

SUMMARY
The signal transduction by G Protein-Coupled Receptors (GPCRs) is mediated by heterotrimeric G proteins composed from one of the 16 Gα subunits and the inseparable Gβγ complex assembled from a repertoire of 5 Gβ and 12 Gγ subunits. However, the functional role of compositional diversity in Gβγ complexes has been elusive. Using optical biosensors, we examined the function of all Gβγ combinations in living cells and uncovered two major roles of Gβγ diversity. First, we demonstrate that the identity of Gβγ subunits greatly influences the kinetics and efficacy of GPCR responses at the plasma membrane. Second, we show that different Gβγ combinations are selectively dispatched from the plasma membrane to various cellular organelles on a timescale from milliseconds to minutes. We describe the mechanisms regulating these processes and document their implications for GPCR signaling via various Gα subunits, thereby illustrating a role for the compositional diversity of G protein heterotrimers.
Graphical Abstract
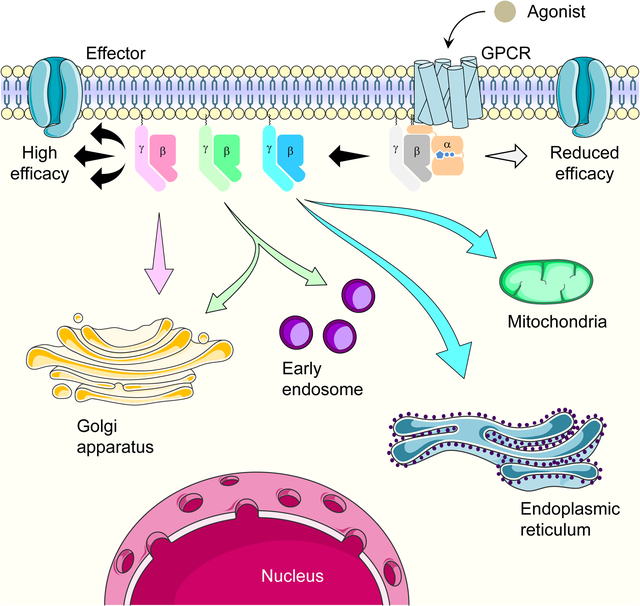
eTOC Blurb
Masuho et al. functionally tested all 60 theoretically possible G protein βγ combinations for their ability to transduce GPCR signals and report that the major role of the Gβγ diversity is in differential signaling to cellular organelles and fine-tuning signaling kinetics and efficacy at the plasma membrane.
INTRODUCTION
The GPCRs are essential for cellular communication governing all critical physiological processes. In humans, more than 800 GPCRs detect a wide range of extracellular stimuli, including hormones, ions, light, and neurotransmitters, and engage a variety of intracellular signaling cascades to trigger cellular responses (Pierce et al., 2002; Wettschureck and Offermanns, 2005). Canonically, the GPCRs transduce their signals by activating heterotrimeric G proteins. They catalyze the binding of GTP to Gα subunits and thereby dissociate GTP-bound Gα from the Gβγ dimer (Hepler and Gilman, 1992; Mahoney and Sunahara, 2016). In their activated state, both GTP-bound Gα and free Gβγ transduce their signals by modulating various downstream effector molecules that generate cellular responses (Gilman, 1987; Neer, 1995; Wettschureck and Offermanns, 2005). Most of the rapid GPCR signaling events occur at the plasma membrane, yet in recent years sustained G protein signaling at intracellular compartments is becoming appreciated (Ferrandon et al., 2009; Irannejad et al., 2013; Vilardaga et al., 2014). However, much of the mechanisms involved in the transduction of signals initiated at the cell surface to the distant intracellular sites remain to be established.
Given a sheer number of different stimuli and a variety of cellular reactions mediated by GPCRs, they need to have diverse signaling characteristics to match physiological demands. This signaling diversification is, in part, achieved by the compositional diversity of G protein heterotrimers. Mammalian genomes encode 16 Gα subunits that differentially couple to individual GPCRs (Inoue et al., 2019; Masuho et al., 2015b; Okashah et al., 2019). The identity of the Gα subunits engaged by a GPCR has a large impact on the nature of the signaling response. For example, activation of Gαi and Gαs alters cAMP production, signaling via Gαq initiates Ca2+ mobilization whereas triggering Gα12/13 leads to cytoskeleton rearrangement (Wettschureck and Offermanns, 2005).
Interestingly, Gβγ dimers show even greater diversity with five Gβ and twelve Gγ subunits, theoretically creating 60 distinct complex configurations. When free from the association with Gα, the inseparable Gβγ dimer also modulates the activity of several effector molecules, including ion channels and phospholipases (Dupre et al., 2009; Smrcka and Fisher, 2019). However, in contrast to intuitively understandable reasons for having many distinct Gα subunits, the role of the Gβγ diversity has been a mystery. Previous studies have shown that Gβ and Gγ can interchangeably form most Gβγ dimer combinations (Hillenbrand et al., 2015; Iniguez-Lluhi et al., 1992; Mervine et al., 2006; Yan et al., 1996). These different dimers show little, if any, differences in Gα association, effector regulation, and GPCR coupling (Hillenbrand et al., 2015; Kanaho et al., 1984; Ueda et al., 1994). However, knockout of individual Gβ and Gγ genes in mice (Schwindinger et al., 2010; Ye et al., 2014) and human genetic disorders associated with mutations in these subunits (Lohmann et al., 2017; Malerba et al., 2019; Stallmeyer et al., 2017) reveal that loss of individual subunits can not be easily compensated and result in detrimental outcomes. These observations suggest that different Gβγ complexes may indeed play distinct roles in cellular signaling. However, the nature of these functional differences in the propagation of GPCR signals is currently unknown.
In this study, we report that the major role of Gβγ compositional diversity is in endowing GPCRs with the plasticity of response generation. We have performed the first functional evaluation of all theoretically possible 60 Gβγ complexes. By examining multiple aspects of Gβγ properties in transducing GPCR signals using a series of live cell Bioluminescence Resonance Energy Transfer (BRET)-based assays, we found that the activity of distinct combinations of Gβγ complexes differ greatly in the kinetics and efficacy across subcellular compartments. We further demonstrate the mechanisms involved in this signaling diversification and its implications for generating distinct spatio-temporal profiles of GPCR signaling bias.
RESULTS
Gβ and Gγ can combine in cells to form all theoretically possible dimers
In order to provide insights into the basis for the diversity of G protein heterotrimers, we examined coexpression of the individual subunits mining recently uncovered large datasets of the human proteome (Kim et al., 2014). This analysis revealed unique expression patterns of Gα, Gβ, and Gγ subunits across 30 different tissues and cell types (Figure S1A). While significantly different expression levels were observed across the samples, most Gα and Gβ subunits were expressed in nearly all tissues and cell types (Figure S1B and S1C). In contrast, Gγ subunits showed a broad spectrum of expression profiles from more confined to more ubiquitous (Figure S1D). Overall, this analysis revealed that many Gβγ combinations are possible in principle, emphasizing the necessity of determining preferences in forming Gβγ complexes experimentally.
We began our functional studies by probing the assembly of all theoretically possible 60 Gβγ combinations using the Bimolecular Fluorescence Complementation (BiFC) with Venus split between Gγ and Gβ subunits (Figure 1A). We grouped Gγ subunits into five classes according to the classification based on sequence homology (Chen et al., 2007). In these experiments, Gβγ dimer formation was assessed by quantifying the fluorescence intensity of reconstituted Venus in HEK239T/17 cells. The cells were additionally co-transfected with GαoA to stabilize the entire complex (Figure S1E and S1F) (Krumins and Gilman, 2006; Li et al., 2013; Schwindinger et al., 2003; Schwindinger et al., 2004). In our previous study, we optimized conditions to ensure stoichiometry of heterotrimer containing Gβ1 and Gγ2 subunits (Masuho et al., 2015b; Masuho et al., 2020b), and this complex was used as a reference for other Gβγ combinations. The assay was validated by competition experiments using untagged Gβ, which substantially reduced fluorescence of BiFC and GPCR-mediated G protein activation, indicating that our measures indeed reflect the assembly of the specific and functional Gβγ complexes (Figure S1G and S1H).
Figure 1. Exaustive functional characterization of all Gβγ dimers in living cells.
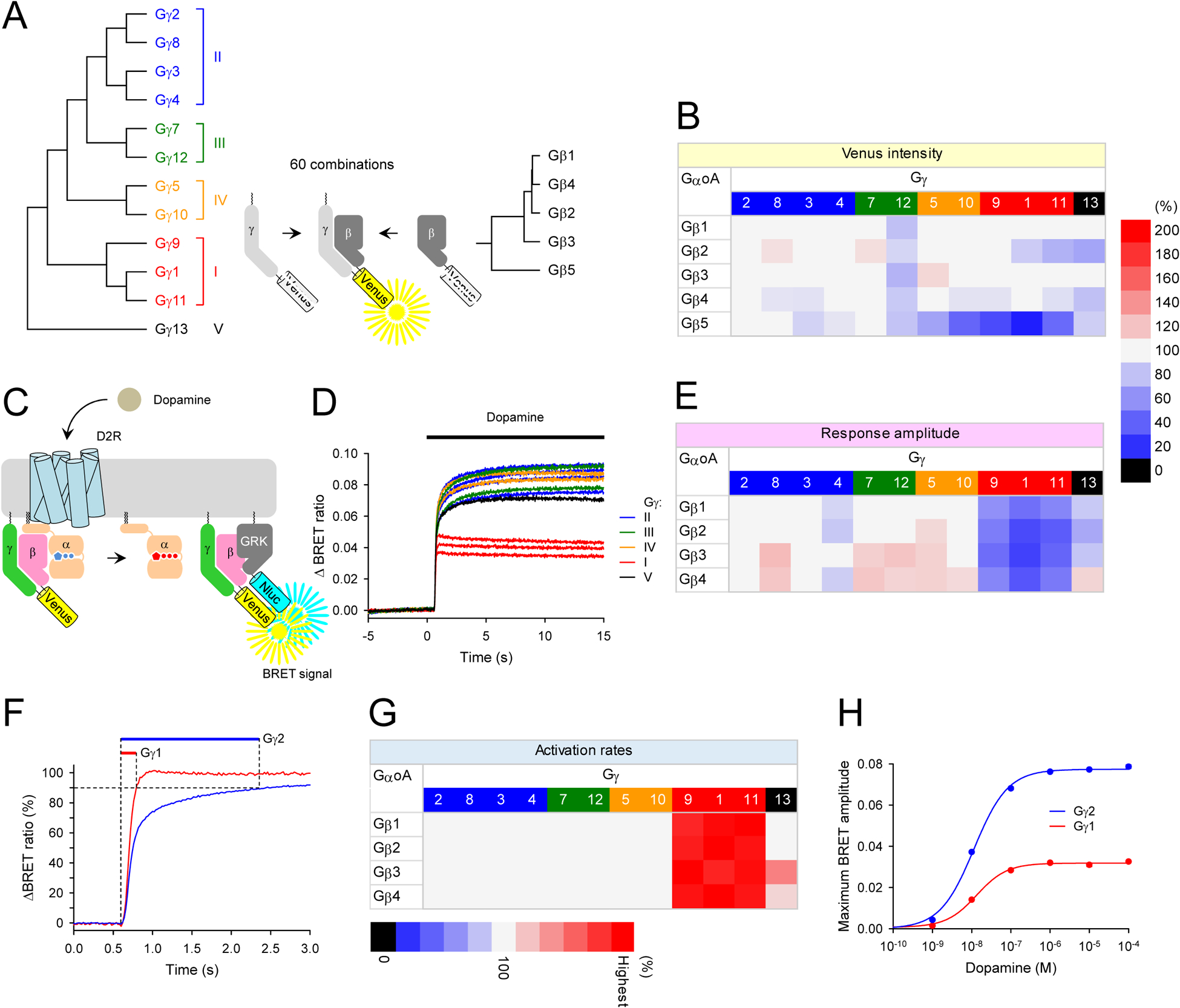
A, Schematic presentation of the BiFC assay to test Gβγ dimer formation. Two non-fluorescent fragments of Venus fused to Gβ and Gγ are brought together by dimer formation between Gβ and Gγ and produces a yellow fluorescent protein, Venus. Phylogenetic trees of Gβ and Gγ subunits are shown. For clarity, Gγ subunits were color-coded according to the classes (class I, red; class II, blue, class III, green; class IV, orange; class V, black). This color-code is used throughout this paper. B, The assessment of sixty possible Gβγ dimer formation by BiFC. The relative Venus signal intensity reflecting expression levels of Gβγ dimers in the presence of exogenous GαoA are shown as a heatmap. The values of Venus intensity were normalized to the Gγ2-containing Gβγ dimers for each Gβ subunit. C, Schematic representation of the BRET assay for real-time monitoring of G protein activity. Activation of a GPCR by an agonist leads to the dissociation of inactive heterotrimeric G proteins into active GTP-bound Gα and Venus-Gβγ subunits. The released Venus-Gβγ can then interact with the Gβγ effector mimetic masGRK3ct-Nluc-HA to produce the BRET signal. Therefore, this assay measure GPCR-induced Gβγ-effector interaction rather than direct measurement of G protein activation. D, Real-time monitoring of G protein activation by the dopamine D2 receptor (D2R). HEK293T/17 cells were transfected with D2R, GαoA, Venus 156–239-Gβ1, and masGRK3ct-Nluc-HA, together with twelve different Venus 1-155-Gγ isoforms individually. Dopamine (100 μM) was applied at 5 second timepoint and the BRET signal was followed across time. E, Functional assessment of GPCR signaling supported by the Gβγ dimers. The values of agonist-induced maximum BRET amplitude were normalized to the Gγ2-containing Gβγ dimers for each Gβ subunit and plotted as a heatmap. F, Quantification of response activation kinetics. Time to reach 90% of maximum amplitude was measured. G, The kinetics of agonist-induced G protein activation of Gβγ dimers. The values of activation kinetics (1/T90% (s−1)) were normalized to the Gγ2-containing Gβγ dimers for each Gβ subunit and plotted as a heatmap. H, Dose-response analysis of Gγ1- and Gγ2-containing Gβγ dimers. Results are expressed as the mean ± SEM (n = 3 biological replicates using independent transfections). Statistics: One-way ANOVA followed by Fisher’s LSD multiple-comparison post hoc test was carried out for panels, B, E, and G (n = 3 biological replicates using independent transfections). Statistically insignificant data (P > 0.05) was colored with gray in the heatmaps.
Using the BiFC assay, we first examined the effect of Gβ subunits on the expression levels of Gβγ dimers with Gγ2 as a representative Gγ subunit. Our experiments showed the rank order of the expression levels, Gβ1 ≈ Gβ5 > Gβ2 ≈ Gβ4 > Gβ3 (Figure S1I). The same rank order was observed when the expression was monitored directly by immunoblotting (Figure S1J), indicating that Gβ subunits significantly influence the expression levels of Gβγ dimers, which is faithfully reported by the BiFC signal. Using these conditions, we examined all 60 combinations of Gβγ dimers. Consistent with previous findings (Hillenbrand et al., 2015), we were able to detect assembly of all Gβγ complexes (Figure 1B). The quantitative analysis showed that conventional Gβ (Gβ1–4) varied in their preferences of Gγ subunits. For instance, the more promiscuous Gβ1 and Gβ3 interacted with most Gγ equally well (11 and 10 out of 12 Gγ subunits, respectively), whereas Gβ2 and Gβ4 showed greater selectivity with significant differences in binding to approximately half of the Gγ subunits. However, with these canonical Gβ subunits, the extent of the Gγ discrimination was rather small, with the changes in Venus intensity not exceeding 30% when compared to standard Gγ2-containing Gβγ dimers. In contrast, the atypical Gβ subunit, Gβ5, exhibited extreme Gγ-selectivity in dimer formation. The Gβ5-containing Gβγ complexes with most classes II and III Gγ exhibited high Venus intensity, but the reconstitution of Gβ5 with class I and IV Gγ produced a substantially lower signal. Since all Gγ subunits could form Gβγ dimers with Gβ1 subunit, the varied Venus intensity with Gβ5 subunit represent its Gγ selectivity rather than the deficiency of class I and IV Gγ expression in the transfected cells. Indeed, a side-by-side comparison of the dimer formation of Gβ1 and Gβ5 with Gγ1 and Gγ2 supported this conclusion (Figure S1K). Overall, these results indicate that all Gβγ complexes, except some composed of Gβ5, can be readily assembled in HEK293 cells in all theoretically possible combinations, yet individual preferences may vary across cell types.
The functional evaluation of all Gβγ subunits reveals key differences in the transduction of GPCR signals at the plasma membrane.
To obtain functional insights, we evaluated all Gβγ dimers for their ability to transmit GPCR signals at the plasma membrane. We used a live-cell BRET assay that monitors GPCR-induced dissociation of Gβγ from Gα in real-time (Figure 1C and 1D). We reconstituted each of the Venus-tagged Gβγ combinations with a plasma-membrane directed reporter, masGRK3ct-Nluc-HA, and a canonical combination of D2 dopamine receptor (D2R) and GαoA in HEK293T/17 cells. Since the trimer formation is required to be activated by agonist-bound GPCRs, we performed co-immunoprecipitation and Western blot analysis of exogenous Gα, Gβ, and Gγ subunits and confirmed the formation of stoichiometric trimers (Figure S1L and S1M). Under these optimized conditions, we measured both the maximum amplitude (Figure 1E) and the onset kinetics (Figure 1F and 1G) of the BRET signal in response to GPCR activation.
We observed that Gβγ complexes formed by Gβ1–4 and Gγ1–13 could generate agonist-induced BRET response, indicating that all canonical Gβγ combinations are functionally competent in transducing GPCR signals (Figure 1D and 1E). We found that the lack of response produced by the Gβ5-containing complexes could be explained by their inability to interact with the masGRK3ct-Nluc-HA sensor (Figure S2A and S2B). Therefore, we examined the function of Gβ5 complexes by an alternative BRET strategy that monitors dissociation of Gβγ from Gα (Figure S2C). We found that Gβγ dimers containing atypical Gβ5 did not produce detectable BRET signals from most of the Gα and Gγ conditions tested in this study (Figure S2D–S2K). Interestingly, we detected the formation of heterotrimers of Gβ5 with GαoA and Gαs in the presence of Gγ2 and Gγ7 judged by the elevation of the baseline BRET ratios (Figure S2D, S2F, S2H, and S2J). No such trimers were evident with Gαq and Gα13 (Figure S2E, S2G, S2I, and S2K). However, only Gβ5γ2-containing trimer with GαoA was able to be activated by D2R (Figure S2D) and by the M4 muscarinic acetylcholine receptor (Figure S2L). Although small, the amplitude of this agonist-induced activation supported by Gβ5-containing complexes was inhibited by PTX, indicating the specificity of the response (Figure S2M). However, the low efficiency of trimer formation (Figure S2D, S2F, S2H, and S2J), the small size of the agonist-induced response (Figure S2N), and poor membrane localization (Figure S2O and S2P) of Gβ5-containing heterotrimers relative to canonical heterotrimers involving Gβ1 raise doubts whether Gβ5 can effectively support GPCR signaling in vivo.
We observed notable differences in the behavior of various canonical Gβγ subunits on the plasma membrane. Amongst rather subtle variations in response properties seen across different complexes, two marked differences stood out. First, we observed a significantly smaller response from Gβγ complexes composed of Gβ1 with Gγ subunits belonging to class I (Gγ1, Gγ9, and Gγ11) (Figure 1D and 1E). Second, we found that these Gβ1γ complexes with class I Gγ subunits supported substantially faster activation kinetics relative to all other combinations (Figure 1F and 1G). The only member of class V, Gγ13, exhibited an intermediate behavior with mostly decreased amplitudes and accelerated kinetics relative to most Gγ of class II, III, and IV but not as pronounced as class I Gγ. These behaviors were observed across all 4 Gβ subunits. Control experiments showed that altering the expression level of Gβγ did not change the differences in amplitudes and kinetics between Gγ1 and Gγ2 within the expression range employed in this study (Figure S3A–S3E). We also tested different Gβ subunits and consistently observed the functional differences between representative Gγ1 and Gγ2 (Figure S3F–S3L). Moreover, when all Gβγ combinations were considered, we detected no significant correlation between the expression levels of Gβγ dimers and the maximum amplitude (r = 0.243) (Figure S2J) or the activation rates (r = −0.193) (Figure S2K). Therefore, we conclude that the apparent differences in kinetics and amplitude are driven largely by the intrinsic functional properties of individual Gγ subunit.
We further tested the function of all Gβγ combinations with another Gα subunit, Gαq (Figure S4A). In these experiments, the M3 muscarinic acetylcholine receptor (M3R) was used for its ability to activate Gαq. These experiments identified subtle variations in kinetics and signaling amplitudes across heterotrimers composed of GαoA vs. Gαq while confirming that the most notable feature of Gβγ complexes containing class I Gγ subunits was their lower signals at the plasma membrane and faster kinetics regardless of the identity of Gα and GPCR. The dose-response analysis revealed that the response mediated by Gγ1 subunits had markedly diminished efficacy without influencing potency (Figure 1H and S3M) and faster G protein activation at higher concentrations (Figure S3N). We also observed this behavior across different combinations of receptors and G proteins and even in a different cell type (Figure S4B and S4C). In summary, we concluded that the most notable differences between the function of various Gβγ complexes at the plasma membrane are the speed and efficacy of agonist-induced G protein activation: the complexes composed of class I Gγ are capable of supporting rapid signaling but generate small signals in contrast to other Gγ-containing complexes which produce slower but larger responses.
Ultra-rapid dissociation of Gβγ dimers shapes GPCR signaling at the plasma membrane.
Previous studies suggested that Gβγ subunits translocate away from the plasma membrane (O’Neill et al., 2012), and that identity of the Gγ subunits may impact the efficacy of the response (Senarath et al., 2018). Therefore, we hypothesized that the dissociation of Gβγ dimer might decrease the amount of Gβγ on the plasma membrane on a timescale of G protein activation, thereby lowering the efficacy and impacting response kinetics. However, previous measurements of Gβγ dissociation from the plasma membrane using confocal microscopy (O’Neill et al., 2012) conflict with this model because the previously observed membrane dissociation rates for the class I Gγ complexes (τ1/2 ~5–38 s) are approximately an order of magnitude slower than the generation of agonist-induced Gβγ response that we observed (τ1/2 ~380 ms). Therefore, we have revisited and quantified the membrane dissociation of Gβγ using a fast kinetic BRET strategy. To monitor the dissociation of Venus-Gβγ dimers from the plasma membrane, we anchored a BRET donor (Nluc-Flag-K-Ras) on the plasma membrane (Figure S5A). In this “proximity” strategy (Lan et al., 2012), a decrease in the BRET signal reports the loss of Venus-Gβγ from the plasma membrane as its distance to the donor increases by more than 10Å due to translocation (Figure 2A).
Figure 2. Impact of Gβγ composition on their dissociation from the plasma membrane and GPCR signaling efficacy.
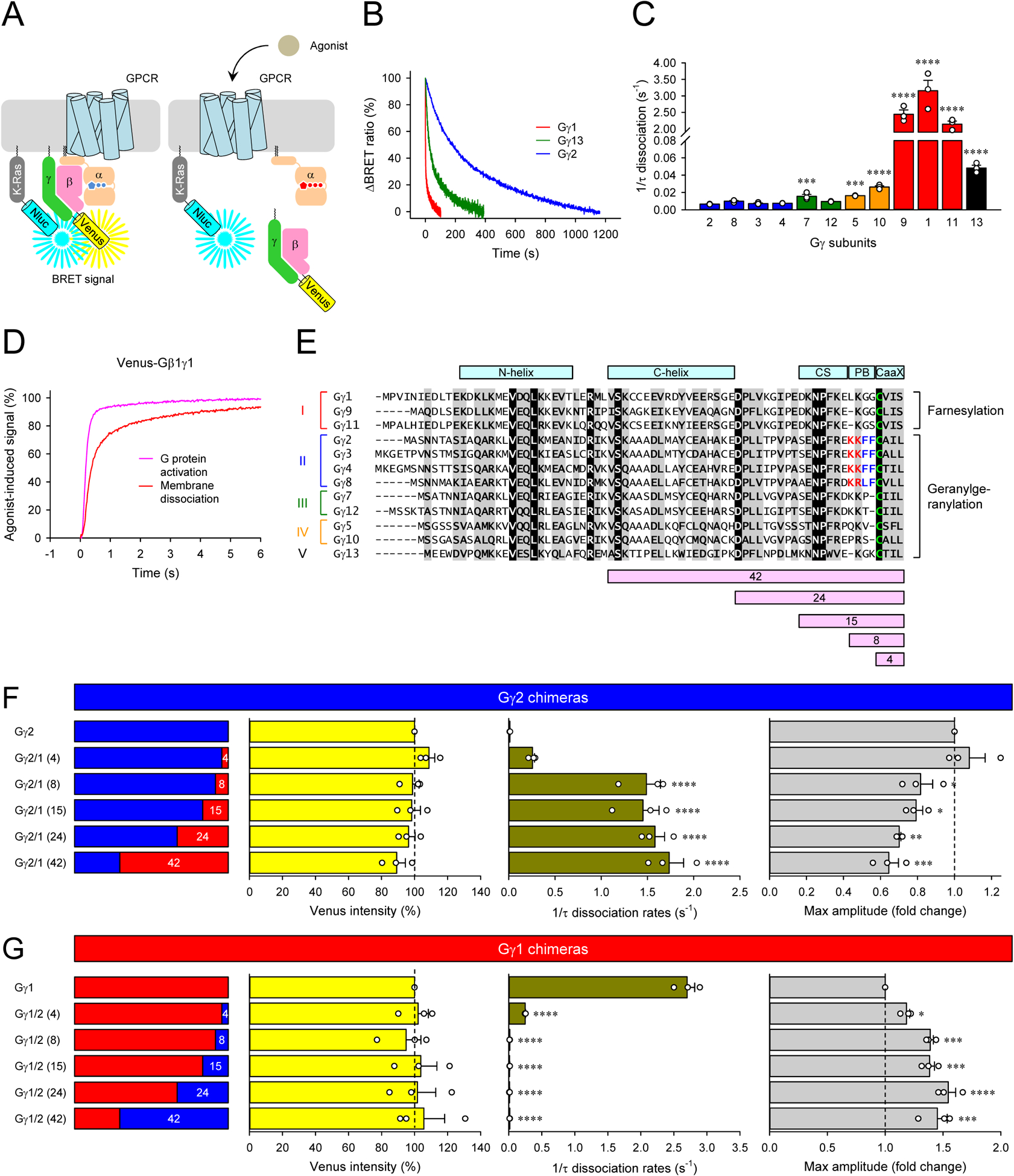
A, Schematic presentation of the BRET assay to monitor the dissociation of Venus-Gβγ from the plasma membrane. The high density of Venus-Gβγ-containing heterotrimer and Nluc-Flag-K-Ras on the plasma membrane causes a high BRET signal. Membrane dissociation of Venus-Gβγ upon G protein activation decreases the density of Venus-Gβγ, lowering the BRET signal. B and C, Real-time monitoring of the membrane dissociation of Venus-Gβγ. HEK293T/17 cells were transfected with D2R, GαoA, Venus 156-239-Gβ1, and Nluc-Flag-K-Ras, together with twelve different Venus 1-155-Gγ isoforms individually. Dopamine (100 μM) was applied, and the BRET signal was followed across time (B). Dissociation rates were plotted as a bar graph (C). D, The time course of G protein activation (dark blue) and Venus-Gβγ dissociation (red) of Gγ1-containing Go. For comparison, the membrane dissociation of Venus-Gβγ was inverted. E, An alignment of all human Gγ subunits. Structural motifs, conformational switch (CS) and poly-basic residues (PB), etc. were highlighted on the alignment. Sequence swapped between Gγ1 and Gγ2 for chimeras were also shown at the bottom of the alignment. F and G, Venus intensity, membrane dissociation rates, and maximum BRET amplitude of Gγ1 and Gγ2 chimeras. Mean ± SEM from three independent experiments are shown as bar graphs (C, F, and G). Statistics: One-way ANOVA followed by Fisher’s LSD multiple-comparison post hoc test was carried out (n = 3 biological replicates using independent transfections) (C, F, and G): * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
Using this BRET-based strategy, we examined the time courses of Gβγ dissociation from the plasma membrane for G proteins composed of GαoA, Gβ1, and all of Gγ subunits introduced one at a time. We found that class II, III, and IV Gγ subunits slowly dissociated from the plasma membrane on a timescale of minutes in quantitative agreement with earlier observations (O’Neill et al., 2012). In contrast, class I Gγ subunits translocated much faster, with class V Gγ exhibiting intermediate behavior (Figure 2B and 2C). Notably, our kinetic measurements indicate that the speed of this process of class I Gγ is about an order of magnitude faster than previously estimated by microscopy (O’Neill et al., 2012). This fast translocation of class I Gγ occurred on the timescale of the GPCR response generation at the plasma membrane, as evidenced by the comparative overlay of G protein activation and membrane dissociation (Figure 2D). These observations suggest that rapid dissociation of Gβγ complexes that occurs as the response develops may limit the response amplitude on the plasma membrane (Figure 1D, 1E, and 1H) due to the depletion of the active G protein. Indeed, confocal imaging of live cells confirms rapid dissociation of Gγ1 but not Gγ2 containing Gβγ from the plasma membrane (Figure S5B). We also examined the dissociation rates of three representatives Gγ subunits, Gγ2, Gγ1, and Gγ13, with different Gβ subunits and GPCRs, and found that the choice of Gβ and GPCR has no impact on observed differences in the dissociation rates (Figure S4D and S4E), indicating that the membrane dissociation rate of Gβγ dimer is defined by the properties of Gγ subunits.
To further probe the relationship of Gβγ translocation with the GPCR response properties and to establish its molecular underpinning, we employed a chimeric mutagenesis approach swapping various structural elements between representative Gγ classes with the largest difference: Gγ1 and Gγ2 (Figure 2E). In particular, we focused on elements previously shown to be involved in controlling the strength of Gγ interaction with the plasma membrane. These included the CaaX box that directs lipidation by either farnesyl in class I Gγ subunits (Gγ1, Gγ9, and Gγ11) or geranylgeranyl in all others (Escriba et al., 2007) and the adjacent poly-basic region (5–8 aa. from C-terminus) (O’Neill et al., 2012). We also swapped a conformational switch region (9–15 aa. from C-terminus) that contacts active Rhodopsin in the form of α-helix (Kisselev and Downs, 2003) and a C-terminal helix that forms one of the two coiled-coil domains for the interaction with Gβ subunit (Wall et al., 1995).
All chimeras formed complexes with the Gβ1 and were expressed at comparable levels (Figure 2F and 2G). We found that the replacement of only CaaX box of Gγ1 with that of Gγ2 slightly accelerated dissociation of the Gβ1γ2 from the plasma membrane but was insufficient to influence the response efficacy (Figure 2F). Additional inclusion of the polybasic region in the swapped sequence was enough to change the property of Gγ2 to Gγ1 type: this chimera exhibited fast dissociation and low efficacy. The replacement of additional elements did not produce any further alterations in response properties. Conversely, the swapping of CaaX box alone or both CaaX motif and the polybasic region from Gγ2 to Gγ1 dramatically slowed the translocation rate of the Gβγ complexes and increased the response amplitude (Figure 2G). Thus, ultra-fast dissociation of the Gβγ complexes is governed by the nature of prenylation on Gγ subunits acting in conjunction with the poly-basic motif. Together, these results indicate that ultra-rapid Gβγ dissociation from the plasma membrane serves as a mechanism for limiting the strength of GPCR signaling at the plasma membrane.
The identity of Gγ subunit regulates the delivery of GPCR-initiated messages from the plasma membrane to intracellular destinations.
The behavior of Gβγ dimers at the plasma membrane raises a provocative possibility that differences in Gβγ may more generally be utilized for adjusting GPCR signaling efficacy and kinetics across cellular compartments. This might be particularly relevant in the context of previously observed translocation of Gβγ complexes to intracellular compartments (Saini et al., 2009; Saini et al., 2007). To explore this hypothesis, we targeted our effector-based GRK3ct-Nluc sensor to various organelles including the cytosolic surface of Golgi apparatus (Golgi), endoplasmic reticulum (ER), early endosome (EE), and mitochondria (Mit) (Figure 3A). Confocal microscopy confirmed that each sensor indeed distributed to characteristic intracellular compartments as expected (Figure S5C). We found that Gβγ containing the representative class I member, Gγ1, was capable of rapidly translocating to intracellular compartments (Figure 3B and 3C). Notably, the speed of the response generation on the organelles occurred on the same timescale as G protein activation at the plasma membrane (Figure 3B). In contrast, Gγ2 containing complexes translocated to intracellular compartments much slower with different rates across individual organelles (Figure 3C and 3D).
Figure 3. Signaling of Gβγ from the plasma membrane to cellular organelles.
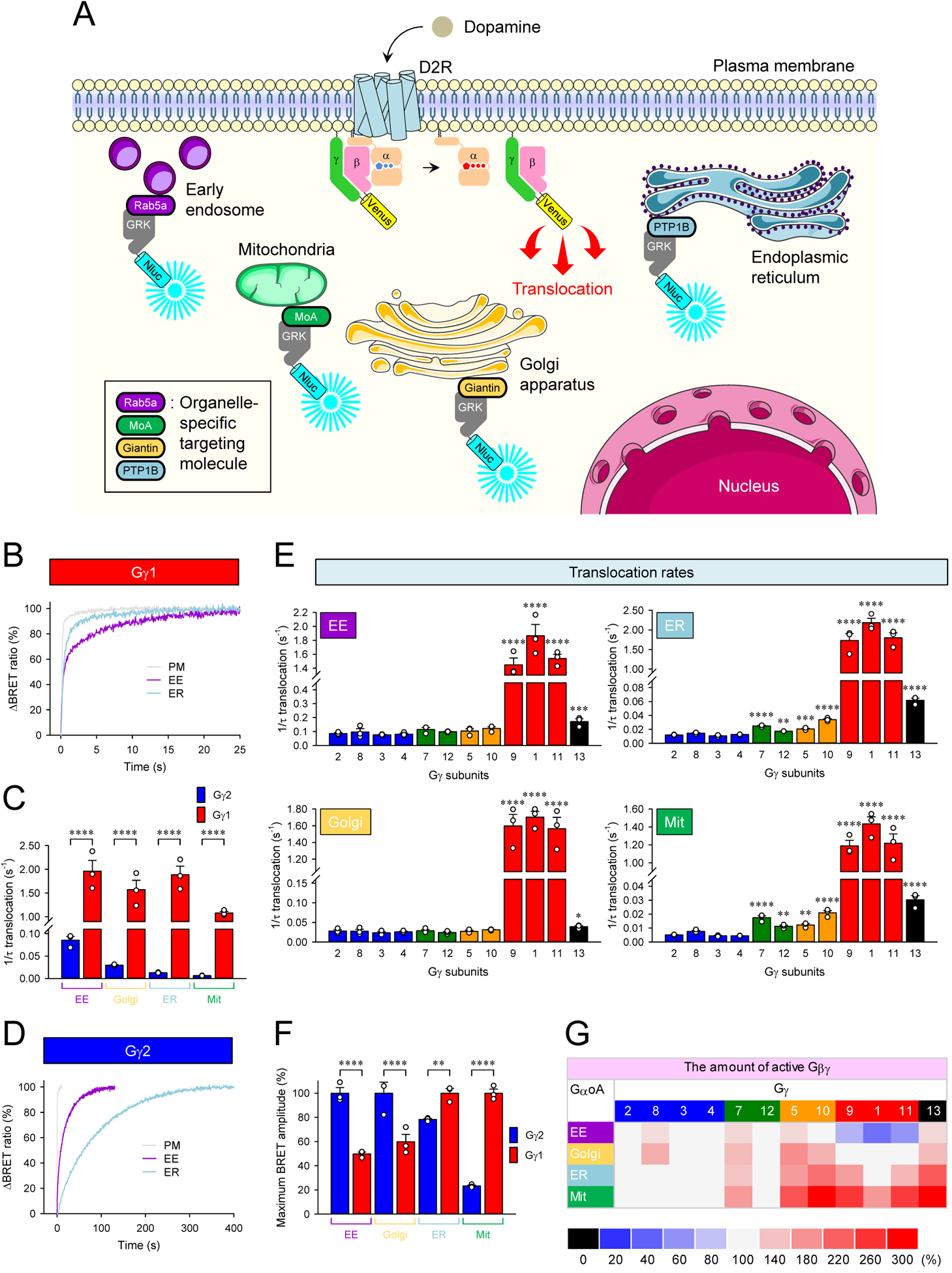
A, Schematic presentation of the BRET assay to monitor the translocation of Venus-Gβγ from the plasma membrane to organelles. GRK3ct-Nluc-HA sensor was recruited to early endosome (EE), mitochondria (Mit), Golgi apparatus (Golgi), and endoplasmic reticulum (ER) by tagging with rab5a, monoamine oxidase A (MoA), giantin, and PTP1B, respectively. Activation of G proteins with dopamine induces dissociation of Venus-Gβγ from the plasma membrane, and it binds with GRK3ct-Nluc-HA sensors at the destination. Graphics were adapted from Servier Medical Art (http://www.servier.com). B, Real-time monitoring of the translocation of Gβ1γ1. HEK293T/17 cells were transfected with D2R, GαoA, Venus 156-239-Gβ1, and Venus 1-155-Gγ1 together with different GRK3ct-Nluc-HA sensors individually. Dopamine (100 μM) was applied, and the BRET signal was followed across time. C, Translocation rates of Gβ1γ1 and Gβ1γ2 to organelles. D, Real-time monitoring of the translocation of Gβ1γ2. E, Effect of Gγ subunits on the translocation rates of Gβγ dimers to organelles. F, Comparison of the amount of Gβ1γ1 and Gβ1γ2 on each organelle by translocation. G, Effect of Gγ subunits on the amount of Gβγ translocated to organelles. Mean ± SEM from three independent experiments are shown as bar graphs (C, E, and F). Statistics: One-way ANOVA followed by Fisher’s LSD multiple-comparison post hoc test was carried out (n = 3 biological replicates using independent transfections) (C, E, and F): * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
We further extended this analysis to all other members of the Gγ family and found that all Gγ subunits were capable of supporting translocation to intracellular compartments (Figure 3E). The rates of such translocation were, for the most part, comparable to the rates of Gβγ dissociation from the plasma membrane for respective complexes (class I > V > III = IV > II) (Figure 2C vs. 3E), indicating that this process is likely driven by the diffusion of Gβγ in the cytoplasm upon their dissociation from the plasma membrane. Deviating from this rule was the speed of signal transfer of geranylgeranylated Gγ subunits to EE and Golgi, which exceeded the speed of their PM dissociation kinetics (Figure 2C vs. 3E), suggesting their reliance on an active process, e.g., endocytosis known to occur on the timescale of seconds (Watanabe and Boucrot, 2017) rather than simple diffusion for translocation to these compartments.
We also observed significant differences in the amplitudes of the intracellular responses driven by the identity of Gγ subunits across compartments (Figure 3F and 3G). These differences did not correlate with the speed of the response generation (Figure 3E and 3G). For example, Gγ2 containing complexes were more efficacious relative to Gγ1 in signaling to EE and Golgi, as they were at the plasma membrane, but the Gγ1 was more efficacious at ER and Mit (Figure 3F). Furthermore, Gβγ dimers composed of Gβ2 or Gβ4 and Gγ1 or Gγ2 exhibited a similar translocation pattern to the EE as Gβ1-containing Gβγ dimers (Figure 3C and 3F vs. S4F), further indicating that the timing and efficacy of Gβγ translocation to intracellular compartments is defined by the identity of Gγ rather than Gβ. Overall, Gβγ complexes are greatly stratified in their propensity to translocate to the intracellular membrane compartments, showing significant differences in both speed and efficiency of process depending on the identity of the Gγ subunits.
Different G protein deactivation mechanisms control the duration of endomembrane signaling.
Observed differences in the kinetics and extent of Gβγ translocation to various endomembrane compartments raise a question about mechanisms that control the lifetime of Gβγ action at these distant sites. In general, at the plasma membrane, the Gβγ signaling is terminated upon re-association with the inactivated Gα-GDP, which is determined by the rate of GTP hydrolysis on the Gα subunit. Thus, one straightforward mechanism to control Gβγ deactivation at endomembrane compartments could be that Gα also dissociates from the plasma membrane and translocates with Gβγ to form inactive trimers at the destination. Therefore, we first investigated the localization of Gα after prolonged GPCR activation when all types of Gβγ complexes translocate (Figure 4A and 4B). Before GPCR activation, the representative Gβ1γ1 and Gβ1γ2 subunits were colocalized with GαoA at the plasma membrane. Following stimulation of D2R with dopamine, both Gβ1γ1 and Gβ1γ2 were present only at the intracellular sites. However, GαoA remained entirely at the plasma membrane, indicating that Gβγ deactivation likely requires its return to the plasma membrane to form an inactive trimer with the Gα-GDP.
Figure 4. Mechanisms of Gβγ signaling deactivation.
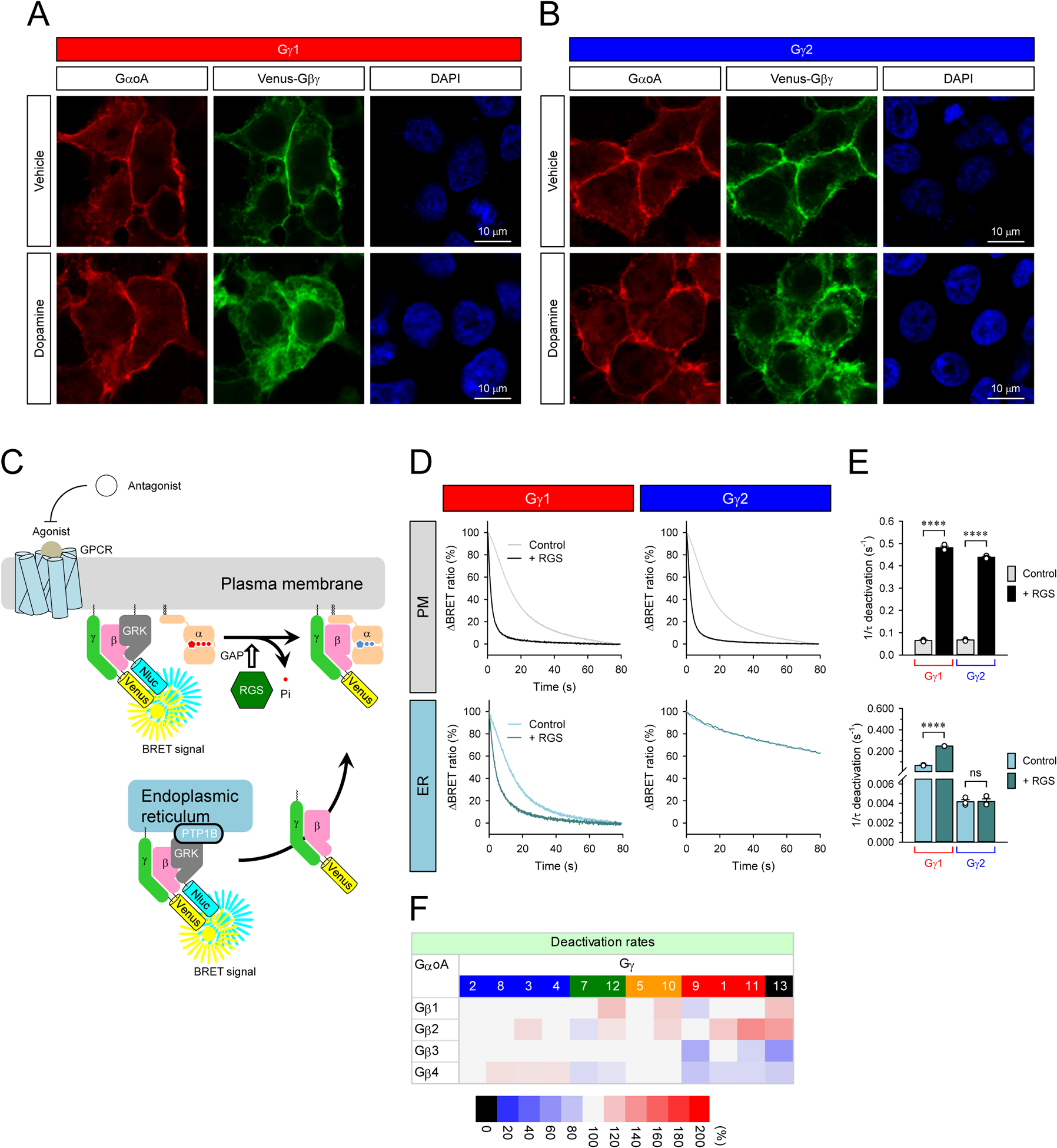
A and B, Subcellular localization of GαoA and β1γ1 and Gβ1γ2 with or without prolonged stimulation of D2R. Transfected cells were stimulated with 100 μM dopamine for 10 min (bottom). Immunocytochemistry was performed with anti-Gαo antibody. The subcellular localization of GαoA and Venus-Gβγ were visualized with confocal microscopy. C, Schematic presentation of the BRET assay to monitor the deactivation of Venus-Gβγ on the plasma membrane and ER. D, Real-time monitoring of the deactivation of Gβ1γ1 and Gβ1γ2 on the plasma membrane and ER. HEK293T/17 cells were transfected with D2R, GαoA, and Venus 156-239-Gβγ with masGRK3ct-Nluc-HA or GRK3ct-Nluc-HA-PTP1B. To accelerate GTP hydrolysis rate, RGS7, Gβ5S, and R7BP were also transfected. The transfected cells were stimulated with 100 μM dopamine for 35 sec (Gγ1 on PM, Gγ2 on PM, and Gγ1 on ER) and 10 min (Gγ2 on ER) to activate G protein. Then, 100 μM haloperidol was applied to inhibit the activity of D2R. E, The deactivation rates of Gβ1γ1 and Gβ1γ2 on the plasma membrane (top) and ER (bottom). Statistics: One-way ANOVA followed by Fisher’s LSD multiple-comparison post hoc test was carried out (n = 3 biological replicates using independent transfections): * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001. F, Effects of Gβγ on G protein deactivation rate on the plasma membrane. Deactivation rates of Go when complexed with 48 different Gβγ dimers are reported as heatmaps. One-way ANOVA followed by Fisher’s LSD multiple-comparison post hoc test was carried out (n = 3 biological replicates using independent transfections). Statistically insignificant data (P > 0.05) are colored in gray.
To understand the mechanisms of Gβγ deactivation, we utilized a similar location-specific BRET strategy and monitored the time course of GPCR signal termination at different locations (Figure 4C). When the sensor was positioned on the plasma membrane and the D2R was stimulated for a short period of time not to allow the appreciable loss of Gβγ from the plasma membrane, we observed no differences in deactivation rates of Gβ1γ1 and Gβ1γ2 (Figure 4D and 4E). Likewise, we found very little effect of the other Gγ subunits on the deactivation rates (Figure 4F), indicating that the identity of the Gγ subunit does not appreciably influence the GTPase activity of Gα. Next, we localized the sensor on the ER and studied the deactivation after prolonged D2R stimulation when all Gβγ complexes are fully translocated to the ER. These experiments revealed that the deactivation rates of Gβ1γ1 and Gβ1γ2 on the ER were markedly different (Figure 4D and 4E). While the deactivation rate of Gβ1γ1 observed with the ER sensor was the same as the deactivation rate on the plasma membrane, the deactivation rate of Gβ1γ2 was substantially slower in the ER than at the plasma membrane. The slow deactivation of Gβ1γ2 in the ER and the lack of GαoA in this compartment suggests that the process is rate limited by the slow dissociation of Gβ1γ2 from the ER and relocation to the plasma membrane for deactivation.
To confirm that the Gβγ activity is terminated at the plasma membrane and is driven by the GαoA deactivation, we performed experiments accelerating the GTPase activity of Gα by expressing Regulator of G protein Signaling (RGS) proteins (Figure 4C). We used RGS7/Gβ5 complex selective for GαoA (Lan et al., 2000; Masuho et al., 2020a; Masuho et al., 2013) and directed it to the plasma membrane by co-expressing their plasma membrane-targeting subunit R7BP (Martemyanov et al., 2005). Expression of the RGS prominently accelerated the deactivation rate of both Gβ1γ1 and Gβ1γ2 at the plasma membrane to a quantitatively indistinguishable extent (Figure 4D and 4E). The RGS also accelerated the deactivation of Gβ1γ1 at the ER, consistent with its ultra-rapid translocation occurring on a much more rapid timescale than GTP hydrolysis rates. This observation indicates that the deactivation rate of Gβ1γ1 in the ER is rate-limited by GTPase activity of Gα remaining on the plasma membrane. In contrast, RGS expression had no effect on deactivation kinetics of slow-translocating Gβ1γ2 complexes, suggesting that its deactivation is rate-limited by translocation and not GTP hydrolysis on Gα. Taken together, these results indicate that Gγ composition of G protein heterotrimers further diversifies the signaling properties of GPCRs by differentially controlling timing of Gβγ deactivation across intracellular compartments.
Translocation of Gβγ generates unique compartment-specific profiles for individual Gα channels.
It is rapidly becoming evident that many GPCRs initiate signaling by multiple Gα species (Inoue et al., 2019; Masuho et al., 2015b; Okashah et al., 2019) and that specific patterns of activated Gα may distinguish the functional properties of individual GPCRs (Anderson et al., 2020; Himmelreich et al., 2017; Masuho et al., 2015b). To determine how the identity of Gγ subunits influences the GPCR engagement of multiple Gα subunits, we studied CCKAR, a promiscuous receptor capable of coupling to a diverse set of nearly all Gα (Hauser et al., 2018). It was paired with the sensor placed in the ER as a compartment that showed significant differences between signaling mediated by Gγ2 and Gγ1 (Figure 5A and 5B). The investigation of Gγ1-containing trimers showed that CCKAR supported the translocation of Gβγ to the ER through all Gα subunits tested, producing a characteristic fingerprint-like G protein-activation profile (Figure 5B). The biggest response amplitude was detected from the Gi/o subfamily, followed by Gq and G12/13 proteins and the small but significant response from the Gs/olf. Activation rates were the fastest for Gq, followed by Gi/o > Gs > G12/13. This G protein-activation profile was markedly different with Gγ2 complexes in both amplitudes and activation rates. Notably, G12/13 supported the signal more prominently, and Gs/olf did not produce a detectable response. All Gα supported very similar Gβγ translocation rates, with a clearly slower timescale relative to Gγ1-supported translocation. These CCKAR-induced Gβ1γ2 translocation rates are consistent with the slow D2R-induced translocation rate of Gβ1γ2 dimers to the ER (Figure 5B vs. 3D), indicating the Gβγ translocation to the ER is independent of the GPCR and Gα types as long as GPCRs can activate G proteins faster than the translocation rate.
Figure 5. Impact of Gβγ identity on location-specific GPCR fingerprints.
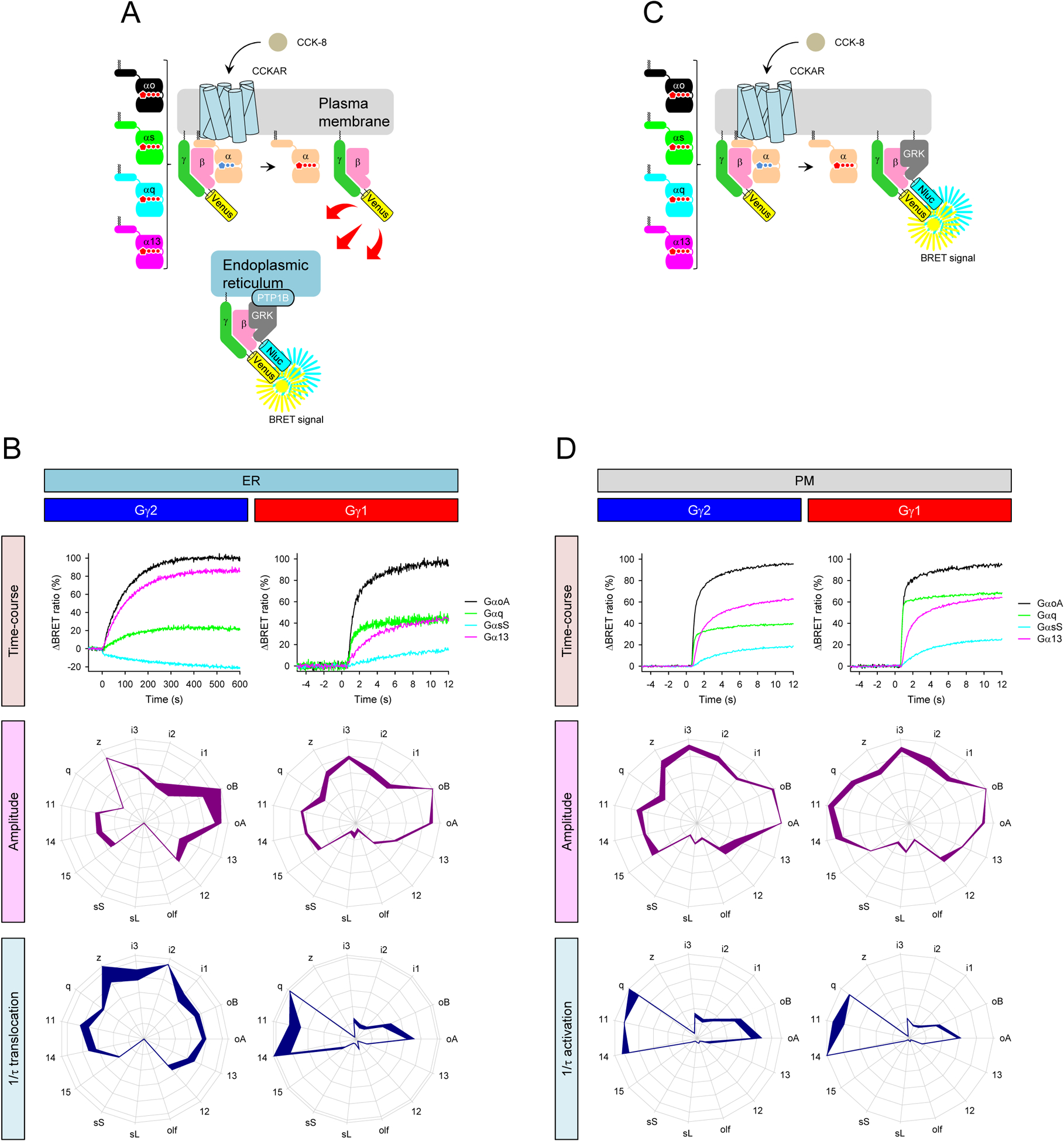
A, Schematic presentation of the BRET assay to monitor the recruitment of Venus-Gβγ to the ER induced by CCKAR which couples to diverse set of G proteins. B, Real-time monitoring of the translocation of Gβ1γ1 (right) and Gβ1γ2 (left) from the plasma membrane and ER (top). Profiling of CCKAR-induced translocation of Gβ1γ1 and Gβ1γ2 to the ER through 15 Gα subunits in extent and speed. The maximum amplitudes (purple) and translocation rate constants (dark blue) from 15 different G proteins were normalized to the largest value and plotted in the wheel diagrams. C, Schematic presentation of the BRET assay to monitor the G protein-coupling profiles of CCKAR on the plasma membrane. D, Real-time monitoring of the CCKAR-induced activity of Gβ1γ1 (right) and Gβ1γ2 (left) on the plasma membrane (top). Profiling amplitudes and kinetics of CCKAR-induced translocation of Gβ1γ1 and Gβ1γ2 to the ER in the presence of 15 Gα different subunits. The maximum amplitudes (purple) and translocation rate constants (dark blue) from 15 different G proteins are normalized to the largest value and plotted in the wheel diagrams. Line thickness represents the SEM of three technical replicates performed independently (B and D).
We further examined the CCKAR-induced G protein activation profile with the PM-localized sensor (Figure 5C and 5D) and found these patterns to be very similar regardless of the Gγ subunit used in the assay. These G protein activation profiles on the PM were also very similar to the Gγ1-mediated profile on the ER, indicating that fast translocation supports equally balanced active Gβγ across the compartments. Taken together, these results show that the composition of different Gγ and Gα subunits in heterotrimers impacts the strength and timing of GPCR-mediated signaling at different organelles.
DISCUSSION
GPCRs are capable of reliably encoding a wide variety of extracellular stimuli and translate them into a unique set of intracellular signaling reactions that stereotypically program cellular responses with incredible plasticity. It is universally accepted that the heterotrimeric G proteins play key roles in transducing GPCR signals. The variety of G protein subunits is thought to underlie the high capacity of GPCRs to generate diverse signals. However, we understand very little about how this signaling diversity is generated. The biggest source of this complexity is provided by the Gβγ subunits that can theoretically form 60 unique combinations, but the role of the differences in the composition of Gβγ complexes has been a long-standing mystery.
The main advance of this study is in the demonstration of the functional role of Gβγ diversity. We show that different Gβγ dimers uniquely translocate to various cellular organelles. This translocation broadcasts signals from the plasma membrane to select intracellular destinations with varying efficiency, kinetics, and persistence. We identified that the elements that dictate the specific migration behavior of individual Gβγ complexes are encoded by the diverse sequences at the carboxy-terminal domain of the Gγ subunit. They rely on both selective lipidation pattern and the adjacent stretch of basic and hydrophobic amino acid residues forming a “destination code”. It is worth noting that the process of Gβγ translocation from the plasma membrane to intracellular compartments has been described before (Ajith Karunarathne et al., 2012; Akgoz et al., 2004; Saini et al., 2007), including observations about its dependence on the identity of the Gγ subunits (Akgoz et al., 2006; O’Neill et al., 2012). Our findings offer key revisions to the prior model by demonstrating the following. First, we find that all canonical Gβγ complexes containing Gβ1–4 and Gγ1–13 subunits translocate from the plasma membrane to the intracellular destinations upon GPCR activation, thereby likely transmitting GPCR signals to organelles. Second, we demonstrate the process of Gβγ translocation to organelles can occur on an extremely rapid millisecond timescale comparable to G protein activation at the plasma membrane. Third, we provide a systematic comparison of destinations accessible by Gβγ subunits showing that they can access virtually all organelles with different kinetics and efficiency. Forth, we show that the identity of Gα and Gγ alters the pattern of agonist-induced Gβγ translocation on the distinct subcellular compartments.
The broadcasting mechanism that we describe endows GPCRs at the cell surface to selectively reach their intracellular targets for signal transduction, depending on the identity of the Gβγ engaged. This mechanism enables encoding different signaling outcomes for the same receptor, thus significantly diversifying signaling and, eventually, cellular responses. Our observations help explaining how GPCRs can signal to non-canonical Gβγ effector molecules increasingly found on organelles (Khan et al., 2016; Smrcka and Fisher, 2019). For instance, Gβγ directly interacts with Mitofusin-1 located in the mitochondria to affect mitochondrial morphology (Zhang et al., 2010). Gβγ can also interact with Rab11a on early and recycling endosomes to promote activation of PI3K-ATK pathway (Garcia-Regalado et al., 2008). Similarly, functions of Gβγ at the Golgi have been noted. The Gβγ dimer can induce the fragmentation of Golgi (Jamora et al., 1997) through PKD- (Jamora et al., 1999) and PLCβ-dependent manner (Saini et al., 2010), regulating anterograde trafficking of cargo proteins through the trans-Golgi network (Khan et al., 2016). Interestingly, this fragmentation can be inhibited by non-translocating Gγ3 (Saini et al., 2010), indicating the importance of efficient translocation of Gβγ to the Golgi. At the Golgi, PAQR3 (also known as RTKG) interacts with Gβγ. Gβγ-binding-deficient PAQR3 mutants inhibits the fragmentation of the Golgi (Jiang et al., 2010). Although in many cases the source of the Gβγ detected in organelles is unclear, it is known that Gβγ translocation to perinuclear Golgi regulates cardiomyocyte hypertrophy (Malik et al., 2015). Therefore, because of the specific subcellular localization of different Gβγ-effector molecules, the timing, strength, and type of signaling mediated by Gβγ are tightly regulated by cell-type-specific expression of Gγ subunits. It is noteworthy that Gβγ released from any Gα we tested could reach the ER, yet the efficiency of this process varied. Thus, we think that the GPCR identity could further fine-tune Gβγ translocation and activation of effector molecules on organelles via differential engagement of Gα. In future studies, it will be important to determine how dynamically regulated Gβγ subcellular distribution that we report in this study affects these cellular functions.
We determined that the translocation of the Gβγ from the plasma membrane is the key driver of signaling to organelles. We described at least two distinct Gβγ translocation mechanisms. In one, fast dissociating farnesylated Gγ rapidly diffuse to all four organelles, EE, Golgi, ER, and Mit, whereas geranylgeranylated Gγ subunits can only reach the ER and Mit through slow diffusion. The second mechanism is utilized by geranylgeranylated Gγ subunits for reaching the EE and Golgi. This process is faster than their membrane dissociation rates, indicating reliance on the active transport mechanism. The difference in time and extent of this translocation across different compartments provides means for spatio-temporal bias of GPCR signals.
Notably, we report that differential translocation of Gβγ complexes to intracellular organelles provides a powerful source for the diversification of GPCR signaling. GPCRs transduce signals from a vast number of stimuli generating unique responses in a context-dependent manner. This capacity requires significant plasticity in signal encoding to match physiological demands. The major source of such signal diversity is the ability of GPCRs to activate multiple G proteins (Bosier and Hermans, 2007; Hermans, 2003). Many GPCRs display rather promiscuous but unique coupling profiles for activation of Gα subunits, which in turn program patterns of effector molecule engagement (Wettschureck and Offermanns, 2005). Apart from selective recognition of particular Gα, a given GPCR activates each G protein with different rates indicative of their preference for G proteins (Masuho et al., 2015b). Further elaborating GPCR signaling complexity, we found that Gγ subunits play an essential role in the spatio-temporal regulation of G protein signaling, further diversifying the functions of GPCR-mediated signaling.
In this study, we further report that another major role played by Gβγ diversity is in providing differential control of signaling kinetics and efficacy at the plasma membrane. We report that Gβγ complexes vary greatly in the rates of their dissociation from the plasma membrane and especially class I Gγ-containing Gβγ dimers are capable of attaining ultra-fast dissociation rates occurring on the timescale of G protein activation. Previous studies observed a much slower translocation of Gβγ complexes from the plasma membrane (O’Neill et al., 2012) and suggested that the identity of the Gγ subunits may serve to adjust the GPCR responsiveness in macrophage-like cells (Senarath et al., 2018). Interestingly, G protein translocation was also demonstrated to occur physiologically in photoreceptors, where it was shown to contribute to light adaptation (Kassai et al., 2005; Majumder et al., 2013; Sokolov et al., 2002). However, previous models based on slow plasma membrane dissociation kinetics suggested that these Gβγ translocation events play a largely adaptive role under the conditions of prolonged or repeated GPCR stimulation. Instead, our observations of ultrafast dissociation suggest that Gβγ identity can greatly impact the kinetics and efficacy of the immediate agonist-induced response. Considering that all canonical Gβγ dimers can dissociate from the plasma membrane, the membrane dissociation of Gβγ, may, in general, provide means of regulating the abundance of Gβγ on the cell surface. Individual cells can then tune the characteristics of their responses by preferentially expressing particular types of the Gγ subunits.
In summary, this study provides a model that the identity of Gγ subunits dictates the kinetics, extent, and location of Gβγ signaling, revealing an additional dimension of diversity in GPCR signaling, essential for supporting broad physiological functions of GPCRs.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents and resources should be directed to, and will be fulfilled by, the Lead Contact, Kirill A. Martemyanov (kirill@scripps.edu).
Materials Availability
Plasmids generated in this study will be distributed upon request without restriction.
Data and Code Availability
The source data for raw traces of GPCR responses used for quantitative analysis reported in this study have not been deposited in a publicly available repository because of their trivial nature and custom format they are generated in. They have been archived locally on the institutional cloud service. To request access, please contact Lead Contact.
This paper does not report any original codes.
Scripts were not used to generate the figures reported in this paper.
Any additional information required to reproduce this work is available from the Lead Contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
This work did not employ animals or human subjects.
METHOD DETAILS
cDNA constructs
Triple HA-tagged M1 muscarinic acetylcholine receptor (3xHA-M1R) (AF498915), M2 muscarinic acetylcholine receptor (M2R) (AF498916), M3 muscarinic acetylcholine receptor (M3R) (AF498917), M4 muscarinic acetylcholine receptor (M4R) (AF498918), adenosine A1 receptor (A1R) (AY136746), Cholecystokinin A receptor (CCKAR) (AY322549), dopamine D1 receptor (D1R) (NM_000794), GαoB (AH002708), Gαz (J03260), Gα11 (AF493900), Gα14 (NM_004297), Gα15 (AF493904), Gαs long isoform (GαsL) (NM_000516), Gαolf (AF493893), Gα12 (NM_007353), Gα13 (NM_006572), RGS7 (AY587875), and Gβ5S (NM_006578) in pcDNA3.1(+) were purchased from cDNA Resource Center (www.cDNA.org). GRK3ct-Nluc-HA-giantin, GRK3ct-Nluc-HA-MoA, GRK3ct-Nluc-HA-PTP1B, and GRK3ct-Nluc-HA-Rab5a in pcDNA3.1(+) were synthesized by GenScript. Flag-tagged dopamine D2 receptors (NM_000795) containing the hemagglutinin signal sequence (KTIIALSYIFCLVFA) at the N-terminus was a gift from Dr. Abraham Kovoor. The pCMV5 plasmids encoding rat GαoA, rat Gαi1, rat Gαi2, rat Gαi3, human Gαq, and bovine Gαs short isoform (GαsS) were gifts from Dr. Hiroshi Itoh. Venus 156-239-Gβ1 (amino acids 156–239 of Venus fused to a GGSGGG linker at the N terminus of Gβ1 without the first methionine (NM_002074)) and Venus 1-155-Gγ2 (amino acids 1–155 of Venus fused to a GGSGGG linker at the N terminus of Gγ2 (NM_053064)) were gifts from Dr. Nevin A. Lambert (Hollins et al., 2009). The other Venus-tagged Gβ and Gγ subunits were constructed in the same way as Venus 156-239-Gβ1 and Venus 1-155-Gγ2. Flag-tagged Ric-8A (NM_053194) in pcDNA3.1 was a gift from Dr. Jean-Pierre Montmayeur (Fenech et al., 2009). Flag-tagged Ric-8B (NM_183172 with one missense mutation (A1586G)) in pcDNA3.1 was a gift from Dr. Bettina Malnic (Von Dannecker et al., 2006). The masGRK3ct-Nluc-HA constructs were constructed by introducing HA tag at the C-terminus of masGRK3ct-Nluc reported previously (Masuho et al., 2015b). PTX-S1 constructs were reported previously (Raveh et al., 2010). Nluc without the first methionine was inserted between residues 91 and 92 of GαoA (NM_020988) with SGGGGSGGGGS (11GS) linker at the N terminus and C terminus of the Nluc to make GαoA-Nluc in pcDNA3.1(+). Nluc without the first methionine was inserted between residues 97 and 98 of Gαq (U0038) with EFMV linker at the N terminus and with LYSS at the C terminus of the Nluc to make Gαq-Nluc in pCMV5. Nluc without the first methionine was inserted between residues 113 and 114 of GαsL (NM_000516) with EFMV linker at the N terminus and with LYSS at the C terminus of the Nluc to make GαsL-Nluc in pcDNA3.1(+). Nluc without the first methionine was inserted between residues 106 and 107 of Gα13 (NM_006572) with EFMV linker at the N terminus and with LYSS at the C terminus of the Nluc to make Gα13-Nluc in pcDNA3.1(+). Flag-Rab5a in pcDNA5/FRT/TO was obtained from Addgene (https://www.addgene.org/). The construct of R7BP in pcDNA3.1/V5-His-TOPO vector was reported previously (Song et al., 2006). GenBank accession number for each sequence is given in parentheses.
Antibodies
Anti-GAPDH antibody (MAB374), anti-HA tag antibody (clone 3F10) (11867423001), anti-GFP antibody (clones 7.1 and 13.1) (11814460001), Anti-GFP N-terminal antibody (G1544), and anti-Flag antibody (F7425) were purchased from MilliporeSigma. Anti-Gαo antibody (551) was purchased from MBL life science. HRP-conjugated anti-rabbit antibody (211-032-171), HRP-conjugated anti-mouse antibody (115-035-174), and HRP-conjugated anti-rat antibody (112-035-175) were purchased from Jackson ImmunoResearch. Alexa Fluor 546-conjugated anti-rabbit antibody (A10040), Alexa Fluor 546-conjugated anti-mouse antibody (A10036), and Alexa Fluor 488-conjugated anti-rat antibody (A21208) were purchased from Thermo Fisher Scientific.
Live cell microscopy
The HEK293T/17 cells transfected with Flag-D2R, GαoA, Venus 156-239-Gβ1, and Venus 1-155-Gγ1 or Venus 1-155-Gγ2 were imaged under a Leica TCS SP8 MP confocal microscope through a 25X objective water-immersion lens. Venus reconstituted by the formation of Gβ1γ2 dimer was excited by 488 nm laser lines, and emission was collected through HyD detectors set to 490–558 nm at 1-sec intervals acquired with a resonant scan speed of 12000 Hz. As previously described (Muntean et al., 2018), coverslips were transferred to a recording chamber and perfused with HBSS-HEPES at 2 ml per minute. A phasic puff of dopamine (100 μM) was rapidly applied (<1 sec) and washed out immediately adjacent to the field of view.
Cell culture and transfection
HEK293T/17 cells and DDT1 MF-2 cells were grown in DMEM supplemented with 10% FBS, minimum Eagle’s medium non-essential amino acids, 1mM sodium pyruvate, and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin) at 37 °C in a humidified incubator containing 5% CO2. For transfection, cells were seeded into 3.5-cm dishes at a density of 2 × 106 cells/dish for HEK293T/17 cells and 1 × 106 cells/dish for DDT1 MF-2 cells. After 2 h, expression constructs (total 5 μg/dish) were transfected into the cells using PLUS (5 μl/dish) and Lipofectamine LTX (6 μl/dish) reagents. GPCR (Flag-D2R (1), 3xHA-M1R (6), M2R (6), M4R (6), M5R (6), or A1R (1)), Gα (GαoA (2), GαoB (1), Gαi1 (1), Gαi2 (2), Gαi3 (1.5), Gαz (1.5), Gαq (2), Gα11 (2), Gα14 (4), Gα15 (2), GαsS (6), GαsL (4), Gαolf (6), Gα12 (3), or Gα13 (4)), Venus 156-239-Gβ (1), Venus 1-155-Gγ (1), and GRK3ct-Nluc-HA sensors (1) were transfected (the number in parentheses indicates the ratio of transfected DNA (ratio 1 = 0.21 μg)). Gα14/15 and Gαolf were transfected with Flag-Ric-8A (1) and Flag-Ric-8B (1), respectively. A construct carrying catalytic subunit of pertussis toxin PTX-S1 were transfected with Gα subunits except for Gαi1–3 and Gαo to inhibit the possible coupling of endogenous Gi/o to GPCRs. An empty vector (pcDNA3.1(+)) was used to normalize the amount of transfected DNA. To monitor the dissociation of Gα and Gβγ, GαoA-Nluc (0.1), Gαq-Nluc (1), GαssL-Nluc (3), or Gα13-Nluc (4) were transfected with Venus 156-239-Gβ (1) and Venus 1-155-Gγ (1). Gαq-Nluc and Gα13-Nluc were transfected with Flag-Ric-8A (1). GαssL-Nluc was transfected with Flag-Ric-8B (1).
Fast kinetic BRET assay to monitor G protein activity in living cells
Cellular measurements of BRET between Venus-Gβγ and GRK3ct-Nluc-HA sensors were performed in living cells (described in detail in (Masuho et al., 2015a; Masuho et al., 2015b)). Sixteen to twenty-four hr post-transfection, HEK293T/17 cells were washed once with BRET buffer (Dulbecco’s Phosphate-Buffered Saline (PBS) containing 0.5mM MgCl2 and 0.1% glucose) and detached by gentle pipetting over the monolayer. Cells were harvested with centrifugation at 500 g for 5 min and resuspended in BRET buffer. Approximately 50,000 to 100,000 cells per well were distributed in 96-well flatbottomed white microplates (Greiner Bio-One). The Nluc substrate, furimazine, were purchased from Promega and used according to the manufacturer’s instruction. BRET measurements were made using a microplate reader (POLARstar Omega or PHERAstar FSX; BMG Labtech) equipped with two emission photomultiplier tubes, allowing us to detect two emissions simultaneously with a highest possible resolution of 20 ms per data point. All measurements were performed at room temperature. The BRET signal is determined by calculating the ration of the light emitted by the Venus- Gβγ (535 nm with a 30 nm band path width) over the light emitted by the Nluc (475 nm with a 30 nm band path width). The average baseline value (basal BRET ratio) recorded prior to agonist stimulation was subtracted from the experimental BRET signal values to obtain the resulting difference (ΔBRET ratio). The rate constants (1/τ) of the activation and deactivation phases were obtained by fitting a single exponential curve to the traces with Clampfit 10.3.
Immunocytochemistry
To detect transfected molecules, HEK293T/17 cells were plated on laminin-coated coverslips coated with natural human laminin on top of the poly-d-lysine layer (Neuvitro). The cells were then transfected with expression plasmids and cultured for 16–24 hrs. The coverslips were transferred into 4% paraformaldehyde in PBS and fixed for 20 min at room temperature. The cells were permeabilized in 0.5% Triton X-100 in PBS for 5 min and were incubated in PBS for 5 min at room temperature to rinse cells. The permeabilized cells were incubated with 5% skim milk in 0.1% Triton X-100 in PBS for 60 min room temperature. After washing with 0.1% Triton X-100 in PBS once, the cells were stained with anti-Gαo antibody (551, MBL) (1:1,000 dilution), anti-HA antibody (11867423001, Roche) (1:100 dilution), or anti-Flag antibody (F7425, MilliporeSigma) (1:100 dilution) in 1% skim milk dissolved in 0.1% Triton X-100 in PBS for 90 min at room temperature. After washing with 0.1% Triton X-100 in PBS three timers, coverslips were incubated with Alexa Fluor 488 conjugated donkey anti-rat IgG (A-21208, Thermo Fisher Scientific) (1:500 dilution) for detecting HA, Alexa Fluor 488 conjugated donkey anti-rabbit antibody (A-21206, Thermo Fisher Scientific) (1:500 dilution) for detecting Nluc-Flag-K-Ras, and Alexa Fluor 546 conjugated donkey anti-rabbit antibody (A10040, Thermo Fisher Scientific) (1:500 dilution) for detecting GαoA and Flag-Rab5a in 1% skim milk dissolved in 0.1% Triton X-100 in PBS for 45 min at room temperature. The coverslips were washed twice with 1% Triton X-100 in phosphate-buffered saline, twice with phosphate-buffered saline, and mounted with VECTASHIELD Vibrance antifade mounting medium with DAPI (Vector Laboratories).
Immunoprecipitation
HEK293T/17 cells in 6-cm plates were transfected with the indicated constructs. Overnight after transfection, cells were washed once with ice-cold PBS and lysed with 1 ml of ice-cold IP buffer (50 mM Tris, pH 7.4, 1% Triton X-100, 300 mM NaCl, and cOmplete Protease Inhibitor Cocktail (MilliporeSigma)) by sonication on ice. After lysis, cell lysates were centrifuged at 14,000 rcf for 15 min at 4°C to remove insoluble debris. A 2 μg/sample of the anti-GFP antibody (clones 7.1 and 13.1) (MilliporeSigma) and 20 μl of Dynabeads Protein G (Thermo Fisher Scientific) were added, and the supernatants were tumbled for 1 h at 4°C. After three washes with 1 ml of ice-cold IP buffer, proteins bound to the beads were eluted with SDS-sample buffer (50 mM Tris, pH 6.8, 1% SDS, 143 mM β-mercaptoethanol, 0.08 mg/ml bromophenol blue, 10% glycerol). Immunoprecipitates were subjected to SDS-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes, and probed with the indicated antibodies.
Western blotting
For each 3.5-cm dish, transfected cells were lysed in 1 ml of sample buffer (62.5 mM tris-HCl, pH 6.8, 2 M urea, 2% SDS, 5% 2-mercaptoethanol, 10% glycerol, bromophenol blue (0.08 mg/ml)). Western blotting analysis of proteins was performed after samples were resolved by SDS–polyacrylamide gel electrophoresis and transferred onto PVDF membranes. Blots were blocked with 5% skim milk in PBS containing 0.1% Tween 20 (PBST) for 30min at room temperature, which was followed by 90 min incubation with specific antibodies diluted in PBST containing 1% skim milk (anti-Gαo antibody (1:1,000), anti-GFP antibody (clones 7.1 and 13.1) (1:1,000), anti-GFP N-terminal antibody (1:5,000), and anti-GAPDH antibody (1:10,000)). Blots were washed in PBST and incubated for 45 min with a 1:10,000 dilution of secondary antibodies conjugated with horseradish peroxidase (HRP) in PBST containing 1% skim milk. Western blotting was performed with BlotCycler automated western blot processor (Precision Biosystems). Proteins were visualized with Kwik Quant imager (Kindle Biosciences).
Quantification and statistical analysis
All experiments were conducted independently, at least three times. Exceptions are noted in Figure Legend. Statistical analyses were performed with GraphPad Prism Ver. 6.07. One-way ANOVA followed y Fisher’s LSD multiple-comparison post hoc test was used for comparing more than three groups and determining statistical significance. To compare two groups, unpaired t-test was performed. The number of biological and technical replicates were reported in figure legends.
Supplementary Material
Highlights.
All theoretically possible Gβγ combinations reconstituted and functionally tested
Gβγ subunits differ in signaling efficacy and kinetics at the plasma membrane
Gβγ subunits differentially translocate from the plasma membrane to organelles
Diversity of the Gβγ subunits contribute to unique signaling profiles of GPCRs
ACKNOWLEDGMENTS
We thank the members of the Martemyanov Lab for the helpful discussions, Ms. Hideko Masuho for technical support and data analyses, and Drs. Stephen Ikeda and Nevin Lambert for sharing Nluc-Flag-K-Ras construct. The authors are also grateful to Dr. Nevin Lambert for valuable insights, help with data interpretation and conceptual direction of the project at the early stages of its development. This work was supported by NIH grants DA036596 and MH105482 to KAM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing conflicts of interest.
REFERENCES
- Ajith Karunarathne WK, O’Neill PR, Martinez-Espinosa PL, Kalyanaraman V, and Gautam N (2012). All G protein betagamma complexes are capable of translocation on receptor activation. Biochem Biophys Res Commun 421, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akgoz M, Kalyanaraman V, and Gautam N (2004). Receptor-mediated reversible translocation of the G protein betagamma complex from the plasma membrane to the Golgi complex. J Biol Chem 279, 51541–51544. [DOI] [PubMed] [Google Scholar]
- Akgoz M, Kalyanaraman V, and Gautam N (2006). G protein betagamma complex translocation from plasma membrane to Golgi complex is influenced by receptor gamma subunit interaction. Cell Signal 18, 1758–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson A, Masuho I, Marron Fernandez de Velasco E, Nakano A, Birnbaumer L, Martemyanov KA, and Wickman K (2020). GPCR-dependent biasing of GIRK channel signaling dynamics by RGS6 in mouse sinoatrial nodal cells. Proc Natl Acad Sci U S A 117, 14522–14531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosier B, and Hermans E (2007). Versatility of GPCR recognition by drugs: from biological implications to therapeutic relevance. Trends in pharmacological sciences 28, 438–446. [DOI] [PubMed] [Google Scholar]
- Chen H, Leung T, Giger KE, Stauffer AM, Humbert JE, Sinha S, Horstick EJ, Hansen CA, and Robishaw JD (2007). Expression of the G protein gammaT1 subunit during zebrafish development. Gene Expr Patterns 7, 574–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre DJ, Robitaille M, Rebois RV, and Hebert TE (2009). The role of Gbetagamma subunits in the organization, assembly, and function of GPCR signaling complexes. Annu Rev Pharmacol Toxicol 49, 31–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escriba PV, Wedegaertner PB, Goni FM, and Vogler O (2007). Lipid-protein interactions in GPCR-associated signaling. Biochimica et biophysica acta 1768, 836–852. [DOI] [PubMed] [Google Scholar]
- Fenech C, Patrikainen L, Kerr DS, Grall S, Liu Z, Laugerette F, Malnic B, and Montmayeur JP (2009). Ric-8A, a Galpha protein guanine nucleotide exchange factor potentiates taste receptor signaling. Frontiers in cellular neuroscience 3, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, Gardella TJ, and Vilardaga JP (2009). Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol 5, 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Regalado A, Guzman-Hernandez ML, Ramirez-Rangel I, Robles-Molina E, Balla T, Vazquez-Prado J, and Reyes-Cruz G (2008). G protein-coupled receptor-promoted trafficking of Gbeta1gamma2 leads to AKT activation at endosomes via a mechanism mediated by Gbeta1gamma2-Rab11a interaction. Molecular biology of the cell 19, 4188–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman AG (1987). G proteins: transducers of receptor-generated signals. Annu Rev Biochem 56, 615–649. [DOI] [PubMed] [Google Scholar]
- Hauser AS, Chavali S, Masuho I, Jahn LJ, Martemyanov KA, Gloriam DE, and Babu MM (2018). Pharmacogenomics of GPCR Drug Targets. Cell 172, 41–54 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler JR, and Gilman AG (1992). G proteins. Trends Biochem Sci 17, 383–387. [DOI] [PubMed] [Google Scholar]
- Hermans E (2003). Biochemical and pharmacological control of the multiplicity of coupling at G-protein-coupled receptors. Pharmacology & therapeutics 99, 25–44. [DOI] [PubMed] [Google Scholar]
- Hillenbrand M, Schori C, Schoppe J, and Pluckthun A (2015). Comprehensive analysis of heterotrimeric G-protein complex diversity and their interactions with GPCRs in solution. Proc Natl Acad Sci U S A 112, E1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelreich S, Masuho I, Berry JA, MacMullen C, Skamangas NK, Martemyanov KA, and Davis RL (2017). Dopamine Receptor DAMB Signals via Gq to Mediate Forgetting in Drosophila. Cell reports 21, 2074–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollins B, Kuravi S, Digby GJ, and Lambert NA (2009). The c-terminus of GRK3 indicates rapid dissociation of G protein heterotrimers. Cell Signal 21, 1015–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iniguez-Lluhi JA, Simon MI, Robishaw JD, and Gilman AG (1992). G protein beta gamma subunits synthesized in Sf9 cells. Functional characterization and the significance of prenylation of gamma. J Biol Chem 267, 23409–23417. [PubMed] [Google Scholar]
- Inoue A, Raimondi F, Kadji FMN, Singh G, Kishi T, Uwamizu A, Ono Y, Shinjo Y, Ishida S, Arang N, et al. (2019). Illuminating G-Protein-Coupling Selectivity of GPCRs. Cell 177, 1933–1947 e1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, et al. (2013). Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamora C, Takizawa PA, Zaarour RF, Denesvre C, Faulkner DJ, and Malhotra V (1997). Regulation of Golgi structure through heterotrimeric G proteins. Cell 91, 617–626. [DOI] [PubMed] [Google Scholar]
- Jamora C, Yamanouye N, Van Lint J, Laudenslager J, Vandenheede JR, Faulkner DJ, and Malhotra V (1999). Gbetagamma-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell 98, 59–68. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Xie X, Zhang Y, Luo X, Wang X, Fan F, Zheng D, Wang Z, and Chen Y (2010). Regulation of G-protein signaling by RKTG via sequestration of the G betagamma subunit to the Golgi apparatus. Molecular and cellular biology 30, 78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaho Y, Tsai SC, Adamik R, Hewlett EL, Moss J, and Vaughan M (1984). Rhodopsin-enhanced GTPase activity of the inhibitory GTP-binding protein of adenylate cyclase. J Biol Chem 259, 7378–7381. [PubMed] [Google Scholar]
- Kassai H, Aiba A, Nakao K, Nakamura K, Katsuki M, Xiong WH, Yau KW, Imai H, Shichida Y, Satomi Y, et al. (2005). Farnesylation of retinal transducin underlies its translocation during light adaptation. Neuron 47, 529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SM, Sung JY, and Hebert TE (2016). Gbetagamma subunits-Different spaces, different faces. Pharmacol Res 111, 434–441. [DOI] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. (2014). A draft map of the human proteome. Nature 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselev OG, and Downs MA (2003). Rhodopsin controls a conformational switch on the transducin gamma subunit. Structure 11, 367–373. [DOI] [PubMed] [Google Scholar]
- Krumins AM, and Gilman AG (2006). Targeted knockdown of G protein subunits selectively prevents receptor-mediated modulation of effectors and reveals complex changes in non-targeted signaling proteins. J Biol Chem 281, 10250–10262. [DOI] [PubMed] [Google Scholar]
- Lan KL, Zhong H, Nanamori M, and Neubig RR (2000). Rapid kinetics of regulator of G-protein signaling (RGS)-mediated Galphai and Galphao deactivation. Galpha specificity of RGS4 AND RGS7. J Biol Chem 275, 33497–33503. [DOI] [PubMed] [Google Scholar]
- Lan TH, Liu Q, Li C, Wu G, and Lambert NA (2012). Sensitive and high resolution localization and tracking of membrane proteins in live cells with BRET. Traffic 13, 1450–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Ponissery-Saidu S, Yee KK, Wang H, Chen ML, Iguchi N, Zhang G, Jiang P, Reisert J, and Huang L (2013). Heterotrimeric G protein subunit Ggamma13 is critical to olfaction. J Neurosci 33, 7975–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann K, Masuho I, Patil DN, Baumann H, Hebert E, Steinrucke S, Trujillano D, Skamangas NK, Dobricic V, Huning I, et al. (2017). Novel GNB1 mutations disrupt assembly and function of G protein heterotrimers and cause global developmental delay in humans. Human molecular genetics 26, 1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney JP, and Sunahara RK (2016). Mechanistic insights into GPCR-G protein interactions. Curr Opin Struct Biol 41, 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder A, Pahlberg J, Boyd KK, Kerov V, Kolandaivelu S, Ramamurthy V, Sampath AP, and Artemyev NO (2013). Transducin translocation contributes to rod survival and enhances synaptic transmission from rods to rod bipolar cells. Proc Natl Acad Sci U S A 110, 12468–12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malerba N, De Nittis P, and Merla G (2019). The Emerging Role of Gbeta Subunits in Human Genetic Diseases. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, deRubio RG, Trembley M, Irannejad R, Wedegaertner PB, and Smrcka AV (2015). G protein betagamma subunits regulate cardiomyocyte hypertrophy through a perinuclear Golgi phosphatidylinositol 4-phosphate hydrolysis pathway. Molecular biology of the cell 26, 1188–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martemyanov KA, Yoo PJ, Skiba NP, and Arshavsky VY (2005). R7BP, a novel neuronal protein interacting with RGS proteins of the R7 family. J Biol Chem 280, 5133–5136. [DOI] [PubMed] [Google Scholar]
- Masuho I, Balaji S, Muntean BS, Skamangas NK, Chavali S, Tesmer JJG, Babu MM, and Martemyanov KA (2020a). A Global Map of G Protein Signaling Regulation by RGS Proteins. Cell 183, 503–521 e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuho I, Martemyanov KA, and Lambert NA (2015a). Monitoring G Protein Activation in Cells with BRET. Methods in molecular biology 1335, 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuho I, Ostrovskaya O, Kramer GM, Jones CD, Xie K, and Martemyanov KA (2015b). Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Science signaling 8, ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuho I, Skamangas NK, and Martemyanov KA (2020b). Live cell optical assay for precise characterization of receptors coupling to Galpha12. Basic Clin Pharmacol Toxicol 126 Suppl 6, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuho I, Xie K, and Martemyanov KA (2013). Macromolecular composition dictates receptor and G protein selectivity of regulator of G protein signaling (RGS) 7 and 9–2 protein complexes in living cells. J Biol Chem 288, 25129–25142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mervine SM, Yost EA, Sabo JL, Hynes TR, and Berlot CH (2006). Analysis of G protein betagamma dimer formation in live cells using multicolor bimolecular fluorescence complementation demonstrates preferences of beta1 for particular gamma subunits. Molecular pharmacology 70, 194–205. [DOI] [PubMed] [Google Scholar]
- Muntean BS, Zucca S, MacMullen CM, Dao MT, Johnston C, Iwamoto H, Blakely RD, Davis RL, and Martemyanov KA (2018). Interrogating the Spatiotemporal Landscape of Neuromodulatory GPCR Signaling by Real-Time Imaging of cAMP in Intact Neurons and Circuits. Cell reports 22, 255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neer EJ (1995). Heterotrimeric G proteins: organizers of transmembrane signals. Cell 80, 249–257. [DOI] [PubMed] [Google Scholar]
- O’Neill PR, Karunarathne WK, Kalyanaraman V, Silvius JR, and Gautam N (2012). G-protein signaling leverages subunit-dependent membrane affinity to differentially control betagamma translocation to intracellular membranes. Proc Natl Acad Sci U S A 109, E3568–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okashah N, Wan Q, Ghosh S, Sandhu M, Inoue A, Vaidehi N, and Lambert NA (2019). Variable G protein determinants of GPCR coupling selectivity. Proc Natl Acad Sci U S A 116, 12054–12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, and Lefkowitz RJ (2002). Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3, 639–650. [DOI] [PubMed] [Google Scholar]
- Raveh A, Cooper A, Guy-David L, and Reuveny E (2010). Nonenzymatic rapid control of GIRK channel function by a G protein-coupled receptor kinase. Cell 143, 750–760. [DOI] [PubMed] [Google Scholar]
- Saini DK, Chisari M, and Gautam N (2009). Shuttling and translocation of heterotrimeric G proteins and Ras. Trends in pharmacological sciences 30, 278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini DK, Kalyanaraman V, Chisari M, and Gautam N (2007). A family of G protein betagamma subunits translocate reversibly from the plasma membrane to endomembranes on receptor activation. J Biol Chem 282, 24099–24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini DK, Karunarathne WK, Angaswamy N, Saini D, Cho JH, Kalyanaraman V, and Gautam N (2010). Regulation of Golgi structure and secretion by receptor-induced G protein betagamma complex translocation. Proc Natl Acad Sci U S A 107, 11417–11422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindinger WF, Betz KS, Giger KE, Sabol A, Bronson SK, and Robishaw JD (2003). Loss of G protein gamma 7 alters behavior and reduces striatal alpha(olf) level and cAMP production. J Biol Chem 278, 6575–6579. [DOI] [PubMed] [Google Scholar]
- Schwindinger WF, Giger KE, Betz KS, Stauffer AM, Sunderlin EM, Sim-Selley LJ, Selley DE, Bronson SK, and Robishaw JD (2004). Mice with deficiency of G protein gamma3 are lean and have seizures. Molecular and cellular biology 24, 7758–7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindinger WF, Mihalcik LJ, Giger KE, Betz KS, Stauffer AM, Linden J, Herve D, and Robishaw JD (2010). Adenosine A2A receptor signaling and golf assembly show a specific requirement for the gamma7 subtype in the striatum. J Biol Chem 285, 29787–29796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senarath K, Payton JL, Kankanamge D, Siripurapu P, Tennakoon M, and Karunarathne A (2018). Ggamma identity dictates efficacy of Gbetagamma signaling and macrophage migration. J Biol Chem 293, 2974–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka AV, and Fisher I (2019). G-protein betagamma subunits as multi-functional scaffolds and transducers in G-protein-coupled receptor signaling. Cell Mol Life Sci 76, 4447–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov M, Lyubarsky AL, Strissel KJ, Savchenko AB, Govardovskii VI, Pugh EN Jr., and Arshavsky VY (2002). Massive light-driven translocation of transducin between the two major compartments of rod cells: a novel mechanism of light adaptation. Neuron 34, 95–106. [DOI] [PubMed] [Google Scholar]
- Song JH, Waataja JJ, and Martemyanov KA (2006). Subcellular targeting of RGS9–2 is controlled by multiple molecular determinants on its membrane anchor, R7BP. J Biol Chem 281, 15361–15369. [DOI] [PubMed] [Google Scholar]
- Stallmeyer B, Kuss J, Kotthoff S, Zumhagen S, Vowinkel K, Rinne S, Matschke LA, Friedrich C, Schulze-Bahr E, Rust S, et al. (2017). A Mutation in the G-Protein Gene GNB2 Causes Familial Sinus Node and Atrioventricular Conduction Dysfunction. Circ Res 120, e33–e44. [DOI] [PubMed] [Google Scholar]
- Ueda N, Iniguez-Lluhi JA, Lee E, Smrcka AV, Robishaw JD, and Gilman AG (1994). G protein beta gamma subunits. Simplified purification and properties of novel isoforms. J Biol Chem 269, 4388–4395. [PubMed] [Google Scholar]
- Vilardaga JP, Jean-Alphonse FG, and Gardella TJ (2014). Endosomal generation of cAMP in GPCR signaling. Nat Chem Biol 10, 700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Dannecker LE, Mercadante AF, and Malnic B (2006). Ric-8B promotes functional expression of odorant receptors. Proc Natl Acad Sci U S A 103, 9310–9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, and Sprang SR (1995). The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell 83, 1047–1058. [DOI] [PubMed] [Google Scholar]
- Watanabe S, and Boucrot E (2017). Fast and ultrafast endocytosis. Curr Opin Cell Biol 47, 64–71. [DOI] [PubMed] [Google Scholar]
- Wettschureck N, and Offermanns S (2005). Mammalian G proteins and their cell type specific functions. Physiological reviews 85, 1159–1204. [DOI] [PubMed] [Google Scholar]
- Yan K, Kalyanaraman V, and Gautam N (1996). Differential ability to form the G protein betagamma complex among members of the beta and gamma subunit families. J Biol Chem 271, 7141–7146. [DOI] [PubMed] [Google Scholar]
- Ye Y, Sun Z, Guo A, Song LS, Grobe JL, and Chen S (2014). Ablation of the GNB3 gene in mice does not affect body weight, metabolism or blood pressure, but causes bradycardia. Cell Signal 26, 2514–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liu W, Liu J, Xiao W, Liu L, Jiang C, Sun X, Liu P, Zhu Y, Zhang C, et al. (2010). G-protein beta2 subunit interacts with mitofusin 1 to regulate mitochondrial fusion. Nature communications 1, 101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source data for raw traces of GPCR responses used for quantitative analysis reported in this study have not been deposited in a publicly available repository because of their trivial nature and custom format they are generated in. They have been archived locally on the institutional cloud service. To request access, please contact Lead Contact.
This paper does not report any original codes.
Scripts were not used to generate the figures reported in this paper.
Any additional information required to reproduce this work is available from the Lead Contact.