

ABSTRACT
Many cytosolic bacterial pathogens hijack the host actin polymerization machinery to form actin tails that promote direct cell-to-cell spread, enabling these pathogens to avoid extracellular immune defenses. However, these pathogens are still susceptible to intracellular cell-autonomous immune responses that restrict bacterial actin-based motility. Two classes of cytosolic antimotility factors, septins and guanylate-binding proteins (GBPs), have recently been established to block actin tail formation by the human-adapted bacterial pathogen Shigella flexneri. Both septin cages and GBP1 microcapsules restrict S. flexneri cell-to-cell spread by blocking S. flexneri actin-based motility. While septins assemble into cage-like structures around immobile S. flexneri, GBP1 forms microcapsules around both motile and immobile bacteria. The interplay between these two defense programs remains elusive. Here, we demonstrate that GBP1 microcapsules block septin cage assembly, likely by interfering with the function of S. flexneri IcsA, the outer membrane protein that promotes actin-based motility, as this protein is required for septin cage formation. However, S. flexneri that escape from GBP1 microcapsules via the activity of IpaH9.8, a type III secreted effector that promotes the degradation of GBPs, are often captured within septin cages. Thus, our studies reveal how septin cages and GBP1 microcapsules represent complementary host cell antimotility strategies.
Keywords: guanylate-binding protein, septin, actin, Shigella flexneri, GBP1, interferon
Guanylate-binding protein 1 microcapsules and septin cages are complementary antimotility factors targeting the enteric pathogen Shigella flexneri.
INTRODUCTION
Microbial pathogens escape extracellular immune responses and cell-autonomous immunity by a variety of evasion strategies, which include hijacking the actin cytoskeleton to promote inter- and intracellular motility. To promote actin-based motility, pathogens evolved different strategies to hijack the host actin polymerization machinery (Welch and Way 2013; Lamason and Welch 2017; Koseoglu and Agaisse 2019; Dowd, Mortuza and Ireton 2021). Actin-based motility of the human-adapted bacterial pathogen Shigella flexneri depends on IcsA, a polar outer membrane protein. IcsA binds to and activates proteins involved in actin polymerization, leading to the formation of comet-like actin structures, referred to as actin tails (Bernardini et al. 1989; Goldberg et al. 1993; Suzuki et al. 1998; Egile et al. 1999; Mauricio et al. 2017).
The force of polymerizing actin propels S. flexneri through the cytosol and enables the formation of bacteria-containing host cell plasma membrane protrusions. These protrusions are engulfed by neighboring cells enabling S. flexneri to spread directly from cell to cell, thus evading extracellular immune defenses while disseminating throughout the colonic epithelium (Agaisse 2016). In order to contain S. flexneri infections, the host requires effective cell-autonomous immune programs targeting cytosolic bacteria. At least two classes of cytosolic proteins interact with S. flexneri inside infected epithelial cells and interfere with actin tail formation: interferon (IFN)-inducible guanylate-binding proteins (GBPs) and ubiquitously expressed septins.
GBPs are dynamin-related large GTPases that show broad antimicrobial functions against viral, protozoan and bacterial pathogens (Daumke and Praefcke 2016; Ngo and Man 2017; Praefcke 2018; Gomes et al. 2019; Kutsch and Coers 2020). Gram-negative bacteria are specifically recognized by the founding member of the GBP family, GBP1, which binds directly to bacterial lipopolysaccharide (LPS) (Kutsch et al. 2020; Santos et al. 2020). LPS consists of a lipid A moiety, inner and outer core sugars, and the O-antigen polysaccharide. LPS molecules are anchored via their lipid A moieties in the bacterial outer membrane, whereas their outward-facing O-antigens form a chemophysical barrier (Simpson and Trent 2019; Giordano, Cian and Dalebroux 2020). Polymerizing GBP1 binds to LPS and forms a uniform protein coat that encapsulates the bacterial cell. This GBP1 microcapsule acts as a surfactant emulsifying the protective LPS barrier and thus interferes with the integrity of the bacterial envelope (Kutsch et al. 2020).
The LPS barrier not only protects Gram-negative bacteria from antimicrobials but also reduces outer membrane fluidity, mainly through lateral interactions of O-antigen side chains (Herrmann et al. 2015; Rojas et al. 2018). The resulting decreased diffusion of outer membrane proteins helps to position IcsA at one bacterial pole, an important prerequisite for efficient actin tail formation and cell-to-cell spread by S. flexneri (Robbins et al. 2001). Furthermore, direct interaction of IcsA with O-antigen has been proposed to favor an IcsA conformation required for interactions with the host actin polymerization machinery (Van den Bosch and Morona 2003). GBP1 blocks S. flexneri actin tail formation and tethers bacteria to immobilized clusters, ultimately blocking bacterial dissemination (Piro et al. 2017; Wandel et al. 2017). Mechanistically, GBP1 as an LPS-binding surfactant interferes with the polar localization and function of IcsA (Kutsch et al. 2020). Shigella flexneri can evade GBP1-mediated immunity via the action of IpaH9.8, an E3 ubiquitin ligase, an effector that is directly secreted via its type III secretion system into the cytosol of infected cells. IpaH9.8 promotes the modification of GBPs, including GBP1, with K48-linked polyubiquitin chains that target them for proteasomal degradation (Li et al. 2017; Piro et al. 2017; Wandel et al. 2017).
Septins are GTP-binding proteins that assemble into non-polar filaments as well as higher order structures on the inner surface of eukaryotic plasma membranes. Septins associate with the actin and microtubule cytoskeleton and play a central role in cell division, membrane remodeling and maintenance of cell polarity (Valadares et al. 2017; Addi, Bai and Echard 2018; Woods and Gladfelter 2020). More recently, septins have been characterized as important executioners of cell-autonomous immunity (Van Ngo and Mostowy 2019; Robertin and Mostowy 2020). Septins, including the ubiquitously expressed Septin 7 (Sept7), form cage-like structures around bacterial and viral pathogens to facilitate antimicrobial autophagy and to block microbial actin-based dissemination (Mostowy et al. 2010; Sirianni et al. 2016; Pfanzelter, Mostowy and Way 2018). Septins bind to anionic lipids present in the outer membrane of immobilized S. flexneri, which leads to the assembly of septin cages in an actin polymerization-dependent manner (Mostowy et al. 2010; Krokowski et al. 2018). Septins also form cage-like structures around bacterial actin tails, yet fail to trap and immobilize already motile bacteria (Mostowy et al. 2010). In contrast to GBPs, septins are not known to be targeted by bacterial E3 ubiquitin ligases. However, S. flexneri is able to break out of its surrounding septin cage, to regain motility, and to escape septin-mediated host defenses by a poorly defined mechanism (Mostowy et al. 2010).
Both GBP1 and septins efficiently block S. flexneri actin-based motility and restrict cell-to-cell spread. Functional interactions between these antimotility factors have not been reported previously. Here, we demonstrate that the assembly of GBP1 microcapsules and septin cages around cytosolic S. flexneri occurs independently. However, we find that GBP1 microcapsules block IcsA-dependent assembly of septin cages. Accordingly, the frequency of septin cage formation is significantly reduced for bacteria that lack bacterially secreted GBP1 inhibitor IpaH9.8 and are therefore efficiently encapsulated by GBP1. Together, our study reveals that GBP1 microcapsules and septin cages function as complementary defense programs to restrict S. flexneri actin tail formation.
MATERIALS AND METHODS
Cell lines
GBP1-deficient HeLa cells (HeLaGBP1-KO) were previously described (Piro et al. 2017). HeLaGBP1-KO cells were stably transduced with an anhydrotetracycline (aTc)-inducible gene expression system to drive the expression of mCherry-GBP1. HeLaGBP1-KO cell lines and parental wild-type HeLa cells (Piro et al. 2017) were cultivated in Dulbecco's modified Eagle medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Corning, Corning, NY, USA), 1% non-essential amino acids (Gibco, Thermo Fisher Scientific) and 55 μM β-mercaptoethanol (Gibco, Thermo Fisher Scientific) at 37°C and 5% CO2.
Bacterial strains and culture conditions
Shigella flexneri 2547T strains were grown on tryptic soy broth (TSB) agar supplemented with Congo Red and antibiotics, as needed (50 µg/ml carbenicillin, 30 µg/ml kanamycin), for 12–16 h at 37°C. For infection experiments, overnight cultures of S. flexneri were diluted 1:30 in fresh TSB and grown for an additional 1–1.5 h to an OD600 of 0.3–0.7. Bacteria were then harvested at 2400 × g for 3 min, washed once with 1× phosphate buffered saline (PBS) (Gibco, Thermo Fisher Scientific) and treated with 100 ng/ml poly-d-lysin hydrobromide (Millipore, Sigma-Aldrich, Burlington , MA, USA) in 1× PBS for 15 min to enhance infectivity.
Shigella flexneri strain construction
Shigella flexneri ΔipaH9.8 was previously reported (Piro et al. 2017). To generate S. flexneri ΔipaH9.8ΔicsA, the KANR cassette from ∆ipaH9.8::FRT-KANR-FRT (Piro et al. 2017) was removed by expressing Flp recombinase via cell transduction with plasmid pCP20 (Cherepanov and Wackernagel 1995). Next, the FRT::KANR::FRT from ∆icsA::FRT-KANR-FRT was PCR amplified and introduced into ∆ipaH9.8 using the lambda red recombination system (Datsenko and Wanner 2000). To transform S. flexneri ΔipaH9.8ΔicsA with a fluorescent plasmid, bacteria were prepared for electroporation and transformed with pEGFPmut2 (Cormack, Valdivia and Falkow 1996), as described previously (Warren 2011).
Bacterial cell culture infections
Cell lines were cultivated on glass coverslips in 24-well plates and were either treated with 200 U/ml IFNγ (Millipore, Sigma-Aldrich) or 1–2 µg/ml aTc (Takara), or left untreated for 20–22 h. Cells were then infected at a multiplicity of infection (MOI) of 6. Poly-d-lysine was used to coat bacteria and thereby enhance bacterial attachment to host cells, as previously described (Enninga et al. 2005). Poly-d-lysine-treated bacteria were resuspended in prewarmed cell culture media and spun on cells for 10 min at 700 × g. Infected cells were incubated for 45 min at 37°C and 5% CO2. Following two washes with 1× Hanks' Balanced Salt Solution (Gibco, Thermo Fisher Scientific), cells were incubated with cell culture media containing 25 µg/ml gentamycin for 2.5 h at 37°C and 5% CO2. At this point, cells were fixed with 4% formaldehyde at room temperature (RT) and washed three times with 1× PBS for immunofluorescence staining.
Immunofluorescence microscopy
Endogenous GBP1 and Sept7 were stained with rabbit monoclonal anti-GBP1 immunoglobulin G (IgG) (Abcam; ab131255, diluted 1:150) or rabbit anti-human Septin 7 (C) IgG (IBL America; catalog no. 18 991, 5 µg/ml final concentration). Cells were permeabilized with 0.25% Triton X-100 in 1× PBS for 10 min at RT and washed three times with 1× PBS. Permeabilized cells were blocked with blocking buffer (1× PBS, 50 mg/ml BSA, 300 mM glycine) for at least 45 min. Cells were treated with primary antibodies diluted in blocking buffer at 4°C overnight. Following incubation with primary antibody, cells were washed with 0.05% Triton X-100 in 1× PBS three times for 5 min and then treated with 1:1000 dilution of anti-rabbit IgG conjugated with Alexa Fluor 568 or Alexa Fluor 660 in blocking buffer for 1 h at RT. F-actin was stained with Alexa Fluor 660 phalloidin (Thermo Fisher Scientific) diluted 1:40 in blocking buffer for 1 h at RT. Cells were washed three times with 0.05% Triton X-100 for 5 min and then mounted on microscopy slides with mounting media (100 mM Tris–HCl, pH 8.5, 25% glycerol, 125 µg/ml Mowiol) diluted 9:1 with 0.1 mg/ml para-phenylenediamine antifading agent. Processed slides were imaged with either a Leica STED and Confocal on Leica DMi8 motorized inverted microscope using a Zeiss Plan-Apochromat 40×/1.3 oil objective or a Zeiss 880 AiryScan Fast Inverted Confocal on AxioObserver Z1 microscope using a Zeiss Plan-Apochromat 63×/1.4 oil objective. Z-stacks were acquired with an interval of 0.5 µm. Images were processed with Fiji.
RESULTS
Both GBP1 encapsulation and septin caging block S. flexneri actin tail formation, but their functional relationship has remained unexplored (Fig. 1A). GBP1-mediated inhibition of actin-based motility is inhibited by S. flexneri IpaH9.8. Confirming previous reports (Li et al. 2017; Piro et al. 2017; Wandel et al. 2017), we observed that a S. flexneri mutant lacking IpaH9.8, ΔipaH9.8 S. flexneri, is more frequently enveloped by IFNγ-induced GBP1 protein than wild-type bacteria (Fig. 1B). Affirming the antimotility effect of the GBP1 microcapsule, we detected substantially reduced actin tail formation by GBP1-susceptible ΔipaH9.8 compared with wild-type S. flexneri at 2.5 h post-infection (hpi) (Fig. 1C and D; Figs S1 and S2, Supporting Information). Actin tail formation by ΔipaH9.8 S. flexneri is restored in GBP1-deficient cells (Fig. 1C and D; Fig. S2, Supporting Information), demonstrating that the IFNγ-induced loss of actin-based motility by a ΔipaH9.8 S. flexneri mutant is due to an effective GBP1-mediated cell-autonomous host defense pathway.
Figure 1.

IFNγ priming reduces septin caging in a GBP1-dependent manner. (A) Septins and GBP1 block S. flexneri actin tail formation by entrapping the bacteria in septin cages or GBP1 microcapsules. (B) HeLa cells either left untreated or treated with IFNγ to stimulate GBP1 expression were infected with either S. flexneri wild type or coisogenic S. flexneri ΔipaH9.8 for 2.5 h, and immunostained for endogenous GBP1. Bacteria targeted by GBP1 were quantified. (C–E) HeLa wild-type and GBP1-deficient (HeLaGBP1-KO) cells were either left untreated or treated with IFNγ and infected for 2.5 h with S. flexneri wild type or ΔipaH9.8 (only representative images of ΔipaH9.8 infections are shown in panel C), and immunostained for filamentous actin and endogenous Sept7. Bacteria with actin tails (D) and Sept7 cages (E) were quantified. (B, D, E) Mean frequencies ± SEM (standard error of the mean) of combined data from three independent experiments are shown. Significance was determined by two-way ANOVA with Tukey's multiple comparison test. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. Actin tails were classified as tails when ≥2.5 µm. Arrows point at actin tails; arrow heads point at septin cages. All scale bars equal 5 µm.
Since the surfactant activity of GBP1 promotes the recruitment of other host factors such as lipid A-binding caspase-4 to the bacterial surface (Fisch et al. 2019, 2020; Kutsch et al. 2020; Santos et al. 2020; Wandel et al. 2020), we hypothesized that GBP1 microcapsules could similarly promote the recruitment of septins to S. flexneri, possibly through the unmasking of bacterial outer membrane-resident anionic lipids such as cardiolipin that were previously shown to serve as septin-binding substrates (Krokowski et al. 2018). To test this hypothesis, we stained cells with antibodies against Sept7, a septin cage subunit required for the incorporation of the additional septins Sept2 and Sept9 (Sirianni et al. 2016). We observed that the frequency of septin cage formation around the ΔipaH9.8 S. flexneri was not increased but rather diminished in IFNγ-primed wild-type cells, thus refuting our initial hypothesis. Furthermore, we found that Sept7 cage formation around the S. flexneri ΔipaH9.8 mutant was restored in GBP1-deficient cells (Fig. 1C and E). Together, these results strongly indicated that, instead of promoting septin cage assembly, GBP1 microcapsules block septin cage formation around S. flexneri. To further test this hypothesis, we ectopically expressed mCherry-GBP1 and monitored co-localization of GBP1 with Sept7 cages. We found that bacteria staining positive for GBP1 mostly lacked Sept7 cages (Fig. 2), lending further support that GBP1 microcapsules surrounding cytosolic S. flexneri impede septin cage assembly.
Figure 2.
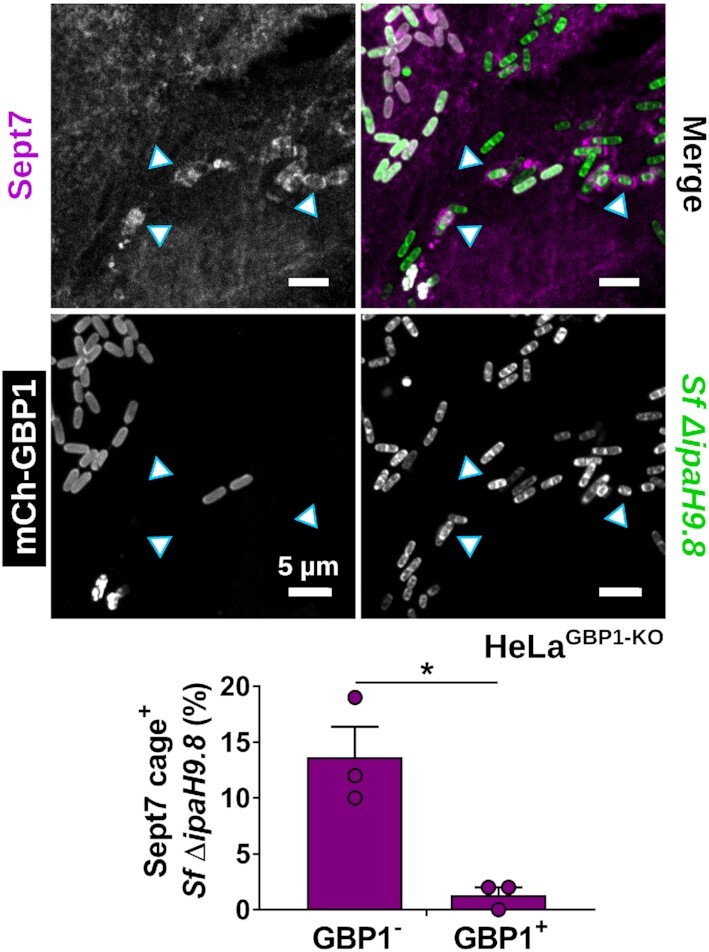
GBP1 microcapsules around S. flexneri interfere with septin caging. HeLaGBP1-KO cells stably expressing mCherry-GBP1 were infected with S. flexneri ΔipaH9.8, fixed after 2.5 hpi, and immunostained for endogenous Sept7. Co-localization of GBP1 and Sept7 on bacteria was quantified. Mean frequencies ± SEM of combined data from three independent experiments are shown. Significance was determined by unpaired two-tailed t-test. *P ≤ 0.05. Arrow heads point at septin cages. All scale bars equal 5 µm.
Next, we investigated whether septin cages block GBP1 microcapsule formation. To address this question, we took advantage of previous observations demonstrating that septin cage formation not only requires septins to interact with bacterial lipids such as cardiolipin but is also dependent on bacterially driven actin recruitment and polymerization (Mostowy et al. 2010). Specifically, it had previously been reported that septins fail to encase S. flexneri in cells treated with actin capping or sequestering factors, and furthermore that septin cages were absent from S. flexneri mutants deficient for the actin polymerization factor IcsA (Mostowy et al. 2010). We therefore revisited the role of IcsA in septin cage and GBP1 microcapsule assembly by using S. flexneri strains deficient for either just the GBP1 inhibitor IpaH9.8 or both IpaH9.8 and IcsA. As expected, based on previous reports (Mostowy et al. 2010), we found that septin cages were not detectable when cells were infected with the IcsA-deficient ΔipaH9.8ΔicsA mutant (Fig. 3). Because S. flexneri ΔipaH9.8ΔicsA lacks the GBP1 inhibitor IpaH9.8 and is deficient for septin cage assembly, we were able to investigate whether the absence of septin cages impacts GBP1 microcapsule assembly. We found that ΔipaH9.8 and ΔipaH9.8ΔicsA S. flexneri were encapsulated by GBP1 at comparable frequencies, therefore indicating that septin cages do not interfere with GBP1 recruitment. Collectively, these data demonstrate that GBP1 microcapsules form independently of septin cages and that GBP1 microcapsules act as the dominant host defense program directed at inhibiting S. flexneri motility in IFNγ-primed human epithelial cells.
Figure 3.

Septin caging but not GBP1 encapsulation of S. flexneri depends on IcsA. HeLaGBP1-KO cells stably expressing mCherry-GBP1 were infected with S. flexneri ΔipaH9.8 or ΔipaH9.8ΔicsA, fixed after 2.5 hpi, and immunostained for endogenous Sept7. Bacteria caged by Sept7 and encapsulated by GBP1 as well as co-localization of Sept7 and GBP1 on bacteria were quantified. Mean frequencies ± SEM of combined data from three independent experiments are shown. Significance was determined by multiple unpaired two-tailed t-tests. **P ≤ 0.01. Arrow heads point at septin cages. All scale bars equal 5 µm.
DISCUSSION
Septins and GBPs have previously been identified as host defense proteins that bind to cytosolic S. flexneri and interfere with the ability of the pathogen to disseminate via actin-based motility. Here, we explored the functional relationship between these two classes of host defense proteins and demonstrate that GBP1 microcapsules around S. flexneri form independent of septin cages, but once formed, block the IcsA-dependent assembly of septin cages. We further show that S. flexneri mutants deficient for the GBP1 inhibitor IpaH9.8 become more susceptible to septin caging in IFNγ-primed cells, thus revealing how a complementary septin-mediated host defense is directed at S. flexneri that escape GBP1-mediated immunity.
Septins assemble into polymeric supramolecular structures, including rings and filaments. Other components of the cytoskeleton such as actin polymers and microtubuli often serve as templates for the formation of long septin filaments (Valadares et al. 2017; Addi, Bai and Echard 2018; Woods and Gladfelter 2020). Previous work by Mostowy et al. (2010) demonstrated that IcsA-mediated actin assembly around the bacterial cell, often referred to as actin halos or actin clouds, is required for septin caging. We confirm these observations and show that septin cages fail to assemble around IcsA-deficient bacteria simultaneously deficient for the GBP1 inhibitor IpaH9.8. Our present study further shows that GBP1 microcapsules block septin cage formation, most likely through interference with IcsA function and the consequential lack of a bacteria-associated actin template needed for septin cage formation. While GBP1 impedes septin caging, the absence of septin cages from ΔipaH9.8ΔicsA S. flexneri mutants does not result in an increase in the number of bacteria trapped inside GBP1 microcapsules, arguing that septins do not interfere with the recruitment of GBP1 to the bacterial surface.
Collectively, our observations reveal that septin- and GBP1-mediated inhibition of actin-based S. flexneri motility function as complementary defense programs (Fig. 4). Septins form cage-like structures around invading S. flexneri, thereby blocking actin tail formation. Following activation of innate immunity signaling and subsequent upregulation of GBP1 expression, the formation of GBP1 microcapsules supersedes septin-mediated defenses. However, bacteria that escape from GBP1-mediated immunity through the activity of the S. flexneri virulence factor IpaH9.8 become targets for the formation of septin cages. These observations underpin the fitness advantages provided by the evolution of functionally overlapping host defense programs.
Figure 4.
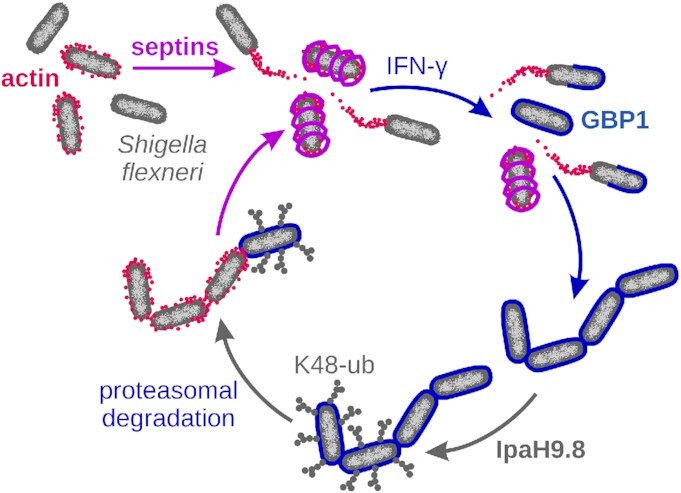
GBP1 microcapsules and septin cages are complementary antimotility factors targeting S. flexneri. Actin halos around immobile S. flexneri promote the assembly of septin cages. In IFNγ-primed human epithelial cells, GBP1 forms microcapsules around both motile and immobile bacteria. These GBP1 microcapsules block IcsA-mediated actin halo and actin tail formation and supersede IcsA-dependent septin caging. Wild-type S. flexneri secretes the type III secreted effector IpaH9.8 that tags GBP1 with K48-linked ubiquitin for proteasomal degradation. The IpaH9.8-mediated evasion of GBP1-driven host defense enables S. flexneri to form actin halos and thereby renders the bacteria again susceptible for septin caging.
ACKNOWLEDGMENTS
This work was performed in part at the Duke University Light Microscopy Core Facility (LMCF). We thank LMCF members Drs Benjamin Carlsen, Yasheng Gao and Lisa Cameron for providing training.
FUNDING
This work was supported by the National Institutes of Health grants AI139425(to JC) and AI128360(to CFL) and by the Deutsche Forschungsgemeinschaft(DFG, German Research Foundation) Research Fellowship 427472513 (to MK). JC received an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.
Supplementary Material
Contributor Information
Miriam Kutsch, Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, NC 27710, USA.
Coral González-Prieto, Center for Bacterial Pathogenesis, Division of Infectious Diseases, Massachusetts General Hospital, Boston, MA 02115, USA; Department of Microbiology, Blavatnik Institute, Harvard Medical School, Boston, MA 02115, USA.
Cammie F Lesser, Center for Bacterial Pathogenesis, Division of Infectious Diseases, Massachusetts General Hospital, Boston, MA 02115, USA; Department of Microbiology, Blavatnik Institute, Harvard Medical School, Boston, MA 02115, USA.
Jörn Coers, Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, NC 27710, USA; Department of Immunology, Duke University Medical Center, Durham, NC 27710, USA.
AUTHOR CONTRIBUTION
MK and JC conceived the project. MK conducted the experiments. MK and JC wrote the manuscript. CG-P and CFL provided reagents. JC supervised the project. JC and CFL acquired funding related to the project.
Conflict of Interest
None declared.
REFERENCES
- Addi C, Bai J, Echard A. Actin, microtubule, septin and ESCRT filament remodeling during late steps of cytokinesis. Curr Opin Cell Biol. 2018;50:27–34. [DOI] [PubMed] [Google Scholar]
- Agaisse H. Molecular and cellular mechanisms of Shigella flexneri dissemination. Front Cell Infect Microbiol. 2016;6:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardini ML, Mounier J, d'Hauteville Het al. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci USA. 1989;86:3867–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. [DOI] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene. 1996;173:33–8. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daumke O, Praefcke GJ. Invited review: mechanisms of GTP hydrolysis and conformational transitions in the dynamin superfamily. Biopolymers. 2016;105:580–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd GC, Mortuza R, Ireton K. Molecular mechanisms of intercellular dissemination of bacterial pathogens. Trends Microbiol. 2021;29:127–141. [DOI] [PubMed] [Google Scholar]
- Egile C, Loisel TP, Laurent Vet al. Activation of the Cdc42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J Cell Biol. 1999;146:1319–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enninga J, Mounier J, Sansonetti Pet al. Secretion of type III effectors into host cells in real time. Nat Methods. 2005;2:959–65. [DOI] [PubMed] [Google Scholar]
- Fisch D, Bando H, Clough Bet al. Human GBP1 is a microbe-specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J. 2019;38:e100926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisch D, Clough B, Domart MCet al. Human GBP1 differentially targets Salmonella and Toxoplasma to license recognition of microbial ligands and caspase-mediated death. Cell Rep. 2020;32:108008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano NP, Cian MB, Dalebroux ZD. Outer membrane lipid secretion and the innate immune response to Gram-negative bacteria. Infect Immun. 2020;88:e00920–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MB, Barzu O, Parsot Cet al. Unipolar localization and ATPase activity of IcsA, a Shigella flexneri protein involved in intracellular movement. Infect Agents Dis. 1993;2:210–1. [PubMed] [Google Scholar]
- Gomes MTR, Cerqueira DM, Guimaraes ESet al. Guanylate-binding proteins at the crossroad of noncanonical inflammasome activation during bacterial infections. J Leukoc Biol. 2019;106:553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann M, Schneck E, Gutsmann Tet al. Bacterial lipopolysaccharides form physically cross-linked, two-dimensional gels in the presence of divalent cations. Soft Matter. 2015;11:6037–44. [DOI] [PubMed] [Google Scholar]
- Koseoglu VK, Agaisse H. Evolutionary perspectives on the moonlighting functions of bacterial factors that support actin-based motility. mBio. 2019;10:e01520–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokowski S, Lobato-Marquez D, Chastanet Aet al. Septins recognize and entrap dividing bacterial cells for delivery to lysosomes. Cell Host Microbe. 2018;24:866–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsch M, Coers J. Human guanylate binding proteins: nanomachines orchestrating host defense. FEBS J. 2020.DOI:10.1111/febs.15662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsch M, Sistemich L, Lesser CFet al. Direct binding of polymeric GBP1 to LPS disrupts bacterial cell envelope functions. EMBO J. 2020;39:e104926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamason RL, Welch MD. Actin-based motility and cell-to-cell spread of bacterial pathogens. Curr Opin Microbiol. 2017;35:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Jiang W, Yu Qet al. Ubiquitination and degradation of GBPs by a Shigella effector to suppress host defence. Nature. 2017;551:378–83. [DOI] [PubMed] [Google Scholar]
- Mauricio RP, Jeffries CM, Svergun DIet al. The Shigella virulence factor IcsA relieves N-WASP autoinhibition by displacing the verprolin homology/cofilin/acidic (VCA) domain. J Biol Chem. 2017;292:134–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostowy S, Bonazzi M, Hamon MAet al. Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe. 2010;8:433–44. [DOI] [PubMed] [Google Scholar]
- Ngo CC, Man SM. Mechanisms and functions of guanylate-binding proteins and related interferon-inducible GTPases: roles in intracellular lysis of pathogens. Cell Microbiol. 2017;19:e12791. [DOI] [PubMed] [Google Scholar]
- Pfanzelter J, Mostowy S, Way M. Septins suppress the release of vaccinia virus from infected cells. J Cell Biol. 2018;217:2911–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piro AS, Hernandez D, Luoma Set al. Detection of cytosolic Shigella flexneri via a C-terminal triple-arginine motif of GBP1 inhibits actin-based motility. mBio. 2017;8:e01979–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praefcke GJK. Regulation of innate immune functions by guanylate-binding proteins. Int J Med Microbiol. 2018;308:237–45. [DOI] [PubMed] [Google Scholar]
- Robbins JR, Monack D, McCallum SJet al. The making of a gradient: IcsA (VirG) polarity in Shigella flexneri. Mol Microbiol. 2001;41:861–72. [DOI] [PubMed] [Google Scholar]
- Robertin S, Mostowy S. The history of septin biology and bacterial infection. Cell Microbiol. 2020;22:e13173. [DOI] [PubMed] [Google Scholar]
- Rojas ER, Billings G, Odermatt PDet al. The outer membrane is an essential load-bearing element in Gram-negative bacteria. Nature. 2018;559:617–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JC, Boucher D, Schneider LKet al. Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat Commun. 2020;11:3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson BW, Trent MS. Pushing the envelope: LPS modifications and their consequences. Nat Rev Microbiol. 2019;17:403–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirianni A, Krokowski S, Lobato-Marquez Det al. Mitochondria mediate septin cage assembly to promote autophagy of Shigella. EMBO Rep. 2016;17:1029–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Miki H, Takenawa Tet al. Neural Wiskott–Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J. 1998;17:2767–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadares NF, d'Muniz Pereira H, Ulian Araujo APet al. Septin structure and filament assembly. Biophys Rev. 2017;9:481–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bosch L, Morona R. The actin-based motility defect of a Shigella flexneri rmlD rough LPS mutant is not due to loss of IcsA polarity. Microb Pathog. 2003;35:11–8. [DOI] [PubMed] [Google Scholar]
- Van Ngo H, Mostowy S. Role of septins in microbial infection. J Cell Sci. 2019;132:jcs226266. [DOI] [PubMed] [Google Scholar]
- Wandel MP, Kim BH, Park ESet al. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat Immunol. 2020;21:880–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandel MP, Pathe C, Werner EIet al. GBPs inhibit motility of Shigella flexneri but are targeted for degradation by the bacterial ubiquitin ligase IpaH9.8. Cell Host Microbe. 2017;22:507–18.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren DJ. Preparation of highly efficient electrocompetent Escherichia coli using glycerol/mannitol density step centrifugation. Anal Biochem. 2011;413:206–7. [DOI] [PubMed] [Google Scholar]
- Welch MD, Way M. Arp2/3-mediated actin-based motility: a tail of pathogen abuse. Cell Host Microbe. 2013;14:242–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods BL, Gladfelter AS. The state of the septin cytoskeleton from assembly to function. Curr Opin Cell Biol. 2021;68:105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.