging from pre-algebra to coursework more advanced than vector calculus. Cognitive performance was evaluated as a standardized score of logic and reasoning questions completed within a two-minute time limit. The cognitive performance measure per participants represented a standardized mean across trials. The UKB phenotype Townsend deprivation index was calculated immediately prior to a participant joining the UKB project using preceding national census information. The measure incorporates four variables including unemployment, non-car ownership, non-home ownership, and household overcrowding. The UKB phenotype average household income before tax was self-reported via touchscreen questionnaire at recruitment to the UKB. Participants were asked to report their household income, in pounds, in categories ranging from “less than £18,000” to “greater than £100,000.”
Psychopathology and psychosocial factors from the Psychiatric Genomics Consortium (PGC), SSGAC, Genetics of Personality Consortium (GPC), UKB, and UKB Brain Imaging Genetics (UKB BIG) were selected based on their genetic correlation with EDU and SES phenotypes (Table S1 and Figs. 1 & S2). To focus our analyses, we predetermined that psychopathology and psychosocial factors would be included if (1) they had heritability (h2) significantly greater than zero, and (2) they were at least nominally genetically correlated with 2/4 EDU and 2/2 SES phenotypes. It is recommended that each phenotype in a genetic correlation pair have h2 z-scores ≥ 4 to produce reliable estimates of genetic overlap21 but mtCOJO and structural equation modeling (see below) only require h2 estimates significantly greater than zero at a nominal level (p<0.05) for each trait included. For this reason, we relaxed the h2 suggestions for genetic correlation analyses with respect to trait inclusion. Genetic correlation estimates should be interpreted in light of this relaxed h2 criteria. In other words, significant genetically correlated trait pairs in which one or both traits exhibited h2 z-scores < 4, such as anxiety, conscientiousness, loneliness, and PTSD, should be interpreted as requiring replication in larger, sufficiently powered data sets. The relaxation of this recommended threshold is not expected to influence results from any other analysis performed herein.
Conditioning
Conditional genome-wide association analysis was performed in Genome-wide Complex Trait Analysis (GCTA) using the mtCOJO feature using the 1000 Genomes Project European ancestry linkage disequilibrium reference panel.18 For case-control GWAS summary statistics, odds ratios and corresponding standard error were converted to log-odds and corresponding standard error.
Causal estimates within mtCOJO were calculated using Generalized Summary-data-based Mendelian Randomization (GSMR). In our main analysis, mtCOJO was used to test the assumption that EDU/SES → psychopathology and psychosocial factors; however, we also tested the reverse scenario in the section Effects of Psychopathology and Psychosocial Factors on Education and Socioeconomic Status. MR relies on three key assumptions about the instrumental variables used to test for causal inference between phenotypes: (1) genetic instruments are associated with the exposure of interest (in our main analysis, EDU/SES were exposure phenotypes), (2) genetic instruments are not associated with the outcome of interest (in our main analysis, psychopathology and psychosocial factors were outcome phenotypes), and (3) genetic instruments do not affect the outcome except through the exposure. In our analyses of psychopathology and psychosocial factors conditioned with the effects of EDU and SES phenotypes (e.g., MR to test the hypothesis that EDU/SES → psychopathology and psychosocial factors), each MR causal inference was performed using genome-wide significant SNPs in the exposure (EDU/SES trait). To test how psychopathology and psychosocial factors influence EDU and SES phenotypes (e.g., MR to test the hypothesis that psychopathology and psychosocial factors → EDU/SES), we relaxed this SNP inclusion threshold where necessary (e.g., when UKB BIG phenotypes served as the exposure phenotype and lacked sufficient numbers of genetic instruments at the level of genome-wide significance) such that at least two SNPs were included in the causal inference. Note that MR-based conditioning with mtCOJO is not sensitive to sample overlap due to the method’s incorporation of sampling covariance between SNP effects into the model.18
Implemented in mtCOJO, heterogeneity in dependent genetic instruments (HEIDI) outlier testing was performed to detect SNPs with outlier effect size estimates assuming SNP effect distributions in both exposure and outcome.18, 22
Heritability and Genetic Correlation
The Linkage Disequilibrium Score Regression (LDSC) method is used for formatting GWAS summary association data and estimating SNP-heritability of a trait (h2) and genetic correlation between traits. LDSC assumes SNPs have not been pruned for linkage disequilibrium and that sample ascertainment of a phenotype was performed in a genetically homogeneous population. With respect to genetic correlation, phenotypes should be ascertained from cohorts representing similar ancestral backgrounds. Genetic correlation with LDSC is not sensitive to sample overlap.
Observed-scale h2 was calculated for each original and conditioned GWAS using the LDSC method with 1000 Genomes Project European reference population.21
Gene-set, Tissue Transcriptomic, and Cell-type Transcriptomic Profile Enrichment
Original and conditioned GWAS were used as standard input for Multi-marker Analysis of GenoMic Annotation (MAGMA v1.06) implemented in FUnctional Mapping and Annotation (FUMA) v1.3.3c with the following parameters: genome-wide significance p < 5×10−8, minor allele frequency ≥ 0.01, and LD blocks merged at < 250kb for LD r2≥0.6 with lead variant.28, 44
SNPs underlying each phenotype of interest were mapped to genes within 10kb physical proximity using FUMA.44 Mapped genes were assessed using the gene-set enrichment feature of FUMA, and gene ontology enrichment analysis with ShinyGO.45
Tissue transcriptomic profile enrichment was performed relative to the GTEx25 v7 53 tissue types with the default 0kb gene window.
Cell-type transcriptomic profile enrichments were performed using eleven human specific transcriptomic profile datasets related to the brain;28 PsychENCODE_Developmental, PsychENCODE_Adult, Allen_Human_LGN_level 1, Allen_Human_MTG_level1, DroNc_Human_Hippocampus, GSE104276_Human_Prefrontal_cortex_all_ages, GSE104276_Human_prefrontal_cortex_per_ages, GSE67835_Human_Cortex, GSE67835_Human_Cortex_woFetal, Linnarson_GSE101601_Human_Temporal_cortex, and Linnarson_GSE76381_Human_Midbrain. Cell-type transcriptomic profiles were assessed in three ways as per the FUMA analysis pipeline. (1) enrichment of cell-type transcriptomic profiles within each selected data set, (2) within data set conditionally independent cell-type transcriptomic profile enrichments, and (3) across data set cell-type transcriptomic profile enrichments.
For analyses within data sets, step-wise conditional significance is evaluated for each cell type in a data set against the p-values for all other cell-types in that same data set. The output from these analyses identify cell types within a data set whose transcriptomic profiles are enriched in a given GWAS independently of the signal from all other cell type transcriptomic profiles in the same data set.
Using within-data-set conditionally independent cell-types identified above, cross-data-set analysis identifies cell-type transcriptomic profiles enriched in a given GWAS independent of all other cell-types from all chosen data sets. Proportional significance (PS) and conditional independence of cell-type pairs indicate that enrichment of these cell-types in a given GWAS are driven by independent genetic signals.
For a given pair of cell types, PS of cell type a given cell type b (PSa,b) ≥ 0.8 and PSb,a ≥ 0.8 indicates independent genetic signals for cell types a. Interpretation of additional PS thresholds for each cell type in a given pair can be seen in detail (https://fuma.ctglab.nl/tutorial#celltype) or in Watanabe, et al.28
Latent Causal Variables
LCV is a method for inferring genetic causal relationships between trait pairs using GWAS summary data using effect size estimates or z-scores.30 The LCV model assumptions are notably weaker than traditional MR assumptions. First, LCV assumes that the distribution of effect sizes for a given trait pair represents one distribution of effect sizes proportional in both traits of interest and a second distribution of SNPs that only affect the outcome trait. LCV therefore assumes that symmetry in shared genetic architectures between traits arises from the action of a latent genetic component rather than a non-genetic confounder commonly elucidated by MR. Second, the model assumes a single genetic LCV mediating trait relationships; however, in simulations of LCV power in the presence of more than one LCV, causal estimates were unlikely to be detected. There are no assumptions of parametric effect size distributions under the LCV model; however, LCV is indeed less well powered for highly polygenic traits.
LCV modeling was implemented in R using the 1000 Genomes Project Phase 3 European reference panel. As recommended, GWAS summary data were filtered to include only SNPs with minor allele frequencies greater than 5% and the major histocompatibility region was removed due to its complex linkage disequilibrium structure. Note that genetic correlation does not imply that shared genetic risks between traits are causal. The LCV model output distinguishes whether genetic correlations support genetic causation and the degree to which (i.e., the genetic causality proportion; gĉp) genetic risk for trait 1 is causal for trait 2. LCV gĉp estimates were only interpreted for trait pairs where both traits exhibit LCV-calculated h2 z-scores ≥ 7.
Genomic Structural Equation Modeling
Genomic structural equation modeling (SEM) was performed using GWAS summary statistics in the genomicSEM and lavaan R packages.46 Because genomic SEM relies on a genetic covariance matrix estimated with LDSC (see Heritability and Genetic Correlation), the same set of assumptions applies here. Though tolerant to deviations form these expectations, genomic SEM is most well-powered when index phenotypes (i.e., those GWAS whose summary statistics are used to model latent factors) are highly heritable, consist of primarily non-overlapping samples, performed in large sample sizes, and have high genetic correlation among them.46
Exploratory factor analyses (EFA) were performed on two groups of phenotypes: (1) all mental health outcomes conditioned with EDU and SES phenotypes and (2) all EDU and SES phenotypes conditioned with all psychopathology and psychosocial factors. EFA were performed for 1 through N factors until the addition of factor N contributed less than 10% explained variance to the model. Confirmatory factor analysis was performed using the diagonally-weighted least squares estimator and a genetic covariance matrix of munged GWAS summary statistics for all phenotypes based on the 1000 Genome Project Phase 3 European linkage disequilibrium reference panel. Munging was performed using LDSC.
Statistical Considerations
Z-tests were used to determine differences in heritability, SNP effects, gene-set enrichments, tissue transcriptomic profile enrichments, cell-type transcriptomic profile enrichments, genomic SEM loadings, and LCV estimates between conditioned and unconditioned GWAS. It should be noted that while much of the data generated for this study relied on one-sided tests (e.g., LDSC tests whether the h2 for Trait X is greater than 0 and MAGMA tests whether the transcriptomic profile of Tissue X is overrepresented relative to all other tissue types), Z-tests reported herein were used to compare the conditioned versus unconditioned versions of a trait GWAS. In other words, two sides were considered – for example, the unconditioned h2 could be greater than or less than the conditioned h2 for a trait. Z-scores for the difference between conditioned and unconditioned measurements were converted to p-values assuming a two-tailed distribution prior to multiple testing correction. P-values were corrected for multiple testing considering a false discovery rate at 5%.
Supplementary Material
Acknowledgements
We would like to thank the research participants and employees of 23andMe Inc for making this work possible. This study was supported by the Simons Foundation Autism Research Initiative (SFARI Explorer Award: 534858 (RP)), the American Foundation for Suicide Prevention (YIG-1-109-16 (RP)), the National Institutes of Health (R21 DC018098 (RP), R21 DA047527 (RP), F32 MH122058 (FRW), and R01 MH117646 (TL)), and the National Center for PTSD of the U.S. Department of Veterans Affairs. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Competing interests
Dr. Krystal reports compensation as the Editor of Biological Psychiatry. He also serves on the Scientific Advisory Boards for Bioasis Technologies, Inc., Biohaven Pharmaceuticals, BioXcel Therapeutics, Inc. (Clinical Advisory Board), Cadent Therapeutics (Clinical Advisory Board), PsychoGenics, Inc, Stanley Center for Psychiatric research at the Broad Institute of MIT and Harvard and the Lohocla Research Corporation. He owns stock in ArRETT Neuroscience, Inc., Biohaven Pharmaceuticals, Sage Pharmaceuticals, and Spring Care, Inc. and stock options in Biohaven Pharmaceuticals Medical Sciences, BlackThorn Therapeutics, Inc. and Storm Biosciences, Inc. He is a co-inventor on multiple patents as listed below: (1) Seibyl JP, Krystal JH, Charney DS. Dopamine and noradrenergic reuptake inhibitors in treatment of schizophrenia. US Patent #:5,447,948.September 5, 1995, (2) Vladimir, Coric, Krystal, John H, Sanacora, Gerard—Glutamate Modulating Agents in the Treatment of Mental Disorders US Patent No. 8,778,979 B2 Patent Issue Date: July 15, 2014. US Patent Application No. 15/695,164: Filing Date: 09/05/2017, (3) Charney D, Krystal JH, Manji H, Matthew S, Zarate C.—Intranasal Administration of Ketamine to Treat Depression United States Application No. 14/197,767 filed on March 5, 2014; United States application or Patent Cooperation Treaty (PCT) International application No. 14/306,382 filed on June 17, 2014, (4): Zarate, C, Charney, DS, Manji, HK, Mathew, Sanjay J, Krystal, JH, Department of Veterans Affairs “Methods for Treating Suicidal Ideation”, Patent Application No. 14/197.767 filed on March 5, 2014 by Yale University Office of Cooperative Research, (5) Arias A, Petrakis I, Krystal JH.—Composition and methods to treat addiction. Provisional Use Patent Application no.61/973/961. April 2, 2014. Filed by Yale University Office of Cooperative Research, (6) Chekroud, A., Gueorguieva, R., & Krystal, JH. “Treatment Selection for Major Depressive Disorder” [filing date 3rd June 2016, USPTO docket number Y0087.70116US00]. Provisional patent submission by Yale University, (7) Gihyun, Yoon, Petrakis I, Krystal JH—Compounds, Compositions and Methods for Treating or Preventing Depression and Other Diseases. U. S. Provisional Patent Application No. 62/444,552, filed on January 10, 2017 by Yale University Office of Cooperative Research OCR 7088 US01, (8) Abdallah, C, Krystal, JH, Duman, R, Sanacora, G. Combination Therapy for Treating or Preventing Depression or Other Mood Diseases. U.S. Provisional Patent Application No. 047162–7177P1 (00754) filed on August 20, 2018 by Yale University Office of Cooperative Research OCR 7451 US01. Dr. Gelernter are named as inventors on PCT patent application #15/878,640 entitled: “Genotype-guided dosing of opioid agonists,” filed January 24, 2018. Drs. Gelernter and Polimanti are paid for their editorial work on the journal Complex Psychiatry. The other authors declare no competing interests.
Data availability
All GWAS association data and analysis materials used in this study are publicly available for download by qualified researchers. All data used to make conclusions discussed in this study are provided as Supplementary Material.
Social Science Genetic Association Consortium: https://www.thessgac.org
Psychiatric Genomics Consortium: https://www.med.unc.edu/pgc/download-results/
UK Biobank: https://www.ukbiobank.ac.uk/register-apply/
23andMe: https://research.23andme.com/research-innovation-collaborations/
Brain Imaging Genetics: http://big.stats.ox.ac.uk
Code availability
Previously developed pipelines were used to produce the results for this study. No custom code was developed.
References
- 1.Keyes KM, Platt J, Kaufman AS, McLaughlin KA. Association of Fluid Intelligence and Psychiatric Disorders in a Population-Representative Sample of US Adolescents. JAMA Psychiatry 74, 179–188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McLaughlin KA, Costello EJ, Leblanc W, Sampson NA, Kessler RC. Socioeconomic status and adolescent mental disorders. Am J Public Health 102, 1742–1750 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.d’Errico A, et al. Socioeconomic indicators in epidemiologic research: A practical example from the LIFEPATH study. PLoS One 12, e0178071 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buniello A, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bulik-Sullivan B, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 47, 1236–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris TT, Davies NM, Hemani G, Smith GD. Why are education, socioeconomic position and intelligence genetically correlated? bioRxiv, 630426 (2019). [Google Scholar]
- 7.Trampush JW, et al. GWAS meta-analysis reveals novel loci and genetic correlates for general cognitive function: a report from the COGENT consortium. Mol Psychiatry 22, 336–345 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marioni RE, et al. Molecular genetic contributions to socioeconomic status and intelligence. Intelligence 44, 26–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hill WD, et al. Molecular Genetic Contributions to Social Deprivation and Household Income in UK Biobank. Curr Biol 26, 3083–3089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anttila V, et al. Analysis of shared heritability in common disorders of the brain. Science 360, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Qi G, Park JH, Chatterjee N. Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat Genet 50, 1318–1326 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Choi SW, O’Reilly PF. PRSice-2: Polygenic Risk Score software for biobank-scale data. Gigascience 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi SW, Mak TS, O’Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avinun R Educational Attainment Polygenic Score is Associated with Depressive Symptoms via Socioeconomic Status: A Gene-Environment-Trait Correlation. bioRxiv, 727552 (2019). [Google Scholar]
- 15.Krapohl E, Plomin R. Genetic link between family socioeconomic status and children’s educational achievement estimated from genome-wide SNPs. Mol Psychiatry 21, 437–443 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards AL, et al. The Relationship Between Polygenic Risk Scores and Cognition in Schizophrenia. Schizophr Bull, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turley P, et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat Genet 50, 229–237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Z, et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat Commun 9, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet Epidemiol 40, 304–314 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat Med 36, 1783–1802 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bulik-Sullivan BK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Z, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 48, 481–487 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Eroglu C, et al. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vergult S, et al. Genomic aberrations of the CACNA2D1 gene in three patients with epilepsy and intellectual disability. Eur J Hum Genet 23, 628–632 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Battle A, Brown CD, Engelhardt BE, Montgomery SB. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crayton JW, Meltzer HY. Degeneration and regeneration of motor neurons in psychotic patients. Biol Psychiatry 14, 803–819 (1979). [PubMed] [Google Scholar]
- 27.Strassnig M, Signorile J, Gonzalez C, Harvey PD. Physical performance and disability in schizophrenia. Schizophr Res Cogn 1, 112–121 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe K, Umicevic Mirkov M, de Leeuw CA, van den Heuvel MP, Posthuma D. Genetic mapping of cell type specificity for complex traits. Nat Commun 10, 3222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lake BB, et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 36, 70–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Connor LJ, Price AL. Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat Genet, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hill WD, et al. Genetic contributions to two special factors of neuroticism are associated with affluence, higher intelligence, better health, and longer life. Mol Psychiatry, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duman RS, Sanacora G, Krystal JH. Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron 102, 75–90 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerhard DM, et al. GABA interneurons are the cellular trigger for ketamine’s rapid antidepressant actions. J Clin Invest, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andersen AM, et al. Polygenic Scores for Major Depressive Disorder and Risk of Alcohol Dependence. JAMA Psychiatry 74, 1153–1160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity Analyses for Robust Causal Inference from Mendelian Randomization Analyses with Multiple Genetic Variants. Epidemiology 28, 30–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol 46, 1985–1998 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Q, Wang J, Hemani G, Bowden J, Small DS. Statistical inference in two-sample summary data Mendelian randomization using robust adjusted profile score. arXiv, (2018). [Google Scholar]
- 38.Clarke TK, et al. Common polygenic risk for autism spectrum disorder (ASD) is associated with cognitive ability in the general population. Mol Psychiatry 21, 419–425 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klein M, et al. Genetic Markers of ADHD-Related Variations in Intracranial Volume. Am J Psychiatry 176, 228–238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abu-Akel A, Allison C, Baron-Cohen S, Heinke D. The distribution of autistic traits across the autism spectrum: evidence for discontinuous dimensional subpopulations underlying the autism continuum. Mol Autism 10, 24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larsson H, Anckarsater H, Rastam M, Chang Z, Lichtenstein P. Childhood attention-deficit hyperactivity disorder as an extreme of a continuous trait: a quantitative genetic study of 8,500 twin pairs. J Child Psychol Psychiatry 53, 73–80 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Gandal MJ, et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Romer AL, et al. Structural alterations within cerebellar circuitry are associated with general liability for common mental disorders. Mol Psychiatry 23, 1084–1090 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ge SX, Jung D. ShinyGO: a graphical enrichment tool for ani-mals and plants. bioRxiv, 315150 (2018). [Google Scholar]
- 46.Grotzinger AD, et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat Hum Behav 3, 513–525 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.