

Abstract
Obesity-driven cardiac lipid accumulation can progress to lipotoxic cardiomyopathy. Soluble epoxide hydrolase (sEH) is the major enzyme that metabolizes epoxyeicosatrienoic acids (EETs), which have biological activity of regulating lipid metabolism. The current study explores the unknown role of sEH deficiency in lipotoxic cardiomyopathy and its underlying mechanism. Wild-type and Ephx2 knock out (sEH KO) C57BL/6J mice were fed with high-fat diet (HFD) for 24 weeks to induce lipotoxic cardiomyopathy animal models. Palmitic acid (PA) was utilized to induce lipotoxicity to cardiomyocytes for in vitro study. We found sEH KO, independent of plasma lipid and blood pressures, significantly attenuated HFD-induced myocardial lipid accumulation and cardiac dysfunction in vivo. HFD-induced lipotoxic cardiomyopathy and dysfunction of adenosine 5’-monophosphate-activated protein kinase-mammalian target of rapamycin complex (AMPK-mTORC) signaling mediated lipid autophagy in heart were restored by sEH KO. In primary neonatal mouse cardiomyocytes, both sEH KO and sEH substrate EETs plus sEH inhibitor AUDA treatments attenuated PA-induced lipid accumulation. These effects were blocked by inhibition of AMPK or autophagy. The outcomes were supported by the results that sEH KO and EETs plus AUDA rescued HFD- and PA- induced impairment of autophagy upstream signaling of AMPK-mTORC, respectively. These findings revealed that sEH deficiency played an important role in attenuating myocardial lipid accumulation and provided new insights into treating lipotoxic cardiomyopathy. Regulation of autophagy via AMPK-mTORC signaling pathway is one of the underlying mechanisms.
Keywords: Autophagy, AMPK-mTORC pathway, cardiac lipid accumulation, lipotoxic cardiomyopathy, soluble epoxide hydrolase
Introduction
Recent national data on obesity prevalence among U.S. adults and youths show that 39.8% of adults and 18.5% of youth were obese [1]. When the caloric intake exceeds the caloric expenditure, obesity initiates. Obesity has many adverse effects on cardiac structure and hemodynamics. The hemodynamic hallmarks of obesity, increased heart rate and stroke volume, are thought to be a compensatory adaptation to increased adipose tissue mass at the expense of left ventricular remodeling, which progress to nonischemic dilated cardiomyopathy [2]. Lipotoxic cardiomyopathy is characterized by functional and structural alterations of the heart, such as echocardiographic changes consistent with systolic dysfunction, increases in myocardial triglyceride (TG) content, lipotoxicity, and enhanced oxidative stress. There is still a lack of effective treatment for obesity-related lipotoxic cardiomyopathy and the mechanism remains under debated.
Soluble epoxide hydrolase (sEH) is the major enzyme that metabolizes epoxyeicosatrienoic acids (EETs) to the less bioactive dihydroxyeicosatrienoic acids (DHET)[3]. EETs are metabolites of arachidonic acid by cytochrome P450 (CYP) [4–6]. sEH inhibition stabilizes EETs and demonstrates cardiac protection effects [7]. sEH gene Ephx2 was identified as a heart failure susceptibility gene in spontaneously hypertensive heart failure rats. sEH deficiency protected mice from pressure overload-induced heart failure and cardiac arrhythmias [8]. sEH inhibition improves coronary endothelial function and prevents cardiac remodeling and diastolic [9]. Our previous study found that sEH knockout (KO) prevented cardiac hypertrophy, not only directly to cardiomyocytes but also to cardiac fibroblasts, by reducing expression of fibroblast growth factor-2[3]. sEH deficiency or inhibition also exhibits diverse and critical biological effects on lipid metabolism such as inhibiting adipogenesis and obesity-associated adipose expansion [10, 11].
Autophagy is a self-conservative cellular mechanism to degrade and recycle unwanted or damaged cell debris including macromolecules, organelles and nutrients [12]. Traditionally, autophagy is implicated in providing amino acids through protein degradation.
Nevertheless, evidence has emerged that autophagy also provides cellular energy through generation of free fatty acids (FFAs) and maintains lipid homeostasis [13, 14]. Adenosine 5’-monophosphate-activated protein kinase (AMPK) is an enzyme that plays a critical role in regulating cellular growth and metabolism, acting as a metabolic sensor [15]. Activation of AMPK can inhibit downstream mammalian target of rapamycin complex (mTORC), which induces an autophagic response [16, 17]. Previous studies found that sEH inhibitor produced antisteatotic action in mice adipose tissue, as well as upregulated hepatic autophagy [18]. It was also established that sEH substrate EETs enhance autophagic responses in HL-1 (cardiac muscle cell line) cells and rat neonatal cardiomyocytes via a pmKATP-AMPK (cardiac plasma-membrane ATP-sensitive K+ channel) dependent pathway during cell starvation [19]. However, whether sEH deficiency attenuates lipotoxic cardiomyopathy through AMPK-mTORC regulated lipid autophagy and contributes to its multiple cardiac protective phenotypes is unclear.
Using sEH gene Ephx2 KO mice, we found sEH KO protected from high fat diet (HFD)-induced myocardial lipid metabolism dysfunction, termed as lipotoxic cardiomyopathy and illustrated AMPK-mTORC signaling pathway-mediated autophagy as one of the underlying mechanisms. The outcomes were echoed by in vitro experiments.
This study broadens the interpretations on the mechanism that sEH deficiency or inhibition protects against cardiac dysfunction and provided a new therapeutic target for the treatment of obesity-related lipotoxic cardiomyopathy.
Materials and methods Reagents
The HFD formula fodder (D12492) was obtained from Guangdong Medical Laboratory Animal Center, (Guangzhou, China). All cell culture reagents were obtained from GibcoBRL (Life Technologies, Inc., Grand Island, NY). mRFP-GFP-LC3 adenoviruses were purchased from Hanbio Co. (Shanghai, China). TRIzol reagent and SuperScript III First-Strand Synthesis Supermix were from Invitrogen (Carlsbad, California). SYBR Green PCR Master Mix was from Applied Biosystems (Foster City, California). TG reagents were from Nanjing Jiancheng Biocompany (Nanjing, China). sEH rabbit mAb was kindly donated by Dr. Bruce D. Hammock from University of California. Species- specific secondary antibodies were from Sigma-Aldrich. Enhanced chemiluminescent substrate was from Pierce (Rockford, IL). Polyvinylidene difluoride (PVDF) membranes were from Merck Millipore (Billerica, MA). All other chemicals and reagents were purchased from Sigma-Aldrich unless otherwise specified.
Mice
Animal experiments were approved by the Committee on Ethics of Animal Experiments in Sun Yat-sen University in China and conducted in accordance with the Guidelines for Animal Experiments, (NIH Publication No. 85-23, revised 1996). Ephx2 gene (coding sEH) knockout male mice with a background of C57BL/6J were obtained from Jackson Laboratory (Bar Harbor, ME). Phenotype of Ephx2 KO mice and genotyping identification procedure have been described previously [3]. In brief, animals were housed under temperature-controlled conditions (21 ± 2 °C), humidity (55 ± 5%), and a 12 h/12 h light–dark circadian cycle with access to food and water ad libitum in the specific pathogen free (SPF) animal facility.
HFD protocol
Eight-week-old male Ephx2 KO and WT mice weighing 18–20 g were subjected to a 1- week adaptation period and then randomly assigned to different treatment groups. Mice were fed with SFD (percentage of kcal from: protein, 20%; fat, 10.0%; and carbohydrates, 70.0%) and HFD (percentage of kcal from: protein, 20.0%; carbohydrate, 20.0%; and fat, 60.0% D12492) for a total of 24 weeks [20].
Blood pressure measurement
The CODA 8 noninvasive blood pressure acquisition system for mice (Kent Scientific, Torrington, CT) was used for all tail-cuff measurements as previously described [21].
Echocardiography
Cardiac geometry and contractile function were evaluated in anesthetized (ether inhalation) mice using the 2-D guided M-mode echocardiography (VEVO 2100, Visual Sonics, Canada). Left ventricular (LV) anterior and posterior wall dimensions during diastole, interventricular septum (IVS), fractional shortening (FS), left ventricular end-diastolic (LVEDD) and end-systolic diameters (LVESD), left ventricular end diastolic posterior wall dimension (LVPWD), LV mass, ejection fraction (EF) and mitral doppler E/A ratio were calculated [3].
TEM
Tissue from the apex of left ventricular for electron microscopy were submerged in cold (4°C) fixative buffer (2% formaldehyde, 2% glutaraldehyde in 0.1 M phosphate buffer (NaPi), pH 7.4) and stored at 4°C for a minimum of 2 h (maximum 6.5 h) before osmication. The heart tissue samples were subdivided into five parallels, ≤1 mm pieces and embedded, cut, and contrasted using standard protocols as previously described [22]. Images were obtained using a Tecnai G2 electron microscope from FEI (Tecnai G2 Spirit Twin, FEI).
Serum and heart lipid measurements
Animals were anesthetized intraperitoneally with 1% pentobarbital (80 mg/kg) to harvest serum samples before sacrifice. Serum TG levels were assessed as described previously [23]. TG levels in heart tissue and primary cultured cardiomyocytes were measured with the Triglycerides Quantification Kit (Nanjing, China). The unit of TG measurement was indicated by microgram per gram protein (mg/g) according to the manufacturer’s instructions.
Cardiac histological analysis and Oil Red O staining
Following anesthesia, hearts were excised and partly placed in 10% neutral-buffered formalin at room temperature for 24 h after a brief rinse with phosphate-buffered saline (PBS). The specimen was embedded in paraffin, cut in 5-μm sections and stained with hematoxylin and eosin. Cardiomyocyte cross-sectional areas were calculated on a digital microscope (×200) using the Image J (ver1.51) software [24]. Oil Red-O staining and quantification have been described previously [25].
Autophagy detection
Detection of autophagosomes using electron microscopy was described previously [26]. Under TEM, autophagosomes (also referred to as initial autophagic vacuoles, AVi) have a double membrane that is usually at least partly visible as two parallel membrane bilayers separated by an electron-lucent cleft [27]. Morphologically, the AVi can be identified by its contents (intact cytoplasm, including ribosomes, and rough endoplasmic reticulum). The late/degradative autophagic vacuoles/autolysosomes (AVd or AL) typically have only one limiting membrane; frequently they contain electron dense cytoplasmic material and/or organelles at various stages of degradation. Autophagic vacuoles were counted from at least 20 random cells, averaged and expressed as the number of autophagic vacuoles per cell.
PA preparation and neonatal CM treatment
PA/BSA (bovine serum albumin) conjugates were prepared as described previously [28]. Primary culture of neonatal mice cardiomyocytes (CMs) were described previously [29]. With sEH deficiency and WT control cardiomyocytes, PA (400 μM, 24h) was employed to induce cardiomyocyte lipotoxicity, and Compound C (5 μM) or 3-MA (10 mM) were incubated with cardiomyocytes. Next, we incubated PA (400 μM), 14,15-EET (1μM), and AUDA (10μM, inhibitor of sEH) with WT cardiomyocytes for 24h. Then, we used Compound C and 3-MA to further illustrate the relationships among sEH and AMPK- mTORC-mediated autophagy.
qRT-PCR
Total RNA was isolated from left ventricles. Quantitative real-time polymerase chain reaction (qRT-PCR) analysis was performed for CD36, ATGL, SREBP1c, LAL, HSL, LPL and GAPDH. The sequences of the primers used for mouse samples were:
CD36, F 5’-ATTGGTCAAGCCAGCT-3’, R 5’- TGTAGGCTCATCCACTAC -3’
GPAT, F 5’-ACTGGGTTGACTGTGGGTTC-3’, and R 5’-GCAGGAATGCAGCACCTTC-3’ ATGL, F 5’-TGTTTCAGACGGAGAGAACG-3’, and R 5’-GGAGGGTGGAGGAATGAGG-3’ SREBP1c, F 5’-AATCAGGACCATGCCG-3’, and R 5’- CTCAACCTATGAAAATAAAGTTTGC-3’ LAL, F 5’-TGGAGGGACAAACCACTGA-3’, and R 5’-AAGGGAATCGGACCACTTG-3’
HSL, F 5’-CTTCTCCCTCTCGTCTGCTG-3’, and R 5’-AATGGTCCTCTGCCTCTGTC-3’ LPL, F 5’-GATCCGAGTGAAAGCCGGAG-3’, and R 5’-TTGTTTGTCCAGTGTCAGCCA-3’ GAPDH, F 5’-GTCCTCGGGCATAATGCGTA-3’, and R 5’-TAACCTCAGATCAGGGCGGA
Western blotting
The protein of left ventricles and neonatal cardiomyocytes was extracted as previously described [5]. Protein were detected with anti-LC3II/3I (1:1,000), anti-p62 (1:1,000), anti- sEH(1:1,000), anti-AMPK (1:1,000), anti-pThr172-AMPK (1:1,000), anti-mTOR (1:1,000), anti-pSer2448-mTOR (1:1,000), and anti-GAPDH (1:1,000) antibodies (Cell Signaling Technology Inc., Beverly, MA). Antigens were detected by the luminescence method. Quantification of band density was determined using Gene Snap software (Syngene, Frederick, MD) and reported in optical density per square millimeter.
Autophagic flux detection
Primary neonatal mouse cardiomyocytes were transfected with mRFP-GFP-LC3 adenoviruses at 50 MOI for 24h as previously described [30]. After indicated treatment, the autophagosomes (merged yellow puncta) and autolysosomes (red puncta) in cardiomyocytes were visualized under a confocal microscope (×400). At least 30 cells were recorded for each group.
Lipidtox red staining and LC3II staining colocalization
C57 primary cells grown on bovine skin collagen-coated coverslips were incubated in the presence or absence of 400 μM of PA, chloroquine chloride (10 μM inhibitor of infusion of autophagosome and lysosome) for 24 h. Immunofluorescence staining were done as previously describe. Stained cells were analyzed by Leica epifluoresence microscope with either a 40×or 60× oil immersion objective [31].
Liquid chromatography tandem mass spectroscopy
All samples were reconstituted in 30 μl of 30% ethanol. Online liquid chromatography was performed with an Agilent 1200 Series capillary HPLC (Agilent Technologies, Santa Clara, CA), which described previously [32].
Statistical analysis
All experiments were repeated at least 3 times, and continuous data were expressed as mean ± standard error of the means (SEMs) when in normal distribution. Homogeneity of the variance was assessed via the F test (2 groups). When comparing results with 2 groups, we applied the standard Student’s t-test if equal variance and normal distribution were satisfied; otherwise, the nonparametric Mann-Whitney U test was used. When there were more than 2 groups and 1 factor was involved, we used one-way analysis of variance (ANOVA) analysis. The P values were adjusted for multiple comparisons where appropriate. Data were analyzed with SPSS 20.0 statistical software package and presented as mean ± SEMs. p<0.05 was considered to be statistically significant.
Results
sEH KO attenuated HFD-induced mouse lipotoxic cardiomyopathy
To determine whether sEH deficiency contributes to protection against HFD-induced lipotoxic cardiomyopathy, we fed WT or sEH KO mice with HFD for 24 weeks. By then, HFD induced a significant increase of sEH expression in WT mice (Fig. 1A). sEH KO mice showed comparable blood pressures in both systolic and diastolic pressures with WT mice (Fig. 1B). Both sEH KO and WT mice developed obesity presenting significant body weight gain with elevated plasma triglyceride compared with mice fed with standard fat diet (SFD) control. The control levels under SFD and increases in body weight and triglyceride after HFD were comparable between sEH KO and WT mice (Fig. 1C). Compared with mice fed with SFD, echocardiographic analysis revealed that HFD significantly decreased left ventricular (LV) fractional shortening (FS%) and increased LV mass in WT mice (Fig. 1D–F, Table 1). Accordingly, HFD significantly increased heart sized in gross observation and heart to body weight ratio (mg/g) in WT mice. Commensurate with the increased heart weight, HFD significantly increased the cardiomyocyte cross-sectional area in WT mice (Fig. 1G–I). These effects were remarkably mitigated in sEH KO mice (sEH KO+HFD vs WT + HFD, p < 0.01 for FS% and LV mass, p < 0.05 for heart/body weight ratio, and p < 0.005 for cell cross-sectional area (Fig. 1D–I). Together, these indicated that sEH KO, independent of plasma lipid or blood pressure, significantly attenuates HFD-induced lipotoxic cardiomyopathy.
Fig. 1. sEH KO attenuated HFD-induced mouse lipotoxic cardiomyopathy.
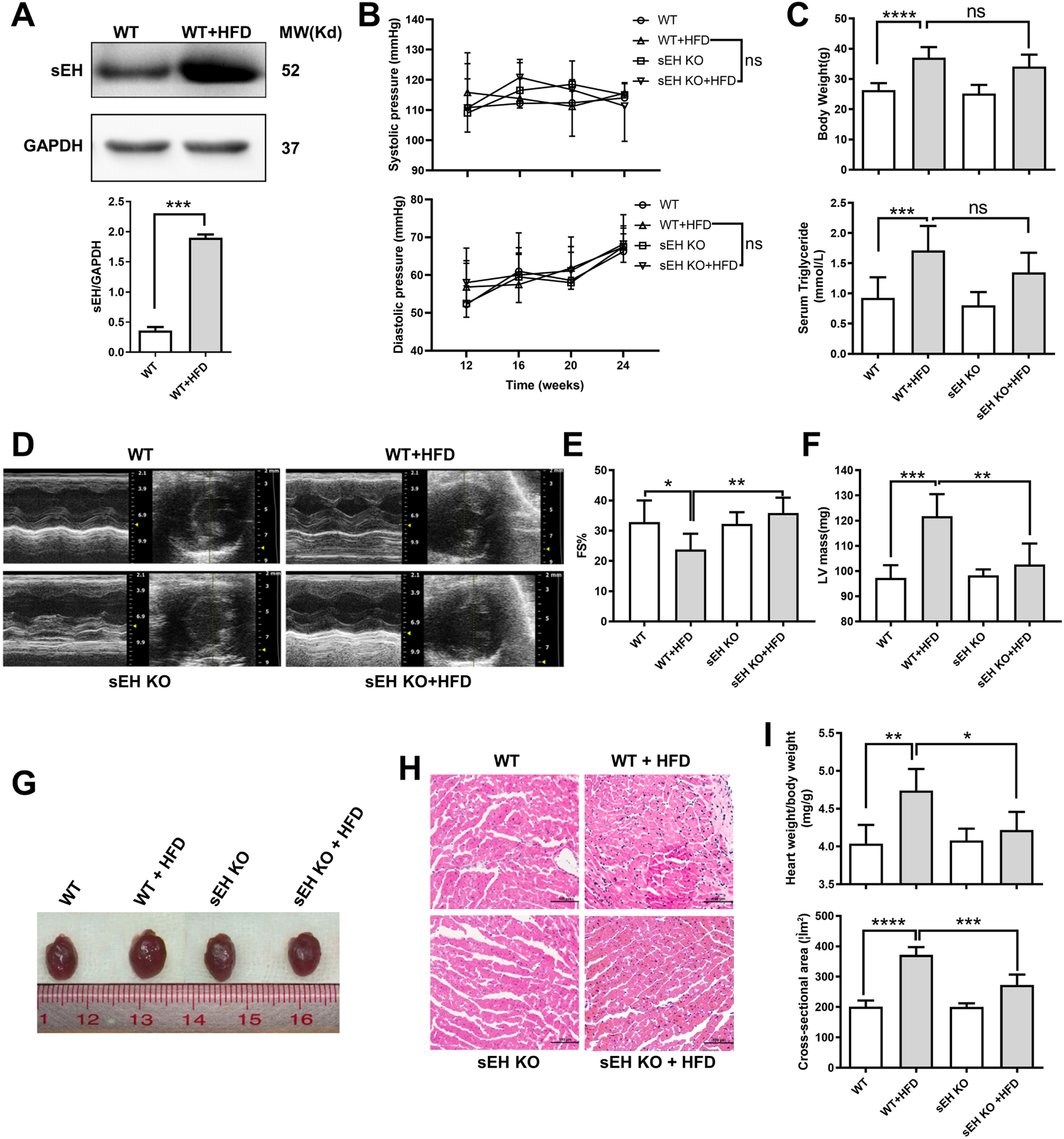
(A) Heart sEH expression levels significantly increased in WT mice at 24 weeks after HFD. (B, C) Blood pressure, body weight gain, and plasm triglyceride level were comparable between sEH KO and WT mice at 24 weeks after HFD. (D-F) Echocardiography showed sEH KO ablated the decrease of fractional shortening (FS) and restored the increase of left ventricle (LV) mass induced by HFD. (G, I-top panel) HFD significantly increased WT mice heart volume in general review and heart to body weight ratio (mg/g), sEH KO attenuated the effects. (H, I-bottom panel) Cardiomyocyte cross-sectional area (H&E staining, ×200) was elevated with HFD, and sEH KO partially reversed the effects. * P<0.05, **P<0.01, *** P<0.005, **** P<0.001. n=8–10 (b, c) and 3–5 (a, d-i) per group.
Table 1.
Echocardiographic parameters of mouse heart in different groups.
| WT | WT+HFD | sEH KO | sEH KO+HFD | |
|---|---|---|---|---|
| IVS d (mm) | 0.92±0.03 | 0.99±0.08 | 0.92±0.02 | 0.91±0.07 |
| IVS s (mm) | 1.42±0.19 | 1.36±0.19 | 1.33±0.11 | 1.52±0.20 |
| LVID d (mm) | 3.53±0.37 | 3.74±0.46 | 3.24±0.36 | 3.31±0.22* |
| LVID s (mm) | 2.09±0.40 | 2.68±0.60* | 2.1±0.16 | 2.10±0.23* |
| LVPW d (mm) | 0.99±0.069 | 1.1±0.047 | 0.95±0.12 | 0.99±0.08 |
| LVPW s (mm) | 1.29±0.23 | 1.35±0.14 | 1.3±0.05 | 1.40±0.06 |
| EF (%) | 55.36±7.31 | 57.16±12.22 | 57.1±2.34 | 67.72±6.28 |
| FS (%) | 36.84±4.45 | 28.56±4.74* | 36.13±2.11 | 35.88±5.06** |
| LV Mass(mg) | 97.27±5.049 | 121.8±8.76*** | 98.33±2.33 | 102.6±8.36** |
| E/A | 1.43±0.05 | 1.21±0.05 | 1.41±0.02 | 1.39±0.10 |
WT+WFD vs. WT, sEH KO+HFD vs. WT+HFD,
p<0.05,
p<0.01,
p<0.005, n=3–5 per group.
IVS, interventricular septum; d, end-diastolic; s, systolic; LVID, left ventricular internal diameter; LVPW, left ventricular posterior wall thickness; EF, ejection fraction; FS, fractional shortening; LV, left ventricular; E/A, mitral doppler peak ratio.
sEH KO attenuated HFD-induced mouse cardiac lipid accumulation
Metabolic perturbations stemming from HFD can lead to accumulation of lipids in the heart[20]. To examine the effect sEH KO on HFD-elicited lipid accumulation in the heart, we analyzed lipid droplet (LD) accumulation in the heart by Oil Red O staining, transmission electron microscope (TEM) and heart triglyceride content measurement in WT and sEH KO mice under SFD or HFD, respectively. Oil Red O staining in myocardium revealed that HFD significantly promoted cardiac lipid accumulation in WT mice. This change was attenuated by sEH KO (Fig. 2A). Heart sections were visualized by TEM at 2,400× and 18,500× magnification. LDs were identified as round light-density structures with homogenous amorphous content and an average diameter of 0.5 μm that increased 10 to 15 fold in response to HFD [14].We found that sEH KO effectively ablated HFD-induced LD accumulation in hearts (sEH KO+HFD vs. WT+HFD, p<0.05, Fig. 2B–C). Moreover, sEH KO mice had lower triglyceride in the heart than WT mice (sEH KO+HFD vs. WT+HFD, p<0.01, Fig. 2D). In sum, these indicated that sEH KO prevents HFD-elicited lipid accumulation in the heart.
Fig. 2. sEH KO attenuated HFD-induced mouse cardiac lipid accumulation and abnormal transcriptions of lipid synthetic and catabolic genes.
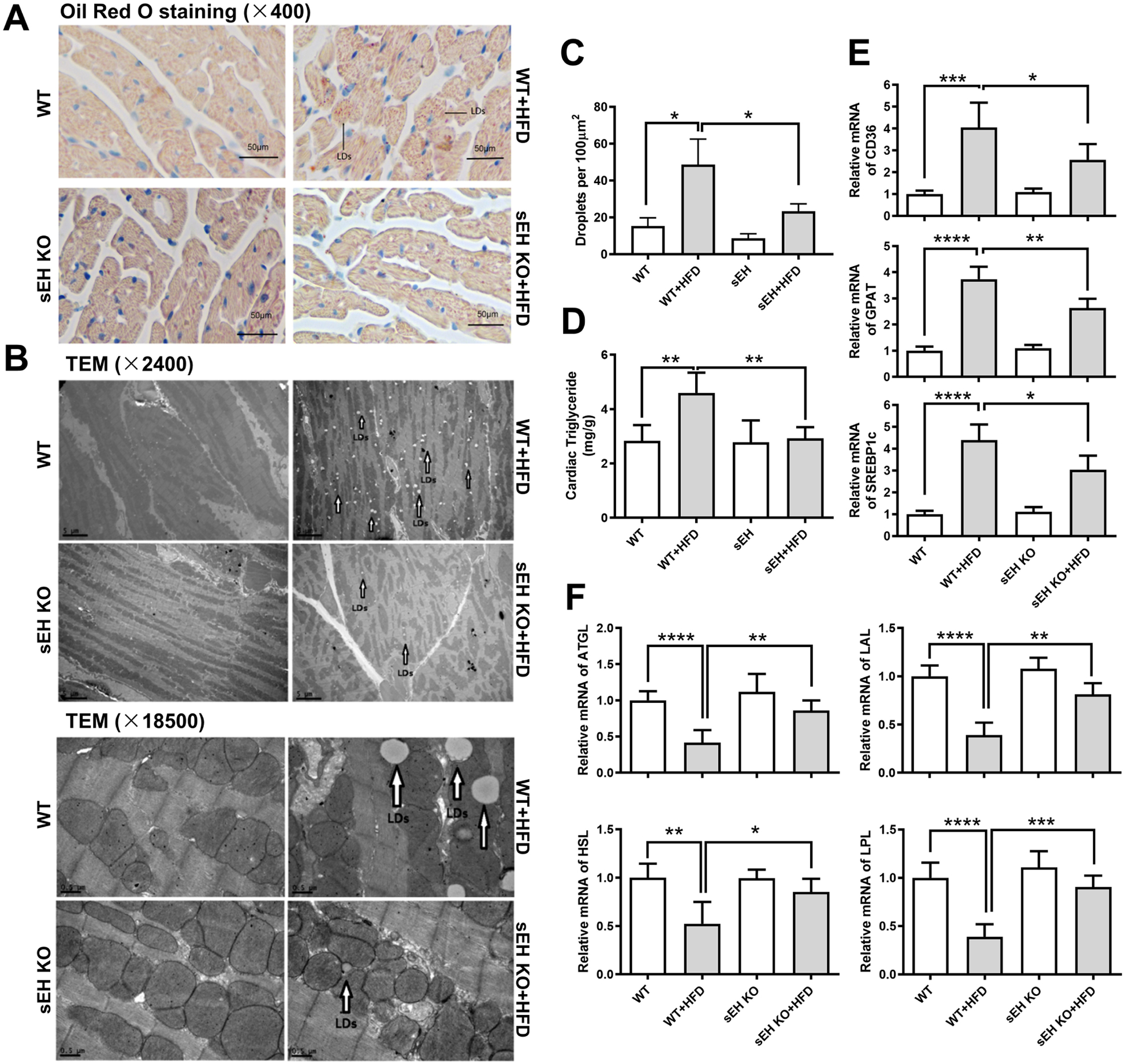
(A) Oil Red O staining in heart sections (400×, Scale bars indicate 50 μm). (B) Heart sections were visualized by transmission electron microscope (TEM) at 2,400× (scale bars indicate 5 μm) and 18,500× (scale bars indicate 0.5 μm) magnification. (C) Quantification of TEM at 18500×, each value represents the average of counts from 10 random fields of each sample. (D) Measurement of triglyceride content in hearts. (E, F) The mRNA expression levels of the lipid anabolic gene CD36, GPAT, and SREBP1c, and the lipid catabolic gene ATGL, LAL, HSL, and LPL were shown. *P<0.05, **P<0.01, ***P<0.005, ****P<0.001. n=3–5 per group. LDs: lipid droplets. LDs were marked with black open arrows.
Effects of sEH KO on HFD-induced alterations of fatty acid metabolism and transportation associated gene expressions
We also measured expressions of key genes involved in fatty acid metabolism. CD36, a transportation protein on the membrane of cardiomyocyte, through which fatty acid mainly enters, dramatically increased in WT mice with HFD intake. Additionally, glycerol-3-phosphate acyltransferases (GPAT), which initiates triglyceride synthesis and sterol regulatory element-binding protein 1c (SREBP1c), the master regulator of fatty acid synthesis, was upregulated accordingly. Meanwhile, genes associated with cardiac fatty acid catabolism, including adipose triglyceride lipase (ATGL), lysosomal acid lipase (LAL), hormone-sensitive lipase (HSL) and lipoprotein lipase (LPL), were downregulated in WT mice with HFD as well. Compared with WT mice, sEH KO failed to elicit any notable effects on these synthetic or catabolic lipid genes under SFD feeding. However, it significantly attenuated HFD-induced elevations in CD36, GPAT, SREBP1c (Fig. 2E) and decreases in ATGL, LAL, HSL, and LPL (Fig. 2F).
sEH KO protected against HFD-induced suppression on AMPK-mTORC mediated autophagy in mouse myocardium
While we were analyzing the LDs accumulation in heart sections under TEM, we surprisingly found plenty of degradative autophagic vacuoles (AVd) inside LDs in HFD feeding sEH KO mice, which conformed to the structures of lipophagy[27]. It encouraged us to ask what the role of the autophagy is during this process. We therefore carefully evaluated the autophagy morphologically as well as the autophagy induction markers. As described in the Methods section, autophagic vacuoles were counted from at least 20 random cells in the heart sections of each group using TEM to define the evidence of autophagy induction, which were averaged and expressed as the number of autophagic vacuoles per cell [26]. The autophagic vacuoles in myocardium of WT mice fed with HFD were significantly depressed. These changes were prevented in HFD feeding sEH KO mice (Fig. 3A–B). Intriguingly, these autophagic vacuoles were inside LDs. On aspect of autophagy markers, the autophagosome adaptor p62 was markedly upregulated, while the LC3II/LC3I ratio decreased significantly in HFD feeding WT mice. These abnormalities were restored in sEH KO mice (Fig. 3C–D top panel).
Fig. 3. sEH KO increased autophagic vacuoles in mouse myocardium and ameliorated HFD-induced suppression on AMPK-mTORC-regulated autophagy.
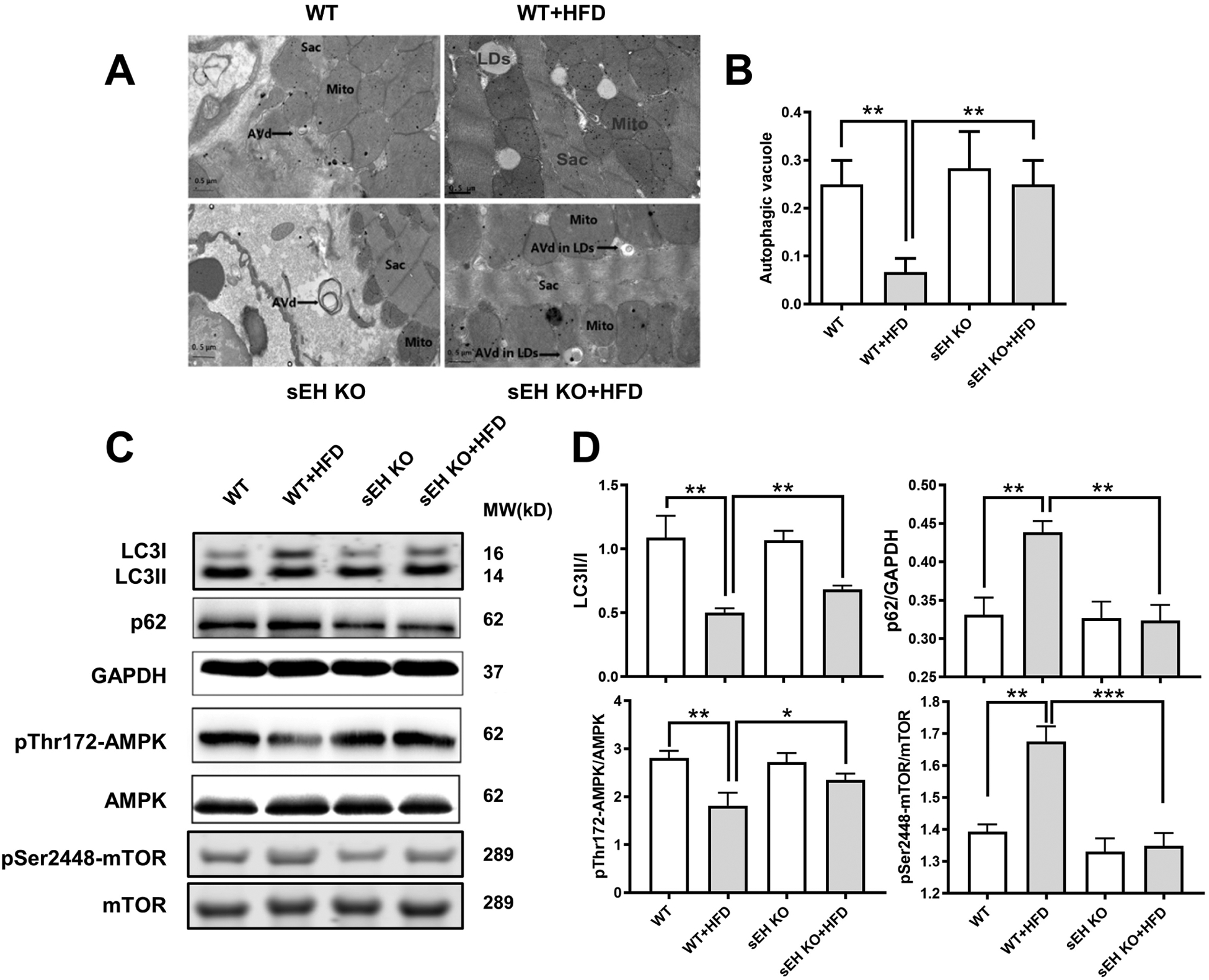
(A) Degradative autophagic vacuoles (AVd) were shown in electron microscopy. Scale bars indicate 0.5 μm. (B) Quantification of AVd counts per cell (n = 20). (C, D) Representative western blots and protein quantifications of LC3II to LC3I, p62 to GAPDH, phosphorylated AMPK to total AMPK, and phosphorylated mTOR to total mTOR ratios. *P<0.05, **P<0.01, ***P<0.005, n =3–5 per group. Sarc: sarcomere. Mito: mitochondria. LDs: lipid droplets. LDs were marked with black solid arrows.
AMPK-mTORC is recognized as a main upstream signaling pathway of autophagy, and previous research has indicated that genetic sEH disruption or sEH inhibition promotes AMPK phosphorylation [33, 34]. We therefore assumed systemic sEH KO promotes phosphorylation of AMPK, then inhibits mTORC and activates autophagy, which ultimately impacts cardiac lipid accumulation. To test this hypothesis, we examined the signaling pathway. Our data showed that HFD intake significantly suppressed phosphorylation of AMPK while it significantly increased phosphorylation of mTORC in WT mice. These effects were ablated by sEH KO (Fig. 3C–D bottom panel).
Inhibition of AMPK or autophagy blocked sEH KO-conferred protection against palmitic acid (PA)-induced lipotoxicity in cardiomyocytes
To further confirm the role of AMPK-mTOR mediated autophagy in protection of sEH KO against cardiac lipotoxicity, we induced lipotoxicity in primary cultured neonatal mouse cardiomyocytes using palmitic acid (PA). AMPK inhibitor Compound C (Com C) and autophagy inhibitor (3-MA) were applied to study their relationships. Cardiomyocyte lipid accumulation, triglyceride content level, sEH expression, AMPK signaling cascade and AMPK-mTORC regulated autophagy markers were assessed. Isolated cardiomyocytes from WT or sEH KO mice were incubated with PA (400 μM for 24 h) with the Com C (5μM) or 3-MA (10 mM), respectively [14]. Consistent with in vivo results, the Oil Red O staining and cardiomyocyte lipid quantification data revealed that PA markedly increased lipid accumulation in WT mouse cardiomyocytes, while sEH KO attenuated lipid accumulation. This sEH KO-conferred protective effect on cardiomyocytes was blocked by Com C or 3-MA, respectively (Fig. 4A–C). Furthermore, PA upregulated levels of sEH and p62 as well as decreased the LC3II/LC3I in WT cardiomyocytes. These effects were restored by sEH KO, but the restoration was reversed by Com C. Regarding AMPK-mTOR regulated autophagy signaling cascade, our data showed that PA inhibited the phosphorylation of AMPK and upregulated the phosphorylation of mTOR in WT mouse cardiomyocytes, sEH KO significantly ablated these effects. The ablation was blocked by Com C (Fig. 4D–F).
Fig. 4. Inhibition of AMPK or autophagy blocked the amelioration of lipid accumulation conferred by sEH KO in primary neonatal cardiomyocytes.
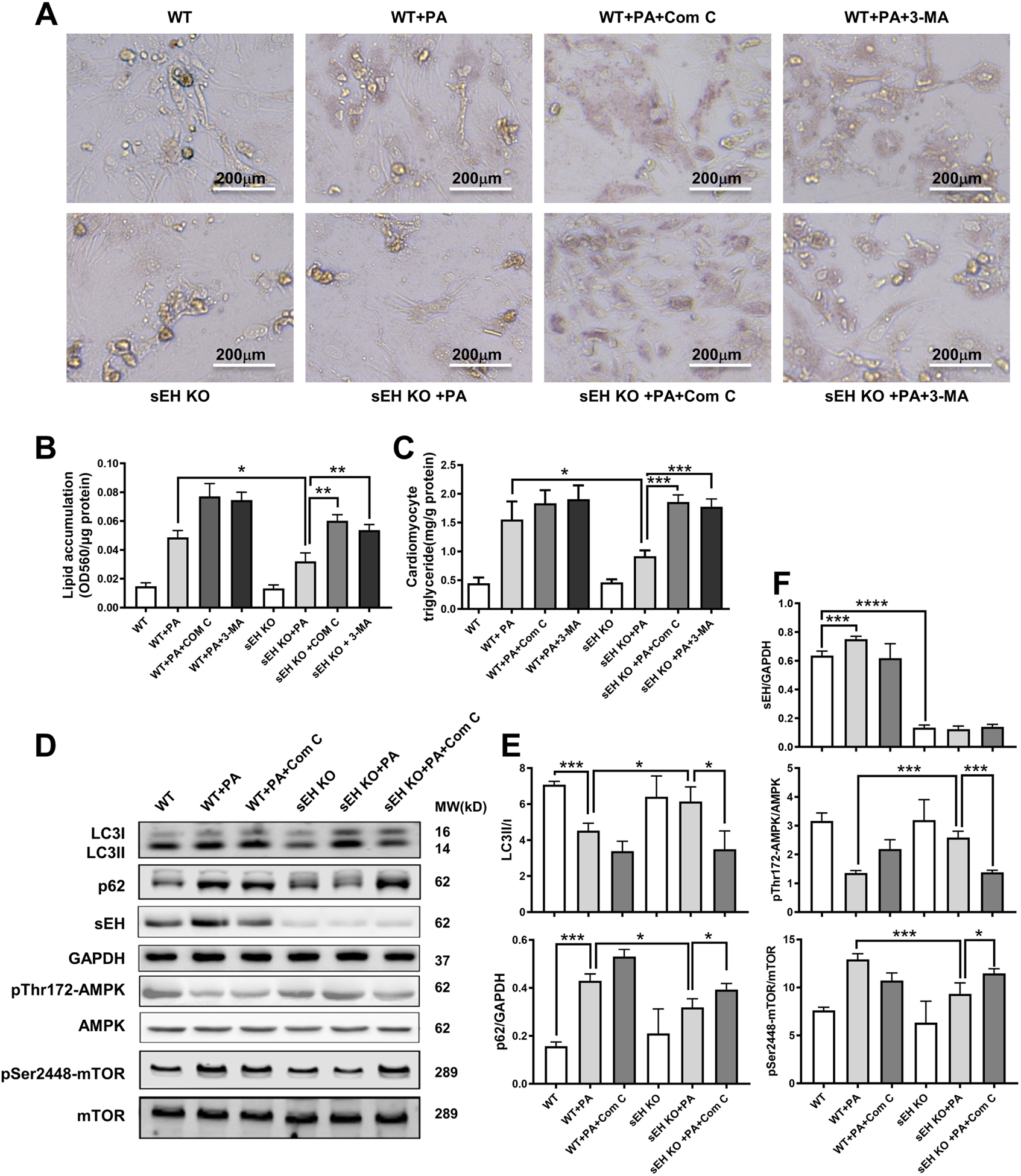
(A) Oil Red O staining of primary neonatal cardiomyocytes from WT and sEH KO mice incubated with palmitic acid (PA) in combination with or without AMPK inhibitor compound C (Com C) or autophagy inhibitor (3-MA). (B) Quantification of lipid accumulation with Oil Red O staining normalized to protein content (OD560/μg protein). (C) Triglyceride content quantification in cardiomyocytes. (D) Representative western blots of sEH and AMPK- mTOR regulated autophagy pathway. (E, F) Protein level quantifications of LC3II to LC3I, p62 to GAPDH, sEH to GAPDH, phosphorylated AMPK to total AMPK, and phosphorylated mTOR to total mTOR ratios. Scale bars indicate 200 μm. *P<0.05, **P<0.01, ***P<0.005, **** P<0.001. n=3 per group.
Inhibition of AMPK or autophagy blocked protection of sEH substrate plus sEH inhibitor against PA-induced lipid accumulation in cardiomyocytes
We have showed that sEH KO prevents cardiac lipid accumulation and upregulates AMPK-mTORC mediated autophagy in myocardium in vivo. These results were recapitulated and blocked with AMPK or autophagy inhibitors in sEH KO cardiomyocytes in vitro. To further confirm this, we performed in vitro experiments in primary neonatal WT cardiomyocytes incubated with PA and sEH substrate 14,15-EET plus sEH inhibitor AUDA. 14,15-EET levels measured by liquid chromatography-mass spectrometry were found to be increased in 14,15-EET plus AUDA treated cardiomyocytes (Fig. 5A). Interestingly, 14,15-EETs plus AUDA treatment significantly reduced PA-induced lipid accumulation in cardiomyocytes, which is comparable to the changes in sEH KO primary cardiomyocytes. Then, we incubated cardiomyocytes with or without Com C or 3-MA. Com C or 3-MA showed a reverse effect on attenuating lipid accumulation (Fig. 5B–C and S1). Nest, we detected the AMPK-mTORC mediated autophagy signaling pathway. As shown in Fig.5D–E and S2, 14,15-EET plus AUDA resulted in upregulation of autophagy revealed by the increase of LC3II to LC3I ratio and decrease of p62. This improvement in autophagy was supported by upregulation of p- AMPK and downregulation of p-mTORC. All these changes could be blocked by Com C or 3-MA.
Fig. 5. Inhibition of AMPK or autophagy blocked the amelioration of lipid accumulation conferred by sEH substrate EET plus sEH inhibitor treatments in primary neonatal cardiomyocytes.
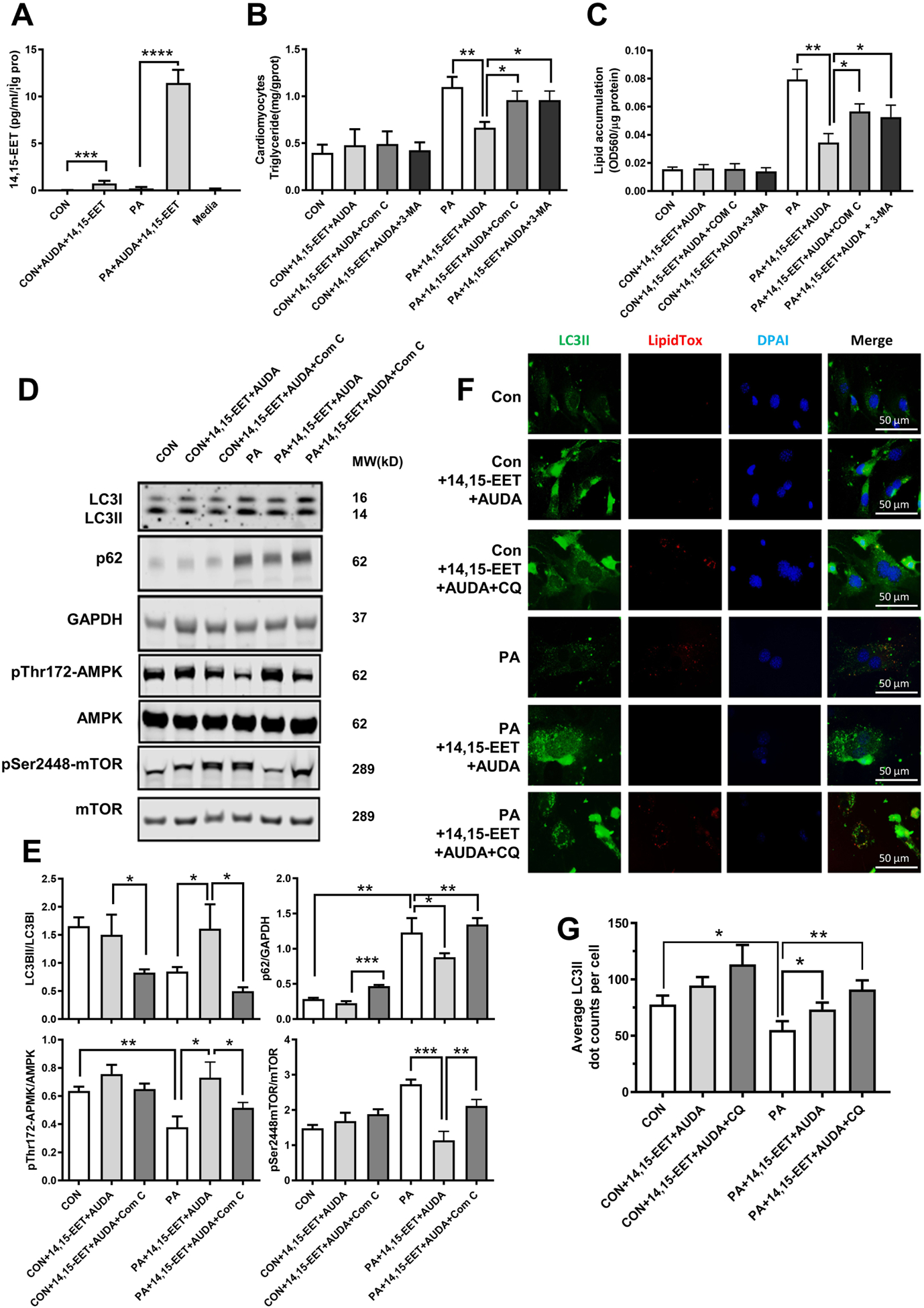
(A) Measurements of 14,15-EET in cardiomyocytes by liquid chromatography-mass spectrometry. (B) Triglyceride content of primary neonatal mouse cardiomyocytes from WT mice incubated with PA, 14,15-EET+AUDA, Compound C (Com C) or 3-MA. (C) Quantification of lipid accumulation with Oil Red O staining normalized to protein content (OD560/ μg protein). (D) Representative western blots of AMPK-mTOR-regulated autophagy pathway under Com C treatment. (E) Protein level quantifications of LC3II to LC3I, p62 to GAPDH, phosphorylated AMPK to total AMPK, and phosphorylated mTOR to total mTOR ratios. (F) Immunofluorescence images of LC3II and lipidTox. Scale bars indicate 50 μm. (G) Quantification of LC3II. *P<0.05, **P<0.01, ***P<0.005, ****P<0.001. n=3 per group. PA, palmitic acid; AUDA, sEH inhibitor; Com C, AMPK inhibitor; 3-MA, autophagy inhibitor; CQ, chloroquine; CON, no PA control; DAPI, 4’,6-diamidino-2-phenylindole; lipidTox, dye for lipid staining.
LC3-II has a crucial role in the formation of autophagosomes, which are subsequently targeted to autolysosomes. An individual autophagosome is represented as a LC3II punctum by immunofluorescence microscope. Chloroquine (CQ), known to prevent the autophagosome-lysosome fusion and result in autophagosome accumulation within the cell, was used as a control treatment to demonstrate morphological hallmarks of autophagosomes during autophagy. In PA-treated primary cardiomyocytes, we found 14,15-EET plus AUDA significantly increased the number of autophagosomes compared to cells without EET plus AUDA demonstrated by only a few LC3II puncta and highly dispersed intracellular fluorescence. Importantly, we observed that the LC3II puncta, indicator of the formation of autophagosomes, was robust and appeared to merge, and there were much fewer lipid droplets in the cells treated with 14,15-EET plus AUDA (Fig. 5F–G).
sEH substrate plus sEH inhibitor reversed PA-induced suppression on autophagic flux in cardiomyocytes
To further illustrate whether sEH deficiency selectively promotes autophagosome formation or it may regulate autophagic flux, we infected primary neonatal WT mouse cardiomyocytes with adenoviruses harboring tandem fluorescent mRFP-GFP-LC3. Then cells were incubated with 14,15-EET plus AUDA with or without PA or chloroquine. We found that sEH deficiency, being simulated with sEH inhibitor AUDA plus sEH substrate 14,15-EET, slightly increased the basal autophagic flux in cardiomyocytes evidenced by the increased number of both autophagosome (yellow) and autolysosome (red) puncta. Intriguingly, PA treatment suppressed the formations of both autophagosome and autolysosome, suggesting a PA-induced impairment of autophagic flux. By way of counter reaction, 14,15-EET plus AUDA treatment significantly increased both yellow and red dots in cardiomyocytes, indicating the autophagic flux was restored. The autophagy was more presented as autophagosomes in the presence of chloroquine, which blocked the autophagic flux by decreasing autophagosome-lysosome fusion. These results certified the pivotal role of 14,15-EET plus AUDA in restoring PA-induced suppression on autophagic flux in cardiomyocytes (Fig. 6A–B).
Fig. 6. sEH substrate EET plus sEH inhibitor alleviated PA-induced suppression on autophagic flux.
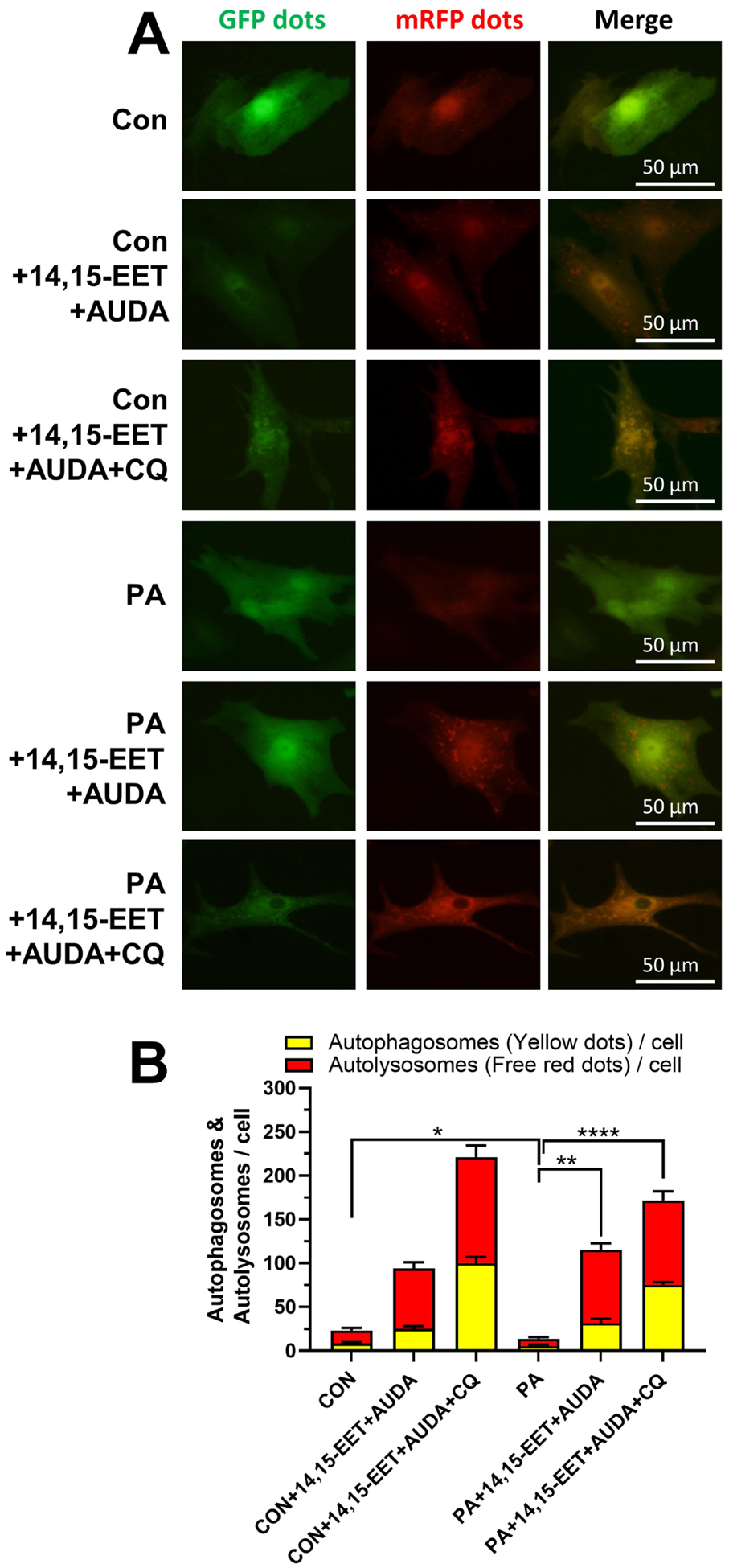
Primary mouse neonatal WT cardiomyocytes were incubated with PA, PA+14,15-EET+AUDA or PA+14,15-EET+AUDA+CQ and without PA as controls. (A) Representative confocal immunofluorescence images of mRFP-GFP-LC3 puncta are shown to present autophagic flux in cardiomyocytes. (B) Quantification of autophagy flux. Graph bars show the mean numbers of autophagosomes (yellow dots) and autolysosomes (red dots) per cell. Scale bars indicate 50 μm. *p < 0.05, **p<0.01, ****p < 0.001. n=3 per group. PA, palmitic acid; AUDA, sEH inhibitor; CQ, chloroquine; CON, no PA control; mRFP, monomeric red fluorescent protein; GFP, green fluorescent protein.
Discussion
In HFD fed mice, we showed that HFD led to obesity and lipotoxic cardiomyopathy, which were attenuated by sEH KO. Dysfunction of the AMPK-mTORC signaling pathway-mediated autophagy due to HFD was restored by sEH KO. In primary neonatal mouse cardiomyocytes, sEH KO or EETs plus AUDA treatment attenuated PA-induced lipid accumulation, which were blocked by inhibition of AMPK or autophagy. These data were further supported by HFD- or PA-induced down-regulation of autophagy markers LC3II/LC3I, as well as p62 accumulation, suggesting the impairment of initiating AMPK-mTORC driven autophagy and autophagic flux. This was rescued by sEH deficiency in vivo and in vitro. Consistent with previous studies [35, 36], sEH KO did not affect bodyweight gain and plasma TG level compared with WT mice under HFD in this study. We also found comparable blood pressures between sEH KO and WT mice fed with HFD. Taken together, these findings revealed that HFD lead to cardiac lipotoxicity, sEH deficiency played a critical role in decreasing lipid accumulation of myocardium by increasing autophagy through AMPK-mTORC signaling pathway (Fig. 7), which highlighted a novel target for treating lipotoxic cardiomyopathy.
Fig. 7. Schematic graph of the mechanism by which the sEH gene regulates lipotoxic cardiomyopathy.
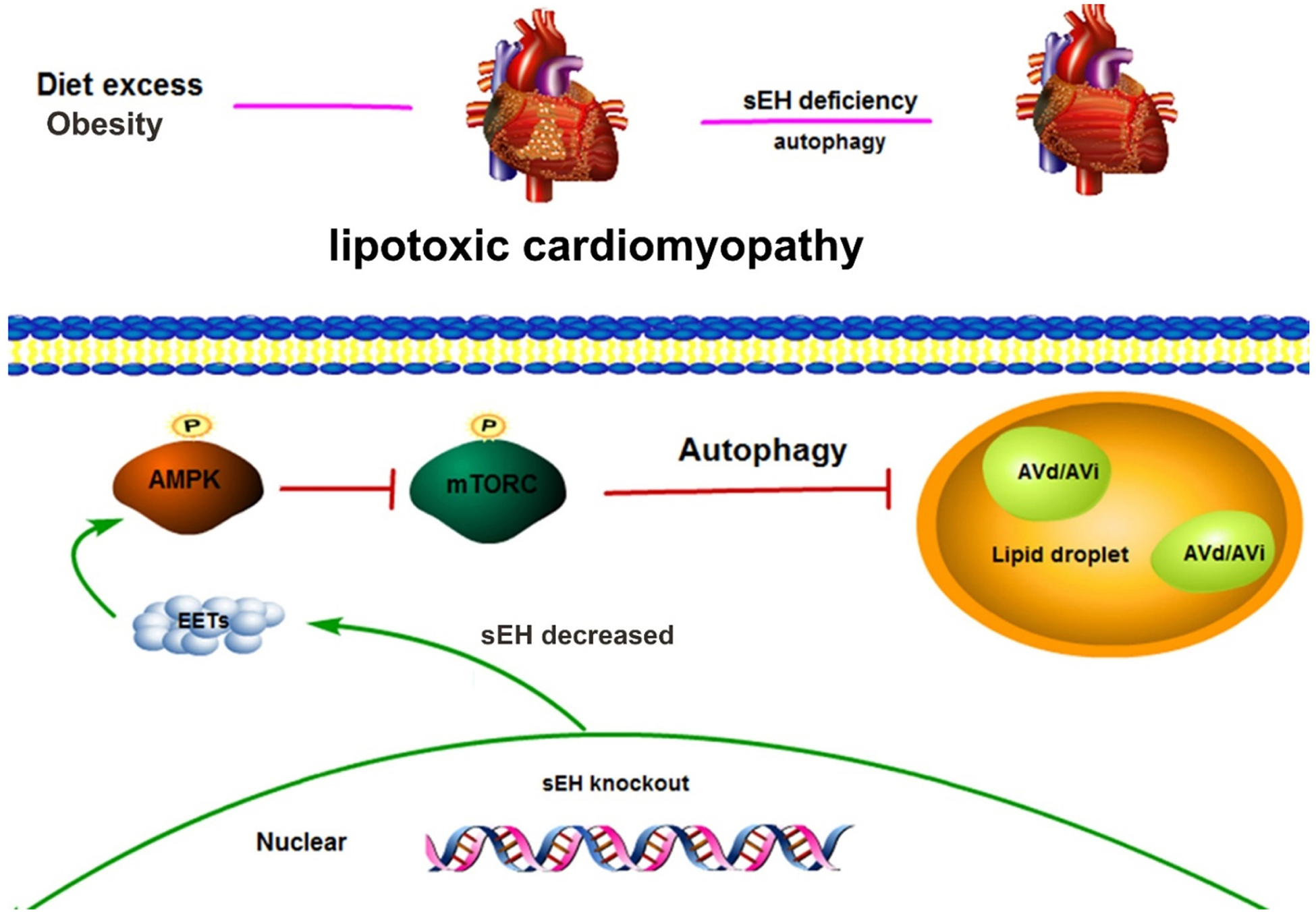
Upper panel shows that HFD led to mouse obesity and lipotoxic cardiomyopathy. These can be attenuated by sEH gene deficiency. Lower panel demonstrates that sEH gene deletion increases EETs, EETs promote AMPK phosphorylation and activates AMPK, activated AMPK decreases mTORC phosphorylation and suppresses mTORC, and suppressed mTORC increases lipid autophagy. This reaction chain finally leads to attenuation of lipotoxic cardiomyopathy resulting from lipid accumulation.
Under normal physiological conditions, autophagy serves as an essential housekeeper to maintain homeostasis for multiple organs including the heart [37]. Recently, a revival of interest in its role has emerged with the discovery that autophagy can mediate lipid metabolism by engulfing lipid in LDs or provide cellular energy through generation of FFAs. Singh et al. termed macrolipophagy as a direct interrelationship between autophagy and lipid metabolism for the first time. They depicted that autophagy regulated lipid content in hepatocytes in vitro and in vivo. Inhibition of autophagy may trap hepatocytes in a harmful cycle in which decreased autophagy promotes lipid accumulation that then further suppresses autophagic function, thereby additionally increasing lipid retention [14]. Lipophagy is ubiquitous as it functions in other cells that do not store lipids in large quantities as hepatocytes including fibroblasts, neurons, and stellate cells[38]. Our study firstly observed some morphological clues and changes of autophagy markers that sEH deficiency upregulated lipophagy in myocardium, which was a mechanism that sEH mitigated cardiac lipid accumulation.
AMPK is a heterotrimeric enzyme composed of a catalytic α subunit tethered to the regulatory β and γ subunits. It is an important regulator of cardiac energy homeostasis. This serine/threonine kinase is activated by adenosine triphosphate (ATP) depletion through phosphorylation of its Thr172 residue by upstream kinases such as liver kinase B1[15]. AMPK responds to increases in the AMP/ATP ratio by switching off energy- consuming pathways. AMPK plays a key role in regulating cellular growth and metabolism, acting as a metabolic sensor, allowing adaptive responses to reduced energy. PA was a widely used reagent to induce cellular model of lipid accumulation in cardiomyocytes[39]. Lipid overload suppresses AMPK activity by impairing AMPK phosphorylation [40]. EETs have been reported to directly interacting with AMPKα2β2γ1 to activate AMPK signaling and protect against cardiac hypertrophy [41]. The most recent research in HFD-mediated renal injury shows 14,15-EET (a major substrate of sEH), but not its sEH-mediated metabolite 14,15-DHET, significantly reversed PA-induced increase in Pax2 (paired box gene 2) and decrease in AMPK activation. In addition, Pax2 may positively regulate AMPK transcription in murine renal mesangial cells [34]. The way EET or sEH activates AMPK in lipotoxic cardiomyopathy may also involve these mechanisms but needs to be further illustrated in the future.
Activation of AMPK can inhibit downstream mTORC signaling, which will induce an autophagic response [42]. AMPK represents the most well characterized regulator of mTORC. In our study, AMPK-mTORC pathway activity status was accompanied by changes of gene expressions involved in lipid metabolism. In the HFD-induced mouse lipotoxic cardiomyopathy model, we detected suppression of the AMPK-mTORC pathway and subsequent increased expressions of lipid transport or anabolic genes of CD36, GPAT, and SREBP1c and decreased expressions of lipid catabolic genes of ATGL, HSL, LAL and LPL (Fig. 2E–F). These changes were consistent with previous reported roles of the AMPK-mTORC pathway in lipid metabolism [43, 44]. sEH KO successfully attenuated the HFD-induced changes of lipid accumulation, lipophagy and lipid metabolism related gene expressions, suggesting that sEH KO protected against cardiac lipotoxicity and mTORC may act as a bridge connecting the crosstalk between the lipid metabolism-related genes and autophagy processes. Although lipophagic responses in other organs were not investigated in this study, considering HFD also induced lipid deposition in liver and kidney and this pathway was involved in the processes [35, 45], the protective effects induced by sEH deficiency or inhibition on lipotoxicity in this study should be non-cardiac-specific and present in kidney and liver as well. The relationship between sEH deficiency conferred protection against lipotoxic cardiomyopathy and AMPK-mTORC mediated autophagy was strongly supported by the in vitro study showing that the protective effects were blocked by AMPK or mTORC inhibitors. However, we believe lack of similar blockade study on the pathway in mice to further consolidate this relation in vivo represents the limitations of this study.
Conclusion
In summary, the in vivo and in vitro experiments demonstrated that sEH deficiency or inhibition attenuated lipid accumulation in cardiomyocytes through upregulation of AMPK-mTORC mediated autophagy. Lipotoxic cardiomyopathy was ameliorated by sEH deficiency in vivo. These findings provided experimental evidence for the potential clinical application of sEH inhibition in treating lipotoxic cardiomyopathy.
Supplementary Material
Fig. S1 Oil Red O staining of primary neonatal cardiomyocytes from WT mice incubated with palmitic acid (PA), 14,15-EET+AUDA, AMPK inhibitor compound C (Com C), or autophagy inhibitor (3-MA). AUDA, sEH inhibitor.
Fig. S2 Representative western blots and quantifications of LC3II/I, p62 under 3-MA treatment on WT mice cardiomyocytes incubated with palmitic acid (PA), 14,15-EET+AUDA, or autophagy inhibitor (3-MA). AUDA, sEH inhibitor. ***P<0.005, ****P<0.001. n=3 per group.
Highlights.
sEH deficiency attenuates lipid accumulation in myocardium in vivo and in vitro
sEH deficiency increases autophagy in myocardium in vitro and in vitro
sEH regulates autophagy in myocardium through AMPK-mTORC pathway
Funding
This work is supported by the National Natural Science Foundation of China (81600236 to L. Wang, 8201101103, 82073408, 81870506, 81670676 and 81422011 to H. Huang) and Project of Traditional Chinese Medicine in Guangdong Province (20201062), Basic Research Project of Shenzhen Science and Technology Innovation Committee (JCYJ20180306174648342 and JCYJ20190808102005602), Shenzhen Futian District Public Health Scientific Research Project (FTWS2019003) and Shenzhen Key Medical Discipline Construction Fund (SZXK002) to H. Huang. Partial support was provided by the NIH-NIEHS (RIVER Award) R35 ES030443-01, the NIEHS Superfund Research Program P42 ES004699 to B. Hammock.
Footnotes
Conflict of interest
The authors have declared that no competing interest exists.
References
- *1+.Hales CM, Carroll MD, Fryar CD, Ogden CL, Prevalence of Obesity Among Adults and Youth: United States, 2015–2016, NCHS Data Brief 288 (2017) 1–8. [PubMed] [Google Scholar]
- *2+.Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, et al. , Obesity and the risk of heart failure, N. Engl. J. Med 347(5) (2002) 305–313. [DOI] [PubMed] [Google Scholar]
- *3+.Zhang H, Wang T, Zhang K, Liu Y, Huang F, Zhu X, et al. , Deletion of soluble epoxide hydrolase attenuates cardiac hypertrophy via down-regulation of cardiac fibroblasts-derived fibroblast growth factor-2, Crit. Care Med 42(5) (2014) e345–354. [DOI] [PubMed] [Google Scholar]
- *4+.Huang H, Morisseau C, Wang J, Yang T, Falck JR, Hammock BD, et al. , Increasing or stabilizing renal epoxyeicosatrienoic acid production attenuates abnormal renal function and hypertension in obese rats, Am. J. Physiol. Renal Physiol 293(1) (2007) F342–349. [DOI] [PubMed] [Google Scholar]
- *5+.Wang L, Liu Y, Wang H, Liu X, Chen J, Wang MH, et al. , Epoxyeicosatrienoic acids attenuating hypotonic-induced apoptosis of IMCD cells via gamma-ENaC inhibition, PLoS One 9(4) (2014) e94400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *6+.Xu X, Zhao CX, Wang L, Tu L, Fang X, Zheng C, et al. , Increased CYP2J3 expression reduces insulin resistance in fructose-treated rats and db/db mice, Diabetes 59(4) (2010) 997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *7+.Wang ZH, Davis BB, Jiang DQ, Zhao TT, Xu DY, Soluble epoxide hydrolase inhibitors and cardiovascular diseases, Curr. Vasc. Pharmacol 11(1) (2013) 105–111. [PubMed] [Google Scholar]
- *8+.Monti J, Fischer J, Paskas S, Heinig M, Schulz H, Gosele C, et al. , Soluble epoxide hydrolase is a susceptibility factor for heart failure in a rat model of human disease, Nat. Genet 40(5) (2008) 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *9+.Roche C, Besnier M, Cassel R, Harouki N, Coquerel D, Guerrot D, et al. , Soluble epoxide hydrolase inhibition improves coronary endothelial function and prevents the development of cardiac alterations in obese insulin-resistant mice, Am. J. Physiol. Heart Circ. Physiol 308(9) (2015) H1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *10+.Bettaieb A, Nagata N, AbouBechara D, Chahed S, Morisseau C, Hammock BD, et al. , Soluble epoxide hydrolase deficiency or inhibition attenuates diet-induced endoplasmic reticulum stress in liver and adipose tissue, J. Biol. Chem 288(20) (2013) 14189–14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *11+.De Taeye BM, Morisseau C, Coyle J, Covington JW, Luria A, Yang J, et al. , Expression and regulation of soluble epoxide hydrolase in adipose tissue, Obesity (Silver Spring) 18(3) (2010) 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *12+.Kandadi MR, Panzhinskiy E, Roe ND, Nair S, Hu D, Sun A, Deletion of protein tyrosine phosphatase 1B rescues against myocardial anomalies in high fat diet-induced obesity: Role of AMPK- dependent autophagy, Biochim. Biophys. Acta 1852(2) (2015) 299–309. [DOI] [PubMed] [Google Scholar]
- *13+.Hashemi HF, Goodman JM, The life cycle of lipid droplets, Curr. Opin. Cell Biol 33((2015) 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *14+.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. , Autophagy regulates lipid metabolism, Nature 458(7242) (2009) 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *15+.Palomer X, Salvado L, Barroso E, Vazquez-Carrera M, An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy, Int. J. Cardiol 168(4) (2013) 3160–3172. [DOI] [PubMed] [Google Scholar]
- *16+.Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, et al. , AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species, Cell Rep 21(1) (2017) 1–9. [DOI] [PubMed] [Google Scholar]
- *17+.Sciarretta S, Forte M, Frati G, Sadoshima J, New Insights Into the Role of mTOR Signaling in the Cardiovascular System, Circ. Res 122(3) (2018) 489–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *18+.Lopez-Vicario C, Alcaraz-Quiles J, Garcia-Alonso V, Rius B, Hwang SH, Titos E, et al. , Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: role for omega-3 epoxides, Proc. Natl. Acad. Sci. U. S. A 112(2) (2015) 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *19+.Samokhvalov V, Alsaleh N, El-Sikhry HE, Jamieson KL, Chen CB, Lopaschuk DG, et al. , Epoxyeicosatrienoic acids protect cardiac cells during starvation by modulating an autophagic response, Cell Death Dis. 4(2013) e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *20+.Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, et al. , Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice, J. Clin. Invest 122(3) (2012) 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *21+.Omarjee L, Roy C, Leboeuf C, Favre J, Henrion D, Mahe G, et al. , Evidence of Cardiovascular Calcification and Fibrosis in Pseudoxanthoma Elasticum Mouse Models Subjected to DOCA-Salt Hypertension, Sci. Rep 9(1) (2019) 16327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *22+.Li Y, Lee S, Langleite T, Norheim F, Pourteymour S, Jensen J, et al. , Subsarcolemmal lipid droplet responses to a combined endurance and strength exercise intervention, Physiol Rep 2(11) (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- *23+.Xu X, Tu L, Feng W, Ma B, Li R, Zheng C, et al. , CYP2J3 gene delivery up-regulated adiponectin expression via reduced endoplasmic reticulum stress in adipocytes, Endocrinology 154(5) (2013) 1743–1753. [DOI] [PubMed] [Google Scholar]
- *24+.Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR, et al. , Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes, J. Clin. Invest 122(11) (2012) 3919–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *25+.Minster RL, Hawley NL, Su CT, Sun G, Kershaw EE, Cheng H, et al. , A thrifty variant in CREBRF strongly influences body mass index in Samoans, Nat. Genet 48(9) (2016) 1049–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *26+.Song YM, Lee YH, Kim JW, Ham DS, Kang ES, Cha BS, et al. , Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway, Autophagy 11(1) (2015) 46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *27+.Klionsky DJ,Abdelmohsen K,Abe A,Abedin MJ,Abeliovich H,Acevedo Arozena A, et al. , Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition), Autophagy 12(1) (2016) 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *28+.Wu J, Wu JJ, Yang LJ, Wei LX, Zou DJ, Rosiglitazone protects against palmitate-induced pancreatic beta-cell death by activation of autophagy via 5’-AMP-activated protein kinase modulation, Endocrine 44(1) (2013) 87–98. [DOI] [PubMed] [Google Scholar]
- *29+.Ehler E, Moore-Morris T, Lange S, Isolation and culture of neonatal mouse cardiomyocytes, J Vis Exp 79) (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- *30+.Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L, et al. , AMPKalpha2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation, Circulation research 122(5) (2018) 712–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *31+.Qiu B, Simon MC, BODIPY 493/503 Staining of Neutral Lipid Droplets for Microscopy and Quantification by Flow Cytometry, Bio Protoc 6(17) (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- *32+.Edin ML, Hamedani BG, Gruzdev A, Graves JP, Lih FB, Arbes SJ 3rd, et al. , Epoxide hydrolase 1 (EPHX1) hydrolyzes epoxyeicosanoids and impairs cardiac recovery after ischemia, J. Biol. Chem 293(9) (2018) 3281–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *33+.Chen G, Xu R, Wang Y, Wang P, Zhao G, Xu X, et al. , Genetic disruption of soluble epoxide hydrolase is protective against streptozotocin-induced diabetic nephropathy, Am. J. Physiol. Endocrinol. Metab 303(5) (2012) E563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *34+.Luo Y, Wu MY, Deng BQ, Huang J, Hwang SH, Li MY, et al. , Inhibition of soluble epoxide hydrolase attenuates a high-fat diet-mediated renal injury by activating PAX2 and AMPK, Proc. Natl. Acad. Sci. U. S. A 116(11) (2019) 5154–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *35+.Liu Y, Dang H, Li D, Pang W, Hammock BD, Zhu Y, Inhibition of soluble epoxide hydrolase attenuates high-fat-diet-induced hepatic steatosis by reduced systemic inflammatory status in mice, PLoS One 7(6) (2012) e39165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *36+.Luria A, Bettaieb A, Xi Y, Shieh GJ, Liu HC, Inoue H, et al. , Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance, Proc. Natl. Acad. Sci. U. S. A 108(22) (2011) 9038–9043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *37+.Woodall BP, Gustafsson AB, Autophagy-A key pathway for cardiac health and longevity, Acta Physiol. (Oxf.) 223(4) (2018) e13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *38+.Cingolani F, Czaja MJ, Regulation and Functions of Autophagic Lipolysis, Trends Endocrinol. Metab 27(10) (2016) 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *39+.Adrian L, Lenski M, Todter K, Heeren J, Bohm M, Laufs U, AMPK Prevents Palmitic Acid-Induced Apoptosis and Lipid Accumulation in Cardiomyocytes, Lipids 52(9) (2017) 737–750. [DOI] [PubMed] [Google Scholar]
- *40+.Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, et al. , AMPK inhibition in health and disease, Crit. Rev. Biochem. Mol. Biol 45(4) (2010) 276–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *41+.Wang B, Zeng H, Wen Z, Chen C, Wang DW, CYP2J2 and its metabolites (epoxyeicosatrienoic acids) attenuate cardiac hypertrophy by activating AMPKalpha2 and enhancing nuclear translocation of Akt1, Aging Cell 15(5) (2016) 940–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *42+.Li Y, Chen Y, AMPK and Autophagy, Adv. Exp. Med. Biol 1206((2019) 85–108. [DOI] [PubMed] [Google Scholar]
- *43+.Mangels N, Awwad K, Wettenmann A, Dos Santos LR, Fromel T, Fleming I, The soluble epoxide hydrolase determines cholesterol homeostasis by regulating AMPK and SREBP activity, Prostaglandins Other Lipid Mediat. 125(2016) 30–39. [DOI] [PubMed] [Google Scholar]
- *44+.Ricoult SJ, Manning BD, The multifaceted role of mTORC1 in the control of lipid metabolism, EMBO Rep 14(3) (2013) 242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *45+.Yamamoto T, Takabatake Y, Takahashi A, Kimura T, Namba T, Matsuda J, et al. , High-Fat Diet- Induced Lysosomal Dysfunction and Impaired Autophagic Flux Contribute to Lipotoxicity in the Kidney, J. Am. Soc. Nephrol 28(5) (2017) 1534–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Oil Red O staining of primary neonatal cardiomyocytes from WT mice incubated with palmitic acid (PA), 14,15-EET+AUDA, AMPK inhibitor compound C (Com C), or autophagy inhibitor (3-MA). AUDA, sEH inhibitor.
Fig. S2 Representative western blots and quantifications of LC3II/I, p62 under 3-MA treatment on WT mice cardiomyocytes incubated with palmitic acid (PA), 14,15-EET+AUDA, or autophagy inhibitor (3-MA). AUDA, sEH inhibitor. ***P<0.005, ****P<0.001. n=3 per group.