

Abstract
Background:
Traumatic brain injury (TBI) is the leading cause of acquired neurologic disability in children. In our model of pediatric TBI, controlled cortical impact (CCI) in rat pups, Docosahexaenoic Acid (DHA) improved lesion volume and cognitive testing as late as Post Injury Day (PID) 50. DHA decreased pro-inflammatory mRNA in microglia and macrophages at PID 3 and 7, but not 30. We hypothesized that DHA affected inflammatory markers differentially relative to impact proximity, early and persistently after CCI.
Methods:
To provide a temporal snapshot of regional neuroinflammation, we measured TSPO (18-kDa Translocator Protein) binding using whole brain autoradiography at PID 3, 7, 30 and 50. Guided by TSPO results, we measured mRNA levels in contused cortex and underlying hippocampus for genes associated with pro-inflammatory and inflammation-resolving states at PID2 and 3.
Results:
CCI increased TSPO binding at all time points, most markedly at PID3 and in regions closest to impact, not blunted by DHA. CCI increased cortical and hippocampal mRNA pro-inflammatory markers, blunted by DHA at PID2 in hippocampus.
Conclusions:
CCI increased TSPO binding in the immature brain in a persistent manner more intensely with more severe injury, not altered by DHA. CCI increased PID2 and 3 mRNA levels of pro-inflammatory and inflammation-resolving genes. DHA decreased pro-inflammatory markers associated with inflammasome activation at PID2. We speculate that DHA’s salutary effects on long term outcomes result from early effects on the inflammasome. Future studies will examine functional effects of DHA on microglia both early and late after CCI.
Level of Evidence:
N/A. This is a not a clinical study but a basic science article that is randomized and controlled using rats. Extrapolating from Evidence Levels for clinical studies, the level of evidence would be II and the study type would be therapeutic.
Keywords: developing brain, traumatic brain injury, nutrition, polyunsaturated fatty acids, neuroprotection
1. Background
Nearly 40,000 children sustain severe traumatic brain injury (TBI) annually in the United States, adversely affecting as many as 1.3 million life-years. (1) Up to half of all children who survive severe TBI sustain major disability. (2) Neurologic disability stems directly from trauma and indirectly from secondary insults, such as free radical production and a pro-inflammatory brain response. Microglia (the brain’s resident immune cells) and infiltrating macrophages play key roles in free radical production and inflammation. Therapies that modulate oxidative stress and inflammation may ameliorate neurologic disability after TBI. (3, 4) For the developing brain, with its limited antioxidant reserve and vigorous inflammatory capacity, such therapies may be particularly neuroprotective. (5–10) At present, effective neuroprotectants for TBI are lacking.
Docosahexaenoic Acid, or DHA, is an appealing candidate therapy for pediatric TBI. DHA is a known antioxidant and anti-inflammatory agent. (11, 12) In vitro and in vivo, DHA modulates the brain’s immune cells away from a pro-inflammatory phenotype towards an anti-inflammatory, reparatory one.(13–15) A structural fatty acid component of brain membranes, DHA is required for normal development, growth and function of the nervous system. DHA is approved as a nutritional supplement for infants and children. (16, 17) Brain DHA loss occurs after TBI, supported by clinical and experimental data. (18–22) In adult mice after TBI, DHA depletion increased the pro-inflammatory response and neurologic dysfunction. (23) Post-TBI DHA loss could be particularly detrimental for the developing, rapidly growing brain after trauma.
Using our model of pediatric TBI, controlled cortical impact (CCI) in rat pups, we showed that rat pups in the DHACCI group (given DHA before or immediately after impact, followed by daily dosing) performed similarly on cognitive testing to those in the SHAMREG group (rats on a regular diet after surgery without injury) at post-injury day 50 (PID50). Similarly, lesion volume at PID 50 decreased in DHACCI rats relative to those treated with regular diet after CCI (REGCCI rats). (24) (25)
The objective of the current study was to test the hypothesis that DHA affected inflammatory markers differentially relative to impact proximity, early and persistently after CCI, as an explanation for benefits on cognitive function and lesion volume observed fifty days after injury. This question was raised because cellular level investigations did not allow us to determine if inflammatory gene expression differed as a function of distance from impact nor if it persisted beyond 7 days after CCI in some brain regions.
Cellular level investigations combined tissue from directly impacted cortex and hippocampus beneath to maximize the RNA obtained from sorted immune cells, such as microglia and macrophages, after flow cytometry processing. Cell counts were high enough to allow mRNA extraction from all target cells in CCI tissues at PID3, 7 and 30, but not for SHAM rats. DHA decreased pro-inflammatory gene mRNA levels in DHACCI rats compared to REGCCI rats at PID3 and 7. We could not determine if inflammation in CCI rats had resolved by PID30, because we lacked comparison SHAM mRNA, though it had greatly decreased. (25)
To provide a temporal and anatomic snapshot of inflammation, we measured TSPO (18-kDa Translocator Protein) binding on coronal sections of whole brain, a well-validated measure of brain inflammation. (26) We studied TSPO binding at PID 3, 7, 30 and 50 and selected three regions of interest (ROIs) for quantitative comparisons between DHACCI, REGCCI SHAMREG brains. We chose to use regular diet in the SHAM group because inflammatory gene mRNA did not differ between SHAMDHA and SHAMREG rat brain in our cellular work. (25)
2. Methods
2.1. Animals
All experimental protocols were approved by the Animal Care and Use Committees at the University of Utah, in accordance with US NIH guidelines and carried out at the University of Utah. All surgical procedures were performed using aseptic technique.
Briefly, male Sprague-Dawley rats were obtained from Charles Rivers Laboratories (Raleigh, NC) on post-natal day (P) 7–10. Rats were housed in litters of 10 with the lactating dam until weaning. After weaning, rats were housed 3—5 per cage and allowed free access to food and water. All cages were kept in a temperature- and light-controlled (12 h on/12 h off) environment.
Rats were fed one of two diets: the 0.1% DHA rodent diet (DHA) or the standard rat chow (REG). The DHA rodent diet (Harlan-Teklad, WI) substitutes 0.1% of the soybean oil in standard chow with purified DHA (U-84-A, Nu-Chek Prep, MN). This substitution results in the same macronutrient content and caloric density (3 kcal/g) as standard rat chow and provides DHA as 1.8% of total fat. All dams and pups were kept on REG diet (Harlan Teklad 8640) except for rats in the DHACCI group. DHA diet was provided to the dam on the day before CCI because 17- day old rat pups depend exclusively on dam milk for their nutrient intake and because gavage feeding rat pups soon after surgery carries a significant risk of pulmonary aspiration. We chose this diet start date guided by data that breast milk DHA peaked at 10hr and lasted 24hrs after ingestion of a DHA supplement. (27) At weaning at P21 rats consumed exclusively the DHA or REG chow as per treatment assigned, for the duration of each experiment.
2.2. CCI procedure
CCI was carried out as previously described. (28) Our model leads to severe injury as assessed by functional testing, imaging and histology in CCI rats, with maintained normal swimming and ambulating abilities. (28–31)
At P17, rats undergoing CCI were anesthetized with 3% Isoflurane for induction. Anesthesia was maintained with 2—2.5% Isoflurane for the duration of surgical preparation using a Vet Equip® Bench Top Isoflurane Anesthesia System (Pleasanton, CA). The rats’ core temperature was monitored via a rectal probe and continuously controlled at 37±0.5 °C using a servo-controlled heating pad. Each rat was placed into a stereotaxic frame (David Kopf, Tujunga, CA). After shaving, prepping with povidone-iodine and incising the scalp, a craniotomy (6-mm×6-mm) was performed over the left parietal cortex (centered at the point 4-mm posterior and 4-mm lateral to bregma). Care was taken not to perforate the dura. Once the craniotomy was complete, anesthesia was reduced to 1% Isoflurane for a 5-min equilibration period. CCI was then delivered (Pittsburgh Precision Instruments, Pittsburgh, PA) to the left parietal cortex using a 5-mm rounded tip to deliver a 2.0 mm deformation at 5 m/s velocity and 100 ms duration. Immediately after CCI, Isoflurane was increased to 2—2.5% and the bone flap was replaced and secured with dental cement (Patterson Dental, Salt Lake City, UT). The scalp incision was sutured and triple antibiotic ointment and bupivacaine 0.5% were applied topically. Isoflurane was stopped and rats allowed to recover in a temperature-controlled chamber. Once fully awake, rats were returned to their dams and littermates. SHAM rats underwent the same surgical craniotomy, equilibration, and closure procedures without CCI.
2.3. Autoradiography for TSPO (18 k Da Translocator Protein) Binding
For TSPO binding assays, eight rats from each of the three groups were used for each assay and time point (PID3, 7, 30 and 50). Rats were anesthetized with intraperitoneal xylazine (8mg/kg) and ketamine (40mg/kg) and euthanized by rapid decapitation. Rat brains were rapidly removed, flash frozen in powdered dry ice, and stored at −80°C until processing. Rat brains were shipped to Dr. Pauly’s laboratory at the University of Kentucky, where frozen tissue was sectioned at 16 μm on a Leica 1850M cryostat (Nussloch, Germany) and was thaw mounted onto Fisher SuperFrost Plus® slides. The slides were stored under desiccation overnight at 4°C then transferred to a −80°C freezer until time of experimentation.
TSPO autoradiography was performed as previously reported with mouse and rat TBI sections in Dr. Pauly’s lab. (32–34) The slides were removed from −80°C and allowed to thaw overnight and loaded into binding racks. The slides were incubated in the following buffers at 4°C: 50 mM Tris HCl (pH 7.4) for 15 minutes, 50 mM Tris HCl and 2 nM [3H]-PK11195 (PerkinElmer, Boston, MA, specific activity = 73.6 Ci/mmol) for 2 hours, 3 washes in 50 mM Tris HCl (pH 7.4) for 3 minutes each, and a brief wash in ddH2O. The slides were left overnight to dry at room temperature, placed into autoradiography cassettes and exposed to Kodak BioMax film for approximately 5 weeks. Brain regions analyzed were selected based on previous studies that have shown increased microglial activation following brain injury including the ipsilateral cortex, hippocampus and thalamus.(33–35). Regions were analyzed in a minimum of 3 sections per animal and averaged. Brain regions were outlined manually according to their anatomical boundaries, and for each brain region SHAMREG rat sections were measured to acquire the background level of intensity level. The threshold intensity value was then set at the highest grayscale value measured for any sham-operated rat (uncalibrated gray scale value of 80), and the percentage of pixels over threshold was recorded for each region.
2.4. Inflammatory Gene mRNA Studies
Rats were anesthetized with intraperitoneal xylazine (8mg/kg) and ketamine (40mg/kg) for tissue collection and euthanized by swift decapitation. Brains were removed and ipsilateral (left) hippocampi and cortices were dissected on ice, snap frozen in liquid nitrogen and stored at −80°C. Eight rats from each of the three groups were used for each assay and time point.
Inflammatory gene mRNA levels were measured by real-time RT-PCR. In brief, total RNA was extracted with Nucleospin II (Macherey-Nagel Inc. Bethlehem, PA), treated with DNase I (Ambion, Austin, TX), and quantified by spectrophotometry (Nano-Drop ND-1000, NanoDrop Technologies, DE). Sample integrity was confirmed by gel electrophoresis. cDNA was synthesized from 1 microgram of DNase-treated total RNA. cDNA- and gene-specific probe and primers were added to Taqman universal PCR master mix (PE Applied Biosystems), and samples were run on the 7900HT Sequence Detection System (Applied Biosystems Foster City, CA). Primers and probes were used from Applied Biosystems. Relative quantification of PCR products was based on value differences between the target and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) control by the comparative threshold cycle method (Taqman Gold RT-PCR manual, PE Applied Biosystems) also known as the 2^delta CT method. Cycle threshold, CT, is the number of cycles needed for fluorescent signal to rise above background; the higher the CT, the lower the expression level. The CT for GAPDH is subtracted from the CT for each gene and expressed logarithmically. (36)
We recognize that inflammatory gene expression alone does not capture the balance between damaging and beneficial effects of inflammation after brain injury. However, increased expression of certain genes is associated with predominantly damaging (pro-inflammatory) or beneficial (anti-inflammatory) effects after TBI. (37)
We assayed mRNA levels of the following pro-inflammatory genes: C-C motif chemokine ligand 2 (CCL2), NM_031530.1; CC motif Chemokine Receptor 2 (CCR2) NM_021866.1; Inducible Nitric Oxide Synthase (iNOS ), NM_012611.3; Tumor Necrosis Factor Alpha (TNFα), NM_012675.3; Interleukin 1 Beta (IL-1β), NM_031512.2; Interleukin 2 (IL-2), NM_053836.1); Interleukin 6 (IL-6), NM_012589.1; Interleukin 18 (IL-18), NM_019165.1); Interleukin 18 Receptor Accessory Protein (IL-18Rap), NM_184047.1; Interferon Gamma (INFγ), NM_ 138880.2.
We assayed mRNA levels of the following inflammation-resolving genes: Arginase 1 (Arg1), NM_012775; Mannose Receptor C, type 1 or Mrc1 (CD206), NM_001106123.2; Interleukin 1 Receptor Alpha (IL-1Rα), NM_013123.3; Interleukin 4 (IL-4), NM_201270.1; Interleukin 10 (IL-10); NM_012854.2; Transforming Growth Factor Beta Receptor 1 (TGFβR1), NM_012775.2.
For the internal control, we used Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), NM_017008. We chose GAPDH because CCI did not alter GAPDH expression neither in sorted cells nor tissue, unlike other housekeeping genes tested in our lab (β-actin and 18S RNA). The importance of using model-specific housekeeping genes has previously been shown. (38)
2.5. Statistics
Investigators blinded to experimental groups performed data acquisition and analyses. All data was distributed normally except for CCR2 and for hippocampal CCL2. For these two genes, we used Kruskal Wallis testing followed by Dunn’s test for multiple comparisons if significance was present. For the remaining, normally distributed, genes we used ANOVA. If standard deviations differed between groups, as it did for a minority of genes assayed, we used Brown-Forsythe one-way analysis of variance (ANOVA) followed by Dunnett’s test for multiple comparisons when p<0.05. Otherwise, we used ordinary ANOVA followed by Tukey’s test for multiple comparisons using GraphPad® Prism 8.43 (GraphPad® Software, CA) for all mRNA data. Comparisons between groups were limited to samples from the same hemisphere ipsilateral to injury and p<0.05 was defined as the cutoff for statistical significance. PK11195 binding data was analyzed using one-way ANOVA followed by Student Neuman-Keuls post hoc test if p was <0.05.
3. Results
3.1. DHA did not Blunt Increased TSPO Binding after CCI
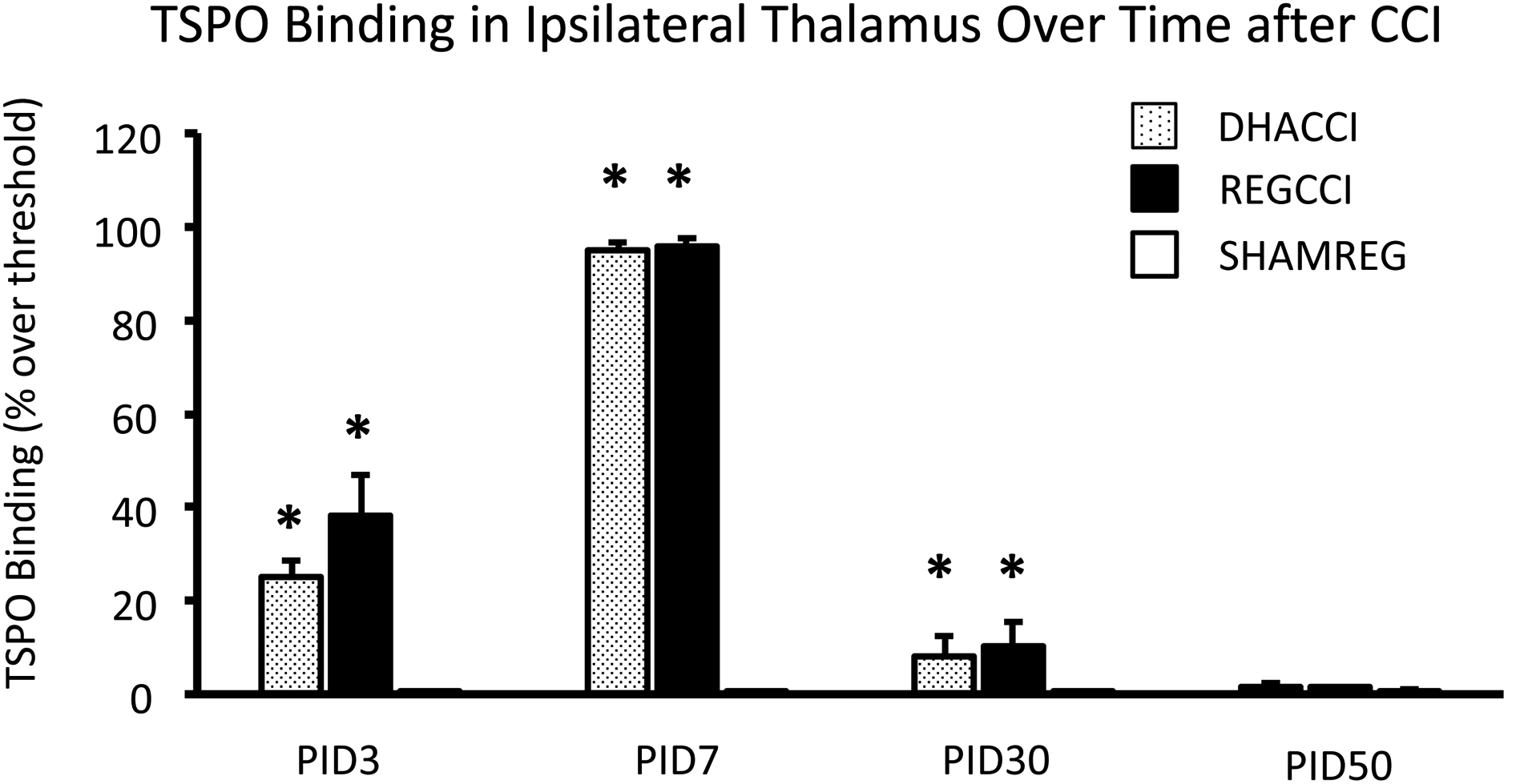
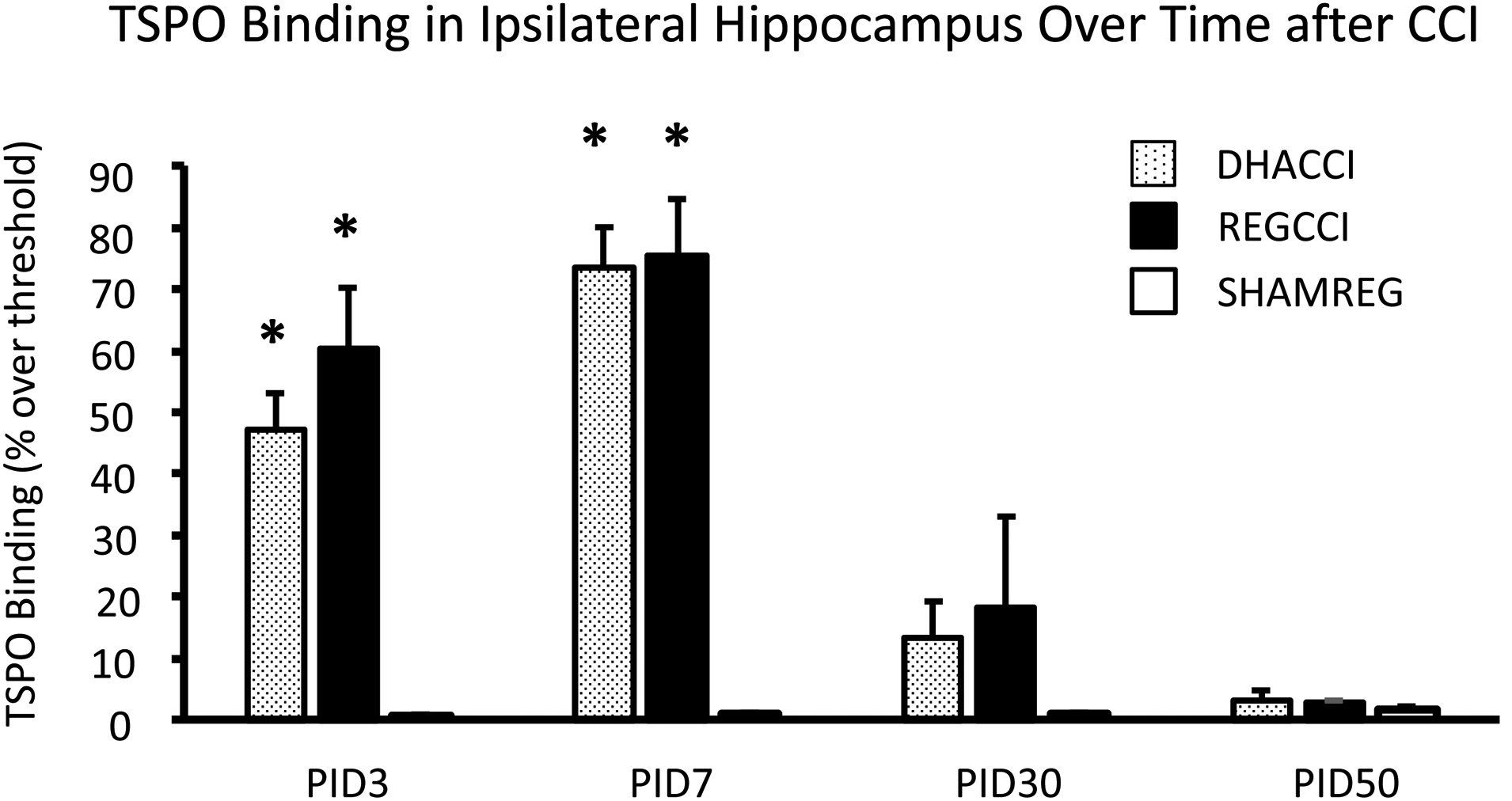
In cortex ipsilateral to impact, as shown in Figure 1A, CCI increased TSPO binding at PID3 (p<0.0001), 7 (p<0.0001), 30 (p<0.001) and 50 (p<0.0001). TSPO binding did not differ between DHACCI and REGCCI at any time point. Injury effect at PID3 was similar to that seen at PID 7; both were greater than at PID30 and 50 (p<0.001). Injury effect did not change between PID30 and 50. In ipsilateral thalamus, as shown in Figure 1B, injury increased TSPO binding at PID3, 7 and 30. Injury effect increased between PID3 and 7 (p<0.001). In ipsilateral hippocampus, as shown in Figure 1C, injury increased TSPO binding at PID3 and 7 (p<0.0001 for both time points) but not at PID30 nor 50. Injury effect was greater at PID7 than at PID3 (p<0.05) but greater at each time point than at PID30 (p<0.001 and p<0.0001, respectively). Representative images of DHACCI, REGCCI and SHAMREG brain autoradiography are shown for all four time points in Supplementary Figure 1.
Figure 1. Increased TSPO binding Ipsilateral to CCI persisted to PID 50 in Cortex, to PID 30 in Thalamus and to PID7 in Hippocampus in DHACCI and REGCCI Rat Brains.
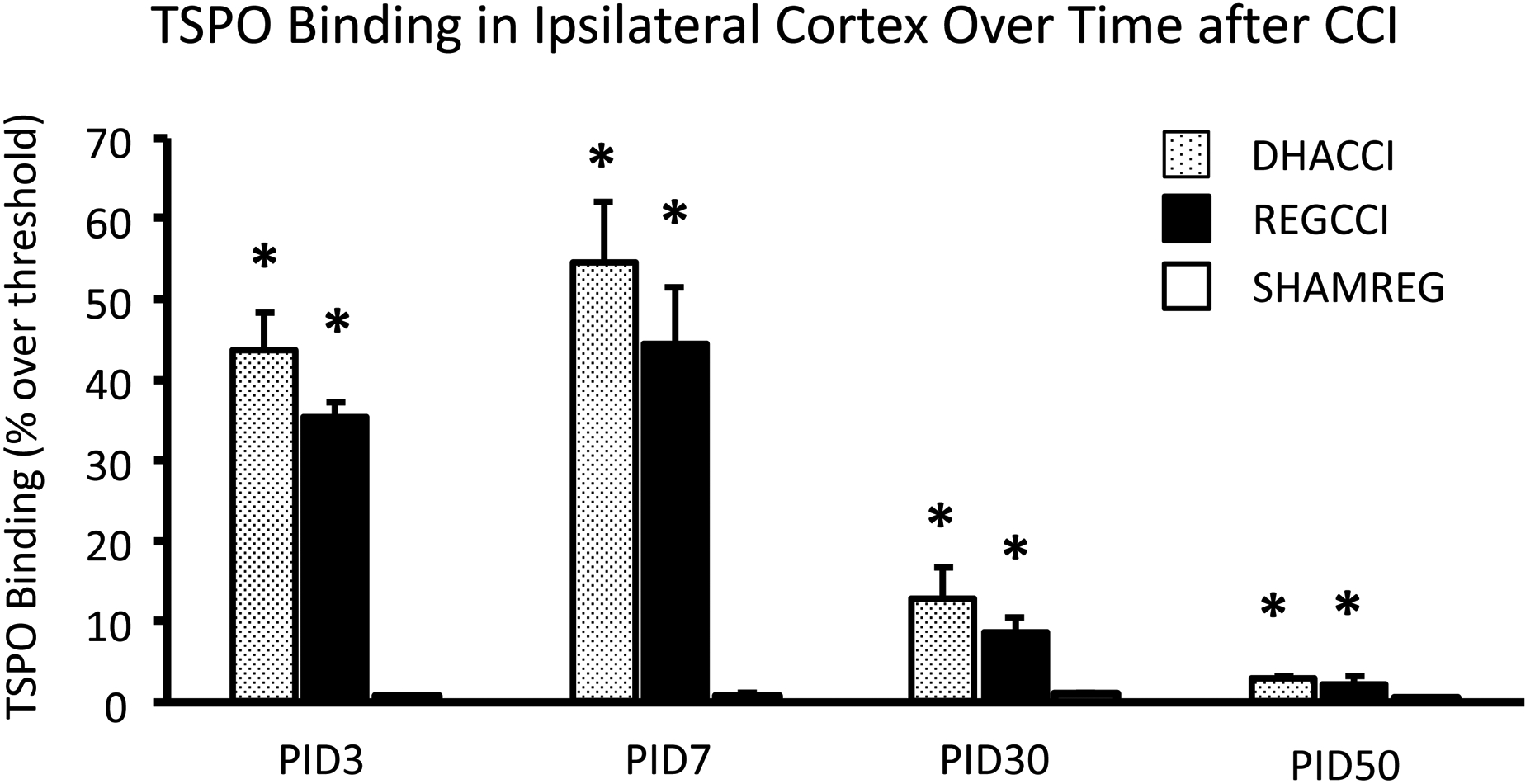
A. Injury increased TSPO binding in ipsilateral Cortex at PID3 (p<0.0001), 7 (p<0.0001), 30 (p<0.001) and 50 (p<0.0001) with no difference between DHACCI and REGCCI at any time point. Injury effect was similar between the two early time points and greater at both these time points than at PID30 and 50 (p<0.001). Injury effect did not change between PID30 and 50.
B. Injury increased TSPO binding in ipsilateral Thalamus at PID3, 7 and 30. Injury effect increased between PID3 and 7 (p<0.001) but for each of these early time points was greater than at PID30.
C. Injury increased TSPO binding in ipsilateral Hippocampus at PID3 and 7 (p<0.0001 for both time points) but not at PID30 nor 50. Injury effect was greater at PID7 than at PID3 (p<0.05) but greater at each time point than at PID30 (p<0.001 and p<0.0001, respectively).
* =p<0.05 relative to SHAMREG. DHACCI is shown in dotted black, REGCCI in black and SHAMREG in white bars.
Abbreviations: TSPO (18 k Da Translocator Protein)
3.2. DHA decreased CCI-induced Elevation in Hippocampal TNFα, IL-1β and Cortical IL-18rap mRNA at PID2
Injury increased hippocampal and cortical mRNA levels of CCL2 and CCR2 on PID2 and 3 (p< 0.001 by Kruskal Wallis tests for all) without differences between CCI groups. ANOVA was performed for the remaining genes. Hippocampal and cortical mRNA levels of pro-inflammatory genes are shown in Figures 2A and 2B, respectively.
Figure 2A. DHA decreased CCI-induced Elevation in Hippocampal TNFα, IL-1β mRNA at PID2.
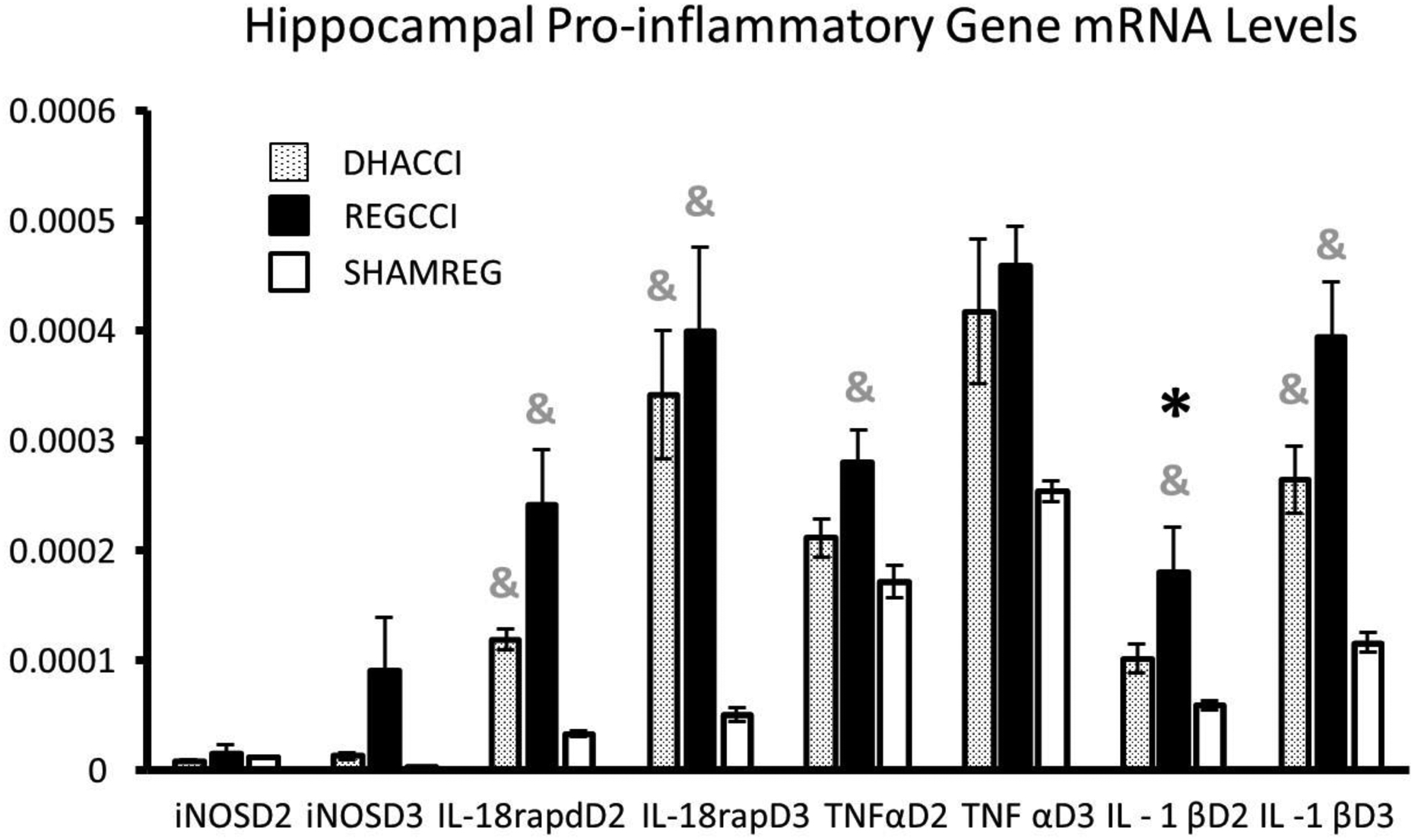
Results for each of the hippocampal pro-inflammatory genes at post injury days (PID) 2 and 3 are shown.
Results are expressed using the 2−ΔCT method relative to GAPDH as mRNA ±SEM, n=8/group.
* =p<0.05 relative to DHACCI and &= p<0.05 relative to SHAMREG. DHACCI is shown in dotted black, REGCCI in black and SHAMREG in white bars.
Abbreviations: TNFα (Tumor Necrosis Factor-α), IL-18rap (Interleukin 18 Receptor Accessory Protein), IL-18 (Interleukin 18), IL-1β (Interleukin 1 β), iNOs (Inducible Nitric Oxide Synthase)
Figure 2B. DHA decreased CCI-induced Elevation in Cortical IL-18rap mRNA at PID2.
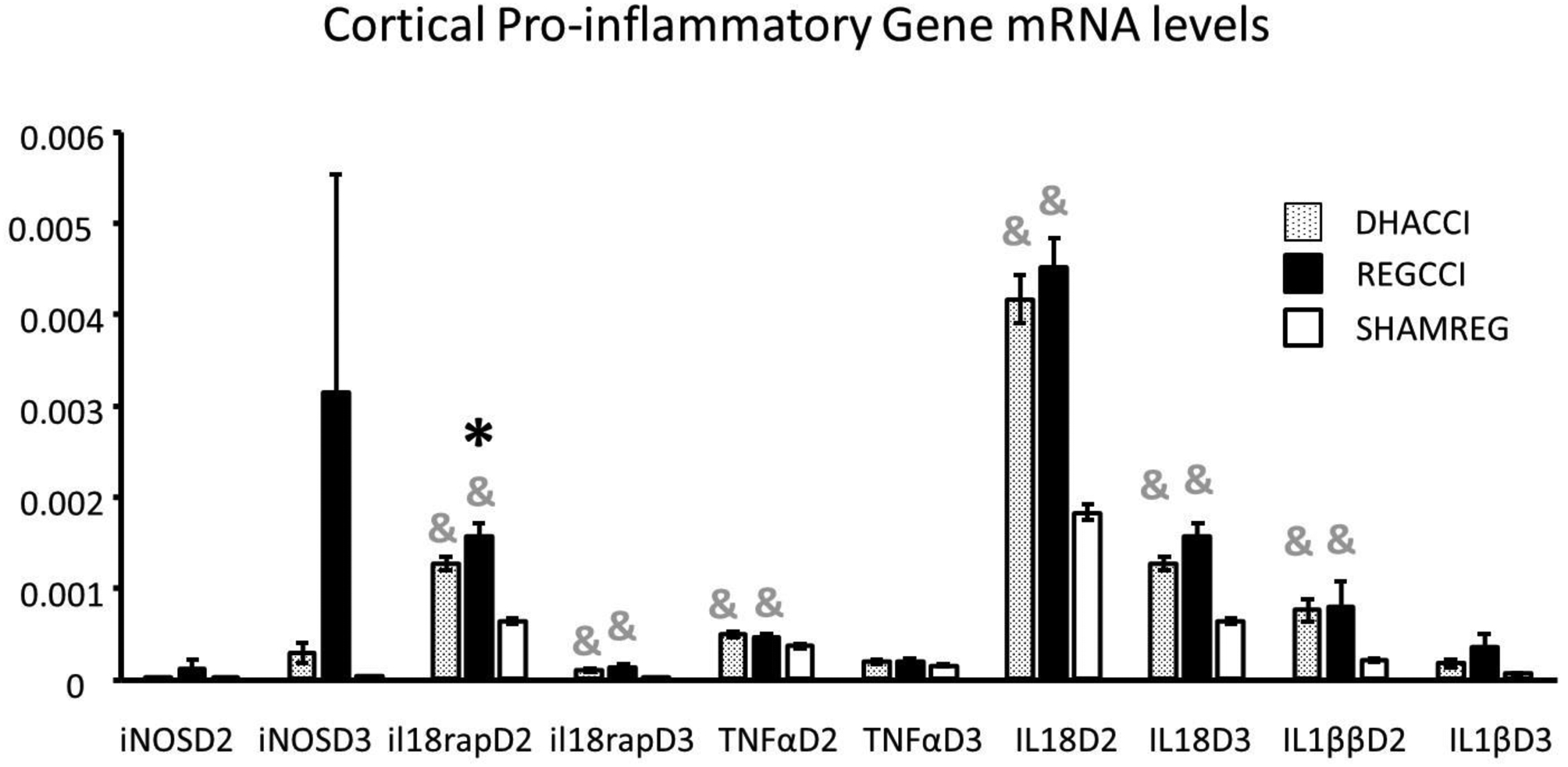
Results for each of the cortical pro-inflammatory genes at post injury days (PID) 2 and 3 are shown.
Results are expressed using the 2−ΔCT method relative to GAPDH as mRNA ±SEM, n=8/group.
* =p<0.05 relative to DHACCI and &= p<0.05 relative to SHAMREG. DHACCI is shown in dotted black, REGCCI in black and SHAMREG in white bars.
Abbreviations: TNFα (Tumor Necrosis Factor-α), IL-18rap (Interleukin 18 Receptor Accessory Protein), IL-18 (Interleukin 18), IL-1β (Interleukin 1 β), iNOs (Inducible Nitric Oxide Synthase)
As noted in Figure 2A, TNFα increased in REGCCI hippocampus (p=0.006) but not in DHACCI relative to SHAMREG on PID2 and trended downwards for DHACCI relative to REGCCI (p=0.09). At PID3, hippocampal TNFα mRNA levels rose in both CCI rats relative to SHAMREG (p<0.05 for each cci group). Hippocampal IL-1 β mRNA levels increased at PID2 in REGCCI relative to DHACCI (p=0.0005) but not in DHACCI relative to SHAMREG. Hippocampal IL-1β and IL-18 mRNA rose in both CCI groups at PID3 (p<0.0001). Hippocampal IL-18 rap increased in both CCI groups at PID2 and 3, with slight blunting by DHA at PID2 (DHACCI trended towards lower than REGCCI, p=0.1).
As noted in Figure 2B, cortical TNFα mRNA rose in both CCI groups at PID2 (p<0.01) but not at PID3. Cortical IL-1β increased in both CCI groups on PID2 (p=0.002) while at PID3 IL-1β trended towards an increase in REGCCI, but not in DHACCI (p=0.06). CCI increased cortical IL-18 on PID2 and 3 (p<0.0001). CCI increased cortical IL-18rap mRNA levels in both groups at PID2 and 3, but in DHACCI decreased relative to REGCCI (p= 0.04) at PID2. CCI injury did not affect iNOS, IL2, IL6 nor INF γ mRNA levels though an upwards trend in REGCCI rats occurred for hippocampal iNOS mRNA levels on PID3 (p=0.09).
3.3. Docosahexaenoic Acid did not affect Anti-inflammatory mRNA levels at PID2 nor PID3
CCI injury did not affect IL-4 mRNA hippocampal nor cortical mRNA levels at PID2 nor PID3, and increased Arg1 only in the PID3 cortex (p<0.001). CCI increased IL-1rα mRNA and CD206 hippocampal and cortical mRNA, and hippocampal TGF β mRNA, on both days (p<0.05 for all) but increased IL10 only in the PID2 hippocampus without differences between CCI groups. In hippocampus, DHACCI TGF β mRNA levels showed a trend towards a decrease relative to REGCCI (p=0.06) at PID2. In cortex, CCI injury increased TGF β mRNA at PID2 and 3 with no differences between CCI groups.
4. Discussion
We hypothesized that DHA’s long term neuroprotective effects were associated with early and persistent effects on inflammatory gene expression in brain regions with different levels of injury. We had previously noted that DHA improved poor performance on cognitive testing and hemispheric tissue loss at 50 days after injury. We had not been able to study spatial nor temporal effects of DHA on inflammation because our mRNA data was limited to cell populations. To address this question, we first used whole hemispheres sliced coronally to investigate TSPO binding at four time points to study temporal effects of DHA on specific anatomic regions important in CCI. (33–35). Injury increased TSPO binding most markedly at day 3, so we studied inflammatory gene expression at day 2 and 3 in dissected tissue from the contused cortex closest to impact and the underlying hippocampus.
We found that CCI increased rat pup brain TSPO binding in a region-specific manner in both treatment groups relative to SHAM control, lasting for up to 7 days in the hippocampus, 30 days in the thalamus and 50 days in the cerebral cortex. To our knowledge, ours is the first study to report TSPO binding after experimental TBI in the immature brain. This finding is important because it suggests that PET (Positron Emission Tomography) could be used for clinical evaluation of inflammation after pediatric TBI. Ligands for TSPO have been used for imaging ex vivo (autoradiography) and in vivo (Positron Emission Tomography or PET) to study experimental and clinical neuroinflammation. (39, 40)
The 18-kDa Translocator Protein (previously known as the peripheral benzodiazepine receptor), is located on the outer mitochondrial membrane. TSPO is expressed in the immature and the mature brain, but under basal conditions expression is limited to the choroid plexus and the ependymal cells that line the cerebral ventricles. (41) Previously thought to reflect only microglial activation, it is now known that TSPO binding is a measure of astrocyte and microglia-driven inflammation. (42, 43) The ligand used in our study, [3H]PK 11195, binds with high specificity in the immature rat brain in vivo and in cultured rat brain cells making it unlikely that lack of DHA effect on TSPO binding could be explained by underestimation of true binding in our study. (41) Others have also reported DHA to confer neuroprotection without affecting TSPO binding after spinal cord injury in a rat model. (44) Alternatively, DHA’s lack of effect upon TSPO binding in our study could result from differential effects on astrocytes and microglia. TSPO binding over time could reflect activity of astrocytes, microglia, or both. (45, 46) We had previously shown that DHA affected histologic microglial, but not astrocytic markers, early after CCI. (25) Finally, DHA may have more potent effects on TSPO binding after mild TBI than after severe, because DHA decreased TSPO binding after experimental concussion. (22) Our model produces severe injury, defined by functional deficits and anatomic parameters. (28–31)
TSPO binding was most intense in the regions closest to impact. CCI increased TSPO binding most persistently in the cortex, followed by thalamus and hippocampus. CCI injury increases with proximity to impact. (47)Therefore, our result is consistent with the concept that inflammation increases with severity of injury after CCI. Similarly, distance from impact did modify inflammatory mRNA levels. In the cortex, more proximal to impact, mRNA levels in CCI brains were much higher relative to SHAM than were hippocampal levels. Possibly because injury was less severe, DHA effects on CCI-induced mRNA changes were more discernable in the hippocampus than in cortex. For example, DHA abrogated CCI effects on hippocampal IL-1 β but only mildly blunted CCI effects on cortical Il-18rap.
At PID2, DHA abrogated CCI-induced elevations in hippocampal IL-1β mRNA and blunted increased hippocampal TNF α and cortical IL-18rap. The cytokines IL-1 β and IL-18 are major products of the inflammasome, a cytosolic protein complex central to TBI pathophysiology. (48) IL18rap is required for cytokine amplification after inflammasome activation. (49) Inflammasome activation after TBI releases IL-1β and IL-18, leading to cell death from pyroptosis and nitric oxide release. (50, 51) Neutralizing antibodies to IL-1 β and IL-18 inhibition improved outcomes in mice in different models of TBI. (52, 53) In a mouse model of pediatric TBI using CCI at day 21 of life, IL-1 β blockade decreased post-traumatic epilepsy, reduced cortical volume loss and improved cognitive deficits after CCI. (54) Finally, TNF α appears to play an important role in inflammasome activation. (55)
DHA and other omega-3 fatty acids inhibit inflammasome activation in macrophages and other cell types, particularly via the NLRP3 pathway, reducing IL-1 β. (56, 57) Semple et al demonstrated powerful, long-term functional and anatomic neuroprotection after acute and relatively short-term IL-1 β inhibition. (54) DHA effects on IL-1 β mRNA suggests that DHA could modulate the inflammasome early after CCI to produce long-term benefits. In future studies, we will explore histologic markers of inflammasome activation including cleaved Caspase-1 and ASC (apoptosis-associated speck-like protein containing a CARD) to evaluate this possibility further.
Our study is not without limitations. For example, we tested males at a single developmental age (17-day old male rats) utilizing one experimental model of TBI, CCI. To address some of these limitations, we are currently conducting studies on DHA effects in female rat pups and in a fluid percussion large-animal model. We did not address effects of DHA on TBI during other time points during brain development. DHA is neuroprotective in adult rats after TBI and in neonatal rats after hypoxic brain injury, which suggests that our results may apply across a wider range of developmental ages. (58, 59) Similarly, we did not directly test for adverse effects of DHA in our model because DHA is designated GRAS (Generally Regarded as Safe) by the Food and Drug Administration (FDA) after extensive research in humans and animals across the lifespan. (60) In addition, randomized placebo-controlled trial of omega-3 fatty acids, including DHA, did not find safety concerns in patients with acute lung injury nor after severe trauma. (61–63) Finally, we did not include a SHAMDHA group in this study because our previous work did not find differences in inflammatory gene expression between SHAMDHA and SHAMREG rats. In addition, we are currently using imaging, cognitive testing and other assays of DHA effect after TBI employing eight groups to include SHAMDHA and SHAMREG rats of both sexes. We acknowledge as another limitation that our results could reflect DHA given before injury, even though DHA was given to the dam and not the pups, because we do not know at what time point DHA levels could have been detectable in the dam milk. Our present and future studies do not use pre-injury DHA but rather post-injury DHA via intraperitoneal injection. (25)
In conclusion, we found that DHA’s long-term neuroprotection is associated with inflammatory gene expression changes suggesting decreased inflammasome activation at day 2 after injury. Studies showing long term benefits of acute and short-term IL-1β inhibition support the possibility that DHA’s long term neuroprotection could stem from early effects on inflammation. We can also conclude that the developing brain mounts a greater inflammatory response as injury severity increases. Finally, we demonstrated for the first time that experimental TBI in the immature brain increases TSPO binding. We speculate that PET imaging using TSPO ligands may be useful to study inflammation after pediatric TBI in the clinical setting. We recognize that inflammation is a highly complex and dynamic response for which markers fall short. We are currently studying DHA effects on inflammation-related proteins and functional assays of the immune response to CCI. Future work will include studying DHA effects on inflammasome activation to uncover potential mechanisms of DHA neuroprotection after developmental TBI. The current results will help us design future mechanism-based clinical trials of DHA in pediatric TBI. Based on our findings, we plan to include measuring IL-1β and IL-18 levels in serum and cerebrospinal fluid and will consider PET imaging to evaluate inflammation after pediatric TBI.
Supplementary Material
Supplementary Figure 1
Representative TSPO Binding Autoradiography Images
[3H]PK11195 autoradiography was performed on rat pup brains at 3, 7, 30 and 50 days after CCI or sham surgery, treated with DHA (DHACCI) or regular diet (REGCCI and SHAMREG). Representative coronal sections from each of the three groups at the four time points are shown, with relative binding densities ranging from minimal (blue), to moderate (yellow, green), to maximal (red). Both CCI groups exhibited focal regions of increased activation, while uninjured sham rats displayed minimal [3H]PK11195 binding.
Abbreviations: TSPO (18 k Da Translocator Protein)
Funding:
This work was supported by the NINDS Grant number 5R21 NS090098-02, National Institutes of Health and by Pediatric Critical Care Medicine at the University of Utah. Neither funding agency had any role in the study design, data collection, analysis, and interpretation, writing of the report nor in the decision to submit the article for publication.
Footnotes
Declarations of interest: none.
REFERENCES
- 1.Popernack ML, Gray N, Reuter-Rice K. Moderate-to-Severe Traumatic Brain Injury in Children: Complications and Rehabilitation Strategies. J Pediatr Health Care. 2015;29(3):e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vavilala MS, Kernic MA, Wang J, Kannan N, Mink RB, Wainwright MS, Groner JI, Bell MJ, Giza CC, Zatzick DF, et al. Acute care clinical indicators associated with discharge outcomes in children with severe traumatic brain injury. Crit Care Med. 2014;42(10):2258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48(6):1393–9; discussion 9–401. [DOI] [PubMed] [Google Scholar]
- 4.Needham EJ, Helmy A, Zanier ER, Jones JL, Coles AJ, Menon DK. The immunological response to traumatic brain injury. J Neuroimmunol. 2019;332:112–25. [DOI] [PubMed] [Google Scholar]
- 5.Bayir H, Kochanek PM, Kagan VE. Oxidative stress in immature brain after traumatic brain injury. Dev Neurosci. 2006;28(4–5):420–31. [DOI] [PubMed] [Google Scholar]
- 6.Ikonomidou C, Kaindl AM. Neuronal death and oxidative stress in the developing brain. Antioxid Redox Signal. 2011;14(8):1535–50. [DOI] [PubMed] [Google Scholar]
- 7.Anthony D, Dempster R, Fearn S, Clements J, Wells G, Perry VH, Walker K. CXC chemokines generate age-related increases in neutrophil-mediated brain inflammation and blood-brain barrier breakdown. Curr Biol. 1998;8(16):923–6. [DOI] [PubMed] [Google Scholar]
- 8.Anthony DC, Bolton SJ, Fearn S, Perry VH. Age-related effects of interleukin-1 beta on polymorphonuclear neutrophil-dependent increases in blood-brain barrier permeability in rats. Brain. 1997;120 ( Pt 3):435–44. [DOI] [PubMed] [Google Scholar]
- 9.Potts MB, Koh SE, Whetstone WD, Walker BA, Yoneyama T, Claus CP, Manvelyan HM, Noble-Haeusslein LJ. Traumatic injury to the immature brain: inflammation, oxidative injury, and iron-mediated damage as potential therapeutic targets. NeuroRx. 2006;3(2):143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Claus CP, Tsuru-Aoyagi K, Adwanikar H, Walker B, Manvelyan H, Whetstone W, Noble-Haeusslein LJ. Age is a determinant of leukocyte infiltration and loss of cortical volume after traumatic brain injury. Dev Neurosci. 2010;32(5–6):454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siriwardhana N, Kalupahana NS, Moustaid-Moussa N. Health benefits of n-3 polyunsaturated fatty acids: eicosapentaenoic acid and docosahexaenoic acid. Adv Food Nutr Res. 2012;65:211–22. [DOI] [PubMed] [Google Scholar]
- 12.Giacobbe J, Benoiton B, Zunszain P, Pariante CM, Borsini A. The Anti-Inflammatory Role of Omega-3 Polyunsaturated Fatty Acids Metabolites in Pre-Clinical Models of Psychiatric, Neurodegenerative, and Neurological Disorders. Front Psychiatry. 2020;11:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fourrier C, Remus-Borel J, Greenhalgh AD, Guichardant M, Bernoud-Hubac N, Lagarde M, Joffre C, Laye S. Docosahexaenoic acid-containing choline phospholipid modulates LPS-induced neuroinflammation in vivo and in microglia in vitro. J Neuroinflammation. 2017;14(1):170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang PK, Khatchadourian A, McKinney RA, Maysinger D. Docosahexaenoic acid (DHA): a modulator of microglia activity and dendritic spine morphology. J Neuroinflammation. 2015;12(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai W, Liu S, Hu M, Sun X, Qiu W, Zheng S, Hu X, Lu Z. Post-stroke DHA Treatment Protects Against Acute Ischemic Brain Injury by Skewing Macrophage Polarity Toward the M2 Phenotype. Transl Stroke Res. 2018;9(6):669–80. [DOI] [PubMed] [Google Scholar]
- 16.Innis SM. Dietary (n-3) fatty acids and brain development. J Nutr. 2007;137(4):855–9. [DOI] [PubMed] [Google Scholar]
- 17.Lauritzen L, Brambilla P, Mazzocchi A, Harslof LB, Ciappolino V, Agostoni C. DHA Effects in Brain Development and Function. Nutrients. 2016;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdullah L, Evans JE, Ferguson S, Mouzon B, Montague H, Reed J, Crynen G, Emmerich T, Crocker M, Pelot R, et al. Lipidomic analyses identify injury-specific phospholipid changes 3 mo after traumatic brain injury. FASEB J. 2014;28(12):5311–21. [DOI] [PubMed] [Google Scholar]
- 19.Requena DF, Maschek JA, Cox J, Parra L, Lolofie A, Schober ME. Effects of Controlled Cortical Impact and Docosahexaenoic Acid on Rat Pup Fatty Acid Profiles. Behav Brain Res. 2019:112295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farias SE, Heidenreich KA, Wohlauer MV, Murphy RC, Moore EE. Lipid mediators in cerebral spinal fluid of traumatic brain injured patients. J Trauma. 2011;71(5):1211–8. [DOI] [PubMed] [Google Scholar]
- 21.Pilitsis JG, Coplin WM, O’Regan MH, Wellwood JM, Diaz FG, Fairfax MR, Michael DB, Phillis JW. Free fatty acids in cerebrospinal fluids from patients with traumatic brain injury. Neurosci Lett. 2003;349(2):136–8. [DOI] [PubMed] [Google Scholar]
- 22.Butt CM, Harrison JL, Rowe RK, Jones J, Wynalda KM, Salem N, Lifshitz J, Pauly JR. Selective Reduction of Brain Docosahexaenoic Acid after Experimental Brain Injury and Mitigation of Neuroinflammatory Outcomes with Dietary DHA. Curr Res Concussion. 2017;04(01):e38–e54. [Google Scholar]
- 23.Desai A, Kevala K, Kim HY. Depletion of brain docosahexaenoic acid impairs recovery from traumatic brain injury. PLoS One. 2014;9(1):e86472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schober ME, Requena DF, Abdullah OM, Casper TC, Beachy J, Malleske D, Pauly JR. Dietary Docosahexaenoic Acid Improves Cognitive Function, Tissue Sparing, and Magnetic Resonance Imaging Indices of Edema and White Matter Injury in the Immature Rat after Traumatic Brain Injury. J Neurotrauma. 2016;33(4):390–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schober ME, Requena DF, Casper TC, Velhorst AK, Lolofie A, McFarlane KE, Otto TE, Terry C, Gensel JC. Docosahexaenoic acid decreased neuroinflammation in rat pups after controlled cortical impact. Exp Neurol. 2019;320:112971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schain M, Kreisl WC. Neuroinflammation in Neurodegenerative Disorders-a Review. Curr Neurol Neurosci Rep. 2017;17(3):25. [DOI] [PubMed] [Google Scholar]
- 27.Lauritzen L, Jorgensen MH, Hansen HS, Michaelsen KF. Fluctuations in human milk long-chain PUFA levels in relation to dietary fish intake. Lipids. 2002;37(3):237–44. [DOI] [PubMed] [Google Scholar]
- 28.Schober ME, Block B, Beachy JC, Statler KD, Giza CC, Lane RH. Early and sustained increase in the expression of hippocampal IGF-1, but not EPO, in a developmental rodent model of traumatic brain injury. J Neurotrauma. 2010;27(11):2011–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adelson PD, Fellows-Mayle W, Kochanek PM, Dixon CE. Morris water maze function and histologic characterization of two age-at-injury experimental models of controlled cortical impact in the immature rat. Childs Nerv Syst. 2013;29(1):43–53. [DOI] [PubMed] [Google Scholar]
- 30.Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39(3):253–62. [DOI] [PubMed] [Google Scholar]
- 31.Dixon CE, Kochanek PM, Yan HQ, Schiding JK, Griffith RG, Baum E, Marion DW, DeKosky ST. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J Neurotrauma. 1999;16(2):109–22. [DOI] [PubMed] [Google Scholar]
- 32.Guseva MV, Hopkins DM, Scheff SW, Pauly JR. Dietary choline supplementation improves behavioral, histological, and neurochemical outcomes in a rat model of traumatic brain injury. J Neurotrauma. 2008;25(8):975–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelso ML, Scheff SW, Pauly JR, Loftin CD. Effects of genetic deficiency of cyclooxygenase-1 or cyclooxygenase-2 on functional and histological outcomes following traumatic brain injury in mice. BMC Neurosci. 2009;10:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelso ML, Wehner JM, Collins AC, Scheff SW, Pauly JR. The pathophysiology of traumatic brain injury in alpha7 nicotinic cholinergic receptor knockout mice. Brain Res. 2006;1083(1):204–10. [DOI] [PubMed] [Google Scholar]
- 35.Raghavendra Rao VL, Dogan A, Bowen KK, Dempsey RJ. Traumatic brain injury leads to increased expression of peripheral-type benzodiazepine receptors, neuronal death, and activation of astrocytes and microglia in rat thalamus. Exp Neurol. 2000;161(1):102–14. [DOI] [PubMed] [Google Scholar]
- 36.Gubern C, Hurtado O, Rodriguez R, Morales JR, Romera VG, Moro MA, Lizasoain I, Serena J, Mallolas J. Validation of housekeeping genes for quantitative real-time PCR in in-vivo and in-vitro models of cerebral ischaemia. BMC Mol Biol. 2009;10:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jha MK, Lee WH, Suk K. Functional polarization of neuroglia: Implications in neuroinflammation and neurological disorders. Biochem Pharmacol. 2016;103:1–16. [DOI] [PubMed] [Google Scholar]
- 38.Rhinn H, Marchand-Leroux C, Croci N, Plotkine M, Scherman D, Escriou V. Housekeeping while brain’s storming Validation of normalizing factors for gene expression studies in a murine model of traumatic brain injury. BMC Mol Biol. 2008;9:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9(12):971–88. [DOI] [PubMed] [Google Scholar]
- 40.Kim SW, Wiers CE, Tyler R, Shokri-Kojori E, Jang YJ, Zehra A, Freeman C, Ramirez V, Lindgren E, Miller G, et al. Influence of alcoholism and cholesterol on TSPO binding in brain: PET [(11)C]PBR28 studies in humans and rodents. Neuropsychopharmacology. 2018;43(9):1832–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Itzhak Y, Roig-Cantisano A, Norenberg MD. Ontogeny of peripheral-type benzodiazepine receptors in cultured astrocytes and brain from rat. Brain Res Dev Brain Res. 1995;84(1):62–6. [DOI] [PubMed] [Google Scholar]
- 42.Chechneva OV, Deng W. Mitochondrial translocator protein (TSPO), astrocytes and neuroinflammation. Neural Regen Res. 2016;11(7):1056–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lavisse S, Guillermier M, Herard AS, Petit F, Delahaye M, Van Camp N, Ben Haim L, Lebon V, Remy P, Dolle F, et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci. 2012;32(32):10809–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tremoleda JL, Thau-Zuchman O, Davies M, Foster J, Khan I, Vadivelu KC, Yip PK, Sosabowski J, Trigg W, Michael-Titus AT. In vivo PET imaging of the neuroinflammatory response in rat spinal cord injury using the TSPO tracer [(18)F]GE-180 and effect of docosahexaenoic acid. Eur J Nucl Med Mol Imaging. 2016;43(9):1710–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ji B, Maeda J, Sawada M, Ono M, Okauchi T, Inaji M, Zhang MR, Suzuki K, Ando K, Staufenbiel M, et al. Imaging of peripheral benzodiazepine receptor expression as biomarkers of detrimental versus beneficial glial responses in mouse models of Alzheimer’s and other CNS pathologies. J Neurosci. 2008;28(47):12255–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maeda J, Higuchi M, Inaji M, Ji B, Haneda E, Okauchi T, Zhang MR, Suzuki K, Suhara T. Phase-dependent roles of reactive microglia and astrocytes in nervous system injury as delineated by imaging of peripheral benzodiazepine receptor. Brain Res. 2007;1157:100–11. [DOI] [PubMed] [Google Scholar]
- 47.Mao H, Jin X, Zhang L, Yang KH, Igarashi T, Noble-Haeusslein LJ, King AI. Finite element analysis of controlled cortical impact-induced cell loss. J Neurotrauma. 2010;27(5):877–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Brien WT, Pham L, Symons GF, Monif M, Shultz SR, McDonald SJ. The NLRP3 inflammasome in traumatic brain injury: potential as a biomarker and therapeutic target. J Neuroinflammation. 2020;17(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheung H, Chen NJ, Cao Z, Ono N, Ohashi PS, Yeh WC. Accessory protein-like is essential for IL-18-mediated signaling. J Immunol. 2005;174(9):5351–7. [DOI] [PubMed] [Google Scholar]
- 50.Voet S, Srinivasan S, Lamkanfi M, van Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med. 2019;11(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wawrocki S, Druszczynska M, Kowalewicz-Kulbat M, Rudnicka W. Interleukin 18 (IL-18) as a target for immune intervention. Acta Biochim Pol. 2016;63(1):59–63. [DOI] [PubMed] [Google Scholar]
- 52.Clausen F, Hanell A, Bjork M, Hillered L, Mir AK, Gram H, Marklund N. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur J Neurosci. 2009;30(3):385–96. [DOI] [PubMed] [Google Scholar]
- 53.Yatsiv I, Morganti-Kossmann MC, Perez D, Dinarello CA, Novick D, Rubinstein M, Otto VI, Rancan M, Kossmann T, Redaelli CA, et al. Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. J Cereb Blood Flow Metab. 2002;22(8):971–8. [DOI] [PubMed] [Google Scholar]
- 54.Semple BD, O’Brien TJ, Gimlin K, Wright DK, Kim SE, Casillas-Espinosa PM, Webster KM, Petrou S, Noble-Haeusslein LJ. Interleukin-1 Receptor in Seizure Susceptibility after Traumatic Injury to the Pediatric Brain. J Neurosci. 2017;37(33):7864–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci. 2014;8:315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dumont A, de Rosny C, Kieu TL, Perrey S, Berger H, Fluckiger A, Muller T, Pais de Barros JP, Pichon L, Hichami A, et al. Docosahexaenoic acid inhibits both NLRP3 inflammasome assembly and JNK-mediated mature IL-1beta secretion in 5-fluorouracil-treated MDSC: implication in cancer treatment. Cell Death Dis. 2019;10(7):485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wierenga KA, Wee J, Gilley KN, Rajasinghe LD, Bates MA, Gavrilin MA, Holian A, Pestka JJ. Docosahexaenoic Acid Suppresses Silica-Induced Inflammasome Activation and IL-1 Cytokine Release by Interfering With Priming Signal. Front Immunol. 2019;10:2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bailes JE, Mills JD. Docosahexaenoic acid reduces traumatic axonal injury in a rodent head injury model. J Neurotrauma. 2010;27(9):1617–24. [DOI] [PubMed] [Google Scholar]
- 59.Berman DR, Mozurkewich E, Liu Y, Shangguan Y, Barks JD, Silverstein FS. Docosahexaenoic acid augments hypothermic neuroprotection in a neonatal rat asphyxia model. Neonatology. 2013;104(1):71–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lien EL. Toxicology and safety of DHA. Prostaglandins Leukot Essent Fatty Acids. 2009;81(2–3):125–32. [DOI] [PubMed] [Google Scholar]
- 61.Stapleton RD, Martin TR, Weiss NS, Crowley JJ, Gundel SJ, Nathens AB, Akhtar SR, Ruzinski JT, Caldwell E, Curtis JR, et al. A phase II randomized placebo-controlled trial of omega-3 fatty acids for the treatment of acute lung injury. Crit Care Med. 2011;39(7):1655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chuntrasakul C, Siltham S, Sarasombath S, Sittapairochana C, Leowattana W, Chockvivatanavanit S, Bunnak A. Comparison of a immunonutrition formula enriched arginine, glutamine and omega-3 fatty acid, with a currently high-enriched enteral nutrition for trauma patients. J Med Assoc Thai. 2003;86(6):552–61. [PubMed] [Google Scholar]
- 63.Bastian L, Weimann A. Immunonutrition in patients after multiple trauma. Br J Nutr. 2002;87 Suppl 1:S133–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Representative TSPO Binding Autoradiography Images
[3H]PK11195 autoradiography was performed on rat pup brains at 3, 7, 30 and 50 days after CCI or sham surgery, treated with DHA (DHACCI) or regular diet (REGCCI and SHAMREG). Representative coronal sections from each of the three groups at the four time points are shown, with relative binding densities ranging from minimal (blue), to moderate (yellow, green), to maximal (red). Both CCI groups exhibited focal regions of increased activation, while uninjured sham rats displayed minimal [3H]PK11195 binding.
Abbreviations: TSPO (18 k Da Translocator Protein)