

Abstract
Hypoxic pulmonary vascular remodelling (PVR) is the major pathological basis of aging-related chronic obstructive pulmonary disease and obstructive sleep apnea syndrome. The pulmonary artery endothelial cell (PAEC) inflammation, and pulmonary artery smooth muscle cell (PASMC) proliferation, hypertrophy and collagen remodelling are the important pathophysiological components of PVR. Endogenous sulfur dioxide (SO2) was found to be a novel gasotransmitter in the cardiovascular system with its unique biological properties. The study was aimed to investigate the role of endothelial cell- (EC-) derived SO2 in the progression of PAEC inflammation, PASMC proliferation, hypertrophy and collagen remodelling in PVR and the possible mechanisms. EC-specific aspartic aminotransferase 1 transgenic (EC-AAT1-Tg) mice were constructed in vivo. Pulmonary hypertension was induced by hypoxia. Right heart catheterization and echocardiography were used to detect mouse hemodynamic changes. Pathologic analysis was performed in the pulmonary arteries. High-performance liquid chromatography was employed to detect the SO2 content. Human PAECs (HPAECs) with lentiviruses containing AAT1 cDNA or shRNA and cocultured human PASMCs (HPASMCs) were applied in vitro. SO2 probe and enzyme-linked immunosorbent assay were used to detect the SO2 content and determine p50 activity, respectively. Hypoxia caused a significant reduction in SO2 content in the mouse lung and HPAECs and increases in right ventricular systolic pressure, pulmonary artery wall thickness, muscularization, and the expression of PAEC ICAM-1 and MCP-1 and of PASMC Ki-67, collagen I, and α-SMA (p < 0.05). However, EC-AAT1-Tg with sufficient SO2 content prevented the above increases induced by hypoxia (p < 0.05). Mechanistically, EC-derived SO2 deficiency promoted HPAEC ICAM-1 and MCP-1 and the cocultured HPASMC Ki-67 and collagen I expression, which was abolished by andrographolide, an inhibitor of p50 (p < 0.05). Meanwhile, EC-derived SO2 deficiency increased the expression of cocultured HPASMC α-SMA (p < 0.05). Taken together, these findings revealed that EC-derived SO2 inhibited p50 activation to control PAEC inflammation in an autocrine manner and PASMC proliferation, hypertrophy, and collagen synthesis in a paracrine manner, thereby inhibiting hypoxic PVR.
1. Introduction
Aging is an important risk factor for a variety of diseases [1–3]. With the increase of the global elderly population, the incidence of aging and age-related diseases such as chronic obstructive pulmonary disease (COPD) and obstructive sleep apnea syndrome (OSAS) has also been gradually increased [4, 5]. Among them, hypoxic pulmonary hypertension (PH) and pulmonary vascular remodelling (PVR) are the critical pathological basis. PVR includes pulmonary artery endothelial cell (PAEC) dysfunction, and pulmonary artery smooth muscle cell (PASMC) proliferation, hypertrophy, and collagen accumulation [6–9]. Previous studies reported that an imbalance among small vasoactive molecules played a critical role in the progression of PVR. The imbalance among protein-derived bioactive molecules, active lipid mediators, small nucleic acids, and gaseous signalling molecules is predominantly involved in the pulmonary artery structural changes and the abnormal vasoconstriction and vasorelaxation [10–16]. However, the mechanisms underlying the excessive PAEC inflammation, PASMC proliferation, hypertrophy, and collagen remodelling remain unclear.
Recent studies have shown that endothelial cells (ECs) play a critical role in maintaining vascular homeostasis, while EC dysfunction leads to various vascular diseases [17, 18]. For example, Xue et al. observed that the overexpression of EC-derived cyclophilin A caused spontaneous PH by promoting PAEC inflammation and PASMC proliferation [19]. Moreover, EC dysfunction and endothelial-to-mesenchymal cell transition enhanced collagen accumulation in the pathogenesis of human fibrotic diseases [20]. These results suggest that ECs might affect the function of ECs and other neighboring cells (primarily smooth muscle cells [SMCs]) in the vascular walls in an autocrine/paracrine manner, by which ECs play an important role in the development of PVR. However, the mechanisms by which ECs affect the behavior of PAECs and PASMCs to play a role in the development of PVR are complex and have not yet been fully elucidated.
In 2008, the endogenous sulfur dioxide (SO2)/aspartate aminotransferase (AAT) pathway was found to exist in the vascular ECs of rats [21]. More recently, the production of endogenous SO2 catalyzed by AAT1 has been identified in human PAECs (HPAECs) [22]. As a gasotransmitter, SO2 has a series of important advantages, including sustained production, rapid diffusion, and free passage through cell membranes with a short half-life. It exerts a variety of biological functions in the cardiovascular system. For example, in vivo, the exogenous SO2 donor showed a protective role in rat atherosclerosis and sepsis-induced cardiac dysfunction via the inhibition of cellular inflammation [23, 24]. In vitro, the deficiency of SO2 contributed to cellular proliferation and collagen accumulation [25]. Moreover, the deficiency of endogenous SO2 might be implicated in the pathogenesis of myocardial hypertrophy [26]. These data strongly suggest that EC-derived SO2 might play a regulatory role in PAEC inflammation in an autocrine manner and in PASMC proliferation, hypertrophy, and collagen deposition in a paracrine manner. As such, in the present study, we further explored whether EC-derived SO2 might control PAEC inflammation in an autocrine manner and control PASMC proliferation, hypertrophy, and collagen deposition in a paracrine manner to reveal a new mechanism for PAEC inflammation and PAEC-PASMC communication in the control of PASMC proliferation, hypertrophy, and collagen remodelling by SO2.
Recent data have suggested that EC inflammation, SMC proliferation, and collagen synthesis are under the control of nuclear factor-κB (NF-κB) [27, 28]. Briefly, some stimuli, such as tumor necrosis factor α (TNF-α), lead to the activation of the IκB kinase complex, allowing IκB to undergo phosphorylation and degradation, and NF-κB dimers (thought to be p50 heterodimers primarily) to translocate to the nucleus to promote gene transcription [29]. The inhibition of p50 evidently restricted E-selectin expression induced by TNF-α in human umbilical vein ECs, and NF-κB activation triggered bladder SMC collagen biosynthesis and proliferation [30, 31]. But the regulatory mechanisms for p50 are unclear so far. Intriguingly, endogenous SO2 exhibited a protective effect by repressing NF-κB activation in adipocytes and macrophages.
Based on these evidences, in this study, we aimed to determine the possible role of EC-derived SO2 in the progression of hypoxic PVR and its possible underlying mechanisms in association with the PAEC inflammatory response, PASMC proliferation, hypertrophy, and collagen production to reveal a new mechanism for PAEC inflammation and PAEC-PASMC communication in the control of PASMC proliferation, hypertrophy, and collagen remodelling by SO2.
2. Materials and Methods
2.1. Animal Model
Endothelial cell-specific AAT1 transgenic (EC-AAT1-Tg) and wild-type (WT) littermate mice (C57BL/6J background) were purchased from Cyagen Biosciences (Suzhou, China). Genotype was confirmed using polymerase chain reaction analysis in 10-day-old mice. Male mice aged 10–12 weeks were randomly divided into WT, hypoxic WT (WT+H), EC-AAT1-Tg, and hypoxic EC-AAT1-Tg (EC-AAT1-Tg+H) groups. The mice of the WT and EC-AAT1-Tg groups breathed in room air (21% O2), whereas hypoxic mice were placed in a small animal hypoxic chamber (9 ± 0.5% O2) (XBS-02B; Hangzhou Aipu Instrument Co., Ltd., China) 8 h daily for 5 weeks to induce hypoxic PH [32]. All institutional and national guidelines for the care and use of animals (fisheries) were followed. The animal experiment was approved by the Laboratory Animal Ethics Committee of Peking University First Hospital (Ethics No. 201526).
2.2. High-Performance Liquid Chromatography (HPLC) Quantitative Detection of SO2 Content in Mouse Lung Tissue
As mentioned earlier, HPLC (Agilent 1200 series; Agilent Technologies, CA, USA) was used to detect the SO2 content in mouse lung tissue. Briefly, 100 μL of the standard sodium sulfite and a sample of mouse lung tissue homogenate were mixed with 70 μL of sodium borohydride (0.212 mol/L) in Tris-HCl (0.05 mol/L, pH 8.5). The mixture was then incubated at room temperature for 30 min. Next, 5 μL of monobromobimane (70 mmol/L) in acetonitrile was mixed with the 170 μL mixture. After the above mixture was incubated at 42°C for 10 min, 40 μL of perchloric acid (1.5 mol/L) was added. To remove the protein precipitates and neutralize the mixture, the mixture was centrifuged (12,400 g) at 25°C for 10 min, and 10 μL of Tris-HCl (2 mol/L, pH 3.0) was added to the supernatant. Finally, the HPLC operation and result analysis were performed as described previously [33].
2.3. Detection of Mouse Right Ventricular Systolic Pressure (RVSP) Changes by Right Heart Catheterization
The RVSP of mice was directly measured by catheterization. The mice were anesthetized by intraperitoneal injection of 0.5% sodium pentobarbital (0.1 ml/10 g). A catheter was inserted through the external jugular vein to reach the right ventricle of the mouse. The BL-420F Biological Function Experimental System (Chengdu Taimeng Instrument Co. Ltd., China) was used to measure the mouse RVSP to predict the changes in pulmonary artery pressure [32].
2.4. Measurement of Mouse Pulmonary Acceleration Time (PAT) and Pulmonary Ejection Time (PET) Using Echocardiography
Echocardiography was performed using the Vevo 2100 high-resolution imaging system (Visualonics, Toronto, Canada). The mice were initially anesthetized by intraperitoneal injection of 0.5% sodium pentobarbital (0.1 ml/10 g). Pulsed-wave Doppler recording of the pulmonary blood flow was obtained from the parasternal short axis view at the aortic valve level. Samples were positioned at the tip of the pulmonary valve leaflets and aligned to maximize laminar flow as described previously. PAT (defined as the time from the onset of flow to peak velocity by pulsed-wave Doppler recording) and PET (the time from the onset to the termination of pulmonary flow) variables were determined. All measured variables were the average of 3–5 cardiac cycles [34].
2.5. Preparation and Morphological Analysis of Mouse Lung Tissue
The mice were killed by cervical dislocation. Their lungs were removed and rinsed with 4°C phosphate-buffered saline (PBS). The lungs were then fixed in 4% paraformaldehyde solution for 24 h to prepare paraffin-embedded sections or frozen sections of lung tissue. The sections were stained with hematoxylin and eosin (HE) and Hart's method for morphological analysis. As previously reported [35], the degree of muscularization can be determined by the ratio of the number of nonmuscular vessels (NMV), partial muscular arteries (PMA), and muscular arteries (MA). PVR can be quantitatively analyzed by calculating the percentage of 15–50 μm pulmonary arteriole muscularization.
2.6. Immunofluorescence
Frozen sections of lung tissue and cells were rinsed with PBS and fixed with 4% paraformaldehyde solution for 20 min. Tissue sections and cells were blocked with bovine serum albumin and incubated with the primary antibodies at 4°C overnight (vWF: 1 : 200, Zhongshan Jinqiao Biotechnology Co., Ltd., China; AAT1 1 : 50, Sigma-Aldrich, USA; p-p50 1 : 50 and ICAM-1 1 : 20, Santa Cruz Biotechnology Company, USA; p50 1 : 100 and Ki-67 1 : 100, Cell Signaling Technology Company USA; F4/80 1 : 300, α-SMA 1 : 300, collagen 1 : 25 and MCP-1 1 : 100, Abcam Company, UK). On the second day, the secondary fluorescent antibodies were incubated in the dark. All sections were counterstained with nuclear 4,6-diamino-2-phenylindole (DAPI; Zhongshan Jinqiao Biotechnology Co., Ltd., China). Immunofluorescence imaging was performed using a confocal laser scanning microscope (Leica, Wetzlar, Germany) for observation and comparison.
2.7. Primary HPAEC Culture
Primary HPAECs and primary EC culture system were purchased from PriCells Biomedical Technology Co., Ltd. (Wuhan, China); cells at 4-6 generation were used for experiments. HPAECs were transfected with lentivirus containing AAT1 shRNA or cDNA (Cyagen Biosciences, Suzhou, China) for 2–3 days to induce AAT1 knockdown or overexpression. 1% O2 was used to induce a hypoxic cell inflammation model.
2.8. Coculture of Primary Human PASMCs (HPASMCs) and HPAECs
Primary HPASMCs and primary SMC culture system were purchased from Lifeline Cell Technology (USA). Transwell plates (Corning, China) were used for the coculture of HPASMCs and HPAECs. We seeded HPASMCs in the lower compartment of the transwell plates by using the SMC culture system. Infected HPAECs were planted in the upper compartment with an EC culture system and were separated from the lower compartments by a microporous membrane. An inhibitor of p50 andrographolide (4 μM, Andro, Selleck, USA) was used for experiments [30].
2.9. SO2 Probe-Based In Situ Detection of SO2 Content in HPAECs
The SO2 fluorescent probe was used to analyze the SO2 content in HPAECs in situ as previously reported [22]. After the unbound SO2 probe was removed and the nuclei were stained with DAPI, we observed the red fluorescence intensity of the SO2 probe under a high-resolution confocal laser microscope.
2.10. Western Blot
Lung tissues or cells were lysed in lysis buffer to obtain total protein. Denatured proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Amerisco, USA). The membranes were incubated with specific primary antibodies (GAPDH 1 : 4000, β-actin 1 : 4000, AAT1 1 : 1000, ICAM-1 1 : 400, MCP-1 : 1000, Ki-67 1 : 500, collagen I 1 : 1000, α-SMA 1 : 1000 and p-p50 1 : 1000) and secondary antibodies (1 : 2000) conjugated with horseradish peroxidase. The FluorChem M MultiFluor System (Proteinsimple, USA) was used to scan the protein bands in grayscale. The ImageJ software was used to quantitatively analyze each band, and each protein band was corrected with its own internal reference GAPDH or β-actin grayscale [33].
2.11. p50 DNA-Binding Activity Detected by Active Motif-Enzyme-Linked Immunosorbent Assay (ELISA)
Active motif-ELISA (Active Motif, USA) was used to determine the DNA-binding activity of p50 in HPAEC nuclear protein. The nuclear protein extraction steps were initially performed as described earlier [14]. The DNA-binding activity of p50 was then evaluated according to manufacturer's instructions. In short, the binding buffer, the sample diluted with lysate, 1x washing buffer, p50 primary antibody (1 : 1000, prepared with 1x antibody binding buffer), secondary antibody, and color developing solution were added in turn. We observed the color change during the experiment. The experiment was stopped when the color turned to moderate dark blue by adding stop solution, and then, the color turned yellow. Finally, we measured the absorbance value at 450 nm as soon as possible.
2.12. Adhesion Test of THP-1 Monocyte Cell Line and HPAECs
After inducing the HPAEC inflammatory response, THP-1 mononuclear cells initially stained with the red fluorescent Dil (Beyotime Biotechnology, China) were added to the HPAECs. HPAECs and THP-1 cells were incubated at 37°C for 1 h. PBS was then used to remove unadhered THP-1 cells. Subsequently, we fixed the cells with 4% paraformaldehyde solution for 20 min. Finally, the mounting medium containing DAPI was added, and fluorescence was observed under an immunofluorescence microscope.
2.13. Data Analysis
Statistical analyses were performed with the SPSS 21.0 software (SPSS Inc., USA). Data were expressed as mean ± SEM and analyzed using one-way ANOVA. p values < 0.05 were considered statistically significant.
3. Results
3.1. Hypoxia Caused Reduction in EC-Derived SO2
Immunofluorescence and HPLC showed decreased AAT1 protein expression in PAECs and SO2 content in the lungs of WT mice under hypoxia compared with control WT mice (Figures 1(a) and 1(b)) (p < 0.05). By Western blot analysis and SO2 fluorescent probe method, we further confirmed that compared with the control vehicle group, the levels of AAT1 protein and SO2 were downregulated in hypoxic HPAECs, whereas overexpression of AAT1 significantly improved the endogenous SO2 level in ECs and prevented the decrease in EC-derived SO2 caused by hypoxia (Figures 1(c) and 1(d)) (p < 0.05).
Figure 1.
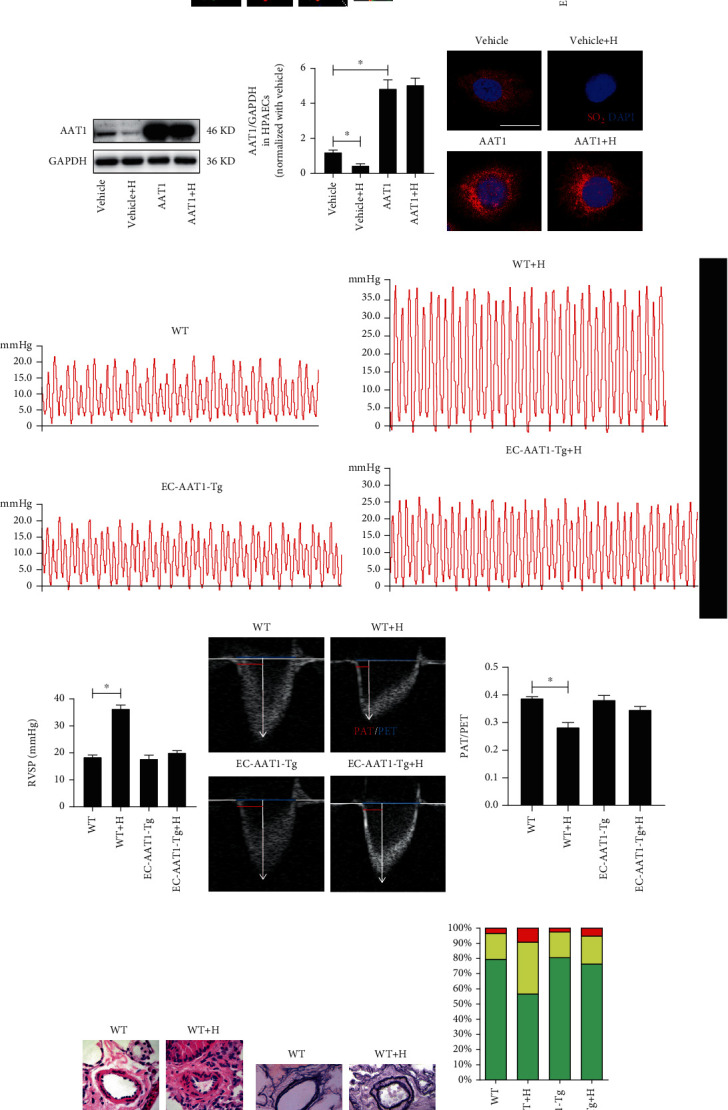
Increased EC-derived SO2 ameliorates hypoxia-induced PH. (a) Immunofluorescence in situ detection of AAT1 protein expression in mouse PAECs. Green fluorescence represents AAT1 protein, and red fluorescence represents vWF, a marker for ECs; the nuclei were stained with DAPI. (b) HPLC method was performed to detect SO2 content in mouse lung tissue (n = 5 − 17). (c) Western blot analysis was carried out to detect the expression level of AAT1 protein in HPAECs (n = 10). (d) Red SO2 fluorescent probe method was used to determine the SO2 level in HPAECs, and the nuclei of HPAECs were stained with DAPI. (e, f) Catheter method was performed to detect mouse right ventricular systolic pressure (RVSP) (n = 6 − 8). (g, h) The ratio of pulmonary acceleration time (PAT) to pulmonary ejection time (PET) of the mouse pulmonary artery blood flow was measured using ultrasound (n = 8). (i, j) HE and Hart's methods were used to detect PVR in mice. (k) Hart's method was conducted to analyze the degree of muscularization of small pulmonary vessels in mice (n = 8); NMV: nonmuscular vessels; PMA: partial muscular arteries; MA: muscular arteries. The data were expressed as mean ± SEM, ∗p < 0.05, scale bar: 20 μm.
3.2. Increased EC-Derived SO2 Ameliorated Hypoxia-Induced PVR and PH In Vivo
To reveal the role of EC-derived SO2 in the pathogenesis of hypoxic PVR, we constructed EC-AAT1-Tg mice to increase SO2 levels in mouse PAECs (Figure S1a). We found an increased expression of AAT1 protein in PAECs and SO2 content in the lung of EC-AAT1-Tg mice, compared with the mice in the WT group (Figures 1(a) and 1(b)) (p < 0.05). After exposure to intermittent hypoxia for 5 weeks, both the RVSP detected by the right heart catheter and the PAT/PET ratio in this experiment suggested that hypoxic WT (WT+H) mice developed significant PH compared with the control WT mice. We also observed that markers of PVR, the thickness of pulmonary artery walls, and the proportion of muscularized arteries were dramatically enhanced in mice of the WT+H group by HE and Hart's methods. However, compared with mice in EC-AAT1-Tg group with increased EC-SO2 content, hypoxia did not induce significant PVR and PH in mice of EC-AAT1-Tg+H group (Figures 1(e)–1(k)) (p < 0.05). The above results suggested that the decrease in SO2 in ECs resulted in increased vascular remodelling and hypoxic PH in mice.
3.3. Increased EC-Derived SO2 Ameliorated Hypoxia-Induced PAEC Inflammation, PASMC Proliferation, Hypertrophy, and Collagen Production
We next sought to determine the role of EC-derived SO2 in PAEC inflammatory reaction, PASMC proliferation, hypertrophy, and collagen synthesis, which dramatically contribute to PVR. In vivo, by immunofluorescence in situ detection of ICAM-1 and MCP-1 expression and macrophage infiltration in pulmonary vasculature, we found that compared with WT mice, hypoxia dramatically increased the expression of ICAM-1 and MCP-1, known markers of EC inflammatory process in mouse PAECs, and macrophage infiltration in the pulmonary arteries of WT+H mice. Moreover, Western blot quantitative determination of ICAM-1 protein in mouse lung showed similar results (Figures 2(a) and 2(b), Figure S2a and S2b) (p < 0.05). But the treatment with EC-AAT1-Tg to increase SO2 level successfully repressed hypoxia-induced increases in the protein expression of ICAM-1 and MCP-1, and macrophage infiltration in mouse lung tissue (Figures 2(a) and 2(b), Figure S2a and S2b) (p < 0.05). Consistent with the in vivo results, we found that AAT1 overexpression to increase the content of EC-derived SO2 evidently inhibited hypoxia-induced increases in ICAM-1 and MCP-1 in HPAECs and THP-1 cell adhesion to HPAECs in vitro (Figure S2c-S2f) (p < 0.05). These data suggested that the increase in SO2 in ECs could inhibit hypoxic vascular inflammation in an autocrine manner.
Figure 2.
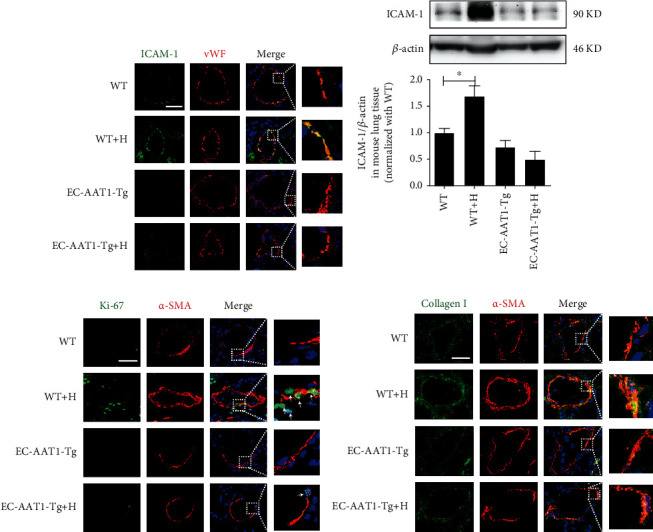
Increased EC-derived SO2 ameliorates hypoxia-induced PAEC inflammation, PASMC proliferation, hypertrophy, and collagen production in vivo. (a) Immunofluorescence in situ detection of ICAM-1 protein expression in mouse PAECs; green fluorescence represents ICAM-1 protein. (b) Western blot analysis was performed to detect the expression level of ICAM-1 protein in mouse lung tissue (n = 10). (c, d) Immunofluorescence in situ detection of Ki-67, α-SMA, and collagen I protein expression in mouse PASMCs; green fluorescence represents Ki-67 and collagen I protein, and red fluorescence represents α-SMA. The data were expressed as mean ± SEM, ∗p < 0.05, scale bar: 20 μm.
We then evaluated the effect of EC-derived SO2 on hypoxic PASMC proliferation, hypertrophy, and collagen deposition in vivo. Compared with the mice in the WT group, immunofluorescence showed enhanced Ki-67 staining in the pulmonary arteries of the WT+H group. While compared with the mice of EC-AAT1-Tg group, no significant increase was found in the expression of Ki-67 induced by hypoxia in the mice of EC-AAT1-Tg+H group (Figure 2(c)). In addition, immunofluorescence showed an increased α-SMA expression, a marker of SMC hypertrophy, in the pulmonary arteries of WT mice under hypoxia compared with WT mice under normoxia, while there was no significant difference in α-SMA expression between EC-AAT1-Tg mice with and without hypoxic exposure (Figures 2(c) and 2(d)). We also observed that the increased collagen I in PASMCs of mice under hypoxia was successfully repressed in EC-AAT1-Tg mice with sufficient SO2 (Figure 2(d)). The above results indicated that increased EC-derived SO2 ameliorated hypoxia-induced PASMC proliferation, hypertrophy, and collagen accumulation in a paracrine manner.
3.4. EC-Derived SO2 Deficiency Activated HPAEC Inflammation, HPASMC Proliferation, Hypertrophy and Collagen Synthesis In Vitro
To directly identify the role of EC-derived SO2 in HPAEC inflammatory process, HPASMC proliferation, hypertrophy, and collagen production, we infected HPAECs with lentivirus containing AAT1 shRNA to reduce the expression of the key enzyme AAT1 for SO2 production. By Western blot analysis and the SO2 fluorescent probe method, we confirmed that the AAT1 protein level and SO2 probe staining significantly decreased in AAT1-knockdown (AAT1 shRNA) HPAECs. However, treatment with SO2 donor restored SO2 level in HPAECs (Figures 3(a) and 3(b)) (p < 0.05).
Figure 3.
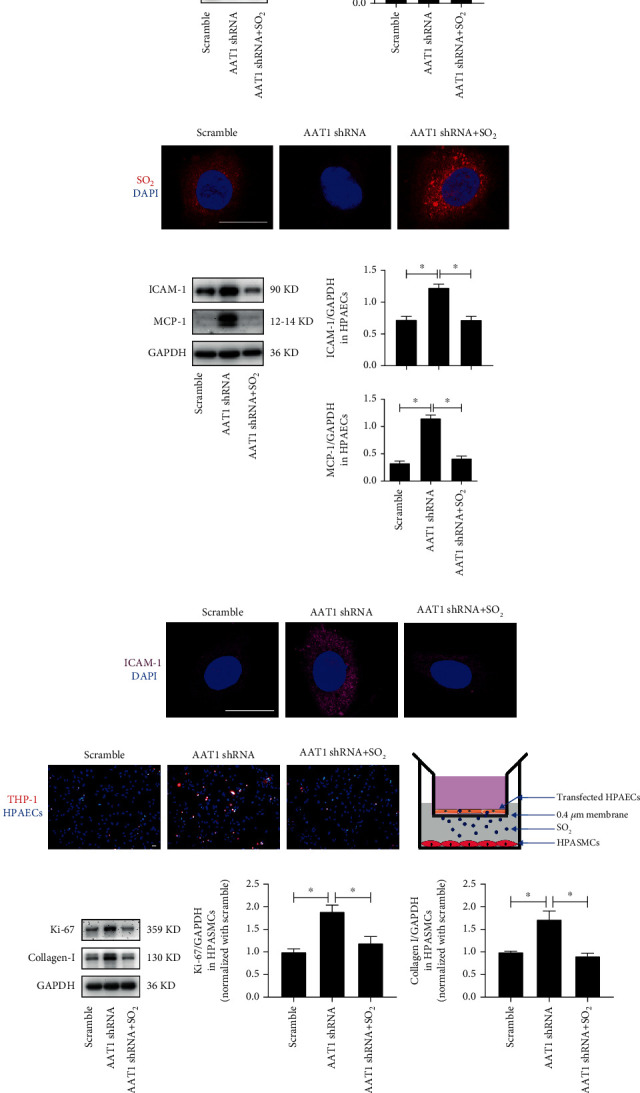
EC-derived SO2 deficiency activates HPAEC inflammation, HPASMC proliferation, and collagen synthesis in vitro. (a) Western blot analysis was performed to detect the expression level of AAT1 protein in HPAECs (n = 12). (b) The red SO2 fluorescent probe method was used to detect the SO2 level in HPAECs; the nuclei were stained with DAPI. (c) Western blot analysis was used to detect the expression level of ICAM-1 and MCP-1 protein in HPAECs (n = 12). (d) Immunofluorescence method was used to detect the expression level of ICAM-1 protein in HPAECs in situ. Purple fluorescence represents ICAM-1 protein. (e) Fluorescence method was conducted to detect the adhesion of THP-1 cells and HPAECs. The red Dil color marks the THP-1 cells, and the DAPI color marks the nuclei of HPAECs. (f) Diagram of the transwell used for the coculture system in vitro. (g) Western blot analysis method to detect the expression level of Ki-67 and collagen I protein in HPASMCs cocultured with HPAECs (n = 10). The data were expressed as mean ± SEM, ∗p < 0.05, scale bar: 20 μm.
Compared with the scramble control group, we further verified that the protein expression of ICAM-1 and MCP-1 and the number of monocyte adhesion to HPAECs in the AAT1 shRNA group all evidently increased. However, supplementation with SO2 donor successfully inhibited these increases in AAT1 shRNA HPAECs (Figures 3(c)–3(e)) (p < 0.05). In addition, HPASMCs cocultured with HPAECs of the AAT1 shRNA group showed increased expression of Ki-67, collagen I, and α-SMA, which were also suppressed by supplementation with SO2 donor in HPAECs (Figures 3(f) and 3(g), Figure S3a and S3b) (p < 0.05). The above studies directly suggested that SO2 derived from ECs regulated the function of HPAECs and HPASMCs. Moreover, the deficiency of EC-derived SO2 activated the inflammatory reaction of HPAECs in an autocrine manner, and the proliferation, collagen biosynthesis, and hypertrophy of HPASMCs in a paracrine manner.
3.5. EC-Derived SO2 Inhibited p50 Activation to Repress PAEC Inflammation, and PASMC Proliferation, and Collagen Deposition
Considering that NF-κB (particularly p50 heterodimers) plays a critical role in cell inflammation, proliferation, and collagen metabolism, we further observed p50 activation in HPAECs and HPASMCs to reveal the mechanism by which EC-derived SO2 played a protective effect. The nuclear translocation of p50 was significantly enhanced in HPAECs of the AAT1 shRNA group compared with the scramble group, but this effect was reversed by the treatment with SO2 donor (Figure 4(a)).
Figure 4.
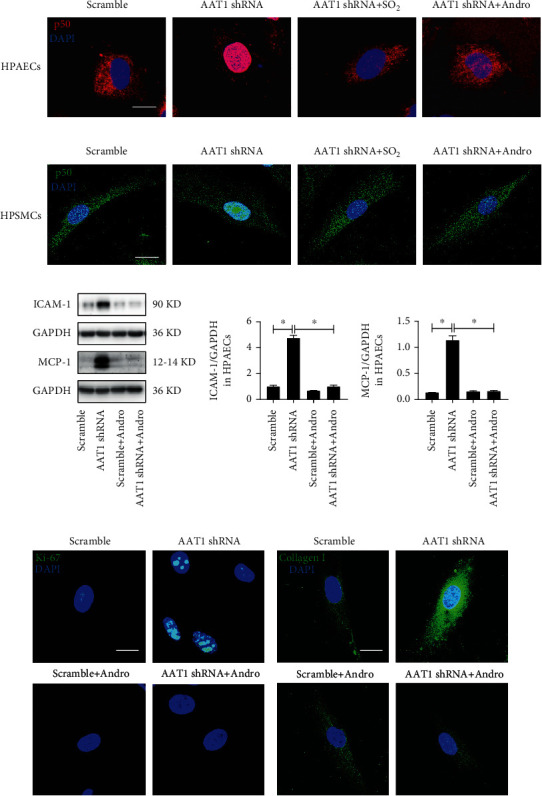
EC-derived SO2 inhibits p50 activation to repress PAEC inflammation, PASMC proliferation, and collagen deposition. (a) Immunofluorescence in situ detection of p50 protein distribution in HPAECs. Red fluorescence represents p50 protein, and DAPI staining labels HPAEC nuclei. (b) Immunofluorescence in situ detection of p50 protein distribution in HPASMCs cocultured with HPAECs. Green fluorescence represents p50 protein, and DAPI staining labels HPASMC nuclei. (c) Western blot analysis was used to detect the expression level of ICAM-1 and MCP-1 protein in HPAECs (n = 12). (d, e) Immunofluorescence in situ detection of Ki-67 and collagen I protein expression in HPASMCs cocultured with HPAECs. Green fluorescence represents Ki-67 and collagen I protein. Andro: andrographolide, an inhibitor of p50. Data were expressed as mean ± SEM, ∗p < 0.05, scale: 20 μm.
Furthermore, in the hypoxia-induced HPAEC inflammatory response, the phosphorylation, nuclear translocation, and activity of p50 evidently increased in HPAECs of the hypoxic vehicle (vehicle+H) group compared with the normoxic vehicle group. However, no significant differences were found in AAT1-overexpressed HPAECs between the normoxic and hypoxic treatment groups (Figure S4a-S4c) (p < 0.05). Moreover, the in vivo EC-AAT1-Tg abolished hypoxia-induced p50 activation (Figure S4d).
Meanwhile, we found that with inhibition of endogenous SO2 generation in HPAECs, nuclear translocation of p50 in HPASMCs cocultured with HPAECs was activated significantly, which was repressed by the treatment with SO2 donor as well (Figure 4(b)). These results suggested that p50 may be the molecular target of SO2.
To investigate whether the inflammatory reaction of HPAECs and the proliferation and collagen accumulation of HPASMCs were mediated by p50, intervention with Andro, an inhibitor of p50, demonstrated that Andro successfully prevented nuclear translocation of p50 induced by the decrease in EC-derived SO2 in HPAECs and HPASMCs (Figures 4(a) and 4(b)). Meanwhile, the enhanced expression levels of ICAM-1 and MCP-1 in HPAECs of the AAT1shRNA group were significantly inhibited by Andro treatment (Figure 4(c)) (p < 0.05). In agreement with the results of HPAECs, the increased expression of Ki-67 and collagen I in HPASMCs cocultured with HPAECs of the AAT1shRNA group was also inhibited by Andro (Figures 4(d) and 4(e)). The above results indicated that Andro completely blocked the augmentation of HPAEC inflammation, HPASMC proliferation, and collagen production caused by the decrease in EC-derived SO2 and p50 was a key molecular target for the control of PAEC inflammation, PASMC proliferation, and collagen remodelling by SO2.
4. Discussion
We demonstrated that EC-derived SO2 was an important endogenous controller of hypoxic PVR. It inhibited PAEC inflammatory process in an autocrine manner and PASMC proliferation, hypertrophy, and collagen accumulation in a paracrine manner. NF-κB p50 signalling might mediate the above effect of EC-derived SO2.
PVR is the main pathological feature of aging-related COPD and OSAS [6, 7]. The key roles of PAEC inflammation, and PASMC proliferation, hypertrophy, and collagen deposition in the pathogenesis of PVR have been suggested by previous studies [8, 9, 11, 25, 36]. However, the mechanisms underlying the endogenous control of PVR have not been identified. Evidence supports that ECs play an important role in the pathogenesis of PVR [18, 19]. However, how PAEC inflammation is regulated and whether PASMC proliferation, hypertrophy, and collagen synthesis are controlled through PAEC-PASMC communication in PVR are not completely clear.
Recently, the gasotransmitter SO2 with a range of properties, including sustained production, rapid diffusion, and broad functions has been shown to be produced endogenously in HPAECs [21, 22]. Previously, our laboratory and others showed the critical roles of SO2 donor in controlling cellular collagen metabolism, inflammation, proliferation, and hypertrophy in the pathogenesis of cardiovascular diseases [23–26]. In the present study, we found that the decrease in endogenous SO2 in hypoxic PH mice with significant PVR and HPAECs, and hypoxic exposure caused the downregulated expression of AAT1 protein in mouse PAECs and HPAECs. This finding indicated that the endogenous SO2/AAT1 pathway in ECs was probably associated with hypoxic PH and PVR.
To investigate the regulatory effects of EC-derived SO2 on the development of hypoxic PH and PVR, and the mechanisms, EC-AAT1-Tg mice with forced expression of AAT1 in ECs were used in the in vivo experiment, and AAT1 cDNA was transfected with a lentivirus vector to hold the AAT1 expression in in vitro experiment. Unlike WT mice, EC-AAT1-Tg mice did not respond to hypoxic stimulation, demonstrated by the results that hypoxic exposure induced an increase in RVSP and PVR in WT mice, but normoxic EC-AAT1-Tg mice exhibited similar RVSP and vascular structure to EC-AAT1-Tg mice exposed to hypoxia. Furthermore, hypoxia-induced the expression of inflammatory factor ICAM-1 and MCP-1 in the PAECs and macrophage infiltration in the pulmonary arteries were alleviated in EC-AAT1-Tg mice. In accordance with the results in vivo, AAT1 overexpression blocked the hypoxia-stimulated expression of ICAM-1 and MCP-1 in HPAECs and monocyte adhesion to HPAECs. It is known that PVR is mainly produced by a thickening of the lamina media due to an increase in PASMC proliferation, hypertrophy, and collagen deposition [8, 11, 25, 36]. Especially, cell hypertrophy is a well-known cell senescence hallmark [2, 37]. Therefore, we observed the effect of EC-derived SO2 on the PASMC behaviors in vivo. The data showed that hypoxia stimulated the increased expression of proliferative marker Ki-67, hypertrophy marker α-SMA, and classical collagen I in PASMCs in WT mice, but not in EC-AAT1-Tg mice. The abovementioned results suggested an important role of EC-derived SO2 in the regulation of hypoxic PVR.
Conversely, to directly investigate the effect of EC-derived SO2 on the HPAEC inflammation, and HPASMC proliferation, hypertrophy, and collagen deposition, HPAECs were transfected with human AAT1 shRNA lentivirus to decrease the level of EC-derived SO2, and SO2 donor was used for rescuing the effect of AAT1 knockdown. As we expected, the deficiency of endogenous SO2 in HPAECs caused spontaneous overexpression of ICAM-1 and MCP-1 in ECs and monocyte adhesion to HPAECs. Recently, the insufficiency of endogenous SO2 in adipocytes and macrophages has been found to induce spontaneous inflammatory responses, strengthening the protective role of endogenous SO2 in the cardiovascular system [38, 39]. Interestingly, HPASMCs cocultured with AAT1 shRNA HPAECs presented a PH phenotype characterized by increased Ki-67 and α-SMA expression, and accumulation of collagen I in association with the decreased SO2 content in HPAECs. These results further supported that EC-derived SO2 was an important endogenous controller of PVR. EC-derived SO2 inhibited the PAEC inflammatory process in an autocrine manner, and the coculture of HPAECs and HPASMCs further verified that EC-derived SO2 inhibited PASMC proliferation, hypertrophy, and collagen accumulation in a paracrine manner.
To demonstrate the mechanism by which EC-derived SO2 controls PAEC inflammation, PASMC proliferation, and collagen accumulation, the treatment with gene knockdown and SO2 donor in HPAECs showed that the deficiency of EC-derived SO2 promoted nuclear translocation of p50 in HPAECs and HPASMCs with significantly augmented PAEC inflammation, PASMC proliferation, and collagen remodelling. By contrast, the increase in EC-derived SO2 by AAT1 overexpression obviously inhibited hypoxia-induced activation of p50 in PAECs, thereby attenuating hypoxic PAEC inflammation, PASMC proliferation, and collagen remodelling in vitro and in vivo. More interestingly, the treatment with Andro, a p50 inhibitor, markedly blocked nuclear translocation of p50, and the subsequent PAEC inflammation, PASMC proliferation, and collagen accumulation induced by the decrease in EC-derived SO2 in HPAECs and HPASMCs. Therefore, these findings clarified that EC-derived SO2 inhibited p50 activation to control the inflammation of PAECs, and PASMC proliferation, and collagen accumulation.
The mechanism by which SO2 inhibits p50 activation has not yet been elucidated. The DNA-binding activity of p50 is regulated by the redox reaction, and NO nitrosylates the cysteine sulfhydryl groups of p50 and significantly inhibits its DNA-binding activity [40, 41]. Intriguingly, SO2 is also implicated in oxidative and reductive modification of proteins, such as AAT1 and p65, to modulate protein function in association with the cysteine sulfhydryl groups [42, 43]. In addition, previous studies also indicated that TGF-β/Smad, Raf-1/MEK-1/Erk/MAPK, AE2, and Dkk1/Wnt signalling pathways were implicated in the regulation of SO2 on PASMC proliferation, hypertrophy, and collagen deposition [35, 44–49]. In the future, further investigating the mechanism by which SO2 regulates p50 and revealing the potential clinical value of application of SO2 gas for the treatment of hypoxic PH are necessary.
5. Conclusions
The obtained data elucidated that endogenous SO2 in ECs exerted autocrine and paracrine effects to control PAEC inflammation, and PASMC proliferation, hypertrophy and collagen remodelling to inhibit PVR induced by hypoxia. The findings revealed a novel PAEC-PASMC communication mechanism in the endogenous control of hypoxic PVR by EC-derived SO2. Moreover, this study identified a potential target for the treatment of hypoxic PVR in aging-related cardiopulmonary diseases.
Acknowledgments
We thank Dr Pan Huang, Wen Yu, Zhizhou Shen, Mengxiang Sun, Qiang Shen, and Yao Song for their expert technical advice and Editage (http://www.editage.cn) for English language editing. This work was supported by the National Natural Science Foundation of China [grant numbers 91439110, 81770422, 82070445].
Contributor Information
Yaqian Huang, Email: yaqianhuang@126.com.
Junbao Du, Email: junbaodu1@126.com.
Hongfang Jin, Email: jinhongfang51@126.com.
Data Availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or Supplementary Materials. Additional data related to this paper may be requested from the authors.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Supplementary Materials
Figure S1: Detection of mouse genotype. The mice numbers are 1-9. Numbers 1, 3, and 7 represent wild-type (WT) mice. Numbers 2, 4-6, and 8-9 represent endothelial cell-specific AAT1 transgenic (EC-AAT1-Tg) mice.
Figure S2: Increased EC-derived SO2 ameliorates hypoxia-induced PAEC inflammation in vivo and in vitro. (a) Immunofluorescence in situ detection of MCP-1 protein expression in mouse PAECs. Red fluorescence represents MCP-1 protein and green fluorescence represents endothelial cell marker CD31. (b) In situ detection of macrophage infiltration around the mouse pulmonary arteries by immunofluorescence. Green fluorescence represents mouse macrophage marker F4/80, and red fluorescence represents vascular smooth muscle marker α-SMA. (c, d) Western blot analysis (n = 9) and immunofluorescence methods were used to detect the expression level of ICAM-1 protein in HPAECs. Yellow fluorescence represents ICAM-1. (e) Western blot analysis was used to detect the expression level of MCP-1 protein in HPAECs (n = 9). (f) Fluorescence method was performed to detect the adhesion of THP-1 cells and HPAECs. Red Dil staining marks THP-1 cells and DAPI color marks the nuclei of HPAECs. The data were expressed as mean ± SEM, ∗p < 0.05, scale: 20 μm.
Figure S3: EC-derived SO2 deficiency stimulated HPASMC hypertrophy in vitro. (a) The expression level of α-SMA protein, a marker of smooth muscle cell hypertrophy, in HPASMCs cocultured with HPAECs was detected by Western blot method (n = 9). (b) The expression level of α-SMA protein in HPASMCs cocultured with HPAECs in situ was observed by immunofluorescence method. The red fluorescence represents α-SMA protein and blue DAPI color marks the nuclei of HPASMCs. The data were expressed as mean ± SEM, ∗p < 0.05, scale bar: 20 μm.
Figure S4: AAT1 overexpression inhibits hypoxia-induced activation of p50 in PAECs. (a) Western blot analysis was used to detect the phosphorylation level of p50 protein in HPAECs (n = 9). (b) The distribution of p50 protein in HPAECs was detected by immunofluorescence in situ. Green fluorescence represents p50 protein. (c) Active motif-ELISA was performed to detect the DNA-binding activity of p50 in HPAECs (n = 9). (d) Immunofluorescence was used to in situ detect p-p50 protein expression in mouse PAECs, green fluorescence represents p-p50 protein. The data were expressed as mean ± SEM, ∗p < 0.05, scale: 20 μm.
References
- 1.De Silva T. M., Modrick M. L., Dabertrand F., Faraci F. M. Changes in cerebral arteries and parenchymal arterioles with aging: role of rho kinase 2 and impact of genetic background. Hypertension. 2018;71(5):921–927. doi: 10.1161/HYPERTENSIONAHA.118.10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.North B. J., Sinclair D. A. The intersection between aging and cardiovascular disease. Circulation Research. 2012;110(8):1097–1108. doi: 10.1161/CIRCRESAHA.111.246876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laurent S. Defining vascular aging and cardiovascular risk. Journal of Hypertension. 2012;30(Suppl):S3–S8. doi: 10.1097/HJH.0b013e328353e501. [DOI] [PubMed] [Google Scholar]
- 4.Fang L., Gao P., Bao H., et al. Chronic obstructive pulmonary disease in China: a nationwide prevalence study. The Lancet Respiratory Medicine. 2018;6(6):421–430. doi: 10.1016/S2213-2600(18)30103-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaspar L. S., Álvaro A. R., Moita J., Cavadas C. Obstructive sleep apnea and hallmarks of aging. Trends in Molecular Medicine. 2017;23(8):675–692. doi: 10.1016/j.molmed.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 6.Sajkov D., McEvoy R. D. Obstructive sleep apnea and pulmonary hypertension. Progress in Cardiovascular Diseases. 2009;51(5):363–370. doi: 10.1016/j.pcad.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Sakao S., Voelkel N. F., Tatsumi K. The vascular bed in COPD: pulmonary hypertension and pulmonary vascular alterations. European Respiratory Review. 2014;23(133):350–355. doi: 10.1183/09059180.00007913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thenappan T., Ormiston M. L., Ryan J. J., Archer S. L. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018;360, article j5492 doi: 10.1136/bmj.j5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Price L. C., Wort S. J., Perros F., et al. Inflammation in pulmonary arterial hypertension. Chest. 2012;141(1):210–221. doi: 10.1378/chest.11-0793. [DOI] [PubMed] [Google Scholar]
- 10.Ban Y., Liu Y., Li Y., et al. S-nitrosation impairs KLF4 activity and instigates endothelial dysfunction in pulmonary arterial hypertension. Redox Biology. 2019;21, article 101099 doi: 10.1016/j.redox.2019.101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falcetti E., Hall S. M., Phillips P. G., et al. Smooth muscle proliferation and role of the prostacyclin (IP) receptor in idiopathic pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine. 2010;182(9):1161–1170. doi: 10.1164/rccm.201001-0011OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng L., Blanco F. J., Stevens H., et al. MicroRNA-143 activation regulates smooth muscle and endothelial cell crosstalk in pulmonary arterial hypertension. Circulation Research. 2015;117(10):870–883. doi: 10.1161/CIRCRESAHA.115.306806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang D., Wang X. L., Chen S. Y., et al. Endogenous hydrogen sulfide sulfhydrates IKKβ at cysteine 179 to control pulmonary artery endothelial cell inflammation. Clinical Science (London, England) 2019;133(20):2045–2059. doi: 10.1042/CS20190514. [DOI] [PubMed] [Google Scholar]
- 14.Feng S. S., Chen S. Y., Yu W., et al. H2S inhibits pulmonary arterial endothelial cell inflammation in rats with monocrotaline-induced pulmonary hypertension. Laboratory Investigation. 2017;97(3):268–278. doi: 10.1038/labinvest.2016.129. [DOI] [PubMed] [Google Scholar]
- 15.Berenyiova A., Dovinova I., Kvandova M., et al. The effect of chronic NO synthase inhibition on the vasoactive and structural properties of thoracic aorta, NO synthase activity, and oxidative stress biomarkers in young SHR. Oxidative Medicine and Cellular Longevity. 2018;2018:10. doi: 10.1155/2018/2502843.2502843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blaha I., López-Oliva M. E., Martínez M. P., et al. Bladder dysfunction in an obese Zucker rat: the role of TRPA1 channels, oxidative stress, and hydrogen sulfide. Oxidative Medicine and Cellular Longevity. 2019;2019:12. doi: 10.1155/2019/5641645.5641645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong B. W., Marsch E., Treps L., Baes M., Carmeliet P. Endothelial cell metabolism in health and disease: impact of hypoxia. The EMBO Journal. 2017;36(15):2187–2203. doi: 10.15252/embj.201696150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun H. J., Wu Z. Y., Nie X. W., Bian J. S. Role of endothelial dysfunction in cardiovascular diseases: the link between inflammation and hydrogen sulfide. Frontiers in Pharmacology. 2020;10:p. 1568. doi: 10.3389/fphar.2019.01568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue C., Sowden M., Berk B. C. Extracellular cyclophilin A, especially acetylated, causes pulmonary hypertension by stimulating endothelial apoptosis, redox stress, and inflammation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2017;37(6):1138–1146. doi: 10.1161/ATVBAHA.117.309212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piera-Velazquez S., Mendoza F. A., Jimenez S. A. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of human fibrotic diseases. Journal of Clinical Medicine. 2016;5(4):p. 45. doi: 10.3390/jcm5040045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.du S. X., JIN H. F., BU D. F., et al. Endogenously generated sulfur dioxide and its vasorelaxant effect in rats. Acta Pharmacologica Sinica. 2008;29(8):923–930. doi: 10.1111/j.1745-7254.2008.00845.x. [DOI] [PubMed] [Google Scholar]
- 22.Liu X., Zhang D., Li K., et al. Effect of endogenous sulfur dioxide on the apoptosis induced by cobalt chloride in the human pulmonary arterial endothelial cells. Journal of Applied Clinical Pediatrics. 2018;13:999–1003. [Google Scholar]
- 23.Li W., Tang C. S., Jin H. F., Du J. B. Regulatory effects of sulfur dioxide on the development of atherosclerotic lesions and vascular hydrogen sulfide in atherosclerotic rats. Atherosclerosis. 2011;215(2):323–330. doi: 10.1016/j.atherosclerosis.2010.12.037. [DOI] [PubMed] [Google Scholar]
- 24.Yang L., Zhang H., Chen P. Sulfur dioxide attenuates sepsis-induced cardiac dysfunction via inhibition of NLRP3 inflammasome activation in rats. Nitric Oxide. 2018;81:11–20. doi: 10.1016/j.niox.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 25.Yu W., Liu D., Liang C., et al. Sulfur dioxide protects against collagen accumulation in pulmonary artery in association with downregulation of the transforming growth factor β1/smad pathway in pulmonary hypertensive rats. Journal of the American Heart Association. 2016;5(10, article e003910) doi: 10.1161/JAHA.116.003910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Q. H., Zhang L. L., Chen S. Y., et al. Downregulated endogenous sulfur dioxide/aspartate aminotransferase pathway is involved in angiotensin II-stimulated cardiomyocyte autophagy and myocardial hypertrophy in mice. International Journal of Cardiology. 2016;225:392–401. doi: 10.1016/j.ijcard.2016.09.111. [DOI] [PubMed] [Google Scholar]
- 27.Afonina I. S., Zhong Z., Karin M., Beyaert R. Limiting inflammation-the negative regulation of NF-κB and the NLRP3 inflammasome. Nature Immunology. 2017;18(8):861–869. doi: 10.1038/ni.3772. [DOI] [PubMed] [Google Scholar]
- 28.Wu W., Zhang W., Choi M., et al. Vascular smooth muscle-MAPK14 is required for neointimal hyperplasia by suppressing VSMC differentiation and inducing proliferation and inflammation. Redox Biology. 2019;22, article 101137 doi: 10.1016/j.redox.2019.101137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perkins N. D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nature Reviews. Molecular Cell Biology. 2007;8(1):49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 30.Xia Y. F., Ye B. Q., Li Y. D., et al. Andrographolide attenuates inflammation by inhibition of NF-κB activation through covalent modification of reduced cysteine 62 of p50. Journal of Immunology. 2004;173(6):4207–4217. doi: 10.4049/jimmunol.173.6.4207. [DOI] [PubMed] [Google Scholar]
- 31.Lai J., Ge M., Shen S., et al. Activation of NFKB-JMJD3 signaling promotes bladder fibrosis via boosting bladder smooth muscle cell proliferation and collagen accumulation. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2019;1865(9):2403–2410. doi: 10.1016/j.bbadis.2019.05.008. [DOI] [PubMed] [Google Scholar]
- 32.Zhang L., Wang Y., Wu G., et al. Blockade of JAK2 protects mice against hypoxia-induced pulmonary arterial hypertension by repressing pulmonary arterial smooth muscle cell proliferation. Cell Proliferation. 2020;53(2, article e12742) doi: 10.1111/cpr.12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang L. L., Jin H. F., Song Y. J., et al. Endogenous sulfur dioxide is a novel inhibitor of hypoxia-induced mast cell degranulation. Journal of Advanced Research. 2020;29:55–65. doi: 10.1016/j.jare.2020.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thibault H. B., Kurtz B., Raher M. J., et al. Noninvasive assessment of murine pulmonary arterial pressure: validation and application to models of pulmonary hypertension. Circulation. Cardiovascular Imaging. 2010;3(2):157–163. doi: 10.1161/CIRCIMAGING.109.887109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Y., Tian Y., Prabha M., et al. Effects of sulfur dioxide on hypoxic pulmonary vascular structural remodeling. Laboratory Investigation. 2010;90(1):68–82. doi: 10.1038/labinvest.2009.102. [DOI] [PubMed] [Google Scholar]
- 36.Zabini D., Granton E., Hu Y., et al. Loss of SMAD3 promotes vascular remodeling in pulmonary arterial hypertension via MRTF disinhibition. American Journal of Respiratory and Critical Care Medicine. 2018;197(2):244–260. doi: 10.1164/rccm.201702-0386OC. [DOI] [PubMed] [Google Scholar]
- 37.Valdés Á., Treuer A. V., Barrios G., et al. NOX inhibition improves β-adrenergic stimulated contractility and intracellular calcium handling in the aged rat heart. International Journal of Molecular Sciences. 2018;19(8, article 2404) doi: 10.3390/ijms19082404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang H., Huang Y., Bu D., et al. Endogenous sulfur dioxide is a novel adipocyte-derived inflammatory inhibitor. Scientific Reports. 2016;6(1, article 27026) doi: 10.1038/srep27026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu Z., Zhang L., Chen Q., et al. Macrophage-derived sulfur dioxide is a novel inflammation regulator. Biochemical and Biophysical Research Communications. 2020;524(4):916–922. doi: 10.1016/j.bbrc.2020.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Taetzsch T., Levesque S., McGraw C., et al. Redox regulation of NF-κB p50 and M1 polarization in microglia. Glia. 2015;63(3):423–440. doi: 10.1002/glia.22762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matthews J. R., Botting C. H., Panico M., Morris H. R., Hay R. T. Inhibition of NF-kappaB DNA binding by nitric oxide. Nucleic Acids Research. 1996;24(12):2236–2242. doi: 10.1093/nar/24.12.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song Y., Peng H., Bu D., et al. Negative auto-regulation of sulfur dioxide generation in vascular endothelial cells: AAT1 S-sulfenylation. Biochemical and Biophysical Research Communications. 2020;525(1):231–237. doi: 10.1016/j.bbrc.2020.02.040. [DOI] [PubMed] [Google Scholar]
- 43.Chen S., Huang Y., Liu Z., et al. Sulphur dioxide suppresses inflammatory response by sulphenylating NF-κB p65 at Cys 38 in a rat model of acute lung injury. Clinical Science (London, England) 2017;131(21):2655–2670. doi: 10.1042/CS20170274. [DOI] [PubMed] [Google Scholar]
- 44.Huang Y., Shen Z., Chen Q., et al. Endogenous sulfur dioxide alleviates collagen remodeling via inhibiting TGF-β/Smad pathway in vascular smooth muscle cells. Scientific Reports. 2016;6(1):p. 19503. doi: 10.1038/srep19503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y., Wang X., Chen S., et al. Sulfur dioxide activates Cl−/HCO3− exchanger via sulphenylating AE2 to reduce intracellular pH in vascular smooth muscle cells. Frontiers in Pharmacology. 2019;10:p. 313. doi: 10.3389/fphar.2019.00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu H. J., Huang Y. Q., Chen Q. H., et al. Sulfur dioxide inhibits extracellular signal-regulated kinase signaling to attenuate vascular smooth muscle cell proliferation in angiotensin II-induced hypertensive mice. Chinese Medical Journal. 2016;129(18):2226–2232. doi: 10.4103/0366-6999.189927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu D., Huang Y., Bu D., et al. Sulfur dioxide inhibits vascular smooth muscle cell proliferation via suppressing the Erk/MAP kinase pathway mediated by cAMP/PKA signaling. Cell Death & Disease. 2014;5(5, article e1251) doi: 10.1038/cddis.2014.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo L., Hong X., Diao B., Chen S., Hei M. Sulfur dioxide attenuates hypoxia-induced pulmonary arteriolar remodeling via Dkk1/Wnt signaling pathway. Biomedicine & Pharmacotherapy. 2018;106:692–698. doi: 10.1016/j.biopha.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 49.Romero M., Ortega A., Izquierdo A., López-Luna P., Bosch R. J. Parathyroid hormone-related protein induces hypertrophy in podocytes via TGF-β1 and p27Kip1: implications for diabetic nephropathy. Nephrology, Dialysis, Transplantation. 2010;25(8):2447–2457. doi: 10.1093/ndt/gfq104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Detection of mouse genotype. The mice numbers are 1-9. Numbers 1, 3, and 7 represent wild-type (WT) mice. Numbers 2, 4-6, and 8-9 represent endothelial cell-specific AAT1 transgenic (EC-AAT1-Tg) mice.
Figure S2: Increased EC-derived SO2 ameliorates hypoxia-induced PAEC inflammation in vivo and in vitro. (a) Immunofluorescence in situ detection of MCP-1 protein expression in mouse PAECs. Red fluorescence represents MCP-1 protein and green fluorescence represents endothelial cell marker CD31. (b) In situ detection of macrophage infiltration around the mouse pulmonary arteries by immunofluorescence. Green fluorescence represents mouse macrophage marker F4/80, and red fluorescence represents vascular smooth muscle marker α-SMA. (c, d) Western blot analysis (n = 9) and immunofluorescence methods were used to detect the expression level of ICAM-1 protein in HPAECs. Yellow fluorescence represents ICAM-1. (e) Western blot analysis was used to detect the expression level of MCP-1 protein in HPAECs (n = 9). (f) Fluorescence method was performed to detect the adhesion of THP-1 cells and HPAECs. Red Dil staining marks THP-1 cells and DAPI color marks the nuclei of HPAECs. The data were expressed as mean ± SEM, ∗p < 0.05, scale: 20 μm.
Figure S3: EC-derived SO2 deficiency stimulated HPASMC hypertrophy in vitro. (a) The expression level of α-SMA protein, a marker of smooth muscle cell hypertrophy, in HPASMCs cocultured with HPAECs was detected by Western blot method (n = 9). (b) The expression level of α-SMA protein in HPASMCs cocultured with HPAECs in situ was observed by immunofluorescence method. The red fluorescence represents α-SMA protein and blue DAPI color marks the nuclei of HPASMCs. The data were expressed as mean ± SEM, ∗p < 0.05, scale bar: 20 μm.
Figure S4: AAT1 overexpression inhibits hypoxia-induced activation of p50 in PAECs. (a) Western blot analysis was used to detect the phosphorylation level of p50 protein in HPAECs (n = 9). (b) The distribution of p50 protein in HPAECs was detected by immunofluorescence in situ. Green fluorescence represents p50 protein. (c) Active motif-ELISA was performed to detect the DNA-binding activity of p50 in HPAECs (n = 9). (d) Immunofluorescence was used to in situ detect p-p50 protein expression in mouse PAECs, green fluorescence represents p-p50 protein. The data were expressed as mean ± SEM, ∗p < 0.05, scale: 20 μm.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or Supplementary Materials. Additional data related to this paper may be requested from the authors.