

Abstract
β1-adrenergic receptors (β1ARs) are the principle mediators of catecholamine action in cardiomyocytes. We previously showed that the β1AR extracellular N-terminus is a target for post-translational modifications that impact on signaling responses. Specifically, we showed that the β1AR N-terminus carries O-glycan modifications at Ser37/Ser41, that O-glycosylation prevents β1AR N-terminal cleavage, and that N-terminal truncation influences β1AR signaling to downstream effectors. However, the site(s) and mechanism for β1AR N-terminal cleavage in cells was not identified. This study shows that β1ARs are expressed in cardiomyocytes and other cells types as both full-length and N-terminally truncated species and that the truncated β1AR species is formed as a result of an O-glycan regulated N-terminal cleavage by ADAM17 at R31↓L32. We identify Ser41 as the major O-glycosylation site on the β1AR N-terminus and show that an O-glycan modification at Ser41 prevents ADAM17-dependent cleavage of the β1-AR N-terminus at S41↓L42, a second N-terminal cleavage site adjacent to this O-glycan modification (and it attenuates β1-AR N-terminal cleavage at R31↓L32). We previously reported that oxidative stress leads to a decrease in β1AR expression and catecholamine responsiveness in cardiomyocytes. This study shows that redox-inactivation of cardiomyocyte β1ARs is via a mechanism involving N-terminal truncation at R31↓L32 by ADAM17. In keeping with the previous observation that N-terminally truncated β1ARs constitutively activate an AKT pathway that affords protection against doxorubicin-dependent apoptosis, overexpression of a cleavage resistant β1AR mutant exacerbates doxorubicin-dependent apoptosis. These studies identify the β1AR N-terminus as a structural determinant of β1AR responses that can be targeted for therapeutic advantage.
Keywords: β-adrenergic receptor, cardiomyocytes, glycosylation, oxidant stress, ADAM17
Graphical Abstract
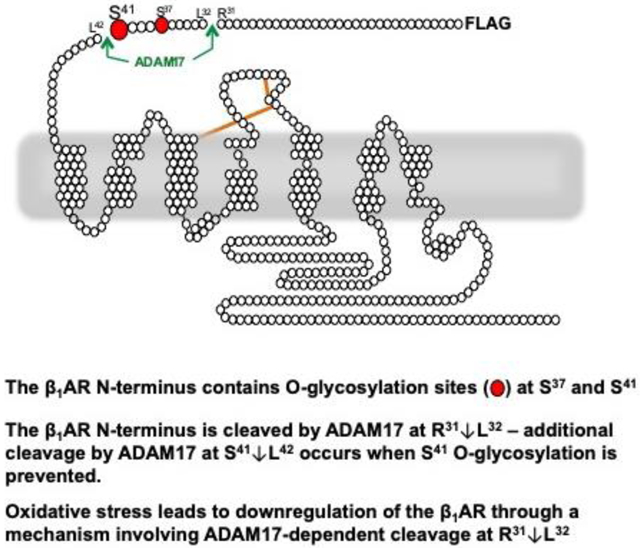
1. INTRODUCTION
Catecholamines enhance the mechanical performance of the heart by activating cardiac β-adrenergic receptors βARs). While cardiomyocytes co-express β1AR and β2AR, the β1AR is the predominant subtype (constituting ~75-80% of the total βAR population) and the principle driver of catecholamine-dependent sympathetic responses in the healthy heart 1. β1ARs provide hemodynamic support in the setting of acute stress by activating a Gs-cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA) pathway that phosphorylates substrates that enhance excitation/contraction coupling 2. Although β1ARs also couple to cardioprotective Gs-independent mechanisms such as extracellular signal-regulated kinase (ERK), chronic/persistent β1AR activation leads to a spectrum of changes (including cardiomyocyte hypertrophy/apoptosis, interstitial fibrosis, and contractile dysfunction) that contribute to the pathogenesis of heart failure 3,4.
Like other G protein-coupled receptor (GPCR) superfamily members, βARs are characterized by an extracellular N-terminus, seven membrane-spanning α-helical segments that are joined by three extracellular loops and three intracellular loops, and an intracellular C-terminus. While βARs have served as prototypes in structural studies designed to elucidate the molecular dynamics of GPCR activation, studies to date have focused on specific amino acid side chains in the transmembrane helices that form the ligand binding pocket and effector docking sites on the βAR’s intracellular surface 5. The relatively short/unstructured βAR N-terminus - a region traditionally viewed as having a negligible role in mechanisms that contribute to receptor activation and regulation - has routinely been removed for these structural studies 5. However, our recent studies challenge the notion that the β1AR N-terminus plays a negligible role in receptor activation/regulation showing that the β1AR extracellular N-terminus is a target for glycan-regulated proteolytic cleavage and that this mechanism influences β1AR signaling responses in cardiomyocytes 6. We showed that the β1AR is detected in cardiomyocytes as both full-length and N-terminally truncated species, that N-terminal cleavage is controlled by an O-glycan modification on the β1AR N-terminus at Ser37/Ser41 (residues in the vicinity of predicted β1AR N-terminal cleavage sites) and that the β1AR extracellular N-terminus functions as a heretofore-unrecognized structural determinant of β1AR activation 6. N-terminally truncated β1ARs are signaling competent, but their signaling properties differ from that of the full-length β1AR they display differences in their signaling bias to cAMP/PKA vs. ERK pathways - and only the N-terminally truncated form of the β1AR constitutively activates AKT and confers protection against doxorubicin-dependent apoptosis in cardiomyocytes 6,7.
Efforts to date to identify the O-glycan modifying enzymes and specific proteases that participate in the maturational processing of the β1AR N-terminus have relied on in vitro approaches. On the basis of in vitro cleavage assays (using short peptides based upon the β1AR N-terminal sequence as substrate and purified matrix metalloproteinase [MMP] or a disintegrin and metalloproteinase [ADAM] enzymes), Goth et al. concluded that the β1AR N-terminus contains two major cleavage sites at R31↓L32 and P52↓L53. They showed that O-glycosylated and unmodified forms of these peptides can serve as substrates for many different MMP and ADAM family enzymes and that O-glycosylation influences the efficiency of peptide cleavage at some sites by some enzymes 8. It is important to note that this previous study interrogated the role of O-glycosylation by subjecting peptides to in vitro O-glycosylation reactions with GalNAc-T2, a reaction that adds a single GalNAc moiety to one or more sites on the peptide. Results obtained in this type of reductionist assay system may not necessarily recapitulate the proteolytic cleavage events that accompany maturational processing of full-length β1ARs in a more physiologically relevant context (such as in differentiated cells such as a cardiomyocyte). First, this approach does not evaluate the role of more physiologically relevant extended and sialylated O-glycan structures that decorate the β1AR N-terminus in a cellular context (and are predicted to undergo remodeling in the setting of inflammation, metabolic disorders, and/or cardiac hypertrophy 9,10). Second, in vitro cleavage assays are not suited to evaluate cleavage events on membrane proteins, a situation where substrate and enzyme may localize to distinct membrane subdomains or (when tethered to the same membrane) be conformationally restrained. Third, the previous study did not resolve the importance of individual O-glycosylation sites in the control of β1AR N-terminal cleavage. Finally, the in vitro approach cannot be used to explore the role of glycan-regulated β1AR N-terminal cleavage events in the response to pathophysiologic cardiac injury. This study addresses these issues by mapping the O-glycosylation sites that regulate β1AR N-terminal cleavage in cardiomyocytes, identifying ADAM17 as the protease required for β1AR N-terminal cleavage in cardiomyocytes, and implicating ADAM17-dependent β1AR N-terminal cleavage in an oxidative stress-dependent mechanism that calibrates cardiomyocyte catecholamine responsiveness.
2. MATERIALS AND METHODS
2.1. Materials
Antibodies were from the following sources: Rabbit polyclonal anti-βAR (ab3442, raised against residues 394-408 in human β1AR) was from Abcam (Cambridge, MA). Mouse monoclonal anti-FLAG M2 and ant-β-actin were from Sigma-Aldrich (Saint Louis, MO). Rabbit monoclonal anti-cleaved caspase-3 (Asp175) (5A1E) was from Cell Signaling Technology (Danvers, MA). IRDye 800CW or 680RD goat anti-rabbit or goat anti-mouse IgG (H+L) were from LI-COR Biosciences (Lincoln, NE). GM6001, GI254023X (ADAM-10 inhibitor), MMP-2/MMP-9 inhibitor II, MMP-9/MMP-13 inhibitor II, doxorubicin, and H2O2 were from Sigma-Aldrich. MMP-2/MMP-3 inhibitor I and TAPI-2 were from Santa Cruz Biotechnology (Dallas, TX). GW280264X (ADAM-10/ADAM-17 inhibitor) was from AOBIOUS Inc (Hopkinton, MA). All other chemicals were reagent grade.
2.2. Chinese hamster ovary (CHO) pro5 and CHO-IdID cell culture and transfection
CHO-pro5 cells (obtained from American Type Culture Collection, Manassas, VA) were cultured in minimum essential medium Eagle-α modification (α-MEM), supplemented with 5% fetal bovine serum (FBS), 100 units/ml penicillin-streptomycin and 100 mM glutamine. CHO-IdID cells (generous gift from Dr. Monty Krieger, Massachusetts Institute of Technology) were cultured in Ham’s F-12 medium, supplemented with 5% FBS, 100 units/ml penicillin-streptomycin, 100 mM glutamine, either with or without galactose (Gal, 20 μM) and N-acetylgalactosamine (GalNAc, 200 μM). Cell transfections were performed in 60 mm culture dishes with 1 μg cDNA of β1AR plasmid using jetOPTIMUS DNA Transfection Reagent (Polyplus Transfection, IIIkirch, France) according to manufacturer’s instructions.
2.3. Plasmids
A plasmid that drives expression of the human S49R389-β1AR harboring an N-terminal FLAG-tag was from Addgene (Watertown, MA). The various single residue substituted β1AR mutant constructs used in this study were generated using the QuikChange mutagenesis system (Agilent Technologies) according to manufacturer’s instructions. The integrity of all constructs was confirmed by DNA sequencing (Genewiz, South Plainfield, NJ).
2.4. Cardiomyocyte culture and adenoviral infections
Cardiomyocytes were isolated from the ventricles of 2-day-old Wistar rats by a trypsin dispersion technique using a differential attachment procedure to enrich for cardiomyocytes followed by irradiation as described previously 11,12. Methods to infect cardiomyocytes with adenoviruses that drive expression of WT or mutant forms of human β1AR (Ad-WT-β1AR and Ad-β1AR-31H32A/52A53A prepared by Welgen Inc. Worcester, MA) were published previously 12.
2.5. Immunoblotting
Immunoblotting was performed on cell extracts according to methods described previously 6,7 or manufacturer’s instructions. Dilutions for primary and secondary antibodies were as follows: Abcam anti-β1AR (ab3442) at 1:3000 followed by IRDye 800CW goat anti-rabbit IgG at 1:5000. Anti-FLAG M2 at 1:700 and anti-β-actin at 1:1000 followed by IRDye 800CW or 680RD goat anti-mouse IgG at 1:5000. Anti-cleaved caspase-3 at 1:700 followed by IRDye 800CW goat anti-rabbit IgG at 1:5000. Each panel in each figure represents results from a single gel exposed for a uniform duration using LI-COR Odyssey CLx imaging system (LI-COR Biosciences Lincoln, NE) for detection and image Studio Lite Ver 5.0 software for quantification of protein expression. All results were replicated in at least three experiments on separate culture preparations.
2.6. Statistical analysis
Results are shown as mean±SEM and were analyzed by using one-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparison post hoc test; p values < 0.05 were considered statistically significant.
3. RESULTS
3.1. Maturational processing of the β1AR N-terminus; proteolytic cleavage at R31↓L32
A previous study concluded that the β1AR N-terminus contains two major cleavage sites at R31↓L32 and P52↓L53 (Fig 1A) based upon in vitro cleavage assays using short peptides corresponding to the β1AR N-terminal sequence and purified ADAM or MMP enzymes 8. Since the proteolytic cleavage events that can be identified in this type of reductionist assay system may not necessarily reflect proteolytic cleavage events that regulate the maturational processing of the full-length β1AR in cells, we used a mutagenesis strategy to map the sites that are targets for proteolytic cleavage of full-length β1ARs in cells.
Figure 1: R31↓L32 is the major site for N-terminal cleavage during maturational processing of the β1AR in cells.

Panel A, Top: The β1AR N-terminal sequence showing a single N-linked glycosylation site in a consensus Nx(T/S) sequence at position 15 (blue), the two known O-linked glycosylation sites at positions 37 and 41 (red) and the two putative cleavage sites at positions 31-32 and 52-53 (green). Panel A, Bottom: Schematic of the N-terminally Flag-tagged β1AR mutants harboring single residue substitutions at one or both putative cleavage sites used in our studies. Panel B: Lysates prepared from CHO-Pro5 cells or cardiomyocytes (CM) that heterologously overexpress the WT-β1AR or β1AR mutants designed to be cleavage resistant were subjected to immunoblot analysis with the anti-β1AR (raised against a β1AR C-terminal epitope that recognizes both full-length and N-terminally-truncated β1ARs) and anti-FLAG (that recognizes the N-terminal FLAG tag on full-length, but not truncated, β1ARs) antibodies. Positions of the full-length and N-terminally truncated β1ARs are denoted by filled and open triangles, respectively. A representative experiment is shown on top with results from 4 (CHO-Pro5 cell) or 7 (CM) separate experiments on separate culture preparations quantified at the bottom. Data are shown as mean±SEM with % of full-length β1AR (upper band) expressed as a percent of total (full length + truncated) β1AR. Data analysis for CHO-Pro5 cells was by ANOVA with post hoc Tukey’s test for multiple comparisons. Data analysis for CMs was by t-test. In each case, * p<0.05 compared with WT-β1AR.
Fig 1B shows that the WT-β1AR heterologously overexpressed in CHO-Pro5 cells is detected as two major bands on immunoblot analysis. Our previous studies established that the upper ~69KDa band (which is recognized by both an anti-β1AR antibody directed against a β1AR C-terminal epitope and anti-FLAG antibody which recognizes the N-terminal FLAG tag) is the full-length β1AR and the ~55KDa lower band (which is recognized by anti-β1AR antibody, but not by anti-FLAG) is an N-terminally cleaved form of the β1AR 6,7. Of note, while β1AR constructs used in this study carry an N-terminal FLAG tag, the electrophoretic motility patterns of β1AR constructs without the N-terminal epitope tag is quite similar (6 and data not shown).
The electrophoretic mobility patterns for the P52A/L53A-β1AR (harboring substitutions designed to prevent cleavage at the P52↓L53 site) and the WT-β1AR are identical, suggesting either that P52↓L53 is not a physiological cleavage site or that the P52A/L53A substitutions are not sufficient to prevent a 52-53 bond cleavage. In contrast, the R31H/L32A-β1AR (or the β1AR harboring double R31H/L32A-P52A/L53A substitutions) is detected as a single band that co-migrates with the full length WT-β1AR. The observation that a R31H/L32A substitution prevents the formation of the N-terminally cleaved form of the β1AR indicates that R31↓L32 is the major β1AR cleavage site during maturational processing of full-length β1ARs in CHO-Pro5 cells. The additional observation that WT-β1ARs are detected as full-length and N-terminally cleaved forms in cardiomyocytes, and that R31H/L32A-P52A/L53 substitutions prevent β1AR cleavage in cardiomyocytes (Fig 1B), indicates that a similar mechanism controls β1AR maturational processing in cardiomyocytes.
3.2. β1AR N-terminal cleavage is prevented by O-glycosylation
We previously reported that glycan modifications at Ser37 and/or Ser41 prevent β1AR N-terminal cleavage 6. β1AR constructs were heterologously expressed in CHO-IdID cells to determine whether the protection afforded by O-glycosylation is specific for the cleavage event at R31↓L32. CHO-IdID cells are UDP-galactose/UDP-N-acetylgalactosamine 4-epimerase-deficient and therefore cannot synthesize UDP-Gal or UDP-GalNAc, the nucleotide sugars required for the addition of galactose (Gal) and N-acetylgalactosamine (GalNAc) to N- or O-linked oligosaccharides under conventional culture conditions with glucose as the sole sugar source 13. The unique advantage of studies in the CHO-IdID cell line is that the 4-epimerase defect can be bypassed and oligosaccharide synthesis can be fully restored by the addition of Gal and GalNAc to the culture medium.
Figure 2 shows that in the presence of Gal/GalNAc (under conditions that permit glycosylation), WT-β1AR and P52A/L53A-β1AR are detected as both full-length and N-terminally truncated species, whereas β1AR constructs harboring single residue substitutions at the R31↓L32 cleavage site (R31H/L32A and R31H/L32A;P52A/L53A) are recovered as a single band corresponding to the full-length fully-glycosylated receptor. These results indicate that the truncated β1AR species identified in CHO-IdID cells grown in the presence of Gal/GalNAc is formed as a result of N-terminal cleavage at R31↓L32.
Figure 2. β1AR N-terminal cleavage at R31↓L32 is prevented by O-glycosylation; the β1AR N-terminus also contains another glycan-regulated cleavage site.

IdID cells that are not transfected (NT) or transfected with WT-β1AR or β1AR mutants harboring single residue substitutions at putative cleavage sites were cultured with or without Gal (20 μM) and GalNAc (200 μM) as indicated. Lysates were probed with the anti-β1AR antibody that recognizes an epitope on the β1AR C-tail (both full-length and truncated forms of the receptor) and anti-FLAG.
Figure 2 also shows the banding patterns of WT and ‘cleavage-resistant’ β1AR constructs in CHO-IdID cells grown without Gal/GalNAc (under conditions that do not support glycosylation). Under these conditions, WT and P52A/L53A-β1ARs are detected by the β1AR antibody (but not by anti-FLAG) primarily as a ~45kDa, non-glycosylated, N-terminally truncated species. While there is a small amount of a somewhat larger β1AR species generated under these conditions, we previously noted that this likely represents a partially glycosylated species that is produced as a result of a low level of sugar scavenging from serum glycoprotein 6. Importantly, β1AR constructs harboring single residue substitutions at the R31↓L32 cleavage site (the R31H/L32A and R31H/L32A;P52A/L53A-β1ARs mutants) are detected as both a full-length un-glycosylated β1AR species (recognized by both anti-β1AR and anti-FLAG antibodies) and ~45kDa un-glycosylated/N-terminally cleaved species (recognized by anti-β1AR, but not anti-FLAG). The observation that 31H32A and 31H32A/52A53A substituted β1ARs can be cleaved under conditions that prevent glycosylation indicates that the β1AR N-terminus also contains an additional glycan-regulated cleavage site that is distinct from the R31↓L32 site.
3.3. An O-Glycan modification at Ser41 prevents β1-AR cleavage at a site that is distinct from R31↓L32
We previously implicated O-glycan modifications at Ser37 and/or Ser41 as post-translational modifications that prevent β1AR cleavage 6. The previous studies did not parse possible independent effects of O-glycan modifications at position 37 vs. 41. This may be relevant since the Ser41 O-glycosylation site (LVPAS37PPAS41L) is the site that conforms more closely to an MMP consensus cleavage sequence (PxS↓L, the hallmark features of an MMP cleavage site consisting of a Pro in the P3 position, small amino acid such as Gly, Ala, or Ser in the P1 position, and a Leu in the P1’ position – the +1 position C-terminal to the scissile peptide bond 14) and in vitro studies show that a short peptide based residues 23-46 of the β1AR N-terminus is cleaved at the Ser41-Leu42 scissile bond by several MMPs (MMP-2, -7, -8, -9, -12, and -13 8). We generated β1AR mutants harboring single residue substitutions at each position (S37A-β1AR and S41A-β1AR; as schematized in Fig 3A) to test the hypothesis that S41 is the major target for the O-glycan modifications that regulate β1AR N-terminal cleavage in cells.
Figure 3. An O-Glycan modification at S41 prevents β1-AR cleavage at an MMP/ADAM consensus motif at S41↓L42.

Panel A: Schematic of the β1AR mutants harboring single residue substitutions at O-glycosylation sites (in red) or cleavage sites (in green). Note: To simplify the cumbersome nomenclature, the 31H32A/52A53A-β1AR construct is labeled β1AR-31/52 in this and subsequent figures. Panel B: Lysates from CHO-Pro5 cells that were not transfected (NT) or that heterologously overexpress the indicated WT or mutant β1AR constructs were subjected to immunoblot analysis with anti-β1AR and anti-FLAG antibodies. A representative experiment is shown on top with results from 6-14 separate experiments on separate culture preparations quantified at the bottom. Data are shown as mean±SEM, with % of full length β1AR (upper band) expressed as a percent of total (full length + truncated) β1AR. Data analysis was by ANOVA with post hoc Tukey’s test for multiple comparisons. * p<0.05 compared with WT-β1AR; ¤ p<0.05 compared with β1AR-S37A; † p<0.05 compared with β1AR-31/52; # p<0.05 compared with β1AR-31/52-S37A. Panels C and D: Lysates from CHO-Pro5 cells that were not transfected (NT) or that heterologously overexpress the indicated WT or mutant β1AR constructs were subjected to immunoblot analysis with anti-β1AR and anti-FLAG antibodies. The data is representative of results in 5-6 separate experiments. Note: Under certain experimental conditions (as in Panel D) a non-specific band that migrates slightly slower than the full-length β1AR can be detected.
Fig 3B shows that an S37A substitution alone leads to a modest increase in β1AR electrophoretic mobility (the effect being most evident when comparing the mobilities of the N-terminally truncated WT- vs. S37A-β1AR species); the S37A substitution also leads to a significant (albeit modest) 21.7% decrease in the abundance of the full-length β1AR species. These results indicate that the Ser at position 37 is a target for an O-glycan modification, but this O-glycan modification plays a relatively minor role to prevent β1AR N-terminal cleavage. Fig 3 also shows that the S37A substitution results in a small but significant decrease in the abundance of full-length receptors when introduced into the 31H32A/52A53-β1AR backbone (the magnitude of this decrease being ~50% of what is observed when the S37A substitution is introduced into the WT-β1AR backbone). These results support the conclusion that an O-glycan modification at Ser37 plays a significant - albeit minor - role to prevent β1AR N-terminal cleavage at both R31↓L32 and some other cleavage site.
The effect of a single S41A substitution is considerably more striking. Fig 3B shows that the S41A substitution results in a marked increase in the electrophoretic mobility of the N-terminal truncated β1AR species and it results in a profound decrease in the abundance of both full-length WT and 31H32A/52A53A-β1ARs (56.8% and 69.7% respectively). These results identify Ser41 as a major β1AR N-terminal O-glycosylation site and implicate the O-glycan modification at Ser41 as the dominant regulator of β1AR N-terminal cleavage at a site that is distinct from the R31↓L32 cleavage site.
We generated an additional set of mutants in which the S41A substitution was introduced along with single residue substitutions designed to disrupt cleavage at a site adjacent to this residue (with PAS41↓LLP mutated to PAA41AAP) as a strategy to more precisely map this second cleavage site. Fig 3C shows that when the S41LLP sequence is replaced by A41AAP in the 31H32A/52A53A-β1AR backbone, the β1AR becomes cleavage resistant; in contrast, a construct with A41AA substitutions in the 52A53A-β1AR backbone is cleaved similar to the WT-β1AR (data not shown). These results support the conclusion that S41↓L42 is a second β1AR N-terminal cleavage site and that an O-glycan modification at Ser41 prevents cleavage at this second site.
Finally, we generated a β1AR mutant that contains alanine substitutions at both the S41 O-glycosylation site and the adjacent cleavage site - but retains the R31↓L32 cleavage site - to determine whether the O-glycan modification at S41 exerts a more long-range effect on β1AR N-terminal cleavage at R31↓L32. Figure 3D shows that the S41A/L42A/L43-β1AR is detected as both a full-length and N-terminally truncated species, but the percent of the S41A/L42A/L43A-β1AR that is recovered as a full-length species is decreased by 25.6 ±3.2% when compared to the WT-β1AR (p<0.05, n=6). Collectively, these results indicate that O-glycosylation at S41 prevents cleavage at the adjacent S41↓L cleavage site and it also exerts a long-range effect to inhibit cleavage at the more distal R31↓L32 site.
3.4. The β1AR N-terminus is cleaved at R31↓L32 and S41↓L42 by ADAM17
The previous in vitro cleavage assays that identified MMP2, MMP3, and ADAM17 as enzymes that cleave a short peptide based upon residues 23-46 in the β1AR N-terminus at R31↓L32 8 and results reported herein that identify PAS41↓LLP (a sequence that conforms to an MMP consensus cleavage motif) as a bone fide cleavage site on the β1AR N-terminus provided the rationale to test the hypothesis that one or both of the cleavage sites on the N-terminus of the full-length β1AR is a target for cleavage by an MMP family enzyme in cells.
Fig 4A shows that treatment with GM6001 (a pan-MMP inhibitor) leads to increased accumulation of the full length WT-β1AR in association with a decrease in the abundance of the N-terminally truncated species; cleavage resistant 31H32A/52A53A-β1ARs are not influenced by the GM6001 treatment. Similar results were obtained in CHO-Pro5 cells and cardiomyocytes, indicating that the R31↓L32 N-terminal cleavage event that accompanies maturational processing of full-length β1ARs in both CHO-Pro5 cells and cardiomyocytes involves an MMP activity.
Figure 4. The β1AR N-terminus is cleaved at R31↓L32 by ADAM17.

Lysates from CHO-Pro5 cells that heterologously overexpress WT-β1AR or the 31H32A/52A53A-β1AR mutant (labeled β1AR-31/52) that were treated for 24 hrs with vehicle or 10 μM GM6001 (Panel A) or vehicle, 10 μM GM6001, 3 μM ADAM10/17 inhibitor, 400 nM ADAM10 inhibitor, or 20 μM TAPI-2 as indicated (Panel B) were subjected to immunoblot analysis with anti-β1AR and anti-FLAG antibodies. For cardiomyocytes (Panels A and B, right), both Ad-WT-β1AR or Ad-β1AR-31/52 infections and drug treatments were initiated on culture day 1, with lysates prepared for immunoblot analysis on culture day 4. In each panel, a representative experiment is shown on top and data are quantified as described in Figure 3 on the bottom (* p<0.05 compared with vehicle control).
Additional experiments with a panel of compounds specific for individual MMP family enzymes identifies a specific role for ADAM17 in CHO-Pro5 cells. Specifically, the effect of GM6001 to increase the accumulation of full length WT-β1ARs (and decrease the abundance of the N-terminally truncated species) is mimicked by an ADAM10/17 inhibitor and TAPI-2, but not a specific inhibitor of ADAM10 (Fig 4B) or MMP-2/3, -2/9, or 9/13 (data not shown). The effect of GM6001 to increase the accumulation of full length WT-β1ARs (and decrease the abundance of the N-terminally truncated species) also is mimicked by the ADAM10/17 inhibitor in cardiomyocytes (Fig 4B).
We used a similar strategy with β1AR constructs harboring a S41A substitution to identify the enzyme activity that cleaves the β1AR N-terminus at PAS41↓LLP – the site protected by the Ser41 O-glycan modification. Fig 5A shows that the cleavage event facilitated by the Ser41 glycosylation defect in the 31H32A/52A53A-β1AR backbone is blocked by GM6001 and Fig 5B shows that it is also blocked by the ADAM10/17 inhibitor and TAPI-2, but not the specific inhibitor of ADAM10; MMP-2/3, -2/9, or 9/13 inhibitors also did not prevent this cleavage (data not shown). These results indicate that the β1AR N-terminus is a target for cleavage by a single enzyme, namely ADAM17, but the precise site on the β1AR N-terminus that is cleaved by ADAM17 is regulated by the type and position of the O-glycan attachment on the β1AR N-terminus (i.e., by substrate); S41↓L42 cleavage is prevented by an O-glycan modification at Ser41.
Figure 5. The β1AR N-terminus is cleaved at S41↓L42 by ADAM17.

Lysates from CHO-Pro5 cells that heterologously overexpress 31H32A/52A53A-β1AR or 31H32A/52A53A-S41A-β1AR mutants (for simplicity labeled as β1AR-31/52 or β1AR-31/52-S41A) that were treated for 24 hr with vehicle or 10 μM GM6001 (Panel A) or vehicle, 10 μM GM6001, 3 μM ADAM10/17 inhibitor, 400 nM ADAM10 inhibitor, or 20 μM TAPI-2 as indicated (Panel B) were subjected to immunoblot analysis with anti-β1AR and anti-FLAG antibodies.
3.5. The mechanism underlying redox-inactivation of cardiomyocyte β1ARs; N-terminal truncation at R31↓L32 by ADAM17.
We recently showed that oxidative stress (resulting from treatment with either H2O2 or doxorubicin) leads to a decrease in β1AR expression and catecholamine responsiveness in cardiomyocytes 7. The mechanism underlying this form of β1AR regulation remains uncertain. Our previous study showed that the H2O2-dependent decrease in β1AR expression can be prevented by pharmacologic inhibitors of novel protein kinase C (PKC) activity, but not by inhibitors of PKA or Src. 7. A nPKC-regulated mechanism that might contribute to the control cardiomyocyte β1AR expression was not identified. The notion that an N-terminal cleavage event might contribute to redox-inactivation of β1ARs also was not considered. On the basis of literature identifying phorbol-12-myristat-13-acetate (PMA, a pharmacologic activator of PKC) as one of the strongest activators of ADAM17 sheddase activity 15, we examined whether redox-inactivation of β1ARs is attributable to an N-terminal cleavage event mediated by ADAM17.
Fig 6 shows that H2O2 decreases the abundance of full-length WT-β1ARs, but the 31H32A/52A53A-β1AR mutant is not affected by this treatment. The H2O2-dependent decrease in full-length WT-β1AR expression is prevented by GM6001, the ADAM10/17 inhibitor, and TAPI-2. These results implicate an ADAM17-dependent β1AR N-terminal cleavage at R31↓L32 in the mechanism underlying the H2O2-dependent downregulation of full-length β1AR expression in cardiomyocytes.
Figure 6. The mechanism for H2O2-dependent downregulation of cardiomyocyte β1ARs; N-terminal cleavage at R31↓L32 by ADAM17.

Cardiomyocytes were infected with adenoviruses that drive expression of WT-β1AR or the 31H32A/52A53A-β1AR mutant (labeled β1AR-31/52) on culture day 1, pretreated for 48 hr (starting on culture day 3) with vehicle, ADAM10/17 inhibitor (3 μM), TAPI-2 (20 μM), or GM6001 (10 μM) as indicated, and then challenged for 1 hr with 0.1 mM H2O2 on culture day 5. A representation experiment is shown on top, with results for 4 (WT-β1AR) or 3 (β1AR-31/52) separate experiments on separate cardiomyocyte culture preparations quantified (as described in Fig 3) at the bottom. *, p<0.05 compared with β1AR; ¤, p<0.05 compared with H2O2+GM6001-treated β1AR or GM6001-treated β1AR.
Figure 7 shows that doxorubicin treatment leads to a decrease in the abundance of the full-length WT-β1AR. Again, this response is attributable to ADAM17-dependent cleavage at R31↓L32 since it is prevented by 31H32A/52A53A substitutions in the β1AR N-terminus or treatment with GM6001, the ADAM10/17 inhibitor, and TAPI-2.
Figure 7. The mechanism for doxorubicin-dependent downregulation of cardiomyocyte β1ARs; N-terminal cleavage at R31↓L32 by ADAM17.

Infections with adenoviruses that drive expression of WT-β1AR or the 31H32A/52A53A-β1AR mutant (labeled β1AR-31/52) was performed on culture day 1, with treatment with doxorubicin (Dox, 10 uM) in the presence of vehicle, ADAM10/17 inhibitor (3 μM), TAPI-2 (20 μM), or GM6001 (10 μM) initiated on culture day 3 and continued for 48 hrs. Lysates were subjected to immunoblot analysis with anti-β1AR and anti cleaved Caspase-3 antibodies (with actin serving as a loading control). Representative immunoblots are depicted at the top with results from 4 or 5 separate experiments on separate cardiomyocyte culture preparations quantified (as described in Fig 3) at the bottom. * p<0.05 compared with β1AR; ¤ p<0.05 compared with Dox+GM6001-treated β1AR or GM6001-treated β1AR. Results for cleaved caspase-3 in doxorubicin-treated β1AR-31/52 cultures are normalized to values in doxorubicin-treated WT-β1AR cultures. n=4, # p<0.05 compared with Dox-treated WT-β1ARs.
We previously reported that heterologous overexpression of an N-terminally truncated form of the β1AR protects against doxorubicin-induced apoptosis in cardiomyocytes 7. Fig 7 shows that heterologous overexpression of 31H32A/52A53A-β1AR (a construct that cannot be cleaved to form the cardioprotective N-terminally truncated β1AR species) exacerbates doxorubicin-dependent apoptosis (tracked as an increase in caspase 3 cleavage). This result lends further support to the conclusion that β1AR N-terminal truncation constitutes a mechanism that confers protection against doxorubicin-induced apoptosis.
4. DISCUSSION
Cardiomyocyte βARs are among the most intensely studied members of the GPCR superfamily primarily because they control a large number of physiologically important processes that impact on the pathogenesis of cardiovascular disease (and hence constitute clinically important targets for drug discovery). Studies to date implicate cardiomyocyte β1ARs in the activation of cellular responses required for normal physiological control of cardiac contractility as well as responses that contribute to the pathogenesis of cardiac arrhythmias, ventricular remodeling, and the evolution of heart failure. However, our current understanding of the molecular basis for βAR signaling responses derives from literature predicated on the assumption that β1ARs signal exclusively as full-length receptor proteins. While evidence that the β1AR N-terminus can serve as a target for glycosylation and proteolytic cleavage was published over 10 years ago 16, the evidence that an O-glycan-regulated cleavage event leads to the generation of distinct molecular species that differ in their coupling to downstream effector responses is very recent 6,7. We showed that β1ARs are detected as both full-length and N-terminally truncated species in model cell lines and cardiomyocytes, that N-terminal cleavage is prevented by O-glycan modifications at Ser37 and/or Ser41 (whereas the N-glycan attachment at Asp15 does not influence β1AR N-terminal cleavage), and that the N-terminally truncated form of the β1AR constitutively activates a Gi-AKT pathway that confers protection against doxorubicin-induced apoptosis 6,7. This study extends the analysis by mapping the precise proteolytic cleavage sites on the β1AR N-terminus and the protease activity responsible for β1AR N-terminal processing in cardiomyocytes.
This study identifies R31↓L32 as the major cleavage site on the β1AR N-terminus; the N-terminally truncated β1AR species generated during the maturational processing of β1ARs in both CHO cells and cardiomyocytes results from cleavage at R31↓L32. While studies in IdID cells indicate that a glycan modification prevents cleavage at this site, the nature of this control remains uncertain. The observation that β1ARs are expressed as a heterogeneous population of both full-length and N-terminally truncated receptor species could suggest that β1ARs (like most glycoproteins) are synthesized as glycoforms – that inherent inefficiencies in the enzymatic pathways for glycoprotein biosynthesis results in the generation of β1ARs variants that differ either in the sites that carry glycan attachments or the composition of the glycan attachment at any individual glycosylation site. Insofar as there is likely to be some specificity in glycan-regulation of β1AR cleavage, this heterogeneity in β1AR O-glycosylation could lead to the generation of receptor subpopulations that differ in their susceptibility to cleavage at the R31↓L32 scissile bond. In this context, chamber-specific differences in glycosylation-associated gene (glycogene) expression and the cardiac glycome 10 (which would result in a single protein, identical in sequence, displaying different O-glycosylation patterns in different cells) could underlie our previous observation that atrial tissue is relatively enriched in the N-terminally truncated form of the β1AR compared to ventricular tissue 6. Our studies also establish that R31↓L32 is not the only glycan-regulated cleavage site on the β1AR N-terminus. The observation that the R31H/L32A-P52A/L53A mutant (that is resistant to cleavage at the R31↓L32 site) is cleaved in IdID cells grown in the absence of Gal/GalNAc provided the rationale for additional studies that used a mutagenesis approach to map a second cleavage site on the β1AR N-terminus to S41↓L42. Of note, cleavage at this site (which is C-terminal to the serine residue identified in this study as the major β1AR N-terminal O-glycosylation site) is completely abrogated by the O-glycan modification at S41.
In vitro studies suggest that the β1AR N-terminus contains as many as four potential cleavage sites that are targets for cleavage by many different MMP and ADAM family enzymes - and that individual sites show differential sensitivities to cleavage by various MMP and ADAM enzymes 8. However, our cell-based experiments indicate that β1AR cleavage is restricted to two sites, R31↓L32 and S41↓L42, that cleavage at both sites requires ADAM17 activity, and that the precise cleavage product generated from the full-length β1AR is influenced by an O-glycan attachment at S41. Of note, our studies assessed the consequences of S41A substitution that completely eliminates O-glycosylation at position 41. It is tempting to speculate that metabolic derangements associated with diabetes or various disease-induced changes in glycogene expression could lead to more subtle changes in β1AR N-terminal O-glycosylation and that this might be sufficient to enhance β1AR N-terminal cleavage at S41↓L42. While there is considerable evidence that glycosylation patterns change in parallel with changes in cellular metabolism in certain cancers (with these altered glycosylation patterns on certain serum or tumor tissue proteins serving as a biomarkers for patient prognosis or response to treatment 17), the notion that the metabolic derangements associated with diabetes also lead to changes in protein O-glycosylation (either globally on membrane proteins in general or specifically on the β1AR) has never been considered.
This study shows that cleavage at both R31↓L32 and S41↓L42 requires ADAM17 activity. The classical paradigm for GPCR signaling places MMP and ADAM family enzymes downstream from GPCR activation in the mechanism for GPCR crosstalk to epithelial growth factor receptors. According to this model, an agonist-activated GPCR stimulates an MMP activity that cleaves an EGFR ligand (such as heparin binding EGF, transforming growth factor α, or ampiregulin) from a membrane precursor, resulting in ligand-mediated autocrine and/or paracrine transactivation of EGFRs. This form of cross-talk has been described for cardiac β1ARs, where a β1AR-dependent EGFR transactivation pathway (involving MMP-dependent release of HB-EGF) confers cardioprotection under conditions of catecholamine toxicity 4,18. Of note, ADAM17 is one of many MMP/ADAM family enzymes that have been implicated in various GPCR-EGFR transactivation pathways 19, but the precise MMP or ADAM family enzyme(s) that acts downstream from the agonist-activated cardiomyocyte β1AR has never been identified. Our study identifies a relationship between the β1AR and ADAM17, but this involves a novel upstream role for ADAM17 in the maturational processing of cardiac β1ARs. While we did not identify a mechanism to account for the specificity in β1AR processing by ADAM17 (and not other MMP or ADAM family enzymes), it is worth noting that the mature form of ADAM17 is reported to localize to cholesterol-enriched membrane subdomains (lipid rafts)20-22. An ADAM17 sheddase activity sequestered in lipid rafts would be in close proximity to the cardiomyocyte β1AR, which also localizes to this subcellular compartment 23. Finally, this study implicates ADAM17 as the enzyme that cleaves the β1AR N-terminus. Studies to determine whether ADAM17 plays a dual role in both the maturational processing of β1ARs and their downstream signaling to EGFR transactivation, or whether these actions are mediated by distinct MMP/ADAM family members that can be pharmacologically discriminated, are the focus of ongoing studies.
We previously reported that oxidative stress leads to β1AR downregulation and decreased catecholamine responsiveness in cardiomyocytes. Our previous studies implicated a nPKC activity in this mechanism, but the exact process underlying the redox-dependent loss in β1AR responsiveness was not identified. This study shows that redox-dependent β1AR downregulation is due to β1AR N-terminal cleavage at R31↓L32 by ADAM17. Several obvious molecular mechanisms might contribute to redox-induced ADAM17-dependent β1AR cleavage and should be considered. First, there is evidence that ADAM17 activity is tightly regulated through changes in its subcellular localization. The bulk of the mature ADAM17 enzyme localizes to an intracellular compartment under basal conditions; only a small pool of the mature enzyme is available at the cell surface 24. The observation that PKC activation leads to an increase in ADAM17 expression and activity at the cell surface 24 (in the context of our previous observation that the H2O2-dependent decrease in β1AR expression requires nPKC activity) could suggest that a ROS-activated nPKC isoform regulates ADAM17 trafficking, enhances its cell surface expression, and thereby triggers β1AR downregulation. Second, ADAM17 is a redox-sensitive enzyme; it contains two evolutionarily conserved cysteinyl sulfhydryl groups in its extracellular disintegrin domain that serve as redox-sensors. The observation that oxidation of these cysteines is required for maximal ADAM17 activity 25 provides a rationale for future studies that focus on whether β1AR cleavage is due to a redox-dependent increase in ADAM17 activity (as has been described in various other cells types 26-28). Finally, while the precise structural requirements for ADAM17 substrate recognition remain uncertain, a redox-dependent change in β1AR secondary structure (or its O-glycosylation) could in theory serve to render the β1AR N-terminus a better substrate for ADAM17-dependent cleavage.
Finally, this study builds upon recent evidence that β1ARs are one of a small number of GPCRs that undergo limited N-terminal proteolysis (i.e., ectodomain shedding). In the case of the β1AR, we previously showed that N-terminal proteolysis provides a mechanism to regulate coupling to downstream signaling pathways; N-terminally truncated (but not full-length) β1ARs constitutively couple to a cardioprotective AKT signaling pathway that protects against doxorubicin-dependent apoptosis 7. Results reported herein showing that heterologous overexpression of a cleavage-resistant form of the β1AR (a maneuver that prevents the formation of the cardioprotective N-terminally truncated β1AR species) exacerbates doxorubicin-dependent apoptosis are consistent with this formation. The observation that β1AR ectodomain cleavage is specifically mediated by ADAM17 exposes a novel role for this enzyme in the control of cardiomyocyte β1AR responsiveness. ADAM17 (or TACE, TNFα converting enzyme) was first identified as a membrane-anchored metalloprotease that releases bioactive TNFα from cells; it subsequently was characterized as a sheddase for a large number of membrane-anchored growth factors, adhesion molecules, cytokines and cytokine receptors that contribute to the pathogenesis of a wide range of clinical disorders, including cancers, various inflammatory states, and neurodegenerative disorders 29. However, the notion that an ADAM17 inhibitor might be useful in the treatment of these disorders must be balanced by a consideration of ADAM17’s cardiac action. Early studies established that ADAM17 is critical for normal heart development and valvulogenesis and more recent studies show that ADAM17 is expressed in the adult heart, its expression is increased in cardiac tissue from patients with myocarditis or dilated cardiomyopathy 30,31, that cardiomyocyte-specific ADAM17 expression is required for post-ischemic angiogenesis and optimal post-MI myocardial repair 32, and that ADAM17 expression affords protection in the setting of pressure overload 33. Our studies identify yet another cardioprotective role for ADAM17, as the enzyme required for the generation of cardioprotective N-terminally truncated β1ARs. This raises yet an additional concern that therapies that inhibit ADAM17 activity for the treatment of cancer or inflammation may have adverse effects in the heart and that off-target cardiotoxicity might limit their usefulness in the clinic.
Highlights:
The β1AR N-terminus functions as a structural determinant of β1AR responses in cardiomyocytes.
The β1AR N-terminus is cleaved at R31↓L32 by ADAM17.
β1ARs contain a second O-glycan-regulated ADAM17-dependent N-terminal cleavage site at S41↓L42.
β1AR inactivation during oxidative stress is due to ADAM17-dependent cleavage at R31↓L32.
N-terminally truncated β1ARs afford protection against doxorubicin-dependent apoptosis
Acknowledgements:
The authors thank HaeJung Chung for technical assistance.
Funding Source: This work is supported by the National Institutes of Health, National Heart, Blood, and Lung Institute grant HL138468.
Footnotes
Declaration of competing interest: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Wang J, Gareri C, Rockman HA. G-protein-coupled receptors in heart disease. Circ Res. 2018; 123: 716–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinberg SF. Beta1-adrenergic receptor regulation revisited. Circ Res. 2018; 123: 1199–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Lucia C, Eguchi A, Koch WJ. New insights in cardiac β-adrenergic signaling during heart failure and aging. Front Pharmacol. 2018; 9: 904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ, Rockman HA. β-Arrestin-mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest. 2007; 117: 2445–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warne T, Edwards PC, Leslie AG, Tate CG. Crystal structures of a stabilized β1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure. 2012; 20: 841–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park M, Reddy GR, Wallukat G, Xiang YK, Steinberg SF. β1-adrenergic receptor O-glycosylation regulates N-terminal cleavage and signaling responses in cardiomyocytes. Sci Rep. 2017; 7: 7890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park M, Steinberg SF. Carvedilol prevents redox inactivation of cardiomyocyte β1-adrenergic receptors. JACC Basic Transl Sci. 2018; 3: 521–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goth CK, Tuhkanen HE, Khan H, Lackman JJ, Wang S, Narimatsu Y, Hansen LH, Overall CM, Clausen H, Schjoldager KT, Petäjä-Repo UE. Site-specific O-glycosylation by polypeptide N-acetylgalactosaminyltransferase 2 (GalNAc-transferase T2) co-regulates β1-adrenergic receptor N-terminal cleavage. J Biol Chem. 2017; 292: 4714–4726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rong J, Han J, Dong L, Tan Y, Yang H, Feng L, Wang Q, Meng R, Zhao J, Wang S, Chen X. Glycan imaging in intact rat hearts and glycoproteomic analysis reveal the upregulation of sialylation during cardiac hypertrophy. J Am Chem Soc. 2014; 136: 17468–17476 [DOI] [PubMed] [Google Scholar]
- 10.Montpetit ML, Stocker PJ, Schwetz TA, Harper JM, Norring SA, Schaffer L, North SJ, Jang-Lee J, Gilmartin T, Head SR, Haslam SM, Dell A, Marth JD, Bennett ES. Regulated and aberrant glycosylation modulate cardiac electrical signaling. Proc Natl Acad Sci USA. 2009; 106: 16517–16522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinberg SF, Robinson RB, Lieberman HB, Stern DM, Rosen MR. Thrombin modulates phosphoinositide metabolism, cytosolic calcium, and impulse initiation in the heart. Circ Res. 1991; 68: 1216–1229 [DOI] [PubMed] [Google Scholar]
- 12.Rybin VO, Sabri A, Short J, Braz JC, Molkentin JD, Steinberg SF. Cross-regulation of novel protein kinase C (PKC) isoform function in cardiomyocytes. Role of PKC epsilon in activation loop phosphorylations and PKC delta in hydrophobic motif phosphorylations. J Biol Chem. 2003; 278: 14555–14564 [DOI] [PubMed] [Google Scholar]
- 13.Kozarsky K, Kingsley D, Krieger M. Use of a mutant cell line to study the kinetics and function of O-linked glycosylation of low density lipoprotein receptors. Proc Natl Acad Sci USA. 1988; 85: 4335–4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eckhard U, Huesgen PF, Schilling O, Bellac CL, Butler GS, Cox JH, Dufour A, Goebeler V, Kappelhoff R, Keller UAD, Klein T, Lange PF, Marino G, Morrison CJ, Prudova A, Rodriguez D, Starr AE, Wang Y, Overall CM. Active site specificity profiling of the matrix metalloproteinase family: Proteomic identification of 4300 cleavage sites by nine MMPs explored with structural and synthetic peptide cleavage analyses. Matrix Biol. 2016; 49: 37–60 [DOI] [PubMed] [Google Scholar]
- 15.Horiuchi K, Le Gall S, Schulte M, Yamaguchi T, Reiss K, Murphy G, Toyama Y, Hartmann D, Saftig P, Blobel CP. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell. 2007; 18: 176–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hakalahti AE, Vierimaa MM, Lilja MK, Kumpula EP, Tuusa JT, Petäjä-Repo UE. Human β1-adrenergic receptor is subject to constitutive and regulated N-terminal cleavage. J Biol Chem. 2010. 285: 28850–28861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reily C, Stewart TJ, Renfrow MB, Novalk J. Glycosylation in health and disease. Nat Rev Nephrol. 2019; 15: 346–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grisanti LA, Talarico JA, Carter RL, Yu JE, Repas AA, Radcliffe SW, Tang HA, Makarewich CA, Houser SR, Tilley DG. β-Adrenergic receptor-mediated transactivation of epidermal growth factor receptor decreases cardiomyocyte apoptosis through differential subcellular activation of ERK1/2 and Akt. J Mol Cell Cardiol. 2014; 72: 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hart S, Fischer OM, Prenzel N, Zwick-Wallasch E, Schneider M, Hennighausen L, Ullrich A. GPCR-induced migration of breast carcinoma cells depends on both EGFR signal transactivation and EGFR-independent pathways. Biol Chem. 2005; 386: 845–855 [DOI] [PubMed] [Google Scholar]
- 20.Tellier E, Canault M, Rebsomen L, Bonardo B, Juhan-Vague I, Nalbone G, Peiretti F. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp Cell Res. 2006; 312: 3969–3980 [DOI] [PubMed] [Google Scholar]
- 21.Ebsen H, Lettau M, Kabelitz D, Janssen O. Subcellular localization and activation of ADAM proteases in the context of FasL shedding in T lymphocytes. Mol Immunol. 2015; 65: 416–428 [DOI] [PubMed] [Google Scholar]
- 22.Takaguri A, Shirai H, Kimura K, Hinoki A, Eguchi K, Carlile-Klusacek M, Yang B, Rizzo V, Eguchi S. Caveolin-1 negatively regulates a metalloprotease-dependent epidermal growth factor receptor transactivation by angiotensin II. J Mol Cell Cardiol. 2011; 50: 545–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rybin VO, Xu X, Lisanti MP, Steinberg SF. Differential targeting of β-adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the cAMP signaling pathway. J Biol Chem. 2000; 275: 41447–41457 [DOI] [PubMed] [Google Scholar]
- 24.Lorenzen I, Lokau J, Korpys Y, Oldefest M, Flynn CM, Künzel U, Garbers C, Freeman M, Grötzinger J, Düsterhöft S. Control of ADAM17 activity by regulation of its cellular localisation. Sci Rep. 2016; 6: 35067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Herrera AH, Li Y, Belani KK, Walcheck B. Regulation of mature ADAM17 by redox agents for L-selectin shedding. J Immunol. 2009; 182: 2449–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brill A, Chauhan AK, Canault M, Walsh MT, Bergmeier W, Wagner DD. Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc Res. 2009; 84: 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pietri M, Schneider B, Mouillet-Richard S, Ermonval M, Mutel V, Launay JM, Kellermann O. Reactive oxygen species-dependent TNF-alpha converting enzyme activation through stimulation of 5-HT2B and alpha1D autoreceptors in neuronal cells. FASEB J. 2005; 19: 1078–1087 [DOI] [PubMed] [Google Scholar]
- 28.Zhang Z, Oliver P, Lancaster JR Jr, Schwarzenberger PO, Joshi MS, Cork J, Kolls JK. Reactive oxygen species mediate tumor necrosis factor alpha-converting, enzyme-dependent ectodomain shedding induced by phorbol myristate acetate. FASEB J. 2001; 15: 303–305 [DOI] [PubMed] [Google Scholar]
- 29.van der Vorst EP, Keijbeck AA, de Winther MP, Donners MM. A disintegrin and metalloproteases: molecular scissors in angiogenesis, inflammation and atherosclerosis. Atherosclerosis. 2012; 224: 302–308 [DOI] [PubMed] [Google Scholar]
- 30.Satoh M, Nakamura M, Satoh H, Saitoh H, Segawa I, Hiramori K. Expression of tumor necrosis factor-alpha--converting enzyme and tumor necrosis factor-alpha in human myocarditis. J Am Coll Cardiol. 2000; 36: 1288–1294 [DOI] [PubMed] [Google Scholar]
- 31.Satoh M, Nakamura M, Saitoh H, Satoh H, Maesawa C, Segawa I, Tashiro A, Hiramori K. Tumor necrosis factor-alpha-converting enzyme and tumor necrosis factor-alpha in human dilated cardiomyopathy. Circulation. 1999; 99: 3260–3265 [DOI] [PubMed] [Google Scholar]
- 32.Fan D, Takawale A, Shen M, Wang W, Wang X, Basu R, Oudit GY, Kassiri Z. Cardiomyocyte a disintegrin and metalloproteinase 17 (adam17) is essential in post-myocardial infarction repair by regulating angiogenesis. Circ Heart Fail. 2015; 8: 970–979 [DOI] [PubMed] [Google Scholar]
- 33.Fan D, Takawale A, Shen M, Samokhvalov V, Basu R, Patel V, Wang X, Fernandez-Patron C, Seubert JM, Oudit GY, Kassiri Z. A disintegrin and metalloprotease-17 regulates pressure overload-induced myocardial hypertrophy and dysfunction through proteolytic processing of integrin β1. Hypertension. 2016; 68: 937–948 [DOI] [PubMed] [Google Scholar]