

Abstract
Sinoatrial node cardiomyocytes (SANcm) possess automatic, rhythmic electrical activity. SAN rate is influenced by autonomic nervous system input, including sympathetic nerve increases of heart rate (HR) via activation of β-adrenergic receptor signaling cascade (β-AR). L-type calcium channel (LTCC) activity contributes to membrane depolarization and is a central target of β-AR signaling. Recent studies revealed that the small G-protein Rad plays a central role in β-adrenergic receptor directed modulation of LTCC. These studies have identified a conserved mechanism in which β-AR stimulation results in PKA-dependent Rad phosphorylation: depletion of Rad from the LTCC complex, which is proposed to relieve the constitutive inhibition of CaV1.2 imposed by Rad association. Here, using a transgenic mouse model permitting conditional cardiomyocyte selective Rad ablation, we examine the contribution of Rad to the control of SANcm LTCC current (ICa,L) and sinus rhythm. Single cell analysis from a recent published database indicates that Rad is expressed in SANcm, and we show that SANcm ICa,L was significantly increased in dispersed SANcm following Rad silencing compared to those from CTRL hearts. Moreover, cRadKO SANcm ICa,L was not further increased with β-AR agonists. We also evaluated heart rhythm in vivo using radiotelemetered ECG recordings in ambulating mice. In vivo, intrinsic HR is significantly elevated in cRadKO. During the sleep phase cRadKO also show elevated HR, and during the active phase there is no significant difference. Rad-deletion had no significant effect on heart rate variability. These results are consistent with Rad governing LTCC function under relatively low sympathetic drive conditions to contribute to slower HR during the diurnal sleep phase HR. In the absence of Rad, the tonic modulated SANcm ICa,L promotes elevated sinus HR. Future novel therapeutics for bradycardia targeting Rad – LTCC can thus elevate HR while retaining βAR responsiveness.
Keywords: L-type calcium channel, cardiomyocyte, heart rhythm, heart rate regulation
Introduction
Spontaneous electrical activity of the sinoatrial node (SAN) initiates cardiac rhythm and is a critical determinant of heart rate (HR). Cardiomyocytes of the SAN (SANcm) are highly specialized cells that generate automatic activity and form the cellular basis for cardiac pacemaking. SANcm exhibit a spontaneous diastolic depolarization. The early phase of diastolic depolarization is dominated by inward cationic funny current (If(1) carried by HCN4(2)). The latter phase of diastolic depolarization is driven by voltage-gated L-type calcium channels (LTCC, with the main pore forming subunit, CaV1.2 or CaV1.3)(3). To a large extent, If and the LTCC complex determine the ‘membrane clock’ that interacts with the calcium clock in the SAN(4, 5). The inter-relation of the membrane clock and calcium clock is underscored by studies of the genetic model of loss of CaV1.3 impairing sarcoplasmic reticulum Ca2+-release(6), and by computational studies linking feedback loops among sarcoplasmic Ca2+-release, NCX, and ICa,L(7). The ability of ICa,L to simultaneously contribute to membrane-and calcium-clocks suggests that targeted regulation of the LTCC is a potentially powerful approach to control HR. Although cellular and molecular mechanisms that contribute to pacemaker activity are well established(8), bradyarrhythmia treatment options are limited mainly to electronic devices that are insensitive to autonomic nervous system regulation. Experimentally, nifedipine (an LTCC antagonist) has a negative chronotropic effect on the leading pacemaker site of the SAN(9); similarly, genetic knockout of CaV1.3 induces bradycardia(10). A fundamental premise of this study is that modulation of LTCC activity conferred by Rad provides a central nodal mechanism for sympathetic nervous system (SNS) control of SAN rhythm.
The LTCC is a heteromultimeric protein complex(11). The pore-forming subunit of the LTCC in SANcm is carried by CaV1.3(10, 12) and CaV1.2(3) channels. Key auxiliary subunits of the LTCC complex include CaVβ2, α2δ, and CaM(11). Recent work (13)highlights the importance of RGK proteins(14) as regulators of ICa,L via association with the β subunits(15–18). Rad is a constituent of the LTCC complex in the myocardium(18, 19). Sympathetic nervous system drive acts via β-AR signaling cascades, resulting in the activation of PKA which in turn modulates LTCC activity(20). β-AR modulation is maintained in transgenic mouse models expressing mutant CaV1.2 and CaVβ that cannot be phosphorylated by PKA(13, 21), suggesting that a non-channel PKA target is central to channel modulation. Using proximity biotinylation to analyze β-adrenergic-dependent changes within the LTCC complex, Rad was recently shown to be depleted from the LTCC complex following acute β-AR stimulation. Rad depletion from the LTCC relieves the constitutive inhibition imposed by Rad association(15, 17, 18, 22). In keeping with its presumptive role as an endogenous inhibitor of LTCC function, we showed that LTCC activity is increased In Rad-null mice (Rad−/−) with properties mirroring β-AR modulation of ICa,L(17). ECG telemetry showed that Rad−/− mice have a complex phenotype that includes differential vascular and inflammatory properties(16). However, this model has multi-organ involvement(16, 23) and possible development-related effects that potentially confound analysis of heart rhythm modification originating from channel modulation in cardiomyocytes. To circumvent these effects, we recently developed an inducible myocardium-restricted Rad knockout (cRadKO) mouse(15). Cardiomyocyte selective Rad deletion was shown to phenocopy β-AR modulated LTCC properties, increasing basal ventricular contractile function(15). Using this same conditional KO model, we now test the hypothesis that myocardial Rad-knockout will modulate sinus heart rate. Here we show that cRadKO mice exhibit an elevated intrinsic heart rate, and elevated sleep phase heart rate. Mechanistically, Rad regulation of HR appears to be driven by modulated ICa,L following Rad loss in SANcm. This work reveals Rad – LTCC interactions as a novel target for future therapeutics for symptomatic bradycardia.
2. Materials & Methods
All experimental procedures and protocols were approved by the Animal Care and Use Committee of the University of Kentucky and conformed to the National Institute of Health “Guide for the Care and Use of Laboratory Animals.”
2.1. Animal Model
Transgenic animals were generated on C57BL/6J background and full description is provided in reference(15). In brief, the conditional Rrad allele was made via a sequential insertion strategy using CRISPR-Cas9 technology, in which exons 2 and 3 of RRAD were flanked by Cre recombinase-dependent loxP (flox:fl) recognition sequences. These Radfl/fl mice were then crossed with mice expressing a tamoxifen-inducible Cre recombinase (MerCreMer) under α-myosin heavy chain promoter(24) to produce Radfl/fl-MHC animal. Rad deficiency was induced by a single intraperitoneal injection in control (Radfl/fl) and experimental mice with tamoxifen dissolved in sunflower seed oil (40 mg/kg body weight), resulting in Rad deficiency in the myocardium of Radfl/fl-MHC mice(15). The single tamoxifen injection protocol minimizes cardiomyopathological effects observed with multi-day administration of tamoxifen(25). For SANcm studies, all mice received tamoxifen and were used >3 weeks after tamoxifen treatment. For telemetry studies, recordings were made before and after tamoxifen treatment in the same mice. Male and female mice were used; the age of the mice was 2–8 months. Radfl/fl-MHC after receiving tamoxifen are abbreviated ‘cRadKO,’ and control mice expressing endogenous Rad are abbreviated, CTRL.
2.2. Single Cell Database Analysis
Fastq files from single-cell RNA-sequencing (scRNA-seq) analysis of isolated mouse sinus atrial node (SAN) tissue, as reported by Linscheid and colleagues(26), were retrieved from NCBI Gene Expression Omnibus (accession number GSE130710, sample H4). Cell Ranger 3.1 pipeline (10X Genomics, USA) was used for read alignment using the mouse mm10 release 93 reference genome modified to include pre-mRNA. Cell calls were made using default parameters for Cell Ranger. Unique molecular identifier counts for all partitions identified as cells were greater than 2150. The re-analyze function in Cell Ranger was used to eliminate cell clusters enriched in mitochondrial reads as described on the 10x website (https://kb.10xgenomics.com). Only one cluster was eliminated because of enrichment in mitochondrial genes (the top most differentially expressed genes were mitochondrial genes indicating that these RNAseq reactions were from dead cells). The remaining 5,472 cells used for analysis. K means clustering was used to define clusters. In Loupe Cell Browser, cells containing RRAD were selected. For cell type determination, we used the genes that were found by Linscheid et al to be the most differentially expressed in each cell type(26). To be defined as a specific cell type cluster, the cluster had to have the following genes as their most deferentially expressed genes: sinus node myocytes: Myh6, Ctnna3, Ryr2, Rbm20, Dmd, Ttn, and Tbx5; fibroblasts: Col1a1, Fbn1, Ddr2, Lama2, Lamc1, Pcsk6, Gpc6, Mecom, Rbms3, and 4930578G10Rik; macrophages: Maf, F13a1, Cd163, C3ar1, P2yr6, Mrc1, Mgl2, Adgre1, and Dab2; vascular endothelial: Ptprb, Icam1, Vwf, Ldb2, Pecam1 and Cdh13; adipocytes: Ucp1, Cidea, Prdm16, Pparg, Lep, Ghr, Slc1a5, Pde3b, Sorbs1, Acsl1, and Adopr2; endocardial: Npr3, Cdh13, Engm Hmcn1 and Gmds and epicardial cells: CTRL1, Rbfox1, Kcnd2, Grip1, Plxna4 and Syne2.
2.3. Quantitative Proteomics Database Analysis
The mass spectrometry proteomics data was downloaded from the ProteomeXchange Consortium via the PRIDE repository (https://www.ebi.ac.uk/pride/). Dataset for mouse SAN cells (PXD008736) include 6 samples with 12 fractions per sample performed in technical duplicate. Raw MS data were analyzed using MaxQuant v1.6.8.0 (Max-Planck Institute of Biochemistry, Department of Proteomics and Signal Transduction, Munich, Germany). Peptide search was performed using the UniProt reference proteome for Mus musculus (Proteome ID UP000000589). False-discovery rate (FDR) was set to 1% for peptide, protein, and side decoy identification with base FDR calculated on delta score. Unmodified, unique and razor peptides were used for protein quantification to address high amino acid sequence similarity between paralogous proteins. All other parameters were kept at default. For positive protein identification the minimum number of peptides was 2. Fixed and variable modifications included in the analysis were carbamidomethylation, methionine oxidation, and N-terminal acetylation with a maximum allowed number of 5 modifications per peptide. To ensure that sensitivity was adequate and that the cells isolated were SAN, we quantified the intensity of Catenin Alpha3 (CTNNA3) and Hyperpolarization-activated cyclic nucleotide gated channel 4 (HCN4) proteins, and eliminated any samples that did not exhibit a significant quantity for each of these proteins. The remaining samples were used to quantify Rad protein.
2.4. RNA In Situ Hybridization
The in situ hybridization experiment was done using RNAscope®(27) (Advanced Cell Diagnostics). Hearts were fixed with 10% neutral buffered formalin for 16–32 hours at room temperature (RT). Washed the tissue in 100% ethanol and embedded in paraffin. Prepared 5μM paraffin sections on a charge slide in RNAase free condition. Sections were cut from the right atrial wall/appendage bounded by inferior and superior vena cava. RNAscope Multiplex Fluorescent Detection Reagent V2 kit (ACD, Cat. No. 323110) was used to probe Rrad and Hcn4 on the section as per the manufacturer’s protocol. A custom designed probe for Rrad was generated (Mm-Rrad-O1, 885451). The Rrad probe recognized sequence corresponding to transcript downstream of the flox site shown to be reduced in heart lysate by qRT-PCR(15). HCN4 probe was from the ACD catalog (Mm-Hcn4-C2, 421271-C2). Background was assessed with negative control probes targeting the bacterial DapB gene (accession #EF191515, (Advanced Cell Diagnostics).
2.5. Cellular electrophysiology
ICa,L was recorded in the whole-cell configuration of the patch-clamp technique. All recordings were performed at room temperature (20° to 22 °C). The pipette solution consisted of (in mmol/L) 125 Cs-methano-sulfonate, 15 TEA-Cl, 1 MgCl2, 10 EGTA, and 5 HEPES, 5 Mg-ATP, 5 phosphocreatine, pH 7.2. Bath solution contained (in mmol/L) 140 NaCl, 5.4 KCl, 1.2 KH2PO4, 5 HEPES, 5.55 glucose, 1 MgCl2, 1.8 CaCl2, pH 7.4. Once a cell was successfully patched, zero sodium bath solution was introduced into the chamber (mmol/L) 150 NMDG, 2.5 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, 5 4-AP, pH 7.2. Recordings of Isoproterenol response were recorded in zero sodium bath solution containing 100 nM isoproterenol.
2.6. ECG radiotelemetry
Mice were anesthetized with isoflurane, and telemetry transmitter units (PhysioTel ETA-F10; Data Sciences International) were implanted in the peritoneal cavity under aseptic conditions. The two ECG leads were secured near the apex of the heart and the right acromion. Mice were housed singly and allowed to recover for at least 2 weeks prior to the start of baseline recordings. ECG data was analyzed for RR interval and PR interval using Ponemah software (Data Sciences International). For intrinsic heart rate atropine (1mg/kg) + propranolol (1mg/kg) was administered ip during the sleep phase. For heart rate variability we used Ponemah Software Platform (Data Sciences International) to calculate SD of HR from normal to normal intervals for data collected from 3 runs of 1–3 minute continuous periods without noise events before and after ISO; for the air jet we limited the analysis to cover the ~5 minutes of sporadic agitation of mice by air jets. Traces were manually examined to verify absence of noise events.
2.7. Statistical Analysis
For statistical analyses performed on all cellular observations the mouse is the primary unit of analysis. Cellular mean ± SEM values are represented in the figures. Investigators were blinded to genotype until data analysis was completed. To plot analyzed data and perform statistical tests, CTRL versus cRadKO data was then decoded and entered into Prism version 8.4. For acute ISO, paired t-tests were used to compare groups. Sample sizes are reported as N=number of hearts, and n=number of cells. For ECG telemetry (Figures 5–7), Students paired t-test was used for baseline versus post-tamoxifen.
Figure 5. Intrinsic heart rate increases after Rad deletion.
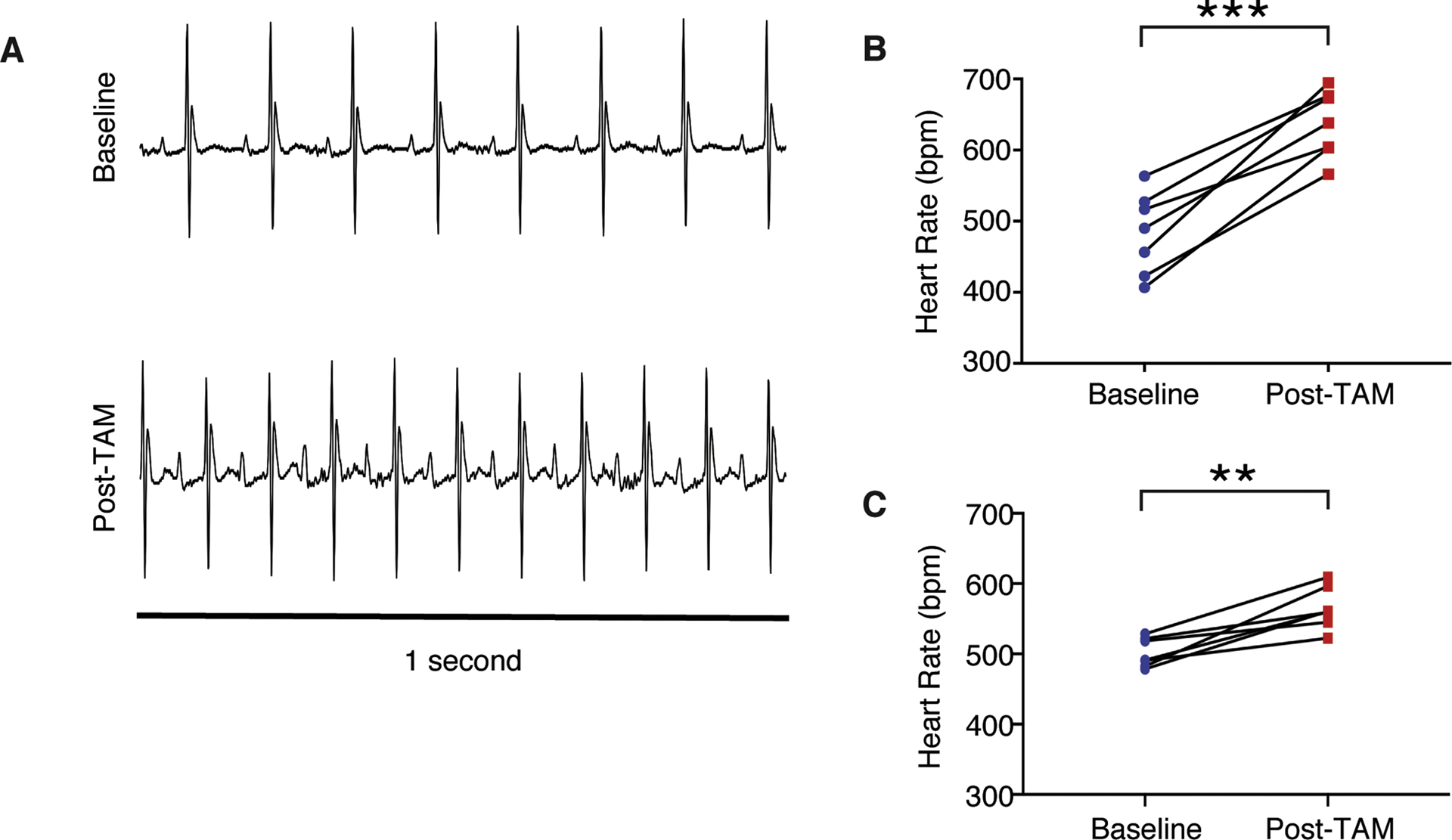
A) Representative ECG signals during the sleep phase demonstrate an increase in intrinsic heart rate after tamoxifen treatment. B),C) Heart rate increases after tamoxifen treatment in B) females (***p<10−3, N = 7 mice) and in C) males (**p = 0.002, N = 7 mice).
Figure 7. Rad deletion demonstrates preserved Heart Rate Variability (HRV).
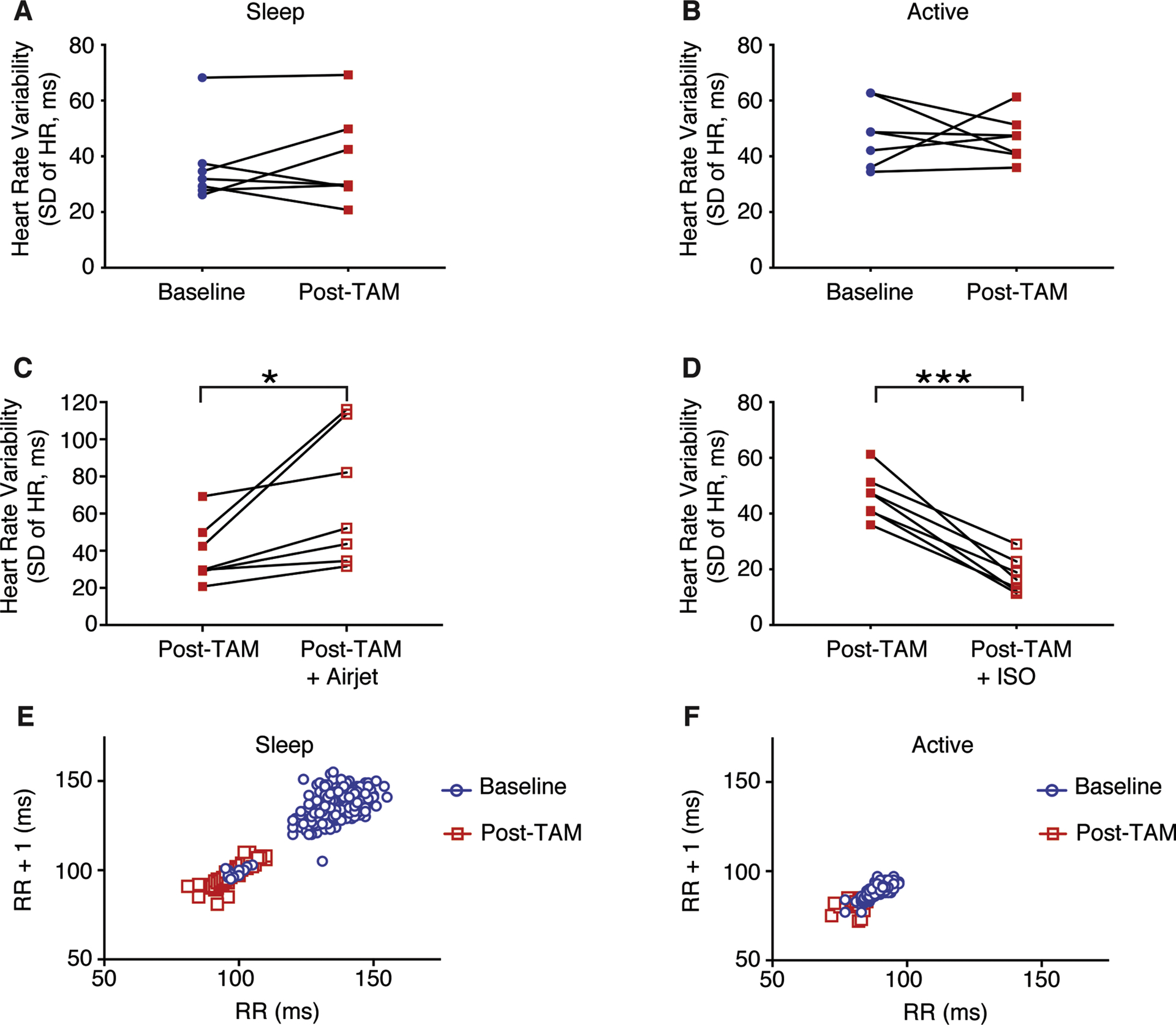
A) HRV is not significantly different after tamoxifen treatment in the sleep phase (p = 0.59), or B) in the active phase (p = 0.80). C) HRV increases Post-TAM after exposed to an airjet (*p = 0.03). D) HRV increases Post-TAM after an injection of isoproterenol (***p<10−3, N = 7 mice). E),F) Poincare plots in which the RR interval is plotted against the subsequent RR interval (RR + 1) in a representative mouse during the sleep phase E) and during the active phase F).
3. Results
3.1. SANcm express Rad
In the cRadKO mouse model cre-recombinase is driven by a myosin heavy chain 6 promoter limiting expression to all cardiomyocytes (reference(24), and personal communication Jeff Molkentin). Following tamoxifen-mediated cre-recombination Rad protein levels are not detected from the whole hearts of cRadKO(15) mice, suggesting cardiomyocyte-restricted expression of Rad in the heart. Similarly, single nucleus RNAseq (snRNAseq) analysis of the mouse SAN showed that RRAD transcript is enriched ~10-fold in SAN cardiomyocytes (SANcm) compared to non-cardiomyocytes from the SAN region(26). However, the SAN contains a heterogeneous mixture of cardiomyocytes(28). To assess Rad expression among SANcm, we re-examined a proteomics and snRNAseq dataset using tissue obtained from a segment of the right atrial wall, containing SAN and surrounding tissue(26). t-distributed stochastic neighbor embedding (t-SNE) shows 203 (of 5,472) cells analyzed can be classified as SANcm (cardiomyocytes) based on the expression of Myh6, Ctnna3, RyR2, Rbm20, Dmd, Ttn, and Tbx5(26, 29). In addition to SANcm, clusters were classified as fibroblasts, epicardial, endocardial, macrophage, adipocyte, and an unidentified cluster(26). Rad RNA was heterogeneously expressed in the SANcm cluster and enriched in a sub-population of SANcm. (Figure 1A). Within the SANcm cluster 38% of cells expressed RRAD transcript (77 of 203 cells). Re-analysis of RNAseq data from van Eif and colleagues(29) also showed RRAD transcript in SAN (data not shown). Analysis of the available proteomic database also support SAN expression of Rad protein. For context we also re-analyzed data for expression of HCN4, RyR2, and CaV1.2 (Figure 1B). To examine Rad expression in SANcm, we performed RNA in situ hybridization in our CTRL and cRadKO mice. HCN4+ cells in our CTRL mice prominently express Rad (Figure 2, upper panels). By contrast, cRadKO hearts show dramatically reduced Rad transcript (Figure 2, middle panels). Three mutually exclusive explanations for the reduced, albeit present signal in cRadKO include: 1) an Rrad transcript with a long ½-life; 2) non-specific background artifact signal; or 3) non-cardiomyocyte signal. The latter possibility is supported by recently published human snRNAseq data(30). These data are consistent with our previously published study, in which tamoxifen induction of the floxed-Rad gene results in loss of Rad from cardiomyocytes across the heart(15). Taken together, these new data suggest that Rad is expressed in SANcm; thus, the loss of Rad might impact SAN function.
Figure 1. RRad is expressed in SANcm.
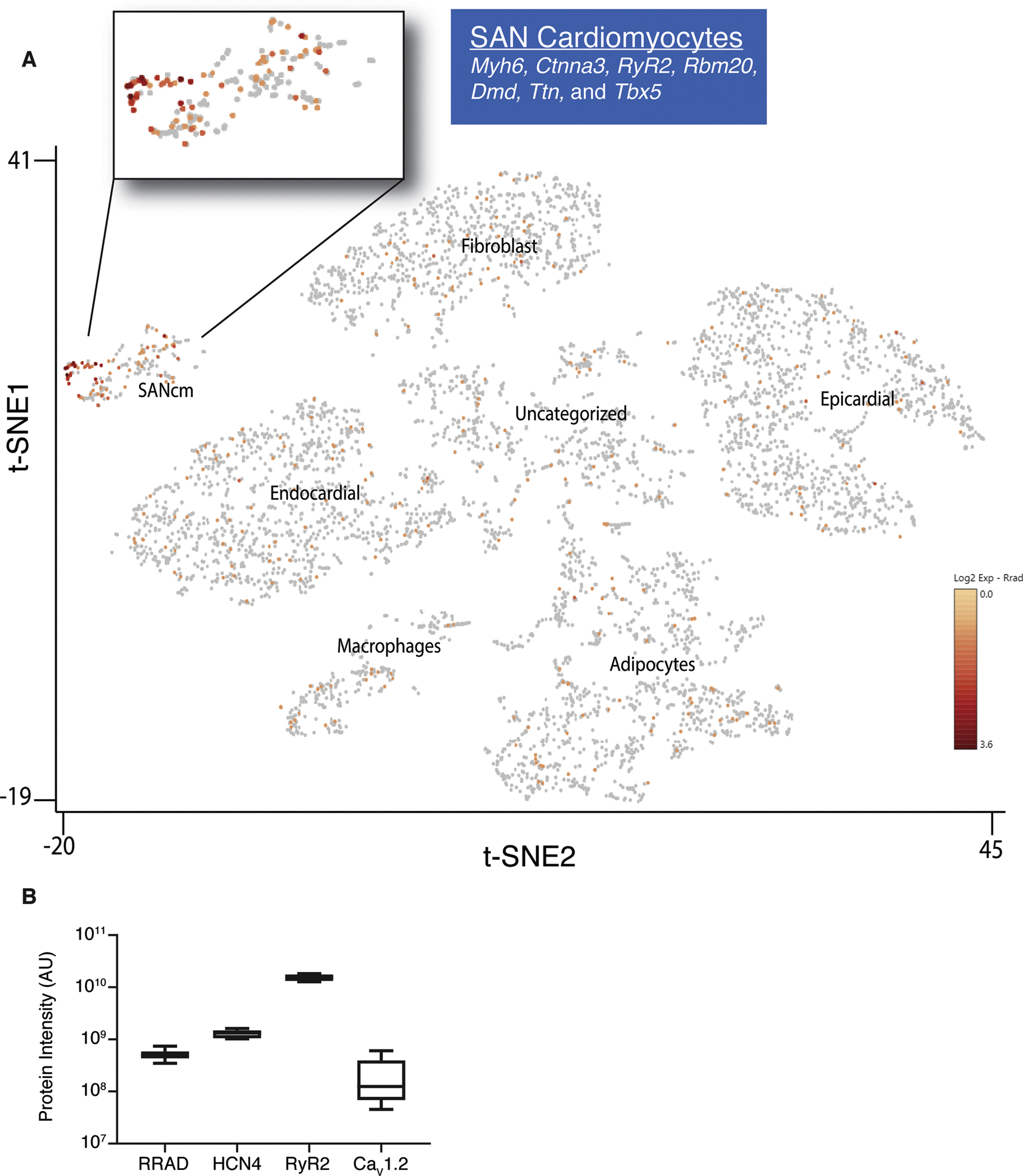
A) Two-dimensional t-SNE (Stochastic Neighbor Embedding) plot outlines the major populations of the sinus node. Analysis of database from Linscheid et al. 2019. Each point represents a single nucleus. Cell populations are colored according to intensity of RRad expression, with an expanded view of the SANcm. B) Quantification of data mined from Linscheid et al. 2019 of the expression of Rad protein in relation to other proteins known to be expressed specifically in SANcm (HCN4) and cardiomyocytes in general (RyR2 and CaV1.2).
Figure 2. Rad expression in SANcm.
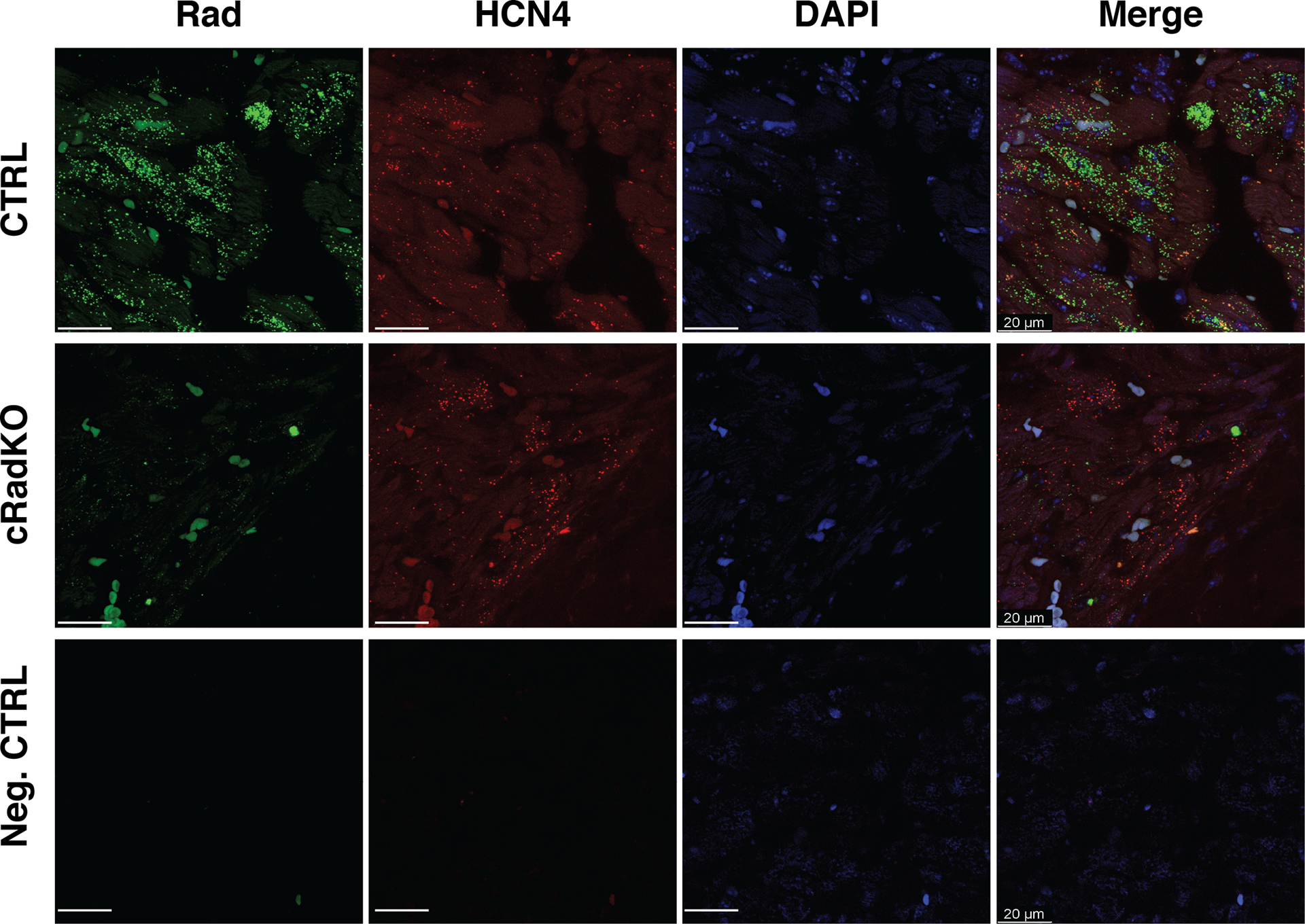
Representative images of single-molecule fluorescence in situ hybridization against Rad (green) and HCN4 (red) in CTRL (top images) and cRadKO (middle images) in SANcm. Lower panel, probe for bacterium DapB gene serves as a background control (bottom images). Heart tissue was collected >2 weeks after tamoxifen treatment. Tissue samples were stained with DAPI (blue). Images of channels merged displayed on the right. Representative of 3 CTRL and 3 cRadKO hearts. Scale bar: 20 μm.
3.2. L-type calcium current (ICa,L) in SANcm
Cells dispersed from the SAN region of cRadKO were morphologically indistinguishable from those of control mice (Figure 3A). Cells selected for recording beat spontaneously in physiological salt solution, were spindle shaped (Figure 3A). Cell capacitance was not different in cells from cRadKO mice (42±4pA/pF, n=17 and 42±4pA/pF, n=21 for CTRL and cRadKO, respectively; Figure 3B). SANcm from cRadKO mice showed larger current density (Figure 3C) with the peak of the peak ICa,L occurring at +5mV for CTRL and 0mV for cRadKO (Figure 3D).A Boltzmann fit of the I(V) curves yielded maximal LTCC conductance showing a significant increase with Rad loss (4.8+0.8pS/pF, n=17, and 10+1.3pS/pF, n=21, p=0.0015 for CTRL and cRadKO, respectively, Figure 3E). The midpoint of current activation (V½) was −7.5±1.4mV for CTRL and −10.1±1.4 for cRadKO (Figure 3F). Steady state inactivation was not different between CTRL and cRadKO ICa,L (data not shown). There was no significant difference in ICa,L properties (current density, maximal conductance, or voltage dependence between females and males, Supplement Figure 1). These data are consistent with alterations observed in ICa,L from ventricular cardiomyocytes following Rad ablation(15), suggesting a major contribution of Rad to basal ICa,L in SANcm.
Figure 3. Rad reduction increases SANcm ICa,L.
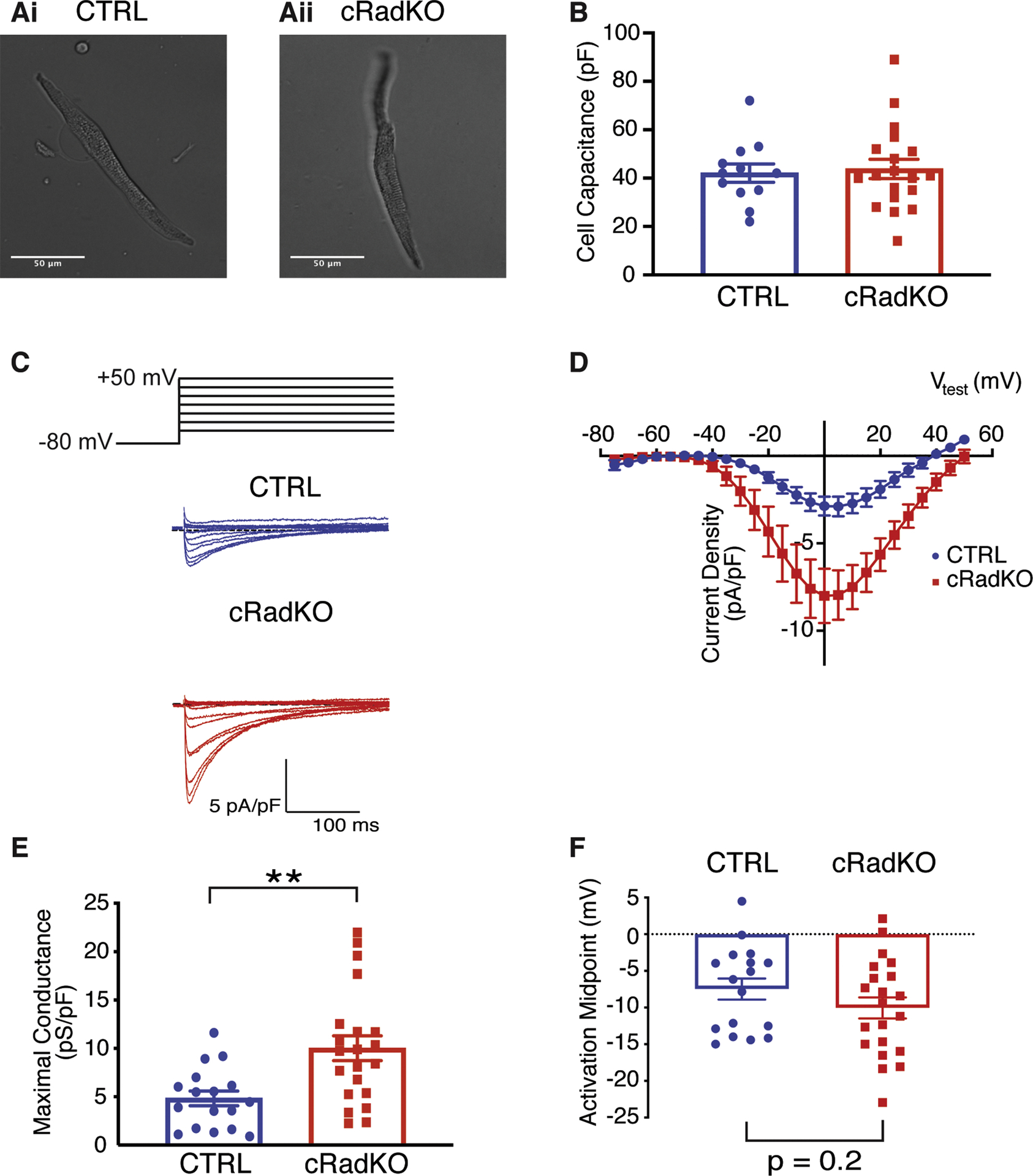
A) Representative photomicrographs of SANcm selected for patching studies from CTRL (Ai) and cRadKO (Aii) hearts. B) Cell capacitance is not significantly different (p = 0.77). C) Family of ICa,L for CTRL (blue) and cRadKO (red). Scale bars: 5 pA/pF, 100 ms. D) Current-voltage relationships for peak ICa,L from CTRL and cRadKO SANcm. E) Maximal conductance is significantly increased (**p<0.01) and F) activation midpoint trends toward a negative shift with Rad deletion (p=0.2). N = 8 mice, n = 17 cells for CTRL; N = 9 mice, n = 21 cells for cRadKO.
A growing literature suggests that Rad, and other members of the RGK GTPase subfamily, play critical roles in calcium-channel modulation, serving as constitutive LTCC inhibitors, whose actions are regulated by protein phosphorylation(14, 18, 19). In agreement with this model, we previously showed that Rad deletion results in ventricular ICa,L that phenocopies the current in wild-type cardiomyocytes following acute β-AR activation by isoproterenol (ISO)(15). We next tested the effect of acute ISO on ICa,L in SANcm from CTRL and cRadKO hearts. ICa,L from CTRL SANcm was significantly increased (Figure 4Ai). Maximal conductance increased (+2.8±0.9pS/pF; Figure 4Bi) and the midpoint of activation shifted negative (−2.1±0.8mV; Figure 4Ci). By contrast, ICa,L in SANcm from cRadKO hearts show no significant increase of conductance (−0.9±1.2pS/pF; Figure 4Bii) and no effect on activation midpoint (−0.2±0.6mV; Figure 4Cii). These results are consistent with our earlier findings from cardiac ventricular cardiomyocytes(15, 17), and support the working hypothesis that Rad functions as a key mediator of β-AR modulation of ICa,L.
Figure 4. β-AR stimulation has no significant effect on cRadKO SANcm ICa,L.
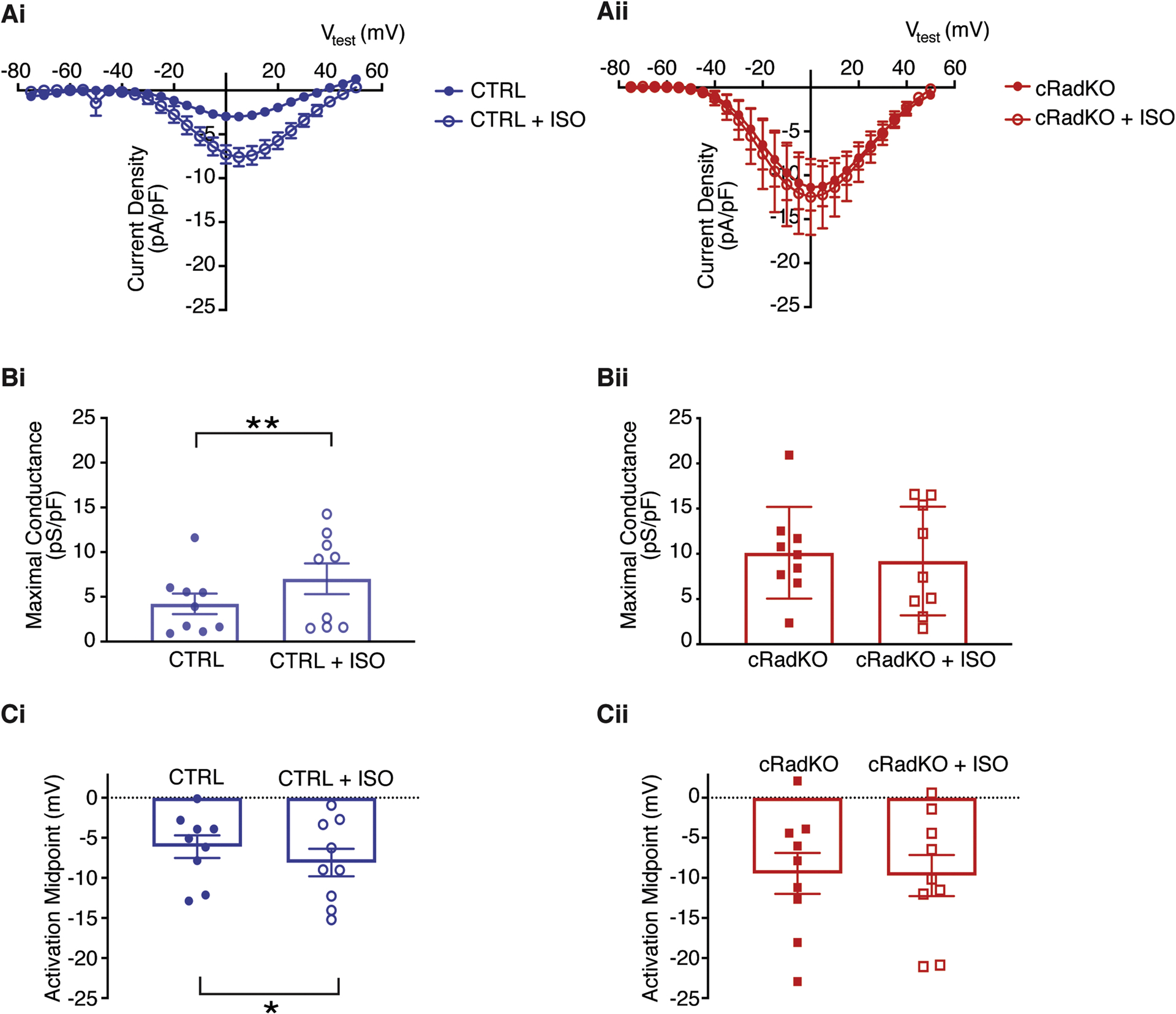
A) Current-voltage relationships for peak ICa,L from CTRL (Ai) and cRadKO SANcm (Aii) for basal (closed symbols) and after 100 nM ISO (open symbols). B) Maximal conductance before and after ISO for CTRL (Bi, **p = 0.01) and cRadKO (Bii, p = 0.45). C) Activation midpoint before and after ISO for CTRL (Ci, *p = 0.05) and cRadKO (Cii, p = 0.68). N = 5 mice, n = 9 cells for CTRL; N = 4 mice, n = 9 cells for cRadKO.
3.3. Rad reduction increases intrinsic heart rate
Rad deletion instills a PKA modulated ICa,L under otherwise unstimulated conditions in a cell autonomous fashion (Figure 3 and(15, 17, 19)). ICa,L contributes to the latter phase of the pacemaker depolarization in SANcm(8, 31–33); thus, modulated-like ICa,L arising from Rad silencing predicts an elevated intrinsic SAN heart rate. To examine the effect of Rad loss on HR in vivo, Radfl/fl.MHC mice were implanted with radiotelemetry units. Measurements were made at baseline and 3 weeks after tamoxifen administration to compare the impact of Rad loss on the same mice. Combined β-AR-block (propranolol) and cholinergic antagonist (atropine), revealed intrinsic HR from ambulatory mice. Induced knockout of cardiomyocyte Rad resulted in a significant elevated intrinsic HR (Figure 5A). In females, intrinsic HR increased 153+19bpm between baseline and >3 weeks after tamoxifen (483±22bpm and 637±18bpm, for baseline and post-tamoxifen, respectively, Figure 5B). Male mice also showed significantly elevated intrinsic HR after tamoxifen (mean difference 63±12bpm; 502±8bpm and 565±11bpm, for baseline and post-tamoxifen, respectively, Figure 5C).
3.4. Rad-deletion effects on HR are greatest when Sympathetic Drive is Relatively Low
The autonomic nervous system (ANS) modulation of heart function is apparent as the diurnal variation of HR. Sympathetic nervous system (SNS) drive is mediated in cardiomyocytes via activation of β-ARs. Figure 6A shows representative ECG traces during the sleep (day) and active phases (night) along with a 48h continuous recording of the moving average of HR at baseline and after Rad loss (Figure 6B). Sleep phase HR was significantly elevated with cRadKO (535±15bpm and 593±13bpm, baseline and cRadKO, respectively; Figure 6C) but the active phase HR was not different (652±8 and 671±10, baseline and post-tamoxifen, respectively; Figure 6D). Similar results were obtained for male mice (sleep phase, 511±11bpm and 598±11bpm; active phase, 647±9 and 671±13bpm, baseline and post-tamoxifen, respectively; Figures 6E and 6F). Diurnal variation remained, but the difference between active and sleeping phase HR diminished after Rad silencing. Taken together these results are consistent with elevated ICa,L in cRadKO SANcm driving more rapid sinus rhythm unmasked by reduced SNS drive during the sleep phase.
Figure 6. Rad deletion increases heart rate in the sleep phase.
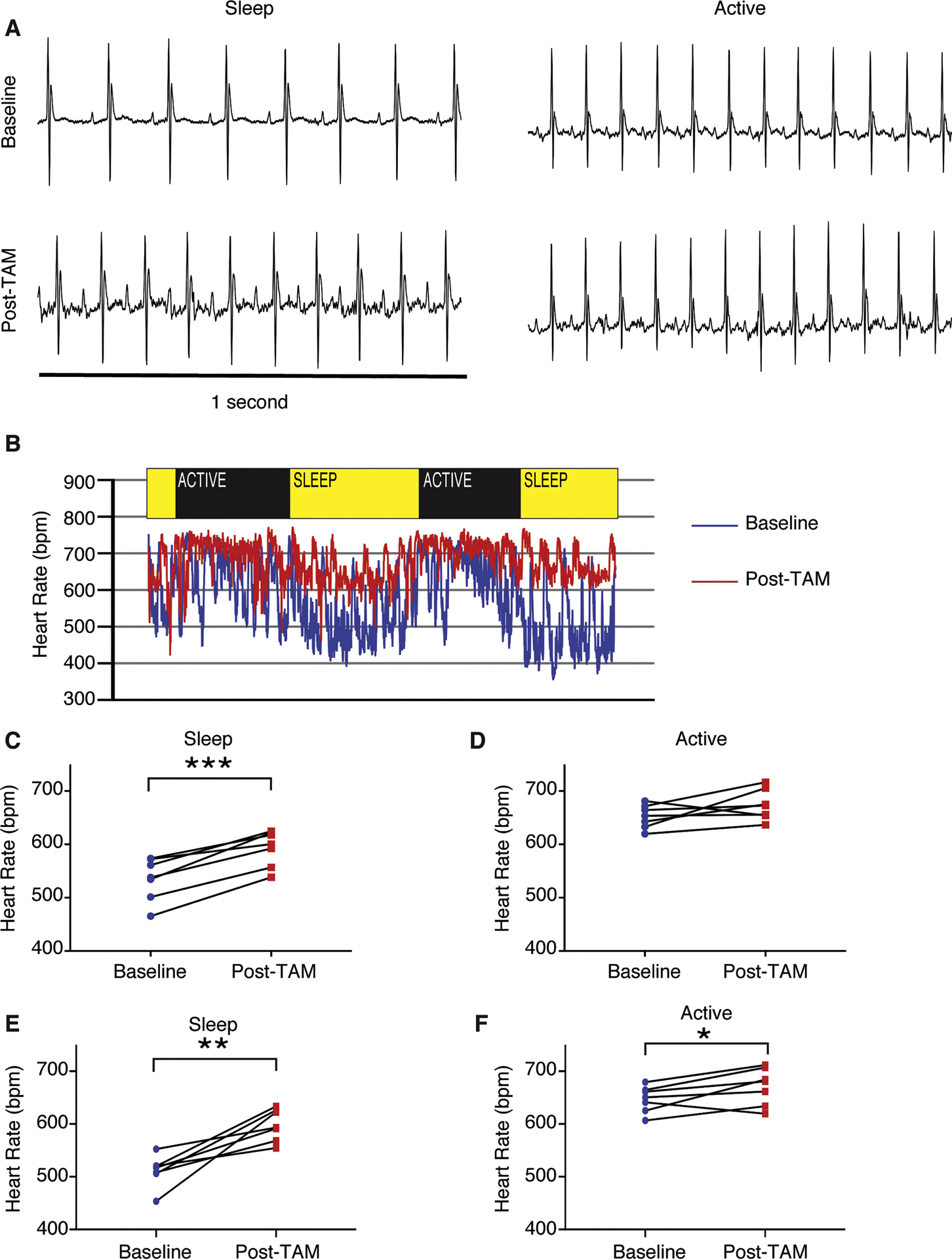
A) Representative ECG signals demonstrate an increase in heart rate during sleep after tamoxifen treatment, but no change while active. B) Continuous recording of heart rate over 48 hours, before tamoxifen (baseline) and after tamoxifen treatment (Post-TAM). C) Heart rate in the sleep phase increases after tamoxifen treatment in females (***p<10−3, N = 7 mice). D) No significant difference in heart rate during the active phase after tamoxifen treatment in females (p = 0.13). E) Heart rate in the sleep phase increases after tamoxifen treatment in males (**p = 0.003, N = 7 mice). F) Heart rate during the active phase increases after tamoxifen treatment in males (*p = 0.05).
We observed no evidence of pathological effects on the heart into senescence with the knockout of Rad(15, 34). Heart rate variability (HRV) reflects ANS input to the heart, and a decrease of HRV is associated with pathologies including progression to heart failure(35). This coupled with a literature indicating that abnormal calcium homeostasis can contribute to heart failure progression(36) motivated us to measure HRV. Cardiac Rad deletion has no effect on HRV in either the active or sleep phases (mean differences before/after tamoxifen 2.2±4ms and −1.5±6ms, for sleep and active phase, respectively; Figure 7A and 7B). In our recently published work we noted that despite tonic modulated ICa,L, the heart in cRadKO mice retains responsiveness to acute β-AR stimulation(15) consistent with the function of a healthy heart. To test for retention of autonomic nervous control of HR in cRadKO we measured the acute responses to a rapid air jet puff and exogenous ISO. The rapid air jet activates the endogenous ANS ‘fight or flight’ response, whereas exogenous ISO acutely over-rides ANS control of HR.The air jet acutely increased HR +33±23bpm (593±13 to 623±24bpm; p=0.18), and HRV (39±6ms and 68±14ms, before and after air-jet, respectively; Figure 7C) whereas exogenous ISO increased HR +59±10bpm (674+11 to 733±8bpm, p<0.01), and reduced HRV (46±3ms and 18±2ms, for post-tam and ISO, respectively; Figure 7D). Also, Poincare plots during the sleep and active phases also showed similar patterns in CTRL and cRadKO (Figures 7E and 7F).
4. Discussion
We showed previously that Rad-deficiency confers a β-AR –modulated phenotype on basal ICa,L without structural or functional remodeling of the heart(37). The main findings of this study are that in SANcm, the deletion of Rad results in elevated ICa,L with properties approximating SNS modulated ICa,L. Second, the absence of Rad elevates intrinsic HR. Third, HR in ambulatory mice is unchanged during the active phase, but is significantly elevated during the sleep/resting phase. Early studies showed that modulated ICa,L is a major contributor to an increased steepness of the diastolic depolarization of SANcm(38). Thus, these results support a key contribution of Rad – LTCC regulation of sinus rhythm in the heart.
SANcm have relatively low membrane capacitance and high input resistances; consequently, in theory, any relatively small inward cation flux, such as Ca2+ current, can accelerate pacemaker depolarizations(39),(33). The contribution of ICa,L to sinus rhythm was experimentally validated by demonstration of the slower HR of CaV1.3 knockout mice(10). There has been some controversy in the field striving to attribute dominant mechanisms to pace-making. For example, some studies suggested that the sarcoplasmic reticulum Ca2+ clock shapes the spontaneous rate, but may not be a dominating factor(40). Here we now show that ICa,L modulation by Rad GTPase regulates the intrinsic SAN firing rate. Our findings do not challenge the primacy of the membrane versus Ca2+ clock; rather, the interdependence of the membrane and Ca2+ clock(8) underscores the critical impact of altered ICa,L on SANcm autonomous firing. Perhaps most importantly, our work shows that Rad-LTCC regulation is a key molecular signaling node for SNS regulation of sinus rhythm.
4.1. Rad is a key contributor to SANmc ICa,L modulation
SANcm express ICa,L carried by pore forming CaV1.2 and CaV1.3 proteins. CaV1.3 contributes to pacemaker activity(10) and regulates the calcium clock in nodal cells(6). CaV1.2 resides mainly in non-junctional plasma membrane where it is thought to mainly contribute to the membrane clock(12). Our approaches do not allow us to dissect whether Rad has biased influence on CaV1.2 or CaV1.3. Liu et al showed that CaV1.2 and CaV1.3 indistinguishably respond to PKA to increase ICa,L(19). There are 3 independent lines of evidence to support the contention that Rad functions in cardiomyocytes to regulate ICa,L modulation. First, our early studies of global, constitutive Rad-KO mice showed Rad deletion phenocopies acute stimulation of the heart(37), and in this model ICa,L was modulated at baseline(17). Second, In the inducible, cardiac-restricted RadKO (cRadKO, as in the present study), ventricular ICa,L was modulated at baseline(15). Third, proximity proteomics evaluation in transgenic mice showed that following acute ISO stimulation, Rad association was reduced with LTCC proteins CaVβ2 and CaV1.2(19). Taken together with the present findings, we conclude that Rad interacts with LTCC; thus, dynamical regulation by PKA of the LTCC heteromultimeric complex is an important contributor to SNS elevation of HR. In native myocardium the signal for Rad-LTCC rearrangements are likely in response to the β-AR –PKA signaling axis. The Rad knockout model provides an extreme, whereby the absence of Rad would represent a maximally depleted CaV1.2 microenvironment, relieving any Rad-dependent inhibition with respect to LTCC function.
There is an imperfect correspondence between intrinsic HR and ICa,L by biological sex in cRadKO (Supplement Figure 1, Figure 3, and Figure 6). Intrinsic HR response in cRadKO is greater in females than males, but there is not a significant difference in ICa,L by sex. This finding raises new questions, principally, ‘What male-specific factors contribute to lower intrinsic HR in the absence of Rad?’ This does not necessarily mean that Rad influences other ion channels or Ca-clock components in a sex-specific manner; rather, this line of reasoning suggests that there are sex-specific intrinsic HR regulatory mechanisms, exclusive of Rad-LTCC signaling. For example, our early work showing Rad – ICa,L regulation established absence of effects on T-type calcium channel current(18). Nevertheless, while Rad might directly influence other channels, to date the only known channel interaction partners with Rad are CaVβ subunits(14) and the C-terminus of the L-type channel(41). There are a large number of signaling systems that sum to setting intrinsic HR(42), any one of which possibly can be altered in a sex specific fashion. The important conclusion is that regardless of sex, a common effect of Rad-deletion is increase ICa,L along with intrinsic HR.
4.2. Relationship among SANcm heterogeneity, Rad expression, and SAN function
In rabbits, peripheral pieces of SAN tissue from regions bordering the crista terminalis have faster firing rates than the central pacemaker site, and pieces bordering the left atrium were quiescent(43). The leading pacemaker site is not, however, a fixed location. During autonomic nervous system signaling different firing rates are driven by different cells in the SAN(44) and depending on external stimuli subsidiary areas can be activated(45). Cell-cell coupling creates complex behavior(46). For this reason, it is important to interpret cellular studies in context of tissue-level, or as in the present case in vivo studies of SAN function. Our finding that 38% of SANcm express enriched Rad transcript suggests an additional, new mechanism of heterogeneity that should be tested in future studies. It follows that the SANcm sub-types that do not express Rad may not possess adaptability to speed heart rate via a modulated ICa,L mechanism, or that other RGK GTPases may play yet to be investigated regulatory roles. Conversely, Rad-expressing SANcm theoretically can increase rate because the fastest firing SANcm should determine rate, which is consistent with the elevated HR in cRadKO mice. Future studies employing sophisticated in situ single cell mapping might help resolve whether Rad-ICa,L contributes to anatomical pacemaker shifts during sympathetic nervous system activation.
Ivabradine slows sinus HR and shows promising effects in heart failure (HF) patients(47) suggesting a possible link between reduced HR and HF progression. β-AR blockers have been employed as a first line of HF treatment(48), also slow HR, but the primary mechanism of action for reduction of re-hospitalization rate of HF patients remains speculative. In the present study we show Rad reduction increases HR, and we previously showed Rad reduction increases cardiac inotropy without pathological consequences(15, 17, 34, 37). Thus, the suggestion that Rad knockout provides beneficial effects in the setting of HF is paradoxical to HR slowing effects of ivabradine and β-AR blockers; however, Rad reduction would directly treat loss of cardiac inotropy in the failing heart, and thus is worthy of further study.
4.3. Diurnal impact and ANS drive with Rad reduction
Rad deletion elevates intrinsic HR consistent with the SANcm origin of the increased sinus rhythm. ANS net input is the sum of SNS drive and parasympathetic input, and the level of ANS drive is regulated by the baroreflex. With elevated HR, hence elevated cardiac output in cRadKO, it follows there should be compensatory reduced SNS drive. More direct measures of sympathetic nerve activity and circulating catecholamines are necessary to examine this issue, yet are highly invasive and can be difficult to interpret secondary to agitation of animals. For example, blood cannot be drawn from conscious mice without provoking a fight or flight response. Anesthesia can be used, but dramatically depresses measurements of circulating catecholamines (for example, compare(49) to(50)). To summarize, our data support the notion that we are not ‘knocking-out’ SNS input to the cRadKO heart, rather we suggest that modulated ICa,L in cRadKO dominant pacemaker cell may usurp the reflexive requirement for added SNS to balance net autonomic drive. In this way, Rad reduction strategies might aid aging related symptomatic bradycardia which has been attributed to depressed SANcm excitability(51, 52), possibly secondary to declines of CaV1.2 protein(9).
4.4. Limitations
Heterogeneity of function at the cellular level is not fully explored here. We did not measure spontaneous AP nor spontaneous firing frequencies. Many of the dispersed SAN cells displayed irregular automatic beating or quiescence. A range of spontaneous activity from fast, regular activity to irregular to quiescence has been noted in dispersed SANmc(53). Heterogeneity among SANcm is also demonstrated in single nucleus(26) and tbx3+ cell-sorted transcriptomics studies(29). It is also possible that differential dispersed cell activity might arise from differential cellular damage artifact during enzymatic dispersal. We also did not consider Rad-deletion effects on non-LTCC targets. While there is no evidence for Rad interactions in the cardiomyocyte beyond protein interactions with LTCC-component proteins, we cannot completely rule out other interactions with Rad. Studies here are limited to the mouse. In future studies it will be important to test the impact of Rad reduction on human or other large mammalian SANcm retained in ex vivo tissue preparations.
4.5. Conclusions
In summary, we have demonstrated that ICa,L contributes to sinus rate by a mechanism including Rad-regulated control of LTCC(15, 19). We previously showed that Rad-ablation promotes a beneficial elevation in Ca2+ dynamics, stimulating heart ventricular function(37), albeit without any apparent pathological consequence(15). Taken together, Rad reduction within all cardiomyocyte types should be explored as a potential therapeutic approach for increasing cardiac output as it simultaneously increases HR and stroke volume. Future novel therapeutics for symptomatic bradycardia targeting Rad – LTCC may elevate HR while retaining βAR responsiveness.
Supplementary Material
Highlights.
SAN cardiomyocytes express Rad
Rad deletion in SAN cardiomyocytes increases ICa,L consistent with Rad’s role as an endogenous LTCC governor
Rad regulates intrinsic HR consistent with βAR modulated ICa,L regulation
HR during the sleep cycle is selectively elevated by Rad deletion
Diurnal variation of HR is preserved, albeit with reduced amplitude
Rad – LTCC association may be a useful target for future therapeutics to treat symptomatic bradycardia
Acknowledgements
The authors would like to thank Andrea Sebastian, Wendy Katz, and Tanya Seward for outstanding technical support including heart dispersal, histology, and ECG telemetry surgical implantations. We are grateful for help from Matthew Hazzard, University of Kentucky Medical Illustrations support team.
Funding
This research was funded by NIH, NHLBI R01 HL131782 (DAA, JS), Department of Defense W81XWH-20-1-0418 (DAA, JS), AHA pre-doctoral fellowship to BMA (19PRE34380909), and Institutional Development Award (IDeA) from NIH NIGMS P30 GM127211.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
We, the authors have no competing interests.
Literature Cited
- 1.DiFrancesco D, Ferroni A, Mazzanti M, Tromba C. Properties of the Hyperpolarizing-Activated Current (i{-f} in Cells Isolated From the Rabbit Sino-Atrial Node. J Physiol. 1986;377:61–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chandler NJ, Greener ID, Tellez JO, Inada S, Musa H, Molenaar P, et al. Molecular Architecture of the Human Sinus Node. Circulation. 2009;119(12):1562–75. [DOI] [PubMed] [Google Scholar]
- 3.Bohn G, Moosmang S, Conrad H, Ludwig A, Hofmann F, Klugbauer N. Expression of T- and L-type calcium channel mRNA in murine sinoatrial node.PG - 73-6. FEBS Lett. 2000;481(1):73–6. [DOI] [PubMed] [Google Scholar]
- 4.Lyashkov AE, Juhaszova M, Dobrzynski H, Vinogradova TM, Maltsev VA, Juhasz O, et al. Calcium Cycling Protein Density and Functional Importance to Automaticity of Isolated Sinoatrial Nodal Cells Are Independent of Cell Size. Circulation Research. 2007;100(12):1723–31. [DOI] [PubMed] [Google Scholar]
- 5.Vinogradova TM, Zhou YY, Maltsev V, Lyashkov A, Stern M, Lakatta EG. Rhythmic ryanodine receptor Ca2+ releases during diastolic depolarization of sinoatrial pacemaker cells do not require membrane depolarization. Circ Res. 2004;94(6):802–9. [DOI] [PubMed] [Google Scholar]
- 6.Torrente AG, Mesirca P, Neco P, Rizzetto R, Dubel S, Barrere C, et al. L-type Cav1.3 channels regulate ryanodine receptor-dependent Ca2+ release during sino-atrial node pacemaker activity. Cardiovasc Res. 2016;109(3):451–61. [DOI] [PubMed] [Google Scholar]
- 7.Lyashkov AE, Behar J, Lakatta EG, Yaniv Y, Maltsev VA. Positive Feedback Mechanisms among Local Ca Releases, NCX, and I(CaL) Ignite Pacemaker Action Potentials. Biophys J. 2018;114(5):1176–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yaniv Y, Lakatta EG, Maltsev VA. From two competing oscillators to one coupled-clock pacemaker cell system. Frontiers in physiology. 2015;6(28). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones SA, Boyett MR, Lancaster MK. Declining into failure: the age-dependent loss of the L-type calcium channel within the sinoatrial node. Circulation. 2007;115(10):1183–90. [DOI] [PubMed] [Google Scholar]
- 10.Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, et al. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci U S A. 2003;100(9):5543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Satin J Regulation of Cardiac Calcium Channels. In: Zipes DP, Jalife J, Stevenson W, editors. Cardiac Electrophysiology: From Cell to Bedside. 7 ed: Elsevier; 2017. p. 1424. [Google Scholar]
- 12.Christel CJ, Cardona N, Mesirca P, Herrmann S, Hofmann F, Striessnig J, et al. Distinct localization and modulation of Cav1.2 and Cav1.3 L-type Ca2+ channels in mouse sinoatrial node. The Journal of Physiology. 2012;590(24):6327–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang L, Katchman A, Kushner J, Kushnir A, Zakharov SI, Chen B-x, et al. Cardiac CaV1.2 channels require β subunits for β-adrenergic–mediated modulation but not trafficking. The Journal of Clinical Investigation. 2019;129(2):647–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Correll RN, Pang C, Niedowicz DM, Finlin BS, Andres DA. The RGK family of GTP-binding proteins: regulators of voltage-dependent calcium channels and cytoskeleton remodeling. Cell Signal. 2008;20(2):292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahern BM, Levitan BM, Veeranki S, Shah M, Ali N, Sebastian A, et al. Myocardial-restricted ablation of the GTPase RAD results in a pro-adaptive heart response in mice. Journal of Biological Chemistry. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manning JR, Chelvarajan L, Levitan BM, Withers CN, Nagareddy PR, Haggerty CM, et al. Rad GTPase Deletion Attenuates Post-Ischemic Cardiac Dysfunction and Remodeling. JACC: Basic to Translational Science. 2018;3(1):83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manning JR, Yin G, Kaminski CN, Magyar J, Feng HZ, Penn J, et al. Rad GTPase Deletion Increases L‐type Calcium Channel Current Leading to Increased Cardiac Contraction. Journal of the American Heart Association. 2013;2(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finlin BS, Crump SM, Satin J, Andres DA. Regulation of voltage-gated calcium channel activity by the Rem and Rad GTPases. PNAS. 2003;100:14469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu G, Papa A, Katchman AN, Zakharov SI, Roybal D, Hennessey JA, et al. Mechanism of adrenergic CaV1.2 stimulation revealed by proximity proteomics. Nature. 2020;577(7792):695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartzell HC, Méry PF, Fischmeister R, Szabo G. Sympathetic regulation of cardiac calcium current is due exclusively to cAMP-dependent phosphorylation. Nature. 1991;351(6327):573–6. [DOI] [PubMed] [Google Scholar]
- 21.Brandmayr J, Poomvanicha M, Domes K, Ding J, Blaich A, Wegener JW, et al. Deletion of the C-terminal phosphorylation sites in the cardiac beta subunit does not affect the basic beta-adrenergic response of the heart and the Cav1.2 channel. J Biol Chem. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang G, Zhu X, Xie W, Han P, Li K, Sun Z, et al. Rad as a novel regulator of excitation-contraction coupling and beta-adrenergic signaling in heart. Circ Res. 2010;106(2):317–27. [DOI] [PubMed] [Google Scholar]
- 23.Withers CN, Brown DM, Byiringiro I, Allen MR, Condon KW, Satin J, et al. Rad GTPase is essential for the regulation of bone density and bone marrow adipose tissue in mice. Bone. 2017;103:270–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, et al. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89(1):20–5. [DOI] [PubMed] [Google Scholar]
- 25.Lexow J, Poggioli T, Sarathchandra P, Santini MP, Rosenthal N. Cardiac fibrosis in mice expressing an inducible myocardial-specific Cre driver. Disease Models and Mechanisms. 2013;6(6):1470–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linscheid N, Logantha SJRJ, Poulsen PC, Zhang S, Schrölkamp M, Egerod KL, et al. Quantitative proteomics and single-nucleus transcriptomics of the sinus node elucidates the foundation of cardiac pacemaking. Nature communications. 2019;10(1):2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. The Journal of molecular diagnostics : JMD. 2012;14(1):22–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyett MR, Honjo H, Kodama I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc Res. 2000;47(4):658–87. [DOI] [PubMed] [Google Scholar]
- 29.van Eif VWW, Stefanovic S, van Duijvenboden K, Bakker M, Wakker V, de Gier-de Vries C, et al. Transcriptome analysis of mouse and human sinoatrial node cells reveals a conserved genetic program. Development. 2019;146(8):dev173161. [DOI] [PubMed] [Google Scholar]
- 30.Litviňuková M, Talavera-López C, Maatz H, Reichart D, Worth CL, Lindberg EL, et al. Cells of the adult human heart. Nature. 2020;588(7838):466–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Protas L, DiFrancesco D, Robinson RB. L-type but not T-type calcium current changes during postnatal development in rabbit sinoatrial node. Am J Physiol. 2001;281(3):H1252–9. [DOI] [PubMed] [Google Scholar]
- 32.Mesirca P, Torrente AG, Mangoni ME. Functional role of voltage gated Ca2+ channels in heart automaticity. Frontiers in physiology. 2015;6(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verheijck EE, van Ginneken AC, Wilders R, Bouman LN. Contribution of L-type Ca2+ current to electrical activity in sinoatrial nodal myocytes of rabbits. Am J Physiol. 1999;276(3):H1064–77. [DOI] [PubMed] [Google Scholar]
- 34.Manning JR, Withers CN, Levitan B, Smith JD, Andres DA, Satin J. Loss of Rad-GTPase produces a novel adaptive cardiac phenotype resistant to systolic decline with aging. American Journal of Physiology.- Heart and Circulatory Physiology 2015;309(8):H1336–H45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandercock GR, Brodie DA. The role of heart rate variability in prognosis for different modes of death in chronic heart failure. Pacing Clin Electrophysiol. 2006;29(8):892–904. [DOI] [PubMed] [Google Scholar]
- 36.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. The Journal of Clinical Investigation. 2013;123(1):37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levitan BM, Manning JR, Withers CN, Smith JD, Shaw RM, Andres DA, et al. Rad-deletion Phenocopies Tonic Sympathetic Stimulation of the Heart. Journal of Cardiovascular Translational Research. 2016;9(5):432–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaza A, Robinson RB, DiFrancesco D. Basal responses of the L-type Ca2+ and hyperpolarization-activated currents to autonomic agonists in the rabbit sino-atrial node. J Physiol.1996;491 ( Pt 2)(Pt 2):347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mangoni ME, Traboulsie A, Leoni A-L, Couette B, Marger L, Le Quang K, et al. Bradycardia and Slowing of the Atrioventricular Conduction in Mice Lacking CaV3.1 α1G T-Type Calcium Channels. Circulation Research. 2006;98(11):1422–30. [DOI] [PubMed] [Google Scholar]
- 40.Honjo H, Inada S, Lancaster MK, Yamamoto M, Niwa R, Jones SA, et al. Sarcoplasmic reticulum Ca2+ release is not a dominating factor in sinoatrial node pacemaker activity. Circ Res. 2003;92(3):e41–4. [DOI] [PubMed] [Google Scholar]
- 41.Pang C, Crump SM, Jin L, Correll RN, Finlin BS, Satin J, et al. Rem GTPase interacts with the proximal CaV1.2 C-terminus and modulates calcium-dependent channel inactivation. Channels (Austin). 2010;4(3):192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacDonald EA, Rose RA, Quinn TA. Neurohumoral Control of Sinoatrial Node Activity and Heart Rate: Insight From Experimental Models and Findings From Humans. Frontiers in physiology. 2020;11:170-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Opthof T, de Jonge B, Jongsma HJ, Bouman LN. Functional morphology of the mammalian sinuatrial node. Eur Heart J. 1987;8(11):1249–59. [DOI] [PubMed] [Google Scholar]
- 44.Mackaay AJ, Op’t Hof T, Bleeker WK, Jongsma HJ, Bouman LN. Interaction of adrenaline and acetylcholine on cardiac pacemaker function. Functional inhomogeneity of the rabbit sinus node. J Pharmacol Exp Ther. 1980;214(2):417–22. [PubMed] [Google Scholar]
- 45.Li N, Hansen BJ, Csepe TA, Zhao J, Ignozzi AJ, Sul LV, et al. Redundant and diverse intranodal pacemakers and conduction pathways protect the human sinoatrial node from failure. Sci Transl Med. 2017;9(400). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verheijck EE, Wilders R, Joyner RW, Golod DA, Kumar R, Jongsma HJ, et al. Pacemaker synchronization of electrically coupled rabbit sinoatrial node cells. J Gen Physiol. 1998;111(1):95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Das D, Savarese G, Dahlström U, Fu M, Howlett J, Ezekowitz JA, et al. Ivabradine in Heart Failure. Circulation: Heart Failure. 2017;10(9):e004112. [DOI] [PubMed] [Google Scholar]
- 48.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Colvin MM, et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Journal of the American College of Cardiology. 2017;70(6):776–803. [DOI] [PubMed] [Google Scholar]
- 49.Pichavaram P, Yin W, Evanson KW, Jaggar JH, Mancarella S. Elevated plasma catecholamines functionally compensate for the reduced myogenic tone in smooth muscle STIM1 knockout mice but with deleterious cardiac effects. Cardiovascular Research. 2018;114(5):668–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mathar I, Vennekens R, Meissner M, Kees F, Van der Mieren G, Camacho Londoño JE, et al. Increased catecholamine secretion contributes to hypertension in TRPM4-deficient mice. The Journal of Clinical Investigation. 2010;120(9):3267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peters CH, Sharpe EJ, Proenza C. Cardiac Pacemaker Activity and Aging. Annual Review of Physiology. 2020;82(1):21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Larson ED, St. Clair JR, Sumner WA, Bannister RA, Proenza C. Depressed pacemaker activity of sinoatrial node myocytes contributes to the age-dependent decline in maximum heart rate. Proceedings of the National Academy of Sciences. 2013;110(44):18011–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim MS, Maltsev AV, Monfredi O, Maltseva LA, Wirth A, Florio MC, et al. Heterogeneity of calcium clock functions in dormant, dysrhythmically and rhythmically firing single pacemaker cells isolated from SA node. Cell Calcium. 2018;74:168–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.